

Abstract
Solid-state CPL measurements were performed for the first time on hybrid, laminar materials based on γ-ZrP pillared with organic diphosphonates. Ad hoc optically pure diphosphonates were synthesized and the luminescence properties of their complexation with Tb(III) were verified in solution. CD and CPL measurements showed that the bistriazolylpyridine chromophores bonded to the metal provided an effective chiral environment that produced significant signals. In the case of the γ-ZrP-derived materials, experimental evidence and simple molecular modeling hinted to the occurrence of supramolecular chirality in the particles, induced by the intrinsic dissymmetry of the organic diphosphonates or by the intercalation of chiral species such as 1-phenethylamine. Chirality at the supramolecular level was revealed in the solid state by the CPL signals measured from reporter Tb(III) ions intercalated in the hybrid matrix.
Keywords: Zirconium phosphate, Laminar materials, Supramolecular chirality, Circularly polarized luminescence, Lanthanide luminescence
1. Introduction
The structure and chemistry of layered materials is fascinating. One excellent example is that of zirconium phosphate and its gamma allotropic form (γ-ZrP). The molecular formula of this salt [Zr(PO4)(H2PO4)] and its laminar structure (Fig. 1) is constituted by two kinds of phosphate groups. Those at the surface of both faces of a layer are bonded to only two Zr metal atoms and are amenable of topotactic exchange reactions with other phosphorus functions such as organic phosphonates. The phosphates interior to the lamellae are no less important. They are responsible for preserving the structure of the layers when reactions occur at their surface. Furthermore, these internal phosphates are bonded to four different Zr atoms and are thus stereogenic centers. The structure of the lamellae of γ-ZrP is therefore intrinsically dissymmetric [1]. Yet, as it happens in racemic mixtures or meso forms, no in-bulk optical activity is expected unless any additional stereocenters of fixed configuration are introduced. In previous work, we have shown that the simple intercalation of an optically pure molecule, such as (+)-1-phenethylamine [(+)-PEA], produces a material showing an optical rotation three orders of magnitude larger than the parent amine in solution [2].
Fig. 1.
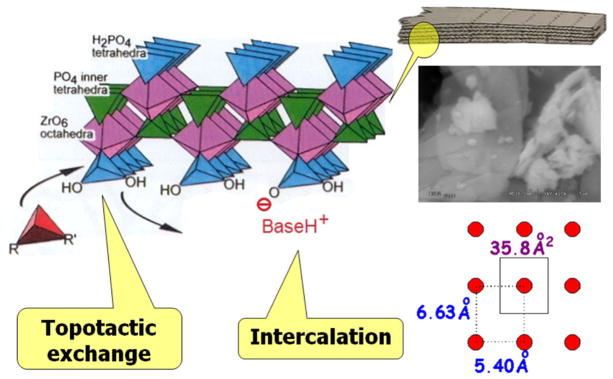
SEM image of γ-ZrP and polyhedral model of a portion of one layer of it, sketching the topotactic exchange and intercalation processes; the scheme at the bottom-right shows the available area around every surface phosphate.
The pillaring of γ-ZrP with either symmetric of dissymmetric diphosphonates has led to interesting stereochemical observations. For instance, the topotactic reaction of γ-ZrP with 1 (Scheme 1) produced a pillared material whose interlaminar distance could be greatly varied in the solid-water interface by simple acid–base reactions. At the maximum basal separation, the ethylenpolyoxa columns are in the usual helicoid conformation. However, no optical rotation was observed because the amine used to cause the elongation was not dissymmetric and the P/M helicity was random. When the base employed to increase the interlayer separation was (+)-PEA a sizeable optical rotation was measured. Interestingly, when (+)-PEA was smoothly replaced with hexylamine, the material still exhibited optical rotation. This suggests that the homohelicity of the columns induced by the chiral (+)-PEA was retained when the latter was replaced by the symmetric hexylamine.
Scheme 1.
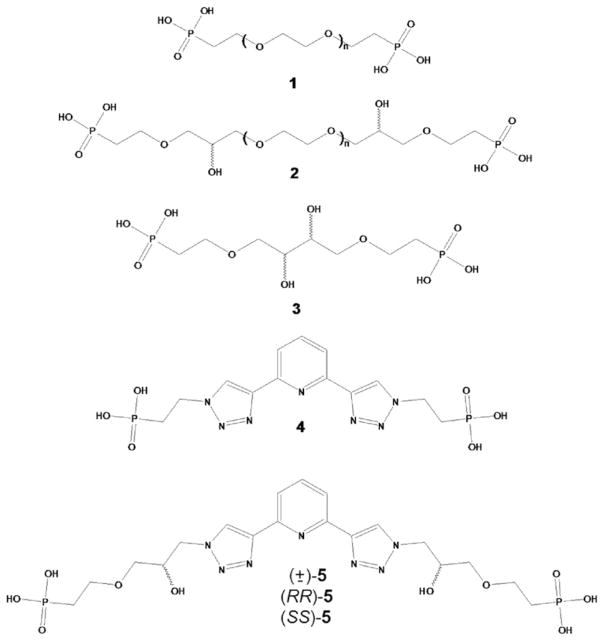
When γ-ZrP was submitted to the topotactic exchange with optically pure 2 (Scheme 1) and the resulting material intercalated with linear, symmetric alkylamines of increasing length, the measured optical rotation passed through a maximum value that occurred at an interlayer distance where molecular modeling suggested the expression of helicity by the columns is maximum [3].
Last but not least, we have observed thermodynamic and kinetic chiral recognition phenomena in materials pillared with optically pure columns derived from tartaric acid (3 in Scheme 1) [4].
To summarize, plenty of evidence shows that laminar γ-ZrP can be developed into a powerful dissymmetric matrix displaying a variety of properties. On the other hand, we have also shown that appropriate γ-ZrP matrices are able to produce the sensitized emission of lanthanides by the well-known antenna effect. For instance, the material bearing diphosphonate 1 (Scheme 1) and intercalated with 2,2′-bipyridyl gave rise to strong Eu(III) and Tb(III) luminescence when these ions were intercalated as well into the layered matrix [5]. The sensitized luminescence was also achieved with γ-ZrP pillared with diphosphonate 4 (Scheme 1) [6].
In this paper, taking advantage of the intrinsic dissymmetry of γ-ZrP and its ability to produce sensitized lanthanide luminescence, we examine the potential of these materials to produce circularly polarized luminescence (CPL). CPL has seldom been measured in the solid state [7–9], and doing so may lead to interesting applications in photonics. To achieve this goal we reacted γ-ZrP with diphosphonate 5 (Scheme 1) that contains the same chromophore of 4 but bears two stereogenic centers.
2. Results and discussion
2.1. Synthesis
The preparation of diphosphonate 5 is outlined in Scheme 2. The fixed stereochemistry of the chiral centers comes from optically pure glycidol whose primary alcohol serves to easily append the phosphonate group (6). Epoxide aperture by sodium azide leads to the appropriate optically pure reagent (8). This reagent is used to perform “click chemistry” on both arms of the diethynylpyridine 7, deprotected in situ, and subsequent hydrolysis of 9 results in the desired diphosphonate 5 with 71% overall yield from glycidol. We measured in methanol [α]D values of −16.1 and +16.4 for (RR)- and (SS)-5 acids respectively (c = 0.01 g mL−1), correspondingly obtained from R- and S-glycidol. It should be noted that when the reaction was performed with (±)-glycidol, a 1:1:2 mixture of (RR)-, (SS)- and meso-9 is produced [and thus of 5; from now on we name these mixtures as (mix)-9 or (mix)-5 for the sake of brevity] whose 1H- and 31P NMR spectra display two sets of different signals, one for the racemate (the same resonances as those of the individual enantiomers) and another for the meso form (Fig. 2). This shows that the diastereomeric species bear slightly different chemical shifts at the phosphonate phosphorous and the triazole and pyridine protons. This finding is quite significant considering the enantiomeric purity of (RR)- and (SS)-5, prepared from commercially pure R- and S-glycidol, respectively. Had some racemization occurred, the meso form would have been produced and signal splitting would have taken place. As this was not observed, we assume that for (RR)- and (SS)-5 a very high degree of enantiopurity was achieved [10].
Scheme 2.
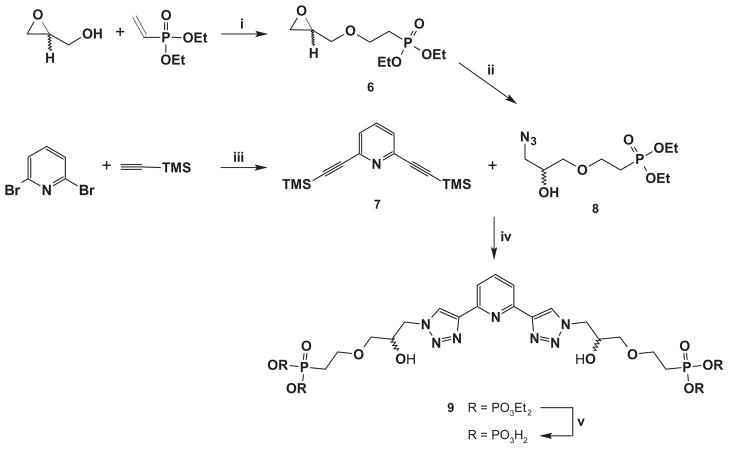
Synthetic route for 5. Reagents and conditions: (i) Cs2CO3, 50 °C, 5 h, 83%; (ii) NaN3, NH4Cl, DMF, 91%; (iii) Pd(PPh3)4, CuI, iPr2NH, toluene, r.t., 72 h, 93%; (iv) K2CO3, CuSO4·5H2O, C6H7NaO6, H2O:tBuOH (1:2), r.t., 48 h, 94%; (v) HCl 35%, 80 °C, 48 h, quant. or TMSI, MeCN, 0 °C→r.t., 24 h, 82%.
Fig. 2.
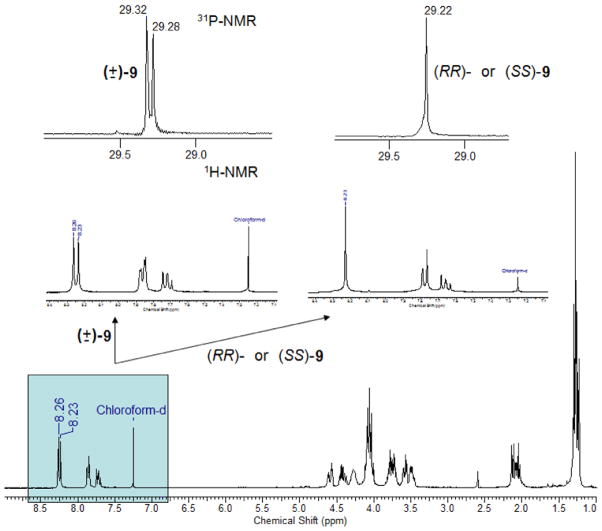
1H- and 31P NMR spectra of the phosphonic diesters 9 precursors of diacids 5 (see Scheme 2).
As mentioned above, γ-ZrP is amenable to topotactic phosphate/phosphonate exchange reactions. The laminar salt has to be exfoliated first in water/acetone at 80 °C for 30 min. The diphosphonic acid is then added to the colloidal dispersion and the new hybrid material flocculates after several minutes. The reaction of γ-ZrP with (RR)-, (SS)- or (mix)-5 (Scheme 2) took place in identical fashion as that with 4 described elsewhere [11]. The molecular formulas of the prepared materials were customarily obtained from solid-state 31P NMR, elemental and thermogravimetry analyses. All the materials were suspended in 0.1 M aqueous solutions of TbCl3·6H2O at r.t. for 48 h and, after the usual work-up, rendered new Tb-containing composites whose metal and chlorine content was determined by TXRF. Table 1 summarizes characterization data for all the created materials.
Table 1.
List of the materials prepared in this work.
| Entry | Reactants | ZrPO4(H2PO4)a[R(H2PO3)2]b(C8H11N)c(Tb)d(Cl)e·zH2O
|
d100 (nm) | |||||
|---|---|---|---|---|---|---|---|---|
| a | b | c | d | e | z | |||
| 1 | (±)-5 | 0.70 | 0.15 | – | – | – | 1.9 | 1.63 |
| 2 | (RR)-5 | 0.68 | 0.16 | – | – | – | 1.9 | 1.61 |
| 3 | (SS)-5 | 0.66 | 0.17 | – | – | – | 2.0 | 1.60 |
| 4 | (±)-5:Tb | 0.70 | 0.15 | – | 0.19 | 0.02 | 2.5 | 1.77 |
| 5 | (RR)-5:Tb | 0.68 | 0.16 | – | 0.21 | 0.01 | 2.5 | 1.68 |
| 6 | (SS)-5:Tb | 0.68 | 0.17 | – | 0.20 | 0.09 | 1.8 | 1.75 |
| 7 | 4 | 0.64 | 0.18 | – | – | – | 1.5 | 1.51–1.69 |
| 8 | 4:(+)-PEA | 0.64 | 0.18 | 0.65 | – | – | 0.2 | 2.19 |
| 9 | 4:(−)-PEA | 0.64 | 0.18 | 0.65 | – | – | 0.2 | 2.20 |
| 10 | 4:(+)-PEA:Tb | 0.72 | 0.14 | 0.05 | 0.30 | 0.02 | 2.0 | 1.81 |
| 11 | 4:(−)-PEA:Tb | 0.72 | 0.14 | 0.05 | 0.30 | 0.02 | 2.0 | 1.82 |
It occurred to us that the materials containing the non-dissymmetric ligand 4 (Scheme 1) could be turned dissymmetric by intercalation of enantiopure 1-phenethylamine (PEA). Therefore, suspensions of the previously reported materials containing ligand 4 were submitted to intercalation of enantiopure 1-phenethylamine (PEA) leading to new crossbred materials containing the phosphonate 4 covalently grafted and the amine intercalated by ionic forces.
Interlayer distances (d100) measured by powder X-ray diffraction deserve some comments (Table 1). Diphosphonate groups in 5 are farther apart than in 4 (Scheme 1). However, the exchanged γ-ZrP materials (compare entries 1–3 and 7) bear similar d100 values (ca. 1.6 nm). This fact can be easily explained considering that the diphosphonates adopt an arrangement parallel to the layers. Simple molecular modeling (Fig. 3) shows how 5 may accommodate in that fashion within the lamellae at the experimental interlayer distance.
Fig. 3.
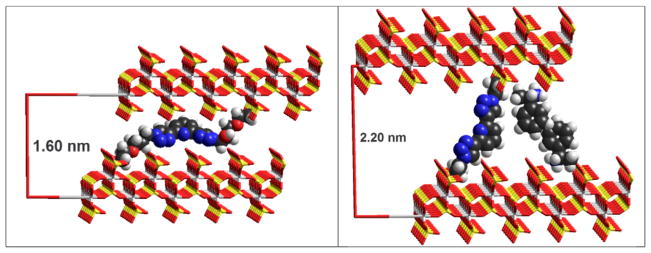
Molecular models at the corresponding experimental interlayer distances of topotactically exchanged γ-ZrP with diphosphonates (SS)-5 (left) and 4 with intercalated PEA (right; see text).
The intercalation of PEA in the interstices left by 4 in γ-ZrP increases the interlayer distance by ca. 0.6 nm and must make the covalently linked ligand stretch. Molecular modeling shows that the stretching is conformationally possible and that there is enough room left for the PEA molecules to form a double layer within the strata of the inorganic salt (Fig. 3).
The inclusion of Tb in the materials bearing 5 increases slightly their interlayer distance whereas in the case of those carrying 4:PEA the opposite situation takes place. The latter observation can be explained because the reaction with TbCl3 expels almost all of the PEA molecules while the former can be the result of a conformational change of the flexible 5 to house the metal ions. That the number of metal ions (TXRF) coincides within experimental error with the number of organic chains – in the case of materials derived from 5 – is significant. It suggests that there is at least one ligand available to interact with every metal. The observed energy transfer between 5 and Tb should be a consequence of this intimate interaction, and will be commented on the next section.
3. Photophysical measurements
3.1. Absorption and emission
A preliminary study in solution of ligand 5 was performed. The UV–vis spectra of (RR)-, (SS)-, and (mix)-5 are shown in Fig. 4 (left). It contains two absorption maxima at 231 and 300 nm. When excited at these maxima, ligands 5 emit fluorescence at 348 nm (Fig. 4, center), the excitation and absorption spectra being coincident. We found that in acidic media the excitation-emission spectra are slightly different to those in neutral or basic conditions. This has been illustrated for (SS)-5 in Fig. 4 (right). Fluorescence and the second excitation band suffer bathochromic shifts of 52 and 30 nm, respectively. This shift to more stable species is probably due to the planarization of the chromophore caused by protonation, a fact previously described in related systems.[12].
Fig. 4.
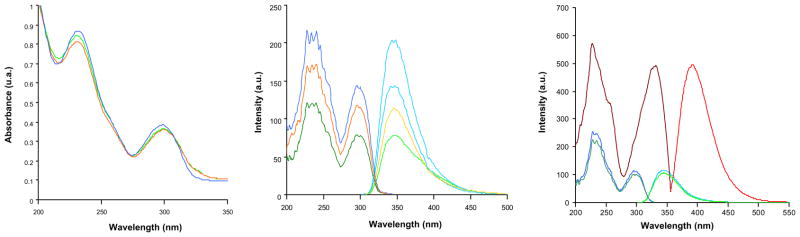
(Left) UV–vis spectra of 10−4 M solutions in water (pH = 7) of the ligands 5. (Middle) Excitation and fluorescence spectra of 10−5 M solutions in water (pH = 7) of the ligands 5. (Right) Excitation and fluorescence spectra of 10−5 Msolutions in water of the ligand (SS)-5 at pH = 2.4 (red), pH = 7.2 (green) and pH = 13 (blue). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In a second step, Terbium(III) complexes of ligands 5 were also studied in solution. They were prepared from commercial TbCl3·6H2O in 10−3 M aqueous solutions with an acidic pH to avoid lanthanide precipitation. Solutions of different 5:Tb ratios (from 10:1 to 1:1) were prepared in order to determine the stoichiometry. The emission spectra of the Tb(III) complexes showed the characteristically well-structured bands of the metal. Fig. 5 displays representative spectra of (SS)-5:Tb at different ligand-to-metal ratios. The strongest emission was attained at the 3:1 (SS)-5:Tb ratio (inset of Fig. 5) suggesting the formation of complexes of stoichiometries 5:Tb 3:1 as previously shown for similar ligands [13]. Excitation spectra show bands at 330 and 250 nm corresponding to the ligand and confirming that 5 de facto sensitizes the metal through the well-known antenna effect.
Fig. 5.
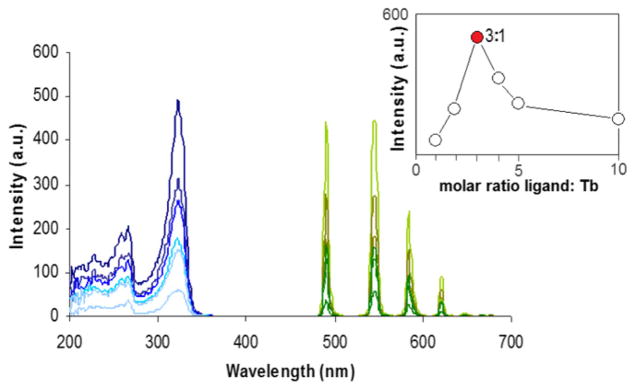
Excitation and luminescence spectra (delay = 0.1 ms) of 10−3 M solutions in water (pH = 2) of (SS)-5:Tb complexes (inset shows that the maximum intensity was attained at the 3:1 5:Tb molar ratio).
The ligand’s excitation and fluorescence spectra when forming the Tb(III) complex can be seen in Fig. 6. When the 5:Tb ratio is 5:1, the spectra resemble those of free 5, no doubt due to excess ligand. In the case of the 1:1 ratio, the excitation spectrum shows a new band centered at 295 nm that can be attributed to the complexed ligand. Excitation of this band causes the 5:Tb species to render a ligand-centered fluorescence at 365 nm implying that intersystem crossing (ISC) is relatively inefficient. Nevertheless, a Tb emission is also seen suggesting that the sought energy transfer to the metal takes place. This latter emission exhibits a longer life-time (ca. 1 ms) characteristic of the triplet-to-lanthanide energy transfer, clearly discriminated when the detection is delayed 0.1 ms.
Fig. 6.
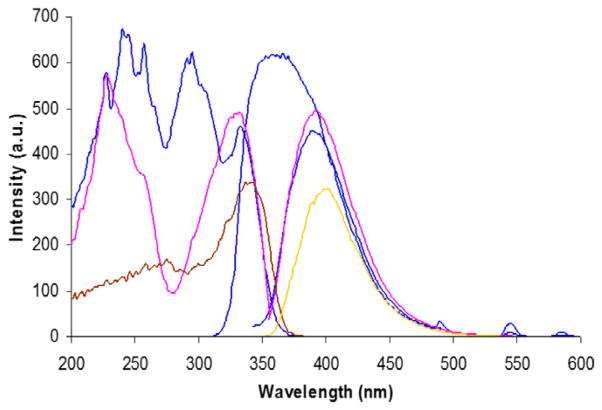
Excitation and fluorescence spectra of the ligand 5 (pink, as taken from Fig. 4 right) and luminescence of 10−3 M solutions in water (pH = 2) of (SS)-5:Tb at 1:1 (blue) and 5:1 (brown/yellow) ratios. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
These results are proof of (i) the formation in water solution of 5:Tb complexes in 3:1 stoichiometry and (ii) the pursued energy transfer between the ligand and the metal that does occur even in an unfriendly aqueous medium that tends to deactivate Tb emission by vibronic coupling of the O-H oscillators.
Circular dichroism (CD) spectroscopy in solution was also quite revealing (Fig. 7). The traces of the free ligands (RR)- (orange) and (SS)-5 (green) show no CD effect suggesting that there is no significant participation of the chromophore in the molecular dissymmetry that the stereogenic centers must exert. In turn, when the 3:1 complexes are formed in the solutions containing excess 10:1 ligand-to-metal ratios, a quite sizable CD effect was observed. The only conclusion to this observation is that the concourse of the metal now induces the chromophore to actively participate in the molecular dissymmetry. This strongly suggests that the chelation of Tb takes place through the heterocyclic core, despite the presence of phosphonic acid groups.
Fig. 7.
CD spectra of 10−3 M acidic solutions in water at 298 K of the free chiral ligands and their Tb(III) complexes.
The idealized model of Fig. 8 (MM+, Hyperchem) [14] depicts a possible triple helical conformation of a hypothetical 3:1 (SS)-5:Tb complex. It shows that the chelated metallic core forms a chiral structure and that the CHOH groups on the stereogenic centers do not participate in the chelation with the metal, but contribute to the high handedness of the molecule.
Fig. 8.
Idealized model of possible 3:1 5:Tb complexes (see text).
We then proceeded to measure the luminescence of the materials in the solid-state for two reasons, (i) one obvious: the solids are not soluble in any solvent (exception made for aqueous HF in which the structure is destroyed) and (ii) because we wished to examine the effect of confinement on active organic molecules within the inorganic layers. Fig. 9 shows the results.
Fig. 9.
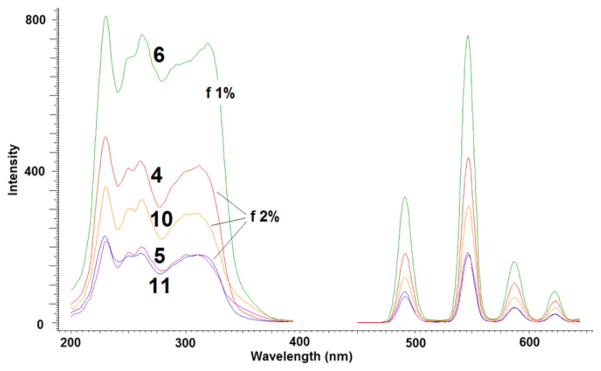
Excitation and emission spectra of Tb time-resolved luminescence spectra (delay = 0.1 ms) of the materials indicated with the entry numerals of Table 1.
A strong Tb luminescence was detected when the solid was excited at 310 nm. In general, it was necessary to use a 2% attenuator to keep the detector from saturation. It is remarkable that the signal from the material of entry 6 of Table 1, containing the (SS)-5 ligand, was even stronger and required a 1% attenuator. In all cases the excitation maxima (230, 250, 260 and 310 nm) correspond to the absorption spectra of the ligands indicating that the antenna effect is taking place within the inorganic layers. It is unclear why the (SS)-5 ligand is most effective in transferring energy to the metal. A reasonable speculation is that the (SS)-5 ligand and the dissymmetric inorganic matrix form a stereochemical matched pair and allows the ligand to adopt a better conformational arrangement than the (RR)-5 counterpart to perform the energy transfer to the metal. Unfortunately, neither X-ray diffraction nor solid-state NMR can provide any evidence of that. It should be noted that within the solid matrix, the organic chains are each attached by covalent phosphonate-Zr bonds to opposing faces of consecutive γ-ZrP layers. Therefore the 3:1 ligand-to-metal stoichiometry detected in solution for ligand 5 is no longer possible within the organic– inorganic solid. This is confirmed by the fact that the materials of entries 4–6 of Table 1 have ca. one metal per organic molecule. Nonetheless, the ligand-to-metal energy transfer takes place and the highly dissymmetric environment around the metal hopefully should give rise to a sizable CPL.
3.2. CPL measurements
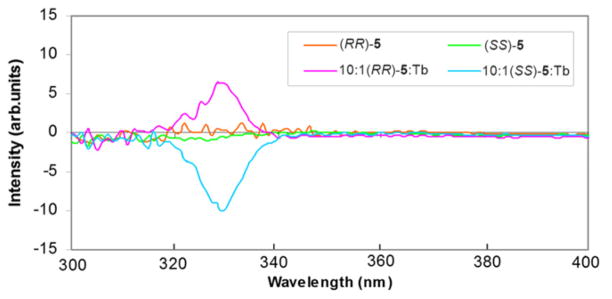
The first measurements were performed in solution to ensure that CPL activity can be detected. Fig. 10 shows the results with solutions of different initial ligand-to-metal ratios (2:1–10:1). Repetitions of every measurement were taken to check for possible instability of the solutions. Regardless of the ratio and the repetition rate, the CPL curves of the 5D4→7F5 transition of Tb(III) (545 nm) are superimposable within experimental error, even after several weeks.
Fig. 10.
CPL (upper curves) and total luminescence (lower curves) spectra of the 5D4→7F5 transition of the chiral Tb(III) complexes in 10−3 M aqueous solutions at 295 K, upon excitation at 323 nm; (SS)-5:Tb (left) at different ratios (see text); (SS)- and (RR)-5:Tb (right).
Due to the short-range nature of f-orbitals and the long distance between the metal and the CHOH stereocenters, the direct contribution of the latter to the CPL of this transition is negligible. The models (Fig. 8) suggested that the stereogenic centers induced a chiral triple-helical configuration of bistriazolylpyridine ligands around the metal. This chiral arrangement surrounding the luminescent metal center must be responsible for the CPL signal observed. The aforementioned CD measurements in solution reinforce this assumption. The mirror image complex (Fig. 8) formed with the (RR)-5 isomer exactly mirrors as expected the CPL signal of its SS counterpart (Fig. 10 right).
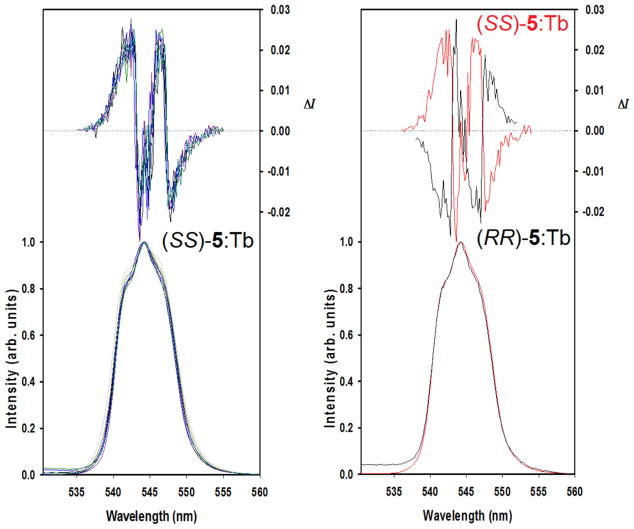
Luminescence measurements in the solid state are much more challenging. The measurements were performed on pellets produced directly from the powder of the synthesized materials or diluted with solid KBr. Samples made from index cards were used to hold and position the pellets for measurements. The use of an empty card holder confirmed that the holder produced no interferences. However, pristine γ-ZrP proved to be heavily reflective and we found it necessary to place the sample-holder plane at 60° with respect to the light source instead of the normal 45°. The purpose behind this change in angle was to obtain a reasonable compromise between minimizing the incidence of stray light and achieving a detectable luminescence signal (Fig. 11). Under these conditions, a strong luminescence at 545 nm from the metal could be easily discriminated. The results of CPL measurements are summarized in Fig. 12.
Fig. 11.
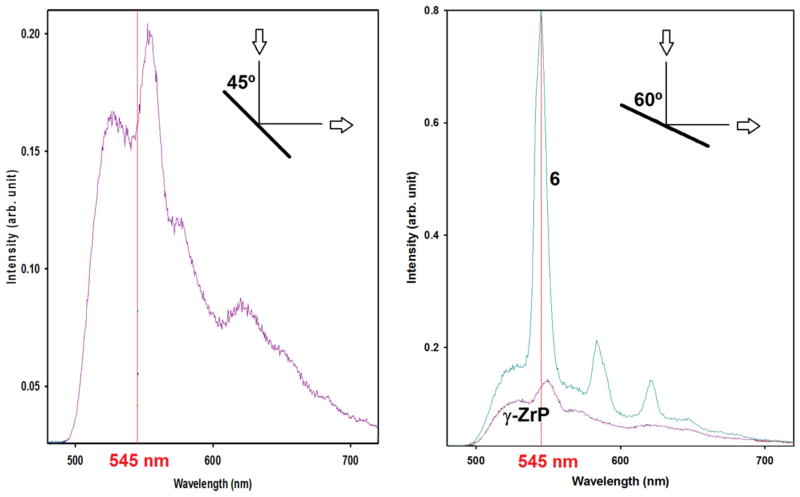
Emission spectra of phosphorescence of pristine γ-ZrP and material of entry 6 in Table 1 [γ-ZrP with (SS)-5:Tb] at different angles of sample holder.
Fig. 12.
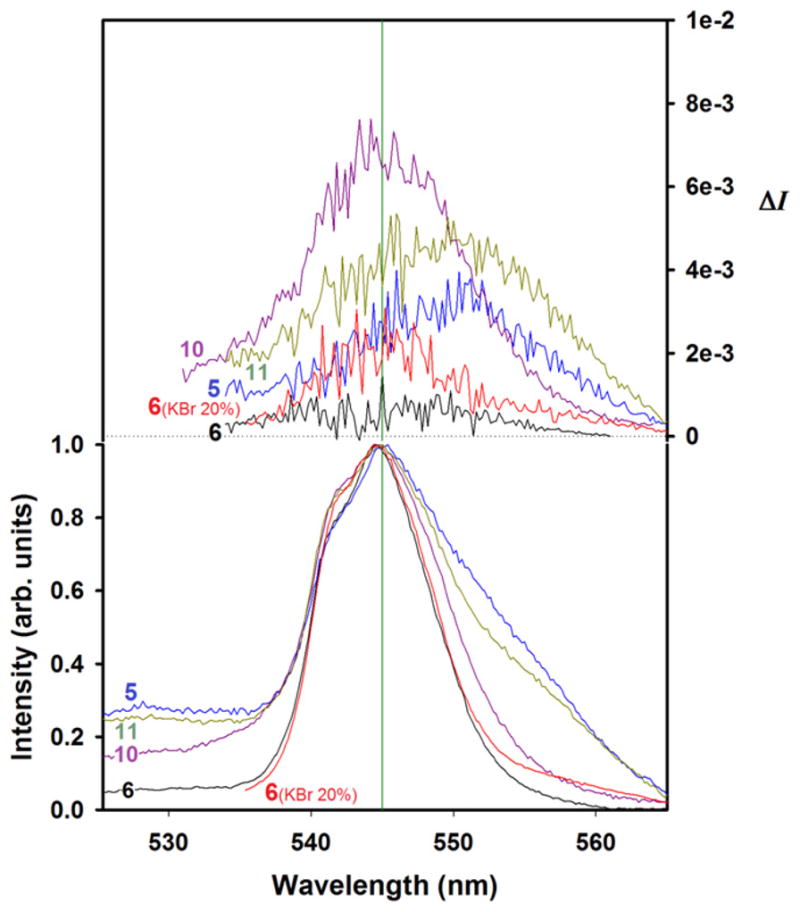
CPL (upper curves) and total luminescence (lower curves) spectra of the 5D4→7F5 transition of the indicated solid materials at 295 K, upon excitation at 330 nm.
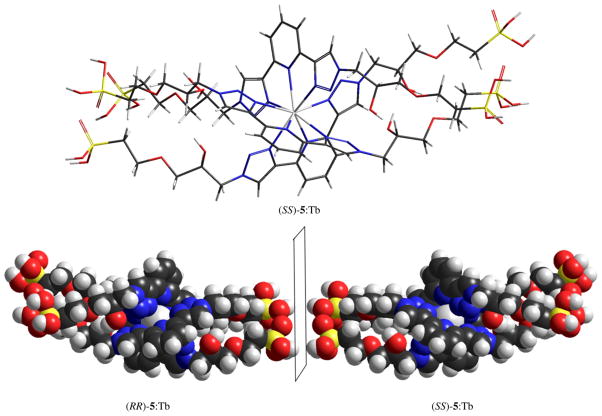
Prior to the discussion of the CPL results, some comments about the organic–inorganic structure may be revealing. Fig. 13 presents models of a small portion of two consecutive layers of pristine γ-ZrP (top), and of this salt with the achiral diphosphonate 4 (middle) and (RR)-5 (bottom) in the interlayer space. Every internal phosphate within a layer of the pure inorganic salt is a stereogenic center to which the R or S configuration can be assigned (see inset in Fig. 13 top). In the absence of any chiral influence, however, the synthesis of γ-ZrP must give rise to a meso-like arrangement of phosphates within the particles as that shown in the upper model. When an achiral diphosphonate like 4 (Scheme 1) is topotactically exchanged, the two phosphonate functions are turned into additional stereogenic centers the meso-like arrangement is broken but, statistically speaking, mirror image particles should be formed as shown in the center model and the material as a whole must not be able to display chiroptical properties. However, when an optically pure diphosphonate, such as (RR)- or (SS)-5 is incorporated into γ-ZrP (or an enantiomerically pure amine as (+)- or (−)-PEA is intercalated in the material bearing the non-chiral ligand 4), the particles can no longer be mirror images. In the case of 5, the configurations of the two stereogenic centers belonging to the organic chiral molecule are preserved and no mirror-image particles can be produced as shown in the lower model.
Fig. 13.
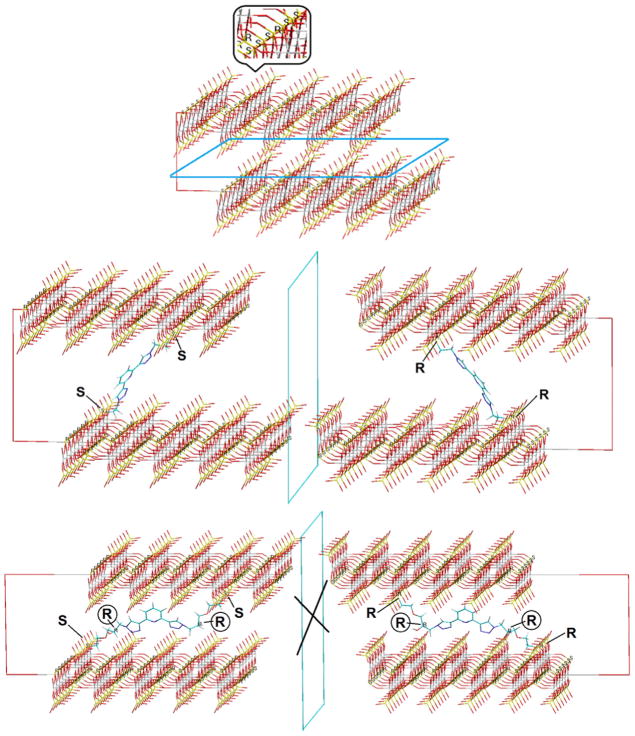
Model of portions and their possible mirror images of pristine γ-ZrP (up) and materials of entries 7 (middle) and 4 (bottom) of Table 1, showing relevant R/S configurations.
This explanation is undoubtedly an oversimplification of reality which is much more complex but we believe it helps understand the macroscopic building of chirality in these γ-ZrP derived materials. As γ-ZrP particles are much larger than those depicted in Fig. 13, the size distribution and diphosphonate content must be statistically treated. Even so, the in-bulk CPL behavior reflected in Fig. 12 can be understood keeping in mind these models. While materials 5 and 6, in one hand, and 10 and 11, in the other, contain enantiomeric pairs of ligand 5 and PEA, respectively, they do not present mirror image CPL curves (cf. Fig. 12). These results can be attributed to the non-mirror image (diastereomeric-like) nature of the particles.
The observed values of ΔI are ca. one order of magnitude smaller than those measured in solution. It should be noted that, for reasons difficult to grasp, material 6 containing (SS)-5 did not show any CPL activity when used neat but exhibited a weak one when diluted at 20% in KBr. A plausible explanation is that crystallites of pure material 6 arranged macroscopically in a way that was harmful for the CPL activity. The fact that CPL activity was small but visible when material 6 was diluted in KBr suggests that the dilution disrupted that deleterious arrangement.
Yet, the most puzzling results are those of materials 10 and 11 (Table 1), consisting of achiral ligand 4 and (+)- or (−)-PEA, respectively. They presented the strongest CPL activity, even though characterization data indicated the amount of the chiral amine is very low, close to our detection limit. The high CPL activity indicates that this combination of components, non-chiral diphosphonate 4 and optically pure PEA, provide the highest dissymmetric environment for the metal in its excited state, even with minute amounts of the chiral amine. The results of CPL activity of materials 10 and 11 might hint to phenomena that would be worth checking in future experiments: the possibility of chiral memory [15] or chirality amplification. The presence of relatively large amounts of chiral PEA in materials 8 and 9 (Table 1) may impinge a conformational handedness of diphosphonate 4 that appears to be preserved when the majority of PEA cations are smoothly displaced by the Tb(III) ions. New experiments should be carefully designed in order to prove that important speculation and exclude that the observations are simply the consequence of uncontrolled artifacts. Against the latter, it may be significant that materials 5 and 11, both containing levorotatory chiral components [(RR)-5 with [α]D = −16.1 and (−)-PEA] bear the CPL maxima shifted ca. +5 nm relative to their dextrorotatory counterparts.
4. Conclusions
We have performed for the first time solid-state CPL measurements in hybrid, laminar materials based on γ-ZrP pillared with organic diphosphonates. In the primary stage of the research, ad hoc optically pure diphosphonates were synthesized. Their complexation with Tb(III) and the luminescence properties of the resulting complexes were checked in solution. It was observed that while the ligand stereocenters were unable to interact directly with the metal, they induced asymmetry over the bistriazolylpyridine ligands bonded to the metal and provided an effective chiral environment, one that led to significant CD and CPL activities. In the case of the γ-ZrP-derived materials it is not possible to know the exact structure of the particles. However, the available experimental data enabled the determination of general compositions and suggested reasonable structures. Simple molecular modeling hints supramolecular chirality of the particles, induced by the intrinsic dissymmetry of the organic diphosphonates or by the intercalation of chiral species such as PEA. The chirality at the supramolecular level was revealed in the solid state by the CPL activity measured from reporter Tb(III) ions intercalated in the hybrid matrix. It is expected that the layered supramolecular scaffolds studied in this work and their optical properties will be valuable in the development of novel solid-state chiral supramolecular organic–inorganic fluorophores.
5. Experimental
5.1. General
All commercial reagents were purchased from Aldrich or Fluka and used without further purification. Liquid 1H, 13C and 31P NMR spectra were recorded on a Bruker AC-300 instrument, in the indicated solvents. All chemical shifts are reported in ppm referenced to residual protonated solvent. Solid state 31P NMR spectra were recorded under MAS and CP-MAS techniques on a Bruker MSL-400 instrument. Elemental analyses were performed on a Perkin Elmer II 2400 CHN analyzer. TXRF analyses were performed by direct solid method using a TXRF 8030C Atomika-FEI spectrometer, equipped with a 3 kW X-ray tube with a Mo/W alloy anode and a double-W/C multilayer monochromator, adjusted to obtain an excitation energy of 17.4 keV (Mo-Ka), for P, and Tb evaluation, and 35 keV (W35), for Zr and Cl evaluation, and a Si(Li) detector with an active area of 80 mm2 with a resolution of 150 eV at 5.9 keV. Optical rotation measurements were performed in a Perkin Elmer 241 MC polarimeter. IR spectra were recorded on a Bruker Vector 22. MS L-SIMS spectra were recorded on a VG Autospec spectrometer. Thermogravimetric analyses were performed in a Mettler Toledo TGA/SDTA 851e analyzer with a heating rate of 5 °C/min. X-ray powder diffraction were recorded on a Phillips PW1710 or Siemens D-5000 diffractometer with CuKα radiation and Ni filter (40 kV, 30 mA). KBr pellets were prepared by grinding 90 mg of the corresponding material (alone or mixed with 80% of dry KBr) and the resultant powders were dried under vacuum for 15 min using a vacuum pump and pressed into a pellet by a hand-operated hydraulic pump keeping it for 20 min at 10 tons. The UV–vis absorption spectra were recorded on a KayaK Hewlett Packard 8453 spectrometer using quartz cells (1 cm). Emission and excitation corrected spectra were acquired on a Varian Cary Eclipse and a Perkin Elmer LS-50 with a red-sensitive photomultiplier Hamamatsu R-928 spectrophotometer, both in solid state and solution. CD spectra were measured on a JASCO J-810 spectropolarimeter using quartz cuvettes (1 cm). Circularly polarized luminescence and total luminescence spectra were recorded on an instrument described previously [16] operating in a differential photon-counting mode. The light source for indirect excitation was a continuous wave 450W xenon arc lamp from a Spex Fluoro-Log-2 spectrofluorometer, equipped with excitation and emission monochromators with dispersions of 4 nm/mm (SPEX, 1681B). Selective excitation of Tb(III) was accomplished with either a Coherent Innova-70 or Coherent saber:sabre TSM 15. The optical detection system consisted of a focusing lens, long pass filter, and 0.22 m monochromator. The emitted light was detected by a cooled EMI-9558B photomultiplier tube operating in photon-counting mode. All measurements were performed with quartz cuvettes with a path length of 1.0 cm.
5.2. Compound 6 (Scheme 2): diethyl [2-(oxiran-2-ylmethoxy)ethyl]-phosphonate
Diethyl-vinylphosphonate (3.39 g, 20.65 mmol) and glycidol (1.53 g, 20.65 mmol) were mixed and Cs2CO3 (1.34 g, 5.34 mmol) was added. The mixture was heated at 50 °C for 5 h under argon. After cooling, the reaction mixture was dissolved in 40 mL of water, extracted with DCM (3 × 25 mL), dried over MgSO4, and evaporated in vacuum. The crude product was purified by flash column chromatography (10% ethanol/ethylacetate, (Rf = 0.48) to afford 4.08 g (83%) of 6 as a colorless oil. [α]D = +8.7° (c = 1.27, chloroform) and [α]D = −8.8° (c = 1.26, chloroform) from (R)- and (S)-glycidol, respectively. 1H NMR (CDCl3) δ (ppm): 1.29 (t, J = 7.1 Hz, 6H); 2.08 (dt, J = 7.5 Hz, JHP = 18.6 Hz, 2H); 2.58 (t, J = 2.7, 2H); 2.75 (t, J = 4.3 Hz, 1H); 3.10 (m, 1H); 3.35 (q, J = 5.9 Hz); 3.73 (m, 3H); 3.95 (m, 4H). 13C NMR (CDCl3) δ (ppm): 16.0 (d, J = 6.1 Hz, 2C); 26.6 (d, J = 140.0 Hz); 43.7, 50.2, 61.2 (d, J = 5.8 Hz, 2C); 64.9, 71.2. 31P NMR (CDCl3) δ (ppm): 28.16 (s, 2P). Elemental analysis (%): calculated: C: 45.8; H: 7.99. Found: C: 45.4; H: 8.04. FAB MS (m/z): 239.1 [M + H]+.
5.3. Compound 7 (Scheme 2): 2,6-bis[(trimethylsilyl)ethynyl]pyridine
2,6-Dibromopyridine (1 g, 4.22 mmol), Pd(PPh3)4 (229 mg, 0.2 mmol) and CuI (38 mg, 0.2 mmol) were introduced into a round-bottomed flask fitted with a septum. After degassing the system (Ar), dry diisopropylamine (4 mL), dry toluene (36 mL) and trimethylsilylacetylene (1.5 mL, 10.5 mmol) were added in that order. The mixture was stirred at room temperature for 4 days and finally filtered. The filtrate was poured into aqueous NH4Cl (15 mL), and extracted with DCM (3 × 30 mL). The extract was washed with brine, dried over MgSO4 and filtered through a thin layer of silica gel. The solvent was evaporated in vacuum to yield 1.1 g of 7 (93%) as a pale-beige solid. 1H NMR (CDCl3) δ (ppm): 0.24 (s, 18H, 6H); 7.37 (d, J = 7.7 Hz, 2H); 7.58 (dd, J = 0.9, 7.7 Hz, 1H). 13C NMR (CDCl3) δ (ppm): −0.4 (2C); 95.3 (2C); 103.1 (2C); 126.6 (2C); 136.2, 143.3 (2C). Elemental analysis (%): calc.: C: 66.3; H: 7.8; N: 5.1. Found: C: 66.0; H: 7.7; N: 5.1. TOF MS EI+: 256.0984.
5.4. Compound 8 (Scheme 2): diethyl [2-(3-azido-2-hydroxypropoxy)ethyl] phosphonates
Diethyl [2-(oxiran-2-ylmethoxy)ethyl]-phosphonate (1.9 g, 4.19 mmol) was dissolved in dry DMF (17 mL) and then sodium azide (0.82 g, 12.6 mmol) and ammonium chloride (0.67 g, 12.6 mmol) were added to the mixture and stirred at 50 °C for 5 h under argon. After removal of DMF under reduced pressure, the residue was dissolved in DCM and filtered to remove insoluble impurities. The solvent was evaporated and the product was dried under vacuum, yielding 91% of the organic azide as a yellowish oil. [α]D = +2.2° (c = 0.94, chloroform) and [α]D = −2.3° (c = 0.97, chloroform) for the compounds obtained from (R)- and (S)-glycidol, respectively. 1H NMR (CDCl3) δ (ppm): 1.31 (t, J = 7.1 Hz, 6H); 2.07 (dt, J = 6.69, JHP = 18.1 Hz, 2H); 3.17 (broad s, 1H); 3.32 (d, J = 5.6 Hz, 2H); 3.49 (m, 2H); 3.75 (m, 2H); 3.92 (m, 1H); 3.95 (m, 4H). 13C NMR (CDCl3) δ (ppm): 16.37 (d, J = 6.1 Hz, 2C); 26.75 (d, J = 141.6 Hz); 53.15, 61.8 (q, J = 3.8 Hz, 2C); 65.1 (d, J = 4.7 Hz); 69.29, 72.29. 31P NMR (CDCl3) δ (ppm): 29.43. IR in Nujol ν (cm−1): 2101 (azide stretching vibration).
5.5. Compound 9 (Scheme 2): tetraethyl {pyridine-2,6-diylbis[1H-1,2,3-triazole-4,1-diyl(2-hydroxypropane-3,1-diyl)oxyethane-2,1-diyl]}bis-phosphonates
2,6-Bis[(trimethylsilyl)ethynyl]pyridine (0.3 g, 1.1 mmol), diethyl[2-(3-azido-2-hydroxypropoxy) ethyl]phosphonate (0.62 g, 2.2 mmol), CuSO4·5H2O (0.054 g, 0.22 mmol), sodium ascorbate (0.113 g, 0.66 mmol) and K2CO3 (0.31 g, 2.2 mmol) were added to a 1:2 mixture of H2O:t-BuOH (6 mL). The mixture was vigorously stirred at r.t. for 36 h. The solvent was then removed in vacuum and the residue was dissolved in 50 mL of DCM and stirred for 20 min. After filtration at reduced pressure in a filter plate, the organic layer was dried over MgSO4 and the solvent evaporated in vacuum to give 0.72 g of a yellow oil (94%). Further purification was performed by column chromatography (20% ethanol, ethylacetate, Rf = 0.82). [α]D = +3.7° (c = 1, chloroform) and +14.8° (c = 1, methanol) for (SS)-9 and [α]D = −3.6° (c = 1, chloroform) and −14.6° (c = 1, methanol) for (RR)-9. 1H NMR (CDCl3) δ (ppm): 1.27 (t, J = 7.01 Hz, 6H); 1.28 (t, J = 7.01 Hz, 6H); 2.08 (dt, J = 6.69, JHP = 18.1 Hz, 4H); 3.46 (m, 4H); 3.76 (m, 4H); 4.07 (m, 8H); 4.26 (broad s, 2H); 4.51 (m, 4H); 7.73 (dd, J = 0.9, 7.4 Hz, 1H); 7.89 (d, J = 7.7 Hz, 2H); 8.23 (s, 1H), 8.26 (s, 1H). 13C NMR (CDCl3) δ (ppm): 16.23 (d, J = 6.1 Hz, 4C); 26.09 (d, J = 141.1 Hz, 2C); 53.29 (2C); 61.66 (d, J = 6.6 Hz, 4C); 65.08 (d, J = 3.02 Hz, 2C); 68.77 (2C); 72.12 (2C); 118.61 (2C); 123.94 (2C); 137.2, 147.46 (2C); 149.43 (2C). 31P NMR (CDCl3) δ (ppm) [sample from (±)-glycidol]: 29.2 (s), 29.3 (s). Elemental analysis (%) for the monohydrated (SS)-9 product: calc.: C: 45.8; H: 6.7; N: 13.8. Found: C: 45.8; H: 6.5; N: 13.2. EM (ESI): 690.2713 ([M + 1]+), ACN + 0.1% formic acid.
5.6. Compound 5 (Scheme 2): {pyridine-2,6-diylbis[1H-1,2,3-triazole-4,1-diyl(2-hydroxypropane-3,1-diyl)oxyethane-2,1-diyl]}bis-phosphonic acid
Method 1: tetraethyl-bis-phosphonate (0.5 g, 0.73 mmol) was heated under reflux at 90 °C in 35% HCl (9.5 mL) for 72 h. The solution was evaporated to dryness under vacuum rendering a yellow oil. The (RR)-5 and (SS)-5 acids were quantified by Q NMR using 2,4,6-trifluoropyridine (99%) as internal standard, obtaining richness of 72 and 91%, respectively.
Method 2: tetraethyl-bis-phosphonate (0.5 g, 0.73 mmol) was dissolved in acetonitrile (30 mL). Iodotrimethylsilane (1.24 mL) was added at 0 °C via syringe under argon. The mixture was maintained at 0–10 °C for 8 h and at r.t. overnight. The solvent was evaporated in vacuum and the residue was stirred for 2 h at r.t. in water (50 mL) and washed with toluene (3 × 15 mL). The solvent was removed, the residue washed with MeOH and aqueous NaHSO3 (10%, 2 × 10 mL). The resultant yellowish solid was filtered yielding 0.35 g of the product (82%). [α]D = +16.4° and −16.2° (c = 1, methanol) for (SS)- and (RR)-5, respectively. 1H NMR (MeOH) δ (ppm): 2.11 (dt, J = 6.8, JHP = 18.3 Hz, 4H); 3.52 (m, 4H); 3.74 (dt, J = 7.04, JHP = 14.9 Hz 4H); 4.22 (broad s, 2H); 4.69 (m, 4H); 8.27 (d, J = 8.14 Hz, 2H); 8.44 (dd, J = 0.9, 7.7 Hz 2H); 9.03 (s); 13C NMR (MeOH) δ (ppm): 29.8 (d, J = 136.4 Hz, 2C); 54.78 (2C); 66.9 (2C); 69.9 (2C); 72.9 (2C); 121.9 (2C); 127.5 (2C), 142.9; 144.73 (2C); 148.2 (2C); 31P NMR (MeOH) δ (ppm): 26.4 (s); elemental analysis (%) for the monohydrated (SS)-5: calc.: C: 38.3; H: 5.2; N:16.4. Found: C: 38.3; H: 5.0; N: 16.2; EM (electrospray): 576.1351 (M−) in MeOH.
5.7. Topotactic exchange of bisphosphonic acids on γ-ZrP and amine intercalation
These reactions were performed as previously described [5]. A certain amount of γ-ZrP or the topotacticaly exchanged solid phase was suspended in an aqueous solution 0.1 M of optically pure 1-phenylethylamine (100 mL of solution per g of solid) and the mixture was vigorously stirred at room temperature for 48 h. The solid was then centrifuged, washed three times with deionized water and ethanol, dried at 100 °C for 3 h and conditioned over saturated solution of BaCl2.
5.8. Material γ-ZrP-4
Interlayer distance 1.51–1.69 nm. Anal. Calcd for ZrPO4(H2PO4)0.64(C13H17N7O6P2)0.18·1.5H2O: C, 8.0; H, 2.1; N, 5.0. Found: C, 8.1; H, 2.3; N, 4.7. MAS 31P NMR δ, ppm (integral): −27.2 (100), −14.2 (65), 14.4 (35). TGA °C (weight loss): 25–150 °C (7.8%), 150–450 °C (7.5%), 450–900 °C (5.6%). MW: 352.6 g mol−1.
5.9. Material γ-ZrP-4:(+)PEA
Interlayer distance 2.19 nm. Amine content:Zr, 0.65:1.00; Anal. Calcd for ZrPO4(H2PO4)0.64(C13H17N7O6P2)0.18 (C8H11N)0.65·0.20H2O: C, 22.2; H, 3.3; N, 6.6. Found: C, 22.4; H, 3.3; N, 6.3. MAS 31P NMR δ, ppm (integral): −25.9 (100), −10.2, −14.1 (70), 10.3 (32). TGA °C (weight loss): 25–150 °C (4.1%), 150–600 °C (22.6%), 600– 900 °C (3.0%). MW: 407.9 g mol−1.
5.10. Material γ-ZrP-4:(−)PEA
Interlayer distance 2.20 nm. Amine content/Zr, 0.65:1.00; Anal. Calcd for ZrPO4(H2PO4)0.64(C13H17N7O6P2)0.18 (C8H11N)0.65·0.20H2O: C, 22.2; H, 3.3; N, 6.6. Found: C, 22.5; H, 3.2; N, 6.4. MAS 31P NMR δ, ppm (integral): −25.9 (100), −10.4, −13.7 (69), 10.7 (33). TGA °C (weight loss): 25–150 °C (4.2%), 150–600 °C (22.6%), 600–900 °C (3.1%). MW: 407.9 g mol−1.
5.11. Material γ-ZrP-(±)-5
Interlayer distance 1.63 nm. Anal. Calcd for ZrPO4(H2PO4)0.70 (C19H29N7O10P2)0.15·1.9H2O: C, 9.3; H, 2.3; N, 3.9. Found: C, 9.1; H, 2.5; N, 3.9. MAS 31P NMR δ, ppm (integral): −27.3 (100), −14.2 (69), 19.5 (31). TGA °C (weight loss): 25–150 °C (3.0%), 150–450 °C (12.2%), 450–900 °C (8.6%). MW: 374.9 g mol−1.
5.12. Material γ-ZrP-(SS)-5
Interlayer distance 1.60 nm. Anal. Calcd for ZrPO4(H2PO4)0.66 (C19H29N7O10P2)0.17·2.0H2O: C, 10.1; H, 2.7; N, 4.3. Found: C, 10.2; H, 2.4; N, 4.5. MAS 31P NMR δ, ppm (integral): −26.9 (100), −14.6 (65), 20.7 (34). TGA °C (weight loss): 25–150 °C (6.9%), 150–450 °C (11.5%), 450–900 °C (8.1%). MW: 374.25 g mol−1.
5.13. Material γ-ZrP-(RR)-5
Interlayer distance 1.61 nm. Anal. Calcd for ZrPO4(H2PO4)0.68 (C19H29N7O10P2)0.16·1.9H2O: C, 9.6; H, 2.6; N, 4.1. Found: C, 9.8; H, 2.5; N, 4.2. MAS 31P NMR δ, ppm (integral): −27.2 (100), −14.3 (67), 19.8 (32). TGA °C (weight loss): 25–150 °C (4.4%), 150–450 °C (11.5%), 450–900 °C (8.1%). MW: 378.7 g mol−1.
5.14. Intercalation of Ln(III)
A certain amount of the topotactically exchanged γ-ZrP solid phase was suspended in an aqueous solution 0.1 M of TbCl3·6H2O (3.2 eq). The mixture was stirred at room temperature for 48 h. The solid was then centrifuged, washed three times with deionized water, dried in the oven at 100 °C for 3 h and the material obtained conditioned over saturated solution of BaCl2.
5.15. Material γ-ZrP-4:(+)PEA:Tb
Interlayer distance 1.81 nm. Amine content/Zr, 0.05; Anal. Calcd. for ZrPO4(H1.1PO4)0.72(C13H17N7O6P2)0.14(C8H11N)0.05(Tb)0.30 (Cl)0.02·2.0H2O: C, 6.6; H, 2.1; N, 3.5. Found: C, 6.6; H, 2.1; N, 3.9. TXRF (%): Zr (100), Tb (31.3), Cl (1.8). TGA °C (weight loss): 25–150 °C (7.5%), 150–700 °C (10.5%), 700–900 °C (1.9%). MW: 406.6 g mol−1.
5.16. Material γ-ZrP-4:(−)PEA:Tb
Interlayer distance 1.82 nm. Amine content/Zr, 0.29; Anal. Calcd. for ZrPO4(H1.1PO4)0.72(C13H17N7O6P2)0.14(C8H11N)0.05(Tb)0.30 (Cl)0.02·2.0H2O: C, 6.6; H, 2.1; N, 3.5. Found: C, 6.6; H, 2.1; N, 4.1. TXRF (%): Zr (100), Tb (31.6), Cl (2.2). TGA °C (weight loss): 25–150 °C (8.7%), 150–700 °C (9.7%), 700–900 °C (1.4%). MW: 406.6 g mol−1.
5.17. Material γ-ZrP-(±)-5:Tb
Interlayer distance 1.77 nm. Anal. Calcd for ZrPO4(H1.5PO4)0.70 (C19H29N7O10P2)0.15(Tb)0.19(Cl)0.02·2.5H2O: C, 8.2; H, 2.6; N, 3.5. Found: C, 8.0; H, 2.5; N, 3.4. TXRF (%): Zr (100), Tb (18.5), Cl (1.5). TGA °C (weight loss): 25–150 °C (8.5%), 150–450 °C (8.9%), 450–900 °C (5.9%). MW: 418 g mol−1.
5.18. Material γ-ZrP-(SS)-5:Tb
Interlayer distance 1.75 nm. Anal. Calcd for ZrPO4(H1.5PO4)0.66 (C19H29N7O10P2)0.17(Tb)0.20(Cl)0.09·1.8H2O: C, 9.3; H, 2.4; N, 4.0. Found: C, 9.3; H, 2.5; N, 4.3. TXRF (%): Zr (100), Tb (19.5), Cl (0.9). TGA °C (weight loss): 25–150 °C (7.3%), 150–450 °C (10.7%), 450–900 °C (5.6%). MW: 415.8 g mol−1.
5.19. Material γ-ZrP-(RR)-5:Tb
Interlayer distance 1.68 nm. Anal. Calcd for ZrPO4(H1.4PO4)0.68(-C19H29N7O10P2)0.16(Tb)0.21(Cl)0.01·2.5H2O: C, 8.6; H, 2.6; N, 3.7. Found: C, 8.7; H, 2.5; N, 3.8. TXRF (%): Zr (100), Tb (20.5), Cl (1.3). TGA °C (weight loss): 25–150 °C (8.3%), 150–450 °C (10%), 450–900 °C (6.3%). MW: 423.8 g mol−1.
Acknowledgments
The Spanish laboratory regrets that the generous government funding received in the near past (MAT2006-00570) ceased to be granted. L.J. expresses her gratitude for a FPI fellowship. This work has been performed under the indirect support of ERCROS-Farmacia S.A. to which we are deeply grateful for the funding of a Sponsored Chair in Chemistry (Cátedra de Patrocinio). G.M. from the American laboratory thanks the National Institute of Health, Minority Biomedical Research Support (1 SC3 GM089589-03 and 3 S06 GM008192-27S1) and the Henry Dreyfus Teacher-Scholar Award for financial support.
References
- 1.Clearfield A, Poojary DM, Shpeizer B. J Chem Soc Dalton Trans. 1995:111. [Google Scholar]
- 2.Brunet E, Mata MJ, Juanes O, Alhendawi HMH, Cerro C, Rodríguez-Ubis JC. Tetrahedron: Asymm. 2006;17:347. [Google Scholar]
- 3.Brunet E, Mata MJ, Alhendawi HMH, Cerro C, Alonso M, Juanes O, Rodríguez-Ubis JC. Chem Mater. 2005;17:1424. [Google Scholar]
- 4.Brunet E, Alhendawi HMH, Juanes O, Rodriguez-Ubis JC. J Mex Chem Soc. 2009;53(3):155. [Google Scholar]
- 5.Brunet E, de la Mata MJ, Juanes O, Rodriguez-Ubis JC. Chem Mater. 2004;16:1517. [Google Scholar]
- 6.Brunet E, Alhendawi HMH, Juanes O, Jiménez L, Rodríguez-Ubis J Mater Chem. 2009;19:2494. [Google Scholar]
- 7.Morita M, Rau D, Kai T. J Lumin. 2002;100:97. [Google Scholar]
- 8.(a) Fukao S, Fujiki M. Macromoecules. 2009;42:8062. [Google Scholar]; (b) Cipparrone G, Pagliusi P, Provenzano C, Shibaev VP. Macromolecules. 2008;41:5992. [Google Scholar]; (c) Lakhwani G, Meskers SCJ, Janssen RAJ. J Phys Chem B. 2007:111. doi: 10.1021/jp067852r. [DOI] [PubMed] [Google Scholar]; (d) Satrijo A, Meskers SCJ, Swager TM. J Am Chem Soc. 2006;128:9030. doi: 10.1021/ja063027c. [DOI] [PubMed] [Google Scholar]; (e) Tang HZ, Novak BM, He J, Polavarapu PL. Angew Chem Int Ed. 2005;44:7298. doi: 10.1002/anie.200501977. [DOI] [PubMed] [Google Scholar]; (f) Saxena A, Guo G, Fujiki M, Yang Y, Ohira A, Okoshi K, Naito M. Macromolecules. 2004;37:3081. [Google Scholar]; (g) Schenning APHJ, Jonkheijm P, Peeters E, Meijer EW. J Am Chem Soc. 2001;123:409. [Google Scholar]; (h) Nakako H, Nomura R, Masuda T. Macromolecules. 2001;34:1496–1502. [Google Scholar]; (i) Nakashima H, Koe JR, Torimitsu K, Fujiki M. J Am Chem Soc. 2001;123:4847–4848. doi: 10.1021/ja010119n. [DOI] [PubMed] [Google Scholar]; (j) Fujiki M. J Am Chem Soc. 2000;122:3336. [Google Scholar]; (k) Chen HP, Katsis D, Mastrangelo JC, Chen SH, Jacobs SD, Hood PJ. Adv Mater. 2000;12:1283. [Google Scholar]; (l) Chen SH, Katsis D, Schmid AW, Mastrangelo JC, Tsutsui T, Blanton TN. Nature. 1999;397:506. [Google Scholar]; (m) Peeters E, Christiaans MPT, Janssen RAJ, Schoo HFM, Dekkers HPJM, Meijer EWJ. Am Chem Soc. 1997;119:9909. [Google Scholar]; (n) Green MM, Peterson NC, Sato T, Teramoto A, Cook R, Lifson S. Science. 1995;1860:268. doi: 10.1126/science.268.5219.1860. [DOI] [PubMed] [Google Scholar]
- 9.(a) Imai Y, Shiota N, Kinuta T, Okuno T, Nakano Y, Harada T, Sato T, Fujiki M, Kuroda R, Matsubara Y. Eur J Org Chem. 2010:1353–1357. [Google Scholar]; (b) Imai Y, Kawano K, Nakano Y, Kawaguchi K, Harada T, Sato T, Fujiki M, Kuroda R, Matsubara Y. New J Chem. 2008;32:1110–1112. [Google Scholar]; (c) Elliott SD, Moloney MP, Gun’ko YK. Nano Lett. 2008;8:2452–2457. doi: 10.1021/nl801453g. [DOI] [PubMed] [Google Scholar]; (d) Yao H, Miki K, Nishida N, Sasaki A, Kimura K. J Am Chem Soc. 2005;127:15536–15543. doi: 10.1021/ja053504b. [DOI] [PubMed] [Google Scholar]
- 10.Unfortunately the enantiomers of 5 could not be separated in our hands by the usual chromatographic techniques.
- 11.Brunet E, Juanes O, Jiménez L, Rodríguez-Ubis JC. Tetrahedron Lett. 2009;50:5361. [Google Scholar]
- 12.Meudtner RM, Ostermeier M, Goddard R, Limberg C, Hecht S. Chem Eur J. 2007;13:9834. doi: 10.1002/chem.200701240. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Huffmanab JC, Flood AH. Chem Commun. 2007:2692. doi: 10.1039/b703301j. [DOI] [PubMed] [Google Scholar]
- 14.The model was built from the core formed by the metal and three bis-triazolylpyridine units taken from crystallography data of a similar complex from Ref. [6]. The phosphonate arms were then left to be minimized using MM+ tool of Hyperchem program while the core was fixed and thus unchanged in the calculation.
- 15.A precedent of a related case of chiral memory can be found in Ref. [4].
- 16.(a) Do K, Muller FC, Muller G. J Phys Chem A. 2008;112(30):6789. doi: 10.1021/jp804463e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bonsall SD, Houcheime M, Straus DA, Muller G. Chem Commun. 2007;35:3676. doi: 10.1039/b704346e. [DOI] [PMC free article] [PubMed] [Google Scholar]