

Abstract
Bis-phenylamides and bis-hydroxyindolamides of DTPA(Gd) are paramagnetic reducing substrates of peroxidases that enable molecular imaging of peroxidase activity in vivo. Specifically, bis-5HT-DTPA(Gd) has been used to image localized inflammation in animal models by detecting neutrophil derived myeloperoxidase (MPO) activity at the inflammation site. However, in other pre-clinical disease models, bis-5HT-DTPA(Gd) presents technical challenges due to its limited solubility in vivo. Here, we report a novel MPO sensing probe obtained by replacing the reducing substrate serotonin (5HT) with 5-hydroxytryptophan (HTrp). Characterization of the resulting probe (bis-HTrp-DTPA(Gd)) in vitro using NMR spectroscopy and enzyme kinetic analysis showed that bis-HTrp-DTPA(Gd): 1) improves solubility in water; 2) acts as a substrate for both HRP and MPO enzymes; 3) induces cross linking of proteins in the presence of MPO; 4) produces oxidation products which bind to plasma proteins and; 5) unlike bis-5HT-DTPA(Gd), does not follow first order reaction kinetics. In vivo MR imaging in mice demonstrated that bis-HTrp-DTPA(Gd) was retained for up to five days in MPO-containing sites and cleared faster than bis-5HT-DTPA(Gd) from MPO-negative sites. In conclusion, bis-HTrp-DTPA(Gd) should offer improvements for MR imaging of MPO-mediated inflammation in vivo especially in high-field MRI, which requires higher dose of contrast agent.
Introduction
Myeloperoxidase (MPO) has emerged as a promising imaging target in molecular imaging 1–5. Myeloperoxidase activity in vivo has a clear outcome-predictive value in several cardiovascular diseases, including myocardial infarction and stroke 6. MPO has also been implicated in the progression of cancer, several CNS pathologies, and in the development of unstable atheroma 7, 8. Because the catalytic activity of MPO is preserved in live tissue either in intact granulocytes or as a component of exocytosed granules, the enzymatic activity of MPO can be used as an imaging marker of localized inflammation in vivo, including cardiovascular disease and vascular wall pathologies 3, 9. We previously synthesized and tested several paramagnetic complexes of mono- and bis- amides of macrocyclic and linear polycarboxylic chelates that “sense” MPO activity 1, 10, 11. These chelates carry heterocyclic hydroxyindole moieties, which donate electrons thereby reducing peroxide-oxidized MPO-I and MPO-II compounds to a catalytically active state 12. The reactions of MPO with hydrogen peroxide and the following reduction of MPO-I and MPO-II can be written as:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where A represents an aromatic (e.g. indole) moiety and P is protein. Reaction schemes (1) and (2) illustrate the commonly observed MPO catalysis of hydrogen peroxide reduction that utilizes chloride anion for generating bactericidal hypochlorite as part of the inflammatory response following neutrophil infiltration and degranulation. Reactions (3) and (4) result in sequential reduction of MPO-I to ground-state MPO by hydroxyindole-containing compounds and tyrosine. Oxidized species generated from these reactions can react with proteins containing tyrosines (6) or can engage in either spontaneous dimerization (5) or oligomerization (7). In the case of plant peroxidases, e.g. horseradish peroxidase (HRP), reaction (2) is not catalyzed since chloride anion is not a reducing substrate of HRP.
Paramagnetic hydroxindole-containing sensors apparently follow the same trend as other reducing organic compounds, i.e. they dimerize, oligomerize and/or form covalent bonds with proteins. Importantly, the latter two reactions result in increased molar relaxivity (r1 and r2) of the paramagnetic sensors 1, 13, 14. This enables direct MR imaging of MPO activity in vivo because the products of the reaction are retained in the tissue for longer periods of time than the initial substrates 2, 3, 5. Although some of the previously developed paramagnetic MPO sensors have very high kinetic stability 14, the thermodynamic stability of bisamides is frequently compromised. Moreover, the solubility of neutrally charged (non-ionic) bis-tryptamides of DTPA 10 and mono-amides of glycyl-MeDOTA 1 in water is limited. Since the amount of injected contrast agent needed to produce detectable contrast in MR images is relatively high (0.1–0.3 mmol/kg), there remains the need to improve the safety, enzymatic specificity and formulation of MPO sensing probes.
The goal of the current study was to synthesize and characterize an alternative charged bifunctional sensor of MPO activity with the same number of donor atoms as in DTPA (i.e. three nitrogen/five oxygen configuration) in anticipation of improved thermodynamic stability and better solubility. We used a natural serotonin precursor, 5-hydroxytryptophan (HTrp), which shares reactive oxygen species (ROS)-scavenging properties with serotonin 15 for synthesis of a novel paramagnetic DTPA diamide-based substrate.
Materials and Methods
Synthesis of bis(5-hydroxytryptophan)-DTPA, (bis-HTrp-DTPA)
2.0 g (5.6 mmol) diethylenetriaminepentaacetic dianhydride was reacted with 2.7 g (12.3 mmol) L-5-hydroxytryptophan in dry DMF in the presence of 1.0 g (12.5 mmol) pyridine. The mixture was stirred at room temperature for 24 hr. The solvent was removed in vacuo. The crude product was solubilized in methanol and precipitated by slow acetone diffusion. The yield of pure product is 50%. MALDI-ITMS - found m/z 796.36 (C36H43N7O14 + H+) m/z expected 795.29. 1H NMR (400 MHz, d6-DMSO), δ (ppm): 10.57 (s, 2H), 8.30 (d, 2H, J = 7.8 Hz), 7.05 (m, 4H, J = 8.6 Hz), 6.83 (s, 2H), 6.53 (d, 2H, J = 8.6), 4.35 (2H), 3.47 (s, 2H), 3.35 (s, 4H), 3.27 (s, 4H), 3.17 (s, 2H), 2.98 (s, 4H), 2.86(s, 4H), 2.71(s, 4H).
Syntheses of gadolinium chelates
4.0 g (5.0 mmol) bis-HTrp-DTPA was dissolved in 100 ml of 1% citric acid solution (pH =6.5). GdCl3.6H2O (1.1 eq.) was added in portions during 1 h and the solution was adjusted to pH = 7.0 with 0.5 M NaOH solution. The mixture was stirred in dark under argon for 48 h. The mixture was lyophilized. Final product was purified by HPLC using a gradient of acetonitrile in water with 35% yield. MS found 951.28 (M + 2H), m/z expected 949.49 (based on formula C36H38N7O14Gd). Gadolinium chelates of bis-5-hydroxytryptamide-DTPA (bis-5HT-DTPA(Gd) and bis-tyramide-DTPA were synthesized as described previously 14, 11. All substrates were purified using C18-HPLC and lyophilized prior to use. Concentrations of gadolinium were measured using ICP-OES (Galbraith Labs.)
Substrate stability measurements
Transmetallation of the gadolinium substrates (thermodynamic and kinetic indices) were measured and calculated as previously described 16. Longitudinal and transverse relaxation rates were measured in phosphate buffer (pH 6.0 and pH 7.0) containing 2.5mM of the substrate and 10μl of 2.5mM zinc chloride at different time points up to three days at 40°C.
Substrate activation studies
Initially, the substrates were dissolved in sterile DPBS, pH 7.4 for preparing 10 mM stock solutions. These stock solutions were further diluted to 0.1–1 mM in DPBS pH 6.8 containing 5 mM glucose. The comparative substrate activation experiments were performed using GOX/MPO or GOX/horseradish peroxidase (HRP, positive control) as complementing activity-coupled enzymatic pairs (3 IU GOX : 1.5 IU MPO or HRP, added directly to the reaction mixture). The enzymes were chemically deglycosylated and capped with hydroxylamine as previously described 17. Glucose oxidase was added to 0.5 ml of pre-heated (40°C) substrate solutions containing HRP or MPO in NMR spectroscopy tubes and the solution was rapidly vortexed. The changes of T1 and T2 relaxation times were measured at 0.47T using an inversion recovery (IR) and Carr-Purcell-Meiboom-Gill (CPMG) pulse sequences respectively. The obtained time-course data was analyzed using Guggenheim approximation for pseudo-first order kinetics 18. The reaction mixtures were further analyzed using 4–15% gradient PAGE (1 μg total protein/lane loaded) and the gels were stained with Silver Stain kit (Thermo-Fisher) to visualize both proteins and polymerized reaction products. Radioactive 111In labeled analogs of bis-HTrp-DTPA and bis-5HT-DTPA ligands were synthesized by transchelating 111In3+ from 111In citrate (obtained by diluting 111InCl3 stock solution (1 mCi, Perkin-Elmer) in 0.01 M sodium citrate, pH 5.5 and adding a 50-fold molar excess of HPLC-purified ligand followed by heating for 5 min at 50°C. The remaining free chelating groups were blocked by adding 1.1-molar excess of GdCl3 and the radiolabeled substrates were purified using C18-HPLC column (Discovery 5μm, Supelco) with an acetonitrile/water gradient. The main radioactive peaks were collected and dried under argon for further experiments.
Protein-binding experiments
Heparinized human blood was centrifuged at 10,000xg for 15 min followed by filtration through 0.4 μm PVDF filter to obtain cell-free total blood plasma. Gradient SDS-PAGE (4–15%) was performed using samples incubated in the presence or absence of the substrates to determine potential protein cross-linking using silver staining.
An aliquot containing 150 μCi of 111In-labeled substrate (0.5μl) was mixed with 20 μl of 1 mM ligand solution in 10 mM sodium acetate/citrate pH 6.0 followed by 10 μl of MPO/GOX or HRP/GOX mixture. Control samples had no enzymes added. The reactions were initiated by diluting the above mixtures with either 10 μl of 4x-Hanks’ balanced saline (with Ca and Mg) pH 8, or with 10μl human plasma. After 30 min incubation at 37°C, 50 μl aliquots of reaction mixtures were loaded on Biospin P-6 columns (Bio-Rad) and the protein fraction was recovered in the spin-through eluate. The bound vs. total radioactivity fractions were determined by counting 111In radioactivity using the eluate and the remaining column using a dose calibrator (247 keV peak, Bicron Instr.). The aliquots of the eluates (20 μg total plasma protein) or equivalent volumes of control samples that did not contain plasma were loaded on 4–15% gradient PAGE and the resultant gels were stained with GelCode Blue stain reagent (Thermo-Fisher), dried and subjected to autoradiography using BioMAX MS film (Carestream Health Inc, Rochester NY).
MRI in vivo and in vitro at 3T
All animal experiments were performed as approved by the Institute Animal Care and Use Committee (IACUC) of the University of Massachusetts Medical School (Worcester, MA, USA). Imaging of MPO activity was performed in a model system as previously described 2 with modifications. Briefly, DBA/2 female mice (NCI Frederick) (n=2), weighing 20–25g, were injected with 200 μl of Matrigel (BD Biosciences) containing MPO (7.5 IU)/GOX (5 IU) mixture in the left femoral muscle region while the contralateral side, which served as the control, received Matrigel (200 μl) containing 5 IU GOX only. The animals were then imaged using MRI. Throughout the imaging sessions, animals were anesthetized using 1% isofluorane in oxygen gas mixture. The lower body of the mouse was placed in custom made holder and then positioned in a 38-mm-diameter, 55-mm-long birdcage RF coil with a nose cone such that the thigh region was in the center of the coil. A circulating water heating blanket was placed under the mouse/RF coil assembly. For administration of contrast agent, a 29-gauge polyurethane catheter (Strategic Applications Inc.) capped with a needle port was placed in the tail vein. A respiratory sensor was placed on the chest area to monitor the respiration rate throughout the imaging session.
All MRI measurements were performed using a Philips Achieva 3.0T/60 cm console equipped with 80mT/m actively shielded gradients. To monitor the temporal evolution of signal enhancement in the right and left femoral muscles following contrast agent delivery, multi-slice, T1-weighted (T1-W) MR images (TR/TE = 700/8 ms) were acquired using spin-echo sequence at various time points after contrast agent administration. Other imaging parameters were: slice thickness = 1 mm; slice separation (center-to-center) = 1.1 mm; field-of-view (FOV) = 30 mm x 30 mm; data acquisition matrix = 320 x 320; 4 NEX. Within an hour after implantation of Matrigel mixtures, pre-contrast images were acquired followed by IV injection of 0.2 mmol/kg bis-HTrp-DTPA(Gd). Several T1-W images were then acquired every fifteen minutes up to the first hour and then at longer time intervals up to 5 days. Pre-contrast T2-W images were acquired prior to contrast administration to corroborate the presence of Matrigel mixture in the T1-W slices. T2-W imaging was performed using multi-slice, multi-spin-echo imaging with TR = 1380 ms, 2 NEX. Eight echoes were acquired with intervening TE values of 25 ms. In vivo T2-W imaging was performed with the same FOV, data acquisition matrix, slice thickness, slice separation, and slice position as the in vivo T1-W images. Imaging of the in vitro phantom samples was done using a spin-echo sequence with TR/TE = 200/30 ms and NEX=1.
Data Analysis
The signal decay in the femoral muscle following administration of bis-HTrp-DTPA(Gd) was evaluated for both MPO-treated and untreated (control) sides. Depending on the size of the implant, five to fourteen slices were selected at each time point covering the entire area of the implant corroborated using the T2 images. ImageJ software package was used to construct a region-of-interest (ROI) around the boundary of the implants. The mean signal intensity (SI) values within each of the ROIs were calculated using ImageJ. Contrast-to-noise ratios (CNRs) were then calculated using:
| (8) |
where CNRMPO/control is the CNR of the MPO or control side, SIMPO/control is the mean signal intensity, SImuscle is the mean signal intensity of muscle, and SDnoise is the standard deviation of noise in the corresponding slice. Analysis of variance (ANOVA) for mixed models was used to determine if there was a significant change in CNR between the MPO-treated and control sides. The Tukey-Kramer HSD multiple comparisons procedure was used to determine if any significant differences existed between the MPO-treated and control sides. Statistical analyses were carried out using the SAS statistical software package (SAS Institute Inc., Cary, NC, USA).
Results
In vitro stability and relaxivity
The gadolinium (III) chelate of bis-HTrp-DTPA(Gd) (substrate I, Fig. 1) showed excellent solubility in water, DPBS or 0.9% NaCl at concentrations upto 40 mg/ml (42 mM) whereas bis-5HT-DTPA(Gd) (substrate II, Fig. 1B) showed a solubility limit of approximately 30 mg/ml in 5% dimeglumine in DPBS, and dissolved only with heating above 60°C (with the compound being precipitated from solution after returning to room temperature). Longitudinal relaxation time measurements at 0.47T, 40°C showed that the r1 of substrates I and II were very similar (4.9 and 5.0 [mM·s]−1, respectively). Purified I and II were further tested as reducing substrates in peroxidase-mediated reactions in the presence of D-glucose and GOX. Endpoint relaxivity measurements of reaction mixtures showed a similar increase of r1 value for both I and II (Table 1 and Fig. 2) in the presence of either horseradish peroxidase (HRP) or MPO (1.7 and 1.8-fold, respectively, at 0.47T). Measurements of kinetic and thermodynamic stability when obtained under conditions of normal tissue pH (pH 7.0) or tissue acidosis (pH 6.0), revealed that substrate II was a more stable chelate of gadolinium when compared to other bis-amides of DTPA(Gd) used in this study (Table 2).
Fig. 1.
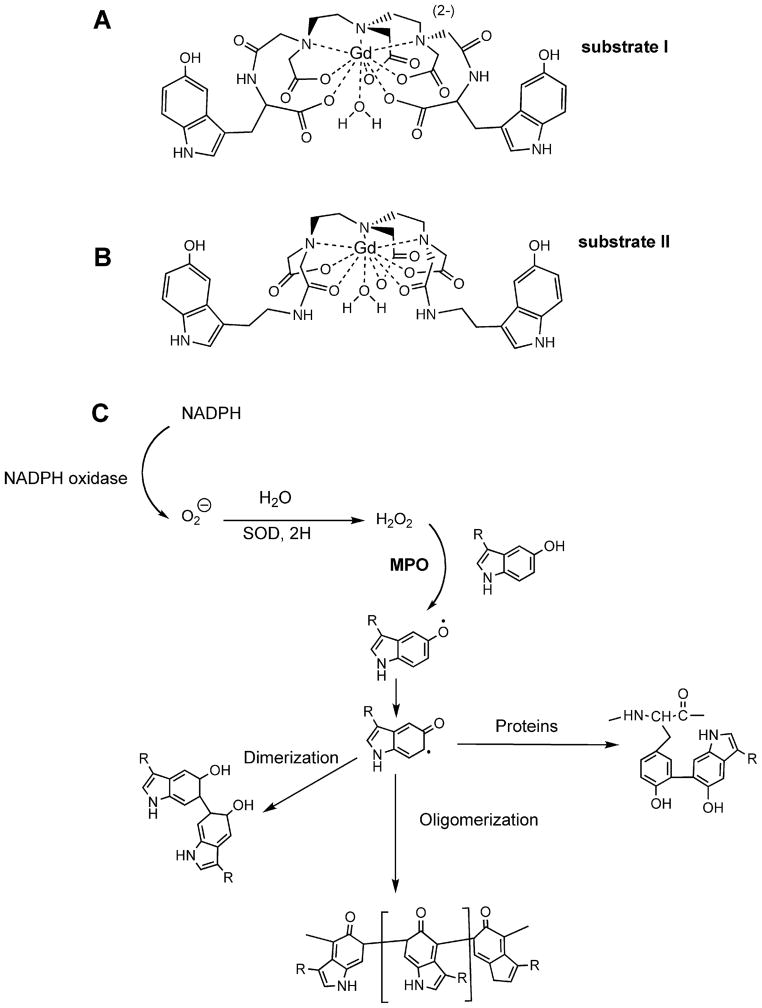
Theoretical structures of MPO substrates built using Gd(III), which has a coordination number of 9. A, - A novel substrate bis-HTrp- DTPA(Gd) (substrate I) synthesized using L-5-hydroxytryptophan. B, - Bis-5HT-DTPA(Gd) (substrate II) synthesized using serotonin (5-hydroxytryptamide). This substrate is also known as MPO-Gd in the literature.2 C,-Hypothetical MPO reaction pathways leading to enzyme-generated products with higher relaxivity. R= paramagnetic moiety.
Table 1.
Measurements of enzyme-mediated changes of r1 and pseudo-first order kinetic constants (keff) in the presence of bis-HTrp-DTPA(Gd) (I), bis-5HT-DTPA(Gd) (II) and bis-Tyr-DTPA(Gd) (III).
| Substrate | Enzyme | r1 [mMs]−1 0.47 T, 40°C | r1enzyme/r1o | keff R1, 10−3 [s]−1 | keff R2, 10−3[s]−1 | b) Effective keff, 10−3 [s]−1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0.25 mM | 0.5 mM | 1 mM | 0.25 mM | 0.5 mM | 1 mM | |||||
| I | - | 4.9 | - | - | - | - | - | - | - | |
| I | MPO | 8.3 | 1.7 | 7.4 | 12.2 | 21.2 | 4.4 | 11.4 | 17.2 | 1.2 |
| I | HRP | 8.8 | 1.8 | 0.4 | 1.6 | 4.3 | 0.2 | 0.9 | 3.0 | 10.5 |
| II | - | 5.0 | - | - | - | - | - | - | - | |
| II | MPO | 8.7 | 1.7 | 0.8 | 0.9 | 0.9 | 0.8 | 0.8 | 0.6 | 3.1 |
| II | HRP | 9.0 | 1.8 | 18.0 | 18.1 | 20.1 | 16.5 | 17.1 | 17.0 | 37.7 |
| IIIa) | HRP | - | - | 7.5 | 17.0 | 24.4 | 6.7 | 16.1 | 30.5 | 83.3 |
bis-Tyr-DTPA(Gd) (substrate III) was tested only in reactions with HRP since it does not react with MPO. x- data does not fit a pseudo-first order model
- determined by measuring initial rates of absorbance increase at 350 nm; 1.01·10−7 M enzyme; 2.5 ·10−4 M H2O2
Fig. 2.
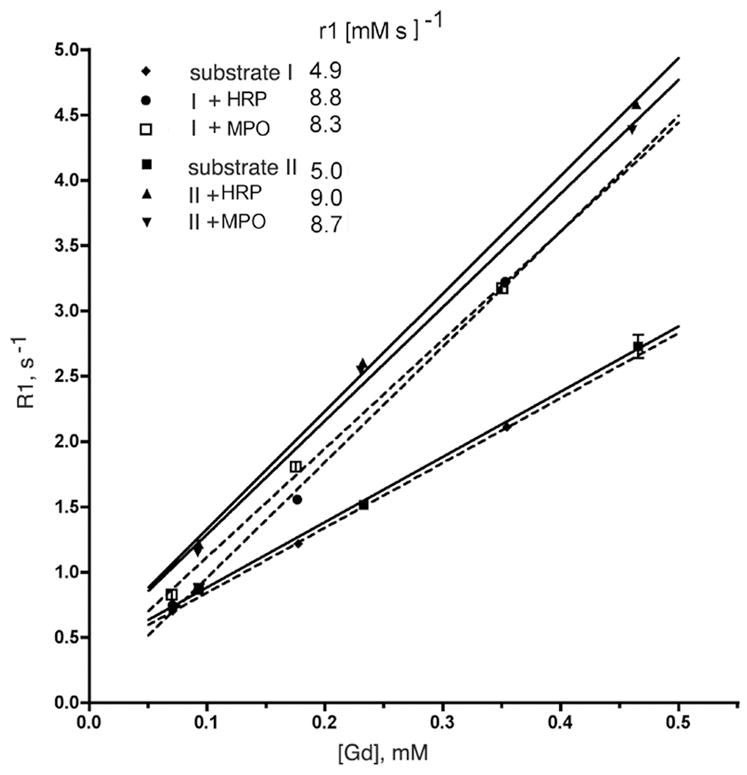
Concentration dependence of proton relaxation rates (R1) in the presence of substrate I (dashed lines) and substrate II (solid lines) solutions after adding MPO/GOX or HRP/GOX enzyme pairs in the presence of glucose. See figure legend for disambiguation of symbols and molar relaxivity values of substrates (r1).
Table 2.
R1 stability measurements in 0.47T at 40°C of bis-HTrp-DTPA(Gd) (I), bis-5HT-DTPA(Gd) (II) and bis-Tyr-DTPA(Gd) (III) at different pH values in DPBS.
| Substrate | pH 6 | pH 7 | ||
|---|---|---|---|---|
| Thermodynamic index: R1(t=4300min)/ R1(t=0) | Kinetic index: Time (mins) required to reach R1(t)/R1(0) = 0.8 | Thermodynamic index: R1(t=4300min)/ R1(t=0) | Kinetic index: Time (mins) required to reach R1(t)/R1(0) = 0.8 | |
| I | 0.22 | 106 | 0.62 | 1504 |
| II | 0.62 | 306 | 0.77 | 3215 |
| III | 0.30 | 82 | 0.56 | 1203 |
Reaction kinetics
Longitudinal and transverse relaxation rate measurements (R1 and R2) at different substrate concentrations showed a concentration dependence of both longitudinal and transverse pseudo-first order kinetic constants for substrate I but not for substrate II (Table 1). We further compared the effective pseudo-first order constants of cumulative relaxivity change reactions under the conditions of continuous enzyme cycling, i.e. steady state kinetics. The reactions were triggered by H2O2 generation via GOX/glucose system that allowed measurements of pseudo-first kinetic constants to be made by tracking the concentration dependence of the reaction rates. In the case of HRP, the enzyme- mediated relaxivity change showed that the pseudo-first order kinetic constants depended on the concentration of bis-tyramide-DTPA(Gd) (substrate III, a positive control for HRP). The reaction of HRP with substrate II showed independence of the effective reaction rate constant on concentration suggesting that the measurements of relaxivity change rates were dependent only from the concentration of the enzyme in the studied range of substrate concentrations. MPO-mediated catalysis of the high-relaxivity products was also independent on the concentration of substrate II. The effective pseudo-first order reaction constants were approximately 20-fold lower in the case of MPO compared to HRP. Conversely, when substrate I was tested in reactions with both enzymes we observed more rapid second order kinetics of R1 and R2 increase in the case of MPO, as compared to HRP.
A direct comparison of the kinetics of relaxivity increase to the kinetics of oxidized products formation was performed by measuring the change in absorbance at 350 nm (Fig 3). The goal was to find out whether changes occurring during the reactions 5, 6 and 7 (see introduction), i.e. downstream from the formation of the total pool of DTPA(Gd)-linked oxidized aromatic groups, happen in concert with the reduction of MPO-II or HRP-II by the paramagnetic substrates. Both MPO and HRP reaction rates followed a linear dependence on substrate concentration (r2 range 0.85–0.9) in the presence of H2O2 (0.2 mM) 19. MPO showed much lower kinetic constant values in the case of substrate II (which reduces both HRP-II and MPO-II), reflecting the overall slow rate of MPO reaction which was about 10-fold slower than that catalyzed by HRP (Table 1).
Fig. 3.
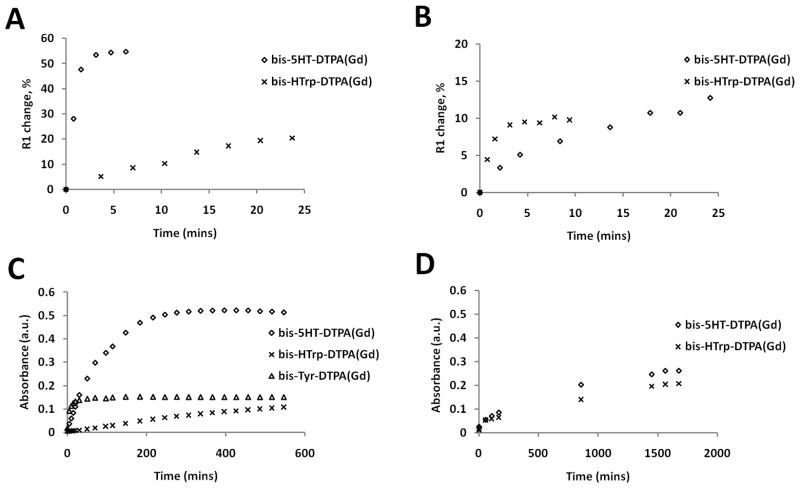
Relaxivity and absorbance measurements of substrates in the presence of HRP/MPO. Longitudinal relaxivity of substrates I and II in HRP/GOX reaction mixture (A) and their absorbance in HRP/H2O2 mixture (C) are shown in the first column. Longitudinal relaxivity of substrates I and II in MPO/GOX reaction mixture (B) and their absorbance in MPO/H2O2 mixture (D) are shown in the second column.
Interaction of the reaction products with proteins
Gradient PAGE analysis of the substrates and reaction mixtures with peroxidases and GOX showed HRP reaction products formed within 15 minutes of incubation. We observed a higher mass spot above the band corresponding to substrate I (Fig. 4, lane 4). The optical density of this spot increased after a 2-h incubation with HRP and GOX mixture (data not shown). The non-reacted substrates migrated with the front of the gel. In the case of MPO there was no apparent formation of higher molecular weight oligomerized products (Fig. 4, lane 5). However, in MPO-containing reaction mixtures we observed the formation of a cross-linked dimer of GOX (165 kD band) (shown with an arrow on Fig. 4). This band became more prominent after a 2 h incubation. The non-crosslinked form of GOX dimer usually completely dissociated in the presence of SDS (see lanes 1,2 and 7,8). No cross-linking was present if substrate I was absent from the reaction mixture (lane 2). In contrast, the analysis of substrate II - containing reaction mixtures (lanes 6–8) showed high-molecular weight condensation products in both HRP and MPO containing reaction mixtures, with the former enzyme catalyzing approximately 8-times the amount of the oligomerized product. No protein cross-linking was present in these reaction mixtures.
Fig. 4.
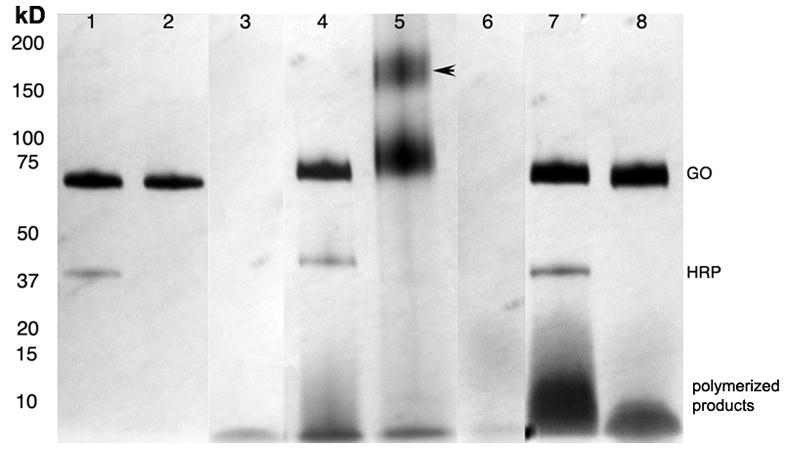
Gradient PAGE (4–15%) of paramagnetic substrates and reaction mixtures with peroxidases and glucose oxidase (silver stain). Reactions were carried out in 5mM D-glucose, DPBS, pH 6.8. Substrates were at 0.5 mM. Lanes: 1 – reaction mixture of HRP and GOX, no substrate; lane 2 – reaction mixture of MPO and GOX, no substrate; 3 – substrate I (20 μl); 4 – reaction mixture of HRP and GOX, substrate I; 5 – reaction mixture of MPO and GOX, substrate I; 6 – substrate II (20 μl); 7 – reaction mixture of HRP and GOX, substrate II; 8 – reaction mixture of MPO and GOX, substrate II. Cross-linked product is marked with an arrow.
In order to determine enzymatic reaction efficacy in the presence of serum proteins and to determine whether the serum proteins bind the chelates directly, we labeled the initial chelates (i.e. bis-5HT-DTPA and bis-HTrp-DTPA) with 111In isotope and measured the accumulation of reaction products in vitro. The radioactivity in eluates of the columns is shown in Table 3. We determined that both substrates I and II bind to plasma proteins at very low levels (not exceeding 0.5%) in the absence of peroxidases. In the absence of plasma proteins, a reaction mixture containing MPO/GOX resulted in 5–6% eluted radioactivity suggesting low levels of oligomerization with the formation of a high molecular-mass fraction. In contrast, HRP resulted in 30% of the substrates converting into a high-molecular mass (>6 kD) fraction of radioactive products. Both MPO and HRP catalyzed the formation of products that were bound to plasma proteins as confirmed by adding plasma proteins to the reactions and separating the obtained products (Table 3). The MPO-catalyzed reaction resulted in approximately 3-times more binding of substrate II products to plasma proteins than substrate I (14% vs. 50%, Table 3), whereas the HRP-catalyzed reaction resulted in approximately equal amounts of radioactivity eluted in the void volume together with plasma proteins (50–60%).
Table 3.
Oligomerization of 111In-labeled substrates I and II (0.5 μM) and binding to plasma proteins
| Substrate | Enzymes | Plasma proteins | Void volume, μCi eluted* | % eluted, average |
|---|---|---|---|---|
| I | - | − | 0.05±0.03 | 0.04 |
| II | - | − | 0.12±0.10 | 0.18 |
| I | - | + | 0.2±0.1 | 0.5 |
| II | - | + | 0.2±0.1 | 0.5 |
| I | MPO/GOX | − | 3.4±0.3 | 5.1 |
| II | MPO/GOX | − | 3.5±0.6 | 5.6 |
| I | HRP/GOX | − | 13.5±0.6 | 27.5 |
| II | HRP/GOX | − | 20.3±0.1 | 20.9 |
| I | MPO/GOX | + | 8.8±0.4 | 14.0 |
| II | MPO/GOX | + | 31.5±3.6 | 48.5 |
| I | HRP/GOX | + | 39.7±8.7 | 62.5 |
| II | HRP/GOX | + | 33.3±4.3 | 52.3 |
Void volume of Biospin P6 columns contains molecules exceeding 6 KDa.
Autoradiography was used to analysis reaction products after labeling the substrates with 111In and low-molecular weight products were resolved by gradient SDS-electrophoresis. The products of MPO in reaction with substrate I resulted in hydrophilic products that were poorly retained in the gel. The main plasma component that was labeled during substrate II oxidation was serum albumin (Fig. 5). HRP showed much more efficient oligomerization of substrate II if compared to substrate I. The distribution of radioactivity associated with plasma proteins in the case of both substrate I and II reactions with HRP was very similar (Fig. 5). Serum albumin and an unidentified 200 kD protein were the main binding partners for oxidized substrates. Both products of substrate I and II bind to plasma proteins, however, the binding is lower in the case of MPO/substrate I combination than in other tested substrate/enzyme combinations.
Fig. 5.
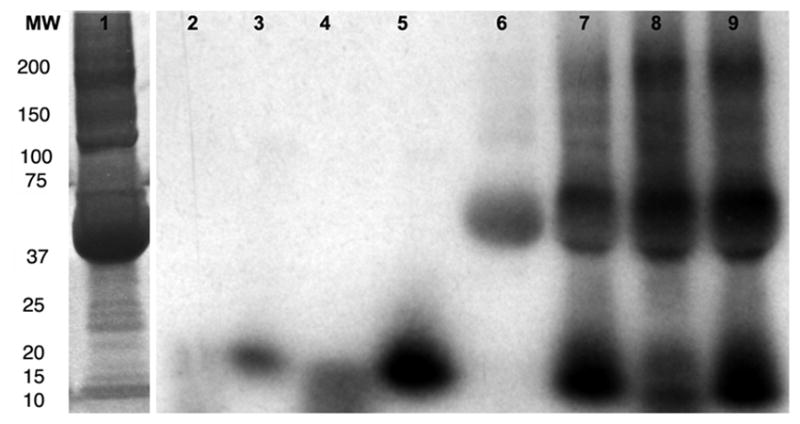
Gradient PAGE (4–15%) analysis of 111In labeled substrate-containing mixtures in the absence and in the presence of blood plasma proteins. 50 μl of Bio-Spin P6 eluates (Table 2) were loaded on a gel. Lane 1 – Coomassie stain of total protein in a serum sample (20 μg total protein); 2 – MPO/GOX reaction mixture, substrate I; 3 – MPO/GOX reaction mixture, substrate II ; 4 – HRP/GOX reaction mixture, substrate I; 5 – HRP/GOX reaction mixture, substrate II; 6 – MPO/GOX reaction mixture, substrate I, 40% plasma; 7 – MPO/GOX reaction mixture, substrate II, 40% plasma; 8 – HRP/GOX reaction mixture, substrate I, 40% plasma; 9 – HRP/GOX reaction mixture, substrate II, 40% plasma.
In vivo imaging of MPO activity using substrate I
The capability of bis-HTrp–DTPA(Gd) to generate MR signal enhancement at sites of localized MPO activity in vivo was assessed in mice. Animals received bilateral femoral muscle implants of basement membrane matrix gel (Matrigel) containing either an MPO (7.5 IU)/GOX (5 IU) mixture (left femoral muscle, denoted by asterisk in Fig. 6A) or containing 5 IU GOX only (right femoral muscle), which served as the control. Fig. 6A shows sequential T1-W images depicting implant enhancement as a function of time post IV injection of bis-HTrp-DTPA(Gd). Strong initial signal enhancement was observed in both MPO active and control femoral muscles implants. However, in control implants, the initial enhancement steadily decreased over time and was barely visible 4 h after contrast injection, while the enhancement of MPO active implants persisted, even after 5 days (p<0.0001) (Fig. 6B). A similar time-dependent course of MR signal enhancement was observed in animals that were injected with a 10-fold lower MPO dose (data not shown).
Fig. 6.
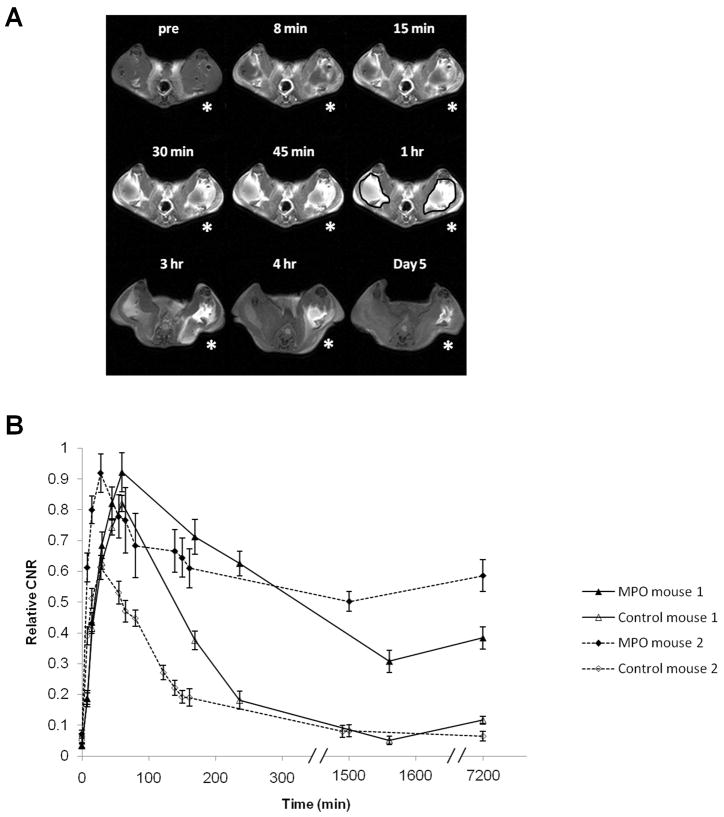
MR imaging of mouse femoral muscle at 3T with implanted Matrigel and MPO/GOX mixture: A) – T1-weighted sequential mouse extremities images are shown post IV injection of bis-HTrp-DTPA(Gd) depicting MPO/GOX enhancement (due to formation of cross linked products) as a function of time in the left femoral muscle region (indicated with asterisk) while the contralateral side implanted with Matrigel served as the control. Time intervals after the injection of bis-HTrp-DTPA(Gd) are shown above each image. Regions of interest were selected for all time points as indicated in the 1 hr time point slice. The signal intensities from all time points were normalized to pre-contrast slice and are thus directly comparable; B) – Relative CNR curves showing enhancement in the femoral muscle of two mice are shown as a function of time post IV injection of bis-HTrp-DTPA(Gd). For each animal the temporal wash-in and washout of bis-HTrp-DTPA(Gd) signal enhancement is shown in the MPO treated muscle (filled triangles and diamonds) and the untreated control side (empty triangles and diamonds). These plots were derived from several slices in each animal at each time point. Within each animal, the CNR is normalized to the maximum value to compute relative CNR.
Discussion
Non-invasive imaging of MPO activity can be performed by using high anatomical resolution MRI 2, high-sensitivity SPECT 22 or bioluminescence 4. MPO activity can be detected and monitored in tissues during the initiation and progression of diseases such as atherosclerosis 3, myocardial infarction 9, stroke 20, and autoimmune encephalitis 21. The precise localization of MPO-containing lesions is enabled by paramagnetic low molecular weight substrates, which are mono- or bis- amides of linear or cyclic chelates 1, 10. Rapid progress in development of high-field MR imaging dictates the need for MPO sensing probes which can be injected at higher doses than previously developed bis-amide based probes which have relatively low solubility and can only be injected at low doses. This study examined the potential benefits of replacing previously used serotonin (5-hydroxytryptamide, 5HT) with 5-hydroxytryptophan (HTrp) in the synthesis of an MR detectable MPO probe. The newly synthesized probe demonstrated greater solubility in aqueous solutions and produced gradual increases in proton relaxation rates (R1 and R2) in the presence of either MPO or HRP with the addition of H2O2. Interestingly, the oxidation of charged bis-HTrp-DTPA(Gd) (substrate I) by HRP resulted in a less efficient reactive intermediate than the non-ionic bis-5HT-DTPA(Gd) (substrate II). Conversely, the reactions catalyzed by MPO resulted in a more rapid proton relaxation rate increase in the presence of substrate I.
The observed substrate (I vs. II) and enzyme (HRP vs. MPO)-dependent differences in the kinetics of R1 and R2 changes raises two important questions: 1) are the observed differences in reaction constants due to the reaction being limited by the formation of oxidized reactive groups (i.e. the efficiency of reduction of oxidized enzymes) and 2) are the observed results related to variations in substrate I and substrate II generated products. In order to address these questions, we determined the kinetics of the enzyme-catalyzed reactions. An analysis of reaction products and the resultant protein reaction mixtures suggests that the reaction products of HRP reduction were not rate-limiting, whereas MPO generated oxidized intermediates at a much slower rate that could be rate limiting for substrate II- mediated rise in relaxivity of the reaction products. In addition we found that the products of HRP vs. MPO reactions were entirely different. While enzyme-mediated oxidation of both substrate I and II resulted in products that showed binding to plasma proteins (Table 3), only MPO oxidized substrate I resulted in detectable cross-linking of GOX which was used as a source of H2O2 in the reaction mixture (Fig. 4). The significance of this finding lies in the fact that cross-linking as opposed to formation of smaller paramagnetic oligomers can potentially result in higher relaxivity increase per reacting Gd-labeled paramagnetic chelate due to the effect of Gd immobilization and the resultant increase in rotational correlation time 23, 24. The formation of such products in blood plasma protein solutions will likely depend on the number of exposed tyrosines and the degree of protein solvation and will require further investigation.
In vivo experiments performed after implantation of MPO positive and MPO negative Matrigel mixtures into the flanks of mice demonstrated that MR signal enhancement on T1-W images was substantially long-lasting in MPO-containing muscle implants. This is likely due to retention of paramagnetic MPO generated reaction products which results from the cross linking of proteins in the Matrigel matrix as characterized by us previously 2, 17, 25. Using bis-5HT-DTPA as the substrate, Nahrendorf et. al. 9 and Chen et. al. 2 observed similar results in vivo in injured myocardium and in extremities implants, respectively, where the wash-in CNR curve reached a maximum at around the 1-hr time point, and was still present after 200 minutes 2. In the current study, we observed an increase in the MR CNR during wash in of bis-HTrp-DTPA(Gd), which reached a maximum during the first hour, and then a gradual decrease (washout) of CNR over the next 5 days. This may be due to a combined effect of less efficient oligomerization of substrate I than II in the presence of MPO and the solubility of the resultant oligomers.
Conclusions
The synthesis of bis-HTrp-DTPA(Gd) resulted in a new MPO sensitive MR imaging probe that produced an increase of Gd-induced proton relaxation rates similar to that of bis-5HT-DTPA(Gd). The new probe has high solubility in aqueous media at room temperature making it preferable for applications requiring higher in vivo probe concentrations (i.e. mouse imaging at higher magnetic fields, reviewed in 26). Kinetic experiments demonstrated that although both MPO and HRP reacted with both substrates there were notable differences in terms of the type of reaction products formed: substrate II and HRP gave insoluble cross-linked final products if the reaction proceeded for several hours while the products of substrate I were soluble. Gradient SDS-PAGE analysis of enzymatic activation of substrate I showed the formation of a 10–20 kDa oligomerized product in the case of HRP, while in the reaction mixture containing MPO we observed the formation of cross-linked proteins. The binding of the reactive products after the substrate activation by MPO was also detected in the presence of plasma proteins. This suggests that protein binding in the case of substrate I reaction with MPO can potentially compensate for the lack of extensive oligomerization. Despite lower stability of substrate II, especially under the conditions modeling tissue acidosis we anticipate it to be an efficient sensor for in vivo imaging of MPO activity due to potential linking of the corresponding reaction products to tissue proteins in inflammatory lesions.
Acknowledgments
This work was supported in part by R01EB000858 and R01AG034901 (A.B.). The authors are grateful to Dr. Mary Mazzanti for editing the text and Jaime O’Callaghan, AS for her assistance with animal experiments.
Abbreviations
- 5HT
5-hydroxytryptamide
- HTrP
5-hydroxytryptophan
- Tyr
tyramide
- MPO
myeloperoxidase
- HRP
horseradish peroxidase
- GOX
glucose oxidase
- MRI
magnetic resonance imaging
References
- 1.Chen JW, Pham W, Weissleder R, et al. Human myeloperoxidase: a potential target for molecular MR imaging in atherosclerosis. Magn Reson Med. 2004;52:1021–8. doi: 10.1002/mrm.20270. [DOI] [PubMed] [Google Scholar]
- 2.Chen JW, Querol Sans M, Bogdanov A, Jr, et al. Imaging of myeloperoxidase in mice by using novel amplifiable paramagnetic substrates. Radiology. 2006;240:473–81. doi: 10.1148/radiol.2402050994. [DOI] [PubMed] [Google Scholar]
- 3.Ronald JA, Chen JW, Chen Y, et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation. 2009;120:592–9. doi: 10.1161/CIRCULATIONAHA.108.813998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross S, Gammon ST, Moss BL, et al. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. 2009;15:455–61. doi: 10.1038/nm.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeLeo MJ, 3rd, Gounis MJ, Hong B, et al. Carotid artery brain aneurysm model: in vivo molecular enzyme-specific MR imaging of active inflammation in a pilot study. Radiology. 2009;252:696–703. doi: 10.1148/radiol.2523081426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. New England J Med. 2003;349:1595–604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama S, Okada Y, Sukhova GK, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Amer J Pathol. 2001;158:879–91. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 9.Nahrendorf M, Sosnovik D, Chen JW, et al. Activatable magnetic resonance imaging agent reports myeloperoxidase activity in healing infarcts and noninvasively detects the antiinflammatory effects of atorvastatin on ischemia-reperfusion injury. Circulation. 2008;117:1153–60. doi: 10.1161/CIRCULATIONAHA.107.756510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Querol M, Chen JW, Weissleder R, et al. DTPA-bisamide-based MR sensor agents for peroxidase imaging. Org Lett. 2005;7:1719–22. doi: 10.1021/ol050208v. [DOI] [PubMed] [Google Scholar]
- 11.Querol M, Chen JW, Bogdanov A., Jr A paramagnetic contrast agent with myeloperoxidase-sensing properties. Org Biomol Chem. 2006;4:1887–95. doi: 10.1039/b601540a. [DOI] [PubMed] [Google Scholar]
- 12.Allegra M, Furtmuller PG, Regelsberger G, et al. Mechanism of reaction of melatonin with human myeloperoxidase. Biochem Biophys Res Commun. 2001;282:380–6. doi: 10.1006/bbrc.2001.4582. [DOI] [PubMed] [Google Scholar]
- 13.Bogdanov A, Jr, Matuszewski L, Bremer C, et al. Oligomerization of paramagnetic substrates result in signal amplification and can be used for MR imaging of molecular targets. Mol Imaging. 2002;1:16–23. doi: 10.1162/15353500200200001. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez E, Nilges M, Weissleder R, et al. Activatable magnetic resonance imaging agents for myeloperoxidase sensing: mechanism of activation, stability, and toxicity. J Am Chem Soc. 2009;132:168–77. doi: 10.1021/ja905274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Betten A, Dahlgren C, Hermodsson S, et al. Serotonin protects NK cells against oxidatively induced functional inhibition and apoptosis. J Leukoc Biol. 2001;70:65–72. [PubMed] [Google Scholar]
- 16.Laurent S, Vander Elst L, Henoumont C, et al. How to measure the transmetallation of a gadolinium complex. Contrast Media Mol Imaging. 2010;5:305–8. doi: 10.1002/cmmi.388. [DOI] [PubMed] [Google Scholar]
- 17.Shazeeb MS, Sotak CH, Deleo M, 3rd, et al. Targeted signal amplifying enzymes enhance magnetic resonance imaging of EGRF expression in an orthotopic model of human glioma. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-10-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornish-Bowden A. Analysis of enzyme kinetic data. Oxford Univ Press; 1995. [Google Scholar]
- 19.Ghibaudi E, Laurenti E. Unraveling the catalytic mechanism of lactoperoxidase and myeloperoxidase. Eur J Biochem. 2003;270:4403–12. doi: 10.1046/j.1432-1033.2003.03849.x. [DOI] [PubMed] [Google Scholar]
- 20.Breckwoldt MO, Chen JW, Stangenberg L, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584–9. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JW, Breckwoldt MO, Aikawa E, et al. Myeloperoxidase-targeted imaging of active inflammatory lesions in murine experimental autoimmune encephalomyelitis. Brain. 2008;131:1123–33. doi: 10.1093/brain/awn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Querol Sans M, Chen JW, Weissleder R, et al. Myeloperoxidase activity imaging using (67)Ga labeled substrate. Mol Imaging Biol. 2005;7:403–10. doi: 10.1007/s11307-005-0020-5. [DOI] [PubMed] [Google Scholar]
- 23.Toth E, Helm L, Merbach AE. Relaxivity of MRI contrast agents. Topics Curr Chem. 2002;221:61–101. [Google Scholar]
- 24.Caravan P, Cloutier NJ, Greenfield MT, et al. The Interaction of MS-325 with Human Serum Albumin and Its Effect on Proton Relaxation Rates. J Am Chem Soc. 2002;124:3152–62. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 25.Querol M, Bogdanov A., Jr Amplification strategies in MR imaging: activation and accumulation of sensing contrast agents (SCAs) J Magn Reson Imaging. 2006;24:971–82. doi: 10.1002/jmri.20724. [DOI] [PubMed] [Google Scholar]
- 26.Sosnovik DE, Nahrendorf M, Weissleder R. Targeted imaging of myocardial damage. Nat Clin Pract Cardiovasc Med. 2008;5 (Suppl 2):S63–70. doi: 10.1038/ncpcardio1115. [DOI] [PMC free article] [PubMed] [Google Scholar]