

Abstract
Background
PKCδ is generally known as a pro-apoptotic and anti-proliferative enzyme in human prostate cancer cells.
Methods
Here, we investigated the role of PKCδ on the growth of PC-3 human prostate cancer cells in vivo and in vitro.
Results
We found that sustained treatment with a specific PKCδ activator (ψδ receptor for active C kinase, ψδRACK) increased growth of PC-3 xenografts. There was increased levels of HIF-1α, vascular endothelial growth factor and CD31-positive cells in PC-3 xenografts, representative of increased tumor angiogenesis. Mechanistically, PKCδ activation increased the levels of reactive oxygen species (ROS) by binding to and phosphorylating NADPH oxidase, which induced its activity. Also, PKCδ-induced activation of NADPH oxidase increased the level of HIF-1α.
Conclusions
Our results using tumors from the PC-3 xenograft model suggest that PKCδ activation increases angiogenic activity in androgen-independent PC-3 prostate cancer cells by increasing NADPH oxidase activity and HIF-1α levels and thus may partly be responsible for increased angiogenesis in advanced prostate cancer.
Keywords: angiogenesis, HIF-1α, NADPH oxidase, prostate cancer, protein kinase C
Introduction
In the US, prostate cancer is the second leading cause of cancer-related deaths in males (1,2). The androgen-independent and advanced type of human prostate cancer cells, PC-3, shows higher levels of reactive oxygen species (ROS) as compared with the androgen-dependent and less invasive types (LNCap and DU 145) and inhibition of ROS generation reduces the invasiveness of the prostate cancer cell lines as shown by matrigel assay. More importantly, human prostate tumor tissues have elevated ROS levels and this was shown to correlate with increased levels of ROS-generating enzymes, like NADPH oxidase (3).
ROS are generated from a number of sources including NADPH oxidase, the mitochondrial electron transport system, xanthine oxidase, cytochrome p-450, and uncoupled nitric oxide synthase (4,5). Among these, the family of NADPH oxidase and the mitochondria systems have emerged as major sources of ROS production (6,7). NADPH oxidase is a major extra-mitochondrial cellular source for producing ROS in a wide variety of non-phagocytic tissues and conditions, including inflammation against injurious stimulus, tissue fibrosis, atherosclerosis and several cancers including prostate cancer (8).
NADPH oxidase consists of membrane-bound subunits, gp91phox and p22phox which form the flavocytochrome b558 complex, together with the cytosolic subunits p40phox, p47phox, and p67phox, as well as the small GTPase, Rac (4). Superoxide production is induced by assembly of these cytosolic and membrane-bound subunits, which is mediated through the phosphorylation of p47phox (9). Because ROS mediates angiogenic signaling, NADPH oxidase is emerging as an important signaling mediator of angiogenesis, especially in cancer, and has been shown to increase in correlation with tumorigenic activity in various cancers (4). For example, Nox1, one of the homologues of the NADPH oxidase catalytic subunit gp91phox, is highly expressed in human prostate cancer in correlation with increased hydrogen peroxide levels (10).
Phosphorylation of several serine residues of p47phox subunit enables it to bind to membrane phospholipids and to interact with p22phox, and brings the p67phox subunit to the complex. The p67phox binds and stabilizes an interaction of the complex with the small GTPase Rac, and the fully formed NADPH complex is able to generate superoxide radical (11,12). Therefore, phosphorylation of p47phox is a critical step in the activation of NADPH oxidase. PKC isozymes are the major kinases responsible for inducing NADPH oxidase activation. PKCβII phosphorylates p47phox and p67phox cytosolic subunits in monocytes, inducing NADPH oxidase activity (12,13). PKCδ phosphorylates p67phox in monocytes when treated with ZOP (an NADPH oxidase activator) (13). Also, p47phox was found to be a substrate for PKCδ in human neutrophils (14) and PKCδ increased mRNA levels of the Nox1 present in vascular smooth muscle cells (15). However, the regulation of NADPH oxidase in the growth of PC-3 human prostate cancer in relation to angiogenesis has not been studied in detail in vivo.
Here, we determined the novel role of PKCδ and the mechanism involved in the growth of PC-3 tumor xenografts using PKCδ-selective peptide regulators. Our data from xenografts suggest that PKCδ can be a target in anti-cancer treatment for prostate cancer against angiogenesis in PC-3 human prostate tumors.
Materials and methods
Cell lines and cell culture
PC-3 human prostate cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM media with 10% fetal bovine serum (FBS, Gibco, NY) with 1% antibiotics (penicillin and streptomycin, Gibco, NY).
Materials
For Western blot analyses, rabbit antibodies directed against Gαi-3 (C-10), anti-phospho threonine (9381), anti-phospho threonine-X-arginine (2351S) antibodies, anti-p47phox(D-10) mouse monoclonal antibodies and PKCδ and ε antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and anti-GAPDH antibody (clone 6C5) was from Advanced Immunochemical (Long Beach, CA). HIF-1α antibodies were from Bethyl Laboratories (Montgomery, TX).
Peptide synthesis and administration
The PKCδ-selective inhibitor (δV1-1, amino acids 8-17 (SFNSYELGSL)) and activator (ψδRACK, amino acids 74-81 (MRAAEDPM)) were derived from the PKCδ V1 region and synthesized by American Peptide Company and conjugated to a membrane-permeable TAT carrier peptide (residues 47-57, [YGRKKRRQRRR]) as previously described (16). TAT carrier peptide was used as a control. Peptides were delivered in vivo using Alzet osmotic mini-pumps (Alzet model 2001) as described (17). The peptides were dissolved in saline and administered at a constant rate (0.5μl/hr) corresponding to 2.4 or 24 mg/day/kg (3mM or 30mM of TAT), 1.4 mg/day/kg (1mM of δV1-1) and 3.8 or 38 mg/day/kg (3mM or 30mM of ψδRACK). Pumps were replaced every 2 weeks because of the t1/2 (about 2 weeks) of the peptides in the pump (17). Peptides were delivered for up to 4 weeks.
Xenograft tumor studies
Six week old male nude mice were purchased from Harlan (Indianapolis, IN) and housed at the animal care facility at Stanford University Medical Center (Stanford, CA). All mice were kept under standard temperature, humidity, and timed lighting conditions and provided mouse chow and water ad libitum. All animal experimentation was conducted in accordance with the Guide for Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council, and published by the National Academy Press (revised 1996) and was approved by the Stanford University Animal Care and Use Committee. Five million PC-3 tumor cells were injected subcutaneously in the flank of male, 7-8 week old, athymic nude mice in a mixture of 1:1 serum-free medium and Matrigel (Beckton Dickinson, Bedford, MA). Peptide treatment began when the tumors reached a group average of 100mm3 after about 1 week. Tumor volume (mm3) was calculated using the equation 0.52 × (width (cm)2 × length (18)).
Immunofluorescence microscopy
Immunofluorescence was performed on fresh-frozen PC-3 tumor sections fixed in O.T.C. compound (Torrance, CA) using FITC-linked anti-mouse CD31 antibodies (1:50, BD Pharmingen, San Diego, CA).
Western blot analysis
Protein concentrations were determined using the Bradford assay. Ten micrograms of protein from tumors were resuspended in Laemmli buffer, loaded on SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were probed with the indicated antibody followed by visualization with ECL.
Preparation of total cell lysates and fractionation
Immunoprecipitation
Proteins (300-500μg) were solubilized in lysis buffer and the whole cell lysate was incubated with primary antibody against p47phox overnight at 4 °C. Then the samples were precipitated with protein-A/G sepharose (Santa Cruz Biotech) at 4 °C for 1 hour and the immunoprecipitates were washed twice in HEPES buffer as previously described (20). The proteins in the immunoprecipitates were separated using SDS-PAGE and analyzed using immunoblotting.
NADPH oxidase activity assay
Cells were serum starved for 14 to 48 hours prior to the measurement and stimulated with 1% fetal bovine serum and/or chemicals as indicated. After washing twice with 1x PBS, cells were collected with 1x PBS containing protease inhibitor cocktail and phosphatase inhibitor cocktail I and II (Sigma, St. Louis, MO) using a cell scraper. The lysate was further homogenized using syringes with 25G needles. Measurement was performed by adding 10μl of lysate into 200μl of reaction buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1mM EGTA, 5μM lucigenin and 100μM NADPH). Chemiluminescence derived from superoxide and lucigenin were detected by a luminometer within 30 minutes after the addition of reaction buffer (Veritas, Turner Byosystems, Sunnyvale, CA).
RNA interference
Small interfering RNA (siRNA) duplexes targeting PKCδ were obtained from Santa Cruz Biotech (human, sc-36253, Santa Cruz, CA). PC-3 cells at ~60% confluency were transfected with control siRNA and PKCδ siRNA using transfection reagents from Gene Silencer (San Diego, CA) according to the manufacturer’s instructions. The cells were collected 48 hours after transfection.
Measurement of vascular endothelial growth factor (VEGF) levels in serum
The concentration of VEGF in the serum was measured using an ELISA kit (human and mouse VEGF immunoassay, Quantikine, R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as mean±SE. Unpaired Student’s t test for differences between two groups, repeated ANOVA for differences over time and 1-factor ANOVA for differences among >2 groups were used to assess significance (p<0.05).
Results
Figure 1. PKCδ activation increases PC-3 prostate tumor growth
Fig. 1. PKCδ activation increases PC-3 prostate tumor growth.
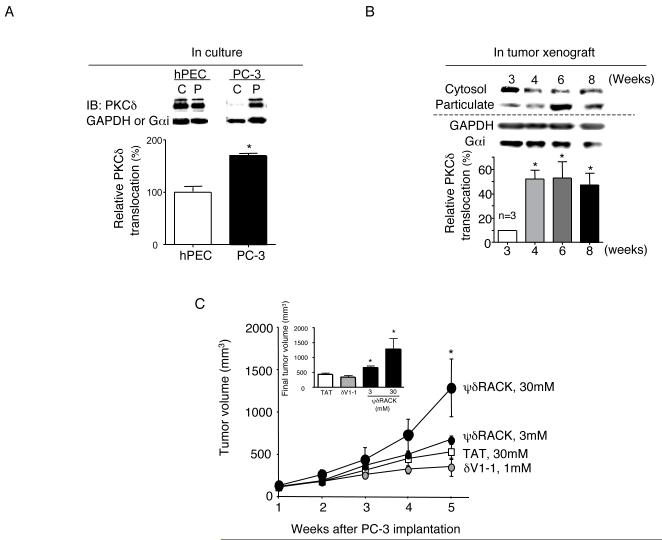
(A) The level of the active form of PKCδ was determined by Western blot analyses of cytosolic (C) and particulate (P) fractions from primary normal human prostate epithelial cells (hPEC) and PC-3 cells grown in culture. Cells were fractionated into cytosolic and particulate fractions as described in Methods. Quantification of the active forms of PKCδ (translocation; expressed as percentage of PKC isozyme in the particulate fraction over sum of cytosolic and particulate fraction enzymes, i.e., total cellular enzyme) is provided in the graph below (n=3, *; p<0.05). A 2-tailed Student’s t test was used to determine significance. Loading controls for cytosolic and particulate fractions (GAPDH and Gαi) are shown in the lower bands. IB; immunoblot. (B) PC-3 xenografts were grown s.c. on nude mice for up to 8 weeks and tumors were obtained at weeks 3-8. PKCδ translocation was analyzed using Western blot and quantification is shown on the graph below (n=3, *; p<0.05) vs. week 3. (C) One week after PC-3 cell injection, mice were implanted with osmotic pumps with control peptide (TAT) at 24 mg/kg/day (30mM) or δV1-1 conjugated to TAT at 1.4 mg/kg/day (1mM) or ψδRACK at 3.8 or 38 mg/kg/day (3mM or 30mM). The peptides were dissolved in saline and administered at a constant rate (0.5μl/hr) for 2 weeks and were replaced once for the next 2 weeks. Tumor volume was measured weekly. Tumors were excised and weighed at week 5. (White squares, TAT; small gray circles, δV1-1; small black circles, 3mM ψδRACK and large black ovals, 30mM ψδRACK, *; p<0.05, repeated ANOVA, n=5-8 each, TAT vs. 30mM ψδRACK-treated group). An inserted graph shows final tumor volumes of each treatment group.
PKCδ activation has been implicated in the growth of several epithelial cancers including prostate, breast and ovarian cancers (21-23). Here, we first compared the cellular distribution of PKCδ as a measure of its active level in PC-3 cells with that in a primary culture of normal human prostate epithelial cells (hPEC) [Figure 1A, left; cytosolic (C) fraction enzyme represents inactive PKC and particulate fraction enzyme (P) represents active PKC (20,24)]. PKCδ was more active in the PC-3 cells relative to the primary hPEC as evidenced by higher levels of this isozyme in the particulate fraction relative to the cytosolic fraction (Figure 1A, n=3 each). Also, PKCδ translocation increased by about 5 fold during the growth of PC-3 human prostate cancer cells in a xenograft model, in vivo (from 10% to 53%; Figure 1B, p<0.05 vs. week 3). We have recently shown that the volume of PC-3 tumor xenografts increases significantly between week 3 and 5 and that angiogenesis in the tumor increased during that period [as measured by endothelial cell proliferation (20)]. The finding that PKCδ translocation increased significantly between week 3 and 4 and was maintained at similar levels afterwards suggests that PKCδ activation may be involved in angiogenesis and the growth of PC-3 tumors.
To examine the role of PKCδ in PC-3 xenograft tumor growth in vivo, PC-3 cancer cells were injected subcutaneously (5×106 cells) into the flank area of male nude mice. After 1 week, mice were treated with control peptide [TAT47-57 cell-permeable carrier peptide] at 30mM (24 mg/kg/day), a PKCδ-selective inhibitor peptide coupled to TAT47-57; δV1-1 at 1mM (1.4 mg/kg/day) or a PKCδ-selective activator peptide coupled to TAT47-57; ψδRACK (16) at 3mM or 30mM (3.8 or 38 mg/kg/day) for the following 4 weeks using Alzet osmotic pumps for sustained infusion (Figure 1C). Final tumor volume was 23% lower in the δV1-1-treated group (440±40 and 340±80, control vs. δV1-1-treated group, n=5-10 each) and 50% higher in the 3mM ψδRACK-treated group (440±40 and 660±60, control vs. ψδRACK-treated group, Figure 1C, *; p<0.05, n=5-6 each, a 2-tailed Student’s t test). The higher concentration of ψδRACK increased final tumor volume by 220% in the ψδRACK-treated group (440±40 and 1,280±340, TAT vs. ψδRACK-treated group, Figure 1C, *; p<0.05, n=5-8 each, Student’s t test and repeated ANOVA). Final tumor volumes are shown in the insert (Figure 1C). These data suggest that PKCδ activation increases during the angiogenically active period of PC-3 tumor growth and that PKCδ activation increases tumor growth rate.
Figure 2. PKCδ activation increases tumor angiogenesis
Fig. 2. PKCδ activation increases tumor angiogenesis.

(A) The level of angiogenesis at the end of the 5-week growth was measured with immunofluorescence by staining blood vessels with anti-CD31 monoclonal antibodies conjugated with FITC (n=3 each, *; p<0.05, Scale bar: 10μm). (B) One week after PC-3 cell injection, mice were implanted with osmotic pumps with control peptide (TAT) or ψδRACK at 38 mg/kg/day (30mM) for 4 weeks. HIF-1α levels were measured by Western blot analyses using whole cell lysates from tumors (Figure 2B left, *; p<0.05 t test, n=3 each). GAPDH was used as a loading control. (C) Next, human VEGF concentration was measured in the serum from mice treated with control peptide (TAT) or ψδRACK by ELISA (Figure 2C left, *; p<0.05, n=4 each). Mouse VEGF concentration was measured in the serum from mice treated with control peptide (TAT) or ψδRACK by ELISA (Figure 2C, right).
We first observed that there were significantly higher CD31-positive tumor vessels in the ψδRACK-treated (30mM, 38 mg/kg/day) tumors compared to TAT controls (1600±220 vs. 86±3 %, p<0.05, Figure 2A and a graph). ψδRACK treatment did not induce apoptosis in this model as measured by TUNEL staining (data not shown). Based on these results, we focused our study on determining the role of PKCδ and angiogenic events during tumor growth.
We also observed increased HIF-1α protein levels in the ψδRACK-treated tumors compared to the controls. At week 5, after 4 weeks of sustained treatment with ψδRACK (38 mg/kg/day), HIF-1α levels increased by more than 3-fold as compared with the control (TAT-treated) group (Figure 2B, 0.15±0.01 and 0.63±0.70, TAT vs ψδRACK, *; p<0.05, n=3 each). Of all the pro-angiogenic factors induced by HIF1-α, VEGF is particularly noteworthy, because it has potent angiogenic properties and is expressed in a large number of human cancers including prostate tumors (25). With ψδRACK treatment (38 mg/kg/day, 30mM), the human VEGF concentration in the serum increased by ~6-fold as compared with the control mice (Figure 2C, left, *; p<0.05, 330±110 vs. 2,700±920 pg/ml/tumor weight, TAT vs. ψδRACK; n=4). However, there was no difference in the concentration of mouse VEGF between the two treatments (Figure 2C right, 190±70 vs. 280±100 pg/ml/tumor weight, TAT vs. ψδRACK) [VEGF concentration was normalized against tumor weight]. These data indicate that PKCδ activation promotes angiogenesis by increasing HIF1-α and pro-angiogenic growth factor VEGF levels in the human prostate cancer xenograft. Increased VEGF production was from human tumor cells and not from the mouse endothelial cells.
Figure 3. PKCδ regulates NADPH oxidase activity in PC-3 cells
Fig. 3. PKCδ regulates NADPH oxidase activities in PC-3 cells.
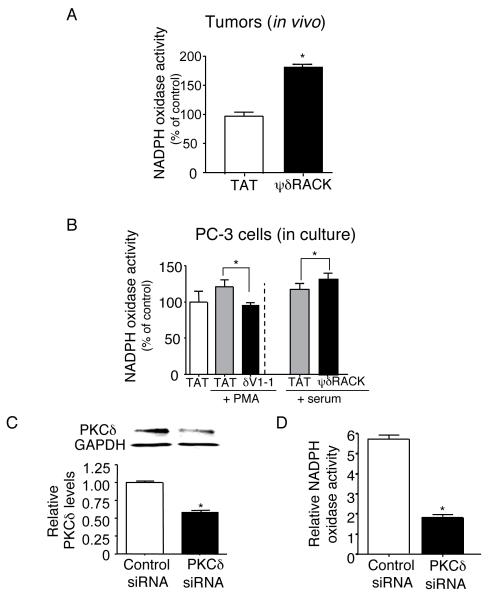
(A) Tumors as described in Figure 1C were analyzed for NADPH oxidase activity. Whole lysates of tumors in PBS with phosphatase and protease inhibitor cocktail were added with lucigenin. Chemiluminescence derived from superoxide and lucigenin was measured using a luminometer (n=5 each). A 2-tailed Student’s t test was used to determine significance. (B) Cells were serum starved for 14 hours and first incubated with PKCδ inhibitor peptide (δV1-1, 1μM) or PKCδ activator peptide (ψδRACK, 1μM) for 15 minutes. After incubation with 1nM PMA or serum for 30 minutes, whole cell lysate was used for the assay. A 2-tailed Student’s t test was used to determine significance (n=5-6 for each treatment). (C) The levels of PKCδ in PC-3 cells were knocked down using siRNA. (D) To test PKCδ regulation of NADPH oxidase activity, NADPH oxidase activities were measured in the cells treated with control siRNA or siRNA of PKCδ. A 2-tailed Student’s t test was used to determine significance (n=3 for each, *; p<0.05, t test).
ROS, which is found in a large number of tumors, has been reported to mediate angiogenic signaling by stimulating endothelial cell growth and migration (4). NADPH oxidase is one of the major cellular sources for continuously producing ROS intracellularly at low levels in a wide variety of cells (8). ROS production by NADPH oxidase can also be further activated by various growth factors and other stimuli (4). Furthermore, NADPH oxidase was shown to be more active in more aggressive prostate cancer cell lines (3). Based on theses results, we examined the molecular mechanisms of increased angiogenesis with PKCδ activation by measuring NADPH oxidase activity in tumors treated with TAT or ψδRACK (Figure 3A). We used tumors obtained by the same protocol as described in Figure 1C, treated with TAT or ψδRACK (30mM). Lucigenin and superoxide-derived chemiluminescence was used as a measure of NADPH oxidase activity in the tissue lysates. ψδRACK treatment resulted in an ~80% increase in the catalytic activity of NADPH oxidase (Figure 3A) relative to TAT treatment.
Next, we observed the regulation of NADPH oxidase activity by PKCδ in vitro, using both the PKCδ isozyme-specific inhibitor and activator peptides in PC-3 cells (Figure 3B). Cells were serum starved and incubated with the PKCδ inhibitory peptide (δV1-1; 1μM) or the PKCδ activator peptide (ψδRACK; 1μM). Treatment with the PKCδ-selective inhibitor decreased NADPH oxidase activity by more than 30% (125±11% and 97±4%, PMA vs. δV1-1+PMA, *; p<0.05, Figure 3B, left panels). Activation of PKCδ with ψδRACK increased NADPH oxidase activity by 22% (100±7% and 122±5%, 1% serum vs. ψδRACK+1% serum, *; p<0.05, Figure 3B, right panels). The difference between in vivo and in vitro data in NADPH oxidase activity with ψδRACK treatment may result from amplified induction of ROS from chronic hypoxia in solid tumors and the contribution of growth factors that may have stimulated PKCδ activity further in vivo. To confirm PKCδ regulation of NADPH oxidase activity, we knocked down the level of PKCδ using siRNA and assayed for the NADPH oxidase activity. Knock down using siRNA decreased the level of PKCδ by ~40% (Figure 3C, *; p<0.05, n=3 each) and decreased NADPH oxidase activity by more than 3 fold (Figure 3D, 6.0±0.2 and 2.0±0.1, control vs. siRNA treated, *; p<0.05, n=3 each). These data show that PKCδ regulates NADPH oxidase activity in vivo and in vitro in PC-3 cancer cells.
Figure 4. PKCδ regulates NADPH oxidase by phosphorylating and inducing the translocation of NADPH oxidase subunit p47phox to the membrane fraction
Fig. 4. PKCδ regulates NADPH oxidase activity by phosphorylating p47phox and inducing its translocation to the membrane fraction.
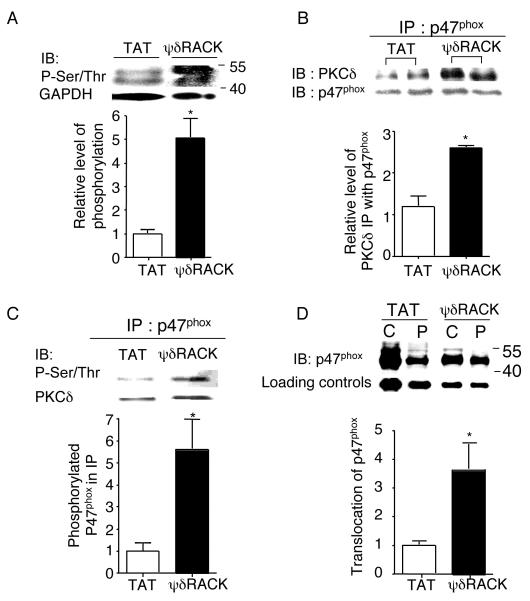
(A) Western blot analyses were performed with total cell lysates of PC-3 tumors treated with TAT or 38 mg/kg/day of ψδRACK for 4 weeks and immunoblotted with antibodies against phospho-serine/threonine (Ser/Thr) (*; p<0.05, n=3 each, Figure 4A). GADPH was used as a loading control. (B) Tumor lysates were immunoprecipitated with antibodies against p47phox and probed for PKCδ. (*; p<0.05, n=3 each, Figure 4B). p47phox was used as a loading control. (C) Phosphorylation levels of the co-immunoprecipitated p47phox were checked by probing with anti-Ser/Thr antibodies (*; p<0.05, n=3 each, Figure 4C). PKCδ was used as a loading control. (D) Finally, tumor lysates were fractionated (as described in the methods) and the translocation of p47phox from the cytosolic to the membrane (particulate) fraction was determined by probing each fraction with anti-p47phox antibodies (Figure 4D, *; p<0.05, n=3 each). GADPH or Gαi was used as a loading control (IB: immunoblot).
We then determined the molecular basis for activation of NADPH oxidase by PKCδ. As mentioned previously, PKC isozymes are the major kinases responsible for inducing NADPH oxidase activation (12,13). Because superoxide production is induced by assembly of the cytosolic and membrane-bound subunits of NADPH oxidase, a process triggered by the phosphorylation of p47phox (12), we hypothesized that PKCδ phosphorylation of p47phox may play a role in the regulation of NADPH oxidase activity in PC-3 cells. Treatment with ψδRACK for 4 weeks (30mM) significantly increased serine/threonine phosphorylation of a ~50kDa band by Western blot analysis of total tumor lysates. The level of the phosphorylated form of putative p47phox was increased by ~5 fold with ψδRACK treatment compared to TAT controls (1.0±0.2 and 5±1, TAT vs. ψδRACK, *; p<0.05, n=3 each, Figure 4A). When tumor lysates were immunoprecipitated (IP) with antibodies against p47phox, the amount of co-immunoprecipitated PKCδ increased by ~150% after ψδRACK treatment as compared with TAT controls (Figure 4B). p47phox was used as a loading control. We next set out to determine the phosphorylation levels of the co-immunoprecipitated p47phox in the tumors. Immunoprecipitated p47phox using anti-p47phox antibodies was probed with anti-serine/threonine antibodies. With ψδRACK treatment, phosphorylation of the immunoprecipitated p47phox increased by 6-fold as normalized by PKCδ (1.0±0.4 and 6.0±1.4, TAT vs. ψδRACK, *; p<0.05, n=3, Figure 4C). Finally, the translocation of p47phox from the cytosolic to the membrane (particulate) fraction increased significantly with ψδRACK treatment (1.0±0.1 and 4.0±0.9, *; p<0.05, n=3, Figure 4D). These data suggest that ψδRACK-induced interaction of NADPH oxidase with PKCδ and phosphorylation of an NADPH oxidase subunit p47phox and induction of its translocation to the cell membrane may partly explain the molecular basis for activation of NADPH oxidase by PKCδ.
Figure 5. PKCδ regulates HIF-1α levels in PC-3 cells via NADPH oxidase
Fig. 5. PKCδ regulates HIF-1α levels in PC-3 cells via NADPH oxidase.
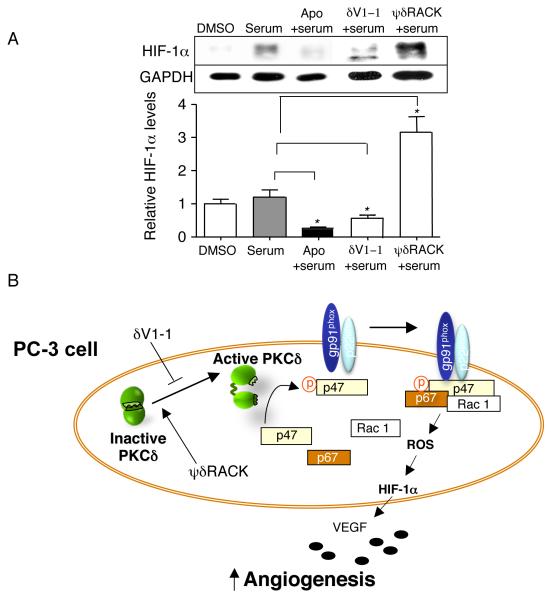
(A) PKCδ regulates HIF-1α levels via NADPH oxidase. PC-3 cells were serum starved for 14 hours and incubated with apocynin (an anti-oxidant and a chemical inhibitor of NADPH oxidase, 1mM) for 5 minutes in the presence of 1% serum (Figure 5A, n=3 each, *; p<0.05). Also, the PC-3 cells were treated with δV1-1 and ψδRACK at 1μM for 4 hours in the presence of 1% serum. The peptides were treated every 1.5 hours and the cells were lysed for Western blot analyses. (B) A schematic diagram summarizes the regulation of tumor-induced angiogenesis via HIF-1α levels by PKCδ and NADPH oxidase in PC-3 cancer cells. p47; p47phox and p67; p67phox.
ROS produced in response to repeated cycles of hypoxia and normoxia was shown to induce angiogenic response by increasing HIF-1α accumulation, which was regulated by NADPH oxidase in PC12 cells (5). Based on this and our data that PKCδ regulates NADPH oxidase activity (Figures 3) and PKCδ activation increased HIF-1α levels (Figure 2), we hypothesized that PKCδ may regulate HIF-1α levels through NADPH oxidase activity. PC-3 cells were serum starved for 14 hours and incubated with apocynin (a chemical inhibitor of NADPH oxidase, 1mM) for 5 minutes. Apocynin decreased HIF-1α levels by more than 4-fold in the presence of 1% serum (Figure 5A, middle panel, n=3 each, *; p<0.05), showing that NADPH oxidase regulates HIF-1α levels in PC-3 cells. Next, we found that PKCδ inhibition decreased HIF-1α levels by almost 50% (1.0±0.03 and 0.6±0.12%, TAT vs. δV1-1, n=3, Figure 5A, second to right panel). On the other hand, PKCδ activation increased HIF-1α levels by more than 3-fold (1.0±0.03% and 3.2±0.5%, TAT vs. ψδRACK, n=3-4, Figure 5A, far right panel). HIF-1α levels were unchanged with δV1-1 or ψδRACK in the presence of apocynin treatment (data not shown). These data suggest that PKCδ regulates HIF-1α levels through NADPH oxidase.
A schematic diagram (Figure 5B) summarizes the regulation of tumor-induced angiogenesis via HIF-1α levels through PKCδ and NADPH oxidase in PC-3 prostate cancer cells. Activated PKCδ can then phosphorylate substrates nearby. PKCδ activation increases phosphorylation of the p47phox subunit of NADPH oxidase, which induces its translocation to the membrane and NADPH oxidase activation. Activated NADPH oxidase then produces large quantities of ROS, increases HIF-1α levels and VEGF production from tumor cells, thus increasing angiogenic activity in tumors.
Conclusions
Identifying the role of specific PKC isozymes in tumors has been hampered by lack of isozyme-specific regulators for each PKC isozyme. Here, using an isozyme-selective inhibitor and an activator peptide for PKCδ (26-28), we show a novel role of PKCδ as an upstream regulator of NADPH oxidase activation leading to increased HIF-1α levels, angiogenesis and tumor growth in PC-3 human prostate cancer. NADPH oxidase activation has been shown to mediate expression of HIF-1α and VEGF and increase tumor growth in ovarian cancer (29). Also, in ~70% of prostate cancer patients, mRNA of catalytic subunit of NADPH oxidase, Nox1, was found only in tumor tissues but not in normal epithelium (10). These upregulated pro-survival signaling pathways in angiogenesis may contribute to the development of resistance to therapy in advanced prostate cancer patients. Development of a safe and targeted molecule to inhibit PKCδ may aid in reducing the resistance experienced by prostate cancer patients.
Previously, LNCaP human prostate cancer cells were shown to undergo apoptosis in response to phorbol esters (30). It was found that the apoptosis induced by phorbol 12-myristate 13-acetate in LNCaP cells was mediated by PKCδ (31). PKCδ was also shown to mediate prostate cancer cell apoptosis induced by chemotherapeutic agents (32). Therefore, PKCδ has been known to be a critical mediator of apoptosis induced by phorbol esters or anticancer drugs. As such, PKCδ was generally found to be pro-apoptotic (21,27,33). However, PKCδ was also reported to be the major kinase that promotes cell survival in mammary and ovarian cancer cells (22,23). Here we revealed a novel role of PKCδ in inducing angiogenesis and growth of PC-3 tumors.
Reactive oxygen species (ROS) mediate angiogenic signaling and NADPH oxidase is one of the major sources of ROS in endothelial cells (4). Increased NADPH oxidase activities correlate with tumorigenic activity in various cancers (8,10). Further, Nox1, a catalytic subunit of NADPH oxidase, was shown to play a critical role in tumor-induced angiogenesis. Inhibition of Nox1 by siRNA or diphenylene iodonium (DPI) inhibited synthesis of VEGF mRNA and protein in K-Ras transformed normal rat kidney cells. Mechanistically, Nox1 inhibition decreased ERK-dependent phosphorylation of Sp1 transcriptional factor and its binding to VEGF promoter (34).
Here, we show that PKCδ co-immunoprecipitates with a subunit of NADPH oxidase critical for its activation and to induce NADPH oxidase activation and promote angiogenesis. These data suggest that disruption of PKCδ-NADPH oxidase inter-molecular protein interactions can negatively modulate tumor-induced angiogenesis. More importantly, PKCδ can be both pro-survival and pro-apoptotic depending on the cell type and binding molecules available as previously mentioned, development of regulators of PKCδ in cancer therapy needs to be approached with caution.
In conclusion, we showed that PKCδ activation can increase NADPH oxidase activity and HIF-1α levels, leading to increased angiogenesis and tumor growth in PC-3 human prostate tumors. Therefore, PKCδ inhibition may provide a useful adjuvant treatment to the current therapy for patients with prostate cancer by inhibiting tumor-induced angiogenesis.
Supplementary Material
Acknowledgements
We thank members of the Mochly-Rosen lab for valuable comments, especially Dr. Alice Vallentin; Dr. Erinn Bruno Rankin at the Department of Radiation Oncology at Stanford Medical School for technical and valuable advice, Dr. Donna Peehl and Ms Rosalie Nolley at the Department of Urology at Stanford Medical School for providing human cells; Dr. Adrienne Gordon for critique on the manuscript. We also thank Ms Marlene Martin for valuable assistance. This study was supported in part by PHS Grant Number CA09151 awarded by the National Cancer Institute, DHHS to J Kim.
This study was supported in part by PHS Grant Number CA09151 awarded by the National Cancer Institute, DHHS to J Kim.
Daria Mochly-Rosen is the founder of KAI Pharmaceuticals, Inc. However, none of the work described in this study was based on or supported by the company.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Mostaghel EA, Montgomery B, Nelson PS. Castration-resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urol Oncol. 2009;27(3):251–257. doi: 10.1016/j.urolonc.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68(6):1777–1785. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 4.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266(1):37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217(3):674–685. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ Res. 2008;103(8):873–880. doi: 10.1161/CIRCRESAHA.108.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol. 2008;30(3):339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 8.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43(3):332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci. 2003;28(9):502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 10.Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, Petros JA, Arnold RS. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62(2):200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- 11.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26(5):675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Siow YL, Au-Yeung KK, Woo CW, O K. Homocysteine stimulates phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase Cbeta activation. Biochem J. 2006;398(1):73–82. doi: 10.1042/BJ20051810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X, Xu B, Bhattacharjee A, Oldfield CM, Wientjes FB, Feldman GM, Cathcart MK. Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukoc Biol. 2005;77(3):414–420. doi: 10.1189/jlb.0504284. [DOI] [PubMed] [Google Scholar]
- 14.Remijsen QF, Fontayne A, Verdonck F, Clynen E, Schoofs L, Willems J. The antimicrobial peptide parabutoporin competes with p47(phox) as a PKC-substrate and inhibits NADPH oxidase in human neutrophils. FEBS Lett. 2006;580(26):6206–6210. doi: 10.1016/j.febslet.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 15.Fan CY, Katsuyama M, Yabe-Nishimura C. PKCdelta mediates up-regulation of NOX1, a catalytic subunit of NADPH oxidase, via transactivation of the EGF receptor: possible involvement of PKCdelta in vascular hypertrophy. Biochem J. 2005;390(Pt 3):761–767. doi: 10.1042/BJ20050287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, 2nd, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98(20):11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111(1):44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 18.McMullin RP, Mutton LN, Bieberich CJ. Hoxb13 regulatory elements mediate transgene expression during prostate organogenesis and carcinogenesis. Dev Dyn. 2009;238(3):664–672. doi: 10.1002/dvdy.21870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Begley R, Liron T, Baryza J, Mochly-Rosen D. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem Biophys Res Commun. 2004;318(4):949–954. doi: 10.1016/j.bbrc.2004.04.121. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Choi YL, Vallentin A, Hunrichs BS, Hellerstein MK, Peehl DM, Mochly-Rosen D. Centrosomal PKCbetaII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008;68(16):6831–6839. doi: 10.1158/0008-5472.CAN-07-6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7(4):281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 22.Grossoni VC, Falbo KB, Kazanietz MG, de Kier Joffe ED, Urtreger AJ. Protein kinase C delta enhances proliferation and survival of murine mammary cells. Mol Carcinog. 2007;46(5):381–390. doi: 10.1002/mc.20287. [DOI] [PubMed] [Google Scholar]
- 23.Lee JW, Park JA, Kim SH, Seo JH, Lim KJ, Jeong JW, Jeong CH, Chun KH, Lee SK, Kwon YG, Kim KW. Protein kinase C-delta regulates the stability of hypoxia-inducible factor-1 alpha under hypoxia. Cancer Sci. 2007;98(9):1476–1481. doi: 10.1111/j.1349-7006.2007.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraft AS, Anderson WB, Cooper HL, Sando JJ. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. J Biol Chem. 1982;257(22):13193–13196. [PubMed] [Google Scholar]
- 25.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15(4):678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bright R, Raval AP, Dembner JM, Perez-Pinzon MA, Steinberg GK, Yenari MA, Mochly-Rosen D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J Neurosci. 2004;24(31):6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279(46):47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 28.Qi X, Mochly-Rosen D. The PKCdelta -Abl complex communicates ER stress to the mitochondria - an essential step in subsequent apoptosis. J Cell Sci. 2008;121(Pt 6):804–813. doi: 10.1242/jcs.024653. [DOI] [PubMed] [Google Scholar]
- 29.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67(22):10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 30.Xiao L, Gonzalez-Guerrico A, Kazanietz MG. PKC-mediated secretion of death factors in LNCaP prostate cancer cells is regulated by androgens. Mol Carcinog. 2009;48(3):187–195. doi: 10.1002/mc.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gavrielides MV, Gonzalez-Guerrico AM, Riobo NA, Kazanietz MG. Androgens regulate protein kinase Cdelta transcription and modulate its apoptotic function in prostate cancer cells. Cancer Res. 2006;66(24):11792–11801. doi: 10.1158/0008-5472.CAN-06-1139. [DOI] [PubMed] [Google Scholar]
- 32.Sumitomo M, Ohba M, Asakuma J, Asano T, Kuroki T, Hayakawa M. Protein kinase Cdelta amplifies ceramide formation via mitochondrial signaling in prostate cancer cells. J Clin Invest. 2002;109(6):827–836. doi: 10.1172/JCI14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murriel CL, Mochly-Rosen D. Opposing roles of delta and epsilonPKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys. 2003;420(2):246–254. doi: 10.1016/j.abb.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 34.Komatsu D, Kato M, Nakayama J, Miyagawa S, Kamata T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene. 2008;27(34):4724–4732. doi: 10.1038/onc.2008.102. [DOI] [PubMed] [Google Scholar]
- 35.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004;384(Pt 3):449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talior I, Tennenbaum T, Kuroki T, Eldar-Finkelman H. PKC-delta-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: role for NADPH oxidase. Am J Physiol Endocrinol Metab. 2005;288(2):E405–411. doi: 10.1152/ajpendo.00378.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.