

Abstract
The regulation of energy metabolism, such as calorie restriction (CR), is a major determinant of cellular longevity. Although augmented gluconeogenesis is known to occur in aged yeast cells, the role of enhanced gluconeogenesis in aged cells remains undefined. Here, we show that age-enhanced gluconeogenesis is suppressed by the deletion of the tdh2 gene, which encodes glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a protein that is involved in both glycolysis and gluconeogenesis in yeast cells. The deletion of TDH2 restores the chronological lifespan of cells with deletions of both the HST3 and HST4 genes, which encode yeast sirtuins, and represses the activation of gluconeogenesis. Furthermore, the tdh2 gene deletion can extend the replicative lifespan in a CR pathway-dependent manner. These findings demonstrate that the repression of enhanced gluconeogenesis effectively extends the cellular lifespan.
Introduction
Aging is a complex process that is associated with the gradual loss of physiological functions, which are regulated by genetic and environmental factors. Recent studies in genetically tractable model systems including yeast, worms, flies and mice demonstrate that longevity can be modulated by single gene mutations [1], [2], [3], [4], [5], [6]. Calorie restriction (CR) is the most effective intervention known to extend lifespan in a variety of metazoan species [7], [8]. CR has also been shown to delay the onset or reduce the incidence of many age-related diseases, including cancer, diabetes and cardiovascular disorders [8], [9], [10]. CR may work by reducing the level of reactive oxygen species (ROS) as a result of the slowed energy metabolism [7], [8]. Although the mechanism by which CR extends longevity and ameliorates age-associated diseases remains unclear, energy metabolism has a key role in longevity via the CR pathway. The central carbon metabolism pathway, which includes the glycolysis/gluconeogenesis and tricarboxylic acid (TCA) pathways, uses various carbon sources, such as glucose and glycerol, to produce ATP molecules. The accumulation of metabolic intermediates produced by enhanced gluconeogenesis has been reported to be an age-induced change in budding yeast [11]. However, the role of augmented gluconeogenesis in cellular aging remains unclear.
Sir2 family proteins (sirtuins) are evolutionally conserved and were originally discovered and studied in yeast as a component of the Sir1/2/3/4 silencing complex [12], [13]. The mammalian orthologs of SIR2 encode nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases and ADP-ribosylases [14]. Previous studies have shown that the sirtuins play important roles in cellular longevity in yeast and in regulating the stress response, cell survival and energy metabolism in multicellular organisms [15]. Earlier studies showed that sirtuins play important roles in CR-induced lifespan extension [16], [17] and in regulating the stress response, cell survival and energy metabolism, suggesting a role for sirtuins in age-related metabolic diseases [15], [18], [19]. The Sir2 homologs in yeast include Hst1, Hst2, Hst3 and Hst4 [20]. Of these, Hst1 exhibits the highest homology with Sir2 and mediates transcriptional regulation independent of the SIR silencing complex [20]. Hst2 functions in concert with Hst1 to downregulate subtelomeric gene expression [21] and plays a role in regulating rDNA silencing and recombination [22], [23]. Hst3 and Hst4 together maintain telomeric silencing and cell cycle progression [20]. Hst3 and Hst4 deacetylate histone H3 on lysine 56 (H3-K56) in chromatin during S phase to the next G1 phase to prevent the genomic instability caused by the continuous acetylation of H3-K56 [24].
Recent studies have shown that several longevity factors and pathways are highly conserved among eukaryotes [3]. The budding yeast S. cerevisiae provides an efficient model for exploring the molecular mechanism of longevity regulation. Budding yeast propagate by asymmetric cell division, in which the partitioning between the two resulting cells is unequal morphologically and molecularly. The larger cell is designated as the mother cell, and each yeast cell can only undergo a certain number of cell divisions, known as the replicative life span (RLS). The process of approaching the limit of cell divisions is recognized as replicative aging. Similar to higher eukaryotic cells, aged yeast cells exhibit a declining division potential and reduced fitness. Another type of yeast lifespan is chronological lifespan (CLS), which measures the length of time that cells remain viable in a non-dividing state. Yeast cells enter the non-dividing stationary phase (or post-diauxic phase) when nutrients are limited. This quiescent state has been suggested to resemble the G0 state in higher eukaryotes [25]. Several longevity factors have been identified through RLS and CLS studies [26], [27], [28], [29], [30]. The yeast rDNA loci consist of a stretch of approximately 9 kb rDNA repeats. Homologous recombination between adjacent repeats is known to result in the excision of repeat units and the formation of extrachromosomal rDNA circles known as ERCs. ERCs are autonomously replicating and preferentially accumulate in the aging mother cell [31]. Sir2 counteracts ERC formation via the establishment and/or maintenance of sister-chromatid cohesion at the rDNA loci [32]. Sir2 also plays a role in stress resistance and the regulation of newborn cell fitness by conferring a superior ROS management to the daughter cell to protect the progeny from aging [33]. Similar to cells harboring deficiencies in DNA damage repair, cells with deletions of both the HST3 and HST4 genes display short RLSs due to the genome instability that accumulates with each cell division [34], [35], [36]. Interestingly, hst3Δ cells reduce CLS [37]. It is unclear why the quiescent hst3Δ cell exhibits a reduced chronological lifespan under nutrient starvation.
In this study, we determined that the short CLS of hst3Δ hst4Δ cells was due to age-enhanced gluconeogenesis. We isolated several genes encoding metabolic enzymes to restore the CLS of hst3Δ hst4Δ cells. Among these isolated genes, the deletion of the TDH2 gene, which encodes an isoform of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is essential for glycolysis/gluconeogenesis, reduced the amount of accumulated metabolites from glucose metabolism in hst3Δ hst4Δ cells and specifically repressed gluconeogenesis. Furthermore, the RLS was extended in cells in which the TDH2 gene was deleted in a manner similar to that induced by the CR pathway. Our data suggest that Hst3 and Hst4 coordinately regulate age-enhanced gluconeogenesis to maintain the CLS, and that a reduction in age-enhanced gluconeogenesis can extend the cellular lifespan.
Materials and Methods
Replicative Lifespan (RLS) Assay (Pedigree Analysis)
The RLS assay, known as pedigree analysis, was performed as described previously [38]. Typically, a minimum of 50 mother cells was counted for each strain tested. To compare the difference of replicative lifespans between strains on statistics, we performed the Wilcoxon Rank-Sum test.
Chronological Lifespan (CLS) Assay
CLS assays were conducted in 0.5% glucose synthetic complete (SC) media [39] supplemented with a fourfold excess of the amino acids for which the strains were autotrophic in the absence (“unbuffered”) of 50 mM citrate-phosphate buffer [pH6.0]. Yeast cells were cultured overnight in YPD at 25°C. The cells (5×106 cells/ml) were suspended in SC medium (0.5% glucose) and cultured at 30°C. Cell viability was determined every 2 days. To determine viability, the cells were washed with 500 µl of PBS pH 7.3 and mixed with 500 µl of 15 µg/ml phloxin B solution [40]. Cell viability was calculated from the number of red-stained cells among the total cell number (n = 100). Almost all BY4742 cells were stained with phloxin B at the start in the presence of 2% glucose, but not in 0.5% glucose. Therefore, we used SC medium containing 0.5% glucose to perform the CLS assay by phloxin B staining.
Growth Rate Assay in Liquid Culture
After being cultured overnight in YPD at 25°C, the cells (5×106 cells/ml) were suspended in SC medium (0.5% glucose) and cultured at 25°C without shaking. The cell number was determined every 24 h using a Z-1 Coulter Counter (Beckman-Coulter Co., IN. USA). At least three replicates were analyzed for each strain.
Preparation of Yeast Cell Extract
The cell extract was prepared according to a previously described method [11], [41]. Briefly, mid-log growth yeast cells (1×108 cells in total) cultured in YPD were harvested and washed three times with PBS buffer (pH 7.3). The cells were suspended in 500 µl of metabolic enzyme extraction buffer (20 mM sodium phosphate pH 7.5, 0.02% bovine serum albumin [BSA], 0.5 mM ethylenediaminetetraacetic acid [EDTA], 5 mM ß-mercaptoethanol, 25% glycerol, 0.5% Triton X-100). The cell suspension was mixed with same volume of acid-washed glass beads (diameter of less than 0.5 mm), and the cells were disrupted by vortexing. Soluble proteins were obtained by collecting the supernatant after centrifugation of the broken cell suspension at 14,000 rpm for 5 min at 4°C. The protein concentration was determined by the Bradford protein assay with BSA as the standard.
GAPDH Glycolytic Assay
The GAPDH glycolytic assay was based on a previously described method [42]. The assay was conducted at 30°C by adding the cell extract (40 µg) to a substrate solution containing 100 mM Tris/HCl (pH 8.5), 1.5 mM NAD+, 5.0 mM sodium phosphate (pH 7.0) and 3.2 mM D-glyceraldehyde-3-phosphate. Changes in absorbance (340 nm) were monitored spectrophotometrically (DU 800, Beckman Coulter Co.).
GAPDH Gluconeogenic Assay
The gluconeogenic substrate of GAPDH, 1,3-diphosphoglycerate, was generated by the following previously described method [42]. The reaction mixture was composed of 80 mM triethanolamine (pH 8.5), 8.0 mM MgSO4, 0.25 mM NADH, 2.4 mM ATP, 12 mM 3-phosphoglycerate and 18.8 µg/ml budding yeast phosphoglycerol kinase (Sigma-Aldrich, USA). The reaction was conducted at 37°C for 30 min and stopped by heating at 100°C for 5 min. The assay was performed at 30°C by mixing the cell extract (4 µg), 0.05 mM NADH and 200 µl of the reaction mixture containing 1,3-diphosphoglycerate. The changes in absorbance (340 nm) were monitored with a spectrophotometer.
Glycogen Quantification Assay
The cell pellet harboring glycogen was prepared according to the following previously described method [43]. The cells were cultured at 30°C (25°C was used for hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ cells) for 3 h in either YPD or YPE liquid media. After harvesting and washing three times with PBS buffer (pH 7.3), the cells (1×108) were suspended in 300 µl of 30% KOH and incubated at 100°C for 2 h. The cell suspension was mixed with 2 volumes of 99% ethanol and centrifuged at 14000 rpm for 5 min at 4°C. The cell pellet was dried and suspended in 100 µl of H2O. The cell suspension (10 µl) was assayed with a Glycogen Assay Kit (Biovision Co., USA) according to the manufacturer’s instructions. The changes in absorbance (570 nm) were monitored with a microplate reader (iMark, BioRad Co., USA).
Reactive Oxygen Species (ROS) Detection
The protocol was modified as previously described [44]. The cells (5×107) were washed with PBS (pH 7.3) and resuspended in 100 µl of PBS containing 5 µg/ml dihydroethidium (DHE). After incubation at room temperature for 5 min, the cells were harvested and suspended in 100 µl of PBS (pH 7.3). DHE-positive cells were viewed under rhodamine fluorescence using a Leica CTR6500 microscope with LAS AF software. The ratio was calculated from the number of DHE-positive cells among the total number of cells (n = 100).
Results
hst3Δ hst4Δ Cells Exhibit Reduced Chronological Lifespan in a Manner Independent of the Regulation of Histone H3-K56 Acetylation
As has been described previously [34], [35], [36], hst3Δ hst4Δ cells exhibited a short RLS (Figs. 1A and S1). Interestingly, hst3Δ hst4Δ cells also exhibited a short CLS following the accumulation of reactive oxygen species (ROS) (Figs. 1B and S2). Recent research shows that pH of the medium affects the cell viability in the CLS assay [45], and we routinely employed SD medium in the absence (“unbuffered”) of 50 mM citrate-phosphate buffer [pH6.0]. To exclude the possibility that the pH alteration would cause to reduce the CLSs of hst3Δ hst4Δ cells, we confirmed that hst3Δ hst4Δ cells reduced the viability even in CLS assay using SC medium even in the presence (“buffered”) of 50 mM citrate-phosphate buffer [pH6.0] (Fig. S3). The short RLS of hst3Δ hst4Δ cells is due to the persistence of H3-K56 acetylation in chromatin that can trigger the genomic instability [24], [36]. Next, we examined whether the short CLS of the hst3Δ hst4Δ cells also depended on histone H3-K56 acetylation. Rtt109 is a histone acetyltransferase that acetylates histone H3-K56 [46], [47], [48], and Asf1 is a histone chaperone that is necessary for the acetylation of K56 of histone H3 [49], [50]. We confirmed that the acetylation of histone H3-K56 was not detected in asf1Δ, rtt109Δ, asf1Δ hst3Δ hst4Δ and rtt109Δ hst3Δ hst4Δ cells (Fig. 1C). Cells containing deletions of either RTT109 or ASF1 did not exhibit reduced CLS (Fig. 1D). However, both asf1Δ hst3Δ hst4Δ and rtt109Δ hst3Δ hst4Δ triple deletion mutants exhibited a reduced CLS identical to that of the hst3Δ hst4Δ double deletion mutant (Fig. 1D). Thus, these data suggest that the short CLS of hst3Δ hst4Δ cells is not due to histone H3-K56 acetylation.
Figure 1. hst3Δ hst4Δ cells reduce CLS independent of the continuous acetylation of histone H3-K56.
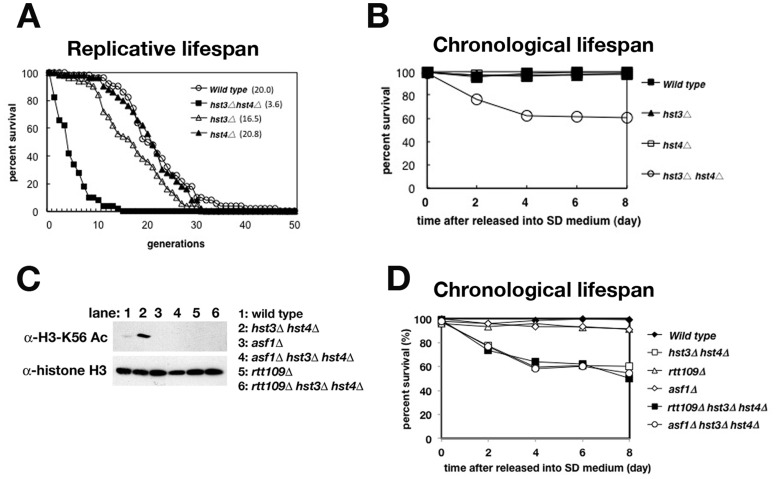
(A) Replicative lifespan (RLS) of wild type (BY4742), hst3Δ, hst4Δ and hst3Δ hst4Δ cells. The median lifespan is given next to each genotype. The difference between strains was performed on statistical calculations (Fig. S1) (B) Chronological lifespan (CLS) analysis of wild type, hst3Δ, hst4Δ and hst3Δ hst4Δ cells. (C) Immunoblot analysis of whole-cell protein extracts using antibodies either against acetylation of histone H3 on K56 or total histone H3 as a loading control. Cell extracts were prepared from asynchronized wild type, asf1Δ, rtt109Δ, hst3Δ hst4Δ, asf1Δ hst3Δ hst4Δ and rtt109Δ hst3Δ hst4Δ cells. Each cell extract was resolved by 15% SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by immunoblot using antibodies to detect K56-acetylated H3, and total histone H3. (D) CLS analysis of wild type, hst3Δ hst4Δ, rtt109Δ, asf1Δ, rtt109Δ hst3Δ hst4Δ and asf1Δ hst3Δ hst4Δ cells.
Metabolic Intermediates from Glucose Metabolism Accumulate in hst3Δ hst4Δ Cells in a Pattern that is Similar to that of Aged Cell
The CLS of yeast cells is monitored as cellular viability under the starvation of nutrient, especially glucose. This observation prompted us to hypothesize that the glucose metabolism might not respond to nutrient starvation, and lead to reduce CLSs of the hst3Δ hst4Δ cells. To evaluate this hypothesis, we analyzed the profiles of the metabolites of glucose metabolism in wild type and hst3Δ hst4Δ cells using capillary electrospray ionization time-of-flight mass spectrometry (CE-TOFMS) [51]. Wild type and hst3Δ hst4Δ cells were cultured under the same conditions employed in the CLS assay. The metabolites were prepared from these cells, and analyzed by CE-TOFMS. All intermediates of glycolysis/gluconeogenesis and the TCA cycle that measured in this study accumulated in the hst3Δ hst4Δ cells more than in the wild type cells (Fig. 2A and Table S1). Several metabolites connecting with glucose storage: glucose-1-phosphate (G1P), glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P), were accumulated in the hst3Δ hst4Δ cells (Fig. 2A). The accumulation of these metabolites is also observed in rapidly aging sip2Δ and aged wild type cells [11]. To confirm the profiles of metabolites in hst3Δ hst4Δ cells are reminiscent of those of aged cells, we analyzed the profiles of the major energy metabolites in young and aged cells using CE-TOFMS. Aged cells (median 16–20 cell divisions) and young cells (zero or one cell division) were isolated using biotin-streptavidin magnetic sorting [31], [52]. Similar to the metabolic profile of hst3Δ hst4Δ cells (Fig. 2A), the metabolites involved in glucose storage (G1P, G6P and F6P) and many metabolites of the TCA cycle accumulated in aged cells (Fig. 2B). The pronounced enhancement of glucose storage in aged cells is brought by the augment gluconeogenesis and the reduced glycolysis [11]. The accumulation of the metabolites in the TCA cycle supports that the gluconeogenesis pathway was enhanced, because these metabolites can be utilized to synthesize glycogen via the gluconeogenesis pathway (Figure 4C). Other metabolic processes, such as nucleotide synthesis and amino acid synthesis, did not exhibit global differences in the accumulation of metabolic intermediates in aged and young cells (Fig. S4 and Table S2). Together, these data suggest that the glucose metabolism of the hst3Δ hst4Δ cells, which has been similar to those of aged cells, might be a cause to reduce the CLSs.
Figure 2. hst3Δ hst4Δ cells accumulate a number of metabolic intermediates from glucose metabolism in a pattern similar to that of aged cells.
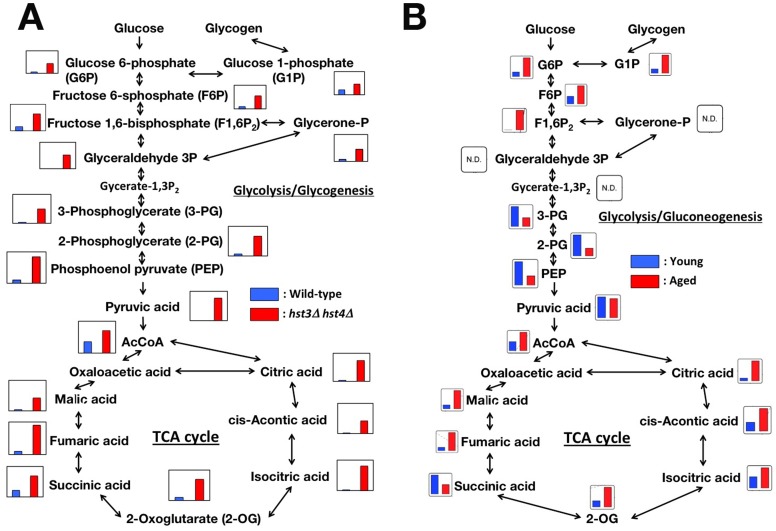
(A) The metabolic profiling data of glucose metabolites in wild type and hst3Δ hst4Δ cells were mapped onto known pathways. Each graph was derived from the calculated amount of metabolic intermediates shown in Table S1. The cells were cultured in SC medium at 30°C for 3 days and analyzed by CE-TOFMS. (B) The metabolic profiling data of central carbon metabolism in both young and aged cells was mapped as in (A). Each graph was derived from the calculated amount of metabolic intermediates shown in Table S2.
Figure 4. Deletion of the TDH2 gene can selectively repress the activation of gluconeogenesis.
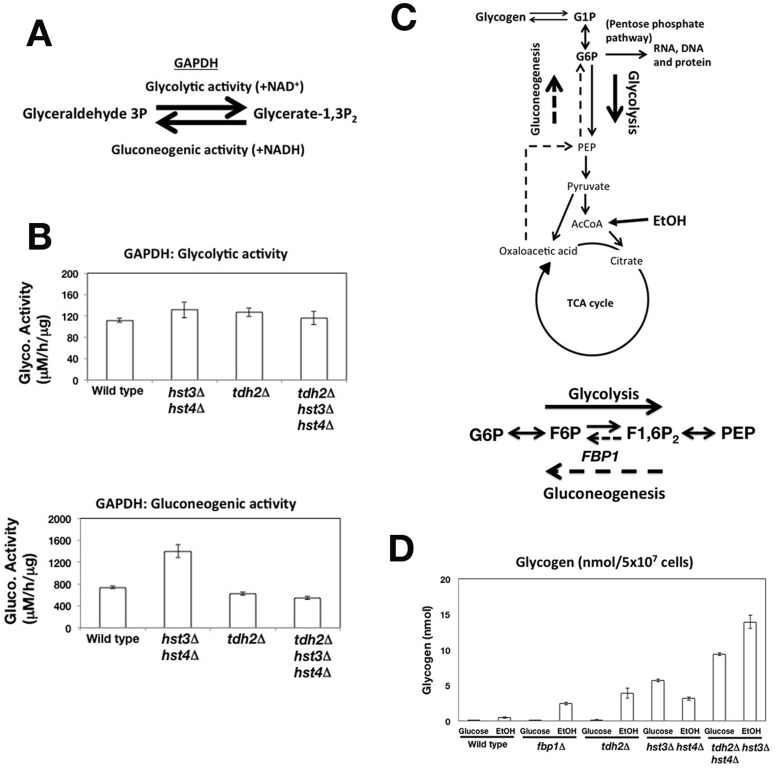
(A) The roles of GAPDH in both glycolysis and gluconeogenesis. (B) Measurement of GAPDH activity. The glycolytic and gluconeogenic activities of GAPDH were monitored using cell lysates prepared from asynchronous cells. (C) A cartoon showing the utilization of ethanol (EtOH) as a non-fermentative carbon source via gluconeogenesis. (D) Glycogen accumulates in gluconeogenesis-deficient cells (fbp1Δ) and tdh2Δ cells. The amount of glycogen prepared from cells cultured in liquid medium containing either glucose or EtOH was quantified. All graphs show the mean values of measurements performed in triplicate. The error bars denote the standard deviation. Note that certain error bars are too small to be visible.
Deletion of the TDH2 Gene Restored both the CLS and Vegetative Growth of hst3Δ hst4Δ cells by Decreasing the Metabolic Intermediates of Glucose Metabolism
Given that the accumulation of glucose metabolites resulted in a shortened chronological lifespan in hst3Δ hst4Δ cells, we hypothesized that reducing the accumulation of metabolic intermediates within the hst3Δ hst4Δ cells could extend this lifespan. One approach to reducing the amount of metabolites is to delete the gene encoding a metabolic enzyme (Fig. S5; gene map of glucose metabolism). We screened a glucose metabolism gene deletion, which was able to restore the CLS of hst3Δ hst4Δ cells. As shown in Figure 3A, the deletion of the TDH2 gene, which encodes a yeast GAPDH [53], was able to completely restore the lifespan of hst3Δ hst4Δ cells. We tested whether tdh2Δ deletion could extend the RLS of hst3Δ hst4Δ cells. tdh2Δ could slightly, but significantly on statistics, extended the RLS of hst3Δ hst4Δ cells (Figs. S1 and S6). Similar to TDH2, we identified two other genes, IDP1 and YAT1, whose deletion rescued the CLS of the hst3Δ hst4Δ cells (Fig. 3B and C). Idp1 is a mitochondrial NADP-specific isocitrate dehydrogenase that catalyzes the oxidation of isocitrate to α-ketoglutarate [54]. Yat1 is a carnitine acetyltransferase that localizes to the outer membrane of the mitochondrion and is involved in the transport of acetyl-CoA from the cytosol into the mitochondrion [55]. The decreasing pH of media reduces the cell viability in CLS assay [45]. We confirmed that any tdh2, idp1 or yat1 gene deletion did not increase the pH of medium to restore the viability of hst3Δ hst4Δ cell in CLS assay (Table S4). Because the biochemical analysis of GAPDH has been already established, we mainly analyzed Tdh2 in this study, as described later. Aged cells usually grow poorly even though the surrounding environment is suitable for cell growth. Similar to aged cells, the hst3Δ hst4Δ cells grew poorly in synthetic complete (SC) medium, which permitted normal growth of wild type cells (Fig. 3D). Interestingly, tdh2Δ hst3Δ hst4Δ cells grew in SC medium as well as wild type cells (Fig. 3D). Additionally, idp1Δ hst3Δ hst4Δ and yat1Δ hst3Δ hst4Δ cells grew as well as wild type cells (Fig. S7A and B). Thus, the deletion of genes involved in glucose metabolism not only restored the CLS of hst3Δ hst4Δ cells but also the poor growth.
Figure 3. Deletion of the TDH2 gene restores both CLS and vegetative growth and reduces the metabolic intermediates of glucose metabolism in hst3Δ hst4Δ cells.
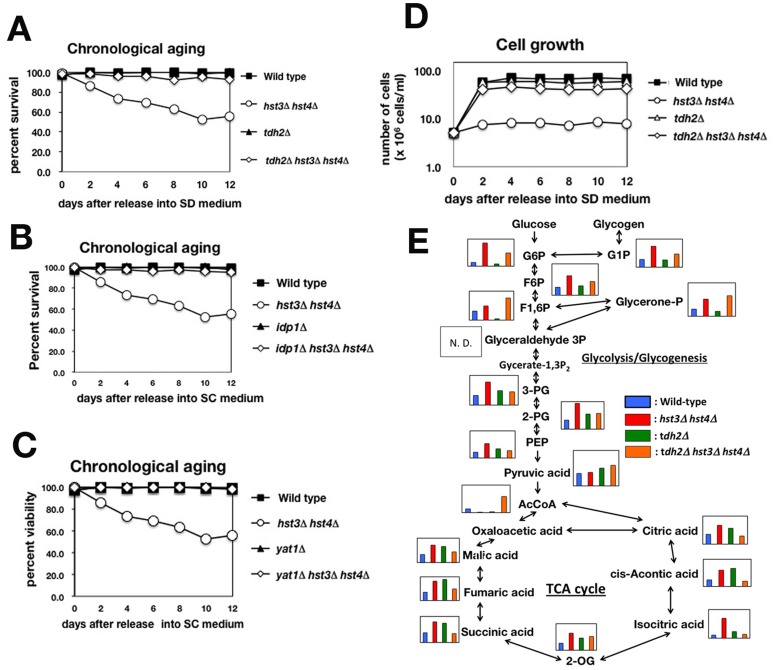
(A) The CLSs of wild type, hst3Δ hst4Δ, tdh2Δ and tdh2Δ hst3Δ hst4Δ cells. (B) The CLSs of wild type, hst3Δ hst4Δ, tdh2Δ and tdh2Δ hst3Δ hst4Δ cells. (C) The CLSs of wild type, hst3Δ hst4Δ, tdh2Δ and tdh2Δ hst3Δ hst4Δ cells. (D) Growth curves of wild type, hst3Δ hst4Δ, tdh2Δ and hst3Δ hst4Δ tdh2Δ cells in SC medium. (E) The metabolic profiling data of central carbon metabolism in wild type, hst3Δ hst4Δ, tdh2Δ and hst3Δ hst4Δ tdh2Δ cells were mapped onto known pathways. Each graph was derived from the calculated amount of metabolic intermediates shown in Table S3.
We analyzed whether the deletion of the TDH2 gene could reduce the accumulation of metabolic intermediates of glucose metabolism in hst3Δ hst4Δ cells. Using CE-TOFMS, we compared the profiles of metabolic intermediates among wild type, hst3Δ hst4Δ, tdh2Δ and tdh2Δ hst3Δ hst4Δ cells (Figs. 3E, S8 and Table S3). As shown in Figure 3E, many metabolites that specifically accumulated in hst3Δ hst4Δ cells (G6P, G1P, and F6P from glycolysis/gluconeogenesis and many metabolites of the TCA cycle) were indeed decreased in tdh2Δ hst3Δ hst4Δ cells. These data suggest that the tdh2 gene deletion contributed to the decrease in the accumulation of metabolic intermediates of glucose metabolism in tdh2Δ hst3Δ hst4Δ cells.
Deletion of the TDH2 Gene Specifically Repressed the Activation of Gluconeogenesis
Next, we investigated whether the tdh2 gene deletion could specifically repress the gluconeogenesis pathway. We initially examined the influence of the tdh2 gene deletion on cellular GAPDH activities that play a pivotal role in glycolysis/gluconeogenesis. In the presence of NAD+, GAPDH promotes glycolysis to catalyze the conversion of glyceraldehyde-3-phosphate (glyceraldehyde-3P) to glycerate-1,3-bisphosphate (glycerate-1,3P2) (Fig. 4A: glycolytic activity). In the presence of NADH, GAPDH promotes gluconeogenesis to catalyze the conversion of glycerate-1,3P2 to glyceraldehyde 3P (Fig. 4A: gluconeogenic activity). We prepared cell extracts from wild type, tdh2Δ, hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ cells cultured in medium containing glucose as the carbon source and compared the glycolytic and gluconeogenic activities of GAPDH among the cell extracts. The glycolytic activities of GAPDH were similar in all of the cell extracts (Fig. 4B: glycolytic activity). The gluconeogenesis pathway is usually repressed in the presence of glucose, and therefore the gluconeogenic activity of GAPDH should be low. However, the gluconeogenic activity of GAPDH in the hst3Δ hst4Δ cell extract was significantly higher than that in the wild type cell extract (Fig. 4B: gluconeogenic activity). Interestingly, the gluconeogenic activity in the tdh2Δ hst3Δ hst4Δ cell extract was approximately the same as that observed in both the wild type and tdh2Δ cell extracts (Fig. 4B: gluconeogenic activity). Thus, the gluconeogenic activity of GAPDH was increased in hst3Δ hst4Δ cells. In addition, the gluconeogenic activity of GAPDH in the hst3Δ hst4Δ cells was repressed by the deletion of the TDH2 gene without affecting the glycolytic activity.
We next addressed whether the tdh2 gene deletion repressed the entire gluconeogenesis pathway in the hst3Δ hst4Δ cells by suppressing the gluconeogenic activity of GAPDH. Budding yeast cells employ gluconeogenesis to utilize non-fermentable carbon sources such as glycerol or ethanol [56]. Ethanol is metabolized in the TCA cycle, and the hexose phosphates produced by gluconeogenesis are used for the biosynthesis of DNA, RNA and proteins via the pentose phosphate pathway (Fig. 4C: cartoon). Because gluconeogenesis-deficient mutant cells are unable to utilize ethanol as a carbon source, we expected that the mutant cells would respond similarly to glucose starvation in the presence and absence of ethanol. Under glucose starvation, yeast cells rapidly accumulate glycogen, a branched polymeric glucose reserve generated from G1P (Fig. 4C) [57], [58]. Previous research reported that tdh2Δ cell grows slower than wild type cell in the presence of ethanol [59], which suggest that the gluconeogenesis pathway does not work well in tdh2Δ cell. We analyzed the accumulation of glycogen to determine if the gluconeogenesis pathway was intact or deficient. When wild type cells were cultured in the presence of ethanol, the amount of glycogen that accumulated increased slightly compared to that in the presence of glucose (Fig. 4D: wild type). Cells lacking the FBP1 gene encoding fructose-1,6-bisphosphatase, which irreversibly converts F1,6P2 to F6P, exhibit a defect in gluconeogenesis (Fig. 4C) [60]. As expected, fbp1Δ cells accumulated a considerable amount of glycogen in the presence of ethanol (Fig. 4D: fbp1Δ). Similar to fbp1Δ cells, tdh2Δ cells also accumulated glycogen in the presence of ethanol but not in the presence of glucose (Fig. 4D: tdh2Δ and fbp1Δ). We confirmed that the expression of the FBP1 gene involved in gluconeogenesis was normally induced in response to non-fermentable carbon sources in the tdh2Δ cells (Fig. S9). These findings indicate that the tdh2Δ cells were deficient in the activation of gluconeogenesis in a manner similar to that observed in fbp1Δ cells, although the gene transcription involved in gluconeogenesis was induced normally.
Next, we examined whether the deletion of tdh2 could repress gluconeogenesis in the hst3Δ hst4Δ background. Like hst3Δ hst4Δ cells, tdh2Δ hst3Δ hst4Δ cells accumulated glycogen even in the presence of glucose (Fig. 4D: hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ). Because several metabolites connected with glycogen synthesis (G6P, G1P and F6P) were accumulated in tdh2Δ hst3Δ hst4Δ cells more than in wild type cells (Fig. 3E), glycogen may be synthesized. Similar to tdh2Δ cells, tdh2Δ hst3Δ hst4Δ cells accumulated glycogen in the presence of ethanol, whereas hst3Δ hst4Δ cells did not (Fig. 4D: tdh2Δ, hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ). Taken together, these data suggest that the tdh2 gene deletion specifically represses gluconeogenesis in hst3Δ hst4Δ cells.
TDH2 Gene Deletion Extends the RLS in a CR-dependent Manner
CR reduces the augmentation of gluconeogenesis in aged yeast cells [11]. We examined whether the reduction of gluconeogenesis by the deletion of the tdh2 gene contributed to the extension of the cellular lifespan in a CR-dependent manner. The RLS of tdh2Δ mother cells was significantly extended compared with that of wild type cells (Figs. 5A and S1). To investigate the genetic pathway of tdh2 gene deletion, we compared the lifespan of the tdh2 gene deletion in CR-mimicked mutant cells. In one CR model, the deletion of the HXK2 gene encoding hexokinase reduces the availability of glucose for glycolysis and extends the RLS [61]. In another model of CR, the deletion of TOR1, which encodes a phosphatidylinositol kinase (PIK)-related kinase and a subunit of the TORC1 complex, drives cells into a nutrient-starvation mode, extending their RLS [29], [62]. Both hxk2Δ and tor1Δ single deletion cells significantly had long lifespans (Figs. 5B, C and S1). However, either the RLSs of hxk2Δ tdh2Δ or tor1Δ tdh2Δ deletion cell were almost same as that of tdh2Δ cell (Figs. 5B, C and S1). Thus, tdh2 gene deletion can extend the RLS in a CR-dependent manner. These suggest that CR extends RLS by suppressing age-enhanced gluconeogenesis.
Figure 5. TDH2 gene deletion extends the RLS in a manner similar to that of the CR pathway.
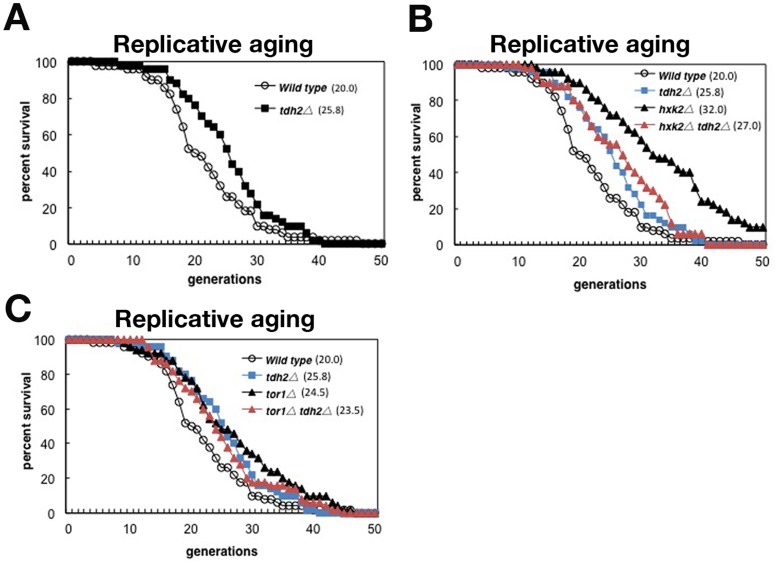
(A) The RLSs of wild type and tdh2Δ cells. (B) The RLSs of wild type, tdh2Δ, hxk2Δ and hxk2Δ tdh2Δ cells. (C) The RLSs of wild type, tdh2Δ, tor1Δ and tor1Δ tdh2Δ cells. The difference between strains was performed on statistical calculations (Fig. S1).
Discussion
Our study suggests that the metabolites in glucose metabolism accumulated in hst3Δ hst4Δ cells are due to the enhanced gluconeogenesis. However, hst3Δ hst4Δ tdh2Δ cells have still accumulated several metabolites in glucose metabolism (Fig. 3E), even though deletion of TDH2 gene repressed the activation of gluconeogenesis. To fix DNA damages occurred in the progression of DNA replication forks which are caused by persistent acetylation of histone H3 in K56 in chromatin, hst3Δ hst4Δ cells grow slowly [24]. Therefore, hst3Δ hst4Δ cell results in expansion of the cell volume by accumulating various metabolites, which are common to cells mutated in chromosomal DNA replication and cell cycle progression (data not shown). Accumulation of several metabolites in hst3Δ hst4Δ tdh2Δ cells may be derived from a kind of side effect that hst3Δ hst4Δ tdh2Δ cells has expanded to the same cell volume of hst3Δ hst4Δ cells (data not shown).
Why the gluconeogenesis pathway is activated in hst3Δ hst4Δ cells is still unclear. Many metabolic enzymes in glucose metabolism, the TCA cycle, the urea cycle, and fatty acid metabolism are acetylated in human liver tissue [63]. In Salmonella, metabolic enzymes in glucose metabolism are acetylated extensively and differentially in response to different carbon sources, and the relative activities of key enzymes controlling the balance between glycolysis and gluconeogenesis are particularly regulated by acetylation [42]. Persistent acetylation of metabolic enzymes that should be deacetylated by Hst3 and Hst4 may lead to a defect in the control of the flux between glycolysis and gluconeogenesis. Target proteins of Hst3 and Hst4 that control glucose metabolism and chronological lifespan should be identified in future studies.
In this study, we isolated Idp1 and Yat1 together with Tdh2, which restored the CLS and poor growth of hst3Δ hst4Δ cells when deleted. It is unclear if either Idp1 or Yat1 can suppress augmented gluconeogenesis in the same manner as Tdh2, although a less direct mechanism may be possible. For example, the deletion of either the Idp1 or Yat1 gene has been proposed to reduce the amount of metabolites to be supplied in glycogen synthesis via gluconeogenesis, which would effectively lead to a reduction in gluconeogenesis. The roles of Idp1 and Yat1 in gluconeogenesis remain to be elucidated.
We also demonstrated that the tdh2 gene deletion was able to restore the RLS of hst3Δ hst4Δ cells on statistics, but the effect was marginal (Figs. S1 and S6). Because the short RLS of the hst3Δ hst4Δ cells is due to the genomic instability [36], the reduction in gluconeogenesis mediated by the tdh2 gene deletion may not be sufficient to restore the shortened RLS caused by the genomic instability.
It will be interesting to extend our findings to other species, particularly multicellular organisms. In C. elegans, a null allele of Clk-1, which encodes a protein with homology to an activator of gluconeogenesis in budding yeast (Cat5p), leads to lifespan extension [64]. This finding is consistent with the correlation between reduced gluconeogenesis and increased lifespan observed in yeast ([11] and our study). CR extends lifespan in many species and has been shown to ameliorate many age-associated disorders, such as diabetes and cancer [17]. Testing whether the inactivation of a metabolic enzyme suppresses augmented gluconeogenesis instead of CR would be valuable, and the results could be applied to extend lifespan and ameliorate age-associated disorders.
Supporting Information
P-value matrices for each figure (Fig. 1A, 5 and S6). Each matrix contains the Wilcoxon Rank-Sum p-value for a 2-tailed test in which the lifespan data for the strain in the corresponding column. Significant p-value (p<0.05) is colored yellow. P-values were calculated using ystat2008 software.
(TIFF)
ROS accumulate specifically in hst3Δ hst4Δ cells. The number of DHE-positive cells was counted by fluorescence microscopy, and the distribution of DHE-positive cells was calculated (n = 100).
(TIF)
hst3Δ hst4Δ cells reduce CLS in medium “buffered”. Wild type and hst3Δ hst4Δ cells cultured in SC medium in the presence “buffered” of 50 mM citrate-phosphate buffer [pH6.0] and 2% glucose. Viability (cultured for 4 days) was measured as colony formation units and calculated as the ratio for the start (0 day). Data represent averages and standard deviations of more than four biological replicas.
(TIFF)
The entire metabolic profiling data set, including central carbon metabolism, for aged cells relative to young cells was mapped onto the network. Each graph was derived from the calculated amount of metabolic intermediates listed in Table S1. The blue and red columns indicate young and aged cells, respectively.
(TIFF)
Map of the central carbon metabolic pathway in budding yeast, based on the information from the Saccharomyces genome database (http://www.yeastgenome.org/). The gene(s) involved in each metabolic reaction are shown in red.
(TIF)
The deletion of the TDH2 gene results in the inability of hst3Δ hst4Δ cells to extend their RLS. The RLSs of wild type, tdh2Δ, hst3Δ hst4Δ and hst3Δ hst4Δ tdh2Δ cells. The median lifespan is given next to each genotype. The difference between strains was performed on statistical calculations (Figure S1).
(TIFF)
The deletion of either the IDP1 or YAT1 gene can restore the growth of hst3Δ hst4Δ cells. (A) The growth curve of wild type, hst3Δ hst4Δ, idp1Δ, and hst3Δ hst4Δ idp1Δ cells in SD medium. (B) The growth curve of wild type, hst3Δ hst4Δ, yat1Δ, and hst3Δ hst4Δ yat1Δ cells in SD medium.
(TIF)
The entire metabolic profiling data set, including central carbon metabolism, for hst3Δ hst4Δ cells relative to wild type cells was mapped onto the network. Each graph was derived from the calculated amount of metabolic intermediates listed in Table 3. Blue column: wild type; red: hst3Δ hst4Δ; green: tdh2Δ; orange: tdh2Δ hst3Δ hst4Δ.
(TIFF)
FBP1 expression is induced in both wild type and tdh2Δ cells in response to ethanol (EtOH). RNA was prepared from cells treated with either glucose or EtOH as a carbon source. Reverse transcription polymerase chain reaction (RT-PCR) was employed to measure the ratio of FBP1 mRNA to ACT1 mRNA. All graphs represent the mean of triplicate experiments. The error bars denote the standard deviation.
(TIFF)
(DOCX)
The metabolic intermediates in aged and young cells. The signal peaks of the metabolic intermediates were converted and normalized to the resultant relative area values, including glucose metabolism intermediates, TCA cycle intermediates, and amino acids. The ratio of the resultant relative area values for aged and young cells is also given.
(XLSX)
The metabolic intermediates in wild type and hst3Δ hst4Δ cells. The calculated amounts of the metabolic intermediates of central carbon metabolism are given. The ratio of the resultant relative area values for wild type and hst3Δ hst4Δ cells is also given.
(XLSX)
The metabolic intermediates among wild type, tdh2Δ, hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ cells. The signal peaks of the metabolic intermediates were converted and normalized to the resultant relative area values, including glucose metabolism intermediates, TCA cycle intermediates, and amino acids. The ratio of the resultant relative area values for aged and young cells is also given.
(XLSX)
The pH of media of wild type, hst3Δ hst4Δ, tdh2Δ, yat1Δ, idp1Δ, hst3Δ hst4Δ tdh2Δ, hst3Δ hst4Δ yat1Δ and hst3Δ hst4Δ idp1Δ. Cells were cultured in SD medium in the absence “unbuffered” of 50 mM citrate-phosphate buffer. The pH of fresh medium is set as pH6.0. Cells were cultured at 30°C for 8 days, and pH of media was measured.
(TIFF)
Acknowledgments
We thank Professors J. Yanagisawa, K. Kimura, A. Murayama, S. Yoshida, T. Usui and K. Irie and members of the Yanagisawa lab for reagents and advice. We also thank Profs. H. Araki and A. Gunjan for their critical reading of this manuscript.
Funding Statement
This work was supported by Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government and Japan Science and Technology (JST) (Grant number: 23131502). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kim S, Benguria A, Lai CY, Jazwinski SM (1999) Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell 10: 3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jazwinski SM (2000) Aging and longevity genes. Acta Biochim Pol 47: 269–279. [PubMed] [Google Scholar]
- 3. Kenyon C (2001) A conserved regulatory system for aging. Cell 105: 165–168. [DOI] [PubMed] [Google Scholar]
- 4.Bitterman KJ, Medvedik O, Sinclair DA (2003) Longevity regulation in Saccharomyces cerevisiae: linking metabolism, genome stability, and heterochromatin. Microbiol Mol Biol Rev 67: 376–399, table of contents. [DOI] [PMC free article] [PubMed]
- 5. Easlon E, Tsang F, Dilova I, Wang C, Lu SP, et al. (2007) The dihydrolipoamide acetyltransferase is a novel metabolic longevity factor and is required for calorie restriction-mediated life span extension. J Biol Chem 282: 6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fontana L, Partridge L, Longo VD (2010) Extending healthy life span–from yeast to humans. Science 328: 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weindruch W. and Walford RL (1998) The retardation of aging and diseases by dietary restriction, Charles C. Thomas.
- 8. Roth GS, Ingram DK, Lane MA (2001) Caloric restriction in primates and relevance to humans. Ann N Y Acad Sci 928: 305–315. [DOI] [PubMed] [Google Scholar]
- 9.Weindruch W. and Walford RL (1998) The retardation of aging and diseases by dietary restriction. Charles C, Thomas.
- 10. Bordone L, Guarente L (2005) Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 6: 298–305. [DOI] [PubMed] [Google Scholar]
- 11. Lin SS, Manchester JK, Gordon JI (2001) Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem 276: 36000–36007. [DOI] [PubMed] [Google Scholar]
- 12. Ivy JM, Klar AJ, Hicks JB (1986) Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol 6: 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rine J, Herskowitz I (1987) Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sauve AA, Wolberger C, Schramm VL, Boeke JD (2006) The biochemistry of sirtuins. Annu Rev Biochem 75: 435–465. [DOI] [PubMed] [Google Scholar]
- 15. Guarente L (2006) Sirtuins as potential targets for metabolic syndrome. Nature 444: 868–874. [DOI] [PubMed] [Google Scholar]
- 16. Lin SJ, Defossez PA, Guarente L (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289: 2126–2128. [DOI] [PubMed] [Google Scholar]
- 17. Lu SP, Lin SJ (2010) Regulation of yeast sirtuins by NAD(+) metabolism and calorie restriction. Biochim Biophys Acta 1804: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dilova I, Easlon E, Lin SJ (2007) Calorie restriction and the nutrient sensing signaling pathways. Cell Mol Life Sci 64: 752–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murayama A, Ohmori K, Fujimura A, Minami H, Yasuzawa-Tanaka K, et al. (2008) Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133: 627–639. [DOI] [PubMed] [Google Scholar]
- 20. Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, et al. (1995) The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9: 2888–2902. [DOI] [PubMed] [Google Scholar]
- 21. Halme A, Bumgarner S, Styles C, Fink GR (2004) Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell 116: 405–415. [DOI] [PubMed] [Google Scholar]
- 22. Perrod S, Cockell MM, Laroche T, Renauld H, Ducrest AL, et al. (2001) A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J 20: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, et al. (2005) HST2 mediates SIR2-independent life-span extension by calorie restriction. Science 309: 1861–1864. [DOI] [PubMed] [Google Scholar]
- 24. Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, et al. (2006) The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 16: 1280–1289. [DOI] [PubMed] [Google Scholar]
- 25. Werner-Washburne M, Braun E, Johnston GC, Singer RA (1993) Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol Rev 57: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun J, Kale SP, Childress AM, Pinswasdi C, Jazwinski SM (1994) Divergent roles of RAS1 and RAS2 in yeast longevity. J Biol Chem 269: 18638–18645. [PubMed] [Google Scholar]
- 27. Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD (2001) Regulation of longevity and stress resistance by Sch9 in yeast. Science 292: 288–290. [DOI] [PubMed] [Google Scholar]
- 28. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, et al. (2002) Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418: 344–348. [DOI] [PubMed] [Google Scholar]
- 29. Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, et al (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310: 1193–1196. [DOI] [PubMed] [Google Scholar]
- 30. Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S (2006) Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev 20: 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sinclair DA, Guarente L (1997) Extrachromosomal rDNA circles–a cause of aging in yeast. Cell 91: 1033–1042. [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M (2004) SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117: 441–453. [DOI] [PubMed] [Google Scholar]
- 33. Erjavec N, Nystrom T (2007) Sir2p-dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 104: 10877–10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsuchiya M, Dang N, Kerr EO, Hu D, Steffen KK, et al. (2006) Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell 5: 505–514. [DOI] [PubMed] [Google Scholar]
- 35. Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, et al. (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hachinohe M, Hanaoka F, Masumoto H (2011) Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae. Genes Cells 16: 467–477. [DOI] [PubMed] [Google Scholar]
- 37. Smith DL Jr, McClure JM, Matecic M, Smith JS (2007) Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell 6: 649–662. [DOI] [PubMed] [Google Scholar]
- 38. Kennedy BK, Austriaco NR Jr, Guarente L (1994) Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol 127: 1985–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amberg DCB, D. J Strathern, J N. (2005) Methods in Yeast Genetics 2005 Edition.
- 40. Ritchie KB, Mallory JC, Petes TD (1999) Interactions of TLC1 (which encodes the RNA subunit of telomerase), TEL1, and MEC1 in regulating telomere length in the yeast Saccharomyces cerevisiae. Mol Cell Biol 19: 6065–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cupp JR, McAlister-Henn L (1992) Cloning and characterization of the gene encoding the IDH1 subunit of NAD(+)-dependent isocitrate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem 267: 16417–16423. [PubMed] [Google Scholar]
- 42. Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, et al. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327: 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gunja-Smith Z, Patil NB, Smith EE (1977) Two pools of glycogen in Saccharomyces. J Bacteriol 130: 818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klinger H, Rinnerthaler M, Lam YT, Laun P, Heeren G, et al. (2010) Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells. Exp Gerontol 45: 533–542. [DOI] [PubMed] [Google Scholar]
- 45. Burhans WC, Weinberger M (2009) Acetic acid effects on aging in budding yeast: are they relevant to aging in higher eukaryotes? Cell Cycle 8: 2300–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, et al. (2007) Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315: 653–655. [DOI] [PubMed] [Google Scholar]
- 47. Driscoll R, Hudson A, Jackson SP (2007) Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315: 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A (2006) Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem 281: 37270–37274. [DOI] [PubMed] [Google Scholar]
- 49. Han J, Zhou H, Li Z, Xu RM, Zhang Z (2007) Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem 282: 28587–28596. [DOI] [PubMed] [Google Scholar]
- 50. Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, et al. (2006) Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci U S A 103: 6988–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ohashi Y, Hirayama A, Ishikawa T, Nakamura S, Shimizu K, et al. (2008) Depiction of metabolome changes in histidine-starved Escherichia coli by CE-TOFMS. Mol Biosyst 4: 135–147. [DOI] [PubMed] [Google Scholar]
- 52. Smeal T, Claus J, Kennedy B, Cole F, Guarente L (1996) Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84: 633–642. [DOI] [PubMed] [Google Scholar]
- 53. McAlister L, Holland MJ (1985) Differential expression of the three yeast glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem 260: 15019–15027. [PubMed] [Google Scholar]
- 54. Haselbeck RJ, McAlister-Henn L (1993) Function and expression of yeast mitochondrial NAD- and NADP-specific isocitrate dehydrogenases. J Biol Chem 268: 12116–12122. [PubMed] [Google Scholar]
- 55. Schmalix W, Bandlow W (1993) The ethanol-inducible YAT1 gene from yeast encodes a presumptive mitochondrial outer carnitine acetyltransferase. J Biol Chem 268: 27428–27439. [PubMed] [Google Scholar]
- 56. Ronne H (1995) Glucose repression in fungi. Trends Genet 11: 12–17. [DOI] [PubMed] [Google Scholar]
- 57. Lillie SH, Pringle JR (1980) Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bacteriol 143: 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilson WA, Roach PJ (2002) Nutrient-regulated protein kinases in budding yeast. Cell 111: 155–158. [DOI] [PubMed] [Google Scholar]
- 59. McAlister L, Holland MJ (1985) Isolation and characterization of yeast strains carrying mutations in the glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem 260: 15013–15018. [PubMed] [Google Scholar]
- 60. Klein CJ, Olsson L, Nielsen J (1998) Glucose control in Saccharomyces cerevisiae: the role of Mig1 in metabolic functions. Microbiology 144 (Pt 1): 13–24. [DOI] [PubMed] [Google Scholar]
- 61. Kaeberlein M, Kirkland KT, Fields S, Kennedy BK (2004) Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol 2: E296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martin DE, Hall MN (2005) The expanding TOR signaling network. Curr Opin Cell Biol 17: 158–166. [DOI] [PubMed] [Google Scholar]
- 63. Zhao S, Xu W, Jiang W, Yu W, Lin Y, et al. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, et al. (1997) Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275: 980–983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P-value matrices for each figure (Fig. 1A, 5 and S6). Each matrix contains the Wilcoxon Rank-Sum p-value for a 2-tailed test in which the lifespan data for the strain in the corresponding column. Significant p-value (p<0.05) is colored yellow. P-values were calculated using ystat2008 software.
(TIFF)
ROS accumulate specifically in hst3Δ hst4Δ cells. The number of DHE-positive cells was counted by fluorescence microscopy, and the distribution of DHE-positive cells was calculated (n = 100).
(TIF)
hst3Δ hst4Δ cells reduce CLS in medium “buffered”. Wild type and hst3Δ hst4Δ cells cultured in SC medium in the presence “buffered” of 50 mM citrate-phosphate buffer [pH6.0] and 2% glucose. Viability (cultured for 4 days) was measured as colony formation units and calculated as the ratio for the start (0 day). Data represent averages and standard deviations of more than four biological replicas.
(TIFF)
The entire metabolic profiling data set, including central carbon metabolism, for aged cells relative to young cells was mapped onto the network. Each graph was derived from the calculated amount of metabolic intermediates listed in Table S1. The blue and red columns indicate young and aged cells, respectively.
(TIFF)
Map of the central carbon metabolic pathway in budding yeast, based on the information from the Saccharomyces genome database (http://www.yeastgenome.org/). The gene(s) involved in each metabolic reaction are shown in red.
(TIF)
The deletion of the TDH2 gene results in the inability of hst3Δ hst4Δ cells to extend their RLS. The RLSs of wild type, tdh2Δ, hst3Δ hst4Δ and hst3Δ hst4Δ tdh2Δ cells. The median lifespan is given next to each genotype. The difference between strains was performed on statistical calculations (Figure S1).
(TIFF)
The deletion of either the IDP1 or YAT1 gene can restore the growth of hst3Δ hst4Δ cells. (A) The growth curve of wild type, hst3Δ hst4Δ, idp1Δ, and hst3Δ hst4Δ idp1Δ cells in SD medium. (B) The growth curve of wild type, hst3Δ hst4Δ, yat1Δ, and hst3Δ hst4Δ yat1Δ cells in SD medium.
(TIF)
The entire metabolic profiling data set, including central carbon metabolism, for hst3Δ hst4Δ cells relative to wild type cells was mapped onto the network. Each graph was derived from the calculated amount of metabolic intermediates listed in Table 3. Blue column: wild type; red: hst3Δ hst4Δ; green: tdh2Δ; orange: tdh2Δ hst3Δ hst4Δ.
(TIFF)
FBP1 expression is induced in both wild type and tdh2Δ cells in response to ethanol (EtOH). RNA was prepared from cells treated with either glucose or EtOH as a carbon source. Reverse transcription polymerase chain reaction (RT-PCR) was employed to measure the ratio of FBP1 mRNA to ACT1 mRNA. All graphs represent the mean of triplicate experiments. The error bars denote the standard deviation.
(TIFF)
(DOCX)
The metabolic intermediates in aged and young cells. The signal peaks of the metabolic intermediates were converted and normalized to the resultant relative area values, including glucose metabolism intermediates, TCA cycle intermediates, and amino acids. The ratio of the resultant relative area values for aged and young cells is also given.
(XLSX)
The metabolic intermediates in wild type and hst3Δ hst4Δ cells. The calculated amounts of the metabolic intermediates of central carbon metabolism are given. The ratio of the resultant relative area values for wild type and hst3Δ hst4Δ cells is also given.
(XLSX)
The metabolic intermediates among wild type, tdh2Δ, hst3Δ hst4Δ and tdh2Δ hst3Δ hst4Δ cells. The signal peaks of the metabolic intermediates were converted and normalized to the resultant relative area values, including glucose metabolism intermediates, TCA cycle intermediates, and amino acids. The ratio of the resultant relative area values for aged and young cells is also given.
(XLSX)
The pH of media of wild type, hst3Δ hst4Δ, tdh2Δ, yat1Δ, idp1Δ, hst3Δ hst4Δ tdh2Δ, hst3Δ hst4Δ yat1Δ and hst3Δ hst4Δ idp1Δ. Cells were cultured in SD medium in the absence “unbuffered” of 50 mM citrate-phosphate buffer. The pH of fresh medium is set as pH6.0. Cells were cultured at 30°C for 8 days, and pH of media was measured.
(TIFF)