

Abstract
Purpose.
To evaluate the effect of lysosomal destabilization on NLRP3 inflammasome activation in RPE cells and to investigate the mechanisms by which inflammasome activation may contribute to the pathogenesis of age-related macular degeneration (AMD).
Methods.
Human ocular tissue sections from patients with geographic atrophy or neovascular AMD were stained for NLRP3 and compared to tissues from age-matched controls. Expression of the IL-1β precursor, pro-IL-1β, was induced in ARPE-19 cells by IL-1α treatment. Immunoblotting was performed to assess expression of NLRP3 inflammasome components (NLRP3, ASC, and procaspase-1) and pro-IL-1β in ARPE-19 cells. Lysosomes were destabilized using the lysosomotropic agent L-leucyl-L-leucine methyl ester (Leu-Leu-OMe). Active caspase-1 was detected using FAM-YVAD-FMK, a fluorescent-labeled inhibitor of caspases (FLICA) specific for caspase-1. IL-1β was detected by immunoblotting and ELISA, and cytotoxicity was evaluated by LDH quantification.
Results.
RPE of eyes affected by geographic atrophy or neovascular AMD exhibited NLRP3 staining at lesion sites. ARPE-19 cells were found to express NLRP3, ASC, and procaspase-1. IL-1α dose-dependently induced pro-IL-1β expression in ARPE-19 cells. Lysosomal destabilization induced by Leu-Leu-OMe triggered caspase-1 activation, IL-1β secretion, and ARPE-19 cell death. Blocking Leu-Leu-OMe–induced lysosomal disruption with the compound Gly-Phe-CHN2 or inhibiting caspase-1 with Z-YVAD-FMK abrogated IL-1β release and ARPE-19 cytotoxicity.
Conclusions.
NLRP3 upregulation occurs in the RPE during the pathogenesis of advanced AMD, in both geographic atrophy and neovascular AMD. Destabilization of RPE lysosomes induces NLRP3 inflammasome activation, which may contribute to AMD pathology through the release of the proinflammatory cytokine IL-1β and through caspase-1-mediated cell death, known as “pyroptosis.”
Several lines of evidence have suggested that inflammation plays a role in the pathogenesis of AMD. This study demonstrates that the destabilization of RPE lysosomes activates the NLRP3 inflammasome, a multiprotein complex that regulates inflammatory cytokine maturation.
Introduction
Age-related macular degeneration (AMD) is the leading cause of blindness affecting the elderly in industrialized nations.1,2 The hallmark of AMD is the appearance of drusen between the retinal pigment epithelium (RPE) and Bruch's membrane (BM) and within BM.3 Greater size, number, and confluency of drusen are risk factors for advanced AMD, but their origin and precise contribution to AMD etiology are unclear.4,5 AMD is also characterized by the accumulation of lipofuscin in RPE lysosomes. Lipofuscin is a collection of autofluorescent substances, such as the bisretinoid A2E, that are believed to be nondegradable compounds from phagocytosed photoreceptor outer segment discs.6
Despite intensive study of the changes that occur during AMD progression, the pathogenesis underlying AMD has not been elucidated; however, several lines of evidence indicate a role for inflammation. Drusen contain a number of proteins associated with inflammation,5 and polymorphisms in certain complement pathway genes are correlated with increased risk of developing AMD.7,8 Additionally, autoantibodies against retinal antigens have been found in the sera of AMD patients.9
In light of the evidence for inflammation in AMD pathogenesis, we and others10–13 have hypothesized a role for the NLRP3 inflammasome in AMD. Inflammasomes are a class of multiprotein complexes that activate caspase-1 by facilitating the cleavage of its zymogen precursor, procaspase-1.14,15 Caspase-1 catalyzes the proteolytic maturation of the proinflammatory cytokines IL-1β and IL-18, which are synthesized as the inactive, cytosolic precursors pro-IL-1β and pro-IL-18, respectively. Upon cleavage, the mature cytokines are secreted.16 Pro-IL-1β expression is a prerequisite for production of mature IL-1β and can be achieved by “priming” cells with a proinflammatory stimulus that activates the IL1B promoter via nuclear factor kappa B (NF-κB) signaling.14,17,18 A second signal, usually associated with the presence of pathogens or host tissue damage, activates inflammasome assembly.
Assembly of the NLRP3 inflammasome involves interaction of the scaffolding protein NLRP3 with the adaptor protein “apoptosis-associated speck-like protein containing a caspase-recruitment domain” (ASC), which in turn recruits procaspase-1 into the complex, leading to its autocatalytic conversion into active caspase-1.19 The NLRP3 inflammasome can be activated by a wide array of structurally diverse stimuli.14 Many of these substances activate NLRP3 by destabilizing lysosomes. Crystalline or insoluble materials such as cholesterol crystals20,21 and fibrillar amyloid-β22 activate NLRP3 in phagocytic myeloid-derived cells by disrupting phagolysosomes following internalization. Additionally, chemical agents that permeabilize the lysosomal membrane activate the NLRP3 inflammasome.23
While NLRP3 has been studied primarily in myeloid cells, some epithelia have also been shown to exhibit NLRP3 inflammasome activity.24–27 More recent work indicates that RPE cells also express the NLRP3 inflammasome.13 Given that lysosomal destabilization activates NLRP3 in myeloid cells, we evaluated whether it also triggers NLRP3 inflammasome activation in RPE cells. A number of AMD-associated insults may disrupt RPE lysosomes. For example, the lipofuscin component A2E has been shown to permeabilize RPE lysosomes.28,29 Furthermore, drusen and individual drusen components activate NLRP3 in myeloid cells by damaging phagolysosomes.12,22 We also assessed potential mechanisms by which inflammasome activity could lead to RPE degeneration, focusing on the release of the potent proinflammatory cytokine IL-1β and the occurrence of a form of inflammasome-mediated programmed cell death called pyroptosis, which is dependent on caspase-1 rather than apoptotic caspases. Because IL-1β secretion requires induction of pro-IL-1β expression via priming, we evaluated the effects of priming on expression of pro-IL-1β and inflammasome components, and then assessed the effects of lysosomal destabilization on primed RPE cells.
Materials and Methods
Immunohistochemistry of Human Retina
De-identified specimens from AMD and control subjects were obtained from a tissue repository established in one of our laboratories at Schepens Eye Research Institute under Institutional Review Board approval and in accordance with the ethical standards of the Declaration of Helsinki. Specimens were fixed in 10% buffered formalin, embedded in paraffin, and sectioned. For immunohistochemistry, sections were deparaffinized, and antigen retrieval was performed with citrate buffer (10 mM citric acid, 0.05% Tween-20, pH 6.0, 95–100°C) for 10 minutes. Following two washes in phosphate-buffered saline (PBS; Sigma-Aldrich, St. Louis, MO), slides were incubated with a mouse monoclonal anti-human NLRP3 antibody (1:100; clone Nalpy3-b; Enzo Life Sciences, Farmingdale, NY) or a mouse IgG1 isotype control antibody (1:100; Caltag, Carlsbad, CA) overnight at 4°C. The secondary antibody, a biotinylated horse anti-mouse IgG (1:200; Vector Laboratories, Burlingame, CA), was visualized via the avidin-biotin-alkaline phosphatase complex (ABC-AP) method (Vectastain ABC-AP Kit; Vector Laboratories) using the Vector Red Substrate (Vector Laboratories). Slides were counterstained with hematoxylin, dehydrated, and mounted with Permount medium (Fisher Scientific, Pittsburgh, PA). For positive controls, human conjunctival tissue corresponding to each eye was also stained with the same antibodies. Uniform expression for NLRP3 was detected (data not shown).
ARPE-19 Cell Culture
Human ARPE-19 cells (American Type Culture Collection, Manassas, VA) were propagated as described previously.30 Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 medium (Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 2 mM L-glutamine (Lonza), and 100 U/mL penicillin–100 μg/mL streptomycin (Lonza) (complete media) and passaged at a ratio of 1:2 to 1:4 using trypsin-versene (Lonza). For experiments, cells were maintained in either 1% FBS or serum-free medium, and transfections were performed in antibiotic-free medium.
Immunocytochemistry
Immunocytochemistry was performed on ARPE-19 cells grown on transwell membranes to allow subcellular localization of NLRP3 in polarized cells. Cells were cultured on transwell membranes for 4 weeks in 1% FBS medium, as described elsewhere,30 to induce RPE polarization and tight junction formation. Monolayers were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), then incubated with a mouse monoclonal anti-human NLRP3 primary antibody (1:100, clone Nalpy3-b; Enzo Life Sciences) or a mouse IgG isotype control antibody (1:100; Invitrogen, Carlsbad, CA) overnight at 4°C. The secondary antibody, a biotinylated horse anti-mouse IgG (1:200; Vector Laboratories), was visualized via the ABC-AP method (Vectastain ABC-AP Kit; Vector Laboratories) using the Vector Red Substrate. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). Transwell membranes with attached monolayers were excised from their supports and mounted on glass slides for confocal microscopy (Leica Microsystems, Wetzlar, Germany).
Priming of ARPE-19 Cells with NF-κB–Inducing Agents
ARPE-19 cells were seeded into 12-well plates (BD Biosciences, San Jose, CA) at a density of 2.6 × 105 cells/well in complete media. At confluence, the culture medium was changed to serum-free, and cells were treated with ultrapure lipopolysaccharide (LPS) from Escherichia coli 0111:B4 strain (InvivoGen, San Diego, CA), recombinant human IL-1α (R&D Systems, Minneapolis, MN), or recombinant human TNFα (PeproTech, Rocky Hill, NJ). Each agent was tested at 4 ng/mL or 50 ng/mL for 24 or 48 hours. Whole cell lysates were immunoblotted for pro-IL-1β.
NLRP3 Knockdown
ARPE-19 cells were grown as described above and seeded into six-well plates (BD Biosciences) at a density of 1.5 × 105 cells/well in antibiotic-free media. The following day, at approximately 40% confluence, cells were transfected with ON-TARGETplus SMARTpool siRNA against human NLRP3 or a nontargeting control SMARTpool (Dharmacon, Lafayette, CO). The target sequences of the siRNAs in each pool are provided in the Table. The control siRNA pool was transfected at a total siRNA concentration of 100 nM using DharmaFECT 4 (Dharmacon), and the NLRP3 siRNA pool was used at 50 nM and 100 nM. At 24 hours posttransfection, cells were washed once with PBS, and complete medium containing 4 ng/mL IL-1α was added to the wells to prime the cells. Cells were lysed at 72 hours posttransfection and immunoblotted for NLRP3.
Table. .
Target Sequences of siRNAs Used in This Study
|
siRNA Pool |
Target Sequences |
| NLRP3 | No. 1: 5′-GCAAGACCAAGACGUGUGA-3′ |
| No. 2: 5′-GAAGUGGGGUUCAGAUAAU-3′ | |
| No. 3: 5′-UGCAAGAUCUCUCAGCAAA-3′ | |
| No. 4: 5′-GGAUCAAACUACUCUGUGA-3′ | |
| Control | No. 1: 5′-UGGUUUACAUGUCGACUAA-3′ |
| No. 2: 5′-UGGUUUACAUGUUGUGUGA-3′ | |
| No. 3: 5′-UGGUUUACAUGUUUUCUGA-3′ | |
| No. 4: 5′-UGGUUUACAUGUUUUCCUA-3′ |
Pooled siRNA against NLRP3 and a nontargeting control siRNA pool were obtained from Dharmacon. Each pool contains four siRNAs, whose target sequences are presented.
NLRP3 Overexpression and Immunoblotting
HEK293T lysates overexpressing the full-length transcript variant of NLRP3 fused to a DDK tag (identical to FLAG, registered trademark of Sigma-Aldrich) and myc tag or transfected with a mock vector were purchased from OriGene (Rockville, MD). Immunoblotting for NLRP3 was performed on ARPE-19 and THP-1 lysates, using the NLRP3- and mock-transfected HEK293T lysates as controls. Blotting for NLRP3 was followed by stripping and reprobing first for α-tubulin and secondly for DDK.
Effect of Priming on Expression of Pro-IL-1β and Inflammasome Components
ARPE-19 cells were seeded into six-well plates at a density of 4.0 × 105 cells/well in complete media. At confluence, the cells were switched to serum-free medium. Recombinant human IL-1α was used for priming. For the dose curve, IL-1α was added to cells at concentrations of 1.56, 3.13, 6.25, 12.5, and 25 ng/mL IL-1α and incubated for 48 hours. For the time course, 4 ng/mL IL-1α was added to wells, and cells were lysed after 3, 6, 12, 24, or 48 hours. Cells were washed with ice-cold PBS, lysed, and subjected to immunoblotting for pro-IL-1β, NLRP3, ASC, or caspase-1.
Acridine Orange Staining of RPE Lysosomes
ARPE-19 cells were seeded on sterile coverslips placed in a six-well plate at a density of 3.0 × 105 cells/well in complete media. The following day, cells were incubated with complete medium containing 5 μM acridine orange for 30 minutes. Cells were washed twice with PBS, then treated with 1 mM L-leucyl-L-leucine methyl ester (Leu-Leu-OMe; Chem-Impex International, Wood Dale, IL) or control buffer for 30 to 45 minutes. Cells were then fixed with 4% paraformaldehyde for 30 minutes and washed three times with PBS. Coverslips were then mounted on glass slides using Vectashield Mounting Medium for Fluorescence (Vector Laboratories) and imaged on an Axioskop 2 mot plus fluorescent microscope (Carl Zeiss, Thornwood, NY).
Fluorescent Detection of Active Caspase-1
ARPE-19 cells were seeded into 24-well plates (BD Biosciences) at a density of 5.0 × 104 cells/well in complete medium, grown to confluence, and then changed to serum-free medium with 4 ng/mL IL-1α. After 48 hours, wells were pretreated with the dipeptidyl peptidase I inhibitor Gly-Phe-CHN2 (MP Biomedicals, Solon, OH) at a concentration of 10 μM, the cathepsin B and L inhibitor Z-FF-FMK (EMD Biosciences, San Diego, CA) at a concentration of 50 μM, or an equal volume of dimethyl sulfoxide (DMSO) vehicle. After 30 minutes, the fluorescent-labeled inhibitor of caspases (FLICA) probe specific for caspase-1 (FAM-YVAD-FMK; Immunochemistry Technologies, Bloomington, MN) was added to each well at the concentration recommended by the manufacturer, followed by the addition of 1 mM Leu-Leu-OMe. After a 2-hour incubation at 37°C, 5% CO2, cell nuclei were stained with Hoechst 33342 (Immunochemistry Technologies). Cells were imaged using a Nikon Eclipse TE2000-S microscope. Quantification of green FLICA signal was performed using Adobe Photoshop (Adobe, San Jose, CA). Blue nuclei in each image were counted manually, and the amount of green signal was normalized to the number of nuclei.
Immunoblotting for Mature IL-1β in Concentrated ARPE-19 Conditioned Media
ARPE-19 cells grown to confluence in T75 flasks were primed with 15 ng/mL IL-1α for 48 hours, then treated with 1 mM Leu-Leu-OMe or control buffer for 3 hours. Conditioned media were harvested and concentrated using 15 mL Amicon centrifugal filter units (Millipore, Billerica, MA) by spinning in an Avanti J-25I centrifuge (Beckman Coulter, Indianapolis, IN) using a JA25.50 rotor (Beckman Coulter) at 5000g for 1 hour at room temperature. Following concentration, conditioned media were immunoblotted for IL-1β alongside a standard dilution of 0, 25, 50, 100, and 200 pg recombinant human mature IL-1β (National Cancer Institute, Rockville, MD) as positive control. Goat anti-IL-1β (R&D Systems) was used as primary antibody at a 1:200 dilution, and horseradish peroxidase (HRP)-linked rabbit anti-goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA) secondary antibody was used at a 1:5000 dilution.
Quantification of IL-1β Secretion and Cytotoxicity
ARPE-19 cells were seeded onto 12-well plates at a density of 1.0 × 105 cells/well in complete media, grown to confluence, and then changed to serum-free medium with 4 ng/mL IL-1α. After 48 hours, Gly-Phe-CHN2 (5 μM), Z-FF-FMK (50 μM), the caspase-1 inhibitor Z-YVAD-FMK (10 μM; BioVision, Mountain View, CA), or DMSO vehicle was added. After 30 minutes, 1 mM Leu-Leu-OMe was added to appropriate cells. Conditioned media were collected after 3 hours.
IL-1β was quantified via ELISA (BD Biosciences), and cytotoxicity was assessed by measuring lactate dehydrogenase (LDH) in conditioned media using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI). Each experimental condition was assayed in triplicate in three independent experiments. Percent LDH release was calculated as 100% × (experimental LDH − spontaneous LDH) / (maximum LDH − spontaneous LDH). Maximum LDH was represented by the LDH levels in wells completely lysed by two freeze–thaw cycles.
Immunoblot Analysis of Whole Cell Lysates
Cells were treated with lysis buffer (Cell Signaling Technology, Danvers, MA) containing a dissolved Complete Mini EDTA-free Protease Inhibitor Cocktail Tablet (Roche, Indianapolis, IN) and 2 mM phenylmethanesulfonyl fluoride (PMSF). Protein concentration was measured via the BCA assay (Thermo Scientific, Waltham, MA). Equal quantities of protein were separated via SDS-PAGE under reducing conditions and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked overnight at 4°C. NLRP3 blots were blocked in Tris-buffered saline with 0.1% Tween (TBS-T) containing 5% (w/v) milk and 0.5% (w/v) BSA; all other blots were blocked in 5% milk in TBS-T.
Membranes were then incubated in primary antibody diluted 1:1000 in their respective blocking solution for 2 hours at room temperature. Primary antibodies used were mouse anti-NLRP3 (Nalpy3-b; Enzo Life Sciences), rabbit anti-caspase-1 (Cell Signaling Technology), goat anti-IL-1β (R&D Systems), and rabbit anti-ASC (Enzo Life Sciences). The caspase-1 and IL-1β antibodies also recognize their uncleaved precursors. After three 10-minute washes in TBS-T, membranes were incubated for 1 hour at room temperature in secondary antibody diluted 1:10,000 in blocking buffer. HRP-linked secondary antibodies included sheep anti-mouse IgG (GE Healthcare, Pittsburgh, PA), donkey anti-rabbit IgG (GE Healthcare), and donkey anti-goat IgG (Santa Cruz Biotechnology). Following four more washes in TBS-T, proteins were visualized by enhanced chemiluminescence using SuperSignal substrates (Thermo Scientific). Membranes were stripped by incubation in 62.5 mM Tris-HCl (pH 6.8), 2% (w/v) SDS, and 0.1 M β-mercaptoethanol for 30 minutes at 55°C to 60°C; reblocked overnight at 4°C in 5% BSA in TBS-T; and reprobed with rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology) to evaluate loading. NLRP3 blots were reprobed with mouse anti-α-tubulin (EMD Biosciences). To detect DDK-tagged NLRP3, blots were reprobed using mouse anti-DDK tag (OriGene).
Statistical Analysis
Data are presented as the mean ± SEM of three independent experiments. To evaluate statistical significance, one-way analysis of variance was performed, followed by the Tukey-Kramer multiple comparisons test using the Prism 4 software package (GraphPad, La Jolla, CA). A P value of <0.05 was considered statistically significant.
Results
NLRP3 Is Present in RPE and Drusen of Patients with AMD
We first sought to determine whether NLRP3 is expressed by human RPE in vivo, and whether there are qualitative differences between its expression in patients with AMD, in the form of geographic atrophy (GA) or neovascular AMD, and age-matched individuals without AMD. Outer retinal sections were collected from two donors with GA, two donors with neovascular AMD, and three donors unaffected by AMD. The sections were stained with an antibody against NLRP3 or an isotype control (Fig. 1). NLRP3 (red, arrowheads) was detected in the RPE of eyes affected by either GA or neovascular AMD; there was no staining in sections from the same donors with the control antibody. Several sections from GA-affected eyes contained drusen within the transitional zone, which exhibited substantial extracellular NLRP3 staining both in choroidal cells behind BM and on the basal side of RPE cells adjacent to areas of drusen (Figs. 1A, 1B). NLRP3 was also detected extracellularly in the vicinity of thickened BM (Fig. 1D) and basal linear deposits (Fig. 1E) in eyes with neovascular AMD outside the area of choroidal neovascularization as well as in basal RPE (Fig. 1D). NLRP3 was detected only at sites of GA or neovascular lesions in AMD eyes and was not observed in the RPE of lesion-free areas of the retina. In contrast to AMD-affected eyes, outer retinal sections from eyes of age-matched donors unaffected by AMD did not exhibit detectable NLRP3 staining.
Figure 1. .
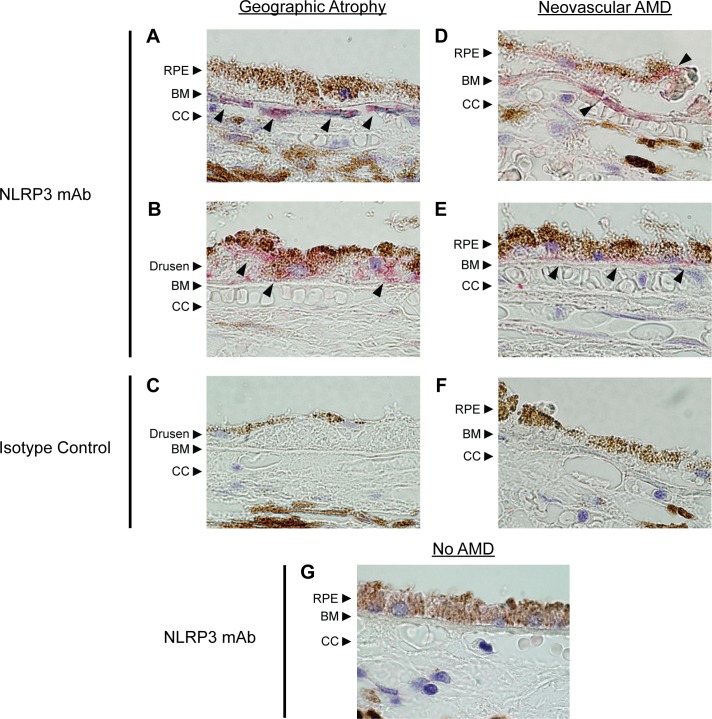
Expression of NLRP3 in AMD-affected eyes. Retinas from donors with GA (A–C), neovascular AMD (D–F), or unaffected eyes (G) were stained for NLRP3. NLRP3 (red, arrowheads) was detected in the RPE of eyes affected with either form of AMD. (A, B) Representative sections of the transitional zone in eyes with GA show areas of staining in the choroidal cells just behind BM ([A], arrowheads), as well as in the basal side of the RPE adjacent to confluent drusen ([B], arrowheads). Certain drusen deposits in GA eyes also showed NLRP3 staining. (D, E) Representative sections just outside the area of choroidal neovascularization show staining in the vicinity of thickened BM as well as in basal RPE ([D], arrowheads). NLRP3 staining was also seen within the thickened BM representing basal linear deposits ([E], arrowheads). There was no detectable signal in sections from the same donors stained with an isotype control (C, F). A representative section from age-matched donors unaffected by AMD (G) did not exhibit any NLRP3 expression. CC, choriocapillaris.
NLRP3 Is Expressed in Human RPE Cells In Vitro
Motivated by the observation of NLRP3 in RPE of AMD patients, we investigated NLRP3 expression in RPE cells in vitro using the human cell line ARPE-19. ARPE-19 cells were cultured on transwell membranes for 4 weeks to allow them to polarize and form tight junctions, characteristics of differentiated RPE in vivo. Immunocytochemical localization revealed NLRP3 distributed in a punctate pattern throughout the cells (Fig. 2A). Confocal microscopy indicated that NLRP3 was preferentially localized toward the basal side of the cells (Fig. 2A).
Figure 2. .
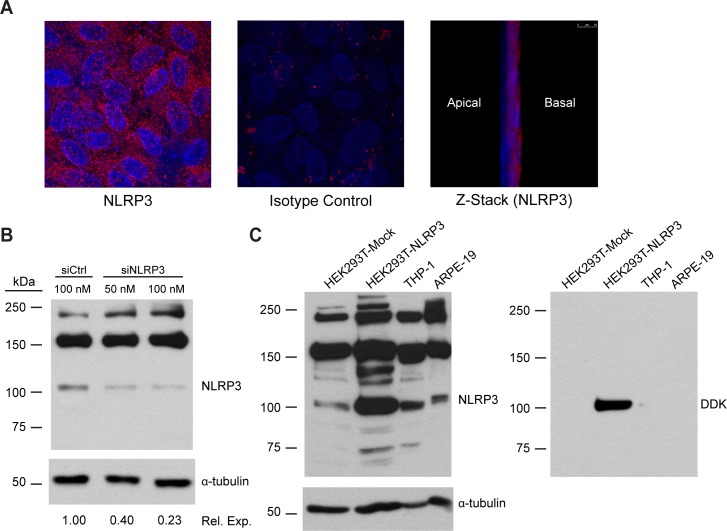
Expression of NLRP3 in human RPE cells in vitro. (A) ARPE-19 cells were cultured on transwell membranes for 4 weeks to form a polarized monolayer. Monolayers were fixed, permeabilized, and stained for NLRP3 (red) and with DAPI to reveal nuclei (blue). Z-stacks of polarized monolayers were generated using confocal microscopy. (B) ARPE-19 cells grown on plastic wells were transfected with a siRNA pool against human NLRP3 or a control siRNA pool. ARPE-19 lysates were subjected to immunoblotting for NLRP3, with α-tubulin as a loading control. Densitometry was performed using NIH ImageJ. Tubulin-normalized NLRP3 band densities are presented as their value relative to the control. (C) ARPE-19 cells were compared to a HEK293T cell lysate overexpressing DDK-tagged NLRP3 (HEK293T-NLRP3) and a mock-transfected HEK293T lysate (HEK293T-Mock) to identify the NLRP3 band. THP-1 cells served as a reference for NLRP3 levels in a cell type with known NLRP3 inflammasome activity. Lysates were immunoblotted for NLRP3, with α-tubulin as a loading control, and reprobed for the DDK tag fused to the NLRP3 overexpressed in HEK293T cells.
We then evaluated NLRP3 expression in ARPE-19 cells by immunoblotting (Fig. 2B). ARPE-19 cells on plastic wells were transfected with a pool of four siRNAs against NLRP3 (siNLRP3) or a nontargeting control siRNA pool in order to validate that the detected band was indeed NLRP3. The Table lists the target sequences of the siRNAs in each pool. A band migrating slightly above 100 kDa, which is consistent with the predicted molecular weights of known NLRP3 isoforms, was detected in the control-transfected cells and was reduced by 77% using siNLRP3 (Fig. 2B).31 We further confirmed the identity of this band as NLRP3 using a lysate of HEK293T cells overexpressing NLRP3 tagged with DDK (identical to FLAG). The ∼100 kDa ARPE-19 band comigrates with a protein that is heavily enriched in the NLRP3-overexpressing lysate compared to mock-transfected HEK293T lysate (Fig. 2C). A band at this molecular weight is also present in the human monocytic leukemia cell line THP-1, which has been demonstrated to express NLRP3 and is known to have NLRP3 inflammasome activity.21,32,33 Reprobing with an anti-DDK tag antibody revealed that the enriched band in the NLRP3-overexpressing HEK293T lysate was indeed NLRP3, which demonstrated the presence of NLRP3 in ARPE-19 cells.
Priming of RPE Cells with NF-κB Agonists Induces Pro-IL-1β Expression
We next sought to “prime” ARPE-19 cells to express pro-IL-1β, a classical inflammasome substrate. In myeloid cells, pro-IL-1β expression is induced by priming cells with a NF-κB–activating stimulus such as LPS.14,18 To determine if NF-κB agonists prime ARPE-19 cells, the cells were treated with LPS, IL-1α, or TNFα at concentrations of 4 ng/mL and 50 ng/mL for 24 or 48 hours. Immunoblotting of ARPE-19 lysates revealed that while almost no pro-IL-1β was detectable in the absence of NF-κB agonists, treatment with IL-1α or TNFα induced pro-IL-1β expression, with IL-1α the most potent agent tested (Fig. 3A). On the other hand, LPS did not upregulate pro-IL-1β under the conditions tested, but this is consistent with the markedly reduced levels of TLR4 in ARPE-19 compared to primary RPE.34
Figure 3. .
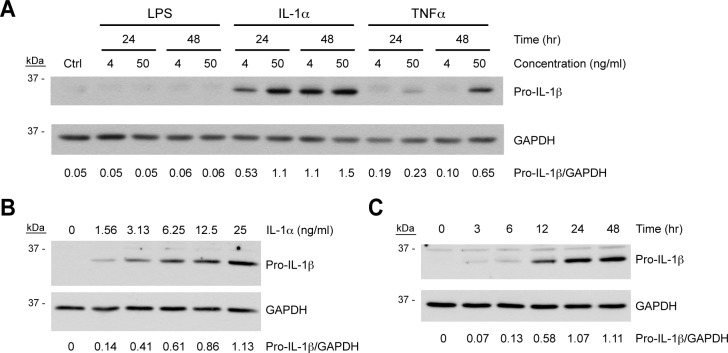
Priming of ARPE-19 cells by inducers of NF-κB. (A) ARPE-19 cells grown to confluence on plastic wells were treated with the NF-κB–activating agents LPS, IL-1α, or TNFα at a concentration of 4 ng/mL or 50 ng/mL for 24 or 48 hours, or were left untreated as a negative control (Ctrl). (B, C) Confluent ARPE-19 cells were treated with increasing concentrations of IL-1α for 48 hours (B) or 4 ng/mL IL-1α for increasing lengths of time (C). ARPE-19 cell lysates were immunoblotted for pro-IL-1β, after which blots were stripped and reprobed for GAPDH as a loading control. Densitometry was performed using NIH ImageJ, and GAPDH-normalized band intensities are displayed for each condition.
Due to its potency, we selected IL-1α for use in subsequent inflammasome experiments. To this end, we further evaluated the dose and time dependency of its induction of pro-IL-1β. Confluent ARPE-19 cells were treated with an IL-1α dose curve for 48 hours (Fig. 3B) or with 4 ng/mL IL-1α for a range of times up to 48 hours (Fig. 3C). Pro-IL-1β was induced by IL-1α in a dose-dependent manner, and treatment of ARPE-19 cells with 4 ng/mL IL-1α upregulated pro-IL-1β expression to a maximal level by 24 hours.
Expression of NLRP3 Inflammasome Components in Human RPE Cells
We next evaluated ARPE-19 cells for expression of the critical NLRP3 inflammasome components ASC and procaspase-1, and we assessed the effect of priming on NLRP3, ASC, and procaspase-1 expression. In addition to pro-IL-1β, priming can also upregulate other inflammasome components, including NLRP3, in some cell types such as mouse macrophages.35 ARPE-19 cells were primed with a dose curve of IL-1α ranging up to 25 ng/mL, and lysates were subjected to immunoblotting.
While priming upregulates NLRP3 levels in various other cell types, NLRP3 is present in ARPE-19 cells even in unprimed conditions, and its expression was largely unresponsive to IL-1α priming (Fig. 4A). The highest NLRP3 induction achieved was only 1.6-fold at 25 ng/mL IL-1α, the highest IL-1α dose used (Fig. 4A). Similarly, ASC was also expressed under basal conditions and was not dose-dependently upregulated by IL-1α priming (Fig. 4B). On the other hand, procaspase-1 was expressed under unprimed conditions but was further upregulated by priming in a dose-dependent manner, reaching a maximal induction of 5- to 6-fold when stimulated by 12.5 ng/mL IL-1α or higher (Fig. 4C). The time dependency of procaspase-1 upregulation was evaluated through treatment of ARPE-19 cells with 4 ng/mL IL-1α for a range of times up to 48 hours. Procaspase-1 levels increased throughout the time course, reaching a 5-fold induction at 48 hours (Fig. 4D).
Figure 4. .
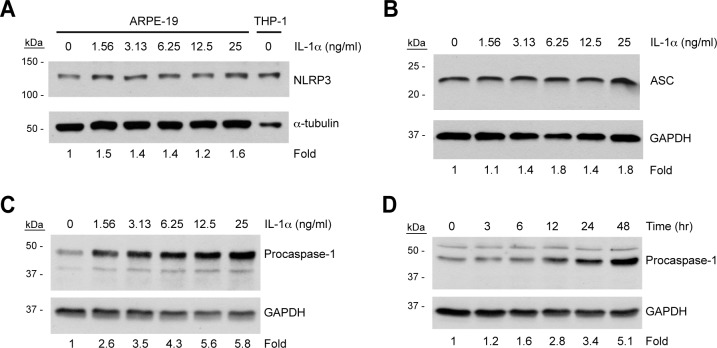
Effect of priming on expression of NLRP3 inflammasome components in human RPE cells. ARPE-19 cells were treated with increasing concentrations of IL-1α for 48 hours (A–C) or 4 ng/mL IL-1α for increasing lengths of time (D). Lysates were immunoblotted for NLRP3 (A), ASC (B), or procaspase-1 (C, D). All blots were stripped and reprobed for GAPDH as a loading control. Densitometry was performed using NIH ImageJ, and band intensities were normalized to GAPDH. Densitometry results are expressed as fold change compared to unprimed cells.
Lysosomal Destabilization in RPE Cells Activates Caspase-1
Since ARPE-19 cells expressed the components of the NLRP3 inflammasome, we tested whether the destabilization of their lysosomes would lead to inflammasome activation as measured by the activation of caspase-1, and if lysosomal enzymes could be involved in NLRP3 inflammasome activation. The lysosomotropic agent Leu-Leu-OMe, which is converted within the lysosome to a membranolytic derivative by dipeptidyl peptidase I (DPP-I),36 was used to disrupt lysosomes. Staining with acridine orange, which fluorescently labels DNA and RNA as green and lysosomes as red, was used to assess the effects of Leu-Leu-OMe on ARPE-19 lysosomal integrity (Fig. 5A). Whereas control ARPE-19 cells contained red punctate structures characteristic of intact lysosomes, treatment with 1 mM Leu-Leu-OMe triggered a loss of lysosomal staining, indicative of lysosomal destabilization.
Figure 5. .
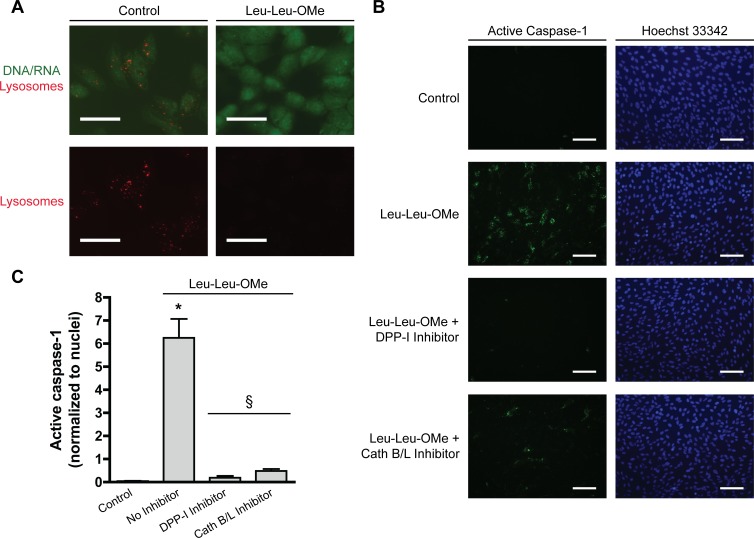
Lysosomal destabilization activates caspase-1 in ARPE-19 cells. (A) ARPE-19 cells were stained with 5 μM acridine orange for 30 minutes and treated for 30 to 45 minutes with 1 mM Leu-Leu-OMe or control buffer. Fluorescence microscopy was used to detect acridine orange sequestered in lysosomes (red) or bound to DNA or RNA (green). Scale bars, 50 μm. (B) ARPE-19 cells were primed with 4 ng/mL IL-1α for 48 hours. Cells were treated with 1 mM Leu-Leu-OMe for 2 hours to disrupt lysosomes, or left untreated (Control). Lysosomal destabilization induced by Leu-Leu-OMe was blocked by inhibiting DPP-I via addition of 10 μM Gly-Phe-CHN2 to cells 30 minutes before addition of Leu-Leu-OMe. The lysosomal proteases cathepsins B and L were inhibited using 50 μM Z-FF-FMK. Active caspase-1 was detected by the FLICA probe FAM-YVAD-FMK (green). Nuclei were labeled by staining with Hoechst 33,342 (blue). Scale bars, 100 μm. (C) Green active caspase-1 signal was quantified and normalized to number of nuclei. Numerical data are represented as mean ± SEM; n = 3. *P < 0.01 versus Control; §P < 0.01 versus Leu-Leu-OMe with no inhibitor.
Caspase-1 activation was evaluated using the FLICA probe FAM-YVAD-FMK that specifically labels active caspase-1. While no active caspase-1 was detected in untreated ARPE-19 cells, treatment with 1 mM Leu-Leu-OMe for 2 hours induced significant caspase-1 activation (Figs. 5B, 5C). To control for the possibility that effects of Leu-Leu-OMe unrelated to lysosomal destabilization were responsible for inflammasome activation, a DPP-I inhibitor (Gly-Phe-CHN2) was used to block the disruption of lysosomes by Leu-Leu-OMe. Addition of Gly-Phe-CHN2 30 minutes prior to Leu-Leu-OMe abrogated its ability to activate the inflammasome (Figs. 5B, 5C), supporting the conclusion that lysosomal destabilization triggers the NLRP3 inflammasome in ARPE-19 cells.
Previous reports demonstrate that leakage of lysosomal enzymes, particularly cathepsins B and L, following lysosomal destabilization plays a critical role in NLRP3 inflammasome induction in myeloid cell types.20,21,23 Therefore, the cathepsin B and L inhibitor Z-FF-FMK37,38 was used to assess the involvement of these lysosomal enzymes in NLRP3 activation in ARPE-19 cells. Pretreatment with Z-FF-FMK significantly inhibited activation of caspase-1 by Leu-Leu-OMe (Figs. 5B, 5C), indicating a role for these lysosomal enzymes in NLRP3 inflammasome induction in RPE cells.
Lysosomal Destabilization in RPE Cells Induces IL-1β Secretion and Cytotoxicity
We next investigated whether lysosomal disruption can induce secretion of mature IL-1β from primed cells. Specific detection of the mature, secreted form of IL-1β can be achieved by immunoblotting of conditioned media, as it clearly distinguishes between mature IL-1β (17 kDa) and its precursor (31 kDa) by virtue of their molecular weights. Although IL-1β ELISAs are much less sensitive for pro-IL-1β than for the mature form,39 pro-IL-1β released from dying cells can be mistaken for low levels of cleaved IL-1β. Immunoblotting demonstrated that Leu-Leu-OMe triggered the release of mature IL-1β from IL-1α-primed ARPE-19 cells that comigrated with recombinant human mature IL-1β (Fig. 6A).
Figure 6. .
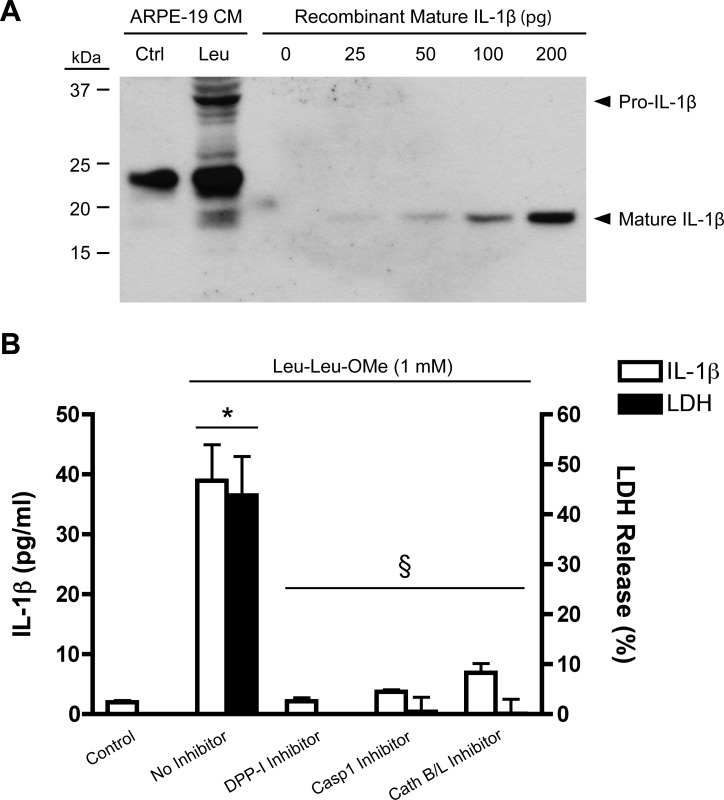
Human RPE cells secrete IL-1β and undergo pyroptotic cell death in response to lysosomal destabilization. (A) ARPE-19 cells primed with 15 ng/mL IL-1α for 48 hours were treated with 1 mM Leu-Leu-OMe (Leu) to disrupt lysosomes or received control buffer (Ctrl). After 3 hours, conditioned media (CM) were concentrated and immunoblotted to detect mature IL-1β (17 kDa) and distinguish it from its precursor (31 kDa). A serial dilution of 0 to 200 pg recombinant human IL-1β was used as reference. (B) IL-1β ELISA was performed on CM from ARPE-19 cells treated with 1 mM Leu-Leu-OMe. LDH release was quantified to evaluate cytotoxicity. The selective inhibitors Gly-Phe-CHN2 (5 μM), Z-YVAD-FMK (10 μM), and Z-FF-FMK (50 μM) were used to block the activity of DPP-I, caspase-1, and cathepsins B and L, respectively. Data represent mean ± SEM of three experiments. *P < 0.01 versus Control; §P < 0.01 versus Leu-Leu-OMe with no inhibitor.
After demonstrating that mature IL-1β is secreted by ARPE-19 cells treated with Leu-Leu-OMe, we used ELISA to quantify the amount of IL-1β secreted and to investigate the mechanisms involved. Leu-Leu-OMe induced the release of approximately 40 pg/mL IL-1β after 3 hours; this was completely blocked by DPP-I inhibition, indicating that IL-1β secretion was caused by lysosomal destabilization (Fig. 6B). The selective caspase-1 inhibitor Z-YVAD-FMK also significantly reduced IL-1β secretion, showing that the lysosomal damage-induced IL-1β release from ARPE-19 cells is mediated by caspase-1 and the inflammasome. Additionally, inhibition of cathepsins B and L using Z-FF-FMK blocked IL-1β secretion, supporting a role for these lysosomal enzymes in inflammasome activation in ARPE-19 cells.
Treatment with Leu-Leu-OMe also caused substantial cytotoxicity, as assessed by lactate dehydrogenase (LDH) release. ARPE-19 cells treated with Leu-Leu-OMe for 3 hours exhibited 40% to 50% cell death (Fig. 6B). Like IL-1β secretion, cytotoxicity was induced by lysosomal destabilization, as it was completely blocked by the DPP-I inhibitor, and was mediated by cathepsins B and/or L. Furthermore, the cell death caused by lysosomal disruption was found to be dependent on caspase-1, indicating an inflammasome-mediated death mechanism.
Discussion
The NLRP3 inflammasome, a regulator of mature IL-1β and IL-18 secretion, is a key mediator of the innate immune response. We detected NLRP3 protein in the RPE of donor human eyes affected by AMD, but not in eyes of age-matched controls. NLRP3 expression was associated with both GA and neovascular AMD and was detected intracellularly in the RPE as well as extracellularly in drusen and in the vicinity of BM, likely released from dying RPE cells. Studies in myeloid cells have shown that NF-κB–mediated priming signals induce the expression of both NLRP3 and pro-IL-1β,35 and we speculate that insults involved in the pathogenesis of AMD may induce these changes in RPE. It is unclear whether the association of NLRP3 expression with both GA and neovascular AMD is due to common or distinct events in the pathogenesis of these advanced forms of AMD.
Unlike normal RPE in vivo, ARPE-19 cells expressed NLRP3 without proinflammatory stimulation, likely due to culture conditions. However, unstimulated ARPE-19 cells did not express pro-IL-1β, indicating that the cells had not completely escaped the requirement for priming. Priming ARPE-19 cells with IL-1α induced pro-IL-1β expression and upregulated procaspase-1. Our finding that LPS did not induce pro-IL-1β expression in ARPE-19 cells is consistent with previous reports that primary RPE cells express the LPS receptor TLR4, whereas TLR4 is downregulated in ARPE-19 cells.34,40,41
It is not clear what molecule(s) might prime the RPE in the context of AMD. As IL-1β expression is downstream of NF-κB, any molecule that activates NF-κB is a potential priming agent.14,18 Although IL-1α was the most potent priming agent evaluated in our system, TNFα also induced RPE cells to express IL-1β. TNFα has been found in choroidal neovascular membranes obtained from AMD-affected eyes,42,43 regulates several cellular activities in RPE cells,44,45 and has been implicated in laser-induced choroidal neovascularization (CNV).46,47 Additionally, higher levels of proteins adducted to carboxyethylpyrrole (CEP) have been found in eyes from donors with AMD than in age-matched controls.48 CEP-adducted human serum albumin has been shown to prime murine and human macrophages and mononuclear cells,12 so it is plausible that CEP adducts act similarly on RPE cells.
Chemical disruption of lysosomes using Leu-Leu-OMe triggered inflammasome activation in RPE cells as evidenced by the detection of active caspase-1, which mediated IL-1β release and RPE cell death. Inflammasome activation was blocked by the addition of Gly-Phe-CHN2, which inhibits the DPP-I-dependent conversion of Leu-Leu-OMe to a membranolytic derivative inside the lysosome. This abolished caspase-1 activation, IL-1β release, and RPE cytotoxicity. These results demonstrate that inflammasome activation and its downstream effects were induced by lysosomal destabilization. Furthermore, inhibition of the lysosomal enzymes cathepsins B and L blocked lysosomal damage-induced inflammasome activation, IL-1β secretion, and pyroptosis, implicating one or both of these enzymes as mediators of NLRP3 inflammasome assembly in the RPE. One potential mechanism may involve the leakage of cathepsins B and/or L, and possibly other lysosomal enzymes, into the cytosol where they act on downstream proteins, leading to NLRP3 activation.
Intracellular pro-IL-1β has been reported in RPE cells,49 but our findings are the first to demonstrate inflammasome-mediated processing of mature IL-1β from RPE cells. As expected, levels of IL-1β released from RPE cells are lower than those released from myeloid cells in response to lysosomal disruption.23 The term “para-inflammation” has been coined to describe a response to tissue stress that is intermediate between inflammation and the basal state.50 Such chronic, low-level inflammation is hypothesized to play a role in age-related and inflammatory disorders such as AMD and atherosclerosis.50–52 We speculate that the IL-1β released by RPE cells in response to lysosomal insults mediates para-inflammation contributing to AMD, and that infiltrating myeloid cells may play a role as well. A number of AMD-associated insults could be responsible for RPE lysosomal destabilization and inflammasome activation during the pathogenesis of AMD. Isolated drusen deposits12 as well as the drusen components amyloid-β and complement factor C1Q12,22,53 have been shown to activate the NLRP3 inflammasome in myeloid cells. Additionally, the lipofuscin component A2E, which accumulates in RPE lysosomes, has been shown to have detergent-like properties that disrupt lysosomal membranes.11,28,29
We demonstrated that the RPE cytotoxicity resulting from lysosomal destabilization is caspase-1 dependent. This is characteristic of “pyroptosis,” a mode of programmed cell death mediated by the inflammasome and caspase-1 rather than apoptotic caspases such as caspase-3. Although apoptosis and pyroptosis are both processes of regulated cell death, pyroptosis is a proinflammatory mode of cell death, whereas apoptosis is noninflammatory. While the plasma membrane remains intact during apoptosis, pyroptosis involves plasma membrane rupture and release of intracellular contents, allowing for detection by LDH quantification.54–56 Thus, the inflammasome may contribute to AMD via both cytokine release and pyroptotic RPE death.
Our data support a two-signal model of NLRP3 inflammasome induction by disruption of RPE lysosomes (Fig. 7). Priming signals (signal 1), such as IL-1α, TNFα, or CEP adducts, activate NF-κB, and possibly other factors, to induce expression of pro-IL-1β and NLRP3. Disruption of RPE lysosomes (signal 2), which may be caused by lipofuscin, drusen components, or other insoluble lysosomal contents, triggers NLRP3 inflammasome assembly via lysosomal enzymes such as cathepsins B and L. These enzymes may leak into the cytosol following lysosomal permeabilization and trigger pathways leading to NLRP3 activation. The assembled inflammasome activates caspase-1, which cleaves pro-IL-1β to form mature IL-1β and also mediates pyroptotic RPE cell death.
Figure 7. .
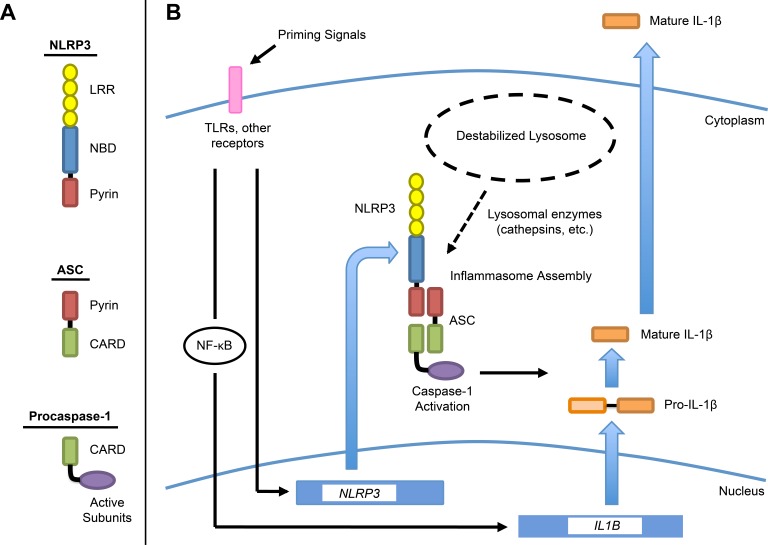
Schematic diagram representing proposed model of NLRP3 inflammasome activation in RPE cells by lysosomal destabilization. (A) Domain architecture of the components of the NLRP3 inflammasome. Inflammasome assembly is mediated by homotypic interactions between pyrin domains on NLRP3 and ASC, and between caspase-recruitment domains (CARDs) on ASC and caspase-1. (B) Two-signal model in which priming signals (signal 1) induce expression of NLRP3 and pro-IL-1β, with pro-IL-1β upregulated via NF-κB. Lysosomal destabilization (signal 2) causes leakage of lysosomal enzymes into the cytosol. These enzymes, such as cathepsins B and L, mediate NLRP3 inflammasome assembly, resulting in caspase-1 activation. Caspase-1 processes pro-IL-1β into mature IL-1β, which is then secreted, and also drives pyroptosis. LRR, leucine-rich repeat; NBD, nucleotide-binding domain.
Two recent studies present different hypotheses regarding the function of the NLRP3 inflammasome pathway in AMD. One study, using a mouse model of laser-induced CNV, suggests that NLRP3 has a protective role in neovascular AMD by inducing IL-18 release from infiltrating macrophages,12 whereas a second study using a mouse model of Alu RNA-induced GA suggests that NLRP3 plays a destructive role in dry AMD by inducing IL-18 secretion from RPE cells.13 These studies suggest that NLRP3 inflammasome may have distinct roles in wet and dry advanced AMD, or even influence the development of one form over the other.
Our data demonstrate that lysosomal destabilization can activate the NLRP3 inflammasome in RPE cells, inducing the secretion of the potent proinflammatory cytokine IL-1β from primed cells and pyroptosis. These processes may constitute novel mechanisms for AMD pathogenesis. The activators and effectors of the NLRP3 inflammasome are consistent with the phenotype of AMD. Taken together with convincing genetic data that implicate a role for inflammation,3,57–59 our findings suggest a mechanism by which insults such as drusen deposition and lipofuscin accumulation can contribute to AMD pathology.
Acknowledgments
We thank Eicke Latz for valuable discussions and advice, as well as Jinling Yang for helpful assistance with fluorescent microscopy.
Footnotes
Supported by NIH Grants EY05435 (PAD), GM07226, and AG039245 (WAT); and a Macular Degeneration Research grant from the American Health Assistance Foundation (BRK).
Disclosure: W.A. Tseng, None; T. Thein, None; K. Kinnunen, None; K. Lashkari, None; M.S. Gregory, None; P.A. D'Amore, None; B.R. Ksander, None
References
- 1. Resnikoff S, Pascolini D, Etya'ale D, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004; 82: 844–851 [PMC free article] [PubMed] [Google Scholar]
- 2. Kaarniranta K, Salminen A. Age-related macular degeneration: activation of innate immunity system via pattern recognition receptors. J Mol Med. 2009; 87: 117–123 [DOI] [PubMed] [Google Scholar]
- 3. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000; 14: 835–846 [PubMed] [Google Scholar]
- 5. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001; 20: 705–732 [DOI] [PubMed] [Google Scholar]
- 6. Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005; 80: 595–606 [DOI] [PubMed] [Google Scholar]
- 7. Chen W, Stambolian D, Edwards AO, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A. 2010; 107: 7401–7406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heurich M, Martinez-Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci U S A. 2011; 108: 8761–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004; 122: 598–614 [DOI] [PubMed] [Google Scholar]
- 10. Masters SL, O'Neill LA. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol Med. 2011; 17: 276–282 [DOI] [PubMed] [Google Scholar]
- 11. Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol. 2012; 90: 299–309 [DOI] [PubMed] [Google Scholar]
- 12. Doyle SL, Campbell M, Ozaki E, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012; 18: 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tarallo V, Hirano Y, Gelfand BD, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012; 149: 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140: 821–832 [DOI] [PubMed] [Google Scholar]
- 15. Bauernfeind F, Ablasser A, Bartok E, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011; 68: 765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007; 7: 31–40 [DOI] [PubMed] [Google Scholar]
- 17. Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010; 40: 620–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hiscott J, Marois J, Garoufalis J, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993; 13: 6231–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009; 119: 3502–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010; 464: 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010; 5: e11765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008; 9: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008; 9: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe H, Gaide O, Petrilli V, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007; 127: 1956–1963 [DOI] [PubMed] [Google Scholar]
- 25. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007; 17: 1140–1145 [DOI] [PubMed] [Google Scholar]
- 26. Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. 2010; 12: 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010; 32: 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schutt F, Bergmann M, Holz FG, Kopitz J. Isolation of intact lysosomes from human RPE cells and effects of A2-E on the integrity of the lysosomal and other cellular membranes. Graefes Arch Clin Exp Ophthalmol. 2002; 240: 983–988 [DOI] [PubMed] [Google Scholar]
- 29. Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999; 40: 2988–2995 [PubMed] [Google Scholar]
- 30. Ford KM, Saint-Geniez M, Walshe T, Zahr A, D'Amore PA. Expression and role of VEGF in the adult retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011; 52: 9478–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007; 55: 443–452 [DOI] [PubMed] [Google Scholar]
- 32. Niemi K, Teirila L, Lappalainen J, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011; 186: 6119–6128 [DOI] [PubMed] [Google Scholar]
- 33. Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010; 10: 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gnana-Prakasam JP, Martin PM, Mysona BA, Roon P, Smith SB, Ganapathy V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem J. 2008; 411: 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009; 183: 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thiele DL, Lipsky PE. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A. 1990; 87: 83–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwata A, Nishio K, Winn RK, Chi EY, Henderson WR Jr, Harlan JM. A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. J Immunol. 2003; 170: 3386–3391 [DOI] [PubMed] [Google Scholar]
- 38. Ravanko K, Jarvinen K, Helin J, Kalkkinen N, Holtta E. Cysteine cathepsins are central contributors of invasion by cultured adenosylmethionine decarboxylase-transformed rodent fibroblasts. Cancer Res. 2004; 64: 8831–8838 [DOI] [PubMed] [Google Scholar]
- 39. Herzyk DJ, Berger AE, Allen JN, Wewers MD. Sandwich ELISA formats designed to detect 17 kDa IL-1 beta significantly underestimate 35 kDa IL-1 beta. J Immunol Methods. 1992; 148: 243–254 [DOI] [PubMed] [Google Scholar]
- 40. Kindzelskii AL, Elner VM, Elner SG, Yang D, Hughes BA, Petty HR. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J Gen Physiol. 2004; 124: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar MV, Nagineni CN, Chin MS, Hooks JJ, Detrick B. Innate immunity in the retina: toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J Neuroimmunol. 2004; 153: 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oh H, Takagi H, Takagi C, et al. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1999; 40: 1891–1898 [PubMed] [Google Scholar]
- 43. Wang XC, Jobin C, Allen JB, Roberts WL, Jaffe GJ. Suppression of NF-kappaB-dependent proinflammatory gene expression in human RPE cells by a proteasome inhibitor. Invest Ophthalmol Vis Sci. 1999; 40: 477–486 [PubMed] [Google Scholar]
- 44. Jin M, He S, Worpel V, Ryan SJ, Hinton DR. Promotion of adhesion and migration of RPE cells to provisional extracellular matrices by TNF-alpha. Invest Ophthalmol Vis Sci. 2000; 41: 4324–4332 [PubMed] [Google Scholar]
- 45. Yang P, Wiser JL, Peairs JJ, et al. Human RPE expression of cell survival factors. Invest Ophthalmol Vis Sci. 2005; 46: 1755–1764 [DOI] [PubMed] [Google Scholar]
- 46. Lichtlen P, Lam TT, Nork TM, Streit T, Urech DM. Relative contribution of VEGF and TNF-alpha in the cynomolgus laser-induced CNV model: comparing the efficacy of bevacizumab, adalimumab, and ESBA105. Invest Ophthalmol Vis Sci. 2010; 51: 4738–4745 [DOI] [PubMed] [Google Scholar]
- 47. Jasielska M, Semkova I, Shi X, et al. Differential role of tumor necrosis factor (TNF)-alpha receptors in the development of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010; 51: 3874–3883 [DOI] [PubMed] [Google Scholar]
- 48. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008; 14: 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Planck SR, Huang XN, Robertson JE, Rosenbaum JT. Retinal pigment epithelial cells produce interleukin-1 beta and granulocyte-macrophage colony-stimulating factor in response to interleukin-1 alpha. Curr Eye Res. 1993; 12: 205–212 [DOI] [PubMed] [Google Scholar]
- 50. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008; 454: 428–435 [DOI] [PubMed] [Google Scholar]
- 51. Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009; 28: 348–368 [DOI] [PubMed] [Google Scholar]
- 52. Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011; 95: 14–25 [DOI] [PubMed] [Google Scholar]
- 53. Isas JM, Luibl V, Johnson LV, et al. Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthalmol Vis Sci. 2010; 51: 1304–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009; 458: 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009; 7: 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suzuki T, Franchi L, Toma C, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007; 3: e111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006; 51: 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006; 38: 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]