

Abstract
Background
Fetal Alcohol Spectrum Disorders (FASD) are a collection of disorders resulting from fetal ethanol exposure, which causes a wide range of physical, neurological and behavioral deficits including heightened susceptibility for alcoholism and addictive disorders. While a number of mechanisms have been proposed for how ethanol exposure disrupts brain development, with selective groups of neurons undergoing reduced proliferation, dysfunction and death, the induction of a new neurotransmitter phenotype by ethanol exposure has not yet been reported.
Principal Findings
The effects of embryonic and larval ethanol exposure on brain development were visually monitored using transgenic zebrafish expressing cell-specific green fluorescent protein (GFP) marker genes. Specific subsets of GFP-expressing neurons were highly sensitive to ethanol exposure, but only during defined developmental windows. In the med12 mutant, which affects the Mediator co-activator complex component Med12, exposure to lower concentrations of ethanol was sufficient to reduce GFP expression in transgenic embryos. In transgenic embryos and larva containing GFP driven by an oxytocin-like (oxtl) promoter, ethanol exposure dramatically up-regulated GFP expression in a small group of hindbrain neurons, while having no effect on expression in the neuroendocrine preoptic area.
Conclusions
Alcohol exposure during limited embryonic periods impedes the development of specific, identifiable groups of neurons, and the med12 mutation sensitizes these neurons to the deleterious effects of ethanol. In contrast, ethanol exposure induces oxtl expression in the hindbrain, a finding with profound implications for understanding alcoholism and other addictive disorders.
Introduction
Fetal Alcohol Syndrome (FAS) is the most widely recognized consequence of prenatal alcohol exposure and is a leading cause of mental retardation [1]. FAS is characterized by three developmental anomalies: 1) prenatal and/or postnatal growth retardation; 2) a distinct facial appearance; and 3) central nervous system (CNS) dysfunction [2]. However, FAS is not the only result of prenatal alcohol exposure. Prenatal ethanol exposure results in a continuum of developmental anomalies leading to a spectrum of physical, mental and behavioral deficits, often with lifelong implications. The term Fetal Alcohol Spectrum Disorder (FASD) is used to describe these disorders. The total prevalence of FASD in the United States has been estimated to be as high as 2–5% [3]. In addition, low levels of fetal alcohol exposure may result in subtle neurodevelopmental consequences that are not necessarily identified as FASD, yet result in susceptibility to a range of neuropsychiatric and behavioral disorders including attention deficit hyperactivity disorder, depression, psychosis, drug addiction and alcoholism [2], [4]–[9].
An individual’s increased susceptibility to these neuropsychological and behavioral disorders is perhaps due in part to fetal ethanol exposure and to the individual’s genetic background. Genetics plays a significant role in an individual’s sensitivity to ethanol, and fetal exposure may exacerbate neurodevelopmental deficits resulting from certain genetic disorders [10], [11]. Genetic disorders associated with mutations in the MED12 gene provide an interesting example. MED12, also known as TRAP230, is a component of Mediator, an evolutionarily conserved multi-protein complex that bridges gene regulatory regions with RNA polymerase, affecting the regulation of a large number of genes [12]. One potential example of fetal ethanol exposure influencing the prognosis of a genetic disorder is Opitz-Kaveggia syndrome, an X-linked mental retardation disorder resulting from mutations in MED12 [13]–[15]. Fetal alcohol exposure could aggravate the neuropsychiatric disorders associated with this syndrome, although the mechanisms involved are not currently understood.
Several hypotheses have been advanced for mechanisms by which fetal ethanol exposure leads to the reduced proliferation, neuronal dysfunction and cell death suggested to be causes of the clinically observed behavioral phenotype. These include: 1) depriving neurons of activity-dependent trophic factors by antagonizing NMDA receptors and potentiating GABAA receptors, thus, triggering apoptosis; 2) disruption of midline serotonergic neurons; 3) interference with L1 CAM function; 4) oxidative stress and free radical damage; 5) disruption of growth factor signaling (e.g., IGF); and 6) interference with neuronal metabolic enzymes [16]–[26]. The deleterious effect of ethanol on fetal brain development may proceed by any one or more of these mechanisms, or others yet to be discovered. In all scenarios, fetal alcohol exposure leads to reduced proliferation, neuronal dysfunction and/or cell death, with some groups of neurons being more sensitive to ethanol than others. The spectrum of mental and behavioral abnormalities associated with FASD is thought to arise as a result of this differential sensitivity, which could produce a pathogenic or suggestive phenotype for diagnosis [20].
Recently, improvements in imaging technologies have enabled investigators and clinicians to visualize structural changes in the brain, such as agenesis of the corpus callosum, thought to arise from high levels of fetal alcohol exposure [27], [28]. While these new technologies are exciting, higher resolution is required to answer critical questions regarding mechanisms and consequences of differential cellular responses to ethanol.
We are particularly interested in the potential effects of fetal alcohol exposure on the development of oxytocin expressing cells of the neuroendocrine hypothalamus, and the impact of oxytocin system dysfunction on the susceptibility to neurobehavioral disorders. This interest arises from the fact that the oxytocin system is known to mediate a wide range of social interactions, many of which are perturbed in individuals with FASD [2], [8], [29]–[33]. However, a direct link between the oxytocin system and FASD has not been established. Zebrafish are being utilized for these experiments because unlike other vertebrate models their anatomy and development allows for direct visualization of alterations in neuronal development in an organism that can be subsequently tested for resulting changes in behavior. The zebrafish oxytocin-like (oxtl) gene produces the neurohormone Isotocin, which is closely related to mammalian oxytocin [34]. Thus, we hypothesized that embryonic ethanol exposure would disrupt development of the oxtl system in zebrafish and lead to subsequent alterations in behavior.
In order to directly visualize the effect of ethanol exposure in intact developing brains at the cellular level, we utilized transgenic zebrafish that express green fluorescent protein (GFP) in specific subsets of neurons. Beyond their external fertilization and optical clarity, zebrafish present several additional experimental advantages for investigating perturbations of brain development. First, a large collection of transgenic lines that express fluorescent proteins in highly restricted neuronal populations has been generated by several laboratories [35]–[42]. Second, zebrafish are amenable to genetic analyses, and a number of mutant strains are readily available (http://zebrafish.org/zirc). Third, the larva and adults display a wide range of basal behaviors that are relevant for alcohol studies including memory, addiction and social behavior [43]–[47]. Finally, due to the small size of the zebrafish embryo, the entire nervous system can be observed as it develops.
In this study, four well-characterized transgenic lines were utilized to evaluate the hypothesis that specific subsets of neurons are sensitive to ethanol exposure. Our results demonstrate that zebrafish are well suited for visual detection of sensitive developmental periods of specific groups of ethanol-sensitive neurons. Accordingly, zebrafish provide an excellent model in which to comprehensively map ethanol dose, duration and stage of exposure to alterations in brain development at a cellular level. In addition, the effects of ethanol were evaluated in med12y82 embryos, which carry a mutation in the transcriptional Mediator component, Med12 [48]. The results of these experiments suggest a highly productive new avenue for investigating the interaction between genetic factors and ethanol exposure. Finally, in order to test the idea that ethanol perturbs the development of the oxytocinergic system, we generated a transgenic line, which expresses GFP from the zebrafish oxtl promoter. Contrary to our expectations, ethanol treatment did not disrupt oxytocinergic system development but instead induced oxtl expression in the posterior hindbrain.
Results
Transgenic Zebrafish Reveal Neuronal Populations that are Particularly Sensitive to Ethanol Treatment
Four transgenic zebrafish lines expressing GFP in specific subsets of neurons were used to directly visualize the effect of ethanol exposure in intact developing brains.
The goosecoid reporter line, Tg(−1.8gsc:GFP)ml1, expressed GFP primarily in two neuronal groups in the neuroendocrine preoptic area (POA). There were also several scattered ectopic cells expressing GFP in olfactory epithelia, epiphysis, tectum, and the eye [49]. The response of gsc:GFP-expressing neurons, particularly in the POA, was examined using a wide temporal range of ethanol exposures (Figure 1A). At least ten embryos were examined at multiple developmental stages from each experimental group. The initial experiments utilized a 20 hr exposure of 2% ethanol starting at 4 hours post fertilization (hpf) because this treatment is known to induce apoptosis without grossly perturbing brain morphology or patterning [50], [51]. However, gsc:GFP expression was unaffected by these early ethanol treatments.
Figure 1. Ethanol exposure reduces the number of gsc:GFP expressing neurons.
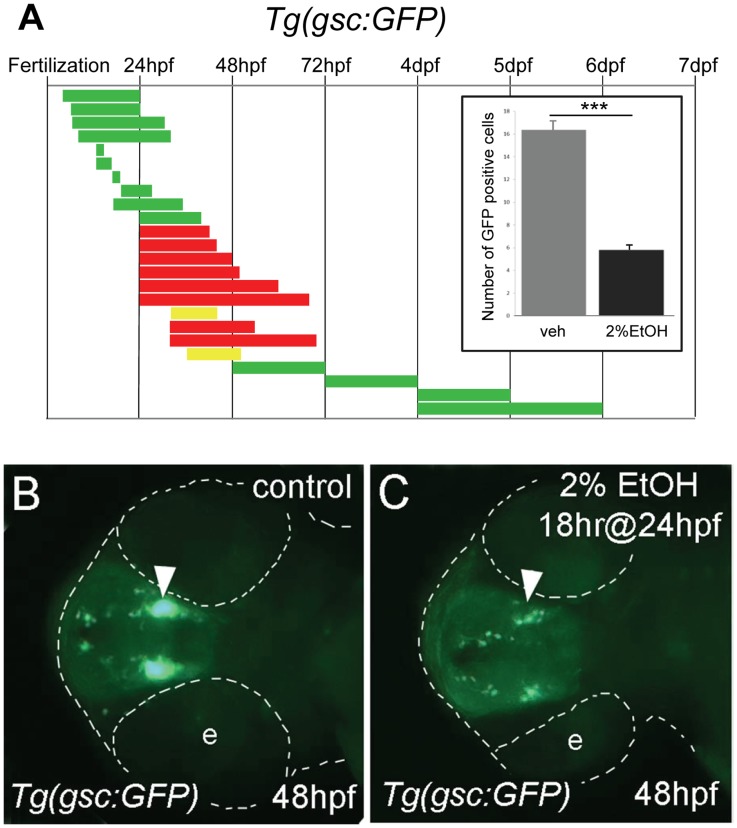
A, the duration and embryonic stage of 2% ethanol treatments in Tg(gsc:GFP) embryos are illustrated with solid bars. Colors indicate the treatment had no effect (green), slightly reduced (yellow), or reduced (red) GFP expression. Inset, the average numbers of GFP positive cells in the preoptic area (POA) of 72 hpf embryos treated with 2% ethanol for 24 hrs at 24 hpf (n = 12) and vehicle control (n = 10). Ethanol treatment resulted in a highly significant reduction of GFP-expressing cells in the POA, ***p<0.001. Error bars represent SEM. B, C, ventral views of Tg(gsc:GFP) embryos at 48 hpf. B, is the control. C, treated with 2% ethanol for 18 hrs at 24 hpf. GFP expression in the neuroendocrine preoptic area (arrowheads) was dramatically reduced in the embryos exposed to ethanol. The embryo and eyes (e) are outlined (dashed white line).
In contrast, ethanol exposure for 18 or 24 hrs starting at 24 hpf resulted in significantly reduced gsc:GFP expression in the neuroendocrine POA (Figure 1B–E). The possibility that ethanol interferes with the differentiation of gsc:GFP-expressing neurons was examined using a series of short, developmental time periods covering stages from 19.5 hpf (22 somite) to 25 hpf (prim 8). However, no reduction of gsc:GFP expression was observed when examined shortly after exposure or at any stage up to 7 days-post-fertilization (dpf) (Figure S1). Finally, a series of chronic exposures were investigated covering the first six days of embryonic and larval development. These studies demonstrated that gsc:GFP-expressing neurons are sensitive to ethanol exposure within a developmental window starting after 24 hpf and ending around 48 hpf. Ethanol treatment after 48 hpf had no effect on gsc:GFP expression (Figure S2). Based on our observation of extensive gsc:GFP positive tracts by 48 hpf, the developmental window of ethanol sensitivity of gsc:GFP-expressing cells is consistent with the predicted period of synaptogenesis of these neurons in the POA.
To determine the effect of ethanol exposure on other neurons, we used isl1:GFP and pax2a:GFP transgenic lines [52], [53]. Tg(isl1:GFP)rw0 embryos express GFP in select forebrain nuclei, a mesencephalic cluster, in cranial motor neurons III, IV, V, VII and X, in facial, glossopharyngeal and vagal sensory ganglia, and in Rohan-Beard primary sensory neurons and motor neurons of the spinal cord [52]. A series of chronic exposures covering the first six days of development was used to identify ethanol-sensitive periods (Figure 2A). Ethanol exposure for 24 hrs starting at 70% epiboly abolished isl1:GFP expression in the two forebrain nuclei and the mesencephalic cluster, and reduced expression in cranial motor nuclei III and IV (Figure 2B–F). In contrast, ethanol treatment for 24 hrs at 24 hpf only resulted in a slight and variable reduction of isl1:GFP expression in the forebrain nuclei, and treatment later than 48 hrs had no effect on isl1:GFP expression (Figure S3). This indicates that isl1:GFP-expressing forebrain neurons are sensitive to ethanol during an earlier developmental period than the gsc:GFP-expressing POA neurons.
Figure 2. Different groups of neurons are sensitive to ethanol exposure at specific developmental stages.
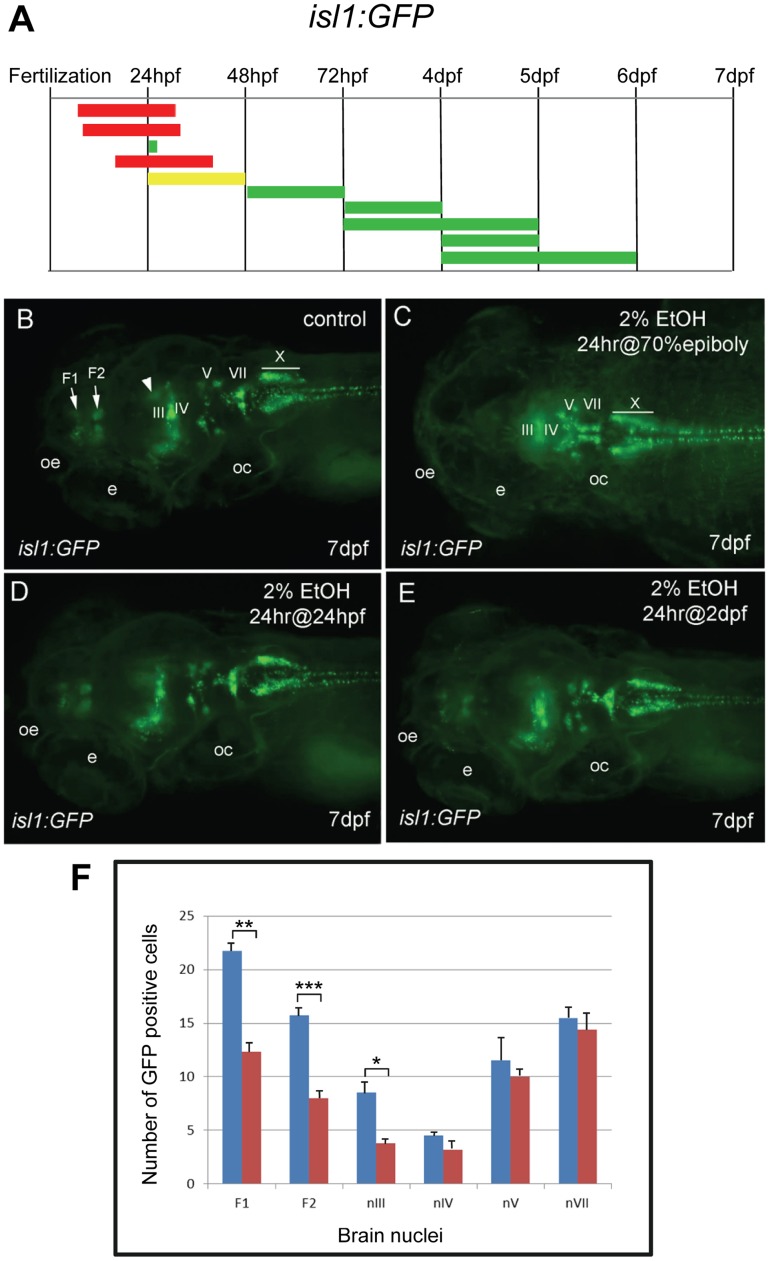
A, the duration and embryonic stage of 2% ethanol treatments illustrated with colored bars for Tg(isl1:GFP) embryos, as described in Fig. 1. B−E, dorsal views of Tg(isl:GFP) larva at 7 dpf. B, is the control. GFP expression was evident in the forebrain nuclei (F1 and F2 arrows), the mesencephalic cluster (arrowhead) and cranial motor nuclei III, IV, V, VII, and X (labeled). C, GFP expression in the forebrain and mesencephalic cluster was absent, and GFP expression in cranial motor nuclei III and IV was reduced (labeled) after 2% ethanol treatment for 24 hrs at the 70% epiboly stage. D, E, GFP expression was largely normal after treatment with 2% ethanol for 24 hrs at 24 hpf (D) and for 24 hrs at 48 hpf (E). Eyes (e), otic capsule (oc), olfactory epithelium (oe). F, the average numbers of GFP-positive cells in individual brain nuclei of control (n = 10) and ethanol-treated (n = 10) embryos. A highly significant reduction in GFP-expressing cells in the two forebrain nuclei, F1 (p = 0.001) and F2 (p<0.001), and cranial motor nucleus III (p<0.05) was observed. In contrast, a slight reduction in GFP cells in motor nuclei IV, V and VII was not significant. Error bars represent SEM. ***p<0.001, **p<0.01, *p<0.05.
Tg(pax2a:GFP)e1 (pGFP5.3) is expressed in the mid-hindbrain border, rhombomeres 3 and 5, one telencephalic area and three diencephalic areas, as well as in the VIIIth cranial nerve ganglion, the hindbrain and spinal cord interneurons [53]. Early ethanol exposure, from dome stage to 24 hpf, did not appear to affect pax2a:GFP expression, whereas treatment for 24 hrs starting at the 14 somite stage or at 24 hpf resulted in an apparent reduction of pax2a:GFP-expressing neurons in the hindbrain. In addition, a small group of pax2a:GFP neurons were eliminated in the forebrain (Figure 3A–E). Interestingly, isl1:GFP-expressing neurons in the hindbrain were unaffected by ethanol treatment.
Figure 3. Forebrain and hindbrain pax2a:GFP positive cells are sensitive to ethanol exposure.
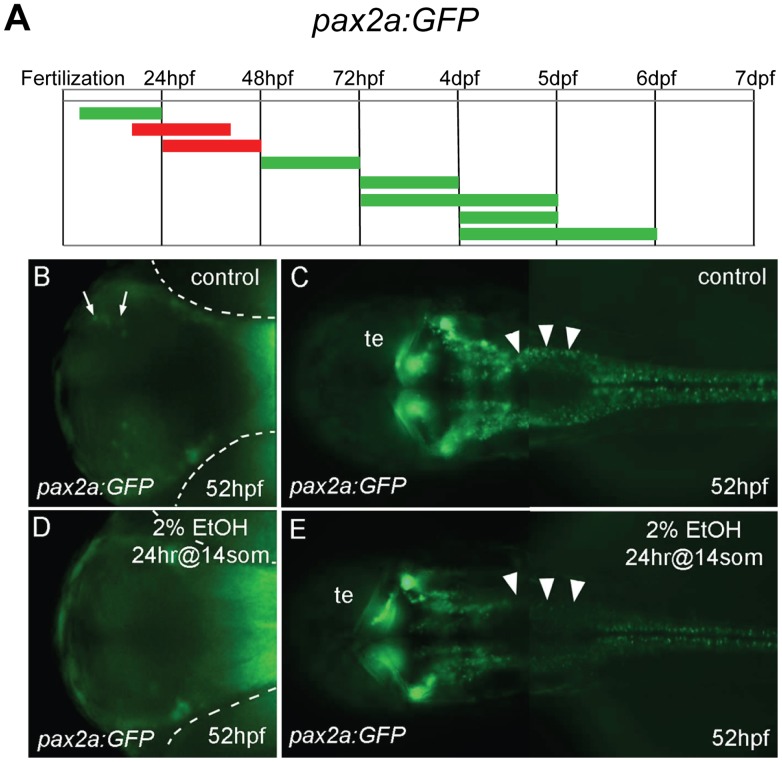
A, the duration and embryonic stage of 2% ethanol treatments illustrated with colored bars for Tg(pax2a:GFP) embryos, as described in Fig. 1. B−E, ventral views (B, D) and dorsal views (C, E) of Tg(pax2a:GFP) embryos at 52 hpf. B, C, are controls. D, E, were treated with 2% ethanol for 24 hrs at the 14 somite stage. B, D, a small, distinct group of neurons in the forebrain (arrows in B) were missing after ethanol exposure (D). The location of the eyes is outlined (dashed white lines). C, E, neurons around the midbrain-hindbrain region were mostly unaffected, hindbrain neurons were drastically reduced (arrowheads), and spinal cord neurons appeared slightly reduced. Tectum (te).
Tg(elavl3:EGFP)knu3 (also known as HuC:EGFP), which expresses GFP in most post-mitotic neurons, was used to observe the effect of ethanol treatments on overall neuronal development [54]. Tg(elavl3:EGFP)knu3 embryos were exposed to a range of ethanol concentrations, from 0.5% to 2.0%, for various durations at several developmental stages. In all cases, no general effects on post-mitotic neurons were observed, suggesting that these ethanol exposures are disrupting development of highly specific subsets of neurons (Figure S4, S5).
Neurons are Sensitized to Ethanol Exposure in med12 Mutant Embryos
As a first step toward understanding the influence of genetic factors on neuronal sensitivity to ethanol, we focused on the zebrafish mutant, med12y82. The med12y82 mutation is a point mutation in the mediator complex gene med12 that creates a premature stop codon, inactivating this protein [48]. Mutations in the human MED12 gene result in a variable mental retardation syndrome with behavioral disturbances [55], [56]. In zebrafish, med12y82/y82 embryos have several features, particularly brain anomalies that appear similar to embryos that have been treated with ethanol (Figure 4A–C). Therefore, we examined ethanol sensitivity in med12y82 mutants that had been crossed into the four transgenic GFP reporter lines.
Figure 4. Neurons are sensitized to ethanol in med12 mutant embryos. A−C.
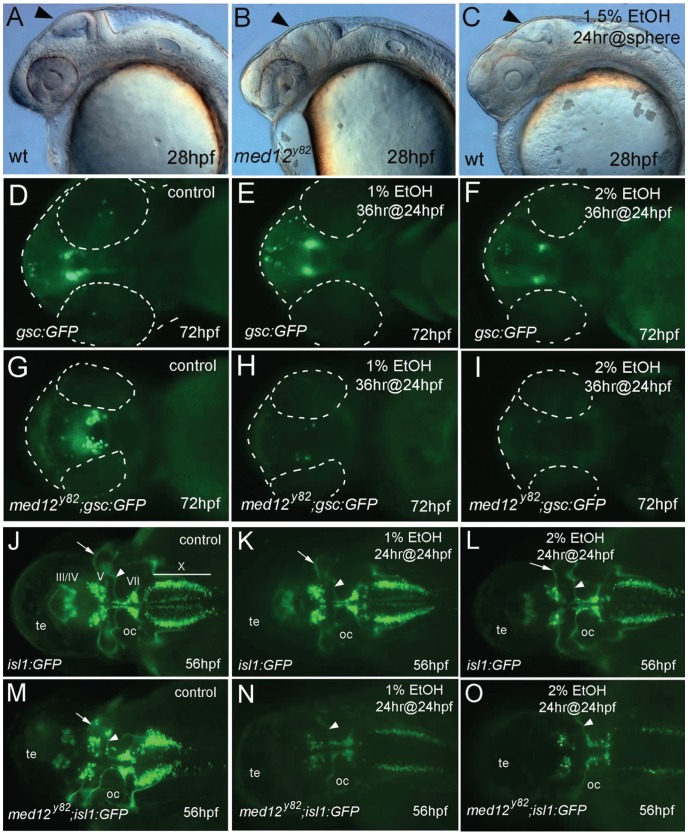
, 28 hpf embryos. A, wild-type (wt), B, med12y82 mutant, and C, wt embryo treated at sphere stage with 1.5% ethanol for 24 hrs. Note the similarly retarded brain development in the med12y82 and ethanol treated embryos (arrows). D−I, ventral views at 72 hpf of Tg(gsc:GFP) embryos (D, E, F) and med12y82; Tg(gsc:GFP) embryos (G, H, I). D, G, control embryos. E, H, treated with 1% ethanol for 36 hrs at 24 hpf. F, I, treated with 2% ethanol for 36 hrs at 24 hpf. Embryos and eyes are outlined (dashed white lines). J−O, Tg(isl1:GFP) embryos (J, K, L) and med12y82; Tg(isl1:GFP) embryos (M, N, O) at 56 hpf, dorsal views. J, M, control embryos. K, N, treated with 1% ethanol for 24 hrs at 24 hpf. L, O, treated with 2% ethanol for 24 hrs at 24 hpf. GFP-expressing cranial motor neurons (labeled) diminished in med12y82; Tg(isl1:GFP) embryos, especially in nuclei III and IV. A reduction of nucleus V neurons in med12y82; Tg(gsc:GFP) embryos treated with ethanol is illustrated by the elimination of the prominent nV axons (arrows). In contrast, the prominent nVII axons remained intact (arrowheads). Otic capsule (oc), tectum (te).
Homozygous med12y82/y82 mutations resulted in severe deficits in several neuronal populations. Ethanol treatment, for the most part, resulted in further neuronal defects in med12 mutant embryos. Interestingly, ethanol exposure at concentrations below those that effect normal embryos resulted in significant enhancement of deficits in med12y82/y82 embryos. In contrast, the ethanol response in heterozygous med12y82/+ embryos was indistinguishable from that in wild-type embryos. In med12y82/y82; Tg(elavl3:EGFP)knu3 embryos, overall numbers of elavl3:GFP-expressing post-mitotic neurons were reduced compared to their wild-type and heterozygous siblings. Ethanol treatment further reduced elavl3:GFP expression in med12y82/y82 embryos, but not in wild-type and heterozygous siblings. However, no obvious region-specific reduction in elavl3:GFP expression was observed in any of the groups (Figure S5A–F). The level of elavl3:GFP expression in 48 hpf embryos was further evaluated by real-time qRT-PCR. The expression of GFP mRNA was normalized to β-actin mRNA levels and relative expression was determined for 1) Tg(elavl3:EGFP) embryos treated with 2% ethanol for 24 hrs at 24 hpf, 2) Tg(elavl3:EGFP) control embryos, 3) med12y82/y82; Tg(elavl3:EGFP) embryos treated with 2% ethanol for 24 hrs at 24 hpf, and 4) med12y82/y82; Tg(elavl3:EGFP) control embryos. The relative expression of elavl3:GFP mRNA was not significantly different among the four groups. However, there appears to be a trend toward higher relative levels of elavl3:GFP mRNA, normalized to β-actin mRNA, in med12 mutant and ethanol-treated embryos (Figure S5G). These results suggest that there is not a specific loss of post-mitotic neurons due to the med12 mutation or to ethanol treatment.
Embryos from med12y82;Tg(−1.8gsc:GFP) incrosses were treated with 1% or 2% ethanol for 36 hrs at 24 hpf. The expression of GFP in the POA of Tg(−1.8gsc:GFP) embryos was reduced by exposure to 2% but unaffected by 1% ethanol. In contrast, exposure of med12y82/y82;Tg(−1.8gsc:GFP) embryos to 1% ethanol resulted in the elimination of gsc:GFP expression in the POA, while untreated med12y82/y82;Tg(−1.8gsc:GFP) embryos had variably reduced and disorganized gsc:GFP-expressing neurons (Figure 4D–I). These results demonstrate sensitization to ethanol exposure in embryos with a med12 mutant genetic background.
GFP expression in med12y82/y82;Tg(isl1:GFP) embryos was absent in the forebrain, highly reduced in cranial motorneuron nuclei III and IV, and displayed moderately reduced expression overall. After exposure to 1% ethanol, which had no effect on isl:GFP expression in wild-type or heterozygous embryos, isl:GFP expression in med12y82/y82 embryos was further reduced. When treated with 2% ethanol, isl:GFP expression in med12y82/y82 embryos was nearly absent (Figure 4J–O).
The early GFP expression in med12y82/y82;Tg(pax2a:GFP) embryos was similar to wild-type Tg(pax2a:GFP) embryos at 24 hpf (Figure S6). However, by 60 hpf, hindbrain and spinal cord neuronal expression was greatly reduced in med12y82/y82;Tg(pax2a:GFP) embryos; 2% ethanol treatment resulted in the near elimination of these neurons, while midbrain-hindbrain border and rhombomeric pattern expression remained fairly normal (Figure S7). These results indicate that while patterning is largely unaffected by med12 mutation or ethanol exposure, differentiating pax2a:GFP neurons are highly sensitive to these perturbations.
In order to investigate cell death as a mechanism by which ethanol potentiates the neuronal deficits of med12 mutants, wild-type and med12y82/y82 embryos were treated with 2% ethanol for 18 hours at 24 hpf, and then stained with acridine orange at 48 hpf to reveal apoptotic and necrotic cells. In addition, embryos were fixed and sectioned, and then stained for apoptotic cells using a TMR based TUNEL assay for quantitative analyses. Untreated wild-type and med12y82/y82 embryos showed relatively low-levels of acridine orange staining [48], [57]. In contrast, ethanol treatment induced high-levels of acridine orange staining in both wild-type and med12y82/y82 embryos. Quantitative analysis of TUNEL stained sections confirmed the acridine orange results. Untreated wild-type and med12y82/y82 embryos showed relatively low levels of apoptosis, however, med12y82/y82 embryos had relatively more apoptotic cells than their wild-type or heterozygous siblings (p<0.05). Nevertheless, it is unlikely that this modest increase in apoptosis could account for the increased sensitivity to ethanol observed in med12 mutant embryos considering the dramatic increase in apoptosis observed following ethanol exposure (Figure 5A–E). Ethanol exposure for 24 hrs starting at 24 hpf resulted in a 5.7 fold increase in apoptotic cells in wild-type embryos (p<0.005) and a 2.8 fold increase in med12 embryos (p = 0.073).
Figure 5. Ethanol exposure causes similar levels of neuronal cell death in wild-type and med12 mutant embryos.
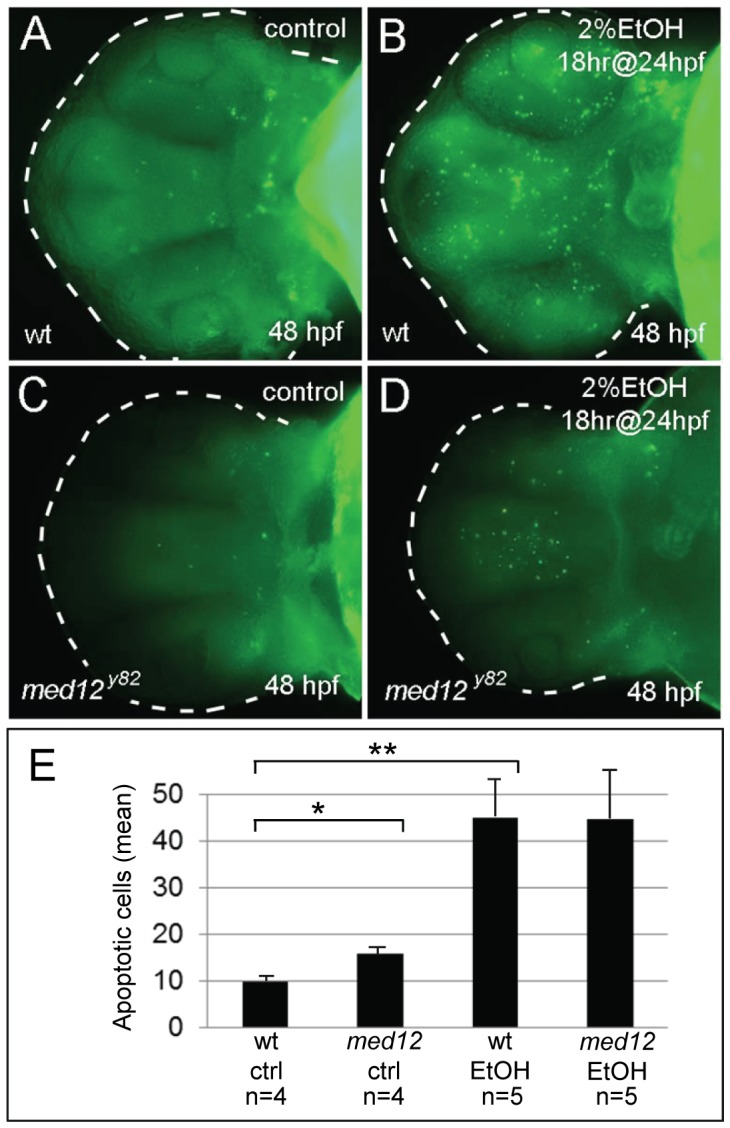
A−D, acridine orange stained embryos at 48 hpf, ventral views. A, untreated wild-type (wt) embryo. B, wt embryo treated with 2% ethanol for 18 hrs at 24 hpf. C, untreated med12y82 embryo. Med12y82 and wt embryos showed similar low levels of apoptosis. D, med12y82 embryo treated with 2% ethanol for 18 hrs at 24 hpf. Med12y82 and wt embryos treated with 2% ethanol showed similar increases in acridine orange staining. E, the average numbers of apoptotic cells in the brains of med12y82 and control embryos treated with ethanol as in panels A−D. There were significantly more apoptotic cells in the brains of med12y82 embryos compared to wild-type embryos (p<0.05), however, following EtOH exposure, the numbers of apoptotic cells in the brains of both med12y82 and wild-type embryos greatly increased. The number of apoptotic cells in the brains of med12 mutant embryos increased 2.8-fold compared to their untreated med12 mutant siblings (p = 0.073), while apoptotic cells increased over 5-fold in ethanol-treated wild-type embryos compared to their untreated siblings (p<0.005). Error bars represent SEM. **p<0.01, *p<0.05.
Transgenic oxtl:GFP Reporter Lines Reveal Ethanol Induction of Oxytocin in the Hindbrain
In order to test the hypothesis that the developing oxytocinergic system would be particularly sensitive to alcohol exposure, we generated transgenic lines that express GFP from the oxtl promoter. In Tg(oxtl:GFP) embryos, GFP expression corresponds exactly with the expression of endogenous oxtl mRNA as determined by ISH at 48 hpf (Figure 6). A more detailed neuroanatomical analysis of the oxytocinergic system based on the Tg(oxtl:GFP) lines will be published elsewhere, though here we highlight the more striking features of these fish. As expected, oxytocinergic cell bodies in the POA prominently express GFP with tracts to the pituitary. A surprising feature of these fish is the presence of extensive tracts and projections throughout the mid- and hindbrain, with tracts continuing down the spinal cord (Figure 7B). While “central” projection patterns are well-known features of the oxytocinergic system, our use of zebrafish allows us to finally visualize and appreciate the full extent of oxytocinergic projections in a living organism.
Figure 6. Expression of GFP in Tg(oxtl:GFP) embryos corresponds exactly with the expression of endogenous oxtl mRNA.
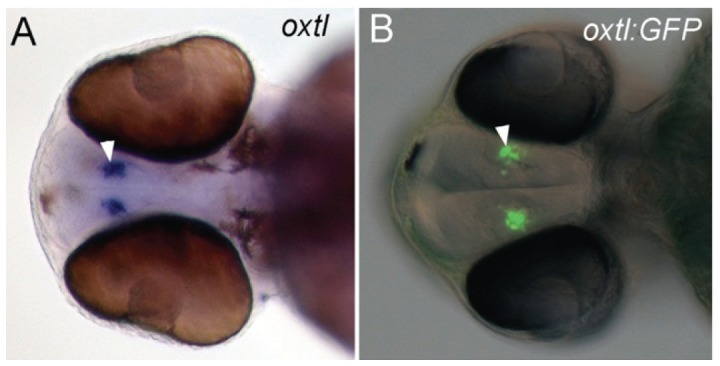
A,B, ventral view of 48 hpf embryos. A, whole-mount in situ hybridization for oxtl mRNA in a 48 hpf embryo. B, GFP expression visualized with epi-fluorescence in a 48 hpf Tg(oxtl:GFP) embryo. Epi-fluorescent and differential interference contrast (DIC) images are overlaid. The oxtl expressing cells are indicated with arrowheads.
Figure 7. Expression of the oxtl:GFP transgene is induced in the hindbrain by exposure to ethanol.
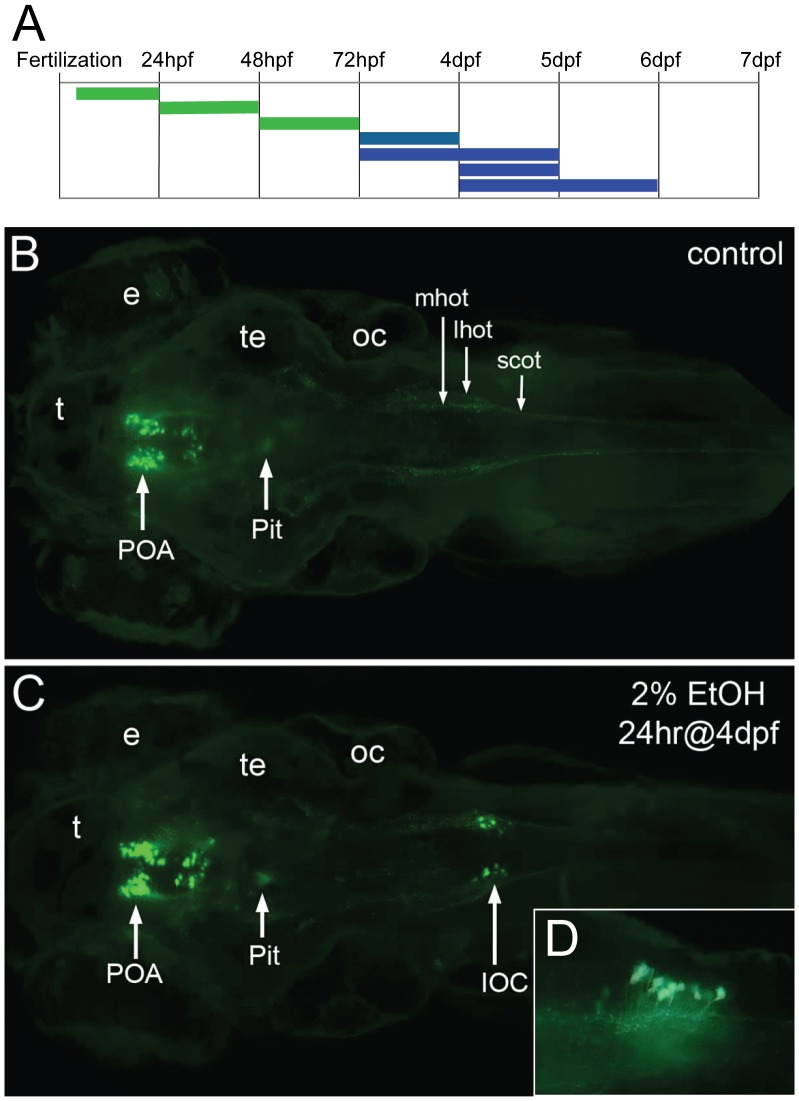
A, the duration and embryonic stage of 2% ethanol treatments are illustrated with solid bars. Oxtl:GFP expression in the neuroendocrine preoptic area was unaffected with all treatments (green), while oxtl:GFP was induced in the hindbrain by later treatments (blue). B, C, Tg(oxtl:GFP) larva shown at 6 dpf, dorsal views. B, control embryo, C, treated with 2% ethanol for 24 hrs at 4 dpf. A small group of large GFP-expressing neurons appears in the hindbrain of the ethanol-treated larva. We call these ethanol-induced GFP-expressing neurons, induced oxytocin cells (IOC). D, inset, a higher magnification view of the IOCs shows processes that appear to enter the hindbrain oxytocinergic tracks, lateral view. Eyes (e), lateral hindbrain oxytocinergic tract (lhot), medial hindbrain oxytocinergic tract (mhot), otic capsule (oc), pituitary (Pit), preoptic area (POA), spinal cord oxytocinergic tract (scot), tectum (te), telencephalon (t).
A series of ethanol exposures was used to investigate developmental windows of alcohol sensitivity in the oxytocinergic system (Figure 7A). Ethanol exposure in Tg(oxtl:GFP) embryos did not alter cell numbers or projection patterns nor projection density of GFP-expressing neurons in the POA. Surprisingly, however, a small group of large GFP-expressing neurons appeared in the posterior hindbrain of Tg(oxtl:GFP) larva treated with 2% ethanol for 24 hrs at 4 dpf (Figure 7B–D). We call these ethanol-induced oxtl:GFP-expressing neurons, Induced Oxtl Cells (IOC). IOCs appear in the posterior region of the hindbrain, dorsal and medial to where at least two hindbrain oxytocinergic tracks converge to form a single tract in the spinal cord (compare 7B and 7C). The position of the IOCs suggest they are in the area postrema, or are, perhaps, part of the posterior most nucleus of the solitary tract, and thus receive afferent visceral innervations. Processes appear to exit the IOCs and join the hindbrain oxytocinergic tracks (Figure 7D).
To confirm that these GFP-expressing IOC’s also express endogenous oxtl, in situ hybridization (ISH) was used to detect GFP, oxtl and tyrosine hydroxylase (th) mRNAs in Tg(oxtl:GFP) larva treated with ethanol for 24 hrs at 4 dpf, and then fixed at 6 dpf (Figure 8A–C). The presence of GFP and oxtl mRNA in the posterior hindbrain of ethanol-treated larva shows that induction of GFP expression coincides with the induction of endogenous oxtl mRNA. th expression in the posterior hindbrain marks the area postrema. The induced GFP/oxtl cells appear to be slightly inferior and medial to the th positive cells. These results demonstrate that oxtl is induced in the hindbrain by ethanol exposure.
Figure 8. Expression of oxtl mRNA is induced in the hindbrain by exposure to ethanol.
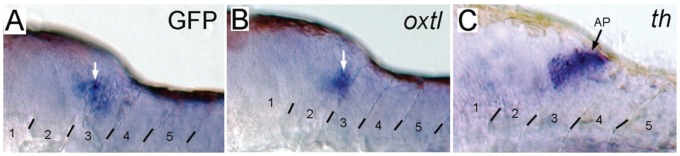
Whole mount in situ hybridization was used to detect: A, GFP; B, oxtl; and C, tyrosine hydroxylase (th) mRNAs in Tg(oxtl:GFP) larva treated with ethanol for 24 hrs at 4 dpf, and then fixed at 6 dpf. th labels dopaminergic and noradrenergic neurons located in the area postrema. Examples of positive staining are indicated by arrows.
Discussion
Alcohol Exposure Results in Complex Changes in Brain Development
It is likely that a number of mechanisms are responsible for the deleterious effects of alcohol. Although several potential mechanisms have been demonstrated in culture systems, the importance and relative contribution of the various mechanisms to lasting damage in the brain remains unclear [16], [19], [25]. Substantial efforts have been devoted to documenting changes in the brains of model organisms that have been exposed to ethanol and these studies have significantly contributed to our understanding of ethanol neurotoxicity [16]–[18], [20]–[24]. However, given the difficulty of controlling alcohol dose and timing and the requirement for static post-mortem analyses, these studies are unable to capture the full range of effects of ethanol concentration, duration of exposure, and embryonic stage of exposure on brain development. Furthermore, the relative importance of various mechanisms of ethanol neurotoxicity may change depending on cell type, dose, duration, and developmental stage of exposure.
By utilizing transgenic zebrafish to monitor brain development, we demonstrate that a wide range of exposures can be rapidly evaluated to identify sensitive subsets of neurons. Several treatment regimens have been used in zebrafish that result in morphologic abnormalities that closely model FAS [58]. Because we are interested in modeling the more subtle neurodevelopmental deficits of FASD, we used ethanol doses that are below those that generate FAS like phenotypes. An advantage of using zebrafish embryos and larva is that dosing is easy and accurate; therefore, it is possible to obtain sensitivity data for any given group of neurons over a wide parameter space consisting of ethanol dose, duration and developmental stage of exposure. Given the large and growing collection of enhancer-specific transgenic lines, it should be feasible to generate a comprehensive map of neuronal sensitivity to ethanol exposure [35]–[41]. Such a map would greatly advance our understanding of the complex effects that alcohol exposure has on brain development. Furthermore, the ability to perform forward genetic screens in zebrafish and the large collection of mutant lines already available will enable investigation of the interaction of ethanol exposure with underlying genetic mutations.
Neurons are Sensitized to Ethanol in med12 Mutant Embryos
Mutations in the human MED12 gene result in X-linked mental retardation disorders, including Opitz-Karreggia syndrome and Lujan syndrome, and these mutations are also associated with schizophrenia [13]–[15], [59], [60]. Some overlapping clinical manifestations of these syndromes, including agenesis of the corpus callosum, behavioral disturbances and cognitive deficits, are reminiscent of FASD [15], [27], [28]. Our results demonstrate a heightened sensitivity to ethanol-induced deficits in select groups of neurons in med12 mutant embryos (Figure 3).
The sensitization to ethanol exposure observed in med12 mutants suggests a synergistic interaction, potentially through an increase in cell death. Several mechanisms of ethanol’s effect on brain development have been proposed to act by inducing apoptosis [21], [61], [62]. Ethanol-induced increases in apoptosis can be observed by acridine orange staining. However, in wild-type embryos, acridine orange staining is not restricted to specific groups of cells. Rather, apoptotic cells appear to be randomly distributed throughout the neuraxis, an observation that argues against increased apoptosis as a mechanism for the ethanol-induced reduction of specific neuronal populations. Nevertheless, ethanol exposure in med12 mutant embryos could sensitize certain neurons to apoptosis-triggering events. In med12 mutant embryos, acridine orange and TUNEL staining shows a modest increase in apoptosis in comparison to wild-type embryos [48], [57]. In contrast, the number of apoptotic cells increase dramatically in both med12 mutant and wild-type embryos following ethanol exposure. In addition, the levels of apoptosis are similar between ethanol treated wild-type and med12 mutant embryos suggesting that the increased sensitivity of subsets of neuronal cells to ethanol exposure in med12 mutant embryos is unlikely to be explained by increased apoptosis.
Novel Oxytocin Expression in the Hindbrain is Induced by Ethanol Exposure
In order to utilize zebrafish for the analysis of the effect of embryonic alcohol exposure on the oxytocinergic system, we generated oxtl:GFP transgenic embryos. The oxytocinergic system was chosen because this system, across a spectrum of vertebrate animals, is closely associated with social behaviors, the disruption of which is one aspect of FASD [29]–[33]. Significantly, zebrafish display defects in social behavior following embryonic ethanol exposure [43], [44], [46], [63]. Thus, we anticipated that this type of exposure would lead to deficits in the oxytocinergic system.
Surprisingly, ethanol exposure induced oxtl:GFP expression in the hindbrain, while not affecting the oxytocinergic system of the POA in oxtl:GFP transgenic larva. The induction of oxytocinergic cells in the hindbrain has not previously been observed in any vertebrate organism. However, ethanol-stimulated induction of hindbrain oxytocinergic neurons in mammals, including humans, would not be an unreasonable phenomenon to expect. The lack of previously observed oxytocin-expressing cells in the mammalian hindbrain may simply be due to the relative difficulty of visualizing specific neuronal phenotypes in the mammalian brain. We were able to observe these oxtl-expressing neurons because the entire nervous system is visible in the zebrafish, a feature of this model that facilitates unexpected discoveries.
The induction of novel neurotransmitter phenotypes as a consequence of fetal alcohol exposure has not been reported previously. In nearly all cases, whether in cell culture, tissue culture or intact animals, ethanol exposure results in decreased proliferation, neuronal dysfunction or cell death [16], [19], [25]. In contrast, we observe robust induction of oxtl expression in the hindbrain of the zebrafish following ethanol exposure.
These observations invite speculation on the potential function of IOCs in the hindbrain and the consequences of their expression in the larva. IOCs arise in the posterior hindbrain, most likely the area postrema. In mammals, the area postrema detects toxins in the blood and is excited by visceral afferent impulses (sympathetic and vagal) arising from the gastrointestinal tract and other peripheral trigger areas [64], [65]. This brain region also contains numerous dopaminergic and serotonergic neurons, which could indicate an interaction between IOCs and these systems, as interactions among the oxytocinergic, dopaminergic and serotonergic systems in the forebrain have been proposed to mediate a host of behaviors [43], [66]–[71]. Furthermore, the area postrema contributes to behavioral responses to blood-borne chemicals, such as taste aversion learning in rats [72]–[74]. Oxytocin has strong effects on behavior through mediating positive feelings of social interactions [29]–[33], [75]. Interestingly, addictive “party” drugs and alcohol increase oxytocin levels, and oxytocin treatment reduces drug self-administration behavior and improves withdrawal symptoms [76], [77]. Based on our observation that alcohol exposure induces oxtl expression in the area postrema, we hypothesize that oxytocin release from area postrema neurons mediates drug-seeking and addictive behavior. Furthermore, we hypothesize that embryonic and/or fetal exposure to ethanol sensitizes IOCs such that they potentiate positive feelings upon re-exposure in later life, and this may partially explain the high incidence of alcoholism and other addictive behaviors observed in FASD [5], [6], [8], [9], [78]–[83].
In conclusion, we have discovered a new phenomenon resulting from ethanol exposure, namely, the induction of oxtl-expressing neurons in the hindbrain. This discovery highlights an under-appreciated advantage of the zebrafish model: The effects of ethanol exposure on brain development, in both wild-type and mutant embryos, can be visualized in the entire embryo at a cellular level. Thus, events occurring at unexpected locations can be discovered in the process of studying seemingly unrelated phenomena. The discovery of induced oxtl expression in the hindbrain may have profound implications for understanding alcoholism and other addictive disorders.
Materials and Methods
Animals
Zebrafish (Danio rerio) were raised, maintained and crossed as described [84]. Development of embryos was at 28°C, and staging was determined by both hpf and morphological characteristics [85]. All procedures were in accordance with NIH guidelines on the care and use of animals and were approved by the Georgetown University Institutional Animal Care and Use Committee, Protocol 11-008.
EtOH Treatments
Ethanol solutions were prepared by diluting 95% ethanol (Molecular Biology Grade, Sigma-Aldrich) in fish water (0.3 gm/L sea salt, Marine Biosystems) with or without 30 mg/L phenylthiourea (Sigma, P7629). Embryos were collected from pair-wise or group mating, debris and unfertilized eggs were removed, and embryos were placed in fish water. Fish water was removed by pipetting or pouring, and the embryos or larva were rinsed once in ethanol solution, and then fresh ethanol solution was added. The number of embryos/larva and the volume of ethanol solution were equivalent for all treatment groups within any given experiment, but varied from experiment to experiment. All culture dishes were sealed with parafilm for the duration of the ethanol treatments. Following treatment, the ethanol solutions were removed, the dish was rinsed once with fish water and the embryos were raised in fish water until observation.
Whole-mount in situ Hybridization
The whole-mount in situ hybridization procedures were performed as previously detailed [86]. Oxtl (ist) antisense riboprobe was synthesized using SP6 RNA polymerase from EcoRI linearized template cDNA [34].
tyrosine hydroxylase (th) was cloned by reverse transcriptase polymerase chain reaction (RT-PCR) using the following primers: th-RT1, 5′-CAACACATTCAGGGCATCTG-3′; th-F1, 5′-CAGCTCCACATCTTCCACAAA-3′; and th-R1, 5′-ACAGAAAACGGTCGCTTGAT -3′. Total RNA was isolated from 3 dpf embryos using TRIzol Reagent (Invitrogen, 15596-026). th RNA was reverse transcribed using the th-RT1 primer with SuperScript III First-Strand Synthesis Supermix (Invitrogen, 18080-400). th cDNA was amplified using 2x PCR Master Mix (Fermentas, #K0171) with primers th-F1 and th-R1. The PCR product was digested with NotI and HindIII, releasing a 1,138 bp internal th fragment, which was gel purified and cloned into pBluescript II (Agilent). th antisense riboprobe was synthesized from NotI linearized template cDNA using T7 RNA polymerase following the protocol in the DIG RNA Labeling Kit (Roche, Indianapolis, IN).
GFP cDNA template was generated by amplification using 2x PCR Master Mix (Fermentas, #K0171) with primers: gfpF1, 5′-GACGGCGACGTAAACGGCCA-3′; and gfpR1-T7, 5′-TAATACGACTCACTATAGGGCTTGCTCAGGGCGGACTGGGT-3′. The T7 sequence is underlined. EGFP antisense riboprobe was synthesized from PCR amplified GFP template using T7 RNA polymerase following the protocol in the DIG RNA Labeling Kit (Roche, Indianapolis, IN).
Isolation and Characterization of Zebrafish oxtl Genomic Clones
Genomic clones representing zebrafish oxtl were isolated by screening an arrayed zebrafish P1-derived Artificial Chromosome (PAC) library obtained from the Resource Center, German Human Genome Project (RZPD library number, 706), and hybridized with a zebrafish [P32]-labeled oxtl cDNA probe. Three clones hybridized strongly with the probe and were therefore obtained from the RZPD. These clones were characterized by restriction endonuclease mapping and Southern hybridization analysis, which shows that they overlap the oxtl transcribed region. We chose the largest oxtl clone (IT2.4) for subcloning and limited sequencing. The sequence from this PAC clone was used to design PCR primers to amplify potential regulatory regions.
Generation of oxtl:EGFP Transgenic Lines
A 1.7 kb region of 5′ oxtl sequence was amplified from the PAC clone IT2.4 using the following primers with restriction sites added at their 5′ ends to facilitate cloning:
Xho-ISTG-F1 5′-GAGACTCGAGCACAGTTGATTCATGTTGAGCA.
ISTG-R1-BamHI 5′-CGGGATCCACTGTGGAGGAAGAGACGTACA.
The amplified region contains 580 bp upstream of the transcriptional start site, the first exon (non-coding) of oxtl, the first intron, and a single bp of the second exon two bp upstream of the translational start site. This region was cloned into the Tol2 vector pT2AL200R150G at unique XhoI to BamHI sites, replacing the Xenopus EF1a enhancer-promoter and rabbit b-globin intron, with the oxtl sequence [87], [88]. The resulting plasmid, pT2-1.7oxtl:GFP, was confirmed by sequencing. Capped Tol2 transposase mRNA was synthesized from the pCS2FA-transposase plasmid as described [89]. About 450 embryos were injected with supercoiled pT2-1.7oxtl:GFP plasmid plus Tol2 transposase RNA as described [90]. Approximately 70% of the 205 surviving embryos expressed GFP in an oxtl pattern [34].
Seven independent lines that expressed GFP in an oxtl pattern were recovered from 32 outcrossed fish. Three lines with the brightest expression were maintained and used for ethanol exposure experiments.
Acridine Orange, TUNEL Staining and Microscopy
Acridine orange staining was done as previously described [91]. Briefly, embryos were manually dechorionated, anesthetized in 0.016% tricaine (Sigma), and then incubated in 50 ng/ml acridine orange (Invitrogen, A1301) for 1hr. Following several washes in fish water, the embryos were mounted in 3% methyl cellulose and imaged. For TUNEL staining, 48 hpf embryos were fixed in 4% paraformaldehyde, embedded in agarose, and then sectioned on a vibratome. Apoptotic cells were visualized using an In Situ Cell Death Detection Kit, TMR red (Roche Applied Science) as per the manufacturer’s instructions. Transgenic embryos were anesthetized then mounted in 3% methyl cellulose or 1% low-melt agarose. Images were acquired using a Zeiss Axioplan2 microscope fitted with an AxioCam camera using AxioVision software.
Statistical Analysis
Statistical analyses were performed using SPSS for Windows (version 20.0.1). Quantitative analyses for GFP-positive cell numbers in specific brain nuclei were performed using unpaired (two-tailed) student’s t test.
Supplemental Methods
RNA Isolation and Real-time Quantitative RT-PCR
RNA was isolated from groups of ten embryos using TRIzol Reagent (Invitrogen). One ug RNA was then converted to cDNA using the High Capacity RNA-to-DNA kit (Applied Biosystems). Reactions containing Fast SYBR Green master mix (Applied Biosystems), cDNA and primers were amplified using an Applied Biosystems 7900HT Fast Real-Time System. The following primers were used: GFP_F1 5′-AGCCGCTACCCCGACCACAT;
GFP_R1 5′-TGCGCTCCTGGACGTAGCCT; bactin2F1 5′-CGAGCTGTCTTCCCATCCA; bactin2F1 5′-CGAGCTGTCTTCCCATCCA.
Relative Quantitation was analyzed in RQ Manager 1.2 software using the Comparative CT method.
Supporting Information
Ethanol exposure during the predicted time period of initial differentiation of gsc -expressing cells had no effect on gsc:GFP expression. A–D, Tg(gsc:GFP) larval at 6 dpf, dorsal views. A, is control. B, treated with 2% ethanol from 11 to 17 somites stage. C, treated with 2% ethanol from 19 to 25 somites stage. D, treated with 2% ethanol from 22 somites to prim8 stage.
(TIF)
Ethanol exposure later than 48 hpf had no effect on gsc:GFP expression. A–F, Tg(gsc:GFP) larval at 7 dpf, dorsal views. A, is control. B, treated with 2% ethanol for 24 hrs at 70% epiboly. C, treated with 2% ethanol for 24 hrs at 24 hpf. D, treated with 2% ethanol for 24 hrs at 2 dpf. E, treated with 2% ethanol for 24 hrs at 3 dpf. F, treated with 2% ethanol for 24 hrs at 4 dpf.
(TIF)
Ethanol exposure later than 48 hpf had no effect on isl1:GFP expression. A–D, Tg(isl1:GFP) larval at 7 dpf, dorsal views. A, is control. B, treated with 2% ethanol for 24 hrs at 2 dpf. C, treated with 2% ethanol for 24 hrs at 3 dpf. D, treated with 2% ethanol for 48 hrs at 4 dpf.
(TIF)
Ethanol exposure had no apparent overall effect on post-mitotic neurons visualized by elavl3:GFP expression. A–C, Tg(elavl3:GFP) larva at 5 dpf, DIC and fluorescent composite photomicrographs, dorsal views. A, is control. B, treated with 1% ethanol for 13 hrs at 35 hpf. C, treated with 2% ethanol for 13 hrs at 35 hpf.
(TIF)
Ethanol exposure had no effect on the overall numbers of post-mitotic neurons in wild-type Tg(elavl3:GFP) embryos, while med12y82 mutant embryos had fewer post-mitotic neurons that became further reduced after ethanol exposure. A–F, lateral view of 36 hpf embryos. A–C, wild-type Tg(elavl3:GFP) embryos. D–F, med12y82;Tg(elavl3:GFP) embryos. A, D, controls. B, E, treated with 1% ethanol from dome stage to 24 hpf. C, F, treated with 2% ethanol for 24 hrs at 3 dpf. G, quantitation of GFP expression by real-time quantitative RT-PCR. GFP expression was normalized to β-actin as an internal control. Relative quantity was compared to control wild-type Tg(elavl3:GFP) embryos (HuC). The relative quantity of GFP in control wild-type Tg(elavl3:GFP) embryos was set to one, which is zero on the log scale. The relative expression of GFP trends higher in med12y82 mutant (kto) embryos and in ethanol-treated embryos as shown by the red bars. Although the overall amount of GFP expression in med12y82 mutant and ethanol-treated embryos appears to be somewhat reduced in panels A–F, when normalized to β-actin GFP expression is essentially equal or even increased relative to control wild-type embryos. This suggests that neither ethanol exposure nor med12 mutation results in a relative reduction in post-mitotic neurons. Error bars represent 95% confidence levels.
(TIF)
Early pax2a:GFP expression was similar in wild-type and med12y82 embryos. A, B, lateral view of 24 hpf embryos. A, wild-type Tg(pax2a:GFP) embryo. B, med12y82;Tg(pax2a:GFP) embryo.
(TIF)
Pax2a:GFP expression was reduced in the hindbrain following ethanol exposure in wild-type, and is severely depleted in med12y82 embryos. A−D, lateral views of 60 hpf embryos. A, B, wild-type Tg(pax2a:GFP) embryos. C, D, med12y82;Tg(pax2a:GFP) embryos. A, C, controls. B, D, treated with 2% ethanol for 24 hrs at 24 hpf.
(TIF)
Acknowledgments
We gratefully acknowledge Jennifer A. Manner, Vivianne E. Greenwood and Nicole C. Johnston for critical comments on the manuscript, Kochi Kawakami for the pT2AL200R150G plasmid, Chi-Bin Chien for the pCS2FA-transposase plasmid, and the Lombardi Comprehensive Cancer Center, Animal Shared Resource, for zebrafish support.
Funding Statement
This project was partially funded by National Cancer Institue grant P30-CA051008, and by the Intramural Research Program of the National Institute of Child Health and Human Development, United States National Institutes of Health. No additional external funding was received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Abel E, Sokol R (1986) Fetal alcohol syndrome is now leading cause of mental-retardation. Lancet 2(8517): 1222–1222. [DOI] [PubMed] [Google Scholar]
- 2.Riley EP, Infante MA, Warren KR (2011) Fetal alcohol spectrum disorders: An overview. Neuropsychol Rev 21(2): 73–80. 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed]
- 3. May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, et al. (2009) Prevalence and epidemiologic characteristics of fasd from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev 15(3) 176–192: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- 4. Baer J, Sampson P, Barr H, Connor P, Streissguth A (2003) A 21-year longitudinal analysis of the effects of prenatal alcohol exposure on young adult drinking. Arch Gen Psychiatry 60(4) 377–385: 10.1001/archpsyc.60.4.377. [DOI] [PubMed] [Google Scholar]
- 5. Barr H, Bookstein F, O'Malley K, Connor P, Huggins J, et al. (2006) Binge drinking during pregnancy as a predictor of psychiatric disorders on the structured clinical interview for DSM-IV in young adult offspring. Am J Psychiatry 163(6) 1061–1065: 10.1176/appi.ajp.163.6.1061. [DOI] [PubMed] [Google Scholar]
- 6. Burd L, Carlson C, Kerbeshian J (2007) Fetal alcohol spectrum disorders and mental illness. International Journal on Disability and Human Development 6(4): 383–396. [Google Scholar]
- 7. Davis K, Desrocher M, Moore T (2011) Fetal alcohol spectrum disorder: A review of neurodevelopmental findings and interventions. Journal of Developmental and Physical Disabilities 23(2) 143–167: 10.1007/s10882–010-9204-2. [Google Scholar]
- 8. Mattson SN, Crocker N, Nguyen TT (2011) Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol Rev 21(2) 81–101: 10.1007/s11065–011-9167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sorensen HJ, Manzardo AM, Knop J, Penick EC, Madarasz W, et al. (2011) The contribution of parental alcohol use disorders and other psychiatric illness to the risk of alcohol use disorders in the offspring. Alcoholism-Clinical and Experimental Research 35(7): 1315–1320. 10.1111/j.1530-0277.2011.01467 x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warren K, Li T (2005) Genetic polymorphisms: Impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res Part A-Clin Mol Teratol 73(4) 195–203: 10.1002/bdra.20125. [DOI] [PubMed] [Google Scholar]
- 11. Bennett B, Downing C, Parker C, Johnson TE (2006) Mouse genetic models in alcohol research. Trends in Genetics 22(7) 367–374: 10.1016/j.tig.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 12. Malik S, Roeder RG (2010) The metazoan mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 11(11) 761–772: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Risheg H, Graham JM Jr, Clark RD, Rogers RC, Opitz JM, et al. (2007) A recurrent mutation in MED12 leading to R961W causes opitz-kaveggia syndrome. Nat Genet 39(4) 451–453: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- 14. Rump P, Niessen RC, Verbruggen KT, Brouwer OF, de Raad M, et al. (2011) A novel mutation in MED12 causes FG syndrome (opitz-kaveggia syndrome). Clin Genet 79(2): 183–188. 10.1111/j.1399-0004.2010.01449.x. [DOI] [PubMed] [Google Scholar]
- 15. Schwartz CE, Tarpey PS, Lubs HA, Verloes A, May MM, et al. (2007) The original lujan syndrome family has a novel missense mutation (p. N1007S) in the MED12 gene. J Med Genet 44(7) 472–477: 10.1136/jmg.2006.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonthius D, Karacay B, Dai D, Pantazis N (2003) FGF-2, NGF and IGF-1, but not BDNF, utilize a nitric oxide pathway to signal neurotrophic and neuroprotective effects against alcohol toxicity in cerebellar granule cell cultures. Dev Brain Res 140(1) 15–28: 10.1016/S0165–3806(02)00549-7. [DOI] [PubMed] [Google Scholar]
- 17. Caldeira J, Wu Y, Mameli M, Purdy R, Li P, et al. (2004) Fetal alcohol exposure alters neurosteroid levels in the developing rat brain. J Neurochem 90(6): 1530–1539. 10.1111/j.1471-4159.2004.02686.x. [DOI] [PubMed] [Google Scholar]
- 18. Dikranian K, Qin Y, Labruyere J, Nemmers B, Olney J (2005) Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev Brain Res 155(1) 1–13: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 19. Fitzgerald DM, Charness ME, Leite-Morris KA, Chen S (2011) Effects of ethanol and NAP on cerebellar expression of the neural cell adhesion molecule L1. PLoS One 6(9) e24364: 10.1371/journal.pone.0024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heaton M, Paiva M, Madorsky I, Shaw G (2003) Ethanol effects on neonatal rat cortex: Comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Dev Brain Res 145(2) 249–262: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 21. Ikonomidou C, Bittigau P, Ishimaru M, Wozniak D, Koch C, et al. (2000) Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287(5455) 1056–1060: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 22. Ikonomidou C, Kaindl AM (2011) Neuronal death and oxidative stress in the developing brain. Antioxid Redox Signal 14(8) 1535–1550: 10.1089/ars.2010.3581. [DOI] [PubMed] [Google Scholar]
- 23. Kane MA, Folias AE, Wang C, Napoli JL (2010) Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: A potential mechanism of ethanol toxicity. FASEB J 24(3) 823–832: 10.1096/fj.09–141572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu J, Yeon J, Chang H, Tison G, Chen G, et al. (2003) Ethanol impairs insulin-stimulated neuronal survival in the developing brain - role of PTEN phosphatase. J Biol Chem 278 (29): 26929–2693710.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]
- 25. Goodlett C, Horn K, Zhou F (2005) Alcohol teratogenesis: Mechanisms of damage and strategies for intervention. Exp Biol Med 230(6): 394–406. [DOI] [PubMed] [Google Scholar]
- 26. Ming Tong, Lisa Longato, Quynh-Giao Ly Nguyen, Chen WC, Spaisman A, et al. (2011) Acetaldehyde-mediated neurotoxicity: Relevance to fetal alcohol spectrum disorders. Oxidative Medicine and Cellular Longevity 213286: 10.1155/2011/213286. [Google Scholar]
- 27. Sowell E, Mattson S, Thompson P, Jernigan T, Riley E, et al. (2001) Mapping callosal morphology and cognitive correlates - effects of heavy prenatal alcohol exposure. Neurology 57(2): 235–244. [DOI] [PubMed] [Google Scholar]
- 28. Lebel C, Roussotte F, Sowell ER (2011) Imaging the impact of prenatal alcohol exposure on the structure of the developing human brain. Neuropsychol Rev 21(2) 102–118: 10.1007/s11065–011-9163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee H, Macbeth AH, Pagani JH, Young WS 3rd (2009) Oxytocin: The great facilitator of life RID A-9333-2009. Prog Neurobiol 88(2) 127–151: 10.1016/j.pneurobio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meyer-Lindenberg A, Domes G, Kirsch P, Heinrichs M (2011) Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nature Reviews Neuroscience 12(9) 524–538: 10.1038/nrn3044. [DOI] [PubMed] [Google Scholar]
- 31. Scantamburlo G, Ansseau M, Geenen V, Legros JJ (2009) Oxytocin: From milk ejection to maladaptation in stress response and psychiatric disorders. A psychoneuroendocrine perspective. Annales D Endocrinologie 70(6) 449–454: 10.1016/j.ando.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 32. Insel TR (2010) The challenge of translation in social neuroscience: A review of oxytocin, vasopressin, and affiliative behavior. Neuron 65(6) 768–779: 10.1016/j.neuron.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donaldson ZR, Young LJ (2008) Oxytocin, vasopressin, and the neurogenetics of sociality. Science 322(5903) 900–904: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]
- 34. Unger J, Glasgow E (2003) Expression of isotocin-neurophysin mRNA in developing zebratish. Gene Expression Patterns 3(1) 105–108: 10.1016/S1567–133X(02)00064-9. [DOI] [PubMed] [Google Scholar]
- 35. Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, et al. (2008) Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A 105(4) 1255–1260: 10.1073/pnas.0704963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robles E, Smith SJ, Baier H (2011) Characterization of genetically targeted neuron types in the zebrafish optic tectum. Frontiers in Neural Circuits 5 1: 10.3389/fncir.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott EK, Baier H (2009) The cellular architecture of the larval zebrafish tectum, as revealed by Gal4 enhancer trap lines. Frontiers in Neural Circuits 3 13: 10.3389/neuro.04.013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, et al. (2007) Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nature Methods 4(4) 323–326: 10.1038/NMETH1033. [DOI] [PubMed] [Google Scholar]
- 39. Amsterdam A, Becker T (2005) Transgenes as screening tools to probe and manipulate the zebrafish genome. Developmental Dynamics 234(2) 255–268: 10.1002/dvdy.20541. [DOI] [PubMed] [Google Scholar]
- 40. Choo B, Kondrichin I, Parinov S, Emelyanov A, Go W, et al. (2006) Zebrafish transgenic enhancer TRAP line database (ZETRAP). Bmc Developmental Biology 6 5: 10.1186/1471–213X-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawakami K, Abe G, Asada T, Asakawa K, Fukuda R, et al. (2010) zTrap: Zebrafish gene trap and enhancer trap database. Bmc Developmental Biology 10 105: 10.1186/1471–213X-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rinkwitz S, Mourrain P, Becker TS (2011) Zebrafish: An integrative system for neurogenomics and neurosciences. Prog Neurobiol 93(2) 231–243: 10.1016/j.pneurobio.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Buske C, Gerlai R (2011) Early embryonic ethanol exposure impairs shoaling and the dopaminergic and serotoninergic systems in adult zebrafish. Neurotoxicol Teratol. 10.1016/j.ntt.2011.05.009. [DOI] [PMC free article] [PubMed]
- 44. Echevarria DJ, Toms CN, Jouandot DJ (2011) Alcohol-induced behavior change in zebrafish models. Rev Neurosci 22(1) 85–93: 10.1515/RNS.2011.010. [DOI] [PubMed] [Google Scholar]
- 45. Egan RJ, Bergner CL, Hart PC, Cachat JM, Canavello PR, et al. (2009) Understanding behavioral and physiological phenotypes of stress and anxiety in zebrafish. Behav Brain Res 205(1) 38–44: 10.1016/j.bbr.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fernandes Y, Gerlai R (2009) Long-term behavioral changes in response to early developmental exposure to ethanol in zebrafish. Alcoholism-Clinical and Experimental Research 33(4): 601–609. 10.1111/j.1530-0277.2008.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kily LJM, Cowe YCM, Hussain O, Patel S, McElwaine S, et al. (2008) Gene expression changes in a zebrafish model of drug dependency suggest conservation of neuro-adaptation pathways. J Exp Biol 211(10) 1623–1634: 10.1242/jeb.014399. [DOI] [PubMed] [Google Scholar]
- 48. Hong S, Haidin C, Lawson N, Weinstein B, Dawid I, et al. (2005) The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney. Proc Natl Acad Sci U S A 102(51) 18473–18478: 10.1073/pnas.0509457102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, et al. (2002) Guidance of primordial germ cell migration by the chemokine SDF-1. Cell 111(5) 647–659: 10.1016/S0092–8674(02)01135-2. [DOI] [PubMed] [Google Scholar]
- 50. Carvan M, Loucks E, Weber D, Williams F (2004) Ethanol effects on the developing zebrafish: Neurobehavior and skeletal morphogenesis. Neurotoxicol Teratol 26(6) 757–768: 10.1016/j.ntt.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 51. Blader P, Strahle U (1998) Ethanol impairs migration of the prechordal plate in the zebrafish embryo. Dev Biol 201(2) 185–201: 10.1006/dbio.1998.8995. [DOI] [PubMed] [Google Scholar]
- 52. Higashijima S, Hotta Y, Okamoto H (2000) Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. Journal of Neuroscience 20(1): 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Picker A, Scholpp S, Bohli H, Takeda H, Brand M (2002) A novel positive transcriptional feedback loop in midbrain-hindbrain boundary development is revealed through analysis of the zebrafish pax2.1 promoter in transgenic lines. Development 129(13): 3227–3239. [DOI] [PubMed] [Google Scholar]
- 54. Park H, Kim C, Bae Y, Yee S, Kim S, et al. (2000) Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons RID C-8194-2009. Dev Biol 227(2) 279–293: 10.1006/dbio.2000.9898. [DOI] [PubMed] [Google Scholar]
- 55. Graham JM Jr, Clark RD, Moeschler JB, Rogers RC (2010) Behavioral features in young adults with FG syndrome (opitz-kaveggia syndrome). American Journal of Medical Genetics Part C-Seminars in Medical Genetics 154C(4) 477–485: 10.1002/ajmg.c.30284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Philiber RA, Madan A (2007) Role of MED12 in transcription and human behavior. Pharmacogenomics 8(8) 909–916: 10.2217/14622416.8.8.909. [DOI] [PubMed] [Google Scholar]
- 57. Hong S, Dawid IB (2011) The transcriptional mediator component Med12 is required for hindbrain boundary formation. Plos One 6(4) e19076: 10.1371/journal.pone.0019076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ali S, Champagne DL, Alia A, Richardson MK (2011) Large-scale analysis of acute ethanol exposure in zebrafish development: A critical time window and resilience. PLoS One 6(5) e20037: 10.1371/journal.pone.0020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Philibert RA, Bohle P, Secrest D, Deaderick J, Sandhu H, et al. (2007) The association of the HOPA(12 bp) polymorphism with schizophrenia in the NIMH genetics initiative for schizophrenia sample. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics 144B(6) 743–747: 10.1002/ajmg.b.30489. [DOI] [PubMed] [Google Scholar]
- 60. Philibert R (2006) A meta-analysis of the association of the HOPA(12 bp) polymorphism and schizophrenia. Psychiatr Genet 16(2) 73–76: 10.1097/01.ypg.0000194443.81813.f0. [DOI] [PubMed] [Google Scholar]
- 61. Miller M (1986) Effects of alcohol on the generation and migration of cerebral cortical-neurons. Science 233(4770) 1308–1311: 10.1126/science.3749878. [DOI] [PubMed] [Google Scholar]
- 62. Gohlke JM, Hiller-Sturmhoefel S, Faustman EM (2008) A systems-based computational model of alcohol's toxic effects on brain development. Alcohol Res Health 31(1): 76–83. [PMC free article] [PubMed] [Google Scholar]
- 63. Kurta A, Palestis BG (2010) Effects of ethanol on the shoaling behavior of zebrafish (danio rerio). Dose Response 8(4) 527–533: 10.2203/dose–response.10-008.Palestis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Price CJ, Hoyda TD, Ferguson AV (2008) The area postrema: A brain monitor and integrator of systemic autonomic state. Neuroscientist 14(2) 182–194: 10.1177/1073858407311100. [DOI] [PubMed] [Google Scholar]
- 65. Johnstone LE, Fong TM, Leng G (2006) Neuronal activation in the hypothalamus and brainstem during feeding in rats RID C-6688-2011. Cell Metab 4(4) 313–321: 10.1016/j.cmet.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 66. Baskerville TA, Douglas AJ (2010) Dopamine and oxytocin interactions underlying behaviors: Potential contributions to behavioral disorders. Cns Neuroscience & Therapeutics 16(3): e92-e123. 10.1111/j.1755-5949.2010.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kastenhuber E, Kratochwil CF, Ryu S, Schweitzer J, Driever W (2010) Genetic dissection of dopaminergic and noradrenergic contributions to catecholaminergic tracts in early larval zebrafish RID B-2169-2010. J Comp Neurol 518(4) 439–458: 10.1002/cne.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McLean D, Fetcho J (2004) Ontogeny and innervation patterns of dopaminergic, noradrenergic, and serotonergic neurons in larval zebrafish. J Comp Neurol 480(1) 38–56: 10.1002/cne.20280. [DOI] [PubMed] [Google Scholar]
- 69. Young KA, Gobrogge KL, Wang Z (2011) The role of mesocorticolimbic dopamine in regulating interactions between drugs of abuse and social behavior. Neurosci Biobehav Rev 35(3) 498–515: 10.1016/j.neubiorev.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Emiliano ABF, Cruz T, Pannoni V, Fudge JL (2007) The interface of oxytocin-labeled cells and serotonin transporter-containing fibers in the primate hypothalamus: A substrate for SSRIs therapeutic effects? Neuropsychopharmacology 32(5) 977–988: 10.1038/sj.npp.1301206. [DOI] [PubMed] [Google Scholar]
- 71. Yoshida M, Takayanagi Y, Inoue K, Kimura T, Young LJ, et al. (2009) Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. J Neurosci 29(7) 2259–2271: 10.1523/JNEUROSCI.5593–08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thiele T, Roitman M, Bernstein I (1996) c-fos induction in rat brainstem in response to ethanol- and lithium chloride-induced conditioned taste aversions. Alcoholism-Clinical and Experimental Research 20(6): 1023–1028. 10.1111/j.1530-0277.1996.tb01941.x. [DOI] [PubMed] [Google Scholar]
- 73. Gallo M, Arnedo M, Aguero A, Puerto A (1990) The functional relevance of the area postrema in drug-induced aversion learning rid B-9714-2011. Pharmacology Biochemistry and Behavior 35(3) 543–551: 10.1016/0091–3057(90)90287-R. [DOI] [PubMed] [Google Scholar]
- 74. Hayes U, Chambers K (2005) Peripheral vasopressin accelerates extinction of conditioned taste avoidance. Physiol Behav 84(1) 147–156: 10.1016/j.physbeh.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 75. MacDonald K, MacDonald TM (2010) The peptide that binds: A systematic review of oxytocin and its prosocial effects in humans. Harv Rev Psychiatry 18(1) 1–21: 10.3109/10673220903523615. [DOI] [PubMed] [Google Scholar]
- 76. McGregor IS, Callaghan PD, Hunt GE (2008) From ultrasocial to antisocial: A role for oxytocin in the acute reinforcing effects and long-term adverse consequences of drug use? Br J Pharmacol 154(2) 358–368: 10.1038/bjp.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sarnyai Z (2011) Oxytocin as a potential mediator and modulator of drug addiction RID A-3283-2009. Addict Biol 16(2): 199–201. 10.1111/j.1369-1600.2011.00332.x. [DOI] [PubMed] [Google Scholar]
- 78. Alati R, Al Mamun A, Williams GM, O'Callaghan M, Najman JM, et al. (2006) In utero alcohol exposure and prediction of alcohol disorders in early adulthood - A birth cohort study RID G-2523-2010 RID F-4414-2010 RID B-1527-2008. Arch Gen Psychiatry 63(9) 1009–1016: 10.1001/archpsyc.63.9.1009. [DOI] [PubMed] [Google Scholar]
- 79. Barbier E, Pierrefiche O, Vaudry D, Vaudry H, Daoust M, et al. (2008) Long-term alterations in vulnerability to addiction to drugs of abuse and in brain gene expression after early life ethanol exposure. Neuropharmacology 55(7) 1199–1211: 10.1016/j.neuropharm.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 80. Chotro MG, Arias C, Laviola G (2007) Increased ethanol intake after prenatal ethanol exposure: Studies with animals RID C-5976-2011. Neurosci Biobehav Rev 31(2) 181–191: 10.1016/j.neubiorev.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 81. McMurray MS, Williams SK, Jarrett TM, Cox ET, Fay EE, et al. (2008) Gestational ethanol and nicotine exposure: Effects on maternal behavior, oxytocin, and offspring ethanol intake in the rat. Neurotoxicol Teratol 30(6) 475–486: 10.1016/j.ntt.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Williams SK, Cox ET, McMurray MS, Fay EE, Jarrett TM, et al. (2009) Simultaneous prenatal ethanol and nicotine exposure affect ethanol consumption, ethanol preference and oxytocin receptor binding in adolescent and adult rats. Neurotoxicol Teratol 31(5) 291–302: 10.1016/j.ntt.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Youngentob SL, Glendinning JI (2009) Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc Natl Acad Sci U S A 106(13) 5359–5364: 10.1073/pnas.0809804106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Westerfield M (1995) The zebrafish book. Eugene, Oregon: University of Oregon Press.
- 85. Kimmel C, Ballard W, Kimmel S, Ullmann B, Schilling T (1995) Stages of embryonic-development of the zebrafish. Developmental Dynamics 203(3): 253–310. [DOI] [PubMed] [Google Scholar]
- 86. Eaton JL, Glasgow E (2006) The zebrafish bHLH PAS transcriptional regulator, single-minded 1 (sim1), is required for isotocin cell development. Developmental Dynamics 235(8) 2071–2082: 10.1002/dvdy.20848. [DOI] [PubMed] [Google Scholar]
- 87. Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N, et al. (2004) A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish RID B-2537-2008. Developmental Cell 7(1) 133–144: 10.1016/j.devcel.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 88. Urasaki A, Morvan G, Kawakami K (2006) Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 174(2) 639–649: 10.1534/genetics.106.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, et al. (2007) The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics 236(11) 3088–3099: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 90. Fisher S, Grice EA, Vinton RM, Bessling SL, Urasaki A, et al. (2006) Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nature Protocols 1(3) 1297–1305: 10.1038/nprot.2006.230. [DOI] [PubMed] [Google Scholar]
- 91. Furutani-Seiki M, Jiang YJ, Brand M, Heisenberg CP, Houart C, et al. (1996) Neural degeneration mutants in the zebrafish, Danio rerio. Development 123: 229–239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ethanol exposure during the predicted time period of initial differentiation of gsc -expressing cells had no effect on gsc:GFP expression. A–D, Tg(gsc:GFP) larval at 6 dpf, dorsal views. A, is control. B, treated with 2% ethanol from 11 to 17 somites stage. C, treated with 2% ethanol from 19 to 25 somites stage. D, treated with 2% ethanol from 22 somites to prim8 stage.
(TIF)
Ethanol exposure later than 48 hpf had no effect on gsc:GFP expression. A–F, Tg(gsc:GFP) larval at 7 dpf, dorsal views. A, is control. B, treated with 2% ethanol for 24 hrs at 70% epiboly. C, treated with 2% ethanol for 24 hrs at 24 hpf. D, treated with 2% ethanol for 24 hrs at 2 dpf. E, treated with 2% ethanol for 24 hrs at 3 dpf. F, treated with 2% ethanol for 24 hrs at 4 dpf.
(TIF)
Ethanol exposure later than 48 hpf had no effect on isl1:GFP expression. A–D, Tg(isl1:GFP) larval at 7 dpf, dorsal views. A, is control. B, treated with 2% ethanol for 24 hrs at 2 dpf. C, treated with 2% ethanol for 24 hrs at 3 dpf. D, treated with 2% ethanol for 48 hrs at 4 dpf.
(TIF)
Ethanol exposure had no apparent overall effect on post-mitotic neurons visualized by elavl3:GFP expression. A–C, Tg(elavl3:GFP) larva at 5 dpf, DIC and fluorescent composite photomicrographs, dorsal views. A, is control. B, treated with 1% ethanol for 13 hrs at 35 hpf. C, treated with 2% ethanol for 13 hrs at 35 hpf.
(TIF)
Ethanol exposure had no effect on the overall numbers of post-mitotic neurons in wild-type Tg(elavl3:GFP) embryos, while med12y82 mutant embryos had fewer post-mitotic neurons that became further reduced after ethanol exposure. A–F, lateral view of 36 hpf embryos. A–C, wild-type Tg(elavl3:GFP) embryos. D–F, med12y82;Tg(elavl3:GFP) embryos. A, D, controls. B, E, treated with 1% ethanol from dome stage to 24 hpf. C, F, treated with 2% ethanol for 24 hrs at 3 dpf. G, quantitation of GFP expression by real-time quantitative RT-PCR. GFP expression was normalized to β-actin as an internal control. Relative quantity was compared to control wild-type Tg(elavl3:GFP) embryos (HuC). The relative quantity of GFP in control wild-type Tg(elavl3:GFP) embryos was set to one, which is zero on the log scale. The relative expression of GFP trends higher in med12y82 mutant (kto) embryos and in ethanol-treated embryos as shown by the red bars. Although the overall amount of GFP expression in med12y82 mutant and ethanol-treated embryos appears to be somewhat reduced in panels A–F, when normalized to β-actin GFP expression is essentially equal or even increased relative to control wild-type embryos. This suggests that neither ethanol exposure nor med12 mutation results in a relative reduction in post-mitotic neurons. Error bars represent 95% confidence levels.
(TIF)
Early pax2a:GFP expression was similar in wild-type and med12y82 embryos. A, B, lateral view of 24 hpf embryos. A, wild-type Tg(pax2a:GFP) embryo. B, med12y82;Tg(pax2a:GFP) embryo.
(TIF)
Pax2a:GFP expression was reduced in the hindbrain following ethanol exposure in wild-type, and is severely depleted in med12y82 embryos. A−D, lateral views of 60 hpf embryos. A, B, wild-type Tg(pax2a:GFP) embryos. C, D, med12y82;Tg(pax2a:GFP) embryos. A, C, controls. B, D, treated with 2% ethanol for 24 hrs at 24 hpf.
(TIF)