

Abstract
We have previously reported chronic low-intensity interval exercise training attenuates fibrosis, impaired cardiac mitochondrial function, and coronary vascular dysfunction in miniature swine with left ventricular (LV) hypertrophy (Emter CA, Baines CP. Am J Physiol Heart Circ Physiol 299: H1348–H1356, 2010; Emter CA, et al. Am J Physiol Heart Circ Physiol 301: H1687–H1694, 2011). The purpose of this study was to test two hypotheses: 1) chronic low-intensity interval training preserves normal myocardial oxygen supply/demand balance; and 2) training-dependent attenuation of LV fibrotic remodeling improves diastolic function in aortic-banded sedentary, exercise-trained (HF-TR), and control sedentary male Yucatan miniature swine displaying symptoms of heart failure with preserved ejection fraction. Pressure-volume loops, coronary blood flow, and two-dimensional speckle tracking ultrasound were utilized in vivo under conditions of increasing peripheral mean arterial pressure and β-adrenergic stimulation 6 mo postsurgery to evaluate cardiac function. Normal diastolic function in HF-TR animals was characterized by prevention of increased time constant of isovolumic relaxation, normal LV untwisting rate, and enhanced apical circumferential and radial strain rate. Reduced fibrosis, normal matrix metalloproteinase-2 and tissue inhibitors of metalloproteinase-4 mRNA expression, and increased collagen III isoform mRNA levels (P < 0.05) accompanied improved diastolic function following chronic training. Exercise-dependent improvements in coronary blood flow for a given myocardial oxygen consumption (P < 0.05) and cardiac efficiency (stroke work to myocardial oxygen consumption, P < 0.05) were associated with preserved contractile reserve. LV hypertrophy in HF-TR animals was associated with increased activation of Akt and preservation of activated JNK/SAPK. In conclusion, chronic low-intensity interval exercise training attenuates diastolic impairment by promoting compliant extracellular matrix fibrotic components and preserving extracellular matrix regulatory mechanisms, preserves myocardial oxygen balance, and promotes a physiological molecular hypertrophic signaling phenotype in a large animal model resembling heart failure with preserved ejection fraction.
Keywords: diastolic heart failure, exercise, MV̇o2, fibrosis, hypertrophic signaling, speckle tracking, coronary flow
prior clinical studies, including the recently completed HF-ACTION trial, indicates exercise training is a safe and effective therapeutic modality in the treatment of patients with stable heart failure (HF) due to left ventricular (LV) systolic dysfunction (9, 25, 37, 58, 63). However, a significant limitation of these studies was the exclusion of patients with symptoms of HF who have preserved ejection fraction (HFpEF) (58). Of an estimated 5 million people in the United States afflicted with HF, ∼50% are diagnosed as having HFpEF (4, 36, 47). The prevalence of HFpEF is increasing at a rate of ≈1%/yr and will soon be the most prevalent HF phenotype (4). Despite having preserved systolic function at rest, these patients display reduced cardiac functional reserve and morbidity and mortality rates similar to values observed in HF patients with reduced systolic function (4, 47, 55). Recent evidence indicates patients diagnosed with HFpEF are a heterogeneous group displaying multiple mechanisms that contribute to its pathology, extending beyond only diastolic dysfunction (4, 47). Conventional HF therapies have failed to improve the prognosis of this HF subgroup over the past 3 decades, illustrating the need for the development of novel treatment strategies (4, 6, 47, 61).
Although the benefits of exercise in HF are becoming apparent, the mechanisms underlying these responses are poorly understood. A significant gap in the literature exists regarding the effects of chronic exercise training in treating developing HF in large-animal models of cardiovascular disease. Recent data from our laboratory demonstrated chronic low-intensity interval exercise training provides significant benefits to the coronary vasculature and cardiac remodeling in miniature swine hearts with LV hypertrophy generated from pressure overload (20, 22). Specifically, we showed chronic exercise limits LV fibrosis, collagen deposition, and mitochondrial dysfunction characterized by increased sensitivity to Ca2+-induced mitochondrial permeability transition. Furthermore, we demonstrated chronic training prevents enhanced coronary vascular sensitivity to endothelin-1 associated with a decrease in smooth muscle Ca2+-sensitive composite K+ currents. The sum of these observations suggests chronic exercise training could be used to effectively treat two critical factors known to be involved in the pathogenesis of HF: 1) myocardial oxygen supply/demand imbalance; and 2) impaired diastolic function often associated with increased fibrotic remodeling of the myocardium.
In the present study, we tested two hypotheses in treadmill-trained aortic-banded Yucatan miniature swine: 1) chronic low-intensity interval training preserves normal myocardial oxygen supply/demand balance; and 2) training-dependent attenuation of LV fibrotic remodeling improves diastolic function. Using pressure-volume (P-V) loops (a gold standard for examining ventricular function) and two-dimensional (2D) speckle tracking echocardiography (a clinical method of assessing LV function in humans), we provide a detailed characterization in vivo of LV systolic and diastolic function, including its relationship to coronary blood flow and myocardial O2 consumption (MV̇o2), under conditions of increasing peripheral mean arterial pressure (MAP) and β-adrenergic stimulation. We also provide new mechanistic insight into exercise-dependent improvements in fibrotic remodeling and diastolic function in HFpEF by examining the expression of several components of the extracellular matrix (ECM) and their regulatory biomarkers, hypertrophic signaling pathways, and passive elements of the cardiomyocyte contractile apparatus.
METHODS
Aortic banding and exercise training.
Before aortic banding, intact male Yucatan miniature swine (27–30 kg; 8 mo old) were assigned into three groups: nonsham sedentary control (Con; n = 4), banded HF sedentary (HF; n = 5), and banded HF exercise trained (HF-TR; n = 5). LV hypertrophy/HF was induced by aortic banding using methods previously published by our laboratory (20) with modifications. These modifications included moving the aortic banding site to the ascending aorta proximal to the brachiocephalic artery. A systolic transstenotic gradient of ∼70 mmHg [70 ± 1 and 71 ± 2 mmHg for HF and HF-TR, respectively, P = nonsignificant (NS)] was achieved while maintaining a distal peripheral vascular MAP of ∼90 mmHg (93 ± 4 and 90 ± 3 mmHg for HF and HF-TR, respectively, P = NS) under anesthesia using phenylephrine (1–3 μg·kg−1·min−1 iv) at a heart rate (HR) of 100 beats/min (101 ± 4 and 97 ± 4 beats/min for HF and HF-TR, respectively, P = NS). Two months postsurgery, transthoracic echocardiography was performed under inhaled isoflurane anesthesia (0.5%) to measure LV end-diastolic dimension and LV diastolic wall thickness utilizing M-mode recordings using a 1.5–4 MHz transducer on a GE Vividi Ultrasound system, as described previously (20). Aortic banding significantly increased LV diastolic wall thickness in HF animals (P < 0.05, 5.6 ± 0.3 and 7.7 ± 0.5 mm for Con and HF, respectively) but did not alter LV end-diastolic dimension (P = NS; 42.7 ± 0.3 and 42.6 ± 1.1 mm for Con and HF, respectively), indicating concentric LV hypertrophy (an observation commonly associated with LV pressure overload) was present before the onset of exercise training. No differences in echocardiographic measures of morphology existed between HF and HF-TR groups at this time point; therefore, data from both aortic-banded groups were combined before the start of exercise training. Following the development of LV hypertrophy, animals began low-intensity interval treadmill training consisting of treadmill running 3 days/wk, 55 min/day, for 15 wk with gradually increasing intensity as tolerated until finally consisting of the following: 1) 5-min warm-up at 2 mph; 2) six 5-min sessions at 3 mph with five 3-min intervals at 4 mph in between; and 3) 5-min cool-down at 2 mph, as previously published (20, 22). Animals were fed a standard diet averaging 15–20 g/kg once daily, and water was provided ad libitum. Dissection of vital tissues and removal of skeletal muscle for analysis of citrate synthase activity (70) occurred at the time of death. All animal protocols were in accordance with the “Principles for the Utilization and Care of Vertebrate Animals Used in Testing Research and Training” and approved by the University of Missouri Animal Care and Use Committee.
Terminal studies.
Animals were initially anesthetized with a telazol (5 mg/kg)/xylazine (2.25 mg/kg) mix and maintained on 100% oxygen using inhaled isoflurane (≈1.75%). Heparin was given with an initial loading dose of 300 U/kg iv, followed by maintenance of 100 U/kg each hour. A median sternotomy was performed, and the pericardium was opened along the left anterior descending coronary artery (LAD) and near the apex for insertion of catheters and flow probes. Great care was taken to leave the pericardium as intact as possible. A 3PSB flow probe (Transonic Systems, Ithaca, NY) was placed around the LAD to measure coronary blood flow. A custom fluid-filled angiocatheter was inserted into the great cardiac vein for coronary venous blood sampling. P-V loops were measured utilizing a calibrated 7F admittance-based ADVantage catheter (SciSense, London, Ontario, Canada) positioned in the LV via a small apical incision. A 14F balloon occlusion catheter was advanced to the inferior vena cava at the level of the apex of the heart via the deep femoral vein. Peripheral systemic MAP was measured, and aortic blood samples collected via a fluid-filled 6F LCB SH guide catheter (Boston Scientific) introduced through a 7F sheath placed in the right femoral artery and positioned in the aorta distal to the aortic band. Catheter placement was visualized and confirmed using angiography (Infimed software) and Visipaque contrast medium.
In vivo cardiovascular function.
Following placement of the catheters and the flow probe, animals were brought to a peripheral MAP of 80 mmHg using phenylephrine (1–5 μg·kg−1·min−1 iv) and allowed to stabilize until a stable coronary blood flow and HR were observed for 5 min. This state of homeostasis was labeled “baseline”. Swine lack coronary vascular α1-receptors (11, 68); therefore, the use of phenylephrine is a valid method to induce systemic vasoconstriction and increase peripheral MAP without directly impacting coronary function in swine. Coronary blood flow and arterial and venous blood samples were collected simultaneously under conditions of increasing pressure (peripheral MAP of 80, 90, 100, and 120 mmHg). Once the target pressure was attained, coronary blood flow and HR were allowed to stabilize until a new homeostasis was established. Following the pressure experiments, phenylephrine was reduced to the baseline dose, and animals were allowed to return to baseline and stabilize for 10 min. P-V loop measures were collected at baseline and following one dose of dobutamine (a β1-adrenergic receptor agonist; 5 μg·kg−1·min−1 iv) administered for 5 min. Coronary blood flow was averaged the last 30 s of each pressure/HR step. Pressure experiments were utilized to examine myocardial oxygen demand/supply balance and cardiac efficiency. β-Adrenergic stimulation was utilized to examine myocardial contractile reserve.
MV̇o2 was determined as previously described (18). Briefly, blood samples were analyzed immediately upon collection (ABL 700; Radiometer, Copenhagen, Denmark) for pH, Po2 (Torr), hemoglobin count (Hb; g/dl), O2 saturation (So2%), and lactate levels (Lactate Scout; Sports Resource Group, Hawthorne, NY). Myocardial blood O2 content [μmol/ml; calculated using the following equation: (Hb × 0.621 × So2%) + (0.00131 × Po2)], O2 delivery (μmol/ml; coronary blood flow × arterial O2 content), and O2 extraction (%; ratio of MV̇o2 to O2 delivery) were determined using methods published previously (18). MV̇o2 was calculated as the product of coronary arterial/venous O2 difference and coronary blood flow and then normalized to the region of the heart perfused by the LAD. Feigl et al. (24) previously estimated LAD perfusion area to be 30% of total heart weight, and this standard has been utilized recently in swine (3).
P-V loops were recorded under conditions of reducing preload achieved through transient occlusion of the inferior vena cava via inflation of the balloon catheter. Indexes of LV function were generated using a minimum of 10 consecutive cardiac cycles with Lab Scribe software (iWorx, Dover, NH), including HR, LV end-systolic and diastolic volume (LVESV and LVEDV, respectively), LV end-systolic and diastolic pressure, ejection fraction (EF%), stroke volume, stroke work (SW), rate-pressure product, and cardiac output. Other previously published indexes of LV function proposed to be less sensitive to myocardial load and/or morphology were also determined using at least 15 consecutive cardiac cycles of constantly reducing preload, including the end-systolic P-V relationship (ESPVR) and preload recruitable SW (PRSW) (28, 66, 72). A quadratic fit (which was judged to more accurately characterize this relationship) was used to determine ESPVR and PRSW. Cardiac efficiency was calculated as the ratio of SW to MV̇o2 (27, 71). The time constant of isovolumic relaxation (τ) was calculated using the method of Raff and Glantz (64).
2D speckle tracking echocardiography.
Transthoracic echocardiography was performed under inhaled isoflurane anesthesia (0.5%) in the supine/right lateral position 6 mo postbanding using a GE Vivid I Ultrasound system, as previously described (20). Analysis was performed offline using GE EchoPac Software. Six segments of the LV and septum were generated from apical four-chamber and short-axis two-dimensional views (acquired at the mitral valve and apex levels) and averaged to determine global strain, strain rate, and displacement in the longitudinal, transverse, radial, and circumferential dimensions over three cardiac cycles (53). Peak mitral annulus velocity at the septum insertion was measured using tissue Doppler from an apical four-chamber view. Torsion was calculated as the difference between mitral and apical end-systolic rotation (degrees) and normalized to both LV hypertrophy (wall thickness) and end-diastolic chamber length, as previously described (65). Normalization to either of these factors did not influence our results; thus only absolute torsion values are reported.
Isolation of cardiac myocytes.
Myocytes were isolated as previously described (33). Briefly, a section of LV free wall (∼10 cm3) near the LAD was removed, and half was rapidly frozen in liquid nitrogen for biochemical analyses, and the other half was placed in ice-cold relaxing solution for myocyte experiments. The piece in relaxing solution was cut into smaller pieces (2–3 mm) and homogenized with a Waring blender. The resultant slurry was centrifuged for 75 s at 165 g, and the pellet was suspended for 3 min in 0.5% ultrapure Triton X-100 (Pierce Chemical) in relaxing solution. The permeabilized myocytes were washed and centrifuged twice with cold relaxing solution with the final suspension kept on ice during the day of the experiment. Relaxing solution in which the ventricles were disrupted, skinned, and suspended contained the following (in mmol/l): 2 EGTA, 5 MgCl2, 4 ATP, 10 imidazole, and 100 KCl at pH 7.0. The compositions of relaxing solution used in passive tension measurements was as follows (mmol/l): 7 EGTA, 5 MgCl2, 20 imidazole, 4 ATP, 14.5 creatine phosphate, pH 7.0, Ca2+ concentrations of 10−9 M (relaxing solution), and sufficient KCl to adjust ionic strength to 180 mM.
Sarcomere length-passive tension measurements.
The experimental apparatus for physiological measurements of myocyte preparations was similar to previous descriptions (49). Myocyte preparations were attached between a force transducer and torque motor by placing the ends of the myocyte preparation into stainless steel troughs (25 gauge). The ends of the myocyte preparations were secured by overlaying a 0.5-mm length of 3–0 monofilament nylon suture (Ethicon) onto each end of the myocyte and then tying the suture into the troughs with two loops of 10–0 monofilament (Ethicon). The attachment procedure was performed under a stereomicroscope (× ∼100 magnification) using finely shaped forceps.
Before mechanical measurements, the experimental apparatus was mounted on the stage of an inverted microscope (model IX-70, Olympus Instrument), which was placed on a pneumatic vibration isolation table with a cutoff frequency of ∼1 Hz. Mechanical measurements were performed using a capacitance-gauge transducer [model 403, sensitivity of 0.5 mN/V (plus a ×10 amplifier) and resonant frequency of 600 Hz; Aurora Scientific, Aurora, ON, Canada]. Length changes were introduced using a DC torque motor (model 308, Aurora Scientific) driven by voltage commands from a personal computer via a 12-bit digital-to-analog converter (AT-MIO-16E-1, National Instruments, Austin, TX). Passive force and length signals were digitized at 1 kHz and stored on a personal computer using LabView for Windows (National Instruments). Sarcomere length was monitored simultaneous with force and length measurements using IonOptix SarcLen system (IonOptix, Milton, MA), which used a fast Fourier transform algorithm of the video image of the myocyte. Microscopy was done using a ×40 objective (Olympus UWD 40) and a ×2.5 intermediate lens.
Passive tension measurements were performed at 14 ± 1°C. For sarcomere length-passive tension measurements, an experimental protocol was performed similar to that previously described (29). Following attachment of myocyte preparation to the apparatus, the relaxed preparation was adjusted over a range of sarcomere lengths from ∼2.15 μm to ∼2.65 μm at ∼0.1-μm increments by manual manipulation of the length micrometer. After each sarcomere length adjustment, ∼20 s were provided to allow for development of steady-state passive force, and then the preparation was rapidly slackened 15–20% of initial length to yield zero force. Passive tension was calculated as the difference in the force transducer signal before and after the slack and divided by the myocyte's cross-sectional area determined from myocyte width measurements.
SDS-agarose gel electrophoresis and autoradiography.
To determine baseline levels of PKA-mediated titin phosphorylation, 100 μg of skinned cardiac myocytes were incubated with the catalytic subunit of PKA (0.1 U/μl) and 50 μCi [γ-32P]ATP for 45 min. The reaction was stopped by the addition of electrophoresis sample buffer and heating at 95°C for 3 min. The samples were then separated by SDS-agarose gel electrophoresis, silver stained, dried, and subsequently exposed to X-ray film (see Fig. 7, inset).
Fig. 7.
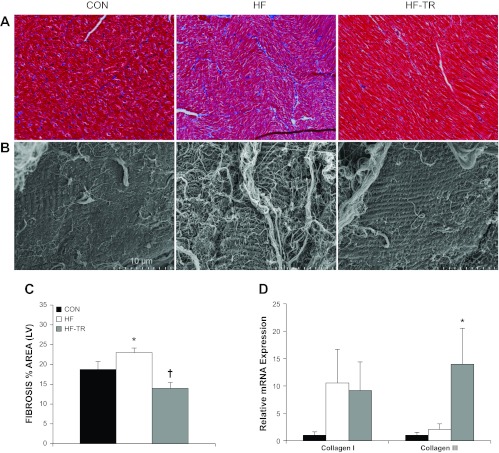
LV fibrosis and relative mRNA expression of collagen I and II isoforms. A: representative histological sections of trichrome-stained LV demonstrating increased fibrosis in HF animals (magnification: ×40). B: representative scanning electron microscopy (SEM) micrographs showing increased quantity and thickness of collagen fibers on the myocardium of the HF group (magnification: ×4,500). C: exercise training prevents increases in LV fibrosis, as indicated by the percent area stained. *P < 0.05, HF vs. Con and HF-TR. †P < 0.05, HF-TR vs. Con. D: collagen III mRNA expression is increased in the LV of HF-TR compared with Con and HF animals. *P < 0.05, HF vs. HF-TR and Con. Values are means ± SE.
Quantitative RT-PCR.
Quantitative real-time-PCR was performed as previously described (21). Pulverized LV in TRIzol solution were quick frozen in liquid nitrogen and stored at −80°C until processed. Total RNA was isolated according to the manufacturer's published protocol for TRIzol. cDNA was transcribed from total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) in a 20-μl reaction. A minus RT reaction was also performed to ensure no genomic DNA contamination. Quantitative RT-PCR was performed on a Bio-Rad MyIQ2 cycler (model no. 170–9790). Each 25-μl reaction contained ×1 SYBER Green Master Mix (Bio-Rad), 0.8 μM forward and reverse primers, and 1 μg of cDNA. The reaction conditions were optimized for each set of primers listed in Table 1. Target gene expression was normalized to 18S ribosomal RNA using the 2−ΔΔ Ct method (45). Linearity and efficiency of each PCR condition were verified by creating a standard curve plotting the critical threshold vs. log of the cDNA dilution.
Table 1.
Primer sequences (5′ to 3′)
| Target Gene | Forward Primer | Reverse Primer |
|---|---|---|
| 18S | CGG CTA CCA CAT CCA AGG AA | AGC TGG AAT TAC CGC GGC |
| BNP | GCA GCA GCC TCT ATC CTC TC | TCC TGT ATC CCT GGC AGT TC |
| Collagen I | GCG TCT CTT CCC TCC CTA GT | GTC TCC CTT GGG TCC CTA TC |
| Collagen III | TCT GAA TTC CCC AGC AAA AG | CCA TTG AGA CAT TTG AAA TTG G |
| MMP-2 | ATG ACG GAG AGG CTG ACA TC | CCA TAC TTC ACA CGC ACC AC |
| MMP-9 | TCG TGG TTC CGA CCT ACT TC | GTT ACC GTC CCG AGT GAA GA |
| TIMP-1 | GTC ATC AGG GCC AAG TTT GT | AAG TAT CCG CAG ACG CTC TC |
| TIMP-4 | GAC GGA AAG GTC TTC ATC CA | ACA TAA TGC TGG GCC TGG TA |
| Fibronectin | TGA AGA ACC CTT GCA GTT CC | GCT TAG GCC TTG GTC AAC AG |
| Elastin | TGC AGT GGT ACC TCA ACT CG | CTT GGC CTT GAC TCC TGT TC |
BNP, brain natriuretic peptide; MMP, matrix metalloproteinase; TIMP, tissue inhibitors of metalloproteinase.
Histology and immunohistochemistry.
Cross sections of LV were formalin fixed, embedded in paraffin, and immunohistochemistry-stained for assessment of fibrosis. Briefly, total fibrosis was visualized from 4-μm-thick sections of LV using Masson's trichrome stain with methods previously established (20). Fibrosis was quantified from four separate fields/animal using Image-Pro Plus analysis software (MediaCybernetics, version 6.2, Bethesda, MD) and expressed as the percent area stained.
Western blotting.
LV tissue was lysed in buffer containing 150 mM NaCl, 10 mM Tris (pH 7.4), 1 mM EDTA, and 1% Triton X-100, as previously described (20). Proteins were resolved by SDS-PAGE using 10–15% acrylamide, transferred onto polyvinylidene difluoride membranes, and blotted using the following commercially available antibodies: all primary (1:500) and secondary (anti-rabbit, 1:1,000) antibodies were from Cell Signaling Technologies. Membranes were incubated with the appropriate alkaline phosphatase or horseradish peroxidase-linked secondary antibody and visualized by enhanced chemifluorescence and chemiluminescence (Amersham).
Scanning and transmission electron microscopy.
LV tissue was collected processed for scanning electron microscopy (SEM) and transmission electron microscopy (TEM), as previously reported (81). Briefly, all LV samples were fixed in a 2% paraformaldehyde, 2% gluteraldehyde in 0.1 M sodium cadodylate solution. SEM samples were rinsed in 0.1 M sodium cadodylate, then incubated at 4°C in a secondary fixative solution of 2% OsO4 in 0.1 M citric acid complex buffer, rinsed three times in 0.1 M Na cacodylate and Milli-Q water, dehydrated in a series of ethanol washes, and dried. After fixation, tissues were attached to Hitachi stubs with carbon adhesive, and silver colloidal paint was applied around tissue edges (to maximize contact with the carbon for optimum imaging) and sputter coated with platinum before imaging on a Hitachi S4700 SEM microscope. TEM samples were rinsed three times in 2-mercaptoethanol and then incubated at 4°C in a secondary fixative 1% osmium tetroxide solution, washed three times in 2-mercaptoethanol and Milli-Q water, dehydrated in a series of acetone solutions and resin infiltration solutions, and polymerized at 60°C. TEM samples were cut with a diamond blade into 85-nm sections and then imaged on a JEOL 1400 Biological TEM microscope.
Three fields from each pig were randomly chosen to review and obtain a minimum of three images from each LV sample at a magnification of ×4,500 (SEM) or ×1,000 (TEM). To prevent sampling bias, the operator was blinded to the experimental condition of each sample, and an independent investigator experienced in electron micrographs analyzed our images postcollection to confirm our findings.
Zymography.
LV was prepared for gelatin zymography, as described previously (76). Active human recombinant matrix metalloproteinase-2 (MMP-2) and MMP-9 (Calbiochem) were used as a positive control. Briefly, 30 mg of LV were homogenized in a glass homogenizer in 500 μl of cold lysis buffer (25 mM Tris·HCl, pH 7.5, 100 mM NaCl, 1% Nonident P-40) with protease inhibitors (10 μg/ml aprotinin, 2 μg/ml leupeptin, 4 mM benzamidine). One hundred micrograms of protein were subjected to electrophoresis on a 10% polyacrylamide gel with gelatin (Bio-Rad) at 125 V for 90 min. Gels were then incubated at room temperature in renaturing solution (2.5% Triton X-100 in dH2O) for 30 min, washed with dH2O, and washed for 30 min at room temperature in developing buffer (50 mM Tris·HCl, pH 7.8, 0.2 M NaCl, 5 mM CaCl2, 0.02% Brij 35). Gels were then incubated for 24 h in developing buffer at 37°C and stained for 1 h with Coomassie blue R-250 (0.5% Coomassie blue R-250, 5% methanol, 10% acetic acid), and then destained (10% methanol, 5% acetic acid). Gels were imaged using a gel-doc system (Bio-Rad) and quantified using ImageJ (National Institutes of Health) software.
Statistical analysis.
All data analysis was performed using SPSS version 19.0 or SigmaStat version 3.5. Group comparisons were made using either one-way or repeated-measures ANOVA or independent samples t-test, as appropriate. A mixed model incorporating linear regression and analysis of covariance was used to compare response variable [coronary blood flow, O2 extraction, coronary venous Po2 (cvPo2) and So2% (cvSo2), change in lactate] slopes plotted vs. MV̇o2, using group as the independent variable. Linear regression was used to examine relationships between ultrasound variables (torsion, early diastolic untwisting rate) and P-V loop measures (τ, LVESV, EF%). Group differences revealed by ANOVA were found using Student Newman-Keuls post hoc analysis. Within-group comparisons were made using paired samples t-test. All data are means ± SE, and significance is reported at P < 0.10 and P < 0.05 levels (10, 83).
RESULTS
LV remodeling.
Postmortem assessment of the heart indicated the presence of cardiac hypertrophy. Body weight was not different between groups (P = NS; 36 ± 2, 39 ± 2, and 38 ± 3 kg for HF, HF-TR, and Con, respectively); therefore, absolute heart morphology measures are reported. Aortic banding significantly increased LV plus septum, atrial, right ventricle, and whole heart weights in HF and HF-TR groups compared with Con (Fig. 1A). Myocardial hypertrophy occurred in all aortic-banded groups, regardless of training status, similar to previous reports from our laboratory (20). LV hypertrophy in the HF group was associated with an increase in the ratio of phosphorylated Akt (p-Akt) to total Akt protein and a less significant decrease in p-JNK/p-SAPK-to-total JNK/SAPK protein ratio (Fig. 1B). The level of Akt phosphorylation was also elevated to a lesser degree in HF-TR animals. Western blots for hypertrophic signaling proteins are show in Fig. 1C.
Fig. 1.
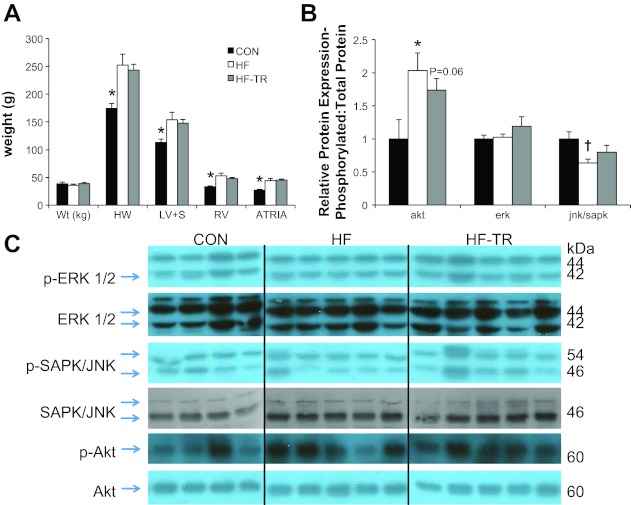
Left ventricular (LV) remodeling and expression of hypertrophic signaling proteins. A: aortic banding generates cardiac hypertrophy, regardless of training status. Global myocardial hypertrophy, as indicated by increased postmortem heart (HW), LV + septum (LV+S), right ventricle (RV), and left + right atrial (Atria) weight, was present in all aortic banded groups. Con, control; HF, heart failure sedentary; HF-TR, heart failure exercise trained. *P < 0.05, Con vs. HF-TR and HF. B: expression of phosphorylated (p)-Akt-to-total Akt protein ratio is increased in the LV of both HF-TR and HF compared with Con animals. *P < 0.05, HF vs. Con. A less significant decrease in p-JNK/p-SAPK-to-total JNK/SAPK ratio was observed only in the HF group. †P = 0.075, HF vs. Con. Values are means ± SE. C: Western blots for all hypertrophic signaling proteins.
LV brain natriuretic peptide mRNA expression.
Aortic banding resulted in a 14-fold increase in LV (P < 0.05; 13.9 ± 6.5, 0.5 ± 0.2, 1.0 ± 0.7 for HF, HF-TR, and Con, respectively) brain natriuretic peptide mRNA expression in HF relative to Con animals. This finding, indicative of the presence of compensatory HF, was attenuated by exercise training and suggests our training protocol was effective in postponing the onset of overt HF.
Citrate synthase activity.
Citrate synthase activity was significantly elevated in the medial head of the triceps in HF-TR animals, indicative of exercise-induced training adaptations to our low-intensity exercise protocol (P < 0.05; 12.6 ± 1.2, 18.0 ± 1.0, and 14.4 ± 1.4 μmol·g wet wt muscle−1·min−1 for HF, HF-TR, and Con, respectively). These data, reproduced separately from those previously published by our laboratory (20), indicate our low-intensity interval training protocol is of sufficient intensity to induce a classic metabolic marker of training.
LV systolic function and contractile reserve.
Representative baseline P-V loops (Fig. 2) and measures (Table 2) are similar to those previously reported in pigs (39). LVESV and SW were increased in HF animals compared with Con. 2D speckle tracking indicated torsion was increased in HF compared with Con animals (Fig. 3A). Cumulative group data, indicated by the regression line, showed torsion was positively correlated with EF% (P < 0.05; Fig. 3B). Despite increased torsion, peak systolic rotation rate at the apex was significantly reduced in HF animals compared with Con (Fig. 3C). Global transverse displacement (Fig. 4A) and strain (Fig. 4B) in the longitudinal view and apical circumferential and radial end-systolic strain rate (Fig. 4C) in the HF group were similar to Con. LV contractility in response to β-adrenergic stimulation is presented in Table 3. Diminished contractile reserve was observed in HF animals, as indicated by the reduced percent increase in ESPVR and PRSW. The percent increase in HR in the HF group was also reduced, suggesting β-adrenergic responsiveness was decreased in these animals.
Fig. 2.
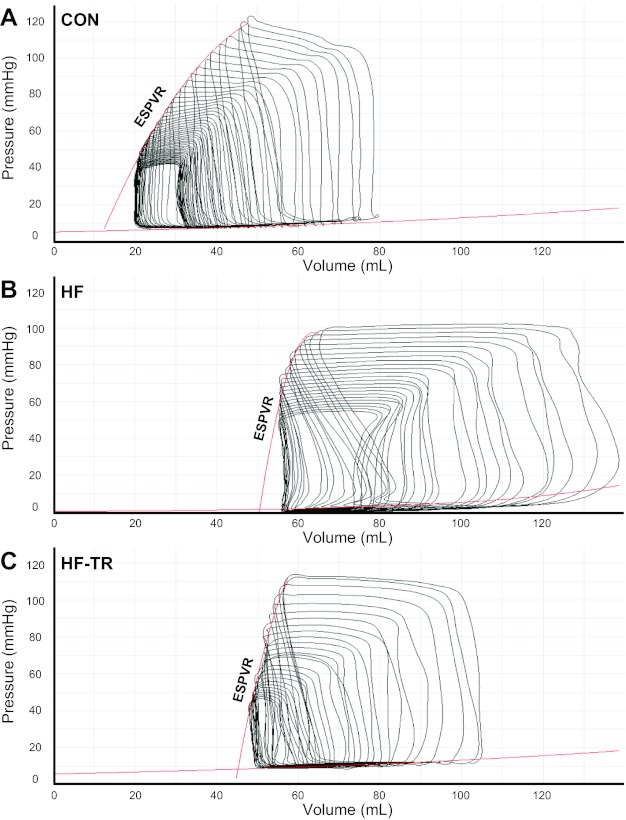
Representative pressure-volume (P-V) loops at baseline (mean arterial pressure = 80 mmHg) from individual Con (A), HF (B), and HF-TR (C) animals. ESPVR, end-systolic P-V relationship.
Table 2.
Pressure-volume analysis of baseline systolic and diastolic function
| Con | HF | HF-TR | |
|---|---|---|---|
| Systolic function | |||
| LVESV, ml | 54 ± 8 | 93 ± 9* | 72 ± 9 |
| LVESP, mmHg | 101 ± 5 | 112 ± 13 | 106 ± 3 |
| SW, J | 0.41 ± 0.10 | 0.69 ± 0.05* | 0.72 ± 0.06* |
| CO, l/min | 3.6 ± 0.8 | 5.9 ± 0.5 | 5.9 ± 1.0 |
| RPP, beats·min−1· mmHg | 8,912 ± 591 | 11,764 ± 1,669 | 9,019 ± 689 |
| SV, ml | 40 ± 9 | 57 ± 3 | 68 ± 10 |
| EF, % | 42 ± 3 | 38 ± 3 | 49 ± 6 |
| Diastolic function | |||
| LVEDV, ml | 94 ± 16 | 150 ± 8* | 140 ± 8* |
| LVEDP, mmHg | 11 ± 1 | 14 ± 2 | 15 ± 2 |
| τ Glantz, ms | 53 ± 3 | 82 ± 6* | 70 ± 6 |
| Cardiac Efficiency | |||
| SW/MV̇o2 | 0.38 ± 0.11 | 0.56 ± 0.12 | 0.97 ± 0.19* |
Values are means ± SE. Con, control; HF, heart failure sedentary; HF-TR, heart failure exercise trained; LVESV, left ventricular end-systolic volume; LVESP, left ventricular end-systolic pressure; SW, stroke work; CO, cardiac output; RPP, rate-pressure product; SV, stroke volume; EF, ejection fraction; LVEDV, left ventricular end-diastolic volume; LVEDP, left ventricular end-diastolic pressure; τ, time constant of left ventricular pressure decay; MV̇o2, myocardial O2 consumption; SW/MV̇o2, ratio of SW to MV̇o2.
Significance is indicated at P < 0.05 vs. Con.
Fig. 3.
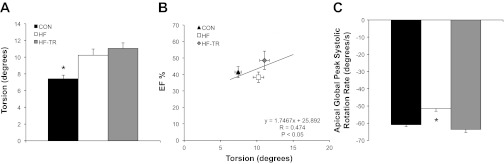
LV torsion and apical systolic rotation rates. A: torsion is increased in aortic-banded animals independent of training status. B: linear regression plot demonstrating a significant correlation between ejection fraction (EF%) and torsion. C: exercise training prevents a reduction in apical global peak systolic rotation rate. Values are means ± SE. *P < 0.05, HF vs. HF-TR and Con.
Fig. 4.
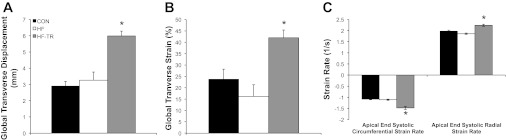
LV systolic transverse strain and apical circumferential and radial strain rates. A and B: exercise training increases global transverse displacement and strain, respectively, in the longitudinal view. C: exercise training increases end-systolic circumferential and radial strain rate at the apex of the heart. Values are means ± SE. *P < 0.05, HF-TR vs. Con and HF.
Table 3.
LV contractility in following β-adrenergic stimulation
| Baseline | 5 μg·kg−1·min−1 Dobutamine | Contractile Reserve, %increase | |
|---|---|---|---|
| HR, beats/min | |||
| Con | 92 ± 4 | 138 ± 6 | 50 |
| HF | 112 ± 5 | 151 ± 7 | 35 |
| HF-TR | 98 ± 8 | 135 ± 4 | 38 |
| ESPVR, mmHg/ml | |||
| Con | 7 ± 3 | 22 ± 3 | 210 |
| HF | 12 ± 3 | 20 ± 4 | 65 |
| HF-TR | 11 ± 4 | 31 ± 6 | 176 |
| PRSW, mmHg | |||
| Con | 48 ± 7 | 99 ± 7 | 107 |
| HF | 71 ± 10 | 129 ± 7* | 82 |
| HF-TR† | 74 ± 7 | 155 ± 13* | 109 |
Values are means ± SE. HR, heart rate; ESPVR, end-systolic pressure-volume relationship; PRSW, preload recruitable stroke work.
Significance is indicated at P < 0.05 vs. Con, same dose value.
P < 0.05 vs. Con (repeated-measures ANOVA; group main effect).
Exercise training prevented the impairments in systolic function observed in HF animals. SW was increased in HF-TR animals, but the parallel increase in LVESV observed in the HF group was attenuated (Table 2). Torsion was also increased in HF-TR animals (Fig. 3A), but, unlike the HF group, was associated with maintenance of apical global peak systolic rotation rate (Fig. 3C). Global transverse displacement (Fig. 4A) and strain (Fig. 4B; longitudinal view) were significantly increased in HF-TR compared with Con and HF animals and likely related to the improved EF%-torsion relationship. Increased torsion in the HF-TR group was also associated with increased apical circumferential and radial end-systolic strain rate (Fig. 4C). Despite similar reductions in β-adrenergic responsiveness (reflected by percent increase in HR similar to the HF group), chronic exercise training effectively preserved contractile reserve, as indicated by the percent increase in ESPVR and PRSW in relation to Con (Table 3). A significant increase in PRSW was observed in HF-TR animals, further suggesting LV contractility was enhanced following training.
Diastolic function.
Diastolic function was impaired in HF animals. P-V data showed increases in baseline LVEDV and τ in HF compared with Con animals (Table 2). 2D speckle tracking indicated LV untwisting rate during early diastole was reduced in HF animals (Fig. 5A). Cumulative data for all groups (indicated by the regression line) showed untwisting rate was negatively correlated to both τ and LVESV (Fig. 5, B and C), reproducing findings from previous work (78). Group mean data from HF animals indicated a rightward shift of this relationship compared with Con, demonstrating decreases in LV untwisting was associated with increases in both τ and LVESV. An increase in tissue Doppler A′ (Fig. 6B) and increased global longitudinal, mitral valve circumferential, and mitral valve radial strain rates during late diastole (Fig. 6C) were observed in the HF group, suggesting an enhanced role of atrial systole compared with Con animals.
Fig. 5.
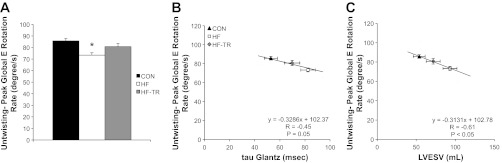
LV untwisting during early diastole. A: exercise training prevents a reduction in peak global LV rotation rate during early diastole. *P < 0.05, HF vs. HF-TR and Con. B and C: linear regression plots demonstrating a significant correlation between untwisting and both the time constant of isovolumic relaxation (τ) and LV end-systolic volume (LVESV). Exercise training attenuates the right downward shift in the relationship between untwisting during early diastole and both τ and LVESV observed in sedentary HF animals. Values are means ± SE.
Fig. 6.
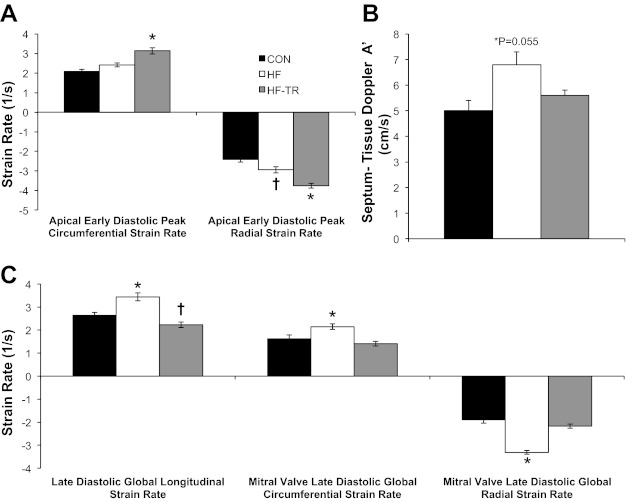
LV early-diastolic apical circumferential and radial strain rates, tissue Doppler, and late-diastolic longitudinal and mitral circumferential and radial strain rates. A: exercise training increases global circumferential and radial strain rate during early diastole at the apex of the heart. *P < 0.05, HF-TR vs. Con and HF. †P < 0.05, HF vs. Con. B: movement of the mitral annulus at the insertion of the septum during atrial systole increased in sedentary HF animals (P = 0.055). C: exercise training prevented increases in global late-diastolic longitudinal and circumferential and radial strain rates at the mitral valve of the heart. *P < 0.05, HF vs. Con and HF-TR. †P < 0.05, HF-TR vs. Con. Values are means ± SE.
Exercise training prevented the diastolic dysfunction observed in the HF group. Baseline LVEDV was increased, however, the significant increase in τ observed in HF animals was prevented (Table 2). Early diastolic untwisting rate was the same as Con animals (Fig. 5A), and rightward shifts in τ and LVESV in relation to early diastolic untwisting rate was attenuated by exercise training (Fig. 5, B and C). Apical circumferential and radial strain rate during early diastole were also increased in HF-TR animals compared with both Con and HF (Fig. 6A). Tissue Doppler A′ (Fig. 6B) and late diastolic strain rates (Fig. 6C) were not different than Con values, indicating the enhanced atrial systole observed in HF animals was not present in the HF-TR group.
LV fibrosis.
Representative histological and SEM sections of the LV from HF, Con, and HF-TR animals are shown in Fig. 7, A and B. Trichrome staining (expressed as the percent area of LV stained) indicated general fibrosis was significantly elevated in HF animals compared with Con (Fig. 7C). The approximate 20% increase in fibrosis is similar to previous reports from our laboratory (20) and demonstrates the reproducibility of this effect in our model. Increased fibrosis in the HF group was associated with increases in MMP-2 and tissue inhibitors of metalloproteinase-4 (TIMP-4) mRNA expression, along with less significant increases in MMP-9 and TIMP-1 mRNA levels relative to Con (Fig. 8A). MMP-2 and MMP-9 activity/abundance levels, determined using gel zymography, and a representative zymography gel are presented in Fig. 8, B and C, respectively. TEM micrographs illustrated distinct collagen bundles surrounding the capillary lumen in HF animals (Fig. 9B), in addition to disruption of the structural organization of mitochondria between sarcomeres and mitochondrial clustering (Fig. 10B) compared with Con (Fig. 10A).
Fig. 8.
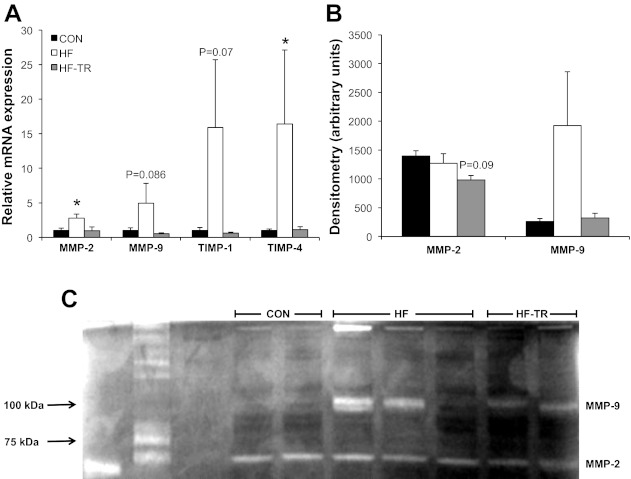
Relative mRNA expression and activity of LV extracellular matrix regulating biomarkers. A: exercise training prevents increases in the relative mRNA expression of multiple metalloproteinase (MMP) and tissue inhibitors of metalloproteinase (TIMP) isoforms. *P < 0.05, HF vs. HF-TR and Con. B: quantification of MMP-2 and MMP-9 activity/abundance as determined by gel zymography. Values are means ± SE. C: representative zymography gel of MMP-2 and MMP-9 activity from all groups.
Fig. 9.
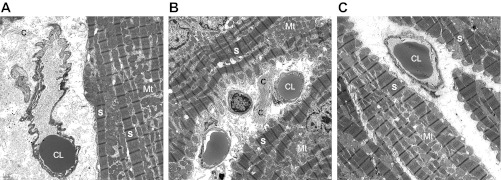
Representative transmission electron microscopy (TEM) micrographs showing pericapillary fibrosis in HF animals. Distinct collagen bundles indicating interstitial pericapillary fibrosis surrounding the capillary lumen are more prominent in HF (B) compared with Con (A) or HF-TR (C) animals and demonstrate coherence in relation to our histology results (magnification: ×4,500). C, collagen bundles; CL, capillary lumen; S, sarcomere; Mt, mitochondria.
Fig. 10.
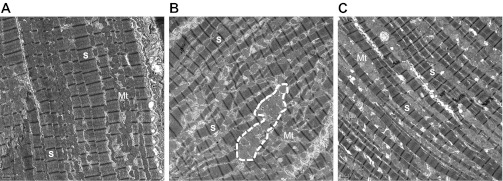
Representative TEM micrographs demonstrating structural disorganization or the myocardium in HF animals. Increased mitochondrial clustering (outlined by white dashed line) and disrupted spatial orientation of mitochondria in relation to the sarcomere is evident in HF (B) compared with Con (A) animals (magnification: ×1,000). Exercise training limits this structural remodeling to a lesser degree than that observed in the HF group (C).
Low-intensity interval exercise training prevented increased myocardial fibrosis in response to pressure overload (Fig. 7C). A reduction in fibrosis compared with Con animals was observed in the HF-TR group and associated with a significant increase in collagen III mRNA expression (Fig. 7D). In contrast to the HF group, MMP-2, MPP-9, TIMP-1, and TIMP-4 mRNA levels were unchanged relative to Con (Fig. 8A). A small decrease in MMP-2 activity/abundance (Fig. 8B) and less significant increase in fibronectin mRNA expression relative to Con (P = 0.068; data not shown) was also observed in HF-TR animals. Analysis of relative elastin mRNA expression showed no significant differences between groups (data not shown). Chronic exercise training limited pericapillary fibrosis, mitochondrial clustering, and sarcomere/mitochondrial disorganization compared with HF animals (Figs. 9C and 10C).
Sarcomere length dependence of passive force and PKA-mediated titin phosphorylation.
Sarcomere length-passive force relationships for permeabilized cardiac myocyte preparations are shown in Fig. 11. Passive force was greater for a given sarcomere length in cardiac myocytes after aortic banding. Increased passive force was associated with greater PKA-mediated back-phosphorylation of titin, indicative of lower baseline levels of titin phosphorylation (autoradiogram inset). Passive force and titin back-phosphorylation were elevated, regardless of training status; consequently, HF and HF-TR data were pooled for this measure.
Fig. 11.
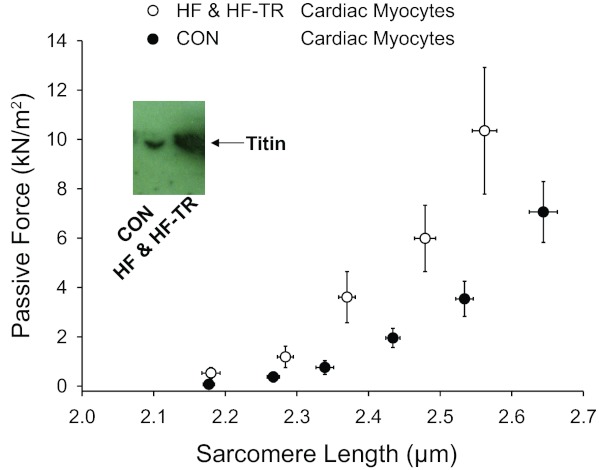
Sarcomere length-passive force relationships for permeabilized cardiac myocyte preparations. Passive force was greater in cardiac myocytes after aortic-banding (P < 0.05 vs. Con) and associated with lower baseline levels of PKA-mediated titin phosphorylation, i.e., greater PKA-mediated back-phosphorylation in lane 1 samples from Con vs. lane 2 HF pigs in autoradiogram inset. Exercise did not affect this finding; therefore, HF-TR and HF data were pooled for this measure. Values are means ± SE.
Myocardial oxygen supply/demand balance in response to increasing peripheral pressure load.
LV hemodynamic and blood gas variables at baseline and during incremental increases in peripheral MAP are summarized in Table 4. Exercise training prevented the increase in HR observed in the HF group. HR was significantly higher in HF compared with both Con and HF-TR animals. Our MV̇o2 (50–175 μmol/min before normalization to heart weight) and CBF (14–45 ml/min before normalization to heart weight) values under sedation are consistent with previously published studies in conscious resting pigs (MV̇o2: 125–225 μmol/min; CBF: 50–75 ml/min) (11–13, 50), verifying the validity of our hemodynamic and blood-gas data. Exercise training improved the slope of the coronary blood flow to MV̇o2 relationship, while coronary blood flow increased at a slower rate in response to increasing oxygen consumption (Fig. 12A) in HF animals. In the HF group, decreased CBF for a given MV̇o2 was associated with increased oxygen extraction (Fig. 12B) and decreased cvSo2 (Fig. 12C). The slope of the cvPo2 (Fig. 12D) to MV̇o2 relationship was also reduced in HF compared with HF-TR animals. In total, these data indicate exercise training was able to inhibit the development of myocardial oxygen supply/demand imbalance following pressure overload, despite the presence of significant LV hypertrophy.
Table 4.
Hemodynamic and blood-gas variables at baseline and during increasing pressure loads
| 80 mmHg | 90 mmHg | 100 mmHg | 120 mmHg | |
|---|---|---|---|---|
| MAP, mmHg | ||||
| Con | 78 ± 3 | 89 ± 2 | 98 ± 1 | 117 ± 1 |
| HF | 83 ± 1 | 91 ± 1 | 100 ± 1 | 117 ± 4 |
| HF-TR | 81 ± 2 | 92 ± 2 | 99 ± 2 | 116 ± 3 |
| HR, beats/min | ||||
| Con | 88 ± 3 | 80 ± 6 | 71 ± 4 | 71 ± 8 |
| HF† | 104 ± 4* | 98 ± 6 | 99 ± 11 | 109 ± 16* |
| HF-TR | 85 ± 6 | 86 ± 6 | 81 ± 6 | 80 ± 7 |
| CBF, ml·min−1·g−1 | ||||
| Con | 0.27 ± 0.02 | 0.31 ± 0.03 | 0.38 ± 0.04 | 0.58 ± 0.11 |
| HF | 0.30 ± 0.07 | 0.30 ± 0.07 | 0.36 ± 0.06 | 0.47 ± 0.11 |
| HF-TR | 0.20 ± 0.04 | 0.26 ± 0.06 | 0.28 ± 0.06 | 0.42 ± 0.10 |
| MV̇o2, μmol O2·min−1·g−1 | ||||
| Con | 1.10 ± 0.06 | 1.36 ± 0.10 | 1.74 ± 0.12 | 1.88 ± 0.21 |
| HF | 1.40 ± 0.22 | 1.35 ± 0.29 | 1.67 ± 0.21 | 2.05 ± 0.52 |
| HF-TR | 0.84 ± 0.12 | 0.95 ± 0.14 | 1.17 ± 0.11 | 1.48 ± 0.27 |
| Arterial Po2, Torr | ||||
| Con | 483 ± 25 | 536 ± 16 | 547 ± 7 | 561 ± 16 |
| HF | 498 ± 30 | 497 ± 37 | 502 ± 39 | 516 ± 20 |
| HF-TR | 527 ± 28 | 540 ± 25 | 521 ± 17 | 555 ± 10 |
| Coronary venous Po2, Torr | ||||
| Con | 25 ± 2 | 23 ± 1 | 27 ± 2 | 31 ± 4 |
| HF | 22 ± 1 | 23 ± 1 | 30 ± 3 | 31 ± 3 |
| HF-TR | 22 ± 1 | 23 ± 2 | 25 ± 2 | 32 ± 3 |
| Coronary venous So2, % | ||||
| Con | 30 ± 2 | 32 ± 1 | 39 ± 4 | 57 ± 7 |
| HF | 27 ± 4 | 29 ± 3 | 38 ± 6 | 50 ± 7 |
| HF-TR | 30 ± 3 | 33 ± 7 | 36 ± 7 | 49 ± 7 |
| Hb, g/100 ml | ||||
| Con | 8.2 ± 0.2 | 9.0 ± 0.2 | 10.1 ± 0.4 | 11.3 ± 0.8 |
| HF | 9.1 ± 0.6 | 9.0 ± 0.5 | 10.8 ± 1.0 | 11.7 ± 0.9 |
| HF-TR | 8.5 ± 0.5 | 8.7 ± 0.5 | 9.7 ± 0.7 | 10.6 ± 1.0 |
Values are means ± SE. MAP, mean arterial pressure; CBF, coronary blood flow; Po2, O2 pressure; So2, O2 saturation; Hb, hemoglobin.
Significance is indicated at P < 0.05 vs. Con, same pressure load.
P < 0.05 vs. Con (repeated-measures ANOVA; group main effect).
Fig. 12.
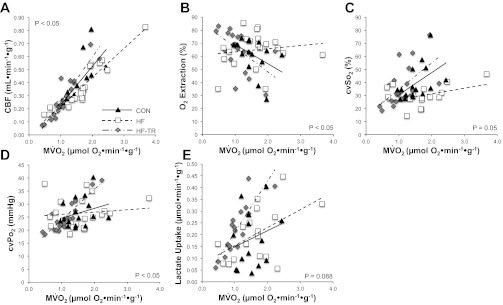
Myocardial oxygen supply/demand balance in response to increasing peripheral mean arterial pressure loads. A: the slope of the relationship between myocardial O2 consumption (MV̇o2) and coronary blood flow (CBF) was reduced in HF compared with HF-TR animals (P < 0.05). B: exercise training prevented the increase in oxygen extraction observed in HF animals, as indicated by the significantly different slope of the O2 extraction- MV̇o2 relationship (P < 0.05). C: increased oxygen extraction in the HF group was complemented by a concurrent decrease in coronary venous O2 saturation (cvSo2), apparent by the reduced slope in the cvSo2- MV̇o2 relationship (P = 0.05). D: the slope for the relationship between coronary venous O2 pressure (cvPo2) and MV̇o2 was increased following exercise training in HF-TR animals (P < 0.05). E: lactate consumption, evident by the positive slope of the relationship between coronary arterial vs. venous lactate difference (lactate uptake) and MV̇o2, was increased in HF-TR animals (P = 0.088).
Cardiac efficiency, expressed as the ratio of SW to MV̇o2 (SW/MV̇o2), was significantly increased under baseline conditions in HF-TR animals (Table 2) and associated with increased myocardial lactate uptake (indicated by the positive slope, P = 0.088) compared with Con (Fig. 12E). Animals in the HF group exhibited lactate uptake similar to Con animals, suggesting, despite impaired myocardial oxygen supply/demand balance, the myocardium was not ischemic, and a metabolic shift to anaerobic glycolysis had not occurred under our experimental conditions.
DISCUSSION
The results of this study illustrate several novel findings. In the presence of existing hypertrophy and developing HF, chronic low-intensity interval exercise training 1) prevents diastolic dysfunction; 2) maintains normal myocardial oxygen supply/demand balance; 3) improves cardiac efficiency; 4) encourages a physiological molecular hypertrophic signaling profile; and 5) promotes expression of more compliant ECM components while preserving normal ECM regulatory mechanisms in a large-animal model resembling HFpEF.
A clinically translational model of HFpEF.
This investigation was primarily designed as a functional study with the purpose of characterizing LV function following chronic exercise training in our swine model of pressure overload and establishing it as a legitimate model of HFpEF using sophisticated in vivo techniques, including simultaneous coronary blood flow, MV̇o2, and P-V measurements in addition to 2D speckle tracking echocardiography. Our previous work suggested diastolic dysfunction and myocardial oxygen supply/demand imbalance were present in our model as a result of pathological interactions between the coronary vasculature, LV remodeling, and myocardial metabolism. Establishment of the existence and nature of these cardiovascular functional deficits was fundamental and substantiates our exploration of molecular mechanisms that we believe underlie exercise-dependent improvements in cardiovascular function in our HFpEF model. Results from the present study indicate our model exhibits many characteristics of HFpEF, including LV hypertrophy, early diastolic dysfunction, increased LV brain natriuretic peptide mRNA levels, elevated HR, increased fibrosis, and diminished LV contractile reserve. Combined with our laboratory's previous work (20, 22), we believe our model accurately reflects the pathophysiology of HFpEF and demonstrates the relevance of our large-animal model for the study of this patient population.
Exercise improves both systolic and diastolic cardiac function.
Patients with HFpEF demonstrate both systolic and diastolic dysfunction, with diastolic impairment long thought to be a key prognostic indicator for diagnosis. Our results show chronic exercise alters mechanical properties of the LV that may benefit systolic emptying and diastolic filling. 2D speckle tracking echocardiography has become increasingly utilized clinically as a diagnostic tool for assessing myocardial function in these patients. A key finding of the present study was increased torsion following chronic exercise, which was associated with a commensurate increase in EF% that occurred in a linear fashion predicted by our regression analysis. The significant relationship between torsion and EF demonstrated in our model has been shown previously in humans (79). Central to this observation in the HF-TR group was prevention of increased LVESV, maintenance of apical systolic rotation rate, increased circumferential/radial strain rates, and enhanced longitudinal transverse strain/displacement, findings not associated with increased torsion in HF animals. Previous studies have demonstrated maintenance or enhancement of ventricular torsion (or systolic twist) and circumferential, radial, and longitudinal systolic strain in HFpEF patients at rest (60, 62, 79), although conflicting data reporting reductions in radial and longitudinal strain exists (54, 75, 79). Data from the HF group emulate these findings, demonstrating compensatory increases in torsion and preserved apical strain rates were associated with maintenance (but not improvement) of EF at rest. Although preservation of resting systolic function is a common feature of HFpEF, decreased cardiac functional reserve is well established in this patient population (55, 75). Similarly, animals in the HF group demonstrated reductions in contractile reserve in response to β-adrenergic stimulation, implying an inability to respond to increasing workloads, despite preserved resting systolic function. Exercise-trained animals exhibited elevated baseline contractility similar to that observed in the HF group. However, in contrast to the HF group, contractile reserve closely matched that seen in the Con group. These findings suggest that diminished contractile reserve in HF animals was not simply the result of less reserve available because of an elevation in baseline contractility, but more so an impairment in β-adrenergic sensitivity. Previous work demonstrated exercise training improves inotropic responsiveness to β-adrenergic activation in a rodent model of hypertension (46) and, although speculative, may be a mechanism underlying exercise-dependent preservation of contractile reserve in response to β-adrenergic stimulation in our study. Increased torsion and strain rate following chronic exercise training were also associated with maintenance of contractile reserve, further demonstrating the utility of chronic exercise in preserving systolic function and functional reserve in a setting of HFpEF.
Chronic exercise training also prevented the development of diastolic impairment. A second key finding of our study was the preservation of early diastolic untwisting rate, enhanced early diastolic strain rates, and prevention of increased τ following 15 wk of chronic training. Subsequently, exercise training was effective in preventing the rightward shift in the relationship between LV untwisting rate and both LVESV and τ observed in Fig. 5, B and C. LV untwisting rate is used as a diagnostic tool for assessment of diastolic function and temporally linked to ventricular relaxation and diastolic filling at rest and in response to exercise (14, 56, 57). The untwisting rate has been correlated with LVESV and indexes of LV relaxation generated from P-V loops, such as τ, in both health and HF (14, 57, 78) and our data confirm these relationships. Previous studies show ventricular untwisting rates during early diastole in HFpEF patients exhibit a great deal of variance, including maintenance during the early stages of disease that gradually declines as HF worsens (60, 62, 74, 75, 78). Consistent with these findings, reductions in early diastolic untwisting rate, increases in LVEDV and τ, and the leftward shift in untwisting rate in relation to both LVESV and τ indicate significant diastolic impairment in the HF group.
A common compensatory mechanism of preserving diastolic filling in HF patients is to augment atrial systole. Increased mitral annulus movement and strain rates during late diastole indicate this is indeed the case in our HF animals. Despite the presence of atrial hypertrophy in both aortic-banded groups, the HF-TR group did not demonstrate indicators of enhanced atrial systole, as tissue Doppler and strain values during late diastole were similar to Con values. In total, our results provide new insight into utilizing 2D speckle tracking to differentiate between pathological and physiological functional characteristics of the heart in HFpEF and elucidate positive adaptations to chronic training in HF that could be utilized to assess the effectiveness of exercise prescription.
Molecular mechanisms of exercise-dependent improvement in diastolic function.
Diastolic dysfunction resulting from increased myocardial stiffness has long been considered a hallmark feature of HFpEF (4). Central to our finding of preserved diastolic function following chronic exercise is a reduction of fibrosis in the HF-TR group. Our laboratory has previously demonstrated this effect of training concurrent with a reduction in collagen deposition (20). Results from the present study suggest chronic exercise attenuates fibrotic LV remodeling via two potential mechanisms: 1) maintenance of normal MMP/TIMP expression; and 2) altering collagen isoform composition. In HF-TR animals, reduced fibrosis was associated with normal mRNA levels of MMP-2, MMP-9, TIMP-1, and TIMP-4. Relative to Con, expression of these regulatory biomarkers of the ECM was substantially increased in the HF group. MMP-to-TIMP ratios are decreased in response to pressure overload (31, 67, 69), and increased plasma expression of these MMP and TIMP isoforms has been proposed as clinical predictors of LV hypertrophy and diastolic HF in humans (86). Recent work by Yarbrough et al. (84) in swine subject to acute and gradual pressure overload over 1 mo demonstrated 1.5- to 2-fold increases in TIMP-1 and TIMP-4 mRNA expression relative to Con and proposed modulation of these biomarkers could be therapeutically relevant. Our data showed an ∼15-fold increase in TIMP isoforms in HF animals, suggesting under our chronically loaded conditions that TIMP expression may continue to increase with the severity of HF. The MMP/TIMP system is extremely complex, and regulation of fibrotic remodeling in HF via manipulation of these molecular components has proven difficult in humans, as evident from the equivocal results reported from studies such as the PREMIER clinical trial (35). Our results demonstrate chronic exercise may be a particularly powerful treatment option in this regard, enabling an integrated response in a diverse system that has proven difficult to manipulate pharmacologically.
Improved diastolic function in HF-TR animals may be linked to improved LV elasticity associated with increased collagen III expression. Collagen type III is more compliant than the type I isoform (80), and the ratio of type III to type I is considered important to determining the elasticity/stiffness of tissue with an increased III-to-I ratio representing increased compliance (5). Burgess et al. previously examined collagen type III-to-I ratio in response to hypertension or exercise in rats (5). Their findings demonstrated a reduction in the type III-to-I ratio that was associated with diminished diastolic function following surgically induced hypertension. Conversely, exercise-trained animals maintained ratios similar to Con in the presence of enhanced diastolic relaxation, demonstrating support for the association between collagen III-to-I ratio and relaxation properties of the heart. Our results showed an increase in collagen III mRNA with no significant change in type I relative to Con in HF-TR animals, suggesting alteration of collagen isoform ratios is a mechanism by which chronic training preserves normal diastolic function and LV elasticity in the presence of existing hypertrophy and developing HF. Similar to previous findings in pressure-overloaded pigs (84), collagen I and III mRNA expression was not altered in the HF group relative to Con.
In addition to ECM remodeling, enhanced intrinsic cardiomyocyte stiffness is a potential factor contributing to diastolic impairment in HFpEF (4). The enhanced passive stiffness in cardiac myofibrils from aortic-banded animals in our study is similar to that observed in human HFpEF patients by Borbely et al. (2). Increased cardiomyocyte stiffness has been linked to titin, a large elastic cytoskeletal protein that has been described as a cardiac “spring”. Previous work has demonstrated an increase in the N2BA (more compliant isoform)-to-N2B (stiffer isoform) ratio in human hearts from patients with HFpEF (2); however, shifts in titin isoform expression were not observed in our study (data not shown). Changes in the phosphorylation status of titin are known to alter passive stiffness in human and pig cardiac myofibrils (2, 32, 40). Our data showed a decrease in baseline PKA-mediated phosphorylation of titin (indicated by an increase in back-phosphorylation), which has been linked to increased passive force in human HF (2) and confirmed in myocytes from our pig model. Importantly, we provide evidence that diminished titin phosphorylation may precede shifting of titin isoforms during the development of HF. Exercise training did not reduce passive stiffness, implying modulation of the ECM plays a more prominent role in exercise-dependent preservation of diastolic function.
Exercise generates a unique hypertrophic signaling phenotype.
Considerable efforts have been made to identify the molecular signaling components that differentiate between pathological and physiological hypertrophy (15). Another central finding of our study is that increases in activated Akt protein expression was not associated with a commensurate decrease in activated JNK/SAPK expression following chronic low-intensity interval training. In his 2007 review, Dorn discusses the “fuzzy logic” of cardiac hypertrophy in the context that hypertrophy typically exhibits both physiological and pathological characteristics (15). Activation of Akt has long been thought to play a role in physiological hypertrophy, but studies in transgenic mice have demonstrated chronic or excessive stimulation of the Akt pathway can also lead to pathological adaptations in the myocardium (48, 59). Exercise studies examining activation of Akt under conditions of pressure overload were mostly done in rodent models of hypertension and show equivocal findings (26, 34, 38, 44, 52). The use of multiple rodent models and varying modes, intensities, durations, frequencies, and overall length of exercise training between these publications make comparison difficult. Results from our large-animal model of HFpEF clearly demonstrate the disparate action of Akt activation in the myocardium, and we speculate the physiological adaptive action of Akt observed following chronic exercise in our study might result from exercise-dependent activation of growth factors such as insulin-like growth factor I (15, 59).
Exercise training also attenuated the decrease in activated JNK/SAPK observed in HF animals. In 2004, Wilkins et al. (82) demonstrated coordinated activation of calcineurin and nuclear factor of activated T cell (NFAT) transcription factors was increased in mice with pathological hypertrophy induced by pressure overload, but not in mice with physiological hypertrophy induced by two different types of exercise modes (wheel running and swim training). Although this is not a consensus finding (23), this study demonstrated calcineurin-NFAT coupling likely plays a role in pathological, but not physiological, growth of the myocardium. Supporting this assertion are more recent studies demonstrating exercise training reduces calcineurin expression in hypertensive rats (26, 38, 44). The role of JNK activation in HF is controversial, as conflicting results have been reported in human failing myocardium (7, 8, 30, 43, 73). However, Liang et al. (42) demonstrated JNK activation reduced cardiac hypertrophy via inhibition of the calcineurin-NFAT signaling pathway, suggesting, under normal conditions, JNK signaling limits cardiac hypertrophy in response to pathological stimuli. Our data imply exercise-dependent preservation of activated JNK/SAPK is a potential mechanism of limiting calcium-mediated pathological remodeling of the myocardium. In total, our results demonstrate chronic exercise results in a unique molecular signaling profile that is associated with physiological hypertrophy, as our laboratory has previously postulated (20).
Exercise preserves myocardial oxygen supply/demand balance and improves LV efficiency.
A critical factor involved in the pathogenesis of HF is development of myocardial oxygen demand/supply imbalance. Another key finding in our study was preservation of normal oxygen supply/demand balance, despite the presence of antecedent pathological LV hypertrophy following chronic low-intensity interval training. Several studies have examined the impact of acute exercise on coronary vascular function and myocardial oxygen supply/demand balance (3, 16, 17, 85), and imbalances in the relationship between coronary blood flow and myocardial metabolism have been observed in porcine and canine models of metabolic syndrome (3, 85) and myocardial infarction (12, 13, 50, 51). To the best of our knowledge, this is the first study to examine and characterize LV function in a swine model of HF and chronic exercise. Key to the interpretation of our results is the impact of exercise on coronary oxygen balance and HR. An enhanced rate of increase in coronary blood flow in response to increasing cardiac metabolic demands, coupled with preservation of coronary oxygen extraction, cvPo2/cvSo2, and conservation of HR similar to that observed in Con animals, demonstrates maintenance of normal resistance vessel function and autonomic regulation are key mechanisms underlying training-induced preservation of myocardial oxygen supply/demand balance in HF. The coronary vasodilatory response to enhanced MV̇o2 stimulated by incrementally increasing peripheral MAP loads was diminished in HF animals and associated with increased coronary oxygen extraction and decreased cvSo2. Changes in oxygen extraction and cvSo2 are a reflection of resistance vessel tone, supporting our assertion that the impaired coronary vascular function our laboratory observed previously (22) is associated with myocardial oxygen/supply demand imbalance in our model approximating HFpEF. HR was also significantly elevated across pressure loads in HF animals compared with Con, potentially limiting coronary blood flow primarily through increases in coronary back pressure brought about by increased extravascular compressive forces (19) or reduced diastolic filling time. However, HR did not change in response to increasing MAP load in HF animals, demonstrating the impaired rate of coronary blood flow increase in response to increasing MV̇o2 was not merely a consequence of elevated HR. The heart rate response in the HF group also suggests the presence of baroreceptor dysfunction as the normal response, a decline in heart rate in reaction to increasing pressure observed in both CON and HF-TR groups, was absent.
Our finding that preservation of oxygen supply/demand balance following chronic exercise was associated with an increase in overall LV efficiency is important. Efficiency in the HF group was similar to that observed in Con; however, it was sustained at a cost of increased myocardial oxygen extraction resulting from the coronary blood flow/metabolic uncoupling described previously. This represents a scenario that the heart is unlikely to maintain long term. Although our data implied the presence of resting myocardial ischemia in HF animals, lactate consumption was similar to Con levels, suggesting increased oxygen extraction was sufficient to prevent the onset of myocardial anaerobic glycolysis. However, the change in resistance vessel tone indicated by enhanced O2 extraction and decreased cvSo2 would suggest that these animals are at a perilous tipping point that, while not ischemic under our experimental conditions of increasing peripheral MAP, may certainly be underperfused and susceptible to ischemia under other conditions of increasing myocardial metabolic demand, such as exercise (which would also increase HR and LV contractility).
Effect of anesthesia and Hb.
A potential confounding variable of this study is the presence of incomplete coronary autoregulation, likely due to the vasodilatory influence of isoflurane. Elevated resting cvPo2 and cvSo2 values compared with those previously observed in conscious resting Yucatan mini-swine (1) suggest vasomotor tone was reduced slightly at baseline. Anesthetic interruption of coronary autoregulation conceivably resulted in modest overperfusion of the myocardium, particularly during pressure loading, leading to a gradual increase in cvPo2 and cvSo2 over the course of our experimental protocol, ultimately resulting in blood-gas/ MV̇o2 relationships deviating from those normally observed in conscious instrumented pigs (11–13, 50). We also explored the influence that elevated and gradually increasing Hb levels over the course of our protocol (likely the result of α-1 mediated splenic contraction from the phenylephrine doses used to increase pressure loads) may have had on our findings. However, previous work by Van Woerkens et al. (77) demonstrated increases in coronary venous oxygen concentrations in response to hemodilution, suggesting the increased Hb levels probably did not contribute significantly to our results. Given that our CBF and MV̇o2 values were consistent with those of previously published studies in conscious resting pigs and all animals were subject to the same conditions (likely affecting the three groups similarly), we consider our results accurate and valid as considered under our experimental conditions.
Limitations.
We demonstrated our large-animal model accurately reflects the pathophysiology of HFpEF. However, there are several limitations to our study. First, hypertension and aortic stenosis are key contributors to the pathogenesis of HFpEF. Although our aortic-banding procedure creates a pressure-overload scenario designed to mimic these disease states, it does not accurately reflect the effects of hypertension systemically nor the comprehensive reduction in arterial distensibility observed along a large portion of the aorta in aortic stenosis. Second, HFpEF is found more often in an aged population, and our study was performed in young, sexually mature swine and limited to only males. Finally, our study design did not include a Con exercise-trained group, limiting our understanding of the pressure overload independent aspect of our training protocol. However, we consider this restriction minimal, given that our training protocol is approximately one-half the intensity of previously used swine training protocols (41) and may not be of sufficient intensity to elicit training adaptations in a healthy animal.
In conclusion, chronic low-intensity interval exercise training attenuates diastolic impairment by promoting compliant ECM fibrotic components and preserving ECM regulatory mechanisms, preserves myocardial oxygen balance, and promotes a physiological molecular hypertrophic signaling phenotype. Our results demonstrate an integrated response in multiple physiological systems following exercise during developing HF and provide novel mechanistic insight into the benefits of training in a clinically translational large-animal model of HFpEF.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute/American Recovery and Reinvestment Act P30 HL101332 (written and awarded to M. Harold Laughlin and managed by C.A. Emter).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.D.M., B.N.M., M.K., L.M.H., K.S.M., and C.A.E. performed experiments; K.D.M., B.N.M., M.K., L.M.H., K.S.M., K.C.D., and C.A.E. analyzed data; K.D.M., B.N.M., M.K., L.M.H., K.S.M., K.C.D., and C.A.E. interpreted results of experiments; K.D.M., B.N.M., M.K., L.M.H., K.S.M., and C.A.E. prepared figures; K.D.M., B.N.M., L.M.H., and C.A.E. drafted manuscript; K.D.M., B.N.M., M.K., L.M.H., K.S.M., K.C.D., and C.A.E. approved final version of manuscript; M.K., L.M.H., K.S.M., K.C.D., and C.A.E. edited and revised manuscript; L.M.H., K.S.M., and C.A.E. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Cory Weimer, Melissa Cobb, Christine Schramm, and Jan Ivey for considerable technical contributions, which were essential to the successful completion of the study, and Dr. Heide Schatten for lending us expertise in electron microscopy and critical independent evaluation of our SEM and TEM images.
REFERENCES
- 1. Bender SB, van Houwelingen MJ, Merkus D, Duncker DJ, Laughlin MH. Quantitative analysis of exercise-induced enhancement of early- and late-systolic retrograde coronary blood flow. J Appl Physiol 108: 507–514, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ. Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 104: 780–786, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Borbouse L, Dick GM, Payne GA, Payne BD, Svendsen MC, Neeb ZP, Alloosh M, Bratz IN, Sturek M, Tune JD. Contribution of BK(Ca) channels to local metabolic coronary vasodilation: effects of metabolic syndrome. Am J Physiol Heart Circ Physiol 298: H966–H973, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 32: 670–679, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burgess ML, Buggy J, Price RL, Abel FL, Terracio L, Samarel AM, Borg TK. Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. Am J Physiol Heart Circ Physiol 270: H151–H159, 1996 [DOI] [PubMed] [Google Scholar]
- 6. Burkhoff D. Mortality in heart failure with preserved ejection fraction: an unacceptably high rate. Eur Heart J 33: 1718–1720, 2012 [DOI] [PubMed] [Google Scholar]
- 7. Communal C, Colucci WS, Remondino A, Sawyer DB, Port JD, Wichman SE, Bristow MR, Singh K. Reciprocal modulation of mitogen-activated protein kinases and mitogen-activated protein kinase phosphatase 1 and 2 in failing human myocardium. J Card Fail 8: 86–92, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol 31: 1429–1434, 1999 [DOI] [PubMed] [Google Scholar]
- 9. Crimi E, Ignarro LJ, Cacciatore F, Napoli C. Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol 6: 292–300, 2009 [DOI] [PubMed] [Google Scholar]
- 10. Curran-Everett D, Benos DJ. Guidelines for reporting statistics in journals published by the American Physiological Society. Am J Physiol Regul Integr Comp Physiol 287: R247–R249, 2004 [DOI] [PubMed] [Google Scholar]
- 11. de Beer VJ, Bender SB, Taverne YJ, Gao F, Duncker DJ, Laughlin MH, Merkus D. Exercise limits the production of endothelin in the coronary vasculature. Am J Physiol Heart Circ Physiol 300: H1950–H1959, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Beer VJ, Sorop O, Pijnappels DA, Dekkers DH, Boomsma F, Lamers JM, Duncker DJ, Merkus D. Integrative control of coronary resistance vessel tone by endothelin and angiotensin II is altered in swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol 294: H2069–H2077, 2008 [DOI] [PubMed] [Google Scholar]
- 13. de Beer VJ, Taverne YJ, Kuster DW, Najafi A, Duncker DJ, Merkus D. Prostanoids suppress the coronary vasoconstrictor influence of endothelin after myocardial infarction. Am J Physiol Heart Circ Physiol 301: H1080–H1089, 2011 [DOI] [PubMed] [Google Scholar]
- 14. Dong SJ, Hees PS, Siu CO, Weiss JL, Shapiro EP. MRI assessment of LV relaxation by untwisting rate: a new isovolumic phase measure of tau. Am J Physiol Heart Circ Physiol 281: H2002–H2009, 2001 [DOI] [PubMed] [Google Scholar]
- 15. Dorn GW., 2nd The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49: 962–970, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Duncker DJ, de Beer VJ, Merkus D. Alterations in vasomotor control of coronary resistance vessels in remodelled myocardium of swine with a recent myocardial infarction. Med Biol Eng Comput 46: 485–497, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duncker DJ, Merkus D. Acute adaptations of the coronary circulation to exercise. Cell Biochem Biophys 43: 17–35, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Duncker DJ, Stubenitsky R, Verdouw PD. Role of adenosine in the regulation of coronary blood flow in swine at rest and during treadmill exercise. Am J Physiol Heart Circ Physiol 275: H1663–H1672, 1998 [DOI] [PubMed] [Google Scholar]
- 19. Duncker DJ, Van Zon NS, Crampton M, Herrlinger S, Homans DC, Bache RJ. Coronary pressure-flow relationship and exercise: contributions of heart rate, contractility, and alpha 1-adrenergic tone. Am J Physiol Heart Circ Physiol 266: H795–H810, 1994 [DOI] [PubMed] [Google Scholar]
- 20. Emter CA, Baines CP. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am J Physiol Heart Circ Physiol 299: H1348–H1356, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emter CA, Bowles DK. Store-operated Ca(2+) entry is not essential for PDGF-BB induced phenotype modulation in rat aortic smooth muscle. Cell Calcium 48: 10–18, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emter CA, Tharp DL, Ivey JR, Ganjam VK, Bowles DK. Low-intensity interval exercise training attenuates coronary vascular dysfunction and preserves Ca2+-sensitive K current in miniature swine with LV hypertrophy. Am J Physiol Heart Circ Physiol 301: H1687–H1694, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eto Y, Yonekura K, Sonoda M, Arai N, Sata M, Sugiura S, Takenaka K, Gualberto A, Hixon ML, Wagner MW, Aoyagi T. Calcineurin is activated in rat hearts with physiological left ventricular hypertrophy induced by voluntary exercise training. Circulation 101: 2134–2137, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Feigl EO, Neat GW, Huang AH. Interrelations between coronary artery pressure, myocardial metabolism and coronary blood flow. J Mol Cell Cardiol 22: 375–390, 1990 [DOI] [PubMed] [Google Scholar]
- 25. Flynn KE, Pina IL, Whellan DJ, Lin L, Blumenthal JA, Ellis SJ, Fine LJ, Howlett JG, Keteyian SJ, Kitzman DW, Kraus WE, Miller NH, Schulman KA, Spertus JA, O'Connor CM, Weinfurt KP. Effects of exercise training on health status in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 301: 1451–1459, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garciarena CD, Pinilla OA, Nolly MB, Laguens RP, Escudero EM, Cingolani HE, Ennis IL. Endurance training in the spontaneously hypertensive rat: conversion of pathological into physiological cardiac hypertrophy. Hypertension 53: 708–714, 2009 [DOI] [PubMed] [Google Scholar]
- 27. Gibbs CL. Cardiac energetics. Physiol Rev 58: 174–254, 1978 [DOI] [PubMed] [Google Scholar]
- 28. Glower DD, Spratt JA, Snow ND, Kabas JS, Davis JW, Olsen CO, Tyson GS, Sabiston DC, Jr, Rankin JS. Linearity of the Frank-Starling relationship in the intact heart: the concept of preload recruitable stroke work. Circulation 71: 994–1009, 1985 [DOI] [PubMed] [Google Scholar]
- 29. Hanft LM, McDonald KS. Length dependence of force generation exhibit similarities between rat cardiac myocytes and skeletal muscle fibres. J Physiol 588: 2891–2903, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103: 670–677, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Heymans S, Schroen B, Vermeersch P, Milting H, Gao F, Kassner A, Gillijns H, Herijgers P, Flameng W, Carmeliet P, Van de Werf F, Pinto YM, Janssens S. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 112: 1136–1144, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 105: 631– 638,, 17 p following 638, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinken AC, Korte FS, McDonald KS. Porcine cardiac myocyte power output is increased after chronic exercise training. J Appl Physiol 101: 40–46, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Huang CY, Yang AL, Lin YM, Wu FN, Lin JA, Chan YS, Tsai FJ, Tsai CH, Kuo CH, Lee SD. Anti-apoptotic and pro-survival effects of exercise training on hypertensive hearts. J Appl Physiol 112: 883–891, 2012 [DOI] [PubMed] [Google Scholar]
- 35. Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, Lyon R, Quinones M, Theroux P, Sydlowski D, Kim HE, Garcia MJ, Jaber WA, Weaver WD. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol 48: 15–20, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 112: e154–e235, 2005 [DOI] [PubMed] [Google Scholar]
- 37. Keteyian SJ, Pina IL, Hibner BA, Fleg JL. Clinical role of exercise training in the management of patients with chronic heart failure. J Cardiopulm Rehabil Prev 30: 67–76, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Kolwicz SC, MacDonnell SM, Renna BF, Reger PO, Seqqat R, Rafiq K, Kendrick ZV, Houser SR, Sabri A, Libonati JR. Left ventricular remodeling with exercise in hypertension. Am J Physiol Heart Circ Physiol 297: H1361–H1368, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol 278: H1345–H1351, 2000 [DOI] [PubMed] [Google Scholar]
- 40. Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 104: 87–94, 2009 [DOI] [PubMed] [Google Scholar]
- 41. Laughlin MH, Overholser KA, Bhatte MJ. Exercise training increases coronary transport reserve in miniature swine. J Appl Physiol 67: 1140–1149, 1989 [DOI] [PubMed] [Google Scholar]
- 42. Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J 22: 5079–5089, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol 35: 1385–1394, 2003 [DOI] [PubMed] [Google Scholar]
- 44. Libonati JR, Sabri A, Xiao C, Macdonnell SM, Renna BF. Exercise training improves systolic function in hypertensive myocardium. J Appl Physiol 111: 1637–1643, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-delta delta C(T)] method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 46. MacDonnell SM, Kubo H, Crabbe DL, Renna BF, Reger PO, Mohara J, Smithwick LA, Koch WJ, Houser SR, Libonati JR. Improved myocardial beta-adrenergic responsiveness and signaling with exercise training in hypertension. Circulation 111: 3420–3428, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Maeder MT, Kaye DM. Heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol 53: 905–918, 2009 [DOI] [PubMed] [Google Scholar]
- 48. Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem 277: 22896–22901, 2002 [DOI] [PubMed] [Google Scholar]
- 49. McDonald KS. Ca2+ dependence of loaded shortening in rat skinned cardiac myocytes and skeletal muscle fibers. J Physiol 525: 169–181, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Merkus D, Houweling B, van den Meiracker AH, Boomsma F, Duncker DJ. Contribution of endothelin to coronary vasomotor tone is abolished after myocardial infarction. Am J Physiol Heart Circ Physiol 288: H871–H880, 2005 [DOI] [PubMed] [Google Scholar]
- 51. Merkus D, Houweling B, van Vliet M, Duncker DJ. Contribution of KATP+ channels to coronary vasomotor tone regulation is enhanced in exercising swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol 288: H1306–H1313, 2005 [DOI] [PubMed] [Google Scholar]
- 52. Miyachi M, Yazawa H, Furukawa M, Tsuboi K, Ohtake M, Nishizawa T, Hashimoto K, Yokoi T, Kojima T, Murate T, Yokota M, Murohara T, Koike Y, Nagata K. Exercise training alters left ventricular geometry and attenuates heart failure in Dahl salt-sensitive hypertensive rats. Hypertension 53: 701–707, 2009 [DOI] [PubMed] [Google Scholar]
- 53. Mondillo S, Galderisi M, Mele D, Cameli M, Lomoriello VS, Zaca V, Ballo P, D'Andrea A, Muraru D, Losi M, Agricola E, D'Errico A, Buralli S, Sciomer S, Nistri S, Badano L. Speckle-tracking echocardiography: a new technique for assessing myocardial function. J Ultrasound Med 30: 71–83, 2011 [DOI] [PubMed] [Google Scholar]
- 54. Morris DA, Perez AV, Blaschke F, Eichstadt H, Ozcelik C, Haverkamp W. Myocardial systolic and diastolic consequences of left ventricular mechanical dyssynchrony in heart failure with normal left ventricular ejection fraction. Eur Heart J Cardiovasc Imaging 13: 556–567, 2012 [DOI] [PubMed] [Google Scholar]
- 55. Norman HS, Oujiri J, Larue SJ, Chapman CB, Margulies KB, Sweitzer NK. Decreased cardiac functional reserve in heart failure with preserved systolic function. J Card Fail 17: 301–308, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Notomi Y, Martin-Miklovic MG, Oryszak SJ, Shiota T, Deserranno D, Popovic ZB, Garcia MJ, Greenberg NL, Thomas JD. Enhanced ventricular untwisting during exercise: a mechanistic manifestation of elastic recoil described by Doppler tissue imaging. Circulation 113: 2524–2533, 2006 [DOI] [PubMed] [Google Scholar]
- 57. Notomi Y, Popovic ZB, Yamada H, Wallick DW, Martin MG, Oryszak SJ, Shiota T, Greenberg NL, Thomas JD. Ventricular untwisting: a temporal link between left ventricular relaxation and suction. Am J Physiol Heart Circ Physiol 294: H505–H513, 2008 [DOI] [PubMed] [Google Scholar]
- 58. O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 301: 1439–1450, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. O'Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest 115: 2059–2064, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park SJ, Miyazaki C, Bruce CJ, Ommen S, Miller FA, Oh JK. Left ventricular torsion by two-dimensional speckle tracking echocardiography in patients with diastolic dysfunction and normal ejection fraction. J Am Soc Echocardiogr 21: 1129–1137, 2008 [DOI] [PubMed] [Google Scholar]
- 61. Paulus WJ, van Ballegoij JJ. Treatment of heart failure with normal ejection fraction: an inconvenient truth! J Am Coll Cardiol 55: 526–537, 2010 [DOI] [PubMed] [Google Scholar]
- 62. Phan TT, Shivu GN, Abozguia K, Gnanadevan M, Ahmed I, Frenneaux M. Left ventricular torsion and strain patterns in heart failure with normal ejection fraction are similar to age-related changes. Eur J Echocardiogr 10: 793–800, 2009 [DOI] [PubMed] [Google Scholar]
- 63. Piepoli MF, Conraads V, Corra U, Dickstein K, Francis DP, Jaarsma T, McMurray J, Pieske B, Piotrowicz E, Schmid JP, Anker SD, Solal AC, Filippatos GS, Hoes AW, Gielen S, Giannuzzi P, Ponikowski PP. Exercise training in heart failure: from theory to practice. A consensus document of the Heart Failure Association and the European Association for Cardiovascular Prevention and Rehabilitation. Eur J Heart Fail 13: 347–357, 2011 [DOI] [PubMed] [Google Scholar]
- 64. Raff GL, Glantz SA. Volume loading slows left ventricular isovolumic relaxation rate. Evidence of load-dependent relaxation in the intact dog heart. Circ Res 48: 813–824, 1981 [DOI] [PubMed] [Google Scholar]
- 65. Russel IK, Gotte MJ, Bronzwaer JG, Knaapen P, Paulus WJ, van Rossum AC. Left ventricular torsion: an expanding role in the analysis of myocardial dysfunction. JACC Cardiovasc Imaging 2: 648–655, 2009 [DOI] [PubMed] [Google Scholar]
- 66. Sagawa K, Suga H, Shoukas AA, Bakalar KM. End-systolic pressure/volume ratio: a new index of ventricular contractility. Am J Cardiol 40: 748–753, 1977 [DOI] [PubMed] [Google Scholar]
- 67. Schubert A, Walther T, Falk V, Binner C, Loscher N, Kanev A, Bleiziffer S, Rauch T, Autschbach R, Mohr FW. Extracellular matrix gene expression correlates to left ventricular mass index after surgical induction of left ventricular hypertrophy. Basic Res Cardiol 96: 381–387, 2001 [DOI] [PubMed] [Google Scholar]
- 68. Schulz R, Oudiz RJ, Guth BD, Heusch G. Minimal alpha 1- and alpha 2-adrenoceptor-mediated coronary vasoconstriction in the anaesthetized swine. Naunyn Schmiedebergs Arch Pharmacol 342: 422–428, 1990 [DOI] [PubMed] [Google Scholar]
- 69. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285–1342, 2007 [DOI] [PubMed] [Google Scholar]
- 70. Srere PA. Citrate synthase. Methods Enzymol 13: 3–5, 1969 [Google Scholar]
- 71. Suga H. Ventricular energetics. Physiol Rev 70: 247–277, 1990 [DOI] [PubMed] [Google Scholar]
- 72. Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res 32: 314–322, 1973 [DOI] [PubMed] [Google Scholar]
- 73. Takeishi Y, Huang Q, Abe J, Che W, Lee JD, Kawakatsu H, Hoit BD, Berk BC, Walsh RA. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc Res 53: 131–137, 2002 [DOI] [PubMed] [Google Scholar]
- 74. Takeuchi M, Borden WB, Nakai H, Nishikage T, Kokumai M, Nagakura T, Otani S, Lang RM. Reduced and delayed untwisting of the left ventricle in patients with hypertension and left ventricular hypertrophy: a study using two-dimensional speckle tracking imaging. Eur Heart J 28: 2756–2762, 2007 [DOI] [PubMed] [Google Scholar]
- 75. Tan YT, Wenzelburger F, Lee E, Heatlie G, Leyva F, Patel K, Frenneaux M, Sanderson JE. The pathophysiology of heart failure with normal ejection fraction: exercise echocardiography reveals complex abnormalities of both systolic and diastolic ventricular function involving torsion, untwist, and longitudinal motion. J Am Coll Cardiol 54: 36–46, 2009 [DOI] [PubMed] [Google Scholar]
- 76. Toth M, Fridman R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol Med 57: 163–174, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Van Woerkens EC, Trouwborst A, Duncker DJ, Koning MM, Boomsma F, Verdouw PD. Catecholamines and regional hemodynamics during isovolemic hemodilution in anesthetized pigs. J Appl Physiol 72: 760–769, 1992 [DOI] [PubMed] [Google Scholar]
- 78. Wang J, Khoury DS, Yue Y, Torre-Amione G, Nagueh SF. Left ventricular untwisting rate by speckle tracking echocardiography. Circulation 116: 2580–2586, 2007 [DOI] [PubMed] [Google Scholar]
- 79. Wang J, Khoury DS, Yue Y, Torre-Amione G, Nagueh SF. Preserved left ventricular twist and circumferential deformation, but depressed longitudinal and radial deformation in patients with diastolic heart failure. Eur Heart J 29: 1283–1289, 2008 [DOI] [PubMed] [Google Scholar]
- 80. Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res 27: 341–348, 1993 [DOI] [PubMed] [Google Scholar]
- 81. Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, Stump C, Ferrario C, Sowers JR. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab 293: E355–E363, 2007 [DOI] [PubMed] [Google Scholar]
- 82. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 94: 110–118, 2004 [DOI] [PubMed] [Google Scholar]
- 83. Williams JL, Hathaway CA, Kloster KL, Layne BH. Low power, type II errors, and other statistical problems in recent cardiovascular research. Am J Physiol Heart Circ Physiol 273: H487–H493, 1997 [DOI] [PubMed] [Google Scholar]
- 84. Yarbrough WM, Mukherjee R, Stroud RE, Rivers WT, Oelsen JM, Dixon JA, Eckhouse SR, Ikonomidis JS, Zile MR, Spinale FG. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. J Thorac Cardiovasc Surg 143: 215–223, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang C, Knudson JD, Setty S, Araiza A, Dincer UD, Kuo L, Tune JD. Coronary arteriolar vasoconstriction to angiotensin II is augmented in prediabetic metabolic syndrome via activation of AT1 receptors. Am J Physiol Heart Circ Physiol 288: H2154–H2162, 2005 [DOI] [PubMed] [Google Scholar]
- 86. Zile MR, Desantis SM, Baicu CF, Stroud RE, Thompson SB, McClure CD, Mehurg SM, Spinale FG. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail 4: 246–256, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]