

Abstract
Background
Flax (Linum usitatissimum L.) is an important crop for the production of bioproducts derived from its seed and stem fiber. Transposable elements (TEs) are widespread in plant genomes and are a key component of their evolution. The availability of a genome assembly of flax (Linum usitatissimum) affords new opportunities to explore the diversity of TEs and their relationship to genes and gene expression.
Results
Four de novo repeat identification algorithms (PILER, RepeatScout, LTR_finder and LTR_STRUC) were applied to the flax genome assembly. The resulting library of flax repeats was combined with the RepBase Viridiplantae division and used with RepeatMasker to identify TEs coverage in the genome. LTR retrotransposons were the most abundant TEs (17.2% genome coverage), followed by Long Interspersed Nuclear Element (LINE) retrotransposons (2.10%) and Mutator DNA transposons (1.99%). Comparison of putative flax TEs to flax transcript databases indicated that TEs are not highly expressed in flax. However, the presence of recent insertions, defined by 100% intra-element LTR similarity, provided evidence for recent TE activity. Spatial analysis showed TE-rich regions, gene-rich regions as well as regions with similar genes and TE density. Monte Carlo simulations for the 71 largest scaffolds (≥ 1 Mb each) did not show any regional differences in the frequency of TE overlap with gene coding sequences. However, differences between TE superfamilies were found in their proximity to genes. Genes within TE-rich regions also appeared to have lower transcript expression, based on EST abundance. When LTR elements were compared, Copia showed more diversity, recent insertions and conserved domains than the Gypsy, demonstrating their importance in genome evolution.
Conclusions
The calculated 23.06% TE coverage of the flax WGS assembly is at the low end of the range of TE coverages reported in other eudicots, although this estimate does not include TEs likely found in unassembled repetitive regions of the genome. Since enrichment for TEs in genomic regions was associated with reduced expression of neighbouring genes, and many members of the Copia LTR superfamily are inserted close to coding regions, we suggest Copia elements have a greater influence on recent flax genome evolution while Gypsy elements have become residual and highly mutated.
Keywords: Transposable elements, Flax, Genome evolution, LTR elements, Gene expression
Background
Transposable elements (TEs) influence the evolution, structure, amplification, gene creation, mutation and transcriptional regulation of genes and genomes [1-6]. They are also useful as genetic markers in basic and applied science [7,8]. TEs occupy a substantial fraction of sequenced plant genomes [9], ranging from over 14% in Arabidopsis [10] to more than 80% in maize [11]. Because of their nature and characteristic patterns of insertion [12], TEs may influence large portions of the genome. A study found that one-sixth of all rice genes had some kind of association with TEs [13]. Some TE insertions occur within or near genes, thereby disrupting normal gene expression [12]. Such insertions may influence phenotypic characteristics, as in petal color of gentians [14], or disruption of vitamin E synthesis in sunflower [15]. However, due to gene redundancy or to insertion in regions of the genome that do not affect gene expression, the majority of TE insertions do not have detectable effects on morphology or physiology. For example, neither the insertion of a Stowaway element in an intron of the manganese superoxide dismutase gene [16], nor the insertion of retrotransposon Vine-1 in one member of the alcohol dehydrogenase multigene family [17] affected plant growth and development. Nevertheless, TEs can influence the evolution of plant gene families, as exemplified by disease resistance genes in several plants [18]. Insertions can also result in the capture of gene fragments by TEs, or the adoption of parts of TEs by genes. Some of the clearest examples of gene capture by TEs involve Pack-MULEs. In rice, over 3000 of these gene-carrying transposon-derived elements were found in 440 Mb of sequence [19], and the acquisition of multiple gene fragments from multiple loci may result in the creation of new genes [20]. Genes such as FAR1 and FHY3 (involved in the phytochrome signalling pathway), have a conserved transposase-derived region, whose DNA binding and regulatory capacities have been adopted for transcriptional control of downstream genes [21,22]. As was first shown by McClintock in the early experiments that uncovered the Ac/Ds TE system in maize [23-26], some types of stress can activate TEs, which can in turn modify gene expression. TE expression triggered by stress has been reported for several elements including: Tnt1 [27,28] and Tto1 [29,30] in tobacco; Tos17 in rice [31,32]; and BARE-1 in barley [33]. However, relatively few active TEs have been identified and several expression studies indicate that transcription and transposition are rare for most elements [12]. While some studies have focused on the expression of individual elements, more recent approaches have compared genome-wide expression data of TEs. These kind of studies have been used to identify TE cassettes in expressed genes in coffee species [34] and Arabidopsis [35], and the activity of different TE families in maize [36] and sugarcane [37]. Flax (L. usitatissimum) is one of over 270 species within the family Linaceae, and is a member of the order Malpighiales along with three other species with published whole genome sequences: poplar (Populus trichocarpa), cassava (Manihot esculenta), and castor (Ricinus communis) [38]. Flax is a predominantly self-polinating annual crop grown in temperate regions [39]. Distinct varieties of flax are cultivated for either seed (i.e. linseed) or bast fibers. We recently reported a whole genome shotgun (WGS) assembly of a linseed variety, CDC Bethune [40]. The assembly contains 302Mb of the estimated 373Mb nuclear genome, in scaffolds with N50=694kb. Flax is considered a diploid (2n=2x=30), although our genome analysis pointed to a recent whole genome duplication 5-9MYa. Flax appears to have originated from its wild relative, L. bienne, with cultivation and domestication probably starting in the Mesopotamian valleys between 8000–10000 years ago [41]. Flax has been studied for decades as a model of genome plasticity [42-45]. In the variety Stormont Cirrus, individuals exposed to certain stresses can produce first generation progeny that show stable changes in several traits including an up to 15% difference in nuclear DNA content. Highly repetitive, tandemly arrayed elements (e.g. 5S rDNA) are among the major contributors to this DNA content variation. A novel, non-TE, low-copy insertion sequence (LIS-1) is also associated with these changes [42,46]. It should be noted that most elite flax varieties, including CDC Bethune, which is the subject of the WGS assembly, do not exhibit this rapid change in genome size. Nevertheless, the study of flax and its repetitive sequences remains of special relevance to understanding genome evolution in general. We previously reported the preliminary identification of TEs as part of the description of the flax WGS assembly [40]. The assembly contained 23.06% TEs as defined by sequence coverage. While the calculated proportion of the genome covered by TEs in flax is slightly lower than other plant species with small genomes, much variation exists in TE content in plants [9]. Only a small proportion of the TEs described in the flax genome could be identified through alignment to previously characterized elements from other species [40]. Instead, most of the TEs were identified only by de novo prediction methods. Here we extend this previous report to present a detailed characterization of the main superfamilies of TEs in flax and to explore their potential influence on genome evolution and gene expression.
Results
TEs in the flax genome
In a previous study, we described a de novo whole genome shotgun (WGS) assembly of flax based on next-generation (Illumina) sequencing [40], including a brief description of the transposable element (TE) component of that assembly. Using various bioinformatics tools to identify repeats de novo, we found a total of 8,162 putative interspersed repeats divided into 456 consensus interspersed repeats for PILER, 5,440 repeats for RepeatScout, 1,977 LTR elements from LTR_finder and 289 LTR elements using LTR_STRUC (Additional file 1). Each of these tools offered certain advantages. For example, PILER was faster and had longer average output sequence length (882.5 bp) than RepeatScout (353.5 bp), but at the same time PILER was more stringent and identified fewer sequences. Furthermore, LTR_finder found more sequences than LTR_STRUC but a few sequences were only found using LTR_STRUC. Although the parameters used with the algorithms for de novo repeat finding were set to find interspersed repeats and to filter out low complexity regions, some of the repeats identified may have nevertheless constituted non-TE gene families, pseudogenes or highly repeated gene domains. We therefore curated the repeats to identify those that most likely represented TEs. After curation, the filtered library had a total of 2142 putative TEs: 85 from PILER, 767 from RepeatScout, 1039 from LTR_finder and 251 from LTR_STRUC (Additional file 1). We combined these annotated de novo repeats with the TEs from the Viridiplantae division of Repbase, to make a database for Repeatmasker, which, when applied to the flax genome assembly, masked a total of 73.8 Mb (23.06% of the assembly) as sequence with high similarity to TEs (Table 1). LTR retrotransposons of the superfamilies Copia and Gypsy were the dominant group with over 69% of the hits and over 74% of the sequence coverage. These superfamilies were followed by the non-LTR retrotransposons of the L1 group and the DNA transposons from the Mutator superfamily. When these results were compared with the analysis of 54.6 Mb of Sanger dideoxy sequence reported for BAC-ends from the same flax variety [47], we found that both data sets showed LTR elements to be the most prevalent group of TEs, with the Copia group as the most abundant type followed by Gypsy elements and LINEs. However, hAT elements were more abundant than Mutator elements in the BAC-end sequences, in contrast to our observations from the WGS assembly. Other smaller groups also differed in their rank order in the BAC and WGS analyses, and the total proportional coverage for the WGS was always higher than for the BAC-end sequencing. This was probably due to differences in methodology; whereas in the present study we used both similarity-based and de novo identification, the BAC-end analysis relied mainly on similarity-based approaches for repeat identification.
Table 1.
Annotation of TE superfamilies in flax WGS assembly determined using a filtered consolidated library produced with de-novo repeats from PILER, RepeatScout, LTR_finder and LTR_STRUC, and a library of TE from the viridiplantae division from Repbase
| Class | Order | Superfamily | No. of matches | Elements percentage (%) | Sequence occupied (bp) | Sequence percentage of TEs (%) | Sequence percentage of genome (%) |
|---|---|---|---|---|---|---|---|
|
Retrotransposons |
LTR |
Copia |
89951 |
38.31 |
29594882 |
40.08 |
9.30 |
| |
|
Gypsy |
72626 |
30.93 |
25123127 |
34.02 |
7.89 |
| |
|
unclassified |
2797 |
1.19 |
902298 |
1.22 |
0.28 |
| |
DIRS |
DIRS |
2 |
0.00 |
102 |
0.00 |
0.00 |
| |
PLE |
Penelope |
548 |
0.23 |
30214 |
0.04 |
0.01 |
| |
LINE |
RTE |
11 |
0.00 |
618 |
0.00 |
0.00 |
| |
|
L1 |
27632 |
11.77 |
6684243 |
9.05 |
2.10 |
| |
SINE |
unclassified |
1 |
0.00 |
49 |
0.00 |
0.00 |
|
DNA transposons |
TIR |
Tc1-Mariner |
191 |
0.08 |
38231 |
0.05 |
0.01 |
| |
|
hAT |
7935 |
3.38 |
1986522 |
2.69 |
0.62 |
| |
|
Mutator |
21124 |
9.00 |
6320424 |
8.56 |
1.99 |
| |
|
P |
2 |
0.00 |
96 |
0.00 |
0.00 |
| |
|
Harbinger |
1384 |
0.59 |
344876 |
0.47 |
0.11 |
| |
|
En-Spm/CACTA |
8330 |
3.55 |
2372592 |
3.21 |
0.75 |
| |
Helitron |
Helitron |
2154 |
0.92 |
434859 |
0.59 |
0.14 |
| |
unclassified |
unclassified |
95 |
0.04 |
8981 |
0.01 |
0.00 |
| TOTALS | 234783 | 100.00 | 73842114 | 100.00 | 23.06 |
Putative expression and abundance of main families of TEs
We compared the TE sequences to EST databases to estimate the relative expression of each type of TE. The majority of ESTs queried were obtained from the same variety as was used for the WGS assembly [48]. To reduce redundancy, all of the putative TE sequences generated by the various de novo algorithms were first aligned to each other to generate clusters. Clustering was performed so that each member of a cluster had ≥80% similarity to every other member of the cluster. Each cluster was referred to as a family in accordance with the standards established by Wicker et al., [49]. A representative TE (usually the longest sequence) from each family was aligned to all available flax ESTs from Genbank. Table 2 shows the number and proportion of families in each major superfamily of TEs that aligned to ESTs. The LTR elements had the largest number of families that aligned to one or more ESTs. This was consistent with their observed abundance and coverage in the genome. LTR elements also had some of the highest proportions of expressed TE families. On the other hand, the hAT superfamily had fewer families than the LTR elements, but had a higher proportion of families with associated ESTs. To establish the copy number of each of the TE families, a representative sequence from each family (as described above) was aligned to the WGS assembly using BLAT. A threshold of 80% was used, following Wicker et al., [49]. Within superfamilies of TEs there were many families with only a few copies in the genome, and a small number of families with a high copy number (Figure 1). When the copy numbers within each family were compared to the relative number of ESTs aligned to that family, no correlation was observed (results not shown). This indicated that elements with a high copy number are not necessarily currently active. This was observed in all families, with the exception of the hAT superfamily, which showed a weak positive correlation (r = 0.66) between copy number and EST counts.
Table 2.
Putative expression of de-novo identified families of TEs. TE families are constituted by sequences with similarity ≥80% among all its members
| superfamily | total number of TE families | Number of families hitting at least one EST with a minimum coverage of 70% EST | proportion of TE families with putative expression |
|---|---|---|---|
|
Copia |
819 |
77 |
9.40 |
|
Gypsy |
263 |
20 |
7.60 |
|
L1 |
115 |
1 |
0.87 |
|
hAT |
68 |
7 |
10.29 |
|
Mutator |
60 |
0 |
0.00 |
|
En-Spm |
38 |
1 |
2.63 |
|
Helitron |
32 |
0 |
0.00 |
|
Harbinger |
15 |
0 |
0.00 |
| Tc1-Mariner | 7 | 0 | 0.00 |
Figure 1.

Copy numbers in TE families within superfamilies. Number of TE families in each one of the major superfamilies having 1–10, 11–20 or more than 20 copies in the genome.
Relationship of TEs to genes
We investigated the distribution of TEs with respect to genes in the WGS scaffolds. We limited our analysis to 108,219,748 bp of genome assembly that was contained in 71 scaffolds of 1Mb or longer, since this allowed analyses of relationships of several contiguous genes and/or TEs. When individual scaffolds were analyzed as units, there was a highly correlated inverse relationship between the coverage of TEs and genes (r = −0.92, p < 0.05 – Figure 2) meaning that overall TE distribution in the flax genome was not completely random. The same trend was found when we analyzed each of the four most abundant superfamilies separately (Copia, Gypsy, L1 and Mutator – Additional file 2). The distribution patterns in the 71 studied scaffolds showed that some had dense coverage of TEs (and few genes), while others had many genes but few TEs, and still other scaffolds had similar proportional coverage of TEs and genes (Additional file 3). We chose 12 representative scaffolds to illustrate the trends of distribution. Four of these were gene-rich, four had a similar proportion of genes and TEs, and four had a higher proportion of TEs than genes (see Figure 2 for the selected 12 scaffolds). We divided the 12 scaffolds into equally sized 50 kb bins and then calculated the proportional coverage of genes, TEs and the four largest superfamilies of transposable elements (Figure 3). Within the scaffolds that were rich in TEs, we observed a few bins in which the frequency of genes was also high (blue line overlapping red line in Figure 3A). The TE rich scaffolds were dominated by Copia and Gypsy superfamilies, with the later having a higher proportion. The L1 and Mutator elements had a lower proportional coverage than the LTR elements. The graphs did not show any apparent clustering pattern of any TE superfamily within each scaffold. A second group of scaffolds with similar proportional coverage of both TEs and genes showed several subregions in which TEs and genes overlapped or alternate in coverage (Figure 3B). Finally the gene-rich scaffolds seemed to be largely devoid of TEs (Figure 3C) and just a very few bins had proportional coverage of TEs close to 25%, while most bins were saturated for genes. We next used Monte Carlo (MC) statistics to test whether there was any scaffold (from our sample of 71 assembly units) in which TEs overlapped genes more frequently than expected by chance. Overlaps did not occur more frequently than expected by chance, whether the scaffolds were analyzed as individual units, or divided into 50 kb bins. Moreover, when the major superfamilies of TEs (Copia, Gypsy, the unclassified LTR elements, L1, Mutator, En-spm/CACTA, hAT, Helitron and Harbinger) were tested individually, none of the groups overlapped genes more than expected by chance in any scaffold (results not shown). Together this shows that that although regions of overlap did occur, and some TEs can be inserted into or close to genes, there were no individual scaffolds or bins with an unexpectedly high proportion of TEs in close association with genes. However, in scaffolds with a larger coverage of TEs, the probability of overlap with genes was higher as judged by a significant positive correlation between the proportion of bases overlapped by TEs in genes and the TE proportional coverage (r = 0.88 p < 0.05), and a significant negative correlation with the gene proportional coverage (r = −0.78 p < 0.05) (Additional file 4). In fact, when we used the 18,129 total predicted genes on these scaffolds to look for EST matches, it was found that the proportion of genes with matching ESTs in each scaffold was negatively correlated (r = −0.77 p < 0.05) with the proportional coverage of TEs (Figure 4). We next performed chi-square tests to determine whether any of the TE superfamilies, as compared to all other TEs, had a higher propensity to insert within or close to genes. Figure 5A shows that while the proportions were not significantly different (p = 0.005) for the Copia, Mutator and Harbinger elements, there were marginally significant differences for the L1 and CACTA TEs, and strong significant differences for the remaining superfamilies. From this later group the hATs, Helitrons and the unclassified LTRs were found inside of genes more often than expected by chance, while Gypsy TEs were less commonly found inside genes than expected. When the analysis was repeated for the regions flanking the genes, three groups of DNA transposons (hAT, CACTA and Mutator) showed significantly higher affinity for the 1 kb of sequence that flanked genes when compared to their overall distributions (Figure 5B). The same was true for the retrotransposons L1 and Copia, while Gypsy elements were significantly underrepresented in this region. Finally the analysis was repeated one last time for the flanking 5 kb of genes (Figure 5C) and in this opportunity only CACTA, L1 and Gypsy showed significant differences with this later one still below the expected numbers on these gene flanking regions.
Figure 2.
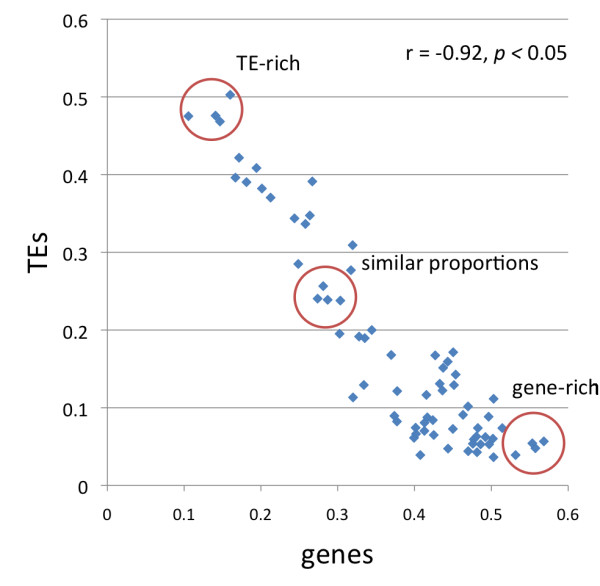
Correlation of the proportional coverage between genes and TEs in scaffolds ≥ 1Mb. Circled points indicate scaffolds selected for further analysis. TE-rich scaffolds (scaffolds 380, 50, 29 and 127); scaffolds with similar proportions of TEs and genes (scaffolds 33, 222, 151 and 132); gene-rich scaffolds (123, 464, 898 and 605).
Figure 3.

Distribution of genes and transposable elements in representative scaffolds. A) TE-rich scaffolds (scaffolds 380, 50, 29 and 127), B) Scaffolds with similar proportions of TEs and genes (scaffolds 33, 222, 151 and 132), C) Gene-rich scaffolds (123, 464, 898 and 605). The line graphs represent the proportional coverage (scale of 0.0 to 1.0) of TEs (red) and genes (blue) in 50kb windows. Each heat map row below each graph represent coverage of the four largest TE superfamilies – top to bottom: Copia, Gypsy, L1, Mutator, in 50kb windows (squares).
Figure 4.
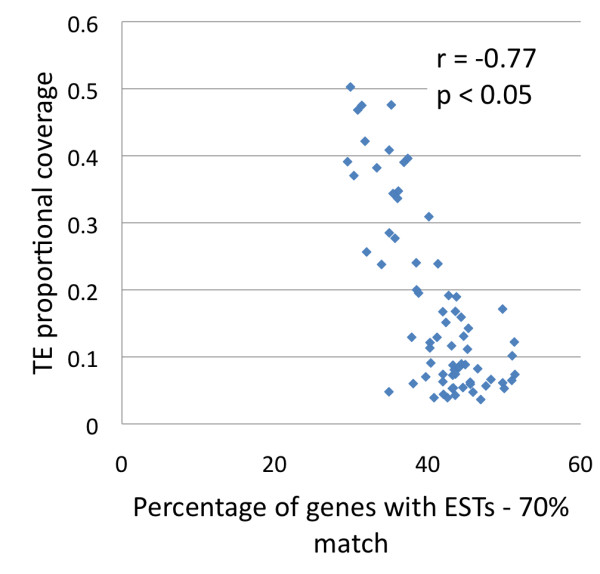
Correlation between the percentage of genes with putative expression and TE proportional coverage. The analysis was performed in scaffolds larger than 1 million bp. Each dot represent the x,y values of one scaffold. The TE proportional coverage is in a 0.0 to 1.0 scale.
Figure 5.
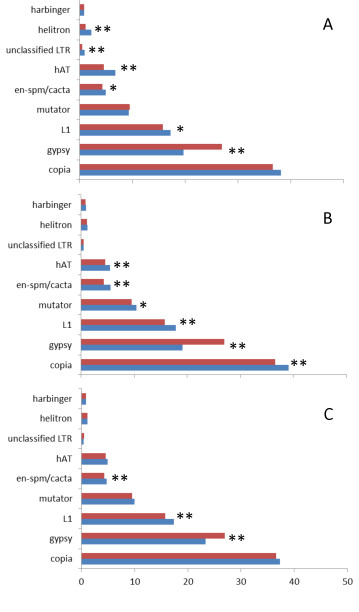
Comparison of TE hits in and around genes to TEs in the genome. A. In all scaffolds over 1 Mb vs. inside genes, B. In all scaffolds over 1 Mb vs. the adjacent 1kb up and downstream of genes, C. In all scaffolds over 1 Mb vs. the adjacent 5kb up and downstream of genes. The bars represent the percentage of the hits of each superfamily among all transposon hits in the scaffolds (red) or inside and in the adjacent gene regions (blue). Asterisks represent significance after Bonferroni correction at: *p < 5 x 10-3 or **p < 5 x 10-4.
Insertions of full LTR elements and evolution
A total of 2,266 putative LTR sequences found by LTR_finder and LTR_STRUC were filtered for redundancy, resulting in 1,767 unique sequences. After curation, some of these sequences were excluded from genome masking, because they contained internal non-TE genes, or other sequences of non-transposon origin, which would have resulted in masking of non-TE genome sections. However, the complete set of 1,767 unique sequences was used in subsequent analyses in order to more fully describe patterns of evolutionary importance like foreign DNA capture by TEs. Among the 1767 unique LTR sequences, 841 sequences corresponded to Copia elements, 207 were Gypsy, 667 were undetermined LTR elements, and the rest were LTRs that flanked other types of TEs. The Copia retrotransposons had an average size of 5.3 kb while the Gypsy TEs were 8.7 Kb on average. The LTR elements that had either regions of undetermined internal size, or regions bearing other types of TEs, were 5.9 kb on average (Additional file 5). When the LTR similarity was compared, the elements within these three groups had a similarity average of 95.4, 88.9 and 90.1 respectively; and the average distance between the TEs and the closest predicted gene was 3.2 kb (Copia), 7.6 (Gypsy) and 7.3 kb (undetermined) (Additional file 6). The divergence of intra-element LTR sequences was used to calculate the age of insertion of the unique elements, and a graph of their distribution in time was built (Figure 6). As seen in the figure, Copia elements had increasing activity in the last 5 million years, with many members active in the very recent past. There were 83 Copia sequences with 100% intra-element LTR similarity, and the average time of insertion of the elements in this superfamily was 1.4 Mya. In the meantime, the less abundant Gypsy elements increased activity around 7–8 Mya ago, but their activity started decreasing 3 to 4 Mya. There was only 1 Gypsy element with 100% LTR similarity and the average date of insertion for these elements was 4.1 Mya. Additionally there were four elements that were inserted more than 15 Mya (not included in Figure 6). Finally, the undetermined LTR elements (elements with internal regions not belonging to Copia or Gypsy domains) increased activity at around the same time as Gypsy elements, and were even more active than Copia elements until around 2.5 Mya, after which time they were still active but in a lesser proportion than Copia. There were 35 undetermined elements with 100% LTR similarity and their average date of insertion was 3.1 Mya. When the 119 retrotransposon sequences bearing 100% LTR pair intra-element similarity were mapped back to all scaffolds in the genome, we found a total of 147 insertion sites with 100% similarity to the original sequences. Only eight Copia TEs had more than one exact match (ranging from two to eight copies) in different genome regions (Additional file 7). When the match threshold was relaxed to 80% to find the number of copies that may have been related to the recently inserted copy, 19 elements had more than one related copy (ranging from 2 to 17 copies); from these, 18 were again Copia elements and one (with six copies) was an undetermined LTR element. When these recent insertion elements were compared to available ESTs, 19 Copia retrotransposons had a related EST (Additional file 8), and 11 undetermined elements had an EST match. Three of the undetermined elements had numerous EST matches (13, 16 and 92 hits respectively); but since the internal regions of these elements were either undetermined or matching sections of basal genes, the putative expression could correspond to genes located elsewhere in the genome. Finally, the distance between these recent TE insertions and genes showed that most of the recently inserted copies of LTR elements were located close to genes (Table 3). From among these LTR elements, all of those located within the first 1 kb flanking the genes were Copia when analyzing the 71 scaffolds of over 1 Mb, and 29 out of 31 were also Copia when analyzing all available scaffolds. To investigate the internal composition of the LTR sequences, all 1767 unique LTR sequences were used as queries in RepeatExplorer [50], which extracted protein domains of query sequences based on comparisons to a large database of transposons. Domains were identified only in the elements we had classified as Copia or Gypsy. All Copia and Gypsy elements matched domains only from their respective superfamilies with the exception of two TEs that we had classified as Copia elements but were found by RepeatExplorer to contain domains of Gypsy TEs. Whether these two instances represented nested insertions, recombination events, and chimeras or misassembled fragments is yet to be investigated. Overall, these results showed that our annotation of TEs was accurate (Additional file 9). Furthermore, Copia elements had more internal recognizable domains than Gypsy elements (Figure 7A). From a total of 841 complete LTR-copia elements, almost 55% contained four or five recognizable domains, while only 13% of the LTR-gypsy elements (a total of 207) had four recognizable domains, and only one sequence in which all five domains were recognized. When each domain was assessed separately, the proportion of Copia elements with a recognizable domain was always higher than in Gypsy (Figure 7B). These observations provide further evidence for a higher level of conservation and potential activity of Copia as compared to Gypsy.
Figure 6.

Insertion age of LTR elements in millions of years (Mya). Number of TEs inserted in the last 15 Mya for major groups of LTR retrotransposons.
Table 3.
Frequency of recent LTR elements insertions in proximity to predicted genes
|
distance from the closest gene |
scaffolds over 1MB |
all scaffolds |
||
|---|---|---|---|---|
| number of LTR elements | proportion of the number of LTR elements | number of LTR elements | proportion of the number of LTR elements | |
| 0-1000 |
13 |
46.43 |
31 |
43.06 |
| 1001-2000 |
4 |
14.29 |
12 |
16.67 |
| 2001-3000 |
2 |
7.14 |
5 |
6.94 |
| 3001-4000 |
1 |
3.57 |
4 |
5.56 |
| 4001-5000 |
1 |
3.57 |
2 |
2.78 |
| >5000 | 7 | 25.00 | 18 | 25.00 |
Figure 7.

Protein domains in LTR elements. Proportion of LTR elements where the main protein domains (GAG, PROT, INT, RT, RH) were identified. In A, the number of domains that could be identified is shown. In B, the percentage of sequences of each superfamily that have each one of the domains is depicted.
Discussion
TEs in the flax genome
The flax genome is estimated to be 373Mb in length, and we have reported here and previously that at least 23% of its sequence is made of TEs [40]. We expect that the actual TE coverage of the complete genome is higher than the 23% we reported, for the following reasons: (i) unclassified repeated sequences found by our de novo approach could constitute new or highly divergent types of TEs, but these were not used for masking; (ii) numerous LTR elements with unknown or non-TE internal domains were not included in the masking, and we did not use specific algorithms to identify possible TEs that lacked internal recognized domains; (iii) the WGS assembly may be missing some regions that are rich in repeated sequences [40]. If the complete genome sequence could be analyzed, including regions missing from the WGS assembly, we expect not only that the proportion of TEs would increase, but also the relative abundance of the main superfamilies could change since Gypsy elements are rich in heterochromatic regions [51-57], which are usually more difficult to assemble. Nevertheless, our estimate of genome coverage by TEs is comparable to what has been found for other sequenced plant genomes with sizes slightly larger than flax (e.g. Oryza sativa - 35% TEs, Lotus japonicus - 30.8%, Medicago truncatula 38%) [9]; indeed it has been proposed that in angiosperms, approximately one third of the genome is made up of TEs [9], which is in general agreement with our estimate for flax. Although TE content may be more related to genome size variation in plants with larger genomes [58] there is a trend showing that genome size correlates positively with the abundance and expansion of TEs [9]. While there are exceptions to this rule, flax with its smaller genome has a much lower percentage of TEs when compared to larger genomes like maize with over 85% TEs [11]. We found that LTR elements (especially Copia) dominated the population of TEs in the flax genome (Table 1). LTR retrotransposon abundance has been described in numerous plant species including some of the closely related species to flax that have been fully sequenced. In castor bean (Ricinus communis) the length covered by LTR elements accounts for about one third of all repeats while DNA TEs constitute less than 2% [59]; while in poplar (Populus trichocarpa) LTR elements constitute around 17% of the bases of all repeats (including low complexity repeats), and DNA TE content is close to 5% [60]. Although the proportion of sequence covered by LTR elements in flax is larger than in castor or poplar, the predominance of LTR elements is typical in many plant genomes [see supplementary table 7 in [61]. However, in most characterized genomes it is the Gypsy group that outnumbers the Copia group (Additional file 10). Ty3-gypsy elements are dominant in: Brachypodium distachyon [62], Oryza sativa [63], Zea mays [11], Sorghum bicolor [64], Carica papaya [65], Arabidopsis thaliana [10]- [LTR element coverage obtained from 61], Fragaria vesca [66], Malus domestica [61], Glycine max [67], Phaseolus vulgaris (data obtained from Phytozome - http://www.phytozome.net/), Populus trichocarpa [60] - [LTR element coverage obtained from 61] and Ricinus communis [59]. Only Linum usitatissimum (this study), Vitis vinifera [68], Theobroma cacao [69] and Cucumis sativus [70] seem to have higher coverage by the Copia superfamily, although in these last two genomes only the number of elements and not the coverage in bp was shown in the referenced papers and therefore they could not be included in Additional file 10. The prevalence of a superfamily may be related to amplification events of specific groups of TEs and to the activity of such elements, which may depend on just a few active copies of the family [12]. An interesting example is the genus Gossypium[71], in which one of the species with the smallest genomes had a high density of Copia elements. Gossypium species with larger genomes had an increased copy number of Gypsy elements, most of which represented just one subgroup of the Gypsy sequences. Such amplification can be lineage-specific and therefore result in changes in genome size [71-73]. In flax we found that Copia elements were abundant, diverse and some members were recently active (see below), which would explain a higher current influence of such elements. LINEs and Mutator elements were the most abundant after the LTR retrotransposons (Table 1). Although these two types of elements seem to be or have been fairly active, their lesser abundance when compared to LTR elements can be explained at least in part by their transposition mechanisms. For example, the mechanism of non-LTR retrotransposition generally creates truncated copies of the elements, which would largely decrease their coverage in a genome [74]; additionally plant LINEs are very diverse and heterogeneous due to the error-prone mechanism of their reverse transcriptase, and the accumulation of mutations during long evolutionary periods [74], which limits their identification. In the case of the Mutator elements, their cut and paste transposition does not increase copy number as much as retrotransposition. Additionally, non-autonomous gene-carrying Mutators or MULEs (Mu-like elements) can sometimes be difficult to identify by traditional bioinformatics approaches, and seem to be widely divergent [75]. Thus, in flax, identification of such elements may also be influenced by the high mutation rates and transposition mechanisms, resulting in lower percentages of identified Mutator and LINE elements.
Putative expression and abundance of main families of TEs
Besides being abundant, LTR elements were also diverse in the flax WGS assembly. The number of families (Table 2) was probably overestimated, since as a result of the masking process, some of the fragments we found may in fact be different segments of a single element. Nevertheless, there was a general correlation between superfamily genome coverage and the number of families found. Alignment of TEs to EST databases showed that just a small proportion of flax TEs might be active, most of which were Copia LTR elements (Table 2). Our results are in agreement with the survey done in over 200,000 ESTs for sugarcane where Copia elements had more matching ESTs than Gypsy retrotransposons [76], although in sugarcane (but not flax), DNA transposons also seemed to be fairly active. In plants, TE activity depends on regulatory factors including stress-driven transcriptional regulation and epigenetic silencing, which allow activation of just a few elements under specific environmental and developmental circumstances [12,37]. For example in maize, where more than 80% of the genome is made of TEs, a survey of over 2 million ESTs showed that only 1.5% of them matched TEs, and most of the families with putative activity were LTR retroelements. Thus for flax, as well as for most plant species studied, the activity of TEs seems relatively low, and may increase to detectable levels only in response to stress. Additionally, it has been shown that in certain families of TEs the percentage of polyadenylated expressed sequences is low [77]. Because most EST libraries are built by poly-A extension, this may artificially limit the proportion of expressed TEs that can be detected by alignment to ESTs. We also found that across all TEs there were fewer families with high copy numbers throughout the genome and most families within each superfamily had less than 10 copies (Figure 1), which is in agreement with findings in soybean where 78% of LTR families are present at copy numbers below 10 [67]. While low copy number could be related to low transposition rates, mechanisms like high mutation rate [78], recombination [79] and nested insertions [80], create rapid variability in TEs that results in divergence among TEs, and therefore, a low number of similar sequences. Since we did not find a correlation between copy numbers and putative expression (related ESTs), it is more likely that mechanisms of divergence and not the transposition of low copy number families account for the trend we found. This lack of correlation is in agreement with previous findings in maize [36] and contradicts a previous view that low copy number elements are the ones that are predominantly expressed [81].
Relationship of TEs to genes
The location of TEs in the flax genome was not completely random. It was evident that some scaffolds came from genomic regions rich in TEs (especially retrotransposons, which constituted the bulk of flax TEs – Figure 3) and were highly depleted in genes. Conversely, other scaffolds were rich in genes but depleted in TEs, and still others had similar coverage of TEs and genes. A global negative correlation of TE coverage and gene coverage agrees with a model in where there is purifying selection against TEs in coding regions to avoid detrimental effects on genome function; this model was clearly presented for Arabidopsis [82]. In sequenced genomes such as those of Sorghum bicolor [64] or Brachypodium distachyon [62], where the distribution of TEs has been mapped to chromosomes, the bulk of retrotransposons seem to be clustered around the centromeres, while less are close to gene-rich regions probably due to rapid elimination by controlling and selective host mechanisms [64]. When each of the 71 largest scaffolds was analyzed as an individual unit, there was no evidence for an overall pattern in which TEs occurred inside genes more that expected by chance (Additional file 4). However, there were certain superfamilies that were more likely to do so when compared to the rest of TEs (Figure 5). Several DNA TE superfamilies and L1 elements fell inside and close to genes more often than expected, while Gypsy elements were always underrepresented in and around genes, and Copia retrotransposons were only significant in the first 1 kb flanking genes. In Arabidopsis an analysis to find chimeric genes/TEs showed significant differences for Copia, En-Spm, Gypsy and Helitrons [35]. While both for flax and Arabidopsis there was an overrepresentation of En-Spm and underrepresentation of Gypsy TEs, Copia elements were overrepresented inside genes in Arabidopsis but not in flax, and Helitrons were underrepresented in Arabidopsis while this superfamily and hATs were significantly overrepresented in flax. The overrepresentation of class II TEs in flax genes is consistent with reviews describing the close association of genes and these elements including the domestication of transposon proteins into genes [4,83]. For example, TEs like En-Spm/CACTA are closely associated with genes in the Triticaceae and they may even capture gene fragments as they move and recombine in the genome [84]. In the case of Helitrons, extensive gene capture and shuffling mediated by these elements has been reported [85-88]. For hATs, gene shuffling has been reported in maize [89], and experiments with rice have shown that the nDart1 and its relatives belonging to hATs tend to fall within of very close to genes [90]. In the meantime, while Mutator elements were not overrepresented inside genes, they were abundant in the 1 kb of DNA flanking them. The close relationship of Mutator elements with genes allow TE-mediated gene movement, as has been shown for Mutator-like elements (MULEs) in rice [19] and Arabidopsis [20], and relates to fixation of TE enzymes like transposase, which is part of FHY3 and FAR1 genes involved in phytochrome A signalling [21,22]. Putative homologs of these two transposase bearing genes were also found in flax (result not shown). The Gypsy underrepresentation in gene coding regions of flax could be related to their tendency to cluster close to centromeric regions. This has been shown in grass species [52], in plants like sunflower [51,55,56] and has more recently been proposed for plants like poplar [57] and Arabidopsis [91]. It has been speculated that the reason for this insertion bias may be related to a specific domain in the integrase protein [91-93]; and such differences in integrase proteins may also be related to the differing distributions between Gypsy and Copia elements. Finally, Copia elements were overrepresented in the 1 kb of sequence that flanked genes (Figure 5). In Arabidopsis, the random pattern of Copia insertion allows them to insert close to coding regions [91], although in time, the elements are subjected to negative selection. A similar pattern could be true for flax since we found that many recently inserted Copia TEs were close to genes (Table 3). This insertion pattern might have important implications since TEs close to genes can become positive regulators of gene expression via their cis-acting elements (in LTRs) or may become targets for epigenetic silencing, which would affect the adjacent gene regions [94-96]. To test if there was a general pattern of regulation of genes by TEs, we matched available ESTs to the predicted genes of the flax genome (Figure 4). The negative correlation of TE coverage and gene expression found means that genes in regions that are rich in TEs could be affected by their nearby insertion. It is likely that genes in close proximity of TEs are affected negatively, because most often these regions are targeted for heterochromatization (and silencing) [96-98] and TE insertion can also cause disruption of genes.
Insertions of full LTR elements
Most of the non-redundant elements with two identifiable LTRs belonged to the Copia superfamily, but a large proportion of retroelements had non-identifiable internal regions, or regions that corresponded to host genes or other non-LTR TEs as has also been shown for poplar (Additional file 5) [57]. Many of these may constitute either non-autonomous elements, or genes captured by TEs. As it turns out, these two concepts are not mutually exclusive. For example, in soybean (Glycine max), an element has been described with an insertion of 10.5 kb containing a mixture of segments derived from non-coding sequence and disease resistance genes [99]. These elements could still be actively driven by autonomous elements if they conserve their LTRs, polypurine tracts (PPTs), and primer binding sites (PBS’). Many of the undetermined elements in flax had such features (these are part of the recognition algorithm of LTR_STRUC and LTR_FINDER), and therefore may still be active. In fact 32 of these undetermined elements had 100% LTR pair similarity and 69 had at least 99% similarity, meaning these TEs constitute relatively recent insertions. It is likely that at least some of the larger flax LTR elements could be classified as LARDs (Large Retrotransposon Derivatives) which have been characterized in detail in the Triticaceae, rice and Medicago [100-102], while some of the shorter than expected LTR retrotransposons are probably TEs that have lost their internal coding regions and are usually classified as Terminal-repeat Retrotransposons in Miniature (TRIMs) [103]. In terms of TE sizes, calculated estimates for plants range from 2–11.8 kb for Copia elements, and from 4.6-18 kb for Gypsy elements [104]. However, a survey of LTR retroelements in rice using LTR_finder found large variation ranges in LTR retrotransposons [105] which is in agreement with the larger variations found in flax (Additional file 5). Nevertheless our averages agree with the Gypsy elements having larger sizes Copia as is common in most plants. When comparing the activity of the LTR elements (Figure 6), the Copia elements appeared to be increasingly and continuously active in the last 5 million years. In the meantime Gypsy elements have been active for the last 7–8 million years but to a lesser extent than Copia and the undetermined elements. In fact, after a peak of activity 3–4 Mya, Gypsy elements have been less active until the present. In comparison, for poplar, the activity of Copia full length TEs does not seem to overshadow the activity of Gypsy elements, but full Copia elements are more abundant than Gypsy[57]. Although the activity of all these retroelements varies, it is interesting to notice that between 5 to 10 Mya, all of them may have been triggered. It is tempting to speculate that a duplication event of the flax genome may have triggered activity of the retrotransposons, and indeed whole genome duplication in this time frame has been inferred based on molecular phylogenies and analysis of Ks distribution in protein coding genes [38,40]. However, rapid turnover of elements is also common [106,107] and could account for the absence of detection in more ancient evolutionary times since TEs may become unrecognizable. When evaluating only the most recent flax LTR element insertions, it was shown that Copia LTR elements have more copies, putative expression and are located close to genes. The lone, recently inserted Gypsy element had no related ESTs. A similar insertion pattern was seen in Arabidopsis where the number of Copia elements with identical LTRs is higher than in Gypsy elements, and recent Copia insertions are closer to genes than Gypsy [91]. It can not be ruled out that the short read assembly methodology used for the flax WGS [40] is biased towards more efficient identification of regions surrounding genes. Nevertheless we found that Gypsy elements followed the opposite trend of Copia, meaning that both types of elements were detected, whether they were closely associated with genes or not. This observation and the agreement with other studies on this trend [91] supports our conclusions.
Conclusions
We showed that transposable elements in flax occupied more than 23% of the flax WGS assembly and were dominated by LTR elements. The distribution of TEs was not random and there were genomic regions that were enriched by these repetitive sequences, which may constitute heterochromatic sections of the genome. In regions shared by both TEs and genes, transposons may have a repressive effect on gene expression as demonstrated by a negative correlation between TE coverage and gene expression. Overrepresented families in close proximity or overlapping genes were mainly from the DNA transposon group, but the Copia group was also often localized to the flanking regions of genes. Copia retrotransposons have been increasingly active in the last 5 million years and have more members with conserved internal domains that contrast with a lower activity and conservation of Gypsy elements. It is possible, however, that older insertions are more difficult to tag by the high rate of mutations especially for TEs located to heterochromatic regions. Because of their recent activity, abundance and diversity, the Copia elements are potential shapers of the flax genome. Further studies, especially under stress-eliciting conditions, are necessary to understand the regulatory effect on adjacent genes and how their activation patterns may have influenced evolution of other flax species.
Methods
Identification of putative TEs within the flax WGS assembly
An unmasked WGS assembly of flax comprising 318,250,901 bases was used as input for TE detection [40]. De-novo identification of transposable elements was performed using RepeatScout [108], PILER [109], LTR_finder [110] and LTR_STRUC [111]. Repeats identified by RepeatScout, under default parameters, were filtered for low complexity using Tandem Repeats Finder [112] and nseg [113]. Repeats with less than 10 hits in the genome were eliminated from the library. For PILER-DF [109] analysis the full genome was compared to itself using PALS (part of the PILER implementation) using the default parameters. Families of dispersed repeats were created using a minimum family size of 3 members and a maximum length difference of 5% between all family members. The consensus sequence for each family was created after aligning the sequences with MUSCLE [114]. LTR TEs were found using LTR_finder using the option –w 2 to get a table output that could be parsed to obtain the sequences corresponding to the elements. LTR_STRUC was used under default parameters. The sequences output by all of these programs were used to create a unified repeat library that could be compared to previously characterized elements. Annotation of the repeats was performed comparing the library to a Viridiplantae TEs database downloaded from Repbase (http://www.girinst.org/repbase/ - update 20110920) and a Plant Repeat Database (http://plantrepeats.plantbiology.msu.edu/) of TEs created from the families Brassicaceae, Fabaceae, Gramineae and Solanaceae (v2_1_0 update 20112006), using tBLASTx and BLASTn, and to the RepeatPeps database of TEs that comes with RepeatMasker (update 20110920) using BLASTx. To test whether TEs might have captured fragments of other genes or belong to gene families instead of TE families, BLASTx was performed against the Genbank nr database. Repeats were classified in a TE superfamily [49] if they showed E values of at least 1e-5 with a common annotation in at least two of the databases to which they were compared. Repeats characterized as putative TEs by the previous approach were joined to the Viridiplantae database of TEs (update 20110920) to use as a library for comparison to find the distribution and coverage TEs in the genome assembly using RepeatMasker v-3.3.0 (http://www.repeatmasker.org/). RMBlast was used as search algorithm with Smith-Waterman cutoff of 225 (this cutoff was used for all RepeatMasker analyses). To automatically annotate the masked regions (matches of the TEs in the genome) in their respective TE superfamilies a custom Perl script was used (kindly provided by Robert Hubley - Institute for Systems Biology, http://www.systemsbiology.org/ -). A table for TEs abundance and coverage was built after filtering and annotation. The percentages were calculated for the elements based on the total number of bases including runs of Xs and Ns since some elements can also include at times undetermined bases; therefore total percentages may differ a slightly from those reported in the original description of the flax genome [40]. The TE values of the WGS assembly were compared to BAC-end sequencing TEs [47].
Putative expression and distribution of TE families
Clusters of TEs with 80% similarity within each superfamily were created using CD-HIT [115]. Only de-novo identified members were used for this analysis since they represented TE sequences identified from the flax genome. The members of each cluster were said to represent a family of TEs, according to the terminology presented by Wicker et al., [49]. One representative member of each family (longest sequence in each cluster) was used for comparison against 286,252 flax ESTs from Genbank using BLAT [116]. A hit to an EST was classified as positive only if 70% of the EST sequence matched to the query sequence. A family was said to be putatively expressed if it had at least one EST match. The proportion of TE families with expression was calculated for each of the major groups. The same analysis was done comparing the TE family representative sequences to the flax assembly, and a TE was considered as a representative copy of the TE family if it matched in 80% of its sequence to the query. A coefficient of correlation was established between copy number in each family and ESTs matches.
Relationship of TEs to genes
The distribution of TEs relative to predicted genes in the WGS assembly was analyzed for all scaffolds ≥ 1 Mb (71 large scaffolds). The proportional coverage and the statistics applied for both genes and TEs were obtained for both: full scaffolds and windows of 50 kb within each scaffold after mapping the coordinates of predicted genes and TEs to the scaffolds using the Genomic Hyperbrowser [117] (http://hyperbrowser.uio.no/rc/ - candidate version). To test whether the distribution of TEs and genes was correlated, a correlation coefficient was calculated for the proportional coverage of the large scaffolds. Proportional coverage graphs and heat maps for comparison of TEs and genes were built for selected scaffolds divided in 50 kb window units or bins (four scaffolds with large proportions of TEs, four with large proportion of genes and four with similar proportions of TEs and genes). The heat maps were built using with Multi Experiment Viewer [118]. To test whether TEs overlapped genes more than expected by chance in any of the scaffolds over 1 Mb we used Monte Carlo (MC) methods [119], preserving the segment lengths and position of the genes and changing the positions of the TEs to create the random probability, with a minimum of 100 MC samples and unlimited maximum number, a sequential MC threshold of 20 and a MCFDR of 0.05; the analysis was repeated for the scaffolds divided into 50 kb bins (2182 bins in total). For generating the random samples the lengths of genes and TEs were conserved, and only the TE positions were randomized which closely reflects the biological context. The proportion of gene coverage overlapping TE sections was calculated from the total base pairs covered by genes in each scaffold and the total base pairs calculated as being overlapped both by genes and TEs. Scaffolds having high proportion of overlap were further analyzed by calculating the overlap proportion in 50 kb bins. The MC statistical analysis was repeated using TE superfamilies. Putative expression of the genes in the scaffolds over 1Mb was determined by comparing the predicted mRNAs to 286,252 ESTs of flax from Genbank using BLAT [116]. A hit to an EST was classified as positive only if it matched 70% of the EST sequence. A gene was said to be putatively expressed if it had at least one EST match. The proportion of genes with expression was calculated for each one of the large scaffolds, and compared with the proportional coverage of TEs. To find out if any of the superfamilies had a bias to insert within genes when compared to the other superfamilies, the number of TE hits inside genes of each superfamily was determined with the Genomic Hyperbrowser [117] using the middle point of the TE sequences to determine if the TE was inside the gene. Then the number of TE hits inside genes was compared to the number of hits in all the scaffolds over 1 Mb using heterogeneity chi-square tests and a Bonferroni correction [120]. These analyses were then repeated to compare the TEs in the adjacent 1 kb, and in the adjacent 5 kb (upstream or downstream from the genes).
Insertions of full LTR elements and evolution
Since LTRs were the most prevalent elements in the flax genome, they were analyzed in further detail. Results from LTR_finder and LTR_STRUC were filtered for redundant sequences using CD-HIT [115]. Since at the time of insertion both LTRs from LTR retrotransposons are 100% similar, the divergence between LTR pairs in every putative element can be used to determine the age of the elements. We used ClustalW [121] for aligning LTR pairs and used the Kimura two parameter method [122] to estimate the nucleotide substitution (K). To estimate the age of insertion we used the following equation: t = K/2r, where t corresponds to the insertion time in millions of years, K corresponds to the number of nucleotide substitutions per site and r corresponds to the nucleotide substitution rate. In this case we chose a rate of 1.5 X 10-8 as reported for chalcone synthase and alcohol dehydrogenase genes in Arabidopsis and Arabis species; this rate has been previously used for dating LTR retrotransposon insertions in Arabidopsis [91], and it is very close to the estimate used for dating LTR retroelements in rice [123] which assumes at least a 2-fold higher mutation rate in TEs than in coding regions. The library of non-redundant elements with 100% LTR similarity was used to search the flax assembly and the flax ESTs using BLAT [116] to establish the abundance, distribution and overall putative expression of the recent insertions. Only hits that covered 100% of the query sequence were selected (no gaps or miss-matches), as these represented complete elements mapped to the genome. The segment distances between LTR retrotransposons elements and the closest genes were determined using the Genomic Hyperbrowser [117]. Finally, non-redundant LTR element sequences were used as input to extract protein domains from both Copia and Gypsy elements using RepeatExplorer by comparing flax LTR elements to a database of curated LTR retrotransposon domain sequences; the parameters for comparison were: minimum similarity 60%, minimum identity 40% and the proportion of the hit length from the length of the database sequence was set to 0.8 [50]. The domains were tabulated to discover the distribution of conserved domains in each superfamily.
Availability of supporting data
The data sets supporting the results of this article are included within the article (Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10).
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LGG conducted all analyses and wrote the manuscript. MKD supervised the research and edited the final version of the manuscript. All authors read and approved the final manuscript.
Authors' information
LGG: Department of Biological Sciences, University of Alberta, Edmonton, AB Canada T6G 2E9. Centennial Centre for Interdisciplinary Science (CCIS), 5–114.MKD: Department of Biological Sciences, University of Alberta, Edmonton, AB Canada T6G 2E9. Centennial Centre for Interdisciplinary Science (CCIS), 5–114.
Supplementary Material
Annotation of de novo repeats. Annotation of de-novo repeats.
Correlation between TE and gene coverage on scaffolds ≥ 1Mb. Scatter plots of correlation of the proportion of coverage between genes and the four largest transposable elements (TEs) superfamilies in scaffolds larger than 1 million bp.
Proportional coverage of genes and TEs. Proportional coverage of genes and TEs in scaffolds over 1 Mb.
Monte Carlo tests. Monte Carlo test for probability that TEs are overlapping genes more than expected by chance.
Filtered LTR elements. Filtered LTR elements.
Distances of LTR elements to their closest gene. Distances of LTR elements to their closest gene.
BLAT analysis of recent LTR TE insertions in genome. BLAT analysis of recent LTR TE insertions in genome.
BLAT analysis of recent LTR TEs against ESTs. BLAT analysis of recent LTR TEs against ESTs.
Protein domains in LTR elements. Identification of protein domains in LTR elements. The name of the sequences are made using superfamily, algorithm, coordinates and similarity percentage between LTR pairs.
Percentage of bp covered by LTR retrotransposon superfamilies. Percentage of bp covered by LTR retrotransposon superfamilies in characterized genomes. Plant species are organized according to phylogenetic relationships. The figures for each genome correspond to Brachypodium distachyon [62], Oryza sativa [63], Zea mays [11], Sorghum bicolor [64], Vitis vinifera [68], Carica papaya [65], Arabidopsis thaliana [10]- [LTR element coverage obtained from 61], Fragaria vesca [66], Malus domestica [61], Glycine max [67], Phaseolus vulgaris (data obtained from phytozome - http://www.phytozome.net/), Populus trichocarpa [60] - [LTR element coverage obtained from 61], Linum usitatissimum (flax - this study), Ricinus communis [59]. The transposable elements from the genomes of Theobroma cacao [69] and Cucumis sativus [70] have more Copia than Gypsy elements but could not be included in the figure since their actual coverage on the genome was not specified.
Contributor Information
Leonardo Galindo González, Email: galindo@ualberta.ca.
Michael K Deyholos, Email: deyholos@ualberta.ca.
Acknowledgements
The authors thank Robert Hubley (Institute for Systems Biology, Seattle, WA, USA), for providing a custom Perl script for the automatic annotation of masked regions in the flax genome. This research was supported by TUFGEN (Total Utilization of Flax Genomics), project funded by Genome Alberta, Genome Prairie, and Genome Canada. MKD is also supported by NSERC (Natural Sciences and Engineering Research Council) Canada.
References
- Bennetzen JL. Transposable element contributions to plant gene and genome evolution. Plant Mol Biol. 2000;42:251–269. doi: 10.1023/A:1006344508454. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev. 2005;15:621–627. doi: 10.1016/j.gde.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Kumar A, Bennetzen JL. Plant retrotransposons. Annu Rev Genet. 1999;33:479–532. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- Dooner HK, Weill CF. Give-and-take: interactions between DNA transposons and their host plant genomes. Curr Opin Genet Dev. 2007;17:486–492. doi: 10.1016/j.gde.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Kazazian HH Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smykal P, Bacova-Kerteszova N, Kalendar R, Corander J, Schulman AH, Pavelek M. Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. TAG Theoretical and applied genetics Theoretische und angewandte Genetik. 2011;122:1385–1397. doi: 10.1007/s00122-011-1539-2. [DOI] [PubMed] [Google Scholar]
- Kalendar R, Flavell AJ, Ellis THN, Sjakste T, Moisy C, Schulman AH. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity. 2011;106:520–530. doi: 10.1038/hdy.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civan P, Svec M, Hauptvogel P. On the coevolution of transposable elements and plant genomes. J Bot. 2011;2011 doi: 10.1155/2011/893546. [DOI] [Google Scholar]
- The_Arabidopsis_Genome_Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei FS, Pasternak S, Liang CZ, Zhang JW, Fulton L, Graves TA. et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Jiang N, Wessler SR. Plant transposable elements: Where genetics meets genomics. Nat Rev Genet. 2002;3:329–341. doi: 10.1038/nrg793. [DOI] [PubMed] [Google Scholar]
- Krom N, Recla J, Ramakrishna W. Analysis of genes associated with retrotransposons in the rice genome. Genetica. 2008;134:297–310. doi: 10.1007/s10709-007-9237-3. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Nishihara M, Mishiba K, Hirano H, Yamamura S. Two different transposable elements inserted in flavonoid 3 ',5 '-hydroxylase gene contribute to pink flower coloration in Gentiana scabra. Mol Genet Genomics. 2006;275:231–241. doi: 10.1007/s00438-005-0083-7. [DOI] [PubMed] [Google Scholar]
- Tang SX, Hass CG, Knapp SJ. Ty3/gypsy-like retrotransposon knockout of a 2-methyl-6-phytyl-1,4-benzoquinone methyltransferase is non-lethal, uncovers a cryptic paralogous mutation, and produces novel tocopherol (vitamin E) profiles in sunflower. Theor Appl Genet. 2006;113:783–799. doi: 10.1007/s00122-006-0321-3. [DOI] [PubMed] [Google Scholar]
- Baek KH, Skinner DZ, Ling P, Chen XM. Molecular structure and organization of the wheat genomic manganese superoxide dismutase gene. Genome. 2006;49:209–218. doi: 10.1139/G05-102. [DOI] [PubMed] [Google Scholar]
- Verries C, Bes C, This P, Tesniere C. Cloning and characterization of Vine-1, a LTR-retrotransposon-like element in Vitis vinifera L., and other Vitis species. Genome. 2000;43:366–376. doi: 10.1139/g99-139. [DOI] [PubMed] [Google Scholar]
- Richter TE, Ronald PC. The evolution of disease resistance genes. Plant Mol Biol. 2000;42:195–204. doi: 10.1023/A:1006388223475. [DOI] [PubMed] [Google Scholar]
- Jiang N, Bao ZR, Zhang XY, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–573. doi: 10.1038/nature02953. [DOI] [PubMed] [Google Scholar]
- Hoen DR, Park KC, Elrouby N, Yu ZH, Mohabir N, Cowan RK, Bureau TE. Transposon-mediated expansion and diversification of a family of ULP-like genes. Mol Biol Evol. 2006;23:1254–1268. doi: 10.1093/molbev/msk015. [DOI] [PubMed] [Google Scholar]
- Hudson ME, Lisch DR, Quail PH. The FHY3 and FAR1 genes encode transposase-related proteins involved in regulation of gene expression by the phytochrome A-signaling pathway. Plant J. 2003;34:453–471. doi: 10.1046/j.1365-313X.2003.01741.x. [DOI] [PubMed] [Google Scholar]
- Lin RC, Ding L, Casola C, Ripoll DR, Feschotte C, Wang HY. Transposase-derived transcription factors regulate light signaling in Arabidopsis. Science. 2007;318:1302–1305. doi: 10.1126/science.1146281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. Mutable Loci in Maize. Carnegie Institute of Washington Year Book. 1948;47:155–169. [Google Scholar]
- McClintock B. The Origin and Behavior of Mutable Loci in Maize. Proc Natl Acad Sci USA. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. Chromosome Organization and Genic Expression. Cold Spring Harb Symp Quant Biol. 1951;16:13–47. doi: 10.1101/SQB.1951.016.01.004. [DOI] [PubMed] [Google Scholar]
- McClintock B. Controlling Elements and the Gene. Cold Spring Harb Symp Quant Biol. 1956;21:197–216. doi: 10.1101/SQB.1956.021.01.017. [DOI] [PubMed] [Google Scholar]
- Grandbastien MA, Spielmann A, Caboche M. Tnt1, a Mobile Retroviral-Like Transposable Element of Tobacco Isolated by Plant-Cell Genetics. Nature. 1989;337:376–380. doi: 10.1038/337376a0. [DOI] [PubMed] [Google Scholar]
- Pouteau S, Grandbastien MA, Boccara M. Microbial Elicitors of Plant Defense Responses Activate Transcription of a Retrotransposon. Plant J. 1994;5:535–542. doi: 10.1046/j.1365-313X.1994.5040535.x. [DOI] [Google Scholar]
- Hirochika H. Activation of Tobacco Retrotransposons During Tissue-Culture. EMBO J. 1993;12:2521–2528. doi: 10.1002/j.1460-2075.1993.tb05907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sugimoto K, Otsuki H, Hirochika H. Transcriptional activation of the tobacco retrotransposon Tto1 by wounding and methyl jasmonate. Plant Mol Biol. 1998;36:365–376. doi: 10.1023/A:1005911413528. [DOI] [PubMed] [Google Scholar]
- Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M. Retrotransposons of rice involved in mutations induced by tissue culture. Proc Natl Acad Sci USA. 1996;93:7783–7788. doi: 10.1073/pnas.93.15.7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han FP, Liu ZL, Tan M, Hao S, Fedak G, Liu B. Mobilized retrotransposon Tos17 of rice by alien DNA introgression transposes into genes and causes structural and methylation alterations of a flanking genomic region. Hereditas. 2004;141:243–251. doi: 10.1111/j.1601-5223.2004.01808.x. [DOI] [PubMed] [Google Scholar]
- Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci USA. 2000;97:6603–6607. doi: 10.1073/pnas.110587497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes FR, Carazzolle MF, Pereira GAG, Colombo CA, Carareto CMA. Transposable elements in Coffea (Gentianales: Rubiacea) transcripts and their role in the origin of protein diversity in flowering plants. Mol Genet Genomics. 2008;279:385–401. doi: 10.1007/s00438-008-0319-4. [DOI] [PubMed] [Google Scholar]
- Lockton S, Gaut BS. The Contribution of Transposable Elements to Expressed Coding Sequence in Arabidopsis thaliana. J Mol Evol. 2009;68:80–89. doi: 10.1007/s00239-008-9190-5. [DOI] [PubMed] [Google Scholar]
- Vicient CM. Transcriptional activity of transposable elements in maize. BMC Genomics. 2010;11:601. doi: 10.1186/1471-2164-11-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo PG, Rossi M, de Jesus EM, Saccaro NL, Kajihara D, Massa R, de Felix JM, Drummond RD, Falco MC, Chabregas SM. et al. Transcriptionally active transposable elements in recent hybrid sugarcane. Plant J. 2005;44:707–717. doi: 10.1111/j.1365-313X.2005.02579.x. [DOI] [PubMed] [Google Scholar]
- McDill J, Repplinger M, Simpson BB, Kadereit JW. The Phylogeny of Linum and Linaceae Subfamily Linoideae, with Implications for Their Systematics, Biogeography, and Evolution of Heterostyly. Systematic Botany. 2009;34:386–405. doi: 10.1600/036364409788606244. [DOI] [Google Scholar]
- Millam S, Obert B, Pret'ova A. Plant cell and biotechnology studies in Linum usitatissimum - a review. Plant Cell Tissue Organ Cult. 2005;82:93–103. doi: 10.1007/s11240-004-6961-6. [DOI] [Google Scholar]
- Wang Z, Hobson N, Galindo L, Zhu S, McDill J, Yang L, Hawkins S, Neutelings G, Datla R, Lambert G, The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012. [DOI] [PubMed]
- Muir A, Westcott N. Flax, the genus Linum. Taylor & Francis Inc; 2003. [Google Scholar]
- Cullis CA. Mechanisms and control of rapid genomic changes in flax. Ann Bot. 2005;95:201–206. doi: 10.1093/aob/mci013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis CA, Cleary W. Rapidly Varying DNA-Sequences in Flax. Can J Genet Cytol. 1986;28:252–259. [Google Scholar]
- Cullis CA. DNA Differences between Flax Genotrophs. Nature. 1973;243:515–516. doi: 10.1038/243515a0. [DOI] [PubMed] [Google Scholar]
- Cullis CA. DNA-Sequence Organization in the Flax Genome. Biochimica Et Biophysica Acta. 1981;652:1–15. doi: 10.1016/0005-2787(81)90203-3. [DOI] [PubMed] [Google Scholar]
- Chen YM, Schneeberger RG, Cullis CA. A site-specific insertion sequence in flax genotrophs induced by environment. New Phytol. 2005;167:171–180. doi: 10.1111/j.1469-8137.2005.01398.x. [DOI] [PubMed] [Google Scholar]
- Ragupathy R, Rathinavelu R, Cloutier S. Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genomics. 2011;12:217. doi: 10.1186/1471-2164-12-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venglat P, Xiang DQ, Qiu SQ, Stone SL, Tibiche C, Cram D, Alting-Mees M, Nowak J, Cloutier S, Deyholos M. et al. Gene expression analysis of flax seed development. BMC Plant Biology. 2011;11:Art#74. doi: 10.1186/1471-2229-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O. et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- RepeatExplorer. http://galaxy.umbr.cas.cz:8080/
- Staton SE, Ungerer MC, Moore RC. The Genomic Organization of Ty3/Gypsy-Like Retrotransposons in Helianthus (Asteraceae) Homoploid Hybrid Species. Am J Bot. 2009;96:1646–1655. doi: 10.3732/ajb.0800337. [DOI] [PubMed] [Google Scholar]
- Miller JT, Dong FG, Jackson SA, Song J, Jiang JM. Retrotransposon-related DNA sequences in the centromeres of grass chromosomes. Genetics. 1998;150:1615–1623. doi: 10.1093/genetics/150.4.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presting GG, Malysheva L, Fuchs J, Schubert IZ. A TY3/GYPSY retrotransposon-like sequence localizes to the centromeric regions of cereal chromosomes. Plant J. 1998;16:721–728. doi: 10.1046/j.1365-313x.1998.00341.x. [DOI] [PubMed] [Google Scholar]
- Jiang JM, Birchler JA, Parrott WA, Dawe RK. A molecular view of plant centromeres. Trends Plant Sci. 2003;8:570–575. doi: 10.1016/j.tplants.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Santini S, Cavallini A, Natali L, Minelli S, Maggini F, Cionini PG. Ty1/copia- and Ty3/gypsy-like DNA sequences in Helianthus species. Chromosoma. 2002;111:192–200. doi: 10.1007/s00412-002-0196-2. [DOI] [PubMed] [Google Scholar]
- Cavallini A, Natali L, Zuccolo A, Giordani T, Jurman I, Ferrillo V, Vitacolonna N, Sarri V, Cattonaro F, Ceccarelli M. et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor Appl Genet. 2010;120:491–508. doi: 10.1007/s00122-009-1170-7. [DOI] [PubMed] [Google Scholar]
- Cossu RM, Buti M, Giordani T, Natali L, Cavallini A. A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genetics & Genomes. 2012;8:61–75. doi: 10.1007/s11295-011-0421-3. [DOI] [Google Scholar]
- Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002;115:49–63. doi: 10.1023/A:1016072014259. [DOI] [PubMed] [Google Scholar]
- Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010;28:951–U953. doi: 10.1038/nbt.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D. et al. The genome of the domesticated apple (Malus x domestica Borkh.) Nat Genet. 2010;42:833-+. doi: 10.1038/ng.654. [DOI] [PubMed] [Google Scholar]
- he_International_Brachypodium_Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- International_Rice_Genome_Sequencing_Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A. et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- Ming R, Hou SB, Feng Y, Yu QY, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KLT. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452:991–U997. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP. et al. The genome of woodland strawberry (Fragaria vesca) Nat Genet. 2011;43:109–116. doi: 10.1038/ng.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng JL. et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- The_French-Italian_Public_Consortium_for_Grapevine_Characterization. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–U465. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN. et al. The genome of Theobroma cacao. Nat Genet. 2011;43:101–108. doi: 10.1038/ng.736. [DOI] [PubMed] [Google Scholar]
- Huang SW, Li RQ, Zhang ZH, Li L, Gu XF, Fan W, Lucas WJ, Wang XW, Xie BY, Ni PX. et al. The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009;41:1275–U1229. doi: 10.1038/ng.475. [DOI] [PubMed] [Google Scholar]
- Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins JS, Hu GJ, Rapp RA, Grafenberg JL, Wendel JF. Phylogenetic determination of the pace of transposable element proliferation in plants: copia and LINE-like elements in Gossypium. Genome. 2008;51:11–18. doi: 10.1139/G07-099. [DOI] [PubMed] [Google Scholar]
- Hu GJ, Hawkins JS, Grover CE, Wendel JF. The history and disposition of transposable elements in polyploid Gossypium. Genome. 2010;53:599–607. doi: 10.1139/G10-038. [DOI] [PubMed] [Google Scholar]
- Schmidt T. LINEs, SINEs and repetitive DNA: non-LTR retrotransposons in plant genomes. Plant Mol Biol. 1999;40:903–910. doi: 10.1023/A:1006212929794. [DOI] [PubMed] [Google Scholar]
- Lisch D. Mutator transposons. Trends in Plant Science. 2002;7:498–504. doi: 10.1016/S1360-1385(02)02347-6. [DOI] [PubMed] [Google Scholar]
- Rossi M, Araujo PG, Van Sluys MA. Survey of transposable elements in sugarcane expressed sequence tags (ESTs) Genetics and Molecular Biology. 2001;24:147–154. [Google Scholar]
- Chang W, Schulman AH. BARE retrotransposons produce multiple groups of rarely polyadenylated transcripts from two differentially regulated promoters. Plant J. 2008;56:40–50. doi: 10.1111/j.1365-313X.2008.03572.x. [DOI] [PubMed] [Google Scholar]
- Casacuberta JM, Vernhettes S, Grandbastien MA. Sequence Variability within the Tobacco Retrotransposon Tnt1 Population. EMBO J. 1995;14:2670–2678. doi: 10.1002/j.1460-2075.1995.tb07265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol. 2003;20:528–540. doi: 10.1093/molbev/msg055. [DOI] [PubMed] [Google Scholar]
- SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, MelakeBerhan A, Springer PS, Edwards KJ, Lee M, Avramova Z. et al. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274:765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- Meyers BC, Tingley SV, Morgante M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001;11:1660–1676. doi: 10.1101/gr.188201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Agrawal N, Bureau TE. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003;13:1897–1903. doi: 10.1101/gr.1281503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28:913–922. doi: 10.1002/bies.20452. [DOI] [PubMed] [Google Scholar]
- Wicker T, Guyot R, Yahiaoui N, Keller B. CACTA transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiology. 2003;132:52–63. doi: 10.1104/pp.102.015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal SK, Giroux MJ, Brendel V, Vallejos CE, Hannah LC. The maize genome contains a Helitron insertion. Plant Cell. 2003;15:381–391. doi: 10.1105/tpc.008375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Gallavotti A, Stryker GA, Schmidt RJ, Lal SK. A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudogenes. Plant Mol Biol. 2005;57:115–127. doi: 10.1007/s11103-004-6636-z. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA. 2001;98:8714–8719. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fefelova N, Caronna J, He LM, Dooner HK. The polychromatic Helitron landscape of the maize genome. Proc Natl Acad Sci USA. 2009;106:19916–19921. doi: 10.1073/pnas.0904742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JB, Zhang F, Peterson T. Transposition of reversed Ac element ends generates novel chimeric genes in maize. PLoS Genet. 2006;2:1535–1540. doi: 10.1371/journal.pgen.0020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi K, Maekawa M, Tsugane K, Iida S. Transposition and target preferences of an active nonautonomous DNA transposon nDart1 and its relatives belonging to the hAT superfamily in rice. Mol Genet Genomics. 2010;284:343–355. doi: 10.1007/s00438-010-0569-9. [DOI] [PubMed] [Google Scholar]
- Pereira V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004;5:R79. doi: 10.1186/gb-2004-5-10-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeyer S. Integration by design. Proc Natl Acad Sci USA. 2003;100:5586–5588. doi: 10.1073/pnas.1031802100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Eickbush TH. Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J Virol. 1999;73:5186–5190. doi: 10.1128/jvi.73.6.5186-5190.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SE, Habera LF, Wessler SR. Retrotransposons in the flanking regions of normal plant genes: a role for copia-like elements in the evolution of gene structure and expression. Proc Natl Acad Sci USA. 1994;91:11792–11796. doi: 10.1073/pnas.91.25.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet. 2003;33:102–106. doi: 10.1038/ng1063. [DOI] [PubMed] [Google Scholar]
- Hollister JD, Smith LM, Guo YL, Ott F, Weigel D, Gaut BS. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci USA. 2011;108:2322–2327. doi: 10.1073/pnas.1018222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009;19:1419–1428. doi: 10.1101/gr.091678.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed I, Sarazin A, Bowler C, Colot V, Quesneville H. Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis. Nucleic Acids Res. 2011;39:6919–6931. doi: 10.1093/nar/gkr324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrzynski A, Ashfield T, Chen NWG, Mammadov J, Nguyen A, Podicheti R, Cannon SB, Thareau V, Ameline-Torregrosa C, Cannon E. et al. Replication of Nonautonomous Retroelements in Soybean Appears to Be Both Recent and Common. Plant Physiol. 2008;148:1760–1771. doi: 10.1104/pp.108.127910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar R, Vicient CM, Peleg O, Anamthawat-Jonsson K, Bolshoy A, Schulman AH. Large retrotransposon derivatives: Abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics. 2004;166:1437–1450. doi: 10.1534/genetics.166.3.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Chaparro C, Quesneville H, Panaud O. Spip and Squiq, two novel rice non-autonomous LTR retro-element families related to RIRE3 and RIRE8. Plant Sci. 2007;172:8–19. doi: 10.1016/j.plantsci.2006.07.008. [DOI] [Google Scholar]
- Wang H, Liu JS. LTR retrotransposon landscape in Medicago truncatula: more rapid removal than in rice. BMC Genomics. 2008;9:382. doi: 10.1186/1471-2164-9-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte CP, Le QH, Bureau T, Kumar A. Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc Natl Acad Sci USA. 2001;98:13778–13783. doi: 10.1073/pnas.241341898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O. LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res. 2005;110:91–107. doi: 10.1159/000084941. [DOI] [PubMed] [Google Scholar]
- Xu L, Zhang Y, Su Y, Liu L, Yang J, Zhu YY, Li CY. Structure and evolution of full-length LTR retrotransposons in rice genome. Plant Systematics and Evolution. 2010;287:19–28. doi: 10.1007/s00606-010-0285-2. [DOI] [Google Scholar]
- Vitte C, Panaud O, Quesneville H. LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genomics. 2007;8:218. doi: 10.1186/1471-2164-8-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JX, Devos KM, Bennetzen JL. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004;14:860–869. doi: 10.1101/gr.1466204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:I351–I358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21:I152–I158. doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics. 2003;19:362–367. doi: 10.1093/bioinformatics/btf878. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. Statistics of Local Complexity in Amino-Acid-Sequences and Sequence Databases. Comput Chem. 1993;17:149–163. doi: 10.1016/0097-8485(93)85006-X. [DOI] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Niu BF, Gao Y, Fu LM, Li WZ. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT - The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandve GK, Gundersen S, Rydbeck H, Glad IK, Holden L, Holden M, Liestol K, Clancy T, Ferkingstad E, Johansen M. et al. The Genomic HyperBrowser: inferential genomics at the sequence level. Genome Biol. 2010;11:R121. doi: 10.1186/gb-2010-11-12-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Hagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li JW, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. DNA Microarrays, Part B: Databases and Statistics. 2006;411:134-+. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Sandve GK, Ferkingstad E, Nygard S. Sequential Monte Carlo multiple testing. Bioinformatics. 2011;27:3235–3241. doi: 10.1093/bioinformatics/btr568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. Biostatistical analysis. USA: Prentice-Hall, Inc.; 1999. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide-Sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Ma JX, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci USA. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotation of de novo repeats. Annotation of de-novo repeats.
Correlation between TE and gene coverage on scaffolds ≥ 1Mb. Scatter plots of correlation of the proportion of coverage between genes and the four largest transposable elements (TEs) superfamilies in scaffolds larger than 1 million bp.
Proportional coverage of genes and TEs. Proportional coverage of genes and TEs in scaffolds over 1 Mb.
Monte Carlo tests. Monte Carlo test for probability that TEs are overlapping genes more than expected by chance.
Filtered LTR elements. Filtered LTR elements.
Distances of LTR elements to their closest gene. Distances of LTR elements to their closest gene.
BLAT analysis of recent LTR TE insertions in genome. BLAT analysis of recent LTR TE insertions in genome.
BLAT analysis of recent LTR TEs against ESTs. BLAT analysis of recent LTR TEs against ESTs.
Protein domains in LTR elements. Identification of protein domains in LTR elements. The name of the sequences are made using superfamily, algorithm, coordinates and similarity percentage between LTR pairs.
Percentage of bp covered by LTR retrotransposon superfamilies. Percentage of bp covered by LTR retrotransposon superfamilies in characterized genomes. Plant species are organized according to phylogenetic relationships. The figures for each genome correspond to Brachypodium distachyon [62], Oryza sativa [63], Zea mays [11], Sorghum bicolor [64], Vitis vinifera [68], Carica papaya [65], Arabidopsis thaliana [10]- [LTR element coverage obtained from 61], Fragaria vesca [66], Malus domestica [61], Glycine max [67], Phaseolus vulgaris (data obtained from phytozome - http://www.phytozome.net/), Populus trichocarpa [60] - [LTR element coverage obtained from 61], Linum usitatissimum (flax - this study), Ricinus communis [59]. The transposable elements from the genomes of Theobroma cacao [69] and Cucumis sativus [70] have more Copia than Gypsy elements but could not be included in the figure since their actual coverage on the genome was not specified.