

Abstract
Because it is likely that, in healthy human subjects, baroreflex mechanisms operate continuously, independent of experimental interventions, we asked the question, In what ways might study of unprovoked, very infrequent muscle sympathetic bursts inform baroreflex physiology? We closely examined arterial pressure and R-R interval responses of 11 supine healthy young subjects to arterial pressure ramps triggered by large isolated muscle sympathetic bursts. We triggered data collection sweeps on the beginnings of sympathetic bursts and plotted changes of arterial pressure (finger volume clamp or intra-arterial) and R-R intervals occurring before as well as after the sympathetic triggers. We estimated baroreflex gain from regression of R-R intervals on systolic pressures after sympathetic bursts and from the transfer function between cross-spectra of systolic pressure and R-R intervals at low frequencies. Isolated muscle sympathetic bursts were preceded by arterial pressure reductions. Baroreflex gain, calculated with linear regression of R-R intervals on systolic pressures after bursts, was virtually identical to baroreflex gain, calculated with the cross-spectral modulus [mean and (range): 24 (7–43) vs. 24 (8–45) ms/mmHg], and highly significant, according to linear regression (r2 = 0.91, P = 0.001). Our results indicate that 1) since infrequent human muscle sympathetic bursts are almost deterministically preceded by arterial pressure reductions, their occurrence likely reflects simple baroreflex physiology, and 2) the noninvasive low-frequency modulus reliably reproduces gains derived from R-R interval responses to arterial pressure ramps triggered by infrequent muscle sympathetic bursts.
Keywords: vagal, baroreflex sequences, cross-spectral transfer function, sympathetic
there is no reason to expect that human autonomic reflexes operate only in the laboratory and that experimental interventions are necessary to bring them into view. Accordingly, an important literature describes efforts to quantify autonomic reflex responses from changes observed in simple time series, recorded without experimental manipulations. Despite the simplicity of this genre of research, it is beset by complexities. For example, causation cannot be measured in the laboratory; the fact that one measured parameter changes before another does not prove that it caused the change. Indeed, it is not even possible to know with certainty which change precedes the other.
Consider the arterial baroreflex. Although there is strong evidence that muscle sympathetic bursts emerge as baroreflex responses to falling arterial pressures (49), the possibility that such bursts appear as manifestations of an underlying central rhythm cannot be dismissed (3). Again, in resting healthy human subjects, arterial pressures, R-R intervals, and muscle sympathetic nerve activity fluctuate in synchrony with breathing (21). If R-R intervals increase after (or in synchrony with) arterial pressure increases, is this evidence of ongoing vagal baroreflex physiology (19)? If the answer to this simple question is uncertain, perhaps it is necessary after all to rely on experimental interventions to elicit unambiguous baroreflex responses.
Before 1969, there was no reliable method available to quantitate human baroreflex function. In that year, however, Smyth, Sleight, and Pickering (46) proposed that R-R interval responses to drug-induced systolic pressure increases can be correlated by linear regression to derive estimates of human vagal baroreflex gain (the “Oxford method”). Subsequently, Bertinieri and colleagues performed linear regression analysis of sequential R-R interval increases on preceding, spontaneously occurring arterial pressure ramps (6). The first human study of “baroreflex sequences,” as they came to be known, correlated R-R interval increases with preceding arterial pressure ramps caused by single or multiple muscle sympathetic bursts (26). (In the present study, we extend this line of inquiry and analyze events occurring before as well as after sympathetic bursts, which were single, not multiple.)
A variety of time- and frequency-domain methods has been developed to quantify spontaneous ongoing vagal baroreflex responses (1, 36–38, 43). Results obtained with these newer methods have been compared with results obtained after vasoactive drug injections (which are taken as the “gold standard”). In most published comparisons, correlations between spontaneous and pharmacological baroreflex gains are statistically significant. An exception, however, was published by Lipman, Salisbury, and Taylor in an article that unambiguously showcased its main conclusion in the title: “Spontaneous indices are inconsistent with arterial baroreflex gain” (34).
Our study explores human autonomic mechanisms in a group of healthy young subjects who are distinguished by the paucity of their muscle sympathetic bursts. In contrast with all previous studies in this genre, we characterized events occurring before as well as after isolated (not multiple) sympathetic bursts in an effort to determine what new information they might provide regarding basic human autonomic mechanisms.
METHODS
Ethical approval and subjects.
This study was approved by the human research committees of the Hunter Holmes McGuire Department of Veterans Affairs Medical Center, Medical College of Virginia at Virginia Commonwealth University, and Vanderbilt University and conformed with the provisions of the Declaration of Helsinki. All subjects gave their written informed consent. This study is a retrospective analysis of data from 11 healthy men and women from the Medical College of Virginia (6 subjects) and Vanderbilt University (5 subjects). The volunteers were studied to obtain healthy control data, and their measurements were recorded on digital FM tape for off-line analysis. For inclusion in this study, we required only that subjects have widely separated muscle sympathetic bursts at rest in the supine position. The range of subjects' age was 18–35 yr; height, 162–177 cm; weight, 52–79 kg; and body mass index, 18–25 kg/m2. All subjects routinely performed light exercise about twice weekly, and no subject was an endurance athlete.
Measurements.
We recorded the electrocardiogram; arterial pressure [intra-arterial catheter connected to a strain gauge pressure transducer, or finger photoplethysmographic volume-clamped pressure (39), with a cuff on the middle finger of the nondominant hand (Finapres model 2300; Ohmeda, Englewood, CO)]; respiration (pneumobelt or Fleisch pneumotachograph); and muscle sympathetic nerve activity. Recordings were made over 10- to 15-min periods, after subjects rested in the supine position for at least 60 min. Postganglionic sympathetic nerve activity was recorded with a tungsten microelectrode inserted into a peroneal nerve near the fibular head (52). Sympathetic bursts supplying skeletal muscle were identified by their characteristic cardiac and respiratory periodicities (21), their increases during held expiration (27) and Valsalva straining (44), and their failure to respond to arousal. The filtered nerve signal was amplified, rectified, and integrated by a Nerve Traffic Analyzer (model 662C-3; University of Iowa Bioengineering, Iowa City, IA). All data were recorded on strip chart and FM or digital recorders and subsequently digitized at 250 Hz with 14-bit resolution with commercial hardware and software (CODAS; Dataq Instruments, Akron, OH).
One author over read computer detection of R waves and systolic and diastolic pressures, and identified sympathetic bursts with signal-to-noise ratios of >2:1, according to their appearances and their nearly fixed latencies after preceding (one removed) R waves (24). With one important exception (see below), we advanced muscle sympathetic nerve activity individually, according to each subject's measured burst latency (range: 1.1–1.38 s, mean ± SE 1.27 ± 0.03 s), in the time series to compensate for peripheral conduction delays. Individual burst latency was determined as the averaged lag time between corresponding previous R wave peak and burst maxima. Figure 1 shows three large sympathetic bursts, occurring over a 20-s period in one subject. These bursts seemed to be preceded by blood pressure reductions and to be followed by arterial pressure and R-R interval increases. Their timing bore no simple relation to breathing.
Fig. 1.
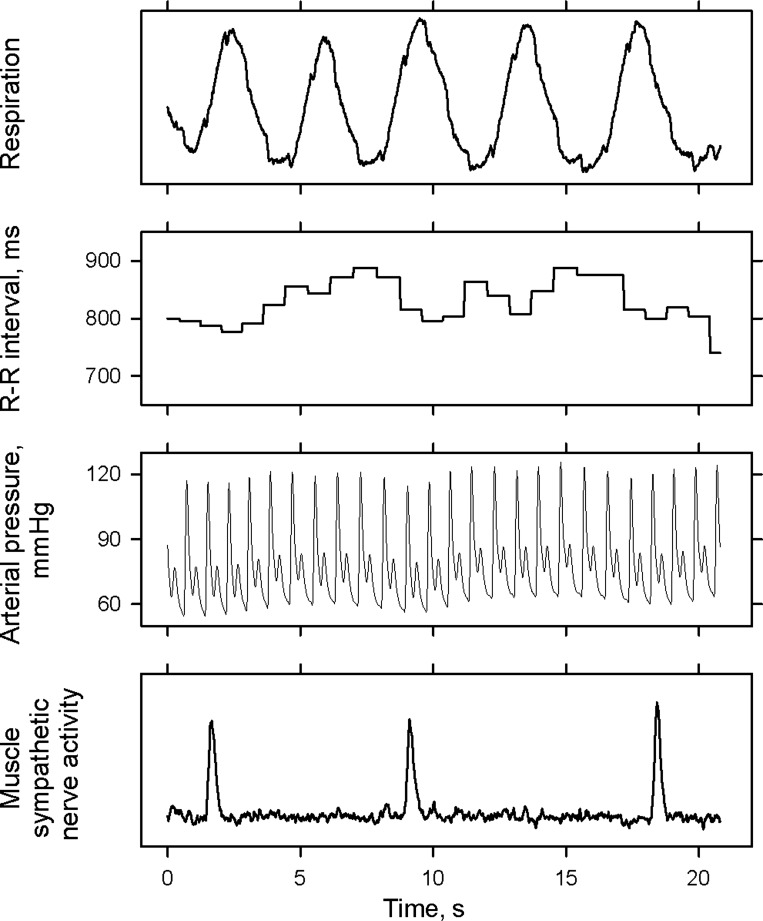
Representative tracing from one subject, showing 3 widely spaced muscle sympathetic bursts.
At the beginning of this research, it was obvious that we would have more bursts to analyze in our group of subjects with very infrequent sympathetic bursts, if we accepted shorter rather than longer burst-free periods before and after reference bursts. Therefore, we studied all data from a different subject to determine the shortest interval between bursts that would preserve discrete arterial pressure and R-R interval oscillations related to the bursts. Figure 2 shows average diastolic pressures (±SE), aligned on sympathetic bursts (vertical dashed lines), from the subject who had the largest number of infrequent sympathetic bursts. This subject had 6 bursts separated by 10 s, 17 separated by 8 s, and 31 separated by 6 s. Diastolic pressure oscillations were broadly concordant, regardless of the number of bursts analyzed. Therefore, for our study, we accepted bursts preceded and followed by 6-s burst-free periods, for all analyses. (This decision was based on our need to have a sufficient number of bursts to analyze; we discuss the consequences of our choice of burst-free intervals in Limitations.) The total number of bursts studied was 125, and the average number of bursts analyzed per subject was 11 (4–30).
Fig. 2.
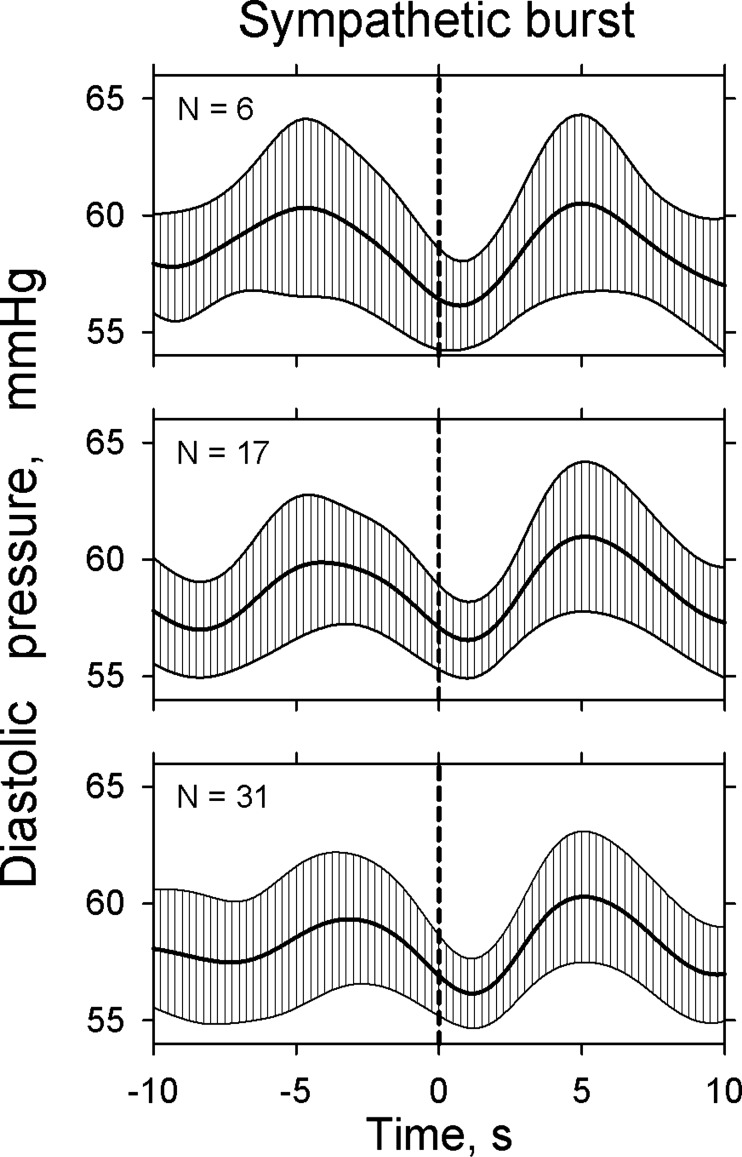
Average (±SE) diastolic pressure, signal-averaged on the leading edge of sympathetic bursts, from another subject. The three panels show analyses of bursts separated by 10 s (top), 8 s (middle), and 6 s (bottom). For the purposes of our study, we chose bursts separated by 6-s burst-free periods and considered these to be “isolated.” This compromise was necessary because very few subjects have muscle sympathetic burst frequencies <10/min.
We set an electronic threshold on the leading edge of each sympathetic burst selected for analysis, about 20% above the baseline level, and recorded the occurrence of each R wave peak, systolic maximum, and diastolic minimum for 10 s before and after this threshold crossing. Subsequently, the measured time between the onset of the burst and the threshold crossing was subtracted (∼0.2 s) to place the timing of the beginning of each burst in its precise location in the data stream.
Signal analysis.
We performed further analyses with locally developed algorithms based on commercial software (PV-Wave Advantage; Visual Numerics, Boulder, CO). We converted R-R intervals into an evenly spaced time series using the algorithm of Berger et al. (5) with modified software created by Daniel Kaplan (http://www.macalester.edu/∼kaplan/hrv/doc/funs/hrtach.html). It used an anti-alias filter, a boxcar, whose width was twice the interval between samples, and then resampled the time series to yield an equidistant output at 4 Hz. We linearly interpolated arterial pressure time series at 4 Hz.
We used a program developed earlier (15) that mathematically determines where curvilinear relations bend. Briefly, this program iteratively performs least-squares linear-regression analyses to the left and right of successive points on a curvilinear relation and adds the sum squared errors of the two regressions. That point in the relation at which the combined sum squared errors is least is taken as the point at which the line bends. (In the examples depicted in Fig. 2, the calculated bending points for the beginnings of the diastolic pressure reductions before the onset of bursts were ∼3–5 s.)
Figure 3 depicts 12 data segments recorded from a different subject than the ones whose data are shown in Figs. 1 and 2. Visual inspection of these data suggests that arterial pressure changes occurring between about 3 s before and after the bursts are deterministic; without exception, each burst was preceded by reductions of diastolic and systolic pressures. Similarly, sympathetic bursts were uniformly preceded by R-R interval reductions. Finally, we aligned and averaged all time series on the beginning of sympathetic bursts for each subject. All latency analyses were performed on these 11 average time series.
Fig. 3.
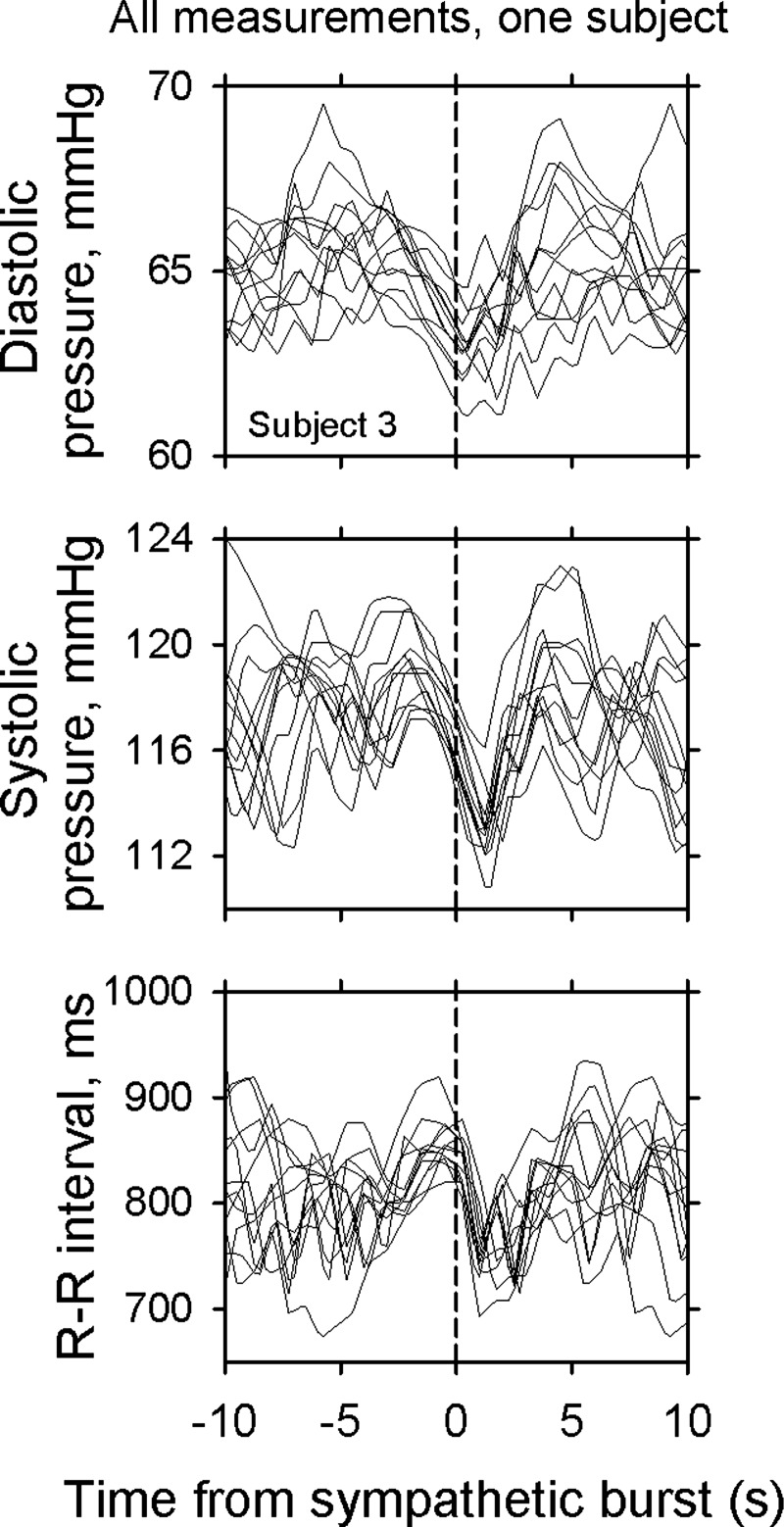
R-R interval and diastolic and systolic pressure responses to bursts from one subject.
We used two approaches to estimate vagal baroreflex gain. First, we used the method described originally for baroreflex estimation after bolus intravenous pressor drug injections (22, 46) and performed linear regression analyses of R-R interval (>4 ms) and systolic pressure (>1 mmHg) increases per beat following sympathetic bursts. Regressions were calculated with R-R interval offsets of 0 and +1 (18), and the slope with the higher correlation coefficient was used for subsequent analyses. We accepted only slopes having correlation coefficients ≥0.85.
Second, we estimated vagal baroreflex gain with cross-spectral transfer function analysis of 300 s of artifact-free data (2, 43). Baroreflex gain was defined as the mean value of the transfer function between systolic pressure and R-R intervals in the range 0.05–0.15 Hz, where the squared coherence between these signals was ≥0.50 and the phase angle was <0. We considered that a negative phase indicates that pressure changes lead R-R interval changes (11, 36). For example, a negative phase angle of 90 degrees at 0.1 Hz was taken to mean that systolic pressure changes lead R-R interval changes by 2.5 s (90°/360° × 10 s). Cross-spectral baroreflex estimates were made over entire recordings, without regard to the presence or absence of sympathetic bursts.
Statistical analyses.
Results are expressed as means and ranges. Correlations between signals were evaluated with least-squares linear-regression and Bland-Altman plots (7, 8). We used the Kolmogorov-Smirnov test to evaluate the distribution of data, Student's paired t-test to determine differences in normally distributed data sets, and the Wilcoxon Signed Rank test to determine differences in nonnormally distributed data sets. We considered differences to be significant when P ≤ 0.05.
RESULTS
Figure 4 documents average systematic relations among arterial pressure and R-R interval changes preceding, as well as following, sympathetic bursts. In this figure, average bending points determined (from all individual relations) are identified by the dashed black vertical lines. The placement of sympathetic bursts (indicated by the vertical gray dashed lines) is different in Fig. 4, left and right. In Fig. 4, left, we followed precedent and moved the sympathetic recording by each subject's latency (range: 1.1–1.38 s) earlier in the data stream to compensate for peripheral conduction delays (49). In Fig. 4, right, the timing of sympathetic bursts was not changed; thus, sympathetic bursts appeared in these analyses at the actual times they arrived to release norepinephrine and trigger hemodynamic changes. The rationale for these different treatments is further elaborated in the discussion.
Fig. 4.
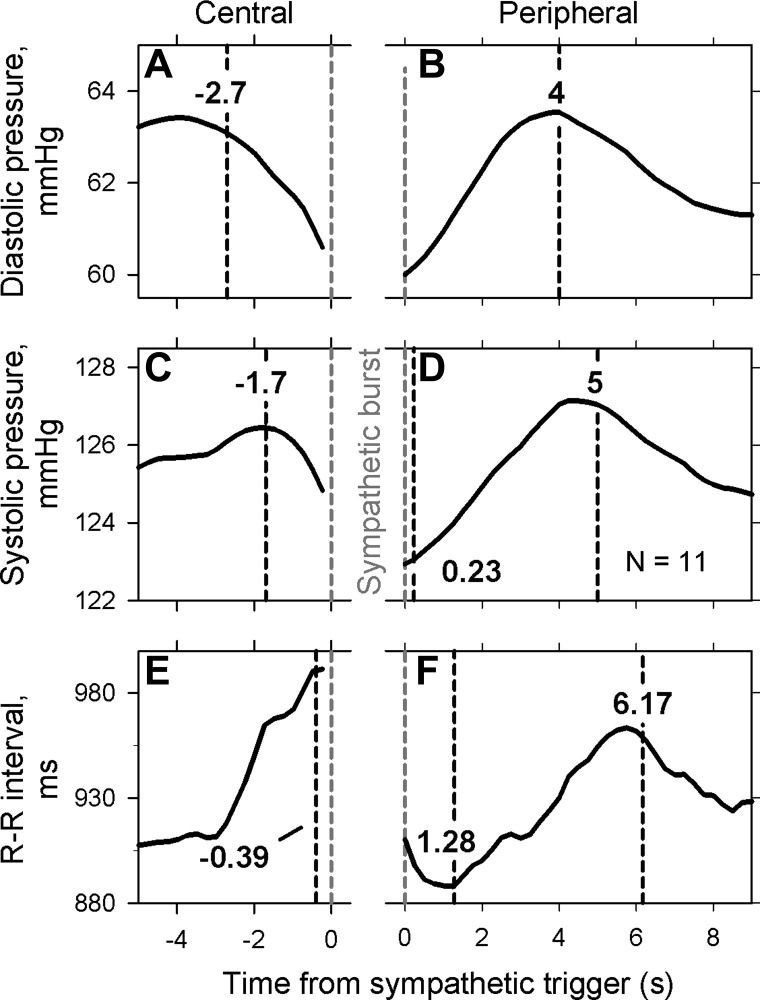
Average signals triggered on sympathetic bursts for all subjects. For measurements shown on left, muscle sympathetic nerve activity was advanced by each subject's measured latency to account for peripheral conduction delays. For measurements shown on the right, the timing of sympathetic nerve activity was not changed (see methods). The beginning of sympathetic bursts is indicated by the vertical dashed gray lines. Latencies, calculated with bending point analyses, are indicated in the vertical dashed black lines.
Diastolic pressure.
Diastolic pressure (Fig. 4A) began to decay −2.7 (−4.7 to −0.7) s before the onset of the reference burst. Diastolic pressures fell before the onset of bursts in 109 of 125 bursts examined (87%). The average reduction of diastolic pressure before the sympathetic burst was 3.8 (1.7–8.6) mmHg. Diastolic pressure peaked 4 (3–6) s after the onset of the sympathetic bursts, with an average increase of 4.3 (2.1–8.8) mmHg (Fig. 4B).
Systolic pressure.
Systolic pressure (Fig. 4C) began to decay −1.7 (−3.9 to 1.7) s before the onset of the reference burst. Systolic pressures fell before the onset of bursts in 107 of 125 bursts examined (86%). The average reduction of systolic pressure before the sympathetic burst was 4.3 (1.4–6) mmHg. Systolic pressure peaked 5.0 (2.5–9.5) s after the onset of the sympathetic bursts, with an increase of 5.2 (2.6–7.2) mmHg (Fig. 4D).
R-R intervals.
R-R intervals (Fig. 4E) began to decay −0.39 (−1.56 to 0.1) s before the onset of the sympathetic bursts and reached a minimum at 1.28 (−0.5 to 2.25) s. The average reduction of R-R intervals was 120 (19–275) ms. R-R intervals peaked 6.17 (3.5–9.25) s after the onset of the sympathetic bursts (Fig. 4F). The average maximum change of R-R intervals was −23 (−107 to 33) ms.
The results of baroreflex gain analysis are summarized in Fig. 5. Figure 5, left, shows all baroreflex sequence slopes for the eight subjects who had at least three discrete pressure and R-R interval ramps following sympathetic bursts. Baroreflex gains (Fig. 5, middle) averaged 23.8 (6.8–42.7) ms/mmHg with the sequence analysis and 23.7 (7.5–45) with cross-spectral analysis (r2 = 0.91, P < 0.001). The Bland-Altman plot [Fig. 5, right (7, 8)] shows the differences between the two estimates of baroreflex gain, plotted as functions of their mean values. The average difference between vagal baroreflex gains derived from the two methods was only 0.06 ms/mmHg. Regression of these data (gray line) indicates that there was no systematic bias favoring one method over the other (r2 = 0.07, P = 0.53). The horizontal dashed lines indicate the limits of agreement (7, 8), calculated as the mean difference between the two measurements ± 1.96 × standard deviation of the differences.
Fig. 5.
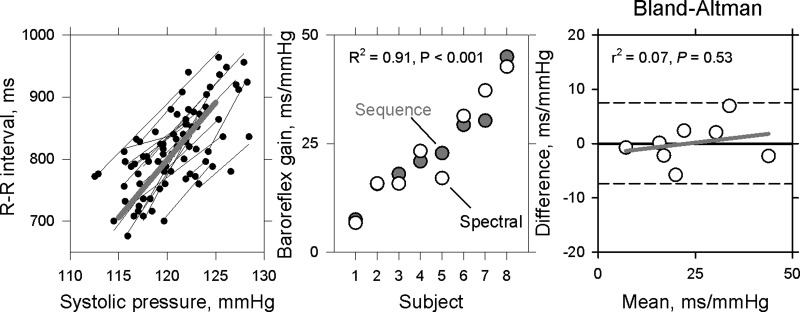
Average spontaneous baroreflex estimates from the eight subjects who had at least three acceptable systolic pressure-R-R interval ramps. Regressions of R-R intervals on systolic pressure increases triggered by sympathetic bursts are shown on left, and average vagal baroreceptor-cardiac gain estimated by cross-spectral and sequence methods are shown in center. The regression coefficients and the slope of the regression (gray line) in the Bland-Altman plot on the right did not show a systematic proportional bias between the two baroreflex measures. The horizontal dashed lines indicate tolerance interval within 95% confidence limits.
DISCUSSION
We studied healthy young subjects who were distinguished by the paucity of their muscle sympathetic bursts and measured hemodynamic events surrounding those bursts. These unique data allowed us to 1) analyze responses to isolated muscle sympathetic bursts, 2) determine what changes precede as well as follow the onset of those bursts, and 3) estimate latencies according to statistical probability. Our study yields two major conclusions. First, the occurrence of isolated muscle sympathetic bursts likely reflects baroreflex physiology, since such bursts are preceded almost deterministically by arterial pressure reductions. Second, following isolated sympathetic bursts, arterial pressures rise, and, after that, R-R intervals increase. R-R interval lengthening likely reflects baroreflex physiology: slopes of postburst systolic pressure/R-R interval relations are virtually identical to baroreflex gains estimated by the low-frequency modulus.
Changes occurring before muscle sympathetic bursts.
Isolated human muscle sympathetic bursts are preceded by falling diastolic pressures, with an average latency of 2.7 s (Fig. 4A), which is consistent with baroreflex causation. This latency is slightly longer than the latency reported by Wallin and Eckberg (52), ∼2.0 s, whose artificial (neck chamber pressure) stimulus to sympathetic firing was much more abrupt (>2,000 mmHg/s) and intense (30 mmHg) than the gradual and small (3.8 mmHg) natural diastolic pressure reduction registered in our study. Our conclusion, that the occurrence of rare sympathetic bursts reflects baroreflex physiology, is supported by other research that documents a close inverse relation between diastolic pressure changes grouped in bins and muscle sympathetic bursts (29). The measured average diastolic and systolic pressure reductions, 3.8 and 4.5 mmHg, are consistent with what is known about baroreflex sensitivity: Landgren documented a clear spike of directly recorded baroreceptor nerve activity in response to a 1-mmHg isolated carotid sinus pressure increase (32), and we reported R-R interval lengthening in response to 5 mmHg of neck suction, a stimulus intensity that is so small that most subjects are not aware that suction was applied (14).
Our study shows that average R-R interval reductions also began before the onset of sympathetic bursts (0.39 s, Fig. 4E). This observation supports the assumption of Eckberg et al. (21) that R-R interval shortening and sympathetic bursting likely reflect autonomic responses to preceding reductions of afferent baroreceptor input, which reciprocally influence vagal and sympathetic neural outflows (31). The tightness of the relation between R-R interval reductions and the emergence of sympathetic bursts is reflected by the narrow range of latencies (−1.56 to 0.1 s). Other studies show that abrupt arterial pressure reductions associated with ectopic beats (23), premature ventricular beats (54), and ventricular tachycardia (45) also trigger muscle sympathetic nerve bursts, with very short latencies.
Changes occurring after muscle sympathetic bursts.
Although our study may be the first to analyze events following isolated muscle sympathetic bursts, there are at least three earlier studies (4, 48, 53) that report hemodynamic changes following multiple sympathetic bursts. We report shorter latencies than these earlier studies because Båth et al. (4) and Wallin and Nerhed (53), and, presumably, Sugiyama and his coworkers (48), advanced sympathetic nerve activity in the time series (in Wallin's reports, by 1.45 s) to subtract peripheral conduction delays. Although this treatment brings sympathetic bursts closer to their times of generation within the central nervous system, it inappropriately increases latencies between the arrival of sympathetic bursts, release of norepinephrine at neuroeffector junctions, and hemodynamic responses. The analyses shown in Fig. 4, right, place referenced bursts at the time they occurred in relation to subsequent hemodynamic changes.
Latencies in published models are at variance with our experimental results. Madwed et al.'s model (35) used a latency of 5 s from the beginning of α-adrenergic stimulation to the beginning of vasoconstriction. We found that diastolic pressure begins to increase at about the time of the onset of the sympathetic burst (Fig. 4B, left). (This very short latency, and that of systolic pressure, likely reflects simply the influence of preceding R-R interval shortening, which began earlier in the time series.) Madwed's model used a latency of 15 s from the beginning of α-adrenergic stimulation to the peak vasoconstrictor response. We found that the average latency from the sympathetic burst to the maximum diastolic pressure elevation is 4 s. This result was similar to one calculated by Brychta and coworkers (10), based on their statistical model.
As discussed, R-R interval shortening likely arrests the arterial pressure decay and shortens the latency between the sympathetic burst and the diastolic pressure rise. Similarly, R-R interval lengthening following the increase of arterial pressure likely contributes to the succeeding fall of arterial pressure. We have no way to quantify these ongoing vagal contributions to pressure changes, which represent complexities inherent in studies of conscious human subjects with intact reflex mechanisms.
Vagal baroreflex gain estimation.
We propose that the hemodynamic changes that occur following large muscle sympathetic bursts trigger a cascade of responses that culminates in a vagal baroreflex sequence. We report that average vagal baroreflex gains, calculated from systolic pressure-R-R interval regressions following sympathetic bursts and low-frequency transfer functions are nearly identical (Fig. 5, middle, r2 = 0.91). The Bland-Altman analysis (Fig. 5, right) shows that 1) the average difference between measurements, 0.06 ms/mmHg, is very small relative to the average baroreflex gain, 23.7 ms/mmHg; 2) there is no significant proportional bias favoring one measurement over the other (regression coefficients, gray line in Fig. 5); and 3) all measured differences fall within the limits of agreement [horizontal dashed lines in Fig. 5 (7, 8)].
The gold standard for human vagal baroreflex measurements.
Since the seminal publication of the Oxford method by Smyth, Sleight, and Pickering in 1969 (46), the gold standard for human vagal baroreflex testing has been R-R interval responses to intravenous bolus injections of vasoactive drugs. We propose that systolic pressure-R-R interval regressions measured after muscle sympathetic bursts provide a more pure provocation of baroreflex responses than the Oxford method. The Oxford method, which has figured so importantly in autonomic research since its introduction, is, withal, an experimental intervention, which therefore, can be challenged on theoretical grounds. The Oxford method involves invasive venous cannulation and injection of drugs with multiple actions (30) into veins, from which they traverse the pulmonary circulation, left heart, and, after they likely alter sinoatrial node automaticity (57), constrict baroreceptive artery smooth muscle (9, 40) and, finally, cause generalized vasoconstriction.
Conversely, a muscle sympathetic burst, which arises spontaneously without experimental provocation, releases the naturally occurring neurotransmitter norepinephrine almost simultaneously in all muscle sympathetic nerve endings (51) which innervate over 40% of human body mass (42). We recognize that our argument is in part philosophical and that muscle sympathetic nerve activity is not recorded in most human research. However, the correlation we document between responses to muscle sympathetic bursts and one noninvasive index, the modulus, provides strong support for use of this noninvasive index to estimate vagal baroreflex gain (37). Although there are many published comparisons of one noninvasive estimate of baroreflex gain with another (for example, see Ref. 33), the present study may be qualitatively different from the others in that our comparison is between a noninvasive index and an arterial pressure increase triggered physiologically.
Spontaneous baroreflex indexes.
We invite comparisons between our study and that of Lipman, Salisbury, and Taylor (34). We confirm Lipman's observation that the low-frequency cross-spectral baroreflex index we used yields responses that are comparable to those derived from R-R interval responses to arterial pressure increases (in their case, induced by sequential nitroprusside and phenylephrine injections and in ours by isolated muscle sympathetic bursts). However, we differ in the confidence we place in the cross-spectral approach: we found much stronger coherence between the two indexes (r = 0.96 in our study vs. r = 0.39 in Lipman's study). There are several disparities between the two studies that may explain this large difference.
First, although Lipman and her coworkers studied a larger group of subjects than we did (97 vs. 11), their volunteers included elderly subjects and patients with coronary artery disease, who are known to have subnormal vagal baroreflex gains (17, 28); our subjects were young and healthy. One consequence of Lipman's subject selection is that her results were strongly skewed toward low levels of baroreflex gain (with ∼75% falling below 10 ms/mmHg, her Fig. 2). Second, the average systolic pressure increase provoked pharmacologically in Lipman's study was large, ∼30 mmHg; ours, which reflected spontaneous pressure variability, was small, 5.2 mmHg. Because humans appear to operate on the “linear” portion of their sigmoid baroreceptor stimulus-R-R interval response relations (41), regressions of small pressure increases from baseline and R-R interval responses should accurately reflect peak baroreflex gains and should not be confounded by inclusion of pressures in nonlinear threshold and saturation ranges (16). More importantly, small spontaneous pressure increases occur over brief time periods, but pressure changes following sequential nitroprusside and phenylephrine injections occur over long periods, minimally tens of seconds. A shorter period allows less time for the occurrence of rapid baroreflex adjustments (13, 47), which oppose and reduce ongoing pressure changes.
Third, Lipman et al. (34) compared their cross-spectral results with responses to two or three sequential injections of nitroprusside and phenylephrine; we compared our results with responses to an average of 11 sympathetic bursts followed by pressure ramps per subject. Fourth, Lipman and her colleagues may have made the tacit assumption that levels of vagal baroreflex gain in resting humans are constant and that, therefore, two or three measurements for each subject, made over a total period of perhaps 6 min, are representative of baroreflex gain made during an entire experiment, lasting hours. Recent studies (2, 20, 55) document very large (as much as 10-fold), ongoing variations of vagal baroreflex gain in resting, ostensibly “steady-state” healthy subjects. The importance of this observation for the present work is that baroreflex gain estimated from many ramps of pressure over an entire experimental session are more likely to reflect average baroreflex gain over time than gain estimated from few ramps measured over a small percentage of the experimental period (56).
Limitations.
Our study has several real or potential limitations. Our study is observational rather than hypothesis-testing research. This limitation may be mitigated by our use of different methods of analysis than those used earlier and by the uniqueness of some of our observations. Our numerical results may enable accurate modeling of human autonomic rhythms without recourse to data obtained from invasive animal experiments (12). It is possible that, by restricting our analysis to data from subjects with infrequent muscle sympathetic bursts, we biased our results. This seems unlikely, since [if we advance sympathetic activity as earlier authors did (4, 48, 53), see discussion] our measurements made after isolated sympathetic bursts are nearly identical to those reported by these authors, who signal-averaged responses on multiple sympathetic bursts. We studied only 11 subjects; however, we characterized baroreflex ramps more fully than Lipman's study [11 vs. 2–3 baroreflex ramps/subject (34)]. We did not compensate for differences in pulse transmission time in Finapres and intra-arterial recordings; although such differences must exist, they are likely to be small, perhaps 100 ms.
We arbitrarily defined “isolated” bursts as being separated by >6.0 s so that we would have sufficient material to perform a meaningful analysis. It would have been preferable to require longer interburst intervals; however, in our experience, subjects with >6 s separating bursts are rare. [Fagius and Wallin (25) studied 15 healthy subjects on two separate occasions; of the 30 recordings, only one had fewer than 10 muscle sympathetic bursts/min.] Based upon our data (Fig. 4), it is likely that bursts occurring somewhat earlier than 6 s before the sympathetic trigger exerted some effect on our results. Although we view the isolated sympathetic bursts we analyzed as relatively pure “impulse” functions, succeeding pressure changes surely were modulated by R-R interval changes, whose quantitative influence we are unable to gauge. Because we did not filter our data to exclude respiratory-frequency fluctuations, low-frequency modulus baroreflex gains may have been compared with baroreflex sequences occurring at low as well as respiratory frequencies. This omission should not have influenced our conclusions, since cross-spectral baroreflex gains calculated at low and respiratory frequencies are similar (33). A final potential limitation is that, although our study treats vagal baroreflex mechanisms, we have not excluded sympathetic opposition to vagally mediated changes. This omission is unlikely to have influenced our conclusions: sympathetic stimulation opposes R-R interval fluctuations at all frequencies (50), and there is no reason to expect that sympathetic mechanisms would affect one baroreflex method more than another.
In summary, we triggered arterial pressure and R-R interval data collections on isolated muscle sympathetic bursts and measured changes occurring before as well as after those bursts. Our study shows that arterial pressure falls almost deterministically before the occurrence of isolated sympathetic bursts, with latencies consistent with baroreflex physiology. We regard postsympathetic burst arterial pressure elevations as consequences of those sympathetic bursts and suggest that such pressure sequences and their R-R interval responses should be regarded as a new gold standard against which other baroreflex methods are compared. Finally, we show that one method of estimating vagal baroreflex responses, the low-frequency transfer function (20, 36, 43), yields results that, in our sample of healthy volunteers, are nearly identical to those recorded after isolated sympathetic bursts.
GRANTS
This research was supported in part by grants from the Department of Veterans Affairs and the National Institutes of Health (HL-22296, UO1 HL-56417, MO1 RR-00095, and IPO1 HL-56693) and contracts from the National Aeronautics and Space Administration (NAS9-19541 and NAG2-408).
DISCLOSURES
The authors have no conflict of interests, financial or otherwise.
AUTHOR CONTRIBUTIONS
Author contributions: A.D., A.A.C., and D.L.E. conception and design of research; A.D., A.A.C., L.A.B., A.C.E., and D.L.E. performed experiments; A.D., A.A.C., L.A.B., K.U.T., T.A.K., and D.L.E. analyzed data; A.D., A.A.C., L.A.B., K.U.T., T.A.K., and D.L.E. interpreted results of experiments; A.D. and D.L.E. prepared figures; A.D. and D.L.E. edited and revised manuscript; A.D., A.A.C., K.U.T., T.A.K., A.C.E., and D.L.E. approved final version of manuscript; D.L.E. drafted the manuscript.
REFERENCES
- 1. Airaksinen KEJ, Tahvanainen KUO, Kuusela TA, Huikuri HV, Niemela MJ, Karjalainen P, Eckberg DL. Cross spectral analysis in assessment of baroreflex gain in patients with coronary artery disease. Ann Noninvas Electrocardiol 2: 229–235, 1997 [DOI] [PubMed] [Google Scholar]
- 2. Badra LJ, Cooke WH, Hoag JB, Crossman AA, Kuusela TA, Tahvanainen KUO, Eckberg DL. Respiratory modulation of human autonomic rhythms. Am J Physiol Heart Circ Physiol 280: H2674–H2688, 2001 [DOI] [PubMed] [Google Scholar]
- 3. Barman SM, Fadel PJ, Vongpatanasin W, Victor RG, Gebber GL. Basis for the cardiac-related rhythm in muscle sympathetic nerve activity of humans. Am J Physiol Heart Circ Physiol 284: H584–H597, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Båth E, Lindblad LE, Wallin BG. Effects of dynamic and static neck suction on muscle nerve sympathetic activity, heart rate and blood pressure in man. J Physiol 311: 551–564, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berger R, Akselrod S, Gordon D, Cohen RJ. An efficient algorithm for spectral analysis of heart rate variability. IEEE Trans Biomed Eng BME-33: 900–904, 1986 [DOI] [PubMed] [Google Scholar]
- 6. Bertinieri G, di Rienzo M, Cavallazzi A, Ferrari AU, Pedotti A, Mancia G. A new approach to analysis of the arterial baroreflex. J Hyperten 3, Suppl 3: S79–S81, 1985 [PubMed] [Google Scholar]
- 7. Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1: 307–310, 1986 [PubMed] [Google Scholar]
- 8. Bland JM, Altman DG. Comparing methods of measurement: why plotting difference against standard method is misleading. Lancet 346: 1085–1087, 1995 [DOI] [PubMed] [Google Scholar]
- 9. Bonyhay I, Jokkel G, Karlocai K, Reneman R, Kollai M. Effect of vasoactive drugs on carotid diameter in humans. Am J Physiol Heart Circ Physiol 273: H1629–H1636, 1997 [DOI] [PubMed] [Google Scholar]
- 10. Brychta RJ, Shiavi R, Robertson D, Biaggioni I, Diedrich A. A simplified two-component model of blood pressure fluctuations. Am J Physiol Heart Circ Physiol 292: H1193–H1203, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Boer RW, Karemaker JM, Strackee J. Relationships between short-term blood-pressure fluctuations and heart-rate variability in resting subjects 1: a spectral analysis approach. Med Biol Eng Comput 23: 352–358, 1985 [DOI] [PubMed] [Google Scholar]
- 12. de Boer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol Heart Circ Physiol 253: H680–H689, 1987 [DOI] [PubMed] [Google Scholar]
- 13. Eckberg DL. Temporal response patterns of the human sinus node to brief carotid baroreceptor stimuli. J Physiol 258: 769–782, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckberg DL. Baroreflex inhibition of the human sinus node: importance of stimulus intensity, duration, and rate of pressure change. J Physiol 269: 561–577, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eckberg DL. Carotid baroreflex function in young men with borderline blood pressure elevation. Circulation 59: 632–636, 1979 [DOI] [PubMed] [Google Scholar]
- 16. Eckberg DL. Nonlinearities of the human carotid baroreceptor-cardiac reflex. Circ Res 47: 208–216, 1980 [DOI] [PubMed] [Google Scholar]
- 17. Eckberg DL, Drabinsky M, Braunwald E. Defective cardiac parasympathetic control in patients with heart disease. N Engl J Med 285: 877–883, 1971 [DOI] [PubMed] [Google Scholar]
- 18. Eckberg DL, Eckberg MJ. Human sinus node responses to repetitive, ramped carotid baroreceptor stimuli. Am J Physiol Heart Circ Physiol 242: H638–H644, 1982 [DOI] [PubMed] [Google Scholar]
- 19. Eckberg DL, Karemaker JM. Respiratory sinus arrhythmia is due to a central mechanism vs. respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol 106: 1740–1744, 2009 [DOI] [PubMed] [Google Scholar]
- 20. Eckberg DL, Kuusela TA. Human vagal baroreflex sensitivity fluctuates widely and rhythmically at very low frequencies. J Physiol 567: 1011–1019, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eckberg DL, Nerhed C, Wallin BG. Respiratory modulation of muscle sympathetic and vagal cardiac outflow in man. J Physiol 365: 181–196, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eckberg DL, Sleight P. Human Baroreflexes in Health and Disease. Oxford, UK: Clarendon, 1992 [Google Scholar]
- 23. Fagius J. Muscle nerve sympathetic activity following ectopic heart beats: a note on the burst pattern of sympathetic impulses. J Autonom Nerv Syst 22: 243–245, 1988 [DOI] [PubMed] [Google Scholar]
- 24. Fagius J, Wallin BG. Sympathetic reflex latencies and conduction velocities in normal man. J Neurol Sci 47: 433–448, 1980 [DOI] [PubMed] [Google Scholar]
- 25. Fagius J, Wallin BG. Long-term variability and reproducibility of resting human muscle nerve sympathetic activity at rest, as reassessed after a decade. Clin Autonom Res 3: 201–205, 1993 [DOI] [PubMed] [Google Scholar]
- 26. Fritsch JM, Eckberg DL, Graves LD, Wallin BG. Arterial pressure ramps provoke linear increases of heart period in humans. Am J Physiol Regul Integr Comp Physiol 251: R1086–R1090, 1986 [DOI] [PubMed] [Google Scholar]
- 27. Fritsch JM, Smith ML, Simmons DTF, Eckberg DL. Differential baroreflex modulation of human vagal and sympathetic activity. Am J Physiol Regul Integr Comp Physiol 260: R635–R641, 1991 [DOI] [PubMed] [Google Scholar]
- 28. Gribbin B, Pickering TG, Sleight P, Peto R. Effect of age and high blood pressure on baroreflex sensitivity in man. Circ Res 29: 424–431, 1971 [DOI] [PubMed] [Google Scholar]
- 29. Hart EC, Joyner MJ, Wallin BG, Karlsson T, Curry TB, Charkoudian N. Baroreflex control of muscle sympathetic nerve activity: a nonpharmacological measure of baroreflex sensitivity. Am J Physiol Heart Circ Physiol 298: H816–H822, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hogan N, Casadei B, Patterson DJ. Nitric oxide donors can increase heart rate independent of autonomic activation. J Appl Physiol 87: 97–103, 1999 [DOI] [PubMed] [Google Scholar]
- 31. Kollai M, Koizumi K. Reciprocal and non-reciprocal action of the vagal and sympathetic nerves innervating the heart. J Autonom Nerv Syst 1: 33–52, 1979 [DOI] [PubMed] [Google Scholar]
- 32. Landgren S. On the excitation mechanism of the carotid baroceptors. Acta Physiol Scand 26: 1–34, 1952 [DOI] [PubMed] [Google Scholar]
- 33. Laude D, Elghozi JL, Girard A, Bellard E, Bouhaddi M, Castiglioni P, Cerutti C, Cividjian A, di Rienzo M, Fortrat JO, Janssen B, Karemaker JM, Leftheriotis G, Parati G, Persson PB, Porta A, Quintin L, Regnard J, Rudiger H, Stauss HM. Comparison of various techniques used to estimate spontaneous baroreflex sensitivity (the EuroBaVar study). Am J Physiol Regul Integr Comp Physiol 286: R226–R231, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Lipman RD, Salisbury JK, Taylor JA. Spontaneous indices are inconsistent with arterial baroreflex gain. Hypertension 42: 481–487, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Madwed JB, Albrecht P, Mark RG, Cohen RJ. Low-frequency oscillations in arterial pressure and heart rate: a simple computer model. Am J Physiol Heart Circ Physiol 256: H1573–H1579, 1989 [DOI] [PubMed] [Google Scholar]
- 36. Pagani M, Somers V, Furlan R, Dell'Orto S, Conway J, Baselli G, Cerutti S, Sleight P, Malliani A. Changes in autonomic regulation induced by physical training in mild hypertension. Hypertension 12: 600–610, 1988 [DOI] [PubMed] [Google Scholar]
- 37. Parati G, DiRienzo M, Mancia G. How to measure baroreflex sensitivity: from the cardiovascular laboratory to daily life. J Hyperten 18: 7–19, 2000 [PubMed] [Google Scholar]
- 38. Parlow J, Viale JP, Annat G, Hughson R, Quintin L. Spontaneous cardiac baroreflex in humans. Comparison with drug-induced responses. Hypertension 25: 1058–1068, 1995 [DOI] [PubMed] [Google Scholar]
- 39. Peňáz J. Photoelectric measurement of blood pressure, volume and flow in the finger. In: Digest of the 10th International Conference on Medical and Biologic Engineering Dresden, Germany: The Conference Committee of the X International Conference on Medical and Biological Engineering, 1973, p. 104 [Google Scholar]
- 40. Peveler RC, Bergel DH, Robinson JL, Sleight P. The effect of phenylephrine upon arterial pressure, carotid sinus radius and baroreflex sensitivity in the conscious greyhound. Clin Sci 64: 455–461, 1983 [DOI] [PubMed] [Google Scholar]
- 41. Rea RF, Eckberg DL. Carotid baroreceptor-muscle sympathetic relation in humans. Am J Physiol Regul Integr Comp Physiol 253: R929–R934, 1987 [DOI] [PubMed] [Google Scholar]
- 42. Rein H, Schneider M. Einführung in die Physiologie des Menschen. Berlin, Germany: Springer-Verlag, 1956 [Google Scholar]
- 43. Robbe HWJ, Mulder LJM, Ruddel H, Langewitz WA, Veldman JBP, Mulder G. Assessment of baroreceptor reflex sensitivity by means of spectral analysis. Hypertension 10: 538–543, 1987 [DOI] [PubMed] [Google Scholar]
- 44. Smith ML, Beightol LA, Fritsch-Yelle JM, Ellenbogen KA, Porter TR, Eckberg DL. Valsalva's maneuver revisited: a quantitative method yielding insights into human autonomic control. Am J Physiol Heart Circ Physiol 271: H1240–H1249, 1996 [DOI] [PubMed] [Google Scholar]
- 45. Smith ML, Ellenbogen KA, Beightol LA, Eckberg DL. Sympathetic neural responses to induced ventricular tachycardia. J Am Col Cardiol 18: 1015–1024, 1991 [DOI] [PubMed] [Google Scholar]
- 46. Smyth HS, Sleight P, Pickering GW. Reflex regulation of arterial pressure during sleep in man. A quantitative method of assessing baroreflex sensitivity. Circ Res 24: 109–121, 1969 [DOI] [PubMed] [Google Scholar]
- 47. Sprangers RLH, Veerman DP, Karemaker JM, Wieling W. Initial circulatory responses to changes in posture: influence of the angle and speed of tilt. Clin Physiol 11: 211–220, 1991 [DOI] [PubMed] [Google Scholar]
- 48. Sugiyama Y, Matsukawa T, Shamsuzzaman ASM, Okada H, Watanabe T, Mano T. Delayed and diminished pressor response to muscle sympathetic nerve activity in the elderly. J Appl Physiol 80: 869–875, 1996 [DOI] [PubMed] [Google Scholar]
- 49. Sundlöf G, Wallin BG. Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J Physiol 274: 621–637, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taylor JA, Myers CW, Halliwill JR, Seidel H, Eckberg DL. Sympathetic restraint of respiratory sinus arrhythmia: implications for vagal-cardiac tone assessment in humans. Am J Physiol Heart Circ Physiol 280: H2804–H2814, 2001 [DOI] [PubMed] [Google Scholar]
- 51. Wallin BG, Burke D, Gandevia S. Coupling between variations in strength and baroreflex latency of sympathetic discharges in human muscle nerves. J Physiol 474: 331–338, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wallin BG, Eckberg DL. Sympathetic transients caused by abrupt alterations of carotid baroreceptor activity in humans. Am J Physiol Heart Circ Physiol 242: H185–H190, 1982 [DOI] [PubMed] [Google Scholar]
- 53. Wallin BG, Nerhed C. Relationship between spontaneous variations of muscle sympathetic activity and succeeding changes of blood pressure in man. J Autonom Nerv Syst 6: 293–302, 1982 [DOI] [PubMed] [Google Scholar]
- 54. Welch WJ, Smith ML, Rea RF, Bauernfeind RA, Eckberg DL. Enhancement of sympathetic nerve activity by single premature ventricular beats in humans. J Am Col Cardiol 13: 69–75, 1989 [DOI] [PubMed] [Google Scholar]
- 55. Westerhof BE, Gisolf J, Karemaker JM, Wesseling KH, Secher NH, van Lieshout JJ. Time course analysis of baroreflex sensitivity during postural stress. Am J Physiol Heart Circ Physiol 291: H2864–H2874, 2006 [DOI] [PubMed] [Google Scholar]
- 56. Westerhof BE, Gisolf J, Stok WJ, Wesseling KH, Karemaker JM. Time-domain cross-correlation baroreflex sensitivity: performance on the UROBAVAR data set. J Hypertens 22: 1371–1380, 2004 [DOI] [PubMed] [Google Scholar]
- 57. White CW, Eckberg DL, Inasaka T. Direct effects of methoxamine and phenylephrine on sinus mode function (Abstract). Am J Cardiol 31: 164, 1973 [Google Scholar]