

Abstract
The purpose of the present investigation was to develop and evaluate a self-microemulsifying drug delivery system (SMEDDS) for improving the oral absorption of a pranlukast hemihydrate (PLH), a very poorly water-soluble drug. An efficient self-microemulsifying vehicle for PLH was selected and optimized using solubility testing and phase diagram construction. The formulations were characterized by assessing self-emulsification performance, droplet size analysis, in vitro drug release characteristics and formulation stability studies. Optimized formulations for in vitro dissolution and bioavailability assessment were Triethylcitrate (TEC; 10%), Tween 20 (50%), Span 20 (25%), triethanolamine (5%), and benzyl alcohol (10%). The SMEDDS readily released the lipid phase to form a fine oil-in-water microemulsion with a narrow distribution size. Saturated solubilities of PLH from SMEDDS in water, pH 4.0 and 6.8, were over 150 times greater than that of plain PLH. The release of 100% PLH from SMEDDS was considerably greater compared to only 1.12% in simulated intestinal fluid (pH 6.8) from plain PLH after 2 hours. The PLH suspension with 0.5% sodium carboxymethylcellulose or 3% PLH-loaded SMEDDS was administrated at a dose of 40 mg/kg as PLH to fasted rats. The absorption of PLH from SMEDDS resulted in about a threefold increase in bioavailability compared with plain PLH aqueous suspension. Our studies illustrated that the potential use of the new SMEDDS can be used as a possible alternative to oral delivery of a poorly water-soluble drug such as PLH.
Keywords: pranlukast hemihydrates, PLH, SMEDDS, bioavailability, solubility
Introduction
Pranlukast hemihydrate (PLH), 4-oxo-8-[4-(4-phenylbutoxy)bentoxy)benzoylamino]-2-(tetrazol-5-yl)-4H-1-benzopyran 1/2H2O, is a novel, potent, and selective cysteinyl leukotriene receptor antagonist that has exhibited not only antileukotriene activity, but also pharmacological activity including antineosinophilic effects.1,2 It antagonizes or reduces bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens. A PLH product (Onon®; Dong-A Pharm. Inc., Seoul, Korea) is orally administered at a 225 mg twice-daily dose, and it has been used to treat chronic bronchial asthma and allergic rhinitis in pediatric and adult patients. PLH exhibits extremely low aqueous solubility (0.8 μg/mL H2O at 37°C), and the absorption ratio is estimated to be approximately 20% based on excretion ratios in the feces following oral administration.3 Thus, such low bioavailability requires a large amount of the drug for disease treatment. The fact that PLH cannot be dissolved in commonly used organic solvents such as methanol, ethanol, and acetone also poses a limitation to the preparation of a PLH formulation through the crystallization process. There are many techniques to improve the dissolution of poorly water-soluble drugs. One of the most popular approaches is the incorporation of an active lipophilic component into inert lipid vehicles such as oils, surfactant dispersions, self-emulsifying formulations, microemulsions, and liposomes.4–17
A self-microemulsifying drug delivery system (SMEDDS) has recently become a more efficient approach to improve the solubility, dissolution, and oral bioavailability for poorly water-soluble drugs. SMEDDS is an isotropic mixture of an oil, surfactant, and cosurfactant with the drug.18,19 The important principle of this system is its ability to form an oil-in-water microemulsion under mild conditions (for example, gentle agitation following dilution by aqueous phases). The mild agitation mimics the digestive motility of the stomach and intestine, providing the agitation required for self-emulsification in vivo.20 The spontaneous creation of microemulsion in the gastrointestinal tract implies that the drug is in a dissolved form, and the small droplet size provides a large interfacial surface area for drug absorption.21 The selection of a suitable self-emulsifying formulation depends upon the assessment of the solubility of the drug in various components, the area of the self-emulsifying region as obtained in the phase diagram, and the droplet size distribution of the following self-emulsification.22
The aims of this study were to develop and characterize the optimal formulation of SMEDDS containing PLH, and to evaluate the oral bioavailability enhancement through SMEDDS in rats after oral administration. An efficient self-microemulsifying vehicle for PLH was selected and optimized using solubility tests and phase diagram construction. The formulations were characterized by assessing self-emulsification performance, droplet size analysis, in vitro drug release characteristics, and formulation stability studies. Finally, the oral absorption of PLH was evaluated in a rat application, and the effect of SMEDDS was discussed.
Materials and methods
Materials
PLH and montelukast was purchased from EstechPharma (EstechPharma Co, Ltd, Seoul, Korea). Span 20, Tween 20, Tween 80, polyethylene glycol 400 (PEG 400), benzyl alcohol (BA), and triethanolamine (TEA) were purchased from Daejung Chemical and Metal Co, Ltd (Seoul, Korea). Cremophor EL® was purchased from BASF Corporation (Ludwigshafen, Germany). TEC, Transcutol P®, Labrasol ®, Lauroglycol FCC®, Labrafil M1944 CS®, Glyceryl triacetate, propylene glycol, and isopropyl myristate were obtained from Gattefossé (Saint-Priest, France). All other chemicals used to prepare the physiological solution such as simulated gastric juice fluid (SGF), simulated intestinal fluid (SIF), and phosphate-buffered saline (PBS) were of analytical grade.
Solubility studies
The solubility of PLH was determined in various oils, surfactants, and cosurfactants. An excess amount of PLH was added into glass vials containing 3 mL of a vehicle (oil, surfactant), and the mixtures were stirred using magnetic stirrer for 24 hours at 30°C. After stirring, the mixtures were centrifuged at 13,000 rpm for 5 minutes (Hettich Zentrifugen 16R; Andreas Hettich GmbH and Co, Tuttlingen, Germany), and supernatants were filtrated using a membrane filter (0.45 μm, 13 mm, Whatman, Kent, UK). The PLH concentration in the filtrate was determined using a high-performance liquid chromatographic system ([HPLC] Shimadzu Corporation, Kyoto, Japan) consisting of a LC10-AD model isocratic pump (Shimadzu Corporation), a SPD-10 A VP model variable spectrophotometric detector (Shimadzu Corporation), and a Nucleosil 100–5 column (C18, 5 μm, 4.6 × 150 mm; Macherey–Nagel GmbH and Co, Düren, Germany) according to a method developed by Chono et al.3 A mixture of acetonitrile, methanol, and 20 mM of anhydrous potassium phosphate (50:40.9:9.1, v/v%) was used as the mobile phase at a flow rate of 0.8 mL/minute, and the detection wavelength was set to 260 nm. The accuracy and precision for intra- and interday assay were within 10% of the relative error and coefficient of variation.
Preparations and screening of PLH-loaded SMEDDS
According to the solubility and pseudoternary phase diagram, optimal formulations could be selected. In order to find out the composition of SMEDDS with a high solubilizing capacity, various oils, surfactants, and cosurfactants were added to a screw capped glass vial and the components were mixed by gentle stirring. The resulting PLH solubility of the mixture was investigated. Based on the solubility test in SMEDDS and superimposed ternary phase diagrams, the contents of the surfactant, cosurfactant, oil, and drug were chosen at the ranges of 50%–70%, 20%–40%, 10%, and 3%, respectively. In addition, three SMEDDS formulations with a high solubilizing capacity (>20 mg/g) were prepared. Briefly, PLH, the surfactant/cosurfactant (S/CoS) mixture, and oil were accurately weighed and placed in glass vials, and the mixture was gently stirred for homogeneous mixing and heated to 90°C ± 5°C and maintained for 20 minutes. After cooling down to room temperature, SMEDDS was obtained, which was a clear and transparent solution. PLH in SMEDDS had the same peak shape and retention time in the HPLC spectrum compared to the standard substance after the heating process, and additionally the degradation peak of PLH was not found in the chromatogram. Therefore, the PLH-loaded SMEDDS was shown to be stable under the heating condition of 90°C for 20 minutes.
Construction of pseudoternary phase diagram
Pseudoternary phase diagrams were constructed in order to obtain the concentration range of components for the existing region of microemulsions using the water titration method. For each pseudoternary phase diagram at a specific S/CoS weight ratio, the oily mixtures of oil, surfactant, and cosurfactant were prepared with the weight ratio of oil to the mixture of surfactant and cosurfactant at 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, and 90:10. Water was added in a dropwise manner to each oily mixture under proper magnetic stirring at 37°C until the mixture became clear at a certain point. The concentrations of the components were recorded in order to complete the pseudoternary phase diagrams, and then the contents of oil, surfactant, cosurfactant, and water at appropriate weight ratios were selected based on these results. To assess the region for aqueous dispersion, the optical clarity of aqueous dispersion of PLH-loaded SMEDDS was spectrometrically evaluated.
Characterization of PLH-loaded SMEDDS formulation
Droplet size analysis and saturated solubility
The droplet size/distribution of the prepared solution was determined by dynamic light scattering analyzer (NPA 250; Microtact Inc, York, PA, USA). In order to determine the effects of different media on the particle size of microemulsification, 1 g of PLH-loaded SMEDDS (equivalent to PLH 30 mg) was added into the distilled water, SGF (pH 1.2), SIF (pH 6.8), and PBS (pH 4.0), and the resulting solution was slightly shaken before the size analysis. Aliquots of these microemulsions were loaded into cuvettes, and their size was measured after dilution to produce the required count rate to enable accurate measurement. Light scattering was monitored at a 90° angle, and the solution was maintained at 25°C. Each sample was analyzed in triplicate, and data are presented as mean ± standard deviation.
In order to determine the effects of different media on the solubility of PLH, 0.2 g of PLH-loaded SMEDDS was added into 1 mL of distilled water, SGF (pH 1.2), SIF (pH 6.8), and PBS (pH 4.0) and the mixtures were stirred using a magnetic stirrer for 24 hours at 30°C.
In vitro dissolution test
An in vitro release test from the capsules filled with the SMEDDS containing 30 mg of PLH was performed using the USP 30 rotating paddle apparatus. Two capsules of each of the PLH-loaded SMEDDS formulations (A, B, and C) were placed separately in 900 mL of the different media (distilled water, SGF [pH 1.2], PBS [pH 4.0], and SIF [pH 6.8]), as well as in the buffer solutions containing 1% Tween 80 to examine the effect of pH on drug release; the paddles were rotated at 50 rpm. The release profiles from PLH-loaded SMEDDS (30 mg) were compared with those of the PLH powder (30 mg) contained in a hard gelatin capsule. During the release studies, 4 mL of the media were taken at 5, 10, 15, 30, 45, 60, 90, and 120 minutes, and were subjected to drug analysis using HPLC. The removed volume was replaced with 4 mL of fresh media at every sampling time.
Stability test
The optimized PLH-loaded SMEDDS (formulation B) was put in hard gelatin capsules and was subject to stability studies under intermediate conditions (25°C/60% relative humidity [RH]) and accelerated conditions (40°C/75% RH) for 3 months. Samples were stored in a stability chamber (KCL-2000, EYELA; Tokyo Rikakikai Co, Ltd, Tokyo, Japan) with humidity and temperature control. Samples were withdrawn at specified intervals (0, 1, 2, and 3 months) for analysis. Appearance, self-emulsifying properties, emulsion droplet size, drug content, and dissolution rate in SIF (pH 6.8) were each evaluated.
Bioavailability studies
Six male rats (weighing ~200–250 g) were used in this study. Animal experimentation was carried out under local Institutional Animal Care and Use Committee approval in accordance with the institution’s ethical guidelines. Two groups of rats were orally administered either PLH suspension with 0.5% sodium carboxymethylcellulose or 3% PLH-loaded SMEDDS (formulation B) at a dose of 40 mg/kg as PLH. Blood samples (0.5 mL) were collected into heparinized microtubes via the tail vein at predose, as well as at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours postdose (n = 3); the samples were centrifuged at 13,200 rpm for 3 min. The plasma samples (0.2 mL) were stored at −80°C prior to liquid chromatography-mass spectrometry (LC-MS/MS) analysis.
Bioanalysis condition of PLH in rat plasma with LC-MS/MS
The plasma samples were thawed and vortexed adequately. To a 50 μL aliquot of samples, 10 μL of internal standard solution and 400 μL of extraction solvent (the ratio of ethyl acetate to methanol = 1:9) were added and the resulting solution was vortex-mixed for 10 minutes to extract the PLH using liquid-liquid extraction. The centrifuged supernatant was evaporated at 55°C for 50 minutes under a vacuum in a SpeedVac system (Thermo Savant [Thermo Fisher Scientific, Waltham, MA]), and it was reconstituted with 50 μL of mobile phase to inject 10 μL onto the LC-MS/MS.
The LC-MS/MS system consisted of an Agilent 1100 HPLC system (binary pump, online degasser, autosampler; Agilent Technologies, Santa Clara, CA) with an API 3200 Qtrap mass spectrometer (Applied Biosystems [Life Technologies, Carlsbad, CA]), equipped with a turbo ion spray interface in negative ionization mode. The spray needle voltage was −4500 V, and the source temperature was 450°C. The declustering potential energy was −55 V. The collision energy was −24 V for PLH and −28 V for montelukast as the internal standard. The predominant precursor ions produced for the analytes employed the [M−H]−. Transitions at 480.1→424.2 for PLH and 584.2→472.2 for montelukast were simultaneously monitored. Chromatographic separation was achieved on a Fusion-RP column (2.1 mm × 50 mm, 5 μm; Phenomenex, Inc, Torrance, CA). The mobile phase was composed of acetonitrile-purified water (85:15, v/v) and eluted at a flow rate of 0.26 mL/minute. Injection volume was set at 10 μL and the autosampler tray were maintained at 10°C. Data acquisition and processing were performed with Analyst software (version 1.4.2; Applied Biosystems).
Pharmacokinetic parameters
The plasma concentration-time curve was constructed, and pharmacokinetic parameters were calculated with a noncompartmental method using WinNonlin software (version 4.1; Pharsight Corporation, St Louis, MO). The area under the peak corresponding to the pranlukast concentration in the plasma-time curve from time zero to the last quantifiable time point (AUClast) was calculated by a linear trapezoidal method. The maximum concentration (Cmax) and the time to reach Cmax (Tmax) were obtained directly from the profile of PLH concentrations in the plasma.
Results and discussion
Screening of SMEDDS formulation composition
The prevention of drug precipitation in the gastrointestinal tract is an important factor for the SMEDDS formulation development, and the precipitation in lumen is mainly caused by poor solubility of a compound.23 Therefore, the components used in SMEDDS should have the solubilizing capacity for a drug, ensuring the solubility of the drug in the resultant dispersion.
The solubility of PLH in various vehicles is presented in Table 1. TEC and BA showed high solubility for PLH, and its solubility was 0.50 ± 0.01 and 5.84 ± 0.08 mg/g, respectively. The high solubility in BA is probably due to the ability of PLH to form a hydrogen bond with the hydroxyl group. Similarly, surfactants (Tween 20, Cremophor EL®) composed of PEO groups showed high solubilization capacities for PLH. Thus, TEC and BA were selected as an oil phase in SMEDDS of PLH for hydrophilic natures, enhanced drug solubility, and the ability to form microemulsion. As a surfactant, Tween 20 (7.76 ± 0.35 mg/g) and Cremophor EL® (3.58 ± 0.04 mg/g) were selected, because these vehicles had the best solubility for PLH. In order to find out the composition of SMEDDS with a high solubilizing capacity, solubility in various mixtures containing oil and surfactant was evaluated and the results are shown in Table 2.
Table 1.
Solubility of pranlukast hemihydrate in various oils and surfactants at 30°C
| Vehicle | Solubility (mg/g) |
|---|---|
| Oils | |
| Glyceryl triacetate | 0.10 ± 0.00 |
| Isopropyl myristate | 0.11 ± 0.00 |
| Triethyl citrate | 0.50 ± 0.01 |
| Propylene glycol | 0.50 ± 0.02 |
| Labrafil M 1944 CS® | 0.93 ± 0.02 |
| PEG 400 | 2.15 ± 0.01 |
| Lauroglycol FCC® | 3.24 ± 0.06 |
| Triethanolamine | 3.97 ± 0.20 |
| Benzyl alcohol | 5.84 ± 0.08 |
| Surfactants/cosurfactants | |
| Span 20 | 0.66 ± 0.01 |
| Labrasol® | 2.22 ± 1.64 |
| Tween 80 | 2.66 ± 0.38 |
| Transcutol P® | 2.96 ± 0.02 |
| Cremophor EL® | 3.58 ± 0.04 |
| Tween 20 | 7.76 ± 0.35 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviation: PEG, polyethylene glycol.
Table 2.
Solubility of pranlukast hemihydrate in various SMEDDS formulation composition
| No | TEC | Tween 20 | Cremophor EL® | Span 20 | Lauroglycol FCC® | TEA | BA | Solubility (mg/g) |
|---|---|---|---|---|---|---|---|---|
| 1 | 10 | 90 | – | – | 2.65 ± 0.10 | |||
| 2 | 20 | 80 | – | – | 2.48 ± 0.02 | |||
| 3 | 30 | 70 | – | – | 2.10 ± 0.05 | |||
| 4 | 10 | 80 | – | – | 10 | 8.01 ± 0.06 | ||
| 5 | 10 | 70 | – | – | 20 | 6.40 ± 0.06 | ||
| 6 | 10 | 60 | – | – | 30 | 8.42 ± 0.02 | ||
| 7 | 10 | 80 | – | – | – | 10 | 8.48 ± 0.09 | |
| 8 | 10 | 80 | 10 | – | 5.99 ± 0.02 | |||
| 9 | 10 | 80 | – | 10 | 14.68 ± 0.10 | |||
| 10 | 10 | 70 | 10 | – | – | 10 | 9.71 ± 0.12 | |
| 11 | 10 | 45 | – | 45 | 3.46 ± 0.07 | |||
| 12 | 10 | 30 | – | 60 | 8.44 ± 0.14 | |||
| 13 | 10 | 60 | – | 30 | 10.24 ± 0.16 | |||
| 14 | 10 | 55 | 10 | – | 5 | 10 | 10 | 14.68 ± 0.12 |
| 15 | 10 | 60 | 15 | – | 5 | 5 | 10 | 14.34 ± 0.09 |
| 16 | 10 | 50 | 25 | – | 5 | 10 | 22.87 ± 1.06 | |
| 17 | 10 | 50 | – | 25 | – | 5 | 10 | 20.65 ± 0.48 |
| 18 | – | 65 | – | 20 | – | 5 | 10 | 20.62 ± 0.28 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviations: SMEDDS, self-microemulsifying drug delivery system; TEC, triethylcitrate; TEA, triethanolamine; BA, benzyl alcohol.
The maximum solubility in the tested formulations was 22.87 mg/g. A TEA and BA (1:2) mixture showed good solubilizing effects and formulation number ~16–18 of solubilized PLH at over 20 mg/g. The optimal formula was selected based on the solubility of PLH in SMEDDS and those formulations are shown in Table 3.
Table 3.
Composition of SMEDDS formulations containing 3% pranlukast hemihydrate
| Formulation | Composition (W/W%) | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| TEC | Tween 20 | Cremophor EL® | Span 20 | TEA | BA | |
| A | 10 | 50 | 25 | – | 5 | 10 |
| B | 10 | 50 | – | 25 | 5 | 10 |
| C | – | 65 | – | 20 | 5 | 10 |
Note: 1 g of each formulation mixture contained 30 mg of pranlukast hemihydrate.
Abbreviations: SMEDDS, self-microemulsifying drug delivery system; TEC, triethylcitrate; TEA, triethanolamine; BA, benzyl alcohol; W/W, weight/weight.
Construction of phase diagram
Pseudoternary phase diagrams were constructed to identify the microemulsion regions and to optimize the concentration of oil, S/CoS, and cosolvent selected from the results of the solubility experiments mentioned above. The S/CoS mixture preferentially adsorbed at the interface between oil and water with reducing interfacial energy, and it also provided a mechanical barrier to coalescence. The decrease in the free energy required for the emulsion formation consequently improved the thermodynamic stability of the microemulsion formulation.24
The TEA/BA (1:2) mixture was effective for the optimization of the microemulsion region, as shown in Figure 1. PLH-loaded SMEDDS was diluted with water to investigate the spontaneous emulsification without phase separation or coalescence. The optical clarity of aqueous dispersion was quantitatively assessed with an ultraviolet-visible spectrophotometer, which measured the transmittance of light in the given wavelength (450 nm).
Figure 1.
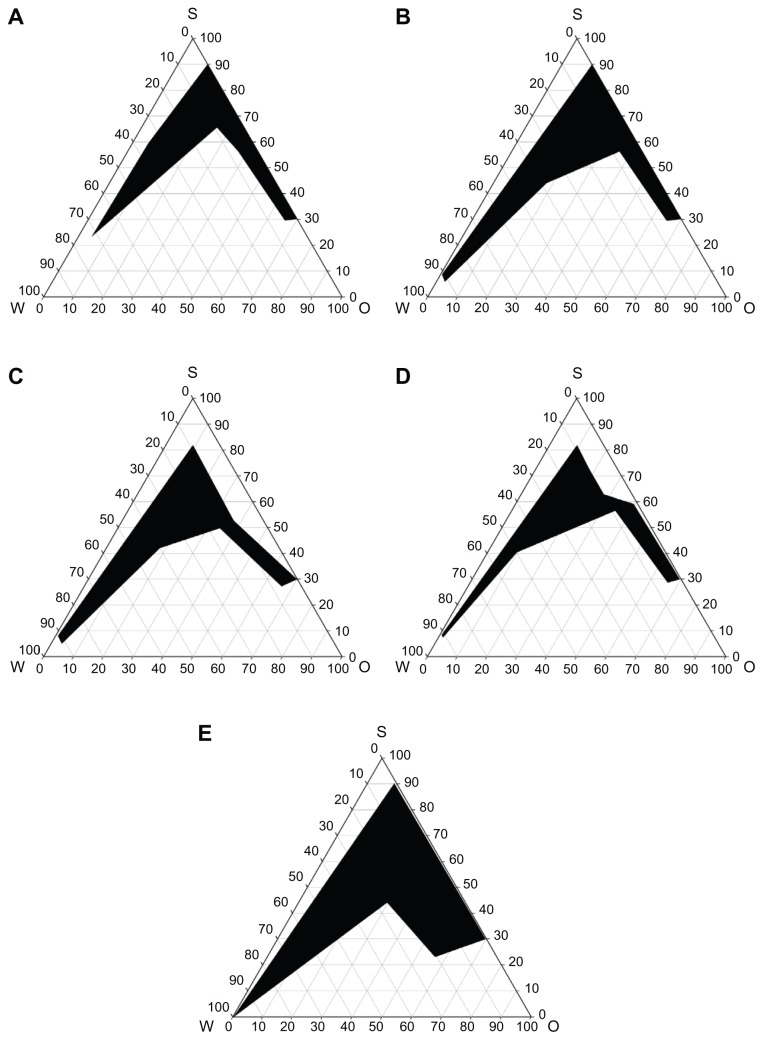
Pseudoternary phase diagrams. Pseudoternary phase diagrams composed of water-TEC-Tween 20/Span 20 (2:1) (A); water-TEC-Tween 20/Cremophor® EL (2:1) (B); formulation A (C); formulation B (D); and formulation C (E). The dark regions represent the microemulsion phase.
Abbreviation: O, oil; S, surfactant; TEC, triethylcitrate; W, water.
An aqueous dispersion with small absorbance is optically clear, and oil droplets are thought to be finely dispersed. The absorbance from the tested aqueous dispersion of PLH-loaded SMEDDS was between 0.0000 and 0.0047.
Characterization of PLH-loaded SMEDDS formulation
The mean particle size of PLH-loaded SMEDDS in different media is presented in Table 4. The change of the mean particle size was not observed after being dispersed in distilled water, PBS (pH 4.0), and SIF (pH 6.8). However, the mean particle size of the three formulations considerably increased in SGF (pH 1.2). It could be thought that a low pH condition might cause the precipitation of PLH because the solubility of PLH in acidic conditions is very low, and that an increased mean particle size might be influenced by precipitated PLH. The particle size of formulation B changed less than that of formulations A and C in SGF (pH 1.2).
Table 4.
Particle size of pranlukast hemihydrate-loaded SMEDDS in aqueous media
| Formulation | Particle size (nm) | |||
|---|---|---|---|---|
|
|
||||
| Water | pH 1.2 | pH 4.0 | pH 6.8 | |
| A | 2.42 ± 0.14 | 888.33 ± 42.90 | 7.71 ± 0.09 | 7.27 ± 0.23 |
| B | 3.62 ± 0.33 | 193.77 ± 8.58 | 7.71 ± 0.01 | 4.56 ± 0.06 |
| C | 1.57 ± 0.22 | 760.33 ± 73.36 | 9.71 ± 0.31 | 6.55 ± 0.25 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
The saturated solubility of PLH in PLH-loaded SMEDDS was increased with respect to the increase of pH values (Table 5). These findings suggest that PLH is an acidic drug, and solubility of the ionic form increases with the release of H+ from the tetrazoleheterocycle on increasing the pH. According to a previous report about the release of H+ from tetrazoleheterocycle, the acid dissociation constant (pKa) of PLH was estimated to be approximately 5.25 Solubility in the three tested formulas was over 150 times that of plain PLH; in particular, the saturated solubility from formulation B in water, buffer pH 1.2, 4.0, and 6.8 media were 5.72 ± 0.17 mg/mL, 3.60 ± 0.18 mg/mL, 4.93 ± 0.22 mg/mL, and 4.26 ± 0.01 mg/mL, respectively. This result shows that the S/Cos ratio (2:1) is a more important factor than type of S or CoS in the solubility for PLH (Table 2).
Table 5.
Saturated solubility of pranlukast from 3% pranlukast hemihydrate-loaded SMEDDS in aqueous media
| Formulation | Solubility (mg/ml) | |||
|---|---|---|---|---|
|
|
||||
| Water | pH 1.2 | pH 4.0 | pH 6.8 | |
| Powder | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 |
| A | 4.73 ± 0.16 | 0.09 ± 0.00 | 4.95 ± 0.10 | 6.07 ± 0.08 |
| B | 5.72 ± 0.17 | 3.60 ± 0.18 | 4.93 ± 0.22 | 4.26 ± 0.01 |
| C | 5.00 ± 0.08 | 0.08 ± 0.00 | 5.04 ± 0.06 | 5.44 ± 0.15 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
In vitro dissolution studies
The effect of different media on the dissolution behavior of PLH in SMEDDS is shown in Figure 2. The dissolution rate and saturated solubility of PLH in SMEDDS increased in association with the increase of pH values, which were similar to the results of a previous report.26 However, PLH from plain PLH was not detected in SGF (pH 1.2) and PBS (pH 4.0). In each test media, the initial dissolution rate of PLH from SMEDDS was significantly faster than that of PLH from plain PLH.
Figure 2.
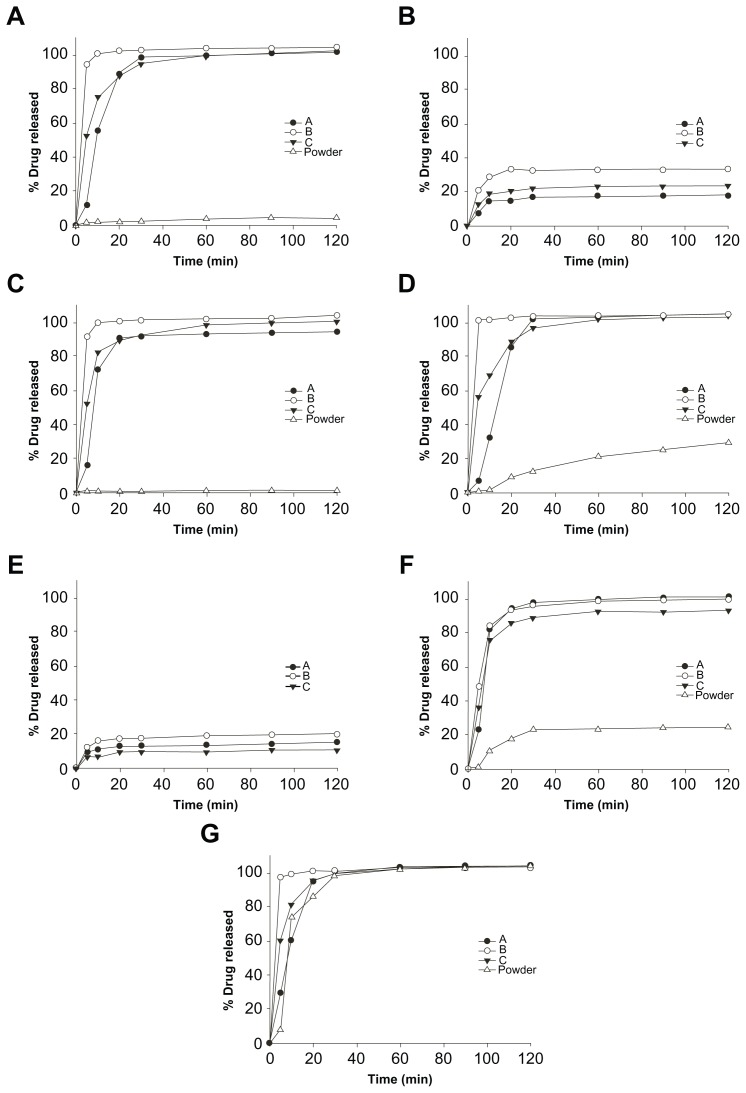
The dissolution profiles of pranlukast hemihydrate from SMEDDS in different media: distilled water (A); pH 4.0 (B); pH 6.8 (C); distilled water + Tween 80 (D); pH 1.2 + Tween 80 (E); pH 4.0 + Tween 80 (F); and pH 6.8 + Tween 80 (G).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
PLH from SMEDDS (formulation B) rapidly released over 95% of the drug within 5 minutes, and dissolution was completed within 30 minutes in distilled water and SIF (pH 6.8), whereas PLH from plain PLH released only 4.12% and 1.12% of the drug after 2 hours, respectively.
As PLH exhibits nonlinear pharmacokinetics, its absorption ratio decreases at high doses, and its absorption ratio after a meal is higher than when fasting; therefore, it would appear that the site of absorption is in the upper part of the gastrointestinal tract.26 Thus, the improvement in the initial dissolution rate at a low pH in the present study is especially significant for the improvement of the gastrointestinal absorption of PLH. It illustrates that perfectly dissolved PLH in SMEDDS can be dispersed in the aqueous phase without concentration loss or precipitation, which permits improved intestinal absorption, and suggests it can enhance the oral bioavailability.
Stability of PLH-loaded SMEDDS
The PLH-loaded SMEDDS was kept at 25°C/60% RH and 40°C/75% RH. The PLH-loaded SMEDDS remained stable for 3 months without crystallization of PLH under the ambient and accelerated storage conditions, and there was no significant change in the drug content, drug release, and particle size of the resultant emulsion (Table 6).
Table 6.
Stability data of capsules filled with 3% pranlukast hemihydrate-loaded SMEDDS
| Period (month) | Storage | Appearance | Drug content (%) | Particle size (nm) | T95% (min) |
|---|---|---|---|---|---|
| 0 | 25°C | Clear yellow liquid | 100.07 ± 0.32 | 3.62 ± 0.33 | |
| 1 | 60% (RH) | Clear yellow liquid | 100.43 ± 0.55 | 3.48 ± 0.24 | <10 |
| 2 | Clear yellow liquid | 100.81 ± 0.67 | 3.43 ± 0.17 | <10 | |
| 3 | Clear yellow liquid | 100.13 ± 0.25 | 3.63 ± 0.04 | <10 | |
| 0 | 40°C | Clear yellow liquid | 100.07 ± 0.32 | 3.62 ± 0.33 | |
| 1 | 75% (RH) | Clear yellow liquid | 100.99 ± 0.55 | 3.58 ± 0.10 | <10 |
| 2 | Clear yellow liquid | 100.32 ± 0.25 | 3.26 ± 0.10 | <10 | |
| 3 | Clear yellow liquid | 100.74 ± 0.30 | 3.41 ± 0.32 | <10 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviations: T95%, time taken for 95% drug release in pH 6.8; RH, relative humidity.
The PLH-loaded SMEDDS was compatible with the hard gelatin capsule shells, which could be illustrated in that there was no deformation of the capsule shells, and no significant changes in their appearance, disintegration time, or microemulsifying properties. Furthermore, the formulation was found to show no phase separation, drug precipitation, or capsule leak. Thus, these studies confirmed the stability of the developed SMEDDS (formulation B) and their compatibility with hard gelatin capsules when used for the in vivo absorption study.
In vivo absorption studies
The plasma concentration versus time profiles are shown in Figure 3, and the pharmacokinetic parameters are summarized in Table 7. Dosing the aqueous suspensions of PLH resulted in the lowest average PLH plasma concentrations. The AUClast and Cmax were 2.7 and 2.8 times greater when PLH was administrated as SMEDDS, respectively, compared with the AUClast obtained for the aqueous PLH suspension. The result reveals that the formulation of PLH as SMEDDS results in a significantly increased absorption of PLH compared with that from the aqueous suspension.
Figure 3.
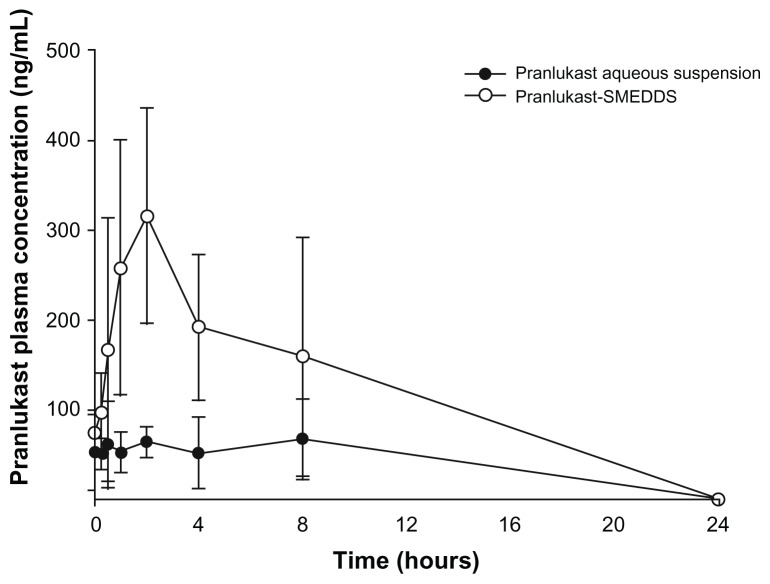
Plasma concentration versus time profiles after oral administration (40 mg/kg) of 3% pranlukast hemihydrate-loaded SMEDDS, compared with pranlukast hemihydrate aqueous suspension (n = 3).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
Table 7.
Pharmacokinetic parameters after oral administration of SMEDDS and an aqueous suspension at a dose of 40 mg/kg
| PLH-loaded SMEDDS | PLH aqueous suspension | |
|---|---|---|
| Cmax (ng/mL) | 384.3 ± 61.8 | 105.2 ± 9.6 |
| Tmax (h) | 4.7 ± 3.0 | 5.0 ± 3.6 |
| AUClast (ng · h/mL) | 2855.3 ± 1129.9 | 967.5 ± 441.4 |
| MRTlast (h) | 5.1 ± 1.8 | 5.6 ± 2.4 |
Note: Each value represents the mean ± SD (n = 3).
Abbreviations: AUC, area under the curve; Cmax, peak plasma concentration; MRTlast, mean residence time; PLH, pranlukast hemihydrate; SMEDDS, self-microemulsifying drug delivery system; Tmax, the time to reach Cmax.
The absorption of PLH was likely improved due to the enhanced solubilization, delay of gastric emptying, increase of mucosal permeability, and the increase of incorporation into lipoproteins. This was then secreted into the lymphatics, which circumvents the liver, thus reducing the hepatic first-pass metabolism.27–30 Enhancement of the bioavailability of PLH loaded-SMEDDS compared with PLH suspension may be due to the effects of a large amount of surfactants and cosurfactants, including improved mucosal permeability, smaller lipid droplets, and greater surface area. PLH dissolved in liquid could be directly absorbed as the microemulsion droplets in the gastrointestinal tract without a dissolution step. The small droplet size of less than 50 nm of oil-in-water microemulsions might penetrate the site of absorption via a transcellular pathway. Nonionic surfactants not only improved the solubility and dissolution of the drug, but may also reduce the interfacial surface tension and enhance penetration of the drug through the epithelial cells. In addition, Tween 20 has been reported to increase tight junction permeability, leading to enhanced drug absorption by the paracellular pathway.31 The pharmacokinetic data in male rats imply that PLH-loaded SMEDDS developed in this study could improve the dissolution and absorption of PLH.
Conclusion
The optimal formulations of the PLH-loaded SMEDDS were successfully developed with an increased dissolution rate, solubility, and bioavailability of a poorly water-soluble drug (PLH). The pseudoternary phase region of PLH-loaded SMEDDS increased with respect to the increase of the content of surfactant and cosurfactant, and the solubility of PLH was increased using 5% TEA and 10% BA. The SMEDDS readily released the lipid phase to form a fine oil-in-water microemulsion with a narrow distribution size. The saturated solubility of PLH from SMEDDS in distilled water, PBS (pH 4.0), and SIF (pH 6.8) was over 150 times than that of plain PLH. Moreover, the saturated solubilities of PLH among formulation B in the distilled water, SGF (pH 1.2), PBS (pH 4.0), and SIF (pH 6.8) media were 5.72 ± 0.17 mg/mL, 3.60 ± 0.18 mg/mL, 4.93 ± 0.22 mg/mL, and 4.26 ± 0.01 mg/mL, respectively. The capsules filled with SMEDDS liquid were found to remain stable over a period of 3 months under limited conditions.
The release of PLH from SMEDDS was faster than plain PLH. Pharmacokinetic studies in rats revealed that SMEDDS showed approximately a threefold greater absorption of PLH compared to the same oral dose of the plain PLH aqueous suspension. Thus, our studies illustrated that the potential use of a new self-microemulsifying system can be a possible alternative to oral delivery of a poorly water-soluble drug such as PLH.
Footnotes
Disclosure
The authors reported no conflicts of interest in this work.
References
- 1.Nakagawa N, Obata T, Kobayashi T, et al. In vivo pharmacologic profile of ONO-1078: a potent, selective and orally active peptide leukotriene (LT) antagonist. Jpn J Pharmacol. 1992;60(3):217–225. doi: 10.1254/jjp.60.217. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi Y, Tamura G, Honma M, et al. The effect of an oral leukotriene antagonist, ONO-1078, on allergen-induced immediate bronchoconstriction in asthmatic subjects. J Allergy Clin Immunol. 1993;92(4):507–512. doi: 10.1016/0091-6749(93)90074-p. [DOI] [PubMed] [Google Scholar]
- 3.Chono S, Takeda E, Seki T, Morimoto K. Enhancement of the dissolution rate and gastrointestinal absorption of pranlukast as a model poorly water-soluble drug by grinding with gelatin. Int J Pharm. 2008;347(1–2):71–78. doi: 10.1016/j.ijpharm.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 4.Aungst BJ. Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J Pharm Sci. 1993;82(10):979–987. [PubMed] [Google Scholar]
- 5.Burcham DL, Maurin MB, Hausner EA, Huang SM. Improved oral bioavailability of the hypocholesterolemic DMP 565 in dogs following oral dosing in oil and glycol solutions. Biopharm Drug Dispos. 1997;18(8):737–742. doi: 10.1002/(sici)1099-081x(199711)18:8<737::aid-bdd59>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 6.Serajuddin AT, Sheen PC, Mufson D, Bernstein DF, Augustine MA. Effect of vehicle amphiphilicity on the dissolution and bioavailability of a poorly water-soluble drug from solid dispersions. J Pharm Sci. 1988;77(5):414–417. doi: 10.1002/jps.2600770512. [DOI] [PubMed] [Google Scholar]
- 7.Khoo SM, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–164. [Google Scholar]
- 8.Craig DQM, Lievens HSR, Pitt KG, Storey DE. An investigation into the physico-chemical properties of self-emulsifying systems using low frequency dielectric spectroscopy, surface tension measurements and particle size analysis. Int J Pharm. 1993;96(1–3):147–155. [Google Scholar]
- 9.Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106(1):15–23. [Google Scholar]
- 10.Wakerly MG, Pouton CW, Meakin BJ, Morton FS. Self-emulsification of vegetable oil-nonionic surfactant mixtures: a proposed mechanism of action. In: Scamehorn JF, editor. Phenomena in Mixed Surfactant Systems. Washington, DC: American Chemical Society; 1986. pp. 242–255. [Google Scholar]
- 11.Kararli TT, Needham TE, Griffin M, Schoenhard G, Ferro LJ, Alcorn L. Oral delivery of a renin inhibitor compound using emulsion formulations. Pharm Res. 1992;9(7):888–893. doi: 10.1023/a:1015896731545. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh PK, Majithiya RJ, Umrethia ML, Murthy RS. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSci Tech. 2006;7(3):77. doi: 10.1208/pt070377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu H, Lu C, Zhou A, Min Z, Zhang Y. Enhanced oral bioavailability of puerarin using microemulsion vehicle. Drug Dev Ind Pharm. 2009;35(2):138–144. doi: 10.1080/03639040801973495. [DOI] [PubMed] [Google Scholar]
- 14.Myers RA, Stella VJ. Systemic bioavailability of penclomedine (NSC-338720) from oil-in-water emulsions administered intraduodenally to rats. Int J Pharm. 1992;78(1–3):217–226. [Google Scholar]
- 15.Schwendener RA, Schott H. Lipophilic 1-beta-D-arabinofuranosyl cytosine derivatives in liposomal formulations for oral and parenteral antileukemic therapy in the murine L1210 leukemia model. J Cancer Res Clin Oncol. 1996;122(12):723–726. doi: 10.1007/BF01209119. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi M, Uechi S, Takara K, Asikin Y, Wada K. Evaluation of an oral carrier system in rats: bioavailability and antioxidant properties of liposome-encapsulated curcumin. J Agric Food Chem. 2009;57(19):9141–9146. doi: 10.1021/jf9013923. [DOI] [PubMed] [Google Scholar]
- 17.Shaikh J, Ankola DD, Beniwal V, Singh D, Kumar MN. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci. 2009;37(3–4):223–230. doi: 10.1016/j.ejps.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Patel D, Sawant KK. Oral bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS) Drug Dev Ind Pharm. 2007;33(12):1318–1326. doi: 10.1080/03639040701385527. [DOI] [PubMed] [Google Scholar]
- 19.Kang BK, Lee JS, Chon SK, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 20.Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. doi: 10.1023/a:1016268311867. [DOI] [PubMed] [Google Scholar]
- 21.Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res. 1992;9(1):87–93. doi: 10.1023/a:1018987928936. [DOI] [PubMed] [Google Scholar]
- 22.Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212(2):233–246. doi: 10.1016/s0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- 23.Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11(Suppl 2):S93–S98. doi: 10.1016/s0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 24.Craig DQM, Barker SA, Banning D, Booth SW. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114(1):103–110. [Google Scholar]
- 25.Welz R, Müller S. 5-(benzylmercapto)-1H-tetrazole as activator for 2′-O-TBDMS phosphoramidite building blocks in RNA synthesis. Tetrahedron Lett. 2002;43(5):795–797. [Google Scholar]
- 26.Ishido M, Shibakawa K, Nakao Y, Sawada M, Aishita H. Studies on the metabolic fate of leukotriene antagonist ONO-1078 (1): absorption, distribution and excretion after single administration to rats. Xenobio Metabol Dispos. 1993;8:2–26. [Google Scholar]
- 27.Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50(1):179–188. doi: 10.1016/s0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 28.Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 29.Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25(1):47–58. [Google Scholar]
- 30.Humberstone AJ, Charman WN. Lipid based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–128. [Google Scholar]
- 31.Dimitrijevic D, Shaw AJ, Florence AT. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J Pharm Pharmacol. 2000;52(2):157–162. doi: 10.1211/0022357001773805. [DOI] [PubMed] [Google Scholar]