

Abstract
Asbestos causes asbestosis (pulmonary fibrosis caused by asbestos inhalation) and malignancies (bronchogenic carcinoma and mesothelioma) by mechanisms that are not fully elucidated. Despite a dramatic reduction in asbestos use worldwide, asbestos-induced lung diseases remain a substantial health concern primarily because of the vast amounts of fibers that have been mined, processed, and used during the 20th century combined with the long latency period of up to 40 years between exposure and disease presentation. This review summarizes the important new epidemiologic and pathogenic information that has emerged over the past several years. Whereas the development of asbestosis is directly associated with the magnitude and duration of asbestos exposure, the development of a malignant clone of cells can occur in the setting of low-level asbestos exposure. Emphasis is placed on the recent epidemiologic investigations that explore the malignancy risk that occurs from nonoccupational, environmental asbestos exposure. Accumulating studies are shedding light on novel mechanistic pathways by which asbestos damages the lung. Attention is focused on the importance of alveolar epithelial cell (AEC) injury and repair, the role of iron-derived reactive oxygen species (ROS), and apoptosis by the p53- and mitochondria-regulated death pathways. Furthermore, recent evidence underscores crucial roles for specific cellular signaling pathways that regulate the production of cytokines and growth factors. An evolving role for epithelial-mesenchymal transition (EMT) is also reviewed. The translational significance of these studies is evident in providing the molecular basis for developing novel therapeutic strategies for asbestos-related lung diseases and, importantly, other pulmonary diseases, such as interstitial pulmonary fibrosis and lung cancer.
Asbestos is a term for a group of naturally occurring hydrated silicate fibers whose tensile strength and resilience are ideal for a variety of construction and insulation purposes. The word “asbestos” is derived from the Greek word for “inextinguishable” or “unquenchable.”1 Industrial production of asbestos began in the 1850s, but by the middle of the 20th century, it was evident that asbestos exposure increased the risk for non-malignant inflammatory (pleural effusions, pleural plaques, rounded atelectasis, and asbestosis) and malignant (mesothelioma and bronchogenic carcinoma) pulmonary diseases.2–4 The first cases of asbestos-associated fibrosis were described in the early 1900s, and the term “asbestosis” was coined by Cooke in 1927.5 Asbestos-associated bronchogenic carcinoma was established by the mid-1950s, whereas the association between asbestos and malignant mesothelioma (MM) was recognized by the 1960s. In the early 1970s, the United States placed a moratorium on asbestos use, and at least 40 other countries have banned or severely restricted asbestos use.6 However, asbestos continues to challenge health care providers and the medico-legal system such that the translational aspects of new studies in this field are urgently needed. The purpose of this review is to summarize the important information reported over the last several years that has increased our understanding of the epidemiology and scientific basis for asbestos-induced lung diseases.
EPIDEMIOLOGY
Despite a dramatic decline in asbestos use in industrialized countries since the 1970s, asbestos-induced lung diseases remain a significant health concern for several reasons. First, more than 30 million tons of asbestos have been mined, processed, and used in the United States since the early 1900s.2 An estimated 27 million workers in the United States were exposed to aerosolized asbestos fibers between 1940 and 1979.7 Globally, Lin et al8 recently demonstrated a direct relationship between the national consumption of asbestos (kg per person per year; 1960–1969) in 33 countries and the number of deaths caused by mesothelioma and asbestosis in 2000–2004. Second, a long interval (latency period) exists between fiber exposure and the development of asbestos-induced lung diseases (30 to 40 years). This requires long-term, careful follow-up of people who were exposed both occupationally and nonoccupationally. A recent study of 18,211 sheet metal workers examined between 1986 and 2004 showed that 9.6% had asbestosis and that 21% had pleural disease; the strongest predictor of both was the calendar year in which the worker began sheet metal work (1940s > 1950s > 1960s > 1970s).9 Third, asbestos exposure from consumer products and from fibers released during structural renovation is a source of morbidity and mortality, especially for occupationally exposed workers.3,10 In the year 2000 in the United States, an estimated 20,000 hospital discharges listed “asbestosis,” and of these discharges, asbestosis was the primary or contributing cause of death in 2000 of them.10 It is estimated that the total number of asbestos-related deaths in the United States may exceed 200,000 by the year 2030.11 Worldwide, asbestos accounts for an estimated 100,000–140,000 lung cancer deaths per year and contributes to nearly 5% to 7% of all lung cancers.12,13 Surveys in Europe and the United States have shown a doubling of the prevalence of asbestos-associated pleural disease, which includes MM, from the early 1970s until 2000.1 The incidence of MM is expected to peak sometime between 2010 and 2020 because of the long latency period.1,14 The prevalence of MM is nearly 2% in asbestos textile workers exposed to approximately 1 fiber/cm3 over 50 years and accounts for approximately 8% of the deaths in asbestos workers.15 In industrial countries, the yearly incidence of MM is 2 cases per million person-years among women and 10 to 30 cases per million person-years among men, but the rate is as high as 270 to 366 cases per million person-years among men exposed to crocidolite asbestos (see below).14,16 Given the above, it is not surprising that asbestos-related diseases have inundated our legal system, which resulted in very large class-action lawsuits—68,000 individual claims in the year 2000 alone—and the bankruptcy of many old-line industrial companies.1 Collectively, these data support the worldwide severe restriction or ban on asbestos importation and use advocated by the World Health Organization in 2005 and the International Labour Organization in 2006.17,18
It is unclear whether a safe threshold level of asbestos exposure exists that does not increase the risk of malignancy. The Occupational Safety and Health Administration (OSHA) established a permissible exposure limit (PEL) for fibers more than 5 μm long with a 3:1 aspect ratio assessed by phase-contrast light microscopy of 0.1 fibers/cm3 over an 8-h period for all fiber types. Two classes of asbestos fibers are serpentine and amphibole fibers. Serpentine fibers (eg, chrysotile) are curly stranded structures, whereas amphiboles (eg, crocidolite, amosite, tremolite, and others) are straight, rod-like fibers. As reviewed elsewhere,3,6,19–22 considerable controversy exists regarding the malignant risks associated with chrysotile exposure, which accounts for more than 95% of asbestos consumption in the United States. It is estimated that chrysotile levels several hundred times those of amphiboles are necessary to induce a similar risk of malignancy.21,22 Well-designed epidemiologic studies that have directly assessed the malignant risk from low-dose asbestos exposure are lacking. However, 1 large-scale, retrospective population study of 405 hospital-based patients and control subjects concluded that 5 years of exposure to the current OSHA PEL would produce a 4-fold excess of pleural MM.23
An important public health issue concerns the risks of developing pulmonary diseases from environmental asbestos exposure from asbestos-containing materials in buildings or materials that affect residents who live near asbestos mines or processing plants. Pleural plaques occur in 20% to 60% of occupationally exposed workers24,25 but in 2% to 6% of nonoccupationally exposed individuals.26 Importantly, no firm evidence suggests that pleural plaques increase the risk of developing an MM or lung cancer. In a review of 13 studies of the relationship between pleural plaques and risk for lung cancer, 10 were deemed appropriate to address this issue, and none of the 10 demonstrated a direct relationship.27 In 1997, an international expert meeting concluded that parietal pleural plaques alone were insufficient for attributing lung cancer to asbestos.28 Although a well-documented case of asbestosis caused by brief inhalation of asbestos has been described, most patients have had significant occupational asbestos exposure over a prolonged period.2,7,29 The development of asbestosis is directly associated with both the magnitude and the duration of asbestos exposure, but low-level asbestos exposure has been linked with the development of a malignant clone of cells.2,6,30 Airborne asbestos levels in public buildings are generally several orders of magnitude below the current OSHA standard, but higher levels occur during renovation and demolition. A recent survey of 3978 indoor samples from 752 buildings that were the subject of litigation stemming from alleged asbestos exposure noted the following: (1) the average concentration of airborne asbestos 5 μm long or longer was 0.00012 fibers/cm3, (2) 99.9% of all samples contained less than 0.01 fibers/cm3, and (3) no asbestos was detected in 90% of the buildings when the analysis was restricted to optically detected fibers (≥5 μm long; ≥0.25 μm wide).31 These findings suggest that intact asbestos-containing materials in buildings pose a negligible risk to occupants being exposed to airborne asbestos levels above the OSHA PEL.
Several recent studies have documented more precisely the risks from environmental asbestos exposure. In 2007, Reid et al32 reported 67 cases of MM among 4,768 residents of Wittenoom, Western Australia, who never worked in the crocidolite asbestos mines or mills that operated between 1943 and 1966. Notably, Wittenoom crocidolite asbestos doubled the risk of MM at a cumulative level of 0.015 fibers/cm3/year, which is about 2 months exposure to OSHA PEL of 0.1 fibers/cm3.32 An increased standardized mortality ratio from MM was also reported in residents who lived as far as 2200 m downwind of an asbestos cement plant in Amagasaki, Japan.33 In the United States, residents of Libby, Montana, have an increased risk of developing asbestos-related lung diseases after exposure to vermiculite that is contaminated with up to 26% amphibole asbestos from a nearby mine that was active between 1920 and 1990.34 A cross-sectional radiographic screening of residents living in Libby conducted on 6668 subjects for the Agency for Toxic Substances and Disease Registry showed the following: (1) 1186 (17.8%) had pleural abnormalities, (2) the prevalence of pleural abnormalities was highest in WR Grace workers in the Libby vermiculite mine (51%) and lowest in residents who reported neither occupational nor domestic asbestos exposure (6.7%), and (3) some risk factors for pleural abnormalities included being a WR Grace worker, having household contact with a WR Grace worker, increasing age, duration of residence in Libby, and recreational contact with the vermiculite piles.35,36 Furthermore, 31 reported cases of MM resulted from nonoccupational exposure while residing in Libby with a latency period ranging from 13 to 67 years.37 A recent study evaluating data from 70 sites in 23 states where Libby vermiculite was used reported no significant increases in asbestosis mortality but detected 11 sites that had excess rates of MM.38 The latter findings represent a challenge because, as noted by Putnam et al,39 the Environmental Protection Agency estimates there are currently nearly 30 million homes in the United States with asbestos-containing vermiculite insulation. Collectively, these data show that environmental asbestos exposure is associated with an excess of asbestos-induced lung diseases, but this finding is about 10 times lower than that observed with occupational asbestos exposure.
Asbestos-induced bronchogenic lung cancer, which is similar to that caused by cigarette smoke, can occur in any lobe of the lung with a comparable distribution of the major histopathologic lung cancer types.40 Currently, no genetic test or biologic marker distinguishes lung cancer caused by asbestos or tobacco smoke. Asbestos is generally attributed as the cause of lung cancer in the setting of asbestosis with the appropriate latency period. A review of 23 studies exploring the causal relationship between asbestos and cigarette smoke in lung cancer concluded that asbestos causes lung cancer in nonsmokers despite the small numbers of such workers available for study.41 Moreover, a multiplicative or synergistic interaction rather than an additive model better described the association between asbestos and cigarette smoke in causing lung cancer. This synergistic interaction, which is not observed with MM, implies that the combined attributable lung cancer risk exceeds the sum of the risk of each agent. The mechanisms underlying this synergistic effect are not established, but implicated mechanisms include impaired lung fiber clearance and enhanced DNA damage.30,42
Controversy persists whether the excess lung cancer risk is limited to asbestos workers with asbestosis.43–47 It is generally agreed that the presence of asbestosis significantly increases the risk of lung cancer in a manner that is similar in patients with other forms of pulmonary fibrosis, especially idiopathic pulmonary fibrosis (IPF). Only 6.5% of 234 patients with lung cancer and asbestos exposure had pleural plaques without histologic asbestosis.40 A review of 34 asbestos cohort studies reported a direct correlation between the rate of asbestosis and lung cancer, which suggests that asbestosis is a better predictor of excess lung cancer risk than measures of asbestos exposure.46 Others have challenged this conclusion, arguing that asbestos exposure, and not asbestosis, causes lung cancer.47 These investigators point out that the presence of emphysema is not required to implicate cigarette smoke as a cause of lung cancer. Asbestos, which is a well-recognized carcinogen, can act on all the critical steps in the formation of a malignant clone of cells (eg, initiation, promotion, and progression) and, as such, is not dependent on the presence of fibrosis. Asbestos-exposed workers can have mutations in the k-ras gene at codon 12 in lung cancers without radiographic asbestosis, which suggests that these 2 events are not necessarily linked.3 Oksa et al48 showed that 11 of 24 patients with progressive asbestosis (46%) developed lung cancer, whereas only 5 of 54 patients with stable asbestosis (9%) developed lung cancer. They postulated that the progression of asbestosis is an independent predictor of lung cancer risk in patients with asbestosis. Thus, it remains unclear whether asbestosis is simply a marker of high-dose asbestos exposure or a necessary requirement for attributing an individual’s lung cancer to asbestos. This controversy is not likely to be resolved soon because of the nonuniform definition of asbestosis used in the various studies (eg, clinical–radiographic vs histopathologic) and the uncertain biologic scenario whereby the molecular mechanisms underlying interstitial fibrosis are required to develop a malignancy. Until more definitive studies have clarified these issues, lung cancer attribution should be based on the merits of each patient’s carcinogen exposure history combined with the appropriate clinical history and radiographic findings.
PATHOPHYSIOLOGY—WHAT’S NEW?
It is well established that the toxic effects of asbestos inhalation depend on the cumulative dose, the elapsed time since the initial exposure, and the physical-chemical properties of the different asbestos fibers.1–3,30 Amphibole fibers, as compared with chrysotile, are generally more toxic in part because amphibole fibers accumulate more readily in the distal lung parenchyma, are not cleared as effectively, and are more durable (estimated half-life in the lungs on the order of decades vs months, respectively).30 The “amphibole hypothesis” implicating that fiber structural characteristics (length, diameter, aspect ratio) are crucial for promoting asbestos-induced lung diseases has been challenged because fiber physical properties alone appear insufficient to account fully for asbestos toxicity.3,30 One confounder that has not been well controlled for is the presence of tremolite amphibole fibers frequently mixed with chrysotile. Although as noted above, chrysotile-induced malignancies are associated with at least a hundred-fold higher lung fiber concentration as compared with amphiboles, chrysotile can promote iron-derived free radical formation in vitro, can injure lung target cells, and can induce asbestosis, lung cancer, and mesothelioma in humans.4,21,22,30 As reviewed in detail elsewhere49–51, numerous studies over the past several years have examined whether biomarkers in asbestos-exposed workers are useful indicators of increased MM risk (eg, mesothelin, megokaryocyte potentiating factor, osteopontin, soluble mesothelin-related protein, and others). To date, none of the biomarkers studied haven proven useful as a screening test for MM because of a significant number of false-positive and false-negative tests. The translational utility of these biomarkers awaits the outcome of ongoing large prospective studies investigating the diagnostic accuracy and the relationship between biomarker levels and mortality.
After asbestos inhalation, alveolar epithelial cells (AEC), and alveolar macrophages (AM) rapidly internalize the fibers that result in an oxidative stress to the lungs. In lung epithelial cells, αvβ5 integrin receptor-mediated endocytosis seems crucial for fiber uptake and subsequent toxicity.52 Accumulating evidence has established that reactive oxygen species (ROS) and reactive nitrogen species (RNS) are important 2nd messengers of asbestos toxicity.3,30,42 The mechanisms underlying free radical generation by asbestos are in part caused by reactions occurring at the surface of mineral dusts, by activation of AMs or neutrophils attempting to take up the fibers and by mitochondrial dysfunction in target cells. Asbestos bodies are formed when the fibers are coated with a mucopolysaccharide as well as iron that can be redox active. Not surprisingly, asbestos-exposed individuals have altered iron homeostasis in the lungs as manifested by increased levels of bronchoalveolar lavage fluid (BALF), iron, transferrin, transferrin receptors, lactoferrin, and ferritin.53 Asbestos fibers induce the expression of ferritin heavy chain (FHC), a core subunit of ferritin, and an iron binding protein in human mesothelial cells and MM cell lines.54 FHC acts as an antiapoptotic protein against oxidative stress. Interestingly, the silencing of FHC using small interfering RNA promotes apoptosis in MM cells. Given the dismal response of MM to chemotherapy, these findings may have an important translational significance. Extracellular superoxide dismutase (EC-SOD), which is expressed in high levels in the lungs and limits the toxic effects of O2−, has recently been shown to be important for preventing both bleomycin- and asbestos-induced murine pulmonary fibrosis.55 More recently, the protective effects of EC-SOD against asbestos were shown to involve inhibiting oxidative fragmentation of hyaluronan in the extracellular matrix and the subsequent inflammatory response.56 Taken together, these studies suggest several novel strategies for limiting the toxic effects of oxidative stress in lungs exposed to asbestos.
DNA damage and apoptosis are important downstream deleterious effects of the ROS and RNS generated by asbestos and occur in all the major lung target cells studied to date.3,30,42 AEC apoptosis is an important early event implicated in the pathogenesis of pulmonary fibrosis from a variety of agents, which include asbestos, as well as in IPF and chronic obstructive pulmonary disease.3,42,57,58 These findings highlight the importance of understanding precisely how AEC apoptosis occurs. Several groups have shown that mitochondria-derived ROS mediate asbestos-induced AEC DNA damage and apoptosis via the mitochondria-regulated (intrinsic) death pathway.59–62 Although the molecular mechanisms are not fully established, 1 implicated mechanism involves protein kinase C-δ (PKC-δ). PKC-δ, which is 1 of at least 12 PKC isozymes, is specifically activated by asbestos, migrates to the mitochondria, and induces AEC intrinsic apoptosis.62 Notably, PKC-δ knockout mice are protected against asbestos-induced peribronchiolar epithelial cell proliferation and cytokine (eg, interleukin-1β(IL-1β), KC, IL-4, IL-6, IL-13, and others) production.63 These findings suggest that the targeted disruption of PKC-δ activation may have a role in the management of asbestos-induced lung diseases.
Another important mechanism that regulates AEC mitochondria-regulated apoptosis after asbestos exposure involves p53 activation.64 p53 is a tumor suppressor protein that is critically involved in the DNA damage cellular response by acting as a transcriptional factor to affect numerous genes that inhibit cell growth to allow time for DNA repair and, if DNA damage is extensive, augment apoptosis in part by the mitochondria-regulated death pathway (see references 65 and 66 for reviews). A normal-functioning p53 response after exposure to DNA damaging agents prevents mutations from accumulating. Not surprisingly, the most common mutations in human tumors involve the p53 gene family members.65,66 The precise mechanisms by which p53 regulates apoptosis are complex and not fully established despite extensive investigation. p53 induces apoptosis in part by activating the mitochondria-regulated death pathway by increasing gene expression of proapoptotic stimuli (eg, BAX, NOXA, PUMA, and others) while inhibiting the expression of antiapoptotic Bcl-2 family members.65,66 DNA damaging agents, which include asbestos, also induce a p53 transcriptional-independent mechanism of apoptosis by stabilizing p53 and promoting rapid mitochondrial p53 translocation.64–66 The DNA binding domain of wild-type p53 protein interacts with the Bcl-xl, which is an antiapoptotic protein, in the cytoplasm to trigger Bax/Bak-induced outer mitochondrial membrane permeabilization. Tumor-derived p53 mutants that block the interaction between p53 and Bcl-xl cause a “double hit” on the mitochondria-regulated apoptotic pathway by preventing both the transcriptional-dependent and direct mitochondrial effects of p53.65,66 A mechanistic link among mitochondria-derived ROS, altered iron homeostasis, and p53-induced apoptosis has been suggested after exposure to various agents including asbestos.64,67 Notably, phytic acid, which is an iron chelator, attenuates asbestos-induced p53 expression in distal alveolar cells and lung fibrosis in a rat model of asbestosis.64
Several lines of evidence support the translational significance of future studies exploring how p53 regulates asbestos-induced AEC apoptosis. First, altered p53 expression is implicated in the pathophysiology of pulmonary fibrosis, which includes that caused by asbestos exposure as well as asbestos-associated malignancies. Increased p53 protein expression occurs in the bronchiolar and alveolar epithelium of humans with IPF as well as rodents exposed to asbestos.64,68–70 p53 levels accumulate in lung cancers of patients with asbestosis.71,72 Second, crocidolite asbestos promotes p53 gene mutations predominantly in exons 9 through 11 in BALB/c-3T3 cells.73 Third, asbestos induces p53 and p21CIP1/WAF1/SD11 (p21) expression in lung epithelial and mesothelial cells that can result in cell cycle arrest.74–76 p21 is a downstream family of proteins (p21, p27, and p57) expressed during a p53 genotoxic stress response that results in cell cycle arrest to allow time for DNA repair.77 The importance of targeting p21 has recently been suggested by studies showing that p21 regulates lifespan in telomerase-deficient mice prone to premature aging, augments pulmonary inflammation caused by oxidative stress, and mediates transforming growth factor-β (TGF-β)–induced pulmonary inflammation and fibrosis that occurs via tumor necrosis factor-α (TNF-α) signaling.78–80 Finally, using microarrays, hierarchical clustering analyses, and a systems biology approach to examine asbestos-induced AEC whole genome expression profiling (~54,000 genes), a crucial role for p53 activation was confirmed along with the activation of nearly 2500 other genes involved with regulating tumor suppression, cell cycle arrest, apoptosis/antiproliferation, and cell survival/antiapoptosis.81 In another gene expression profile study, asbestos-exposed epithelial and mesothelial lung cell lines had increased expression of p53 as well as PKC-δ, thioredoxin, and many other intriguing genes that warrant more investigation.82 Some important targets identified from these and other studies mentioned in this review are shown in Fig 1. Collectively, these data firmly implicate a prominent role for p53 in mediating asbestos-induced AEC mitochondrial dysfunction and apoptosis; these data also suggest that iron-derived ROS from the mitochondria have an important role in activating p53. Studies exploring the genetics of the p53 pathway and its negative regulator MDM2 are identifying novel targeted therapies that may prove useful for managing various cancers.83 Future studies are warranted to define the crucial downstream p53 targets important for mediating the apoptotic effects of p53 after asbestos exposure that, ideally, would not alter the multiple beneficial roles of p53 in DNA repair and cell survival.
Fig 1.
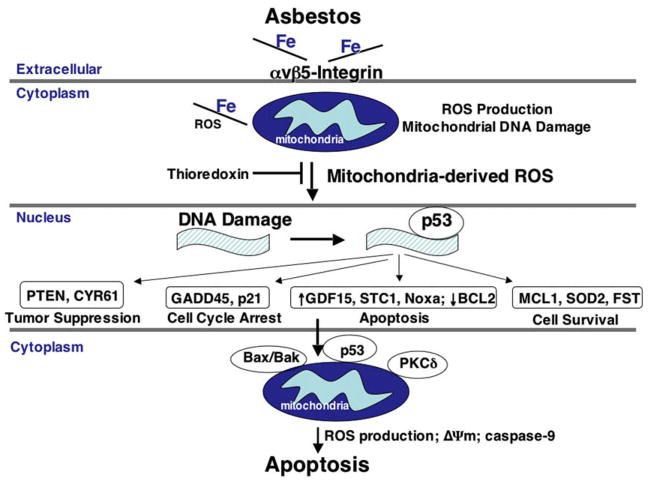
Hypothetical model depicting some of the crucial events that lead to asbestos-induced alveolar epithelial cell mitochondria-regulated apoptosis. Asbestos, which is an iron containing fiber, is rapidly internalized via the αvβ5 integrin receptor and induces mitochondria-derived ROS production. By mechanisms that are still uncertain, mitochondrial ROS signaling that results from asbestos exposure stabilizes p53 and promotes p53-dependent transcription of a variety of important proteins involved with tumor suppression, cell cycle arrest, apoptosis, and cell survival. Asbestos-induced AEC intrinsic apoptosis is augmented by mitochondrial translocation of proapoptotic Bcl-2 family members (eg, Bax and Bak) p53 and PKCδ. The model is a modified version of one initially developed by Hevel et al81 that incorporates their findings as well as the work of others.52,59–62,64,81,82 (Color version of figure is available online.)
It is well known that asbestos-induced mitogen activated protein kinase (MAPK) pathway members are important 2nd messengers crucial for mediating cell surface signals to the nucleus. Recent studies using human monocytes show that increased TNF-α gene expression in patients with asbestosis is caused by constitutively activated p38 and decreased phosphorylated extracellular signal-regulated kinase (ERK) MAPK when compared with normal subjects.84 Furthermore, the reduced ERK signaling was caused by increased lung MAPK phosphatase (MKP)-3 activity. In murine lung epithelium, the disruption of MAPK-1 using a dominant-negative transgene targeted to the bronchiolar epithelium with the CC10 promoter inhibits asbestos-induced proliferation and procollagen gene expression.85 Inactivation of the Akt and MAPK pathways mediates asbestos-induced AEC apoptosis, which is a potentially crucial step for promoting a fibrotic response.86 However, the Akt/mammalian target of rapamycin (mTOR) signaling pathways augment tumor formation in part by promoting the formation of cells that are resistant to apoptosis.87,88 A provocative recent study showed that mTOR increases survival of MM cells and that rapamycin (a mTOR inhibitor) or silencing p–S6K (a major downstream target of mTOR), can each augment cell death of these otherwise apoptosis-resistant MM cells.89 Thus, the targeted disruption of MKP-3 or activated p38 in macrophages to limit TNF-α gene expression or regulating Akt/MAPK/mTOR signaling in the lung epithelium or in MM cells are novel approaches that merit additional study.
A murine laser capture microdissection study has recently documented that asbestos induces gene expression (TNF-α, TGF-β, and others) at the bronchiolar-alveolar duct junctions and first alveolar duct bifurcation; this location is precisely where fiber deposition is greatest.90 In animal models of asbestosis, the development of fibrosis directly correlates with TNF-α levels, and TNF-α receptor knockout mice are protected against asbestosis.91,92 TNF-α is also prominently implicated in the direct association between inflammation and malignancy because it is crucial for mediating tumor promotion and cell transformation in various model systems.93,94 In human mesothelial cells, TNF-α prevents asbestos-induced cell death via a nuclear factor-κB (NF-κB)-dependent mechanism despite the presence of cytogenetic abnormalities.95 Using transgenic (Tg) mice expressing an inhibitory κB (IκB) mutant resistant to phosphorylation-induced degradation and targeted to the bronchiolar epithelium via the CC10 promoter in a murine model of asbestosis, Tg(+) mice exposed to asbestos had less BALF inflammatory cytokine levels (eg, KC, IL-6, and IL-1β) and reduced levels of bronchiolar and distal epithelial proliferation as compared with Tg(−) mice.96 Two groups have recently shown that bone marrow progenitor cells contribute to the inflammatory and fibrogenic effects of asbestos.97,98 These studies underscore the importance of TNF-α, the NF-κB–dependent pathway, and bone marrow progenitor cells in the pathogenesis of asbestos-induced lung disease that may lead to novel therapeutic strategies.
Another recently described mechanism by which asbestos and silica, but not inert particulates, activate pulmonary inflammation and fibrosis is via Nalp3 inflammasome sensing.99,100 Nalp3, which can activate caspase-1 in response to diverse stimuli, is a member of the NLR family of over 20 proteins that contain an N-terminal protein-protein interaction domain that consists of a caspase activation and recruitment domain (CARD), a central nucleotide-binding NACHT, and a C-terminal leucine-rich repeat domain.101 The Nalp3 inflammasome is formed when Nalp3 activation recruits the adaptor molecule apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (ASC) and then caspase-1 by CARD–CARD interactions. Notably, asbestos-induced inflammatory cell lung recruitment, cytokine production (IL-1β and KC), and silicosis are all reduced in mice deficient in Nalp3, ASC, or caspase-1.99,100 Furthermore, using specific inhibitors and select knockout mice, an important role for fiber uptake, an intact actin cytoskeleton, and ROS generated by nicotinamide adenine dinucleotide phosphate oxidase during phagocytosis were shown to be necessary. Thus, asbestos-induced Nalp3 inflammasome activation is an innovative therapeutic target with clear translational significance.
As reviewed in detail elsewhere, increased TGF-β expression and decreased expression of bone morphogenetic proteins (BMPs) are associated with the development of epithelial-mesenchymal transition (EMT) important in the pathogenesis of cancer and fibrosis, which includes IPF.102 A recent study has shown that gremlin, which is a BMP antagonist that is overexpressed in patients with IPF,103 is also overexpressed in patients with asbestosis as well as in a murine model of asbestosis.104 Notably, asbestos-induced gremlin expression could be blocked by inhibitors of the TGF-β receptor or the MAPK pathway as well as by exogenously administered BMP-7, which is known to prevent TGF-β-induced EMT and fibrosis in the kidney and liver.105,106 Collectively, these findings emphasize the importance of the balance between TGF-β and BMP signaling in the development of asbestosis as well as other fibrotic disorders. Also, restoration of lung BMP signaling activity is a novel therapeutic approach worthy of additional investigation.
CONCLUSIONS
Asbestos-related lung diseases remain a significant challenge to health care providers as well as to investigators studying the basic mechanisms that underlie asbestos-induced pulmonary toxicity. Given the long latency between asbestos exposure and disease as well as the direct relationship between asbestos consumption and mortality from asbestos-related lung diseases, a total worldwide asbestos ban is strongly supported.6,8,17,18 If asbestos use continues in countries where it is less regulated, we can expect the asbestos-induced lung disease crisis to continue for most of the 21st century.6 Studies published in the last several years have yielded unique insights into the epidemiologic significance of asbestos exposure as well as the basic mechanisms that account for asbestos-induced pulmonary toxicity. The significance of these investigations is that they provide the molecular basis for developing novel treatment strategies for asbestos-related lung diseases. Importantly, these studies demonstrate that the asbestos paradigm has broad translational implications for our understanding of other pulmonary diseases where comparable mechanistic pathways are implicated and for which innovative management approaches are urgently required.
Acknowledgments
The author acknowledges the helpful comments provided by Dr. David W. Cugell.
Supported by a Merit Review grant from the Department of Veterans Affairs.
Abbreviations
- AEC
alveolar epithelial cell
- AM
alveolar macrophages
- ASC
apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain
- BALF
bronchoalveolar lavage fluid
- BMP
bone morphogenetic protein
- CARD
caspase activation and recruitment domain
- EC-SOD
extracellular superoxide dismutase
- EMT
epithelial-mesenchymal transition
- ERK
extracellular signal-related kinase
- FHC
ferritin heavy chain
- IL
interleukin
- iκB
inhibitory κB
- IPF
idiopathic pulmonary fibrosis
- MAPK
mitogen actvated protein kinase
- MKP-3
MAPK phosphatase-3
- MM
malignant mesothelioma
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor-κB
- OSHA
Occupational Safety and Health Administration
- PEL
permissive exposure limits
- PKC-δ
protein kinase C-δ
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Tg
transgenic
- TGF-β
transforming growth factor-β
- TNF-α
tumor necrosis factor-α
References
- 1.Cugell DW, Kamp DW. Asbestos and the pleura: a review. Chest. 2004;125:1103–17. doi: 10.1378/chest.125.3.1103. [DOI] [PubMed] [Google Scholar]
- 2.Guidotti TL, Miller A, Christiani D, et al. Diagnosis and initial management of nonmalignant diseases related to asbestos. Am J Respir Crit Care Med. 2004;170:691–715. doi: 10.1164/rccm.200310-1436ST. [DOI] [PubMed] [Google Scholar]
- 3.Kamp DW, Mossman BT. Asbestos-associated cancers: clinical spectrum and pathogenic mechanisms. Clin Occup Environ Med. 2002;2:753–78. [Google Scholar]
- 4.Hein MJ, Staynor LT, Lehman E, Dement JM. Follow-up study of chrysotile textile workers: cohort mortality and exposure-response. Occup Environ Med. 2007;64:616–25. doi: 10.1136/oem.2006.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooke WE. Pulmonary asbestosis. Br Med J. 1927;2:1024–5. doi: 10.1136/bmj.2.3491.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner GR. The fallout from asbestos. Lancet. 2007;369:973–4. doi: 10.1016/S0140-6736(07)60472-3. [DOI] [PubMed] [Google Scholar]
- 7.Murphy RL, Becklake MR, Brooks SM, et al. The diagnosis of nonmalignant diseases related to asbestos. Am Rev Respir Dis. 1986;134:363–8. doi: 10.1164/arrd.1986.134.2.363. [DOI] [PubMed] [Google Scholar]
- 8.Lin R-T, Takahashi K, Karjalainen A, et al. Ecological associations between asbestos-related diseases and historical asbestos consumption: an international analysis. Lancet. 2007;369:844–9. doi: 10.1016/S0140-6736(07)60412-7. [DOI] [PubMed] [Google Scholar]
- 9.Welch LS, Haile E, Dement J, Michaels D. Change in the prevalence of asbestos-related disease among sheet metal workers 1986 to 2004. Chest. 2007;131:863–9. doi: 10.1378/chest.06-1155. [DOI] [PubMed] [Google Scholar]
- 10.O’Reilly KMA, McLaughlin AM, Beckett WS, Sime PJ. Asbestos-related lung disease. Am Fam Physician. 2007;75:683–8. [PubMed] [Google Scholar]
- 11.Nicholson WJ, Perkel G, Selikoff IJ. Occupational exposure to asbestos: population at risk and projected mortality; 1980–2030. Am J Ind Med. 1982;3:259–311. doi: 10.1002/ajim.4700030305. [DOI] [PubMed] [Google Scholar]
- 12.LaDou FJ. The asbestos cancer epidemic. Environ Health Perspect. 2004;112:285–90. doi: 10.1289/ehp.6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tossavainen A. Global use of asbestos and the incidence of mesothelioma. Int J Occup Environ Health. 2004;10:22–5. doi: 10.1179/oeh.2004.10.1.22. [DOI] [PubMed] [Google Scholar]
- 14.Robinson BWS, Lake RA. Advances in malignant mesothelioma. N Engl J Med. 2005;353:1591–603. doi: 10.1056/NEJMra050152. [DOI] [PubMed] [Google Scholar]
- 15.Peto J, Doll R, Herman C, Binns W, Clayton R, Goffe T. Relationship of mortality to measures of environmental asbestos pollution in an asbestos textile factory. Ann Occup Hyg. 1985;29:305–55. doi: 10.1093/annhyg/29.3.305. [DOI] [PubMed] [Google Scholar]
- 16.McDonald JC, Sebastien P, McDonald AD, et al. Epidemiological observations on mesothelioma and the implications for nonoccupational exposure. In: Bignon J, Peto J, Saracchi R, editors. Non-occupational exposure to mineral fibers. IARC Science Publication 90. Lyon, France: International Agency for Research on Cancer; 1989. [Google Scholar]
- 17.World Health Organization. Summary consensus report of WHO workshop on mechanisms of fibre carcinogenesis and assessment of chrysotile asbestos substitutes. Lyon France: World Health Organization; Nov-Dec. 2005. [Google Scholar]
- 18.LaDou J, Landrigan P, Bailar JC., III A call for an international ban on asbestos. CMAJ. 2001;164:489–90. [PMC free article] [PubMed] [Google Scholar]
- 19.Yarborough CM. The risk of mesothelioma from exposure to chrysotile asbestos. Curr Opin Pulm Med. 2007;13:334–8. doi: 10.1097/MCP.0b013e328121446c. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein DM, Hoskins JA. The health effects of chrysotile: current perspective based upon recent data. Regul Toxicol Pharmacol. 2006;45:252–64. doi: 10.1016/j.yrtph.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Hodgson JT, Darnton A. The quantitative risk of mesothelioma and lung cancer in relation to asbestos exposure. Ann Occup Hyg. 2000;44:565–601. [PubMed] [Google Scholar]
- 22.Roggli VL. Environmental asbestos contamination: what are the risks? Chest. 2007;131:336–8. doi: 10.1378/chest.06-2649. [DOI] [PubMed] [Google Scholar]
- 23.Iwatsubo Y, Pairon JC, Boutin C, et al. Pleural mesothelioma, dose-response relation at low levels of asbestos exposure in a French population-based case-control study. Am J Epidemiol. 1998;148:133–42. doi: 10.1093/oxfordjournals.aje.a009616. [DOI] [PubMed] [Google Scholar]
- 24.Merchant JA. Human epidemiology: a review of fiber type and characteristics in the development of malignant and nonmalignant disease. Environ Health Perspect. 1990;88:287–93. doi: 10.1289/ehp.9088287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller A, Lilis R, Godbold J, Chan E, Selikoff IJ. Relationship of pulmonary function to radiographic interstitial fibrosis in 2,611 long-term asbestos insulators. Am Rev Respir Dis. 1992;145:263–70. doi: 10.1164/ajrccm/145.2_Pt_1.263. [DOI] [PubMed] [Google Scholar]
- 26.Rogan WJ, Ragan NB, Dinse GE. X-ray evidence of increased asbestos exposure in the US population from NHANES I and NHANES II, 1973–1978. Cancer Causes Control. 2000;11:441–9. doi: 10.1023/a:1008952426060. [DOI] [PubMed] [Google Scholar]
- 27.Weiss W. Asbestos-related pleural plaques and lung cancer. Chest. 1993;103:1854–9. doi: 10.1378/chest.103.6.1854. [DOI] [PubMed] [Google Scholar]
- 28.Henderson DW, Rantanen J, Barnhart S, et al. Asbestos, asbestosis, and cancer: the Helsinki criteria for diagnosis and attribution; a consensus report of an international expert group. Scand J Work Environ Health. 1997;28:311–6. [PubMed] [Google Scholar]
- 29.Barbers RG, Abraham JL. Asbestos occurring after a brief inhalational exposure: usefulness of bronchoalveolar lavage in diagnosis. Br J Ind Med. 1989;46:106–10. doi: 10.1136/oem.46.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157:1666–80. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 31.Lee RJ, Van Orden DR. Airborne asbestos in buildings. Regul Toxicol Phamacol. 2008;50:218–25. doi: 10.1016/j.yrtph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Reid A, Berry G, de Klerk N, et al. Age and sex differences in malignant mesothelioma after residential exposure to blue asbestos (crocidolite) Chest. 2007;131:376–82. doi: 10.1378/chest.06-1690. [DOI] [PubMed] [Google Scholar]
- 33.Kurumatani N, Kumagai S. Mapping the risk of mesothelioma due to neighborhood asbestos exposure. Am J Respir Crit Care Med. 2008;178:624–9. doi: 10.1164/rccm.200801-063OC. [DOI] [PubMed] [Google Scholar]
- 34.Noonan CW. Exposure matrix development for Libby cohort. Inhal Toxicol. 2006;18:963–7. doi: 10.1080/08958370600835021. [DOI] [PubMed] [Google Scholar]
- 35.Peipins LA, Lewin M, Campolucci S, et al. Radiographic abnormalities and exposure to asbestos-contaminated vermiculite in the community of Libby, Montana, USA. Environ Health Perspect. 2003;111:1753–9. doi: 10.1289/ehp.6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price B. Exposure to airborne amphibole structures and health risks: Libby, Montana. Regul Toxicol Pharmacol. 2008;51:S97–109. doi: 10.1016/j.yrtph.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Whitehouse AC, Black CB, Heppe MS, Ruckdeschel J, Levin SM. Environmental exposure to Libby asbestos and mesotheliomas. Am J Indust Med. 2008;51:877–880. doi: 10.1002/ajim.20620. [DOI] [PubMed] [Google Scholar]
- 38.Horton DK, Bove F, Kapil V. Select mortality and cancer incidence among residents in various U.S. communities that received asbestos-contaminated ore from Libby. Montana Inhal Toxicol. 2008;20:767–75. doi: 10.1080/08958370801983240. [DOI] [PubMed] [Google Scholar]
- 39.Putnam EA, Smartt A, Groves A, Schwanke C, Brezinski M, Perhouse MA. Gene expression changes after exposure to six-mix in a mouse model. J Immunotoxicol. 2008;5:139–44. doi: 10.1080/15476910802085772. [DOI] [PubMed] [Google Scholar]
- 40.Roggli VL, Sanders LL. Asbestos content of lung tissue and carcinoma of the lung: a clinicopathologic correlation and mineral fiber analysis of 234 cases. Ann Occup Hyg. 2000;44:109–17. [PubMed] [Google Scholar]
- 41.Lee PN. Relation between exposure to asbestos and smoking jointly and the risk of lung cancer. Occup Environ Med. 2001;58:145–53. doi: 10.1136/oem.58.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla A, Gulumian M, Hei T, Kamp D, Rahman Q, Mossman BT. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Radic Biol Med. 2003;34:1117–29. doi: 10.1016/s0891-5849(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 43.Hessel PA, Gamble JF, McDonald JC. Asbestos, asbestosis, and lung cancer: a critical assessment of the epidemiological evidence. Thorax. 2005;60:433–36. doi: 10.1136/thx.2004.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibbs A, Attanoos RL, Churg A, Weill H. The “Helsinki Criteria” for attribution of lung cancer to asbestos exposure. Arch Pathol Lab Med. 2007;131:181–84. doi: 10.5858/2007-131-181-THCFAO. [DOI] [PubMed] [Google Scholar]
- 45.Henderson DW, Rodelsperger K, Woitowitz HJ, Leigh J. After Helsinki: a multidisciplinary review of the relationship between asbestos exposure, with emphasis on studies published during 1997–2004. Pathology. 2004;36:517–50. doi: 10.1080/00313020400010955. [DOI] [PubMed] [Google Scholar]
- 46.Weiss W. Asbestosis: a marker for the increased risk of lung cancer among workers exposed to asbestos. Chest. 1999;115:536–49. doi: 10.1378/chest.115.2.536. [DOI] [PubMed] [Google Scholar]
- 47.Henderson DW, Roggli VL, Shilkin KB, et al. Is asbestosis an obligate precursor for asbestos-related lung cancer? In: Peters GA, Peters BJ, editors. Asbestos health effects, treatment and control. Charlottesville, VA: The Michie Co; 1995. [Google Scholar]
- 48.Oksa P, Klockars M, Karjalainen A, et al. Progression of asbestosis predicts lung cancer. Chest. 1998;113:1517–21. doi: 10.1378/chest.113.6.1517. [DOI] [PubMed] [Google Scholar]
- 49.Creaney J, Yeoman D, Demelker Y, et al. Comparison of osteopontin, megakaryocyte potentiating factor, and mesothelin proteins as markers in the serum of patients with malignant mesothelioma. J Thorac Oncol. 2008;3:851–7. doi: 10.1097/JTO.0b013e318180477b. [DOI] [PubMed] [Google Scholar]
- 50.Amati M, Tomasetti M, Mariotti L, Tarquini LM, Valentino M, Santarelli L. Assessment of biomarkers in asbestos-exposed workers as indicators of cancer risk. Mutat Res/Gene Toxicol and Environ Mutag. 2008;655:52–8. doi: 10.1016/j.mrgentox.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 51.van Meerbeeck JP, Hillerdal G. Screening for mesothelioma: more harm than good. Am J Respir Crit Care Med. 2008;178:781–2. doi: 10.1164/rccm.200806-955ED. [DOI] [PubMed] [Google Scholar]
- 52.Pande P, Mosleh TA, Aust AE. Role of αvβ5 integrin receptor in endocytosis of crocidolite and its effect on intracellular glutathione levels in human lung epithelial (A549) cells. Toxicol Appl Pharmacol. 2006;210:70–7. doi: 10.1016/j.taap.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 53.Ghio AJ, Stonehuerner J, Richards J, Devlin RB. Iron homeostasis in the lung following asbestos exposure. Antioxid Redox Signal. 2008;10:371–7. doi: 10.1089/ars.2007.1909. [DOI] [PubMed] [Google Scholar]
- 54.Aung W, Hasegawa S, Furukawa T, Saga T. Potential role of ferritin heavy chains in oxidative stress and apoptosis in human mesothelial and mesothelioma cells: implications for asbestos-induced carcinogenesis. Carcinogenesis. 2007;28:2047–52. doi: 10.1093/carcin/bgm090. [DOI] [PubMed] [Google Scholar]
- 55.Fattman CL, Tan RJ, Tobolewski JM, Oury TD. Increased sensitivity to asbestos-induced lung injury in mice lacking extracellular superoxide dismutase. Free Radic Biol Med. 2006;40:601–7. doi: 10.1016/j.freeradbiomed.2005.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao F, Koenitzer JR, Tobolewski JM, et al. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J Biol Chem. 2008;283:6058–66. doi: 10.1074/jbc.M709273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee CG, Cho SJ, Kang MJ, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor B1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–89. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–46. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aljandali A, Pollack H, Yeldandi A, Li Y, Weitzman SA, Kamp DW. Asbestos causes apoptosis in alveolar epithelial cells: role of iron-induced free radicals. J Lab Clin Med. 2001;137:330–9. doi: 10.1067/mlc.2001.114826. [DOI] [PubMed] [Google Scholar]
- 60.Panduri V, Weitzman SA, Chandel N, Kamp DW. The mitochondria-regulated death pathway mediates asbestos-induced alveolar epithelial cell apoptosis. Am J Respir Cell Mol Biol. 2003;28:241–8. doi: 10.1165/rcmb.4903. [DOI] [PubMed] [Google Scholar]
- 61.Panduri V, Weitzman SA, Chandel N, Kamp DW. Mitochondria-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1220–7. doi: 10.1152/ajplung.00371.2003. [DOI] [PubMed] [Google Scholar]
- 62.Shukla A, Stern M, Lounsbury KM, Flanders T, Mossman BT. Asbestos-induced apoptosis is protein kinase Cδ-dependent. Am J Respir Cell Mol Biol. 2003;29:198–205. doi: 10.1165/rcmb.2002-0248OC. [DOI] [PubMed] [Google Scholar]
- 63.Shukla A, Lounsbury KM, Barrett TF, et al. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase Cδ knockout mice. Am J Pathol. 2007;170:140–51. doi: 10.2353/ajpath.2007.060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Panduri V, Surapureddi S, Soberanes S, Weitzman SA, Chandel N, Kamp DW. p53 mediates amosite asbestos induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Cell Mol Biol. 2006;34:443–52. doi: 10.1165/rcmb.2005-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olsson A, Manzi C, Strasser A, Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumor suppression? Cell Death Differ. 2007;14:1561–75. doi: 10.1038/sj.cdd.4402196. [DOI] [PubMed] [Google Scholar]
- 66.Janicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15:959–76. doi: 10.1038/cdd.2008.33. [DOI] [PubMed] [Google Scholar]
- 67.Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaroson SA. Influence of reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003;23:8576–85. doi: 10.1128/MCB.23.23.8576-8585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuwano K, Kunitake R, Kawasaki M, et al. p21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154:477–83. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 69.Mishra A, Liu J-Y, Brody AR, Morris GF. Inhaled asbestos fibers induce p53 expression in the rat lung. Am J Respir Cell Mol Biol. 1997;16:479–85. doi: 10.1165/ajrcmb.16.4.9115760. [DOI] [PubMed] [Google Scholar]
- 70.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–74. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 71.Nuorva K, Makitaro R, Huhti E, et al. p53 protein accumulation in lung carcinomas of patients exposed to asbestos and tobacco smoke. Am J Respir Crit Care Med. 1994;150:528–33. doi: 10.1164/ajrccm.150.2.8049841. [DOI] [PubMed] [Google Scholar]
- 72.Husgafvel-Oursiainen K, Kannio A, Oksa P, et al. Mutations, tissue accumulation and serum levels of p53 in patients with occupational cancers from asbestos and silica exposure. Environ Mol Mutagen. 1997;30:224–30. [PubMed] [Google Scholar]
- 73.Lin F, Liu Y, Liu Y, Keshava N, Li S. Crocidolite induces cell transformation and p53 gene mutation in BALB/c-3T3 cells. Teratog Carcinog Mutag. 2000;20:273–81. doi: 10.1002/1520-6866(2000)20:5<273::aid-tcm3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 74.Johnson NF, Jaramillo RJ. p53, Cip1, and Gadd153 expression following treatment of A549 cells with natural man-made vitreous fibers. Environ Health Persp. 1997;105:1143–5. doi: 10.1289/ehp.97105s51143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levresse V, Renier A, Fleury-Feith J, et al. Analysis of cell cycle disruptions in cultures of rat pleural mesothelial cells exposed to fibers. Am J Respir Cell Mol Biol. 1997;17:660–71. doi: 10.1165/ajrcmb.17.6.2854. [DOI] [PubMed] [Google Scholar]
- 76.Paakko P, Ramet M, Vahakangas K, et al. Crocidolite asbestos causes an induction of p53 and apoptosis in cultured A549 lung carcinoma cells. Apoptosis. 1998;3:203–12. doi: 10.1023/a:1009655007284. [DOI] [PubMed] [Google Scholar]
- 77.Tuder RM, Yun JH, Graham BB. Cigarette smoke triggers code red: p21 switches on danger responses in the lung. Am J Respir Cell Mol Biol. 2008;39:1–6. doi: 10.1165/rcmb.2008-0117TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choudhury AR, Ju Z, Djojosubroto MW, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 79.Yao H, Yang S-R, Edirisinghe I, et al. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol. 2008;39:7–18. doi: 10.1165/rcmb.2007-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamasaki M, Kang HR, Homer RJ, et al. P21 regulates TGF-β1-induced pulmonary responses via a TNFα signaling pathway. Am J Respir Cell Mol Biol. 2008;38:346–53. doi: 10.1165/rcmb.2007-0276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hevel JM, Olson-Buelow LC, Ganesan B, Stevens JR, Hardman JP, Aust AE. Novel functional view of the crocidolite asbestos-treated A549 human lung epithelial transcriptome reveals an intricate network of pathways with opposing functions. BMC Genomics. 2008;9:376. doi: 10.1186/1471-2164-9-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nymark P, Lindholm PM, Korpela MV, et al. Gene expression profiles in asbestos-exposed epithelial and mesothelial cell lines. BMC Genomics. 2007;8:62. doi: 10.1186/1471-2164-8-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 84.Tephly LA, Carter AB. Asbestos-induced MKP-3 expression augments TNF-α gene expression in human monocytes. Am J Respir Cell Mol Biol. 2008;39:113–23. doi: 10.1165/rcmb.2007-0356OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manning CB, Sabo-Attwood T, Robledo RF, et al. Targeting the MEK1 cascade in lung epithelium inhibits proliferation and fibrogenesis by asbestos. Am J Respir Cell Mol Biol. 2008;38:618–26. doi: 10.1165/rcmb.2007-0382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baldys A, Pande P, Mosleh T, Park S-H, Aust AE. Apoptosis induced by crocidolite asbestos in human lung epithelial cells involves inactivation of Akt and MAPK pathways. Apoptosis. 2007;12:433–47. doi: 10.1007/s10495-006-0577-8. [DOI] [PubMed] [Google Scholar]
- 87.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 88.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–42. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Wilson SM, Barbone D, Yamg T-M, et al. MTOR mediates survival signals in malignant mesothelioma grown as tumor fragment spheroids. Am J Respir Cell Mol Biol. 2008;39:576–83. doi: 10.1165/rcmb.2007-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yin Q, Brody AR, Sullivan DE. Laser capture microdissection reveals dose-response of gene expression in situ consequent to asbestos exposure. Int J Exp Path. 2007;88:415–25. doi: 10.1111/j.1365-2613.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lemaire I, Oullet S. Distinctive profile of alveolar macrophage-derived cytokine release induced by fibrogenic and nonfibrogenic mineral dusts. J Toxicol Environ Health. 1996;47:465–78. doi: 10.1080/009841096161618. [DOI] [PubMed] [Google Scholar]
- 92.Liu JY, Brass DM, Hoyle GW, Brody AR. TNF-α receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol. 1998;153:1839–47. doi: 10.1016/s0002-9440(10)65698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suganuma M, Okabe S, Marino MW, Sakai A, Sueoka E, Fujiki H. Essential role of tumor necrosis factor alpha (TNF-α) in tumor promotion as revealed by TNF-α-deficient mice. Cancer Res. 1999;59:4516–8. [PubMed] [Google Scholar]
- 94.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25:409–16. doi: 10.1007/s10555-006-9005-3. [DOI] [PubMed] [Google Scholar]
- 95.Yang H, Bocchetta M, Kroczynska B, et al. TNF-alpha inhibits asbestos-induced cytotoxicity via a NF-kappaB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc Natl Acad Sci U S A. 2006;103:10397–402. doi: 10.1073/pnas.0604008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haegens AB, Gell TF, Shukla J, et al. Airway epithelial NF-κB activation modulates asbestos-induced inflammation and mucin production in vivo. J Immunol. 2007;178:1800–8. doi: 10.4049/jimmunol.178.3.1800. [DOI] [PubMed] [Google Scholar]
- 97.Spees JL, Pociask DA, Sullivan DE, et al. Engraftment of bone marrow progenitor cells in a rat model of asbestos-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:385–94. doi: 10.1164/rccm.200607-1004OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levis J, Loi R, Butnor KJ, et al. Decreased asbestos-induced lung inflammation and fibrosis after radiation and bone marrow transplant. Am J Respir Cell Mol Biol. 2008;38:16–25. doi: 10.1165/rcmb.2007-0249OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:935–40. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 102.Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs. PLoS Med. 2008;5:373–80. doi: 10.1371/journal.pmed.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koli K, Myllarniemi M, Vuorinen K, et al. Bone morphogenetic protein-4 inhibitor gremlin is over-expressed in idiopathic pulmonary fibrosis. Am J Pathol. 2006;169:61–71. doi: 10.2353/ajpath.2006.051263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Myllarniemi M, Lindholm P, Ryynanen MJ, et al. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:321–9. doi: 10.1164/rccm.200706-945OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 106.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derived from hepatocytes in liver fibrosis via epithelial-to-mesenchymal transition. J Biol Chem. 2007;282:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]