

Abstract
Background
Prevalence of colorectal cancer (CRC) in the British Bangladeshi population (BAN) is low compared to British Caucasians (CAU). Genetic background may influence mutations and disease features.
Methods
We characterized the clinicopathological features of BAN CRCs and interrogated their genomes using mutation profiling and high-density single nucleotide polymorphism (SNP) arrays and compared findings to CAU CRCs.
Results
Age of onset of BAN CRC was significantly lower than for CAU patients (p=3.0 x 10-5) and this difference was not due to Lynch syndrome or the polyposis syndromes. KRAS mutations in BAN microsatellite stable (MSS) CRCs were comparatively rare (5.4%) compared to CAU MSS CRCs (25%; p=0.04), which correlates with the high percentage of mucinous histotype observed (31%) in the BAN samples. No BRAF mutations was seen in our BAN MSS CRCs (CAU CRCs, 12%; p=0.08). Array data revealed similar patterns of gains (chromosome 7 and 8q), losses (8p, 17p and 18q) and LOH (4q, 17p and 18q) in BAN and CAU CRCs. A small deletion on chromosome 16p13.2 involving the alternative splicing factor RBFOX1 only was found in significantly more BAN (50%) than CAU CRCs (15%) cases (p=0.04). Focal deletions targeting the 5’ end of the gene were also identified. Novel RBFOX1 mutations were found in CRC cell lines and tumours; mRNA and protein expression was reduced in tumours.
Conclusions
KRAS mutations were rare in BAN MSS CRC and a mucinous histotype common. Loss of RBFOX1 may explain the anomalous splicing activity associated with CRC.
Keywords: Colorectal cancer, Genome analysis, British Bangladeshi, RBFOX1, KRAS, BRAF
Background
Colorectal cancer (CRC) is common [1] and most cases arise sporadically. A small proportion of CRC is associated with inherited syndromes such as familial adenomatous polyposis (FAP; <1% of CRC), MUTYH-associated polyposis (MAP; rare recessive condition, carrier estimated at ~1%) and Lynch syndrome / hereditary non-polyposis colon cancer (LS/HNPCC; 2-4% of CRC) [1]. FAP patients have germline mutations in one allele of the adenomatous polyposis (APC) tumour suppressor gene, MAP patients carry biallelic mutations in the MUTYH oxidative damage repair gene and LS is caused by germline mutations in DNA mismatch repair genes (mostly MLH1 and MSH2, less commonly MSH6 and PMS2) [1]. Clinically, LS differs from FAP in that CRC arises without extensive polyposis. CRC in FAP, MAP and LS are characterised by an early age of onset (FAP, mean age, 40 years [2,3]; MAP, median, 45 years (range, 21–67) [4]; LS, median 43 years (range, 20–79) [5]). In comparison, sporadic CRC is rare until the sixth decade [6].
Sporadic CRCs are defined by their dominant genomic instability pathway and classified into two major groups. Chromosome unstable CRC (CIN) are characterised by gross chromosomal abnormalities and present in approximately 65-70% of CRC [7]. Microsatellite unstable (MSI) CRCs account for another 15%, and are driven by defective mismatch repair, which causes increased mutations of short nucleotide repeats (microsatellites) [8]. These two genomic instability pathways are associated with a difference in prognosis: MSI cancers behave less aggressively [9]. However, the classification of sporadic CRCs as CIN or MSI is not always straightforward, as a significant proportion appears microsatellite and chromosome stable (MACS) [10,11]. The frequent presence of epigenomic instability in sporadic CRC in the form of global hypomethylation or the CpG island methylator phenotype further complicates the picture [11,12].
Over a third of the population in the borough of Tower Hamlets, London, United Kingdom (UK) are Bangladeshi in origin. Interestingly, despite a high incidence of diabetes mellitus in the BAN population group [13], prevalence of CRC is lower at 27/100,000 compared to 342/100,000 in the non-Bangladeshi British population [14]. In an expression array profiling study of a small cohort of BAN CRCs, Ahmed et al. [14] reported that the percentage of cases aged less than 40 was high (61%).
The reduced susceptibility to CRC in the BAN population and the apparent early age of CRC onset are intriguing. Here, we compared the clinicopathological characteristics of BAN CRC patients presenting to our local hospital to those from a group of sporadic CAU CRC patients. We compared molecular genetic profiles between tumours from these patient groups, including genome-wide DNA copy number alterations, KRAS and BRAF mutation to test whether CRCs in the BAN population showed molecular features distinct from CAU CRCs.
Results
Clinicopathological features of patients
Between June 1997 and August 2008, all patients with CRC were invited to participate in research and CRCs were resected from 44 consented BAN patients at our centre. BAN CRC patients were recruited sequentially with patients from the CAU population and there was no prior selection; no BAN patients were missed during this time period. The age of CRC onset in the BAN cohort (median, 58; range, 25–80) was significantly lower (p=3.0 × 10-5) than for sporadic CAU CRC patients (median 71; range, 21–91) presenting at the same hospital. Although the early age of onset could be partly attributed to a relatively young population [15], it can also be an indication of a higher prevalence of inherited CRC syndromes or other predisposing conditions. However, as none of the BAN CRC patients in our cohort had a history of polyposis or chronic gastrointestinal inflammation when they were diagnosed, it is unlikely that their cancers are associated with FAP, MAP or inflammatory bowel disease.
The lower age of onset might reflect a higher prevalence of LS. To investigate this, we performed genotyping with MSI markers BAT25 and BAT26 using DNA extracted from tumour and matched normal tissue samples available from 37 patients (age of onset ≤45, n=17; age of onset >45, n=20; median age, 51). We identified 7 patients (4 early-onset, 3 late-onset) with MSI (all positive at both the BAT25 and BAT26 loci). In addition, immunohistochemistry detected loss of one or more mismatch repair proteins (MLH1, MLH6 or PMS2) in five of the seven cases (Additional file 1: Table S1). No tumour showed loss of MSH2. Two of these patients showed an early-onset of cancer at 40 and 43 years of age whereas the remaining three were older at diagnosis (65, 71 and 73). Hence, genetic features suggestive of potential LS cases were detected in 5 out of 37 patients in our cohort (13.5%), and in 2 out of 17 patients belonging to the early age group (11.8%). Because family histories were usually not available from the BAN patients, supporting evidence for a familial predisposition to CRC could not be obtained. However, additional cancers presented in two of the five potential LS cases: gastric adenocarcinoma in one individual and both an adenocarcinoma of the lung and endometrial cancer in another (Additional file 1: Table S1). In order to obtain a more uniform group of sporadic BAN samples for further investigation, we excluded all seven patients whose tumours were MSI and an additional patient (age, 51 years) whose tumour showed loss of PMS2 protein expression. Importantly, this did not alter the median age of onset (51.6 vs. 51.4 years). The clinicopathological data on these BAN patients with sporadic MSS stable cancers are summarised in Table 1.
Table 1.
Clinical and pathological characteristics of the BAN MSS colorectal cancers
| |
Overall |
Early onset (≤45 years) |
Late onset (>45 years) |
P value |
|---|---|---|---|---|
| (n = 29) | (n = 13) | (n = 16) | (Early Vs Late) | |
|
Age of Onset - Yr | ||||
| Median |
57 |
35 |
64 |
- |
| Range |
25-80 |
25-45 |
51-80 |
|
|
Sex – No (%) | ||||
| Female |
10 (34.5) |
5 (38.5) |
5 (31.3) |
N.S. |
| Male |
19 (65.5) |
8 (61.5) |
11 (68.7) |
|
|
Site of Cancer – No (%) | ||||
| Right |
6 (20.7) |
0 (0) |
6 (38.5) |
0.021 |
| Left |
23 (79.3) |
13 (100) |
10 (62.5) |
|
|
Dukes’ Stage – No (%) | ||||
| A |
3 (10.3) |
1 (7.7) |
2 (12.5) |
N.S. |
| B |
6 (20.7) |
2 (15.4) |
4 (25) |
|
| C |
19 (65.5) |
10 (76.9) |
9 (56.2) |
|
| D |
1 (3.4) |
0 (0) |
1 (6.3) |
|
|
Mucin - No (%) | ||||
| No |
20 (69.0) |
7 (53.8) |
13 (81.2) |
N.S. |
| Yes |
9 (31.0) |
6 (46.2) |
3 (18.8) |
|
|
Vascular Invasion - No (%) | ||||
| No |
16 (55.2) |
8 (61.5) |
8 (50) |
N.S. |
| Yes |
13 (44.8) |
5 (38.5) |
8 (50) |
|
|
Differentiation - No (%) | ||||
| Well |
1 (3.4) |
1 (7.7) |
0 (0) |
N.S. |
| Moderate |
24 (82.8) |
10 (76.9) |
14 (87.5) |
|
| Poor |
4 (13.8) |
2 (15.4) |
2 (12.5) |
|
|
Resection Status - No (%) | ||||
| 0 |
21 (72.4) |
10 (76.9) |
11 (68.7) |
N.S. |
| 1 |
7 (24.1) |
3 (23.1) |
4 (25) |
|
| 2 | 1 (3.4) | 0 (0) | 1 (6.3) | |
All p values were calculated using Fisher’s Exact Test (two-tailed) and p ≤ 0.05 was considered to be significant.
Subsequent comparisons focused on the remaining 29 BAN MSS patients with presence of mismatch repair protein expression: 13 patients diagnosed under the age of 45 were classified as early-onset (median, 34; range, 25–45), while the remaining 16 patients diagnosed over the age of 45 (median, 64; range, 51–80) were classified as late-onset. Overall, BAN MSS cancers were frequently left-sided (79.3%) and more prevalent in males (65.5%). Most of the cancers were moderately differentiated (82.8%). There was a high percentage of mucinous tumours (31%) compared to the background CAU population (5-15%) [16]. Within the BAN MSS group, left-sided location was significantly more prevalent in early-onset (13/13, 100%) than in late-onset cancers (10/16, 63%) (p=0.02, Fisher’s Exact Test) (Table 1). However, early-onset and late-onset BAN MSS cancers had similar male: female ratios and showed no difference in stage, prevalence of mucinous phenotype, frequency of vascular invasion, degree of differentiation or resection (R) status (Table 1).
Sequence analysis of KRAS and BRAF mutation hotspots
We compared the mutation frequencies of KRAS and BRAF between BAN CRC and CAU CRCs by performing sequence analysis of known mutation hotspots (codon 12 and 13 for KRAS and codon 600 for BRAF). In the BAN MSS samples (n=29) we found two mutations at codon 12 of KRAS (2/29, 5.4%), one in an early-onset and another in a late-onset CRC. This mutation rate is lower than in our previously characterised CAU MSS CRCs (KRAS, 33/134, 25%) [11], and the differences between populations were significant (p=0.04). Interestingly, the presence of KRAS mutations has been associated with a non-mucinous histotype [17]. The low prevalence of KRAS mutations in our BAN MSS samples may be correlated to the high percentage of mucinous tumour observed (30% in BAN MSS samples with wildtype KRAS). No mutations were observed in the BRAF gene in BAN. Although a lower frequency of BRAF mutation is expected for MSS CRCs, this frequency is still lower than that observed in our CAU MSS sample set (17/144, 12%).
Genomewide copy number analysis using single nucleotide polymorphism (SNP) beadarray
The pattern of somatic genomic copy number alterations in BAN and CAU CRCs was evaluated using SNP arrays. DNA was obtained from ten matched tumour-normal pairs from the BAN MSS patient group (early-onset, n=4; late-onset, n=6), where fresh frozen tissues were available. For comparison, we extracted DNA from paired tumour and normal tissues from a group of MSS CRC from the CAU population (n=27). We had previously classified these samples as CIN (n=11) and MACS (n=16) CRCs, using flow cytometry ploidy analysis and loss of heterozgosity (LOH) markers specific to chromosome instability [11].
In total, 37-paired CRC samples were hybridised onto Illumina bead arrays (BAN, n=10, Hap550 bead arrays; CIN, n=11, and MACS, n=16, HumanHap370- Duo bead arrays) and analysed using OncoSNP [18]. The use of high-density SNP array allowed a wide range of genomic alterations to be determined, including small amplifications, small deletions, and copy number neutral loss of heterozygosity (2nLOH) events. All tumour samples contained genomic alterations. To obtain an overview of the genetic complexity of the samples using SNP array data, we determined: (i) the average ploidy of the sample, by taking the average copy number over all SNPs; (ii) the percentage of the genome altered, by taking the fraction of SNPs showing somatic differences compared to the matched normal. In order to support our analysis, we also used genome-wide SNP genotyping data from an additional dataset generated from 636 CRC samples to provide a background for visualisation.
The cancer samples fell into three broad, distinct clusters (Figure 1). The first was a diploid group (average ploidy=2) of cancers with a minimal number of genomic aberrations (<10%). This group contained seven of the sixteen MACS samples (44%) and one of the four early-onset cancers (25%) from the BAN cohort. By definition, we would not expect any CIN cancers to be associated with this group, and our SNP genotyping data supported this. The second was the set of chromosomally unstable cancers (>50% genome altered) exhibiting polyploidy (average ploidy >3). As expected, the majority of the CIN cancers, as defined by earlier ploidy analysis (82%, 9/11), was located in this group, and was accompanied by a single MACS (6%) CRC. Also in this set were two early-onset (50%) and four late-onset BAN cancers (67%). A third intermediate group (Figure 1) had an average ploidy >2 and moderate levels of genomic aberrations (10-50%). Within this group there was a concentration of MACS cancers (50%, 8/16) and early- (25%, 1/4) and late-onset (33%, 2/6) BAN MSS cancers along with a small number of CAU CIN CRCs (18%, 2/11)
Figure 1.
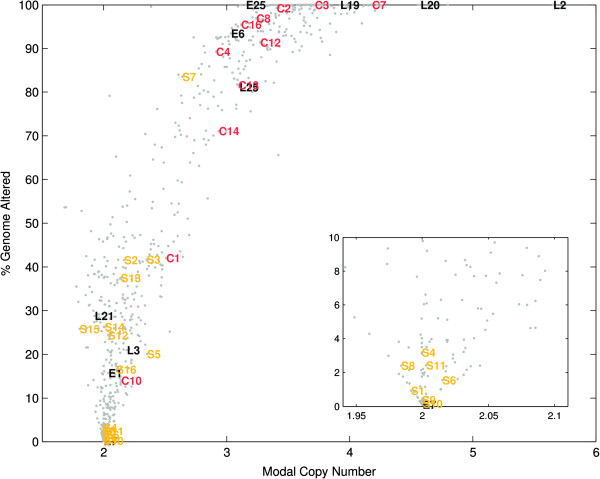
The distribution of genomic complexity for (C)IN, MAC(S), (E)arly and (L)ate-onset colorectal cancer. Data for 636 samples (grey) from the Ludwig Colon Cancer Initiative data set was used as background reference. (Inset) Expanded view showing clustering of samples with few genomic aberrations.
Genome-wide permutation testing was used to assess if any SNPs were significant markers for differentiating between early- and late-onset BAN cancers. In our sample set, no individual marker or set of markers had any stratification effect (p >0.001).
Chromosomal regions frequently altered in Bangladeshi and Caucasian colorectal cancers
In terms of absolute copy number change (Figure 2A, in blue), the most frequently observed gene amplifications in the BAN CRC samples were located on chromosomes 7, 8q and 17q. This pattern overlaps with CAU MSS cancers where amplification in CIN CRCs are mostly observed on chromosomes 2q, 5p, 7p, 13, 17q and 20q, and in MACS cancers on chromosomes 7, 8q and 20q (Figure 2A). After eliminating amplification due to increased ploidy, the relative copy number gains in the BAN samples remained on chromosomes 7 and 8q (Figure 2B). Absolute copy number loss (Figure 2A, in red) was relatively rare compared to gains, with the most noticeable losses in the BAN CRCs located on chromosome 8p.
Figure 2.
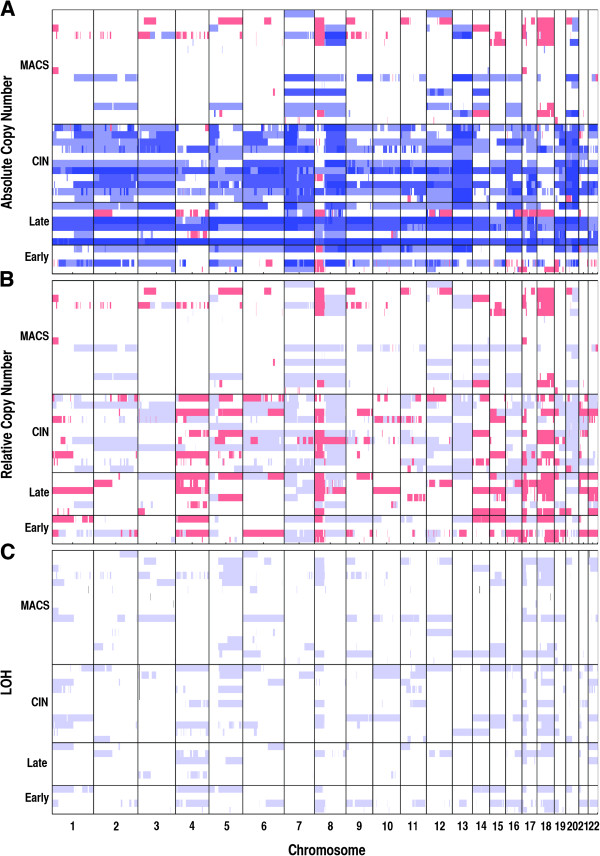
Genome-wide copy number profiles. (A) Genome-wide absolute copy number profiles for the four classes of colorectal cancers. Blue – Copy number gain (>2). Red – Copy number loss (<2). (B) Genome-wide relative (to average ploidy) copy number profiles. Blue – relative gain, Red – relative loss. (C) Genome-wide LOH profiles.
A different picture emerged when deletions relative to average ploidy were analysed (Figure 2B, in red), and regions of frequent relative loss in BAN samples included chromosomes 8p, 17p and 18q (8/10, 80%). These results, again, showed similarities with the alterations detected typically in the CIN and MACS CAU cancers, although few regions of loss were detected in MACs samples (Figure 2B). In the BAN samples, LOH was not observed as frequently as amplification and deletions (Figure 2C), and the regions with the highest frequency of LOH were located on 4q, 17p and 18q (5/10, 50%). The pattern of LOH in BAN samples was again similar to that observed in the CIN and MACS sporadic cancers, where LOH was observed repeatedly on chromosomes 17p (13/27, 48%) and 18q (15/27, 55%).
In total, over 45% of our cases showed deletions and/or LOH at chromosome 17p (BAN: 5/10, 50%; CIN: 5/11, 45%; MACS: 7/16, 44%). The genomic regions altered in most cases were large, encompassing the majority of the short arm of chromosome 17. Importantly, almost all of these alterations involve LOH or allelic deletion at the TP53 locus (17p13.1) (BAN, 4/5, 80%; CIN, 5/5, 100%; MACS, 6/7, 86%). Loss of TP53 is a consistent feature across the BAN, CIN and MACS CRCs and is therefore likely to be the major factor influencing tumour development at this locus. A similar situation was observed on chromosome 18q, where SMAD4 (18q21.2) was lost in six BAN samples (6/10; 60%) six CIN samples (6/11, 55%) and three MACS samples (3/16; 19%). In comparison, gross chromosomal alterations at the APC locus (5q22.2) were less frequent in all subgroups of samples (BAN, 3/10, 30%; CIN, 4/11, 36%; MACS, 3/16, 19%).
A small deletion on chromosome 16p13.2, containing the RBFOX1 gene, was detected in both Bangladeshi and Caucasian CRCs
Interestingly, we detected a small deletion on chromosome 16p13.2 from 6.0 to 7.7 Mb in ten samples (Figure 3A), including five BAN (5/10, 50%), 2 from early-onset and 3 from late-onset, three CIN (3/11, 27%) and one MACS (1/16, 6%). Accordingly, this deletion might be more frequent in BAN CRCs than CAU CRCs (5/10 versus 4/27; p=0.04, Fisher’s Exact Test). According to the University of California Santa Cruz (UCSC) Genome Bioinformatics database (http://genome.ucsc.edu/), this genomic region contains only one known gene, RNA binding protein, fox-1 homolog (RBFOX1). Deletion of RBFOX1 has been detected in 35% of MSI and 21% of MSS colon tumours in a previous study, and fluorescence in situ hybridization was performed to confirm the somatic deletion of this locus in four samples [19]. Deletion of RBFOX1 was also presented in a significant proportion of CRC (106/419) in The Cancer Genome Atlas dataset [20] (Additional file 2: Figure S1). The deletion clusters included hemizygous loss of the entire gene and more focal deletions primarily targeting the regulatory and 5’ exons of RBFOX1.
Figure 3.
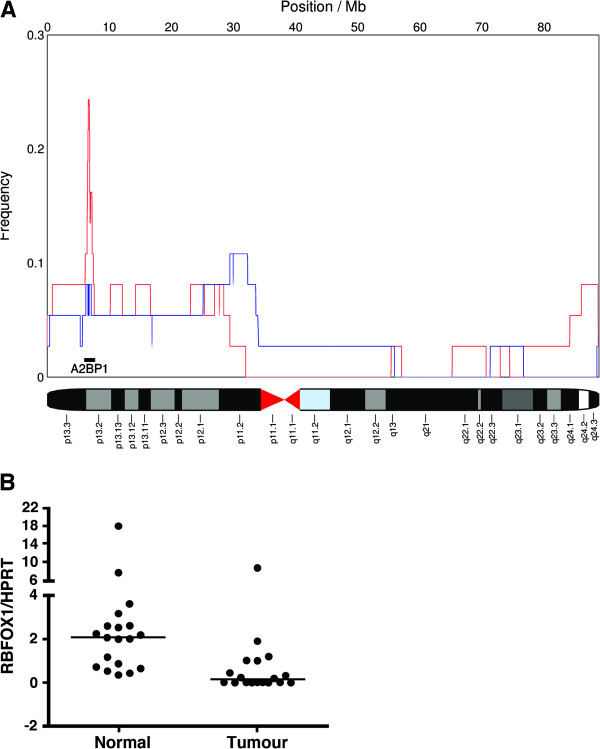
Genomic alterations and expression changes of RBFOX1 in CRC. (A) Analysis of relative copy number (blue) gain and (red) loss across all cancers (CIN, MACS and BAN) shows recurrent deletions on chromosome 16p13 affecting the RBFOX1 gene. (B) Scatter plot showing RBFOX1 gene expression level as detected by quantitative real time PCR. Nineteen tumour / normal sample pairs were tested and mRNA level is expressed as ratio of RBFOX1 and HPRT concentration, [RBFOX1] / [HPRT].
RBFOX1 is an RNA binding protein, which has a role in splicing regulation, and the gene is transcribed into a few alternatively spliced isoforms [21,22]. Although RBFOX1 is highly expressed in the nucleus and / or cytoplasm in the brain, heart and skeletal muscle, its expression in the intestines has not been defined clearly. Therefore, we sought evidence of RBFOX1 expression by quantitative real time PCR (QRT-PCR) using cDNA from 43 colorectal cell lines and 19 paired CAU CRC tumour/normal tissues, where good quality RNA was available. In most cell lines (36/43, 84%), the threshold cycle (Ct) values produced in the QRT-PCR experiment indicated absence or undetectable levels of RBFOX1 mRNA transcript, whereas low level of RBFOX1 mRNA was found in the remaining seven cell lines (Ct range: 33.1-33.8) (Additional file 3: Table S2). In contrast, RBFOX1 mRNA was detected in all normal tissues samples (Ct range: 23.6-36.7, median, 31.6). Furthermore, in most tumour samples the expression level of RBFOX1 was reduced compared to the normal paired tissue samples (Figure 3B, Additional file 3: Table S2). As RBFOX1 appears to be expressed at low level, the possibility that its expression is restricted to particular cell types was considered. The immunohistochemical expression of RBFOX1 in human non-neoplastic colon and colon cancer tissues was assessed (Figure 4). Staining was detected in the cytoplasm of normal colonic epithelium and reduced levels of RBFOX1 expression were observed in the some of the tumour samples we tested (Figure 4C).
Figure 4.
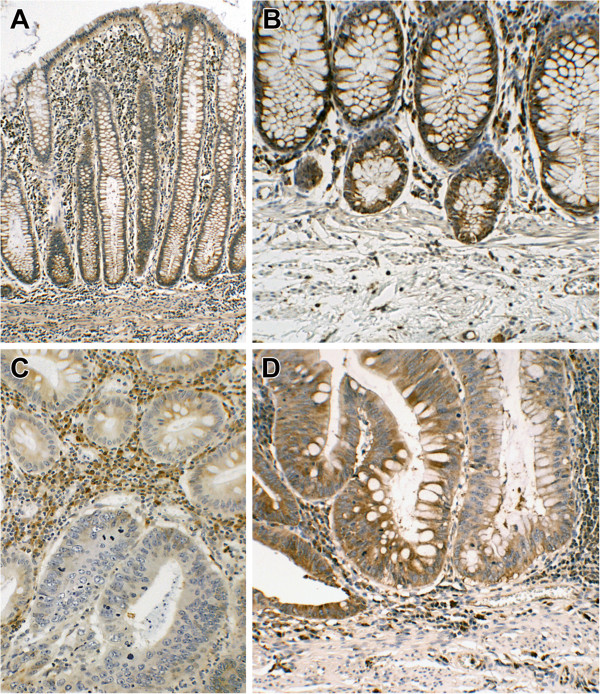
Immunohistochemical detection of RBFOX1. (A) Normal appearing colonic mucosa with diffuse cytoplasmic staining of most epithelium (x100). (B) Normal colon with diffuse staining of the epithelium and some stromal cells and inflammatory cells; the pericryptal sheath fibroblasts appear unstained (x200). (C) Adenocarcinoma showing absence of RBFOX1 staining in the majority of the cells lining the two-neoplastic glands in the lower half of the picture (x200). (D) Neighbouring glands show persistence (left) or reduction/ loss (right) of RBFOX1 immunoreactivity on epithelial cell cytoplasm (x200).
To assess the mutation frequency of RBFOX1 in CRC, we performed high-resolution melt (HRM) analysis on 39 CRC cell lines and 137 tumour and normal samples. All changes were then confirmed by bidirectional Sanger sequencing. In total, we found six novel mutations in five cell lines, (5/39, 12.8%) and three novel mutations in two tumour/normal samples pairs (2/133, 1.5%) (Table 2, Additional file 4: Table S3). The somatic mutations were further analyzed for the potential or likely effect of amino acid substitutions on protein structure and function, using Sorts Intolerant From Tolerant (SIFT) and Polymorphism Phenotyping (PolyPhen-2) (Additional file 4: Table S3). Of the nine novel mutations, two were predicted to be possibly damaging/deleterious by SIFT and PolyPhen-2, two were silent mutations and the remaining five produced contradictory predictions by SIFT and PolyPhen-2. Further investigations will be required to understand the functional effects of these somatic mutations on RBFOX1 function and CRC.
Table 2.
RBFOX1 mutations and single nucleotide polymorphisms found in CRC cell lines and patient tumour samples
| Mutations | Known SNPs | |
|---|---|---|
|
Cell Lines | ||
| CoCM1 |
c.776C>T; p.Ser259Phe |
|
| HCT15 |
c.288C>T; p.Ser96Ser |
|
| LIM2405 |
|
c.393A>G; p.Glu131Glu (rs140174146) |
| LIM2551 |
c.154delC |
|
| LOVO |
1. c.293C>T; p.Ala98Val |
|
| |
2. c.353C>T; p.Thr118Met |
|
| LS411 |
c.293C>T; p.Ala98Val |
|
|
Patient Samples | ||
| 33 |
1. c.851C>T; p.Ala284Val |
|
| |
2. c.1023G>A; |
|
| |
p.Gln341Gln |
|
| 53 |
c.350C>T; p.Pro117Leu* |
|
| 93 | c.598G>A; p.Val200Met | |
*Mutation from patient 53 is identical to known SNP rs139251660, but was present only in tumour but not in the paired normal tissue.
Discussions
Very little research has been done to characterise CRCs arising in Bangladeshi patients, despite the reportedly low frequency of CRC and suggestions of a high proportion of early-onset CRCs in this population [14]. Here we confirmed that BAN patients have a relatively early age of onset of CRC compared to CAU patients presenting at the same hospital and that the age of onset remains low even after potential LS cases have been omitted. This raises the possibility that BAN CRCs have arisen as a result of environmental influences and/or a genetic predisposition that can lead to somatic molecular changes and clinicopathological features dissimilar to those found in tumours from the wider UK population. This study aimed to uncover any clinical or genetic features specific to BAN CRCs.
We observed no major differences in the clinical features between early- and late-onset BAN MSS, and most findings were consistent with CAU MSS cancers reported previously [11]. Survival of the early-onset and the late-onset BAN MSS CRC was similar, even though the early-onset cases were all left-sided unlike the late-onset (62.5% left-sided). We cannot, however, rule out a relationship between age of onset and survival as our cohort was relatively small. It also remains to be seen if BAN CRC characteristics change over time and between generations, particularly as environmental factors such as diet change can impact on the population.
There were, however, an unexpectedly large proportion of mucinous tumours in the BAN sample set. This observation may be linked to the reduced frequency of KRAS mutations in BAN MSS compared with CAU CRCs and CRCs in other populations. Specifically, KRAS is mutated at frequencies between 30%-44% in African American, Chinese and Japanese CRCs [23-25]. It is difficult to ascertain if the high proportion of early-onset cases causes the reduction in the number of KRAS mutations in BAN, as previous studies suggested only marginal differences between early-onset and late-onset CAU CRCs with regard to KRAS mutations (<50 years, 29%; >50 Years, 33%) [26]. These frequencies also concur with those we reported previously for CAU CRCs [11]. On the other hand, there is evidence that KRAS mutation frequency is lower in early-onset CRC in India (≤50 years, 24%; ≥60 Years, 47%) [27]. Further investigation in BAN CRCs will help to explain the aetiology behind the low KRAS mutations and high mucin percentage.
We found no BRAF mutations in BAN MSS CRC, although the absence of MSI cancers in the cohort may contribute to this observation [28]. Overall, it will be interesting to confirm these results in a larger cohort of BAN patients and establish how this may affect clinical decision, particularly in view of the increased use of targeted therapy in CRC.
We utilised high-density SNP bead arrays to detect any genomic alterations that might be specific to BAN patients. The BAN cohort contained both diploid and polyploid cancers, and the copy number changes detected were largely comparable to those seen in the CAU CRCs. We also identified small, heterozygous deletions on chromosome 16p13.2 in a substantial number of BAN and CAU MSS CRCs. Deletion of this part of the genome has been previously described in a number of studies on genomic alterations in CAU and Japanese CRCs [29-33].
In our sample set, the deletion was more frequent in BAN CRCs compared to CAU cancers, although there was no difference in occurrence between early- and late-onset BAN CRCs. This region of the genome encodes RBFOX1, a highly conserved RNA-binding protein that regulates tissue-specific alternative splicing indicating important basic functions in development and differentiation. When we examined RBFOX1 in a large number of CRCs from The Cancer Genome Atlas Network sample set, we found that the regulatory regions and 5’ exons of the gene is often deleted, which may lead to aberrant RBFOX1 expression or isoform distribution. Interestingly, in a recent study the 5’ untranslated region of RBFOX1 was also rearranged in 4/25 melanoma [34], pointing to the possible importance of this part of the gene.
Very little is known about the expression or role of RBFOX1 in the intestine. Using immunohistochemistry, we confirmed that RBFOX1 is expressed at low levels in normal gut tissues and that expression is often lost in CRC. We also identified a small number of novel somatic mutations in CRC. Therefore, RBFOX1 appears to be targeted by various mechanisms in CRC. Functionally, loss of RBFOX1 activity may lead to aberrations in the splicing of a significant number of genes, generating diverse functional products that vary from those found in normal tissue [35]. Alternative splicing is a key feature of cancer [36-38] including CRC [35,39] and the identification of RBFOX1 targets in CRC will help to determine if RBFOX1 deletion is a critical feature of dysfunctional splicing in CRC.
To our knowledge, this is the first report of genomic characterisation of CRC in British Bangladeshi populations. We concluded that there were no genetic copy number alterations unique to BAN MSS CRCs, but that mutations of RAS signalling oncogenes were comparatively rare. The functional role of RBFOX1 mutations in BAN and CAU MSS CRC highlights aberrant alternative splicing in CRC as an important mechanism for further study.
Materials and methods
Patients and tumour samples
CRCs were resected from 44 patients of Bangladeshi origin at Barts and the London NHS Trust between January 1997 and August 2008. Tumour tissue (fresh frozen or formalin fixed paraffin embedded) was obtained from 37 patients. Within this group, 17 (46%) were 45 years old or under at the time of diagnosis.
Fresh-frozen tumor and matched normal tissues were retrieved from 133 stage II and III CRC patients treated at the Royal Melbourne Hospital and Western Hospital Footscray, Australia. A total of 67 individuals were female and 66 were male, with a median age at diagnosis of 73 years (range, 30–92 years). 89 cancers were stage II and 44 were stage III. 57 cancers were from the proximal colon, 64 from the distal colon and 12 from the rectum.
Microsatellite instability genotyping and immunohistochemistry of mismatch repair protein
For microsatellite instability assay, DNA was extracted from tumour tissues using QIAamp DNA mini kit (Qiagen, Hilden, Germany) and genotyping was performed using BAT25 and BAT26 mononucleotide markers [40] on the ABI 3100 DNA Sequencer (Applied Biosystems) and analysed using GeneScan software (Applied Biosystems). For fresh frozen tumour tissues, only samples with >80% tumour were used in experiments, and FFPE tumour tissues were macrodissected before DNA extraction. Tumours were classified as microsatellite unstable if one or more of the markers showed bandshift of 3 base pairs or more. The expression of the mismatch repair proteins was assessed using immunohistochemistry (IHC) [41].
Statistical analysis
GraphPad Prism version 4.03 for Windows (GraphPad Software, San Diego, California, USA) was used for statistical analysis.
Single nucleotide polymorphism beadarrays and data analysis
Single nucleotide polymorphism (SNP) genotyping of Bangladeshi and Caucasian MSS CRCs was performed using the HumanHap 550 Duo and 370 Duo BeadChip (Illumina) respectively, after confirmation of DNA quality using Agilent biochips. For comparison, we also utilised Ilumina 610-Quad SNP genotyping data from 636 cancers obtained from the Ludwig Colon Cancer Initiative, Australia.
For copy number analysis, SNP data for the tumour samples were pre-processed by regressing Log R Ratio values for the tumour against corresponding values for the paired normal to reduce the effect of array artefacts. The SNP data was then examined for copy number and LOH changes in paired tumour/normal tissue using OncoSNP (version 2.25) [PMID: PMC2965384]. We used the intra-tumour heterogeneity option with 10 EM iterations and sub-sampling window size of 30. Two early-onset BAN and 4 CIN samples were excluded on the basis of poor data quality. For comparisons, copy number and LOH calls from the Illumina Human Hap500 and 610-Quad SNP genotyping data were remapped using nearest neighbour interpolation on to the HumanHap370-Duo probe set. FOXRB1 deletions were further investigated in a dataset of 419 colorectal cancers from The Cancer Genome Atlas Project. Affymetrix SNP 6.0 CEL files were preprocessed and converted into the OncoSNP (v1.3) format using the PennCNV-Affy tool [PMID: PMC2045149]. Colorectal tumours showing RBFOX1 deletions were identified and then clustered using a k-means algorithm. Genomic coordinates are in Human Genome Build hg18.
Mutation analysis
Mutation hotspots in KRAS and BRAF were analysed by direct sequencing (Life Technologies). RBFOX1 exons were screened using high resolution melt curve analysis on xthe ABI 7500 Fast Real Time PCR system (Life Technologies) and analysed using the HRMv2.0.1 software (Life Technologies). Somatic mutations were then validated by bidirectional Sanger sequencing twice and confirmed to be absent in paired normal tissues. Primer sequences are available from authors. Mutations were analysed for the effect of amino acid substitutions on protein structure and function, using Sorts Intolerant From Tolerant (SIFT) at http://sift.bii.a-star.edu.sg/ and Polymorphism Phenotyping (PolyPhen-2) at http://coot.embl.de/PolyPhen/.
Quantitative real time PCR and immunohistochemistry on RBFOX1
Quantitative Real-time polymerase chain reaction (QPCR) was performed using TaqMan assays (Applied Biosystems) against RBFOX1 (Hs00251554_m1) and thecontrol housekeeping gene HPRT (hypoxanthine phosphoribosyltransferase 1;HuHPRT_0604009). Immunohistochemistry was carried out using rabbit polyclonal primary antibody against RBFOX1 (sc-135476, Santa Cruz Biotechnology).
Ethics approval
Ethical approval for this study was obtained from the Human Research Ethics Committees of all sites and all patients gave informed consent.
Competing interest
The author(s) declare that they have no competing interests.
Authors’ contributions
NSe, ASa, NSu, GPE, CL performed the experiments and data analysis. CY, DM, JBC carried out bioinformatics and biostatistics. PG, AG, MT, SA, RF, DP, SD provided tissue samples and clinical data. RF performed analysis for immunohistochemistry. OS, ASi and CL contributed to design the studies, interpretion of the data and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1. Loss of mismatch repair proteins MLH1, MLH6 and PMS2 in a subset of British Bangladeshis colorectal cancers. No tumour showed loss of MSH2.
Figure S1. UCSC Genome Browser showing hemizygous deletions/LOH (Red) and homozygous deletions (Yellow) of RBFOX1 in The Cancer Genome Atlas colorectal adenocarcinoma sample set. Deletions are typically hemizygous loss of the entire gene and focal deletions primarily targeting 5’ end of the gene.
Table S2.RBFOX1 mRNA expression in CRC cell lines and paired CRC and normal tissues samples.
Table S3. Somatic mutations and single nucleotide polymorphisms found in CRC cell lines and paired CRC and normal tissues samples.
Contributor Information
Neel Sengupta, Email: neel.sengupta@qmul.ac.uk.
Christopher Yau, Email: c.yau@imperial.ac.uk.
Anuratha Sakthianandeswaren, Email: Anu.Sakthianandeswaren@ludwig.edu.au.
Dmitri Mouradov, Email: Dmitri.Mouradov@ludwig.edu.au.
Peter Gibbs, Email: peter.gibbs@ludwig.edu.au.
Nirosha Suraweera, Email: n.suraweera@qmul.ac.uk.
Jean-Baptiste Cazier, Email: jcazier@well.ox.ac.uk.
Guadalupe Polanco-Echeverry, Email: guadalupita@yahoo.com.
Anil Ghosh, Email: a.ghosh@qmul.ac.uk.
Mohamed Thaha, Email: m.a.thaha@qmul.ac.uk.
Shafi Ahmed, Email: shafi.ahmed@bartsandthelondon.nhs.uk.
Roger Feakins, Email: r.m.feakins@qmul.ac.uk.
David Propper, Email: d.j.propper@qmul.ac.uk.
Sina Dorudi, Email: sinadorudi@me.com.
Oliver Sieber, Email: Oliver.Sieber@ludwig.edu.au.
Andrew Silver, Email: a.r.silver@qmul.ac.uk.
Cecilia Lai, Email: c.lai@qmul.ac.uk.
Acknowledgements
We thank the Victorian Cancer Biobank for the provision of patient specimens and BioGrid Australia for providing de-identified clinical data, Professor Richard Poulsom for preparing and analysis of Figure 4 and Linda Hammond for help with ploidy analysis.
Funding
This work was supported by Barts and The London Charity. Neel Sengupta is supported by a Bowel and Cancer Research clinical fellowship. Christopher Yau is supported by a UK Medical Research Council Special Training Fellowship in Biomedical Informatics (Ref. No. G0701810), Nirosha Suraweera is a Barts and The London Charity UK post-doctoral fellow and Anil Ghosh is a Constance Travis Clinical Fellow.
References
- Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- Bulow S. Clinical features in familial polyposis coli. Results of the Danish Polyposis Register. Dis Colon Rectum. 1986;29:102–107. doi: 10.1007/BF02555389. [DOI] [PubMed] [Google Scholar]
- Caspari R, Friedl W, Mandl M, Moslein G, Kadmon M, Knapp M, Jacobasch KH, Ecker KW, Kreissler-Haag D, Timmermanns G. et al. Familial adenomatous polyposis: mutation at codon 1309 and early onset of colon cancer. Lancet. 1994;343:629–632. doi: 10.1016/S0140-6736(94)92634-4. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Franken PF, Reinards TH, Weiss MM, Wagner A, van der Klift H, Kloosterman S, Houwing-Duistermaat JJ, Aalfs CM, Ausems MG. et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP) J Med Genet. 2005;42:e54. doi: 10.1136/jmg.2005.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai SJ, Xu Y, Cai GX, Lian P, Guan ZQ, Mo SJ, Sun MH, Cai Q, Shi DR. Clinical characteristics and diagnosis of patients with hereditary nonpolyposis colorectal cancer. World J Gastroenterol. 2003;9:284–287. doi: 10.3748/wjg.v9.i2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winawer SJ, Fletcher RH, Miller L, Godlee F, Stolar MH, Mulrow CD, Woolf SH, Glick SN, Ganiats TG, Bond JH. et al. Colorectal cancer screening: clinical guidelines and rationale. Gastroenterology. 1997;112:594–642. doi: 10.1053/gast.1997.v112.agast970594. [DOI] [PubMed] [Google Scholar]
- Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087. doi: 10.1053/j.gastro.2009.12.064. e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Cai G, Xu Y, Lu H, Shi Y, Lian P, Peng J, Du X, Zhou X, Guan Z, Shi D, Cai S. Clinicopathologic and molecular features of sporadic microsatellite- and chromosomal-stable colorectal cancers. Int J Colorectal Dis. 2008;23:365–373. doi: 10.1007/s00384-007-0423-7. [DOI] [PubMed] [Google Scholar]
- Silver A, Sengupta N, Propper D, Wilson P, Hagemann T, Patel A, Parker A, Ghosh A, Feakins R, Dorudi S, Suraweera N. A distinct DNA methylation profile associated with microsatellite and chromosomal stable sporadic colorectal cancers. Int J Cancer. 2012;130:1082–1092. doi: 10.1002/ijc.26104. [DOI] [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D. et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Balarajan R, Raleigh VS. Patterns of mortality among Bangladeshis in England and Wales. Ethn Health. 1997;2:5–12. doi: 10.1080/13557858.1997.9961810. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Banerjea A, Hands RE, Bustin S, Dorudi S. Microarray profiling of colorectal cancer in Bangladeshi patients. Colorectal Dis. 2005;7:571–575. doi: 10.1111/j.1463-1318.2005.00818.x. [DOI] [PubMed] [Google Scholar]
- Joy D, Hazel G, Linda Z, editor. Office for National Statistics. Focus on Ethnicity and Religion. (Newport, South Wales, UK) through Palgrave Macmillan at Basingstoke, Hampshire, UK: The Office of National Statistics; [Google Scholar]
- Symonds DA, Vickery AL. Mucinous carcinoma of the colon and rectum. Cancer. 1976;37:1891–1900. doi: 10.1002/1097-0142(197604)37:4<1891::AID-CNCR2820370439>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Bazan V, Migliavacca M, Zanna I, Tubiolo C, Grassi N, Latteri MA, La Farina M, Albanese I, Dardanoni G, Salerno S. et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13:1438–1446. doi: 10.1093/annonc/mdf226. [DOI] [PubMed] [Google Scholar]
- Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G, Harris A, Ragoussis J, Sieber O, Holmes CC. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol. 2010;11:R92. doi: 10.1186/gb-2010-11-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann K, Terdiman JP, French AJ, Roydasgupta R, Sein N, Kakar S, Fridlyand J, Snijders AM, Albertson DG, Thibodeau SN, Waldman FM. Chromosomal instability in microsatellite-unstable and stable colon cancer. Clin Cancer Res. 2006;12:6379–6385. doi: 10.1158/1078-0432.CCR-06-1248. [DOI] [PubMed] [Google Scholar]
- TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, Yasuda K, Inoue K. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22:905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata S, Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33:2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K, Brim H, Giardiello F, Smoot DT, Nouraie M, Lee EL, Ashktorab H. Distinct BRAF (V600E) and KRAS mutations in high microsatellite instability sporadic colorectal cancer in African Americans. Clin Cancer Res. 2009;15:1155–1161. doi: 10.1158/1078-0432.CCR-08-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Zhou J, Yang Z, Huang Y, Wu X, Shen H, Tang J, Chen Q. KRAS, BRAF and PIK3CA mutations and the loss of PTEN expression in Chinese patients with colorectal cancer. PLoS One. 2012;7:e36653. doi: 10.1371/journal.pone.0036653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi R, Harada J, Tuul M, Zhao Y, Ando K, Saeki H, Oki E, Ohga T, Kitao H, Kakeji Y, Maehara Y. Prognostic relevance of KRAS and BRAF mutations in Japanese patients with colorectal cancer. Int J Clin Oncol. 2012. [Epub ahead of print] [DOI] [PubMed]
- Berg M, Danielsen SA, Ahlquist T, Merok MA, Agesen TH, Vatn MH, Mala T, Sjo OH, Bakka A, Moberg I. et al. DNA sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset. PLoS One. 2010;5:e13978. doi: 10.1371/journal.pone.0013978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman R, Kotapalli V, Adduri R, Gowrishankar S, Bashyam L, Chaudhary A, Vamsy M, Patnaik S, Srinivasulu M, Sastry R, Evidence for possible non-canonical pathway(s) driven early-onset colorectal cancer in India. Mol Carcinog. 2012. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- Andersen CL, Lamy P, Thorsen K, Kjeldsen E, Wikman F, Villesen P, Oster B, Laurberg S, Orntoft TF. Frequent genomic loss at chr16p13.2 is associated with poor prognosis in colorectal cancer. Int J Cancer. 2011;129:1848–1858. doi: 10.1002/ijc.25841. [DOI] [PubMed] [Google Scholar]
- Douglas EJ, Fiegler H, Rowan A, Halford S, Bicknell DC, Bodmer W, Tomlinson IP, Carter NP. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817–4825. doi: 10.1158/0008-5472.CAN-04-0328. [DOI] [PubMed] [Google Scholar]
- Kurashina K, Yamashita Y, Ueno T, Koinuma K, Ohashi J, Horie H, Miyakura Y, Hamada T, Haruta H, Hatanaka H. et al. Chromosome copy number analysis in screening for prognosis-related genomic regions in colorectal carcinoma. Cancer Sci. 2008;99:1835–1840. doi: 10.1111/j.1349-7006.2008.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JF, Gariboldi M, Sokolova V, Capobianco P, Lampis A, Perrone F, Signoroni S, Costa A, Leo E, Pilotti S, Pierotti MA. Integrative approach for prioritizing cancer genes in sporadic colon cancer. Genes Chromosomes Cancer. 2009;48:953–962. doi: 10.1002/gcc.20697. [DOI] [PubMed] [Google Scholar]
- Richter H, Slezak P, Walch A, Werner M, Braselmann H, Jaramillo E, Ost A, Hirata I, Takahama K, Zitzelsberger H. Distinct chromosomal imbalances in nonpolypoid and polypoid colorectal adenomas indicate different genetic pathways in the development of colorectal neoplasms. Am J Pathol. 2003;163:287–294. doi: 10.1016/S0002-9440(10)63652-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P. et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang X, Wu F, Huang R, Xue F, Liang G, Tao M, Cai P, Huang Y. Transcriptome Profiling of the Cancer. Adjacent Non- Tumor and Distant Normal Tissues from a Colorectal Cancer Patient by Deep Sequencing. PLoS One. 2012;7:e41001. doi: 10.1371/journal.pone.0041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Schneider-Poetsch T, Yoshida M. Splicing in oncogenesis and tumor suppression. Cancer Sci. 2012;103(9):1611–6. doi: 10.1111/j.1349-7006.2012.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64:7647–7654. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, Dee S. et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukola A, Eklin K, Laiho P, Salovaara R, Kristo P, Jarvinen H, Mecklin JP, Launonen V, Aaltonen LA. Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC) Cancer Res. 2001;61:4545–4549. [PubMed] [Google Scholar]
- Lipton LR, Johnson V, Cummings C, Fisher S, Risby P, Eftekhar Sadat AT, Cranston T, Izatt L, Sasieni P, Hodgson SV. et al. Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J Clin Oncol. 2004;22:4934–4943. doi: 10.1200/JCO.2004.11.084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Loss of mismatch repair proteins MLH1, MLH6 and PMS2 in a subset of British Bangladeshis colorectal cancers. No tumour showed loss of MSH2.
Figure S1. UCSC Genome Browser showing hemizygous deletions/LOH (Red) and homozygous deletions (Yellow) of RBFOX1 in The Cancer Genome Atlas colorectal adenocarcinoma sample set. Deletions are typically hemizygous loss of the entire gene and focal deletions primarily targeting 5’ end of the gene.
Table S2.RBFOX1 mRNA expression in CRC cell lines and paired CRC and normal tissues samples.
Table S3. Somatic mutations and single nucleotide polymorphisms found in CRC cell lines and paired CRC and normal tissues samples.