

Abstract
Purpose.
To determine the function of tumor necrosis factor receptor-2 (TNFR2) in retinal development and ischemia-induced revascularization in an oxygen-induced retinopathy (OIR) model.
Methods.
Mice with a global deletion of TNFR2 (TNFR2-KO) or with a vascular endothelial cell (EC)–specific TNFR2 transgene (TNFR2-TG) were compared to wild-type C57BL/6 mice (WT). Retinal vasculature development was visualized by whole-mount and cross-sectional isolectin staining. In the OIR model, neonatal mice were subjected to 75% oxygen from postnatal day (P)7 to P12 and then returned to normoxia from P12 to P17. Immunostaining and biochemical analyses were performed to assess the effects of TNFR2 deletion and TNFR2 transgenesis on retinal vascular repair.
Results.
TNFR2 deletion slightly delayed, while TNFR2 transgenesis weakly promoted, intraretinal vascular development and intraretinal vessel growth. TNFR2 deletion enhanced, while TNFR2 transgene reduced, hyperoxia-induced vaso-obliteration. However, hypoxia-induced revascularization and development of deep intraretinal vessels at P17 were reduced in TNFR2-KO but increased in TNFR2-TG mice without significant increase in preretinal neovascularization (NV). Moreover, TNFR2-TG/KO mice in which only vascular EC express TNFR2 sufficiently rescued the vascular defects of TNFR2-KO in the OIR model. Biochemical analyses of retina tissues showed that the phenotypic changes in retina correlated with TNFR2-dependent activation of Nuclear factor–κB (NF-κB) survival and bone marrow kinase (Bmx)-VEGFR2 angiogenic pathways.
Conclusions.
TNFR2 plays a marginal role during retinal vascular development. TNFR2 in vascular EC strongly prevents hyperoxia-induced vaso-obliteration by inhibiting cell apoptosis, and promotes retinal repair by enhancing hypoxia-induced revascularization without increasing pathological neovascular tufts. Therefore, activation of TNFR2 signaling may be an ideal strategy for the treatment of OIR.
TNFR2 plays a marginal role during retinal vascular development; however, TNFR2 strongly prevents hyperoxia-induced vaso-obliteration by preventing apoptosis, and promotes retinal repair by enhancing hypoxia-induced revascularization without increasing pathological neovascular tufts.
Introduction
Angiogenesis, the formation of new blood vessels from existing ones, is essential for embryogenesis and postnatal organ repair under physiological conditions.1,2 It is also tightly associated with pathological conditions such as ischemic and diabetic retinopathy.3 Retinopathy of prematurity continues to be a significant source of morbidity in premature infants.4,5 The mouse model of oxygen-induced retinopathy (OIR) is characterized by an initial phase of retinal vasculature regression under hyperoxia associated with cell death.6,7 When the supplemental oxygen is withdrawn, the second phase occurs and the retina is in a state of relative hypoxia, leading to retinal angiogenesis. This phase is characterized by ischemia-induced revascularization in the central area along with pathological neovascularization (NV) at the peripheral area. While revascularization is beneficial to retinal repair, the peripheral NV forms tufts and is often associated with pathogenesis of retinopathy induced by ischemia and diabetes.6,7 Therefore, promoting revascularization and limiting NV would be an ideal therapeutic strategy for the treatment of ischemia- and diabetes-induced retinopathy.
Tumor necrosis factor-α (TNF) has been associated with ischemic and diabetic retinopathy in both human and animal models.8–11 It elicits a broad spectrum of biological effects including proliferation, differentiation, and apoptosis.12,13 These differences in the TNF-induced response are due, in part, to the presence of two distinct TNF-specific plasma membrane–localized receptors, type I 55 kDa TNFR (TNFR1) and type II 75 kDa TNFR (TNFR2).14,15 TNFR1 is expressed ubiquitously. In contrast, basal TNFR2 expression is detected only in certain cell types, including vascular endothelial cells (EC), cardiac myocytes, and some neuronal cells.16–18 It can also be induced in EC in pathological settings such as ischemia.19,20 In vascular EC, TNFR1 primarily mediates inflammation and apoptosis whereas TNFR2 promotes cell activation, migration, growth, or proliferation.19–23 TNFR1 via formation of TNFR1 signaling complex activates MAP kinase, NF-κB, and apoptotic signaling cascades.24,25 Less is known regarding TNFR2 downstream signaling. TNFR2 through TRAF2 can activate Nuclear factor–κB (NF-κB).26 We have previously identified nonreceptor tyrosine kinase, bone marrow kinase (Bmx) that plays an important role in cell survival and migration, as a TNFR2-specific tyrosine kinase that mediates TNFR2-induced EC migration and angiogenesis.19–21 Mechanistically, we have shown that TNFR2 and Bmx form a complex to transactivate VEGFR2, a potent angiogenic receptor.19–21,27,28
The physiological and pathological functions of TNFRs have been investigated using genetically modified mice. Mice with deficiency of TNFR1 or TNFR2 or both are viable, and studies from these mice suggest that the two receptors play common as well as distinct roles in pathogenesis of diseases.16 TNF signaling, particularly TNFR1, has been shown to be required for the formation of secondary lymphoid organs, as well as host defense and inflammatory responses.29,30 TNF and TNFR1 have also been implicated in ischemic retinopathies.8–10 However, the role of TNFR2 in physiological and pathological retina angiogenesis has not been clearly defined. Here we used mice with TNFR2 deletion or EC-specific transgenesis of TNFR2 to determine the function of TNFR2 in retinal vascular development and OIR. Our data demonstrated that signaling through TNFR2 was not essential for retinal vascular development. However, TNFR2 expression protected retina from hyperoxia-induced damage and enhanced hypoxia-induced angiogenesis in an OIR model.
Materials and Methods
Animal Protocol
All experimental procedures adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Committee for the Use of Animals in Research. Wild-type (C57BL/6J) and TNFR2-KO mice were purchased from Jackson Laboratory (Bar Harbor, ME). Both TNFR2-KO and TNFR2-TG mice were back-crossed to C57BL/6J mice. TNFR2-TG mice were generated as described previously.19,20
Mouse Model of Oxygen-Induced Retinopathy
Oxygen-induced retinopathy was induced in mice using the protocol of Smith and colleagues6,7 as we recently described.31 On postnatal day 7 (P7), pups with their dams were placed in a 75% oxygen (hyperoxia, H) chamber for 5 days and returned to room air (relative hypoxia) from P12 to P17. Age-matched mice raised under normoxia (N) served as controls.
Fluorescein Staining of Whole-Mount Retinas
Eyes from neonatal mice were collected on P3, P5, and P7 and fixed in 4% paraformaldehyde for 4 hours. For dissection, the cornea, lens, sclera, and hyloid vessels were removed. Retinas were permeabilized in 0.5% Triton/PBS with 5% normal horse serum overnight at 4°C, followed by incubation with fluorescein-labeled isolectin B4 (Invitrogen, Carlsbad, CA) diluted 1:50 in 0.1% Triton/PBS overnight at 4°C. Retinas were then washed five times with PBS and mounted by making four incisions in fluorescent mounting medium. Pictures were taken with the same exposure and gain using an fluorescence microscope (Zeiss Axiovert 200M; Carl Zeiss Microscope, LLC, Thornwood, NY). Vessel-obliterated areas on P12 and preretinal NV areas on P17 were outlined using ImageJ software (National Institutes of Health [NIH], Bethesda, MD) and quantified as the percentage of total area of retina analyzed.31
Assessment of Developmental Retinal Angiogenesis
To compare superficial retinal vasculature development, TNFR2-KO, TNFR2-TG, and WT pups were sacrificed, and retinal whole mounts were stained with isolectin B4 (Invitrogen) to visualize EC as described above. The area of the retina covered by vascular bed was outlined using NIH ImageJ software and determined as the percentage of the total retinal area for comparison. In addition, cross sections from eyes harvested on P7, P9, and P11 were stained with isolectin to visualize the vessel formation in the deep retina.
Quantification of Neovascular Nuclei
Following euthanasia, eyes were removed and fixed in 4% paraformaldehyde overnight. Eyes were embedded in paraffin, sectioned into 6 μm thick sagittal sections across the entire retina, and stained with hematoxylin and eosin (H&E). Fifteen sections on each side of the optic nerve, at least 40 μm apart, were used to assess NV in a double-blinded manner via counting of EC nuclei anterior to the internal limiting membrane (on the vitreal side) as previously described.6,7,31 Eyes from six mice of each genotype group were analyzed, and the number of endothelial nuclei per section was averaged for each eye.
TUNEL Staining of Frozen Sections
Eyes were enucleated and embedded in optimal cutting temperature (OCT) compound at −80°C. Sections (8 μm) were cut and placed on glass slides, then fixed in 4% paraformaldehyde at room temperature for 15 minutes, followed by three 5-minute washes with PBS. Sections were permeabilized (0.1% Triton X-100, 0.1% sodium citrate) for 2 minutes on ice and then incubated with TUNEL reaction mix (Roche Diagnostics, Basel, Switzerland) for 1 hour at 37°C in a humidified chamber. After three washes in PBS, sections were mounted in medium with 4′,6-diamidino-2-phenylindole (DAPI). Sections were viewed by fluorescence microscopy, and images were captured in digital format (both using Zeiss Axiovert 200M; Carl Zeiss Microscope, LLC).
Immunofluorescence Analysis
Paraffin-embedded samples were used for immunostaining analysis. Eye sections were deparaffinized, rehydrated through a graded alcohol series, and heated in 10 mM sodium citrate for antigen retrieval. Paraffin sections were preincubated with 5% normal horse serum/PBS for 1 hour and then incubated with anti–proliferating cell nuclear antigen (anti-PCNA) rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti p-P65 rabbit polyclonal antibody (Cell Signaling, Beverly, MA), or isolectin B4 (Invitrogen) overnight at 4°C. Bound antibodies were detected with fluorescence-conjugated second antibody (Vector Labs, Burlingame, CA) and mounted in medium with DAPI.
Gene Expression in the Retina
Total RNA of retinas from P17 pups was isolated using the RNeasy kit with DNase I digestion (Qiagen, Valencia, CA). Reverse transcription (RT) was done by standard procedure (Super Script first-strand synthesis system; Qiagen) using 1 μg total RNA. Quantitative real-time polymerase chain reaction (PCR) was performed using iQ SYBR Green Supermix on iCycler real-time detection system (Bio-Rad Laboratories, Inc., Hercules, CA). RT-PCR with specific primers was performed as previously described.19,20,28
Protein Extraction and Western Blot Analysis
Freshly dissected unfixed retinal tissue was homogenized in lysis buffer. The lysates were centrifuged at 13,000g for 10 minutes at 4°C. Supernatants were collected and determined with a Bradford Protein Assay kit (Bio-Rad Laboratories, Inc.). The cell lysates were subjected to SDS–polyacrylamide gel electrophoresis and then immunoblotting (Immobilon P; Millipore, Milford, MA) with specific antibodies, followed by detection using an enhanced chemiluminescence kit (Amersham Life Science, Arlington Heights, IL). The primary antibodies used were anti-Bmx goat polyclonal (Santa Cruz Biotechnology), anti-HIF-1α rabbit polyclonal (Santa Cruz Biotechnology), and total and phospho-VEGFR2 rabbit antibodies (Cell Signaling), TNFR2 rabbit polyclonal antibody (Abcam, Cambridge, MA), and anti β-actin mouse monoclonal antibody (Sigma, St. Louis, MO). Immunoblotting for Bmx-VEGFR2 signaling was performed as previously described.19,20,28
Statistical Analysis
All results are expressed as the mean ± SEM. After a normal distribution test, the data were compared by student t-test. Differences were considered statistically significant at P < 0.05.
Results
Effects of Deletion or Transgenesis of TNFR2 on Retinal Vascular Development
Wild-type C57BL/6 (WT), TNFR2-KO, and TNFR2-TG mice were verified by genotyping with the specific primers (see Supplementary Material and Supplementary Fig. S1A, http://www.iovs.org/content/54/1/211/suppl/DC1). To determine the effects of TNFR2 on mouse retinal vascular development, the superficial retinal vascularized areas in the ganglion cell layer (GCL) of WT, TNFR2-KO, and TNFR2-TG mice at various postnatal days were measured by isolectin staining and quantified as a percentage of the total retinal surface. As illustrated in a schematic diagram (see Supplementary Material and Supplementary Fig. S1B, http://www.iovs.org/content/54/1/211/suppl/DC1), the vasculature in WT mouse grew from the center toward the edge of the retina during development. The vasculature front reached one-third of the retina on P3, leaving the outer retina completely avascular. The vasculature front reached two-thirds of the retina on P5 and reached the retina edge by P7 (Fig. 1A). TNFR2-KO mice showed delayed vascularization while TNFR2-TG exhibited promoted vascularization compared to WT at P5. However, no significant differences in the pericyte:EC ratio were detected among the three strains (see Supplementary Material and Supplementary Figs. S1C, S1D, http://www.iovs.org/content/54/1/211/suppl/DC1). All three groups showed similar vascularized areas by P7 (Fig. 1A with quantification in Fig. 1B). In addition, we compared development of the deeper intraretinal vessels, which grow from the GCL into the inner plexiform layer (IPL) and outer plexiform layer (OPL) (see schematic diagram in Supplementary Fig. S1B [see Supplementary Material and Supplementary Fig. S1B, http://www.iovs.org/content/54/1/211/suppl/DC1]). Cross-sectional staining indicated that there were fewer intraretinal vessels in the TNFR2-KO mice at P7 and P9 compared to WT and TNFR2-TG pups, but all three groups reached comparable levels at P11 (Fig. 1C with quantification in Fig. 1D). These data suggest that TNFR2 deletion and transgenesis had weak effects on retinal vascular development.
Figure 1. .
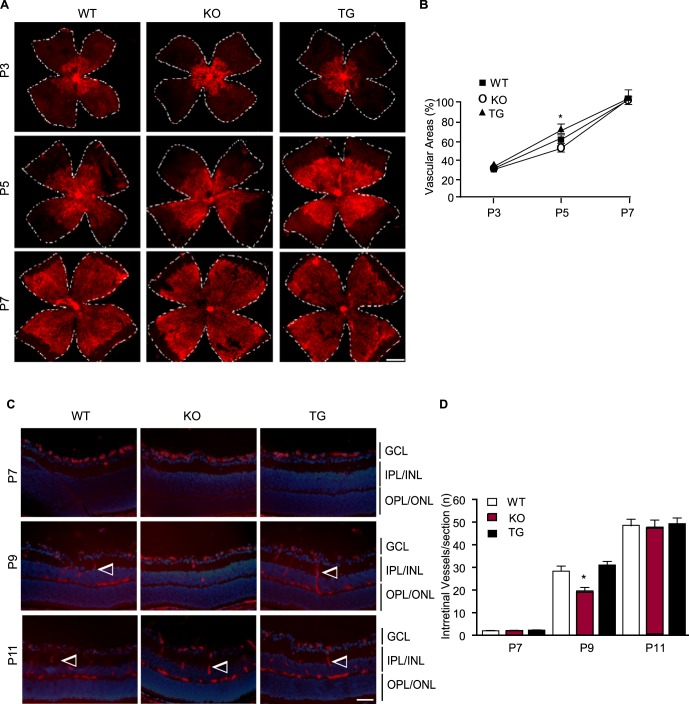
Effects of TNFR2 on retinal vascular development. Retinas from WT, TNFR2-KO, and TNFR2-TG were collected from P3 to P11. (A, B) The superficial retinal vasculatures were visualized by isolectin staining. Images from P3, P5, and P7 are shown (A). Scale bar: 500 μm. Vascularized areas were quantified as a percentage of the total retinal surface (B). n = 8 retinas from four mice for each strain. (C, D) Intraretinal vessels from the GCL into the deeper IPL and OPL layers were examined by isolectin staining of eye cross sections. Images from P7, P9, and P11 are shown, and intraretinal vessels are indicated by arrowheads (C). Scale bar: 50 μm. Intraretinal vessel number/section was quantified (D). Data are mean ± SEM from three sections of each retina and 12 retinas from six mice for each group. *P < 0.05 for TNFR2-KO compared to age-matched WT group.
TNFR2 Protects Oxygen-Induced Vascular Regression and Promotes Hypoxia-Induced Revascularization in the Retina
Neonatal WT, TNFR2-KO, and TNFR2-TG mice were subjected to the OIR model (Fig. 2A shows a scheme of the OIR model). Briefly, P7 mice were exposed to 75% oxygen for up to 5 days (until P12) and then allowed to recover in room air for an additional 5 days (P17). To determine the role of TNFR2 in oxygen-induced vascular regression, whole-mount isolectin staining of P12 retinas was used to visualize the avascular region formation after hyperoxia. Hyperoxia-exposed mice all exhibited central vaso-obliteration at P12 (Fig. 2B). Quantitative analyses indicated that TNFR2-KO increased, whereas TNFR2-TG reduced, the avascular regions as compared to WT mice (Fig. 2B with quantification in Fig. 2C). After the return to normoxia, mouse retina was in a state of relative hypoxia, which induces revascularization of the ischemic central capillary beds. Whole-mount staining of retina showed that TNFR2-KO had delayed revascularization whereas TNFR2-TG had facilitated revascularization in the ischemic central capillary beds on P17 compared to WT mice (Fig. 2B with quantification in Fig. 2D). Ischemia also induces pathological vitreous NV to form tufts, which were quantified with a program we described recently31 (also see Fig. 6). Interestingly, TNFR2-TG did not significantly increase vitreous tufts at P17 compared to WT (Fig. 2B with quantification in Fig. 2E). These data suggest that TNFR2 facilitates vascular repair in the OIR model.
Figure 2. .
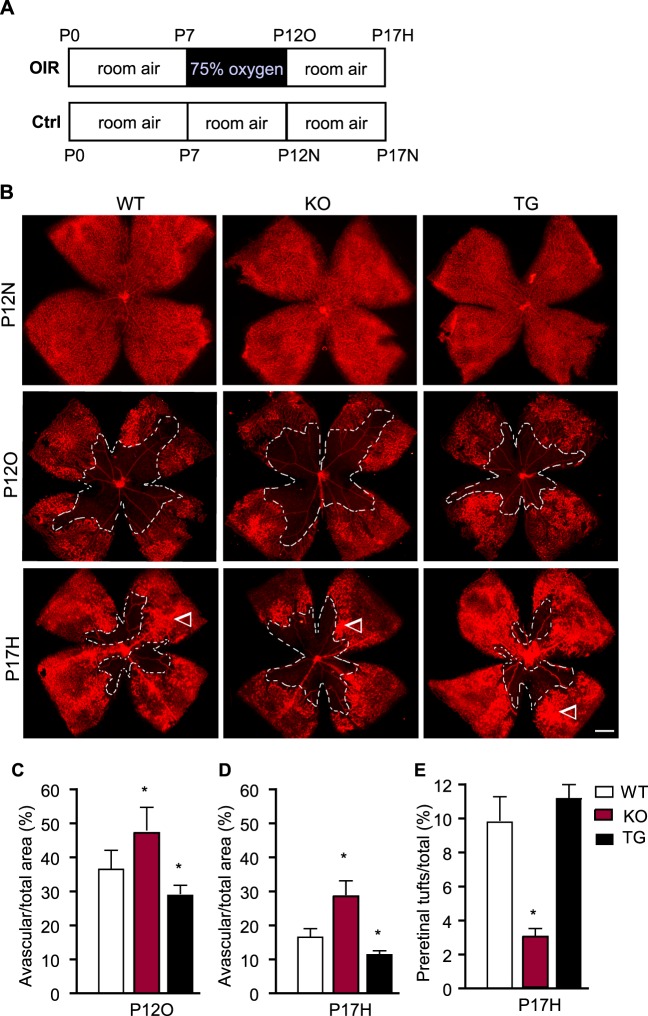
TNFR2 protects retina from oxygen-induced vascular regression and increases neovascularization in an OIR model. (A) A scheme of the OIR model. Neonatal mice were exposed to 75% oxygen for 5 days from P7 to P12. At P12, the mice were returned to room air (21% oxygen) until P17. Mice maintained in room air were used as controls. Retinas from the OIR and control groups were harvested at P12 (P12O and P12N) and P17 (P17H and P17N). (B) Retinal vasculatures were visualized by whole-mount isolectin staining. Arrowheads indicate the pathological vitreous NV tufts. Scale bar: 500 μm. (C, D) Avascular area (%) was quantified as avascular/total retina ([C], P12O; [D], P17H). (E) Areas of preretinal NV (tufts) on P17H retinas were quantified as a percentage of whole retina area. For (C–E), n = 6 for each group. *P < 0.05 for TNFR2-KO or TNFR2-TG compared to WT group.
Figure 6. .
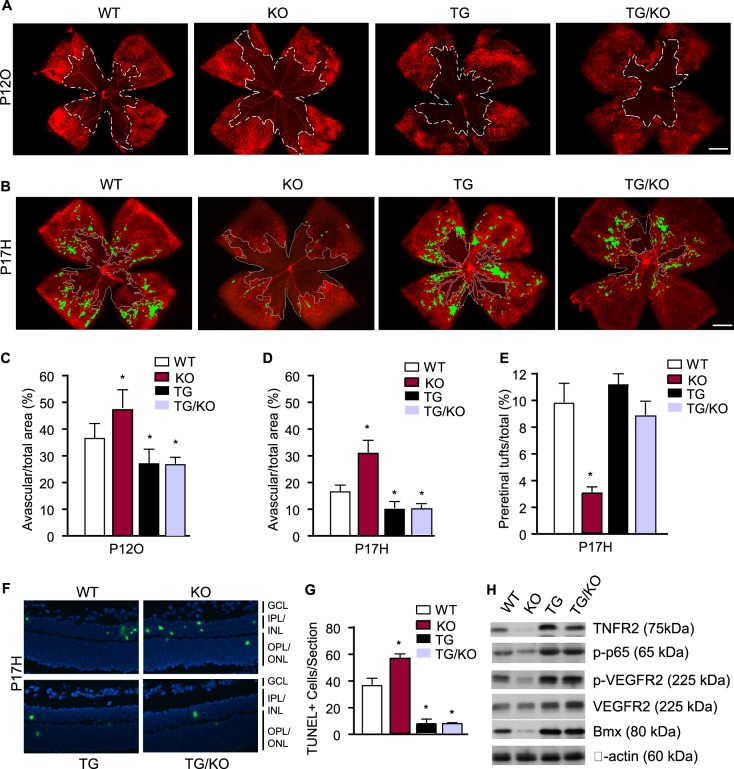
The TNFR2 transgene in EC is sufficient to mediate ischemia-induced responses. WT, TNFR2-KO, TNFR2-TG, and TNFR2-TG/KO mice were subjected to the OIR model. Retinal vasculatures were visualized by whole-mount isolectin staining. (A) Avascular areas at P12O are indicated by white lines. Scale bar: 500 μm. (B) Avascular areas and areas of preretinal NV (tufts) at P17H are indicated by white lines and green color, respectively. (C–E) Avascular areas in P12O (C) and P17H retinas (D) were quantified as a percentage of whole retina area. Areas of preretinal NV (tufts) on P17H retinas were quantified as a percentage of whole retina area (E). n = 6 for each group. *P < 0.05 for TNFR2-KO, TNFR2-TG, or TNFR2-TG/KO compared to WT group. (F) Apoptotic cells in P17H groups were detected by TUNEL staining. Scale bar: 50 μm. (G) Apoptotic cells were counted as mean of TUNEL-positive cells/section from 36 sections of six mice. *P < 0.05 for KO or TG compared to WT mice. (H) TNFR2-dependent protein expression and signaling pathways. Retinas from WT, TNFR2-KO, TNFR2-TG, and TNFR2-TG/KO at P17H were harvested. Protein expression (TNFR2, Bmx, VEGFR2, and β-actin) and activation (phospho-p65 and p-VEGFR2) were determined by immunoblotting as in Figure 5. Representative blots from three independent experiments are shown.
TNFR2 Increases Hypoxia-Induced Formation of Endothelial Tip Cells and Intraretinal Vessel Growth
Similar to retinal development, both surface vascular sprouting and intraretinal vessel growth contribute to vascular recovery in the OIR model. To investigate potential cellular mechanisms for TNFR2-mediated vascular recovery, we analyzed hypoxia-induced tip cell formation toward the avascular area and during intraretinal vessel growth. Endothelial tip cells are vital for the development of new capillaries, which are characterized by specialized apical filopodia.32 During development, these cells are localized at the retinal vascular front. In OIR, these cells were noticeably sparse at the junction between the vascularized and avascular areas of the retina at P17 in WT mice31 (Fig. 3A). In contrast to the situation in developmental retina, TNFR2 deletion reduced, whereas the TNFR2 transgene increased, ischemia-induced formation of tip cells as visualized by high-power images of the revascularization regions in the OIR model (Fig. 3A with quantification in Fig. 3B). Staining of the retina also showed that TNFR2-KO mice had fewer deep intraretinal vessels whereas TNFR2-TG had more at P17 compared to age-matched WT (Fig. 3C with quantification in Fig. 3D). These data indicate that TNFR2 mediates both hypoxia-induced formation of endothelial tip cells and intraretinal vessel growth.
Figure 3. .
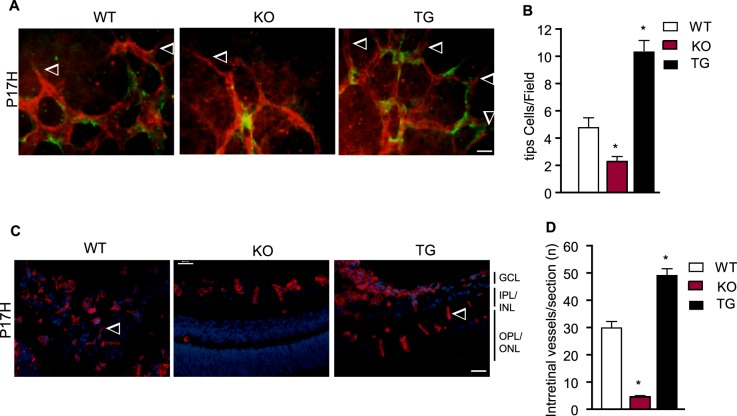
TNFR2 increases EC tip cells and intraretinal vessel growth in an OIR model. (A) P17H retinas were subjected to whole-mount staining with isolectin for EC (red) and neuron-glial antigen 2 (NG2) for pericytes (green). Representative images of retina tip cells (indicated by arrowheads) from WT, TNFR2-KO, and TNFR2-TG groups are shown. Scale bar: 20 μm. (B) Tip cells were quantified. Data are mean ± SEM from six mice for each group. *P < 0.05 for TNFR2-KO or TNFR2-TG compared to WT group. (C) Intraretinal vessels were examined by isolectin staining of P17H retinal cross sections. Intraretinal vessels are indicated by arrowheads. Scale bar: 50 μm. (D) Intraretinal vessel number/section was quantified. Data are mean ± SEM from three sections of each retina and 12 retinas from six mice for each group. *P < 0.05 for TNFR2-KO or TG compared to WT group.
TNFR2 Reduces Hyperoxia/Hypoxia-Induced Cellular Apoptosis and Enhances Cellular Survival and Proliferation of the Retina
Increased revascularization and intraretinal vessel growth could be due to decreased damage from cellular augmented apoptosis, increased EC survival, and/or EC proliferation upon hyperoxia/hypoxia. We first examined the retinal morphology by H&E staining. TNFR2 deletion or transgenesis had no drastic effects on the gross morphology of retinas under normal and OIR conditions, and the three groups showed similar thickness in the central inner nuclear layer (INL), IPL, outer nuclear layer (ONL), and OPL. However, superficial vascular tubes, indicative of revascularization, were clearly increased in TNFR2-TG and reduced in TNFR2-KO compared to WT retinas (arrowheads in Fig. 4A). NV tufts, as measured by the numbers of vascular nuclei extending beyond the inner limiting membrane, were less in TNFR2-KO but similar between WT and TNFR2-TG (indicated by arrows in Fig. 4A with quantification in Fig. 4B).
Figure 4. .
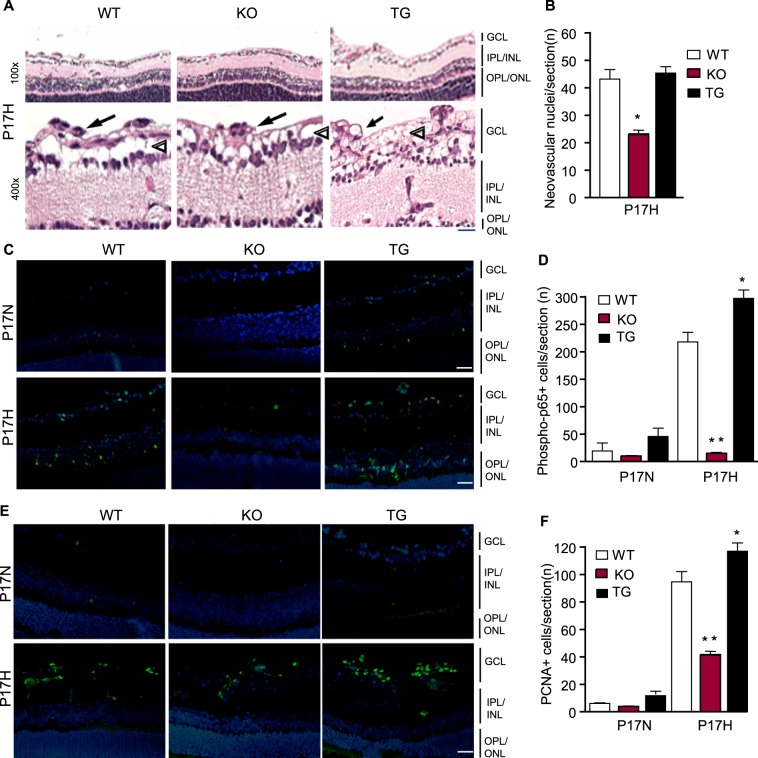
TNFR2 enhances hypoxia-induced angiogenesis by promoting EC proliferation and survival. (A) P17H retinas were subjected to H&E staining. Representative images at low and high power from WT, TNFR2-KO, and TNFR2-TG groups are shown. Scale bar: 10 μm. Arrowheads indicate revascularization whereas arrows indicate NV tufts. (B) Neovascular nuclei were quantified. (C, E) Representative images of P17N and P17H retinal cross sections with phospho-p65 and PCNA immunostaining. Scale bar: 50 μm. (D, F) Phopho-p65-positive nuclei/section and PCNA-positive nuclei/section were quantified and are shown in (D, F) respectively. Data in (B, D, F) are mean ± SEM from three sections of each retina and 12 retinas from six mice for each group. *P < 0.05 and **P < 0.01 for TNFR2-KO or TG compared to WT group.
We then performed TUNEL assay for apoptosis, immunostaining with phospho-p65 for NF-κB survival signaling, and PCNA for cellular proliferation. Apoptosis of capillaries, including both retinal surface and deep layer vessels, is one critical mechanism for hyperoxia-caused vaso-obliteration of the central retinal capillary beds.6,7 Apoptosis as detected by TUNEL staining of retinal sections at P12 was increased in TNFR2-KO but decreased in TNFR2-TG mice. Apoptotic cells were reduced in P17 retinas across all strains (see Supplementary Fig. S2A with quantification in Fig. S2B [see Supplementary Material and Supplementary Figs. S2A, S2B, http://www.iovs.org/content/54/1/211/suppl/DC1]), suggesting that TNFR2 protects cells from hyperoxia-induced apoptosis. Of note, some apoptotic cells were detected in the GCL and more in the INL, consistent with our recent observations that both vascular EC and neuronal cells undergo apoptosis in response to hyperoxia.31
TNFR2, via TRAF2, induces NF-κB activation, which has been implicated in cell survival and proliferation.19,20,27 Phosphorylation of p65 (a factor indicative of NF-κB pathway activation) and PCNA-positive cells in P17 retinal cross sections were significantly reduced in TNFR2-KO but increased in TNFR2-TG compared to WT (Figs. 4C, 4D for p-p65 and Figs. 4E, 4F for PCNA). Phospho-p65-positive nuclei were detected throughout all layers, with strongest staining observed at the GCL and ONL. However, TNF protein expression detected by immunostaining was not significantly different among the three groups (see Supplementary Material and Supplementary Fig. S2C, http://www.iovs.org/content/54/1/211/suppl/DC1), suggesting that NF-κB signaling is primarily controlled by the TNFR2 protein levels. Interestingly, PCNA-positive staining was primarily detected within superficial vasculature and NV tufts at the GCL but not the ONL, suggesting that the vascular EC at the GCL are proliferative and subsequently sprouting into the IPL and OPL layers.
TNFR2 Enhances Specific Angiogenic Pathways
To define the molecular mechanism by which TNFR2 mediates EC survival and proliferation, we determined gene expression and activation of several angiogenic signaling pathways that contribute to retinal vascular repair. The levels of gene expression were determined by quantitative RT-PCR in normoxic (N), hyperoxic (O), and hypoxic (H) retinas, and data represent the fold increase for each gene, taking the value for the normoxia P12N as 1.0. Hyperoxia at P12 (P12N versus P12O) had no effects on most of the angiogenic factors or their cognate receptors other than causing downregulation of VEGF and VEGFR2. Hypoxia at P17 drastically increased hypoxia- and inflammation-responsive genes (HIF-1α, VEGF-A, TNF, TNFR2, Bmx, angiopoietin-2, and Tie2) but had no effects on VEGFR2. Expression of TNFR1 and TRAF2 mRNA was not significantly altered either. More importantly, TNFR2 specifically augmented hypoxia-induced gene expression of Bmx, angiopoietin-2, and Tie-2 (Fig. 5A), suggesting that these genes are TNFR2 dependent.
Figure 5. .
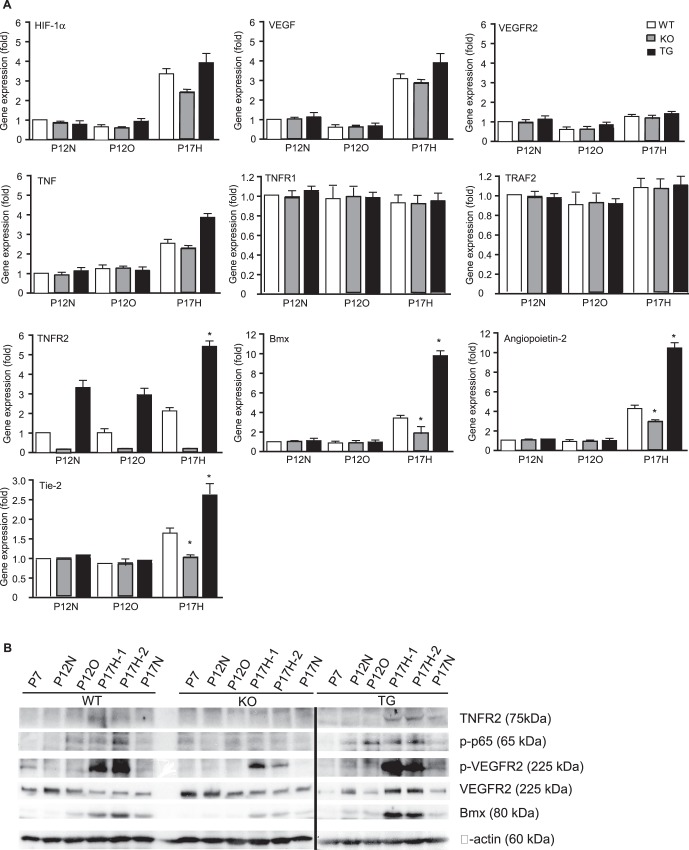
TNFR2 enhances hypoxia-induced angiogenesis by activating inflammatory and angiogenic pathways. (A) Gene expression of angiogenic factors was determined by quantitative RT-PCR. Data represent fold changes with P12N WT retina set as 1.0. Data are mean ± SEM from six retinas for each group. *P < 0.05 for P17H TNFR2-KO or TG compared to WT group. (B) TNFR2-dependent protein expression and signaling pathways. Retinas from WT, TNFR2-KO, and TNFR2-TG at indicated stages were harvested. Protein expression (TNFR2, Bmx, VEGFR2, and β-actin) was determined by immunoblotting with respective antibodies. Activation of NF-κB (phospho-p65) and VEGFR2 was determined by immunoblotting with phospho-specific antibodies. Representative blots from three independent experiments are shown.
TNFR2 protein and TNFR2-dependent signaling, including activation of NF-κB, Bmx, and VEGFR2,19–21,27,28 were further examined by immunoblotting. NF-κB activation (phospho-p65) could be detected in hyperoxia-exposed P12 retinas and was maintained during hypoxia. TNFR2 and Bmx proteins as well as VEGFR2 phosphorylation were upregulated on P17H retinas. The hypoxia-induced responses were reduced by TNFR2 deletion but augmented by the TNFR2 transgene (Fig. 5B). These data are consistent with our previous findings that expression of TNFR2 and Bmx and activation of VEGFR2 are cooperatively regulated.19,20
The TNFR2 Transgene in EC Is Sufficient to Promote Vascular Repair in the Retina
To further determine if the TNFR2 transgene in EC is sufficient to mediate retinal vascular repair in the OIR model, we crossed TNFR2-TG with TNFR2-deficient mice to generate TNFR2-KO/TG mice.19,20 In these mice, the TNFR2 transgene is expressed specifically in EC, as determined by genotyping and immunostaining with anti-TNFR2.20 TNFR2-KO/TG had no apparent changes in retina development compared to WT mice as measured by whole-mount isolectin staining (not shown). We next examined if the TNFR2 transgene in EC could restore the defects in retinal recovery observed in TNFR2-KO. Neonatal WT, TNFR2-KO, TNFR2-TG, and TNFR2-KO/TG mice subjected to OIR and oxygen-induced vascular regression at P12 (P12O) and hypoxia-induced vascular repair at P17 were examined by whole-mount isolectin staining as described in Figure 2. Quantitative analyses indicated that the TNFR2-KO/TG mice exhibited a phenotype similar to that of the TNFR2-TG in reducing hyperoxia-induced vaso-obliteration (Fig. 6A with quantification in Fig. 6C) while promoting hypoxia-induced vascular recovery in comparison to WT mice (Fig. 6B with quantification in Fig. 6D). However, ischemia-induced pathological neovascular tufts were not significantly increased by the TNFR2 transgene (highlighted by yellow color in Fig. 6B with quantification in Fig. 6E). TNFR2-KO/TG mice also had reduced TUNEL-positive apoptotic cells in P17 retina (Fig. 6F with quantification in Fig. 6G). Biochemical analyses indicated that TNFR2-TG/KO showed increased TNFR-dependent activation of NF-κB and VEGFR2 as well as expression of Bmx as observed in TNFR2-TG mice (Fig. 6H). These data support that the TNFR2 transgene in vascular EC was sufficient to activate NF-κB survival and VEGFR2 angiogenic signaling, promoting vascular recovery in the OIR model.
Discussion
The roles of TNF and TNF receptors have been previously examined in ischemic retinopathies.8–10 Increased expression of TNF has been noted during the room air recovery phase in OIR models, thus implicating the involvement of this cytokine in the retinal response to ischemia-induced injury.9 Ablation of the TNF gene or inhibition of TNF by inhibitors significantly improved vascular recovery within ischemic tissue with a concomitant reduction in pathological intravitreal NV in the OIR model. However, mice with deficiency in both TNFR1 and TNFR2 had different phenotypes from the TNF-deleted mice in the same model. Specifically, TNFR1/2 double-knockout mice exhibited a slower development of deep retinal vessels with a prolonged period of hypoxia-induced retinal NV.8 These phenotypic discrepancies could be due to distinct functions of TNFR1 and TNFR2 in the retina. By using TNFR2 knockout mice in the present study, we clearly showed that a global deletion of TNFR2 causes a delay in the physiological development of intraretinal vessel growth, an increase in hyperoxia-induced vaso-obliteration, and a reduction in hypoxia-induced vascular recovery. In contrast, an EC-specific TNFR2 transgene is sufficient to reduce hyperoxia-induced vaso-obliteration and promote hypoxia-induced vascular recovery. Interestingly, the TNFR2 transgene did not significantly increase preretinal NV. Although the role of TNFR1 was not examined in the current study, our previous work demonstrated that TNFR1 and TNFR2 play differential roles in ischemia-mediated angiogenesis in a hindlimb ischemia model, partly due to their opposite effects on EC survival and migration.19,20 Specifically, TNFR1 signaling induces EC apoptosis while TNFR2 activates EC survival and migratory pathways. It is conceivable that TNFR2 mediates EC migration-dependent intraretinal vessel growth while TNFR1 balances EC apoptosis and survival to limit preretinal NV. This would explain the results from our TNFR1/2 double-knockout mice showing a slower development of deep retinal vessels with a prolonged period of hypoxia-induced retinal NV.8 Therefore, specific activation of TNFR2 signaling without disruption of TNFR1 functions in EC could be a therapeutic approach for the treatment of ischemic retinopathy.
One important finding in our study is that vascular EC-specific expression of TNFR2 is sufficient to rescue the defects observed in TNFR2-deficient mice. TNFR2 transgenic mice with a global expression of TNFR2 gene driven by the β-actin promoter have been reported.33 However, expression of TNFR2 in these mice, comparable to disease-relevant levels of human TNFR2 in some tissues, promotes a severe, lethal, multi-organ inflammatory syndrome with elevated NF-κB activation. In contrast, the TNFR2 transgene in our model is driven by a vascular EC-specific VE-cadherin promoter. These mice do not exhibit significant changes in basal vasculature or inflammatory syndromes. However, the TNFR2 transgene accelerates ischemia-mediated angiogenesis in both an ischemic hindlimb model19,20 and the ischemic retinal model described here. The EC-specific TNFR2 transgene, unlike the global TNFR2 transgene, causes no damage to the tissues.19,20 Therefore, the EC-specific TNFR2 transgenic mice provide a useful model to examine the role of TNFR2 in pathological angiogenesis.
TNFR2-dependent activation of NF-κB survival signaling could contribute to TNFR2-mediated protection from hyperoxia-induced vaso-obliteration at P12. This conclusion is supported by the data showing that TNFR2-KO mice had larger and TNFR2-TG mice smaller avascular areas compared with WT mice. The vaso-obliteration correlated with the extent of apoptosis in the retina. Moreover, NF-κB activation could be detected at hyperoxia-exposed P12 retinas, prior to activation of Bmx-VEGFR2 signaling at P17. Our previous data demonstrated that TNFR2 protects EC from apoptosis by activating NF-κB pathway in a TRAF2- but not Bmx-dependent manner.19 NF-κB activity could also contribute to expression of inflammatory and angiogenic genes, and we have identified several TNFR2-specific genes in the retina. TNFR2 itself is a hypoxia-responsive gene, and hypoxia-induced TNF, angiopoietin-2, and Bmx are all shown to be specifically enhanced in TNFR2-TG mice, suggesting that these genes may be TNFR2 dependent. These TNFR2-dependent angiogenic proteins, together with other hypoxia-induced factors such as VEGF and angiopoietins,1,2,34 likely contribute to TNFR2-mediated vascular recovery in the retina.
Bmx (also endothelial/epithelial tyrosine kinase, Etk), a TNFR2-responsive tyrosine kinase, has been implicated in cell migration and proliferation.19,35 Bmx also mediates TNFR2-induced transactivation of VEGFR2, a potent angiogenic receptor normally activated by VEGF.1,2 We have previously shown that formation and activation of the TNFR2–Bmx–VEGFR2 complex could be detected in hindlimb ischemia.19,20 Similarly, increased expression/activation of TNFR2, Bmx, and VEGFR2 was observed in ischemic retinas. It is plausible that TNFR2–Bmx–VEGFR2 complex is formed in response to VEGF in ischemic retinas. This could also explain the phenotypic discrepancy between TNF-KO and TNFR1/2-double-knockout mice, as TNFR2 may have TNF-independent functions. We have previously shown that TNFR2 and Bmx in bone marrow–derived cells play a role in ischemia-induced vascular remodeling.19,20,28 Goukassian et al. have further demonstrated that TNFR2 regulates mobilization of circulating bone marrow–derived endothelial progenitor cells into ischemic hindlimbs.22 The circulating endothelial progenitor cells have also been implicated in repair of ischemic retina.3,36 Further investigation into the function of TNFR2 in activation and recruitment of ischemia-induced endothelial progenitor cells in the OIR model is needed.
Collectively, we have defined the physiological and pathological function of TNFR2 in the retina in mouse models. Our study suggests that specific activation of TNFR2 signaling in vascular EC may be a novel strategy for the treatment of retinopathy of prematurity and other vascular diseases in humans.
Supplementary Material
Footnotes
This work was supported by NIH Grants R0 HL085789, R01HL065978, and R01HL109420; the National Nature Science Foundation of China (81170863 and 30872819); and Guangdong Provincial Science and Technology Foundation (2008B030301095).
Disclosure: T. Wan, None; Z. Xu, None; H.J. Zhou, None; H. Zhang, None; Y. Luo, None; Y. Li, None; W. Min, None
These authors contributed equally to the work presented here and should therefore be regarded as equivalent authors.
References
- 1. Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005; 438: 932–936 [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000; 407: 249–257 [DOI] [PubMed] [Google Scholar]
- 3. Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005; 438: 960–966 [DOI] [PubMed] [Google Scholar]
- 4. Adamis AP, Aiello LP, D'Amato RA. Angiogenesis and ophthalmic disease. Angiogenesis. 1999; 3: 9–14 [DOI] [PubMed] [Google Scholar]
- 5. Heidary G, Vanderveen D, Smith LE. Retinopathy of prematurity: current concepts in molecular pathogenesis. Semin Ophthalmol. 2009; 24: 77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994; 35: 101–111 [PubMed] [Google Scholar]
- 7. Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009; 4: 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ilg RC, Davies MH, Powers MR. Altered retinal neovascularization in TNF receptor-deficient mice. Curr Eye Res. 2005; 30: 1003–1013 [DOI] [PubMed] [Google Scholar]
- 9. Gardiner TA, Gibson DS, de Gooyer TE, de la Cruz VF, McDonald DM, Stitt AW. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am J Pathol. 2005; 166: 637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kociok N, Radetzky S, Krohne TU, Gavranic C, Joussen AM. Pathological but not physiological retinal neovascularization is altered in TNF-Rp55-receptor-deficient mice. Invest Ophthalmol Vis Sci. 2006; 47: 5057–5065 [DOI] [PubMed] [Google Scholar]
- 11. Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002; 16: 438–440 [DOI] [PubMed] [Google Scholar]
- 12. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001; 104: 487–501 [DOI] [PubMed] [Google Scholar]
- 13. Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009; 4: 71–95 [DOI] [PubMed] [Google Scholar]
- 14. Fiers W, Beyaert R, Boone E, et al. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm. 1995; 47: 67–75 [PubMed] [Google Scholar]
- 15. Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999; 17: 331–367 [DOI] [PubMed] [Google Scholar]
- 16. Carpentier I, Coornaert B, Beyaert R. Function and regulation of tumor necrosis factor type 2. Curr Med Chem. 2004; 11: 2205–2212 [DOI] [PubMed] [Google Scholar]
- 17. Vandenabeele P, Declercq W, Beyaert R, Fiers W. Tumor necrosis factor receptors: structure and function. Trends Cell Biol. 1995; 5: 392–399 [DOI] [PubMed] [Google Scholar]
- 18. Bradley JR, Thiru S, Pober JS. Disparate localization of 55-kd and 75-kd tumor necrosis factor receptors in human endothelial cells. Am J Pathol. 1995; 146: 27–32 [PMC free article] [PubMed] [Google Scholar]
- 19. Luo D, Luo Y, He Y, et al. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am J Pathol. 2006; 169: 1886–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo Y, Wan T, He Y, et al. Endothelial-specific transgenesis of TNFR2 promotes adaptive arteriogenesis and angiogenesis. Arterioscler Thromb Vasc Biol. 2010; 30: 1307–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002; 22: 7512–7523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goukassian DA, Qin G, Dolan C, et al. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007; 115: 752–762 [DOI] [PubMed] [Google Scholar]
- 23. Zhang L, Sivashanmugam P, Wu JH, et al. Tumor necrosis factor receptor-2 signaling attenuates vein graft neointima formation by promoting endothelial recovery. Arterioscler Thromb Vasc Biol. 2008; 28: 284–289 [DOI] [PubMed] [Google Scholar]
- 24. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994; 78: 681–692 [DOI] [PubMed] [Google Scholar]
- 25. Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995; 81: 513–523 [DOI] [PubMed] [Google Scholar]
- 26. Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995; 83: 1243–1252 [DOI] [PubMed] [Google Scholar]
- 27. Zhang R, Xu Y, Ekman N, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003; 278: 51267–51276 [DOI] [PubMed] [Google Scholar]
- 28. He Y, Luo Y, Tang S, et al. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Invest. 2006; 116: 2344–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruuls SR, Hoek RM, Ngo VN, et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity. 2001; 15: 533–543 [DOI] [PubMed] [Google Scholar]
- 30. Milicevic NM, Klaperski K, Nohroudi K, et al. TNF receptor-1 is required for the formation of splenic compartments during adult, but not embryonic life. J Immunol. 2011; 186: 1486–1494 [DOI] [PubMed] [Google Scholar]
- 31. Liang X, Zhou H, Ding Y, et al. TMP prevents retinal neovascularization and imparts neuroprotection in an oxygen-induced retinopathy model. Invest Ophthalmol Vis Sci. 2012; 53: 2157–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerhardt H, Golding M, Fruttiger M, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003; 161: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Douni E, Kollias G. A critical role of the p75 tumor necrosis factor receptor (p75TNF-R) in organ inflammation independent of TNF, lymphotoxin alpha, or the p55TNF-R. J Exp Med. 1998; 188: 1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joussen AM, Poulaki V, Tsujikawa A, et al. Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol. 2002; 160: 1683–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000; 19: 5651–5661 [DOI] [PubMed] [Google Scholar]
- 36. Stitt AW, O'Neill CL, O'Doherty MT, Archer DB, Gardiner TA, Medina RJ. Vascular stem cells and ischaemic retinopathies. Prog Retin Eye Res. 2011; 30: 149–166 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.