

Abstract
The Mycobacterium tuberculosis complex (MTBC) consists of closely related species that cause tuberculosis in both humans and animals. This illness, still today, remains to be one of the leading causes of morbidity and mortality throughout the world. The mycobacteria enter the host by air, and, once in the lungs, are phagocytated by macrophages. This may lead to the rapid elimination of the bacillus or to the triggering of an active tuberculosis infection. A large number of different virulence factors have evolved in MTBC members as a response to the host immune reaction. The aim of this review is to describe the bacterial genes/proteins that are essential for the virulence of MTBC species, and that have been demonstrated in an in vivo model of infection. Knowledge of MTBC virulence factors is essential for the development of new vaccines and drugs to help manage the disease toward an increasingly more tuberculosis-free world.
Keywords: Mycobacterium tuberculosis, virulence factors, virulence, pathogen, virulence genes
Introduction
Members of the genus Mycobacterium are characterized by a very complex cell wall envelope that is responsible for the remarkable low permeability of their cells as well as the characteristic differential staining procedure (known as Zhiel-Neelsen acid-fast stain), which specifically stains all members of the genera. Both features are due to the presence of long chain α-alkyl, β-hydroxy fatty acids in their cell wall. The Mycobacterium genus is usually separated into two major groups on the basis of their growth rate. One group includes slow-growing species such as the well-known pathogens Mycobacterium tuberculosis, Mycobacterium bovis and Mycobacterium leprae [ethiological agents of human tuberculosis (TB), bovine tuberculosis (BTB) and leprosy respectively]; the other group gathers fast-growing species such as Mycobacterium smegmatis, which in general are opportunistic or non-pathogenic bacteria.
The Mycobacterium tuberculosis complex (MTBC) refers to group of species (M. tuberculosis, Mycobacterium canettii, Mycobacterium africanum, Mycobacterium microti, M. bovis, Mycobacterium caprae and Mycobacterium pinnipedii) that are genetically very similar. From those species, M. tuberculosis is the most well known member, infecting more than one-third of the world’s human population; it is also able to infect animals that have contact with humans. M. canettii and M. africanum, closely related to M. tuberculosis, can also cause human TB and are usually isolated from African patients or African ancestry. M. bovis displays the broadest spectrum of host infection, affecting humans, domestic or wild bovines and goats. M. caprae has been isolated only from goats. Besides, a laboratory-selected mutant of M. bovis, isolated by Calmette and Guérin and known as M. bovis var BCG, is the only vaccine used in TB prevention during early childhood. M. microti is a rodent pathogen, usually isolated from voles (rodents of the genus Microtus and related genera) that can also cause disease in immunocompromised human patients.1,2 Finally, M. pinnipedii infects seals.3
It has been suggested that MTBC members have evolved from a common ancestor via successive DNA deletions/insertions resulting in the present Mycobacterium speciation and their differences in pathogenicity. Genomic analysis has been fundamental for these studies and helped to identify 14 regions (known as regions of difference or RD1–14). These regions, present in the reference laboratory strain M. tuberculosis H37Rv, are absent from the vaccine strain M. bovis var BCG; thus, helping to pinpoint chromosomal genes related to pathogenicity. In parallel, six regions, known as H37Rv deletion 1 to 5 (RvD1–5) and M. tuberculosis specific deletion 1 (TbD1), are absent from the M. tuberculosis H37Rv genome relative to other members. By contrast, M. canettii contains all of the RD, RvD and TbD1 regions and it is believed that this is the most closely related genome to that of the bacilli’s ancestor. M. africanum strains mainly isolated from West Africa lack the RD9 region, whereas those from East Africa have it preserved but lack the RD3. M. microti lacks a specific region, named RDmic and the regions RD7, RD8, RD9 and RD10. Some strains that have been isolated from voles missed also part of the RD5 region. The most common M. bovis strains, “classical M. bovis,” isolated from bovines in Argentina, the Netherlands, United Kingdom and Spain, as well as from humans, showed the greatest number of RD deletions, lacking regions RD4, RD5, RD6, RD7, RD8, RD9, RD10, RD12 and RD13. M. caprae is closely related to M. bovis except that it contains several nucleotide substitutions in the gyrB gene that are not found in other members of the MTBC.4 In addition, the lack of the regions RD1, RD2 and RD14 in M. bovis var BCG apparently occurred during and after the attenuation process.1 Even the handling of the original BCG vaccine strain (Pasteur), after being distributed to different centers in the world, has translated into specific mutations present in each of M. bovis var BCG strains.
Tuberculosis still remains to be one of the leading causes of mortality throughout the world. The HIV/AIDS pandemic, the deterioration in public health systems in developing countries, and the emergence of multi-drug resistance forms of tuberculosis have contributed further to that spread. The MTBC species infect their mammalian host primarily in the lungs. In this organ, the mycobacteria are engulfed within alveolar macrophages, in which the bacteria are contained in endocytic compartments that can maturate to phagosomes. Under normal circumstances, phagosomes are fused to lysosomes and the phagosomal contents are exposed to lysosomal hydrolases, reactive oxygen and nitrogen species that destroy the intracellular bacteria. MTBC species have evolved several mechanisms to circumvent the hostile environment of the macrophage, such as inhibiting phagosome-lysosome fusion and to escape acidic environments inside the phagolysosome.5
The infection is normally contained in the lung by formation of granulomas where the activated macrophages and other immune cells surround the site of infection to limit tissue damage and restrict mycobacterial dissemination.6,7 Concomitantly, virulent MTBC species have developed strategies to avoid or modulate the immune response in their favor. In the granuloma, some of the bacteria may remain dormant for decades without any active clinical disease (latent tuberculosis). Nevertheless, in any immune-depressing condition the dormant bacteria can become active, replicate and spread into the lung and other tissues.7
In recent years, there have been considerable advances in the understanding of the molecular bases of pathogenicity, virulence and persistence of mycobacteria. One significant contribution has been the identification of essential mycobacterial virulence genes. In particular, the use of transposon mutant libraries in combination with different in vivo screening methods has allowed the massive identification of virulence genes and, therefore, the elucidation of mechanisms that the bacilli employ to survive and persist in the hosts. Most of these virulence genes encode enzymes of several lipid pathways, cell surface proteins, regulators and proteins of signal transduction systems. Another group of relevance is the one involved in mycobacterial survival inside the aggressive microenvironment of the host macrophages. It is noticeable that mycobacteria lack classical virulence factors such as toxins, which are typical of other bacterial pathogens, and that many of the virulence genes of MTBC species are also conserved in non-pathogenic mycobacteria. These findings suggest that pathogenic species have adapted their genomes from a free-lifestyle to the intracellular environment with minimal acquisition of exclusive virulence genes.
There are a wide variety of conditions and parameters to define a virulence gene and the discussion about what constitutes a virulence gene is still unsettled. As a consequence, the definitions have not yet being accepted universally by researchers. The existence of opportunistic pathogens and highly susceptible individuals (i.e., immunodeficient ones) further complicates the task to reach precise definitions. Undoubtedly, one requisite to classify a gene as a virulence factor is that its absence attenuates the virulence of the microorganism in an in vivo model. However, this criterion comprises a large spectrum of genes, including housekeeping genes that have a function in survival in the host. These housekeeping genes are involved in basic cellular metabolism and are not generally considered as virulence factors.
Taking these concepts into consideration, the present review focuses on those genes/proteins whose inactivation in the mycobacterial genome leads to a measurable loss in pathogenicity or virulence in a validated TB model but fails to impair the bacterial growth in all the standard in vitro conditions (excluding stress and starvation) in which the wild-type strain normally replicates. This review also describes genes/proteins such as those required either for expression or transport of virulence factors.
The current review is aimed to update the most recent progress in the identification and characterization of all kind of virulence genes of the MTBC. Previous updates have covered different aspects of the MTBC. The excellent review by Smith fully describes the M. tuberculosis pathogenesis and summarizes the contribution of individual genes to the virulence of pathogenic Mycobacterium species.7 Other revisions have focused on certain aspects of M. tuberculosis-host interaction5,8 such as defense against host-induced stress,9 bacterial carbon metabolism10 and latent tuberculosis11,12; or particular mycobacterial virulence compounds or genes, such as: proteases,13 lipids,14-16 regulators,17 sigma factors,18 secretion systems,19,20 among others.
In the present review, the virulence determinants have been divided into the following categories based on their function, molecular features or cellular localization: (1) Lipid and fatty acid metabolism, including catabolism of cholesterol, (2) cell envelope proteins: including cell wall proteins, lipoproteins and secretion systems, (3) proteins inhibiting antimicrobial efectors of the macrophage, including those involved in responses to oxidative and nitrosative stresses, phagosome arresting and inhibition of apoptosis, (4) protein kinases, (5) proteases, including metalloproteases, (6) metal-transporter proteins, divided into importer and exporters, (7) gene expression regulators, including two component systems, sigma factors and other transcriptional regulators, (8) proteins of unknown function, including PE and PE_PGRS families and (9) other virulence proteins. A brief introduction to each topic is included at the beginning of each section. The information provided here is intended to help readers to better understand the factors that potentially could give rise to a tuberculosis pandemic. Furthermore, it provides and might hopefully reveal a source of potential targets to contain it.
A summary of all virulence genes described in this review is presented in Table 1.
Table 1. Virulence factor of the Mycobacterium tuberculosis complex.
| Category | Gen Name | Rv number | Description | Attenuation evidences |
References | ||
|---|---|---|---|---|---|---|---|
| Model | Result | Complementation | |||||
|
Lipids and Fatty Acid Metabolism |
kasB |
Rv2246 |
3-oxoacyl-[acyl-carrier protein] synthase 2 kasb |
C57BL/6 mice (lda) |
Reduced CFUs in organs and lung pathology Increased animal survival |
Yes |
25 |
|
Mycolic acid synthesis |
mmaA4 |
Rv0642c |
Methoxy mycolic acid synthase 4 |
C57BL/6 mice (lda/iv) |
Reduced CFUs in organs |
Yes |
26 |
| |
pcaA |
Rv0470c |
Mycolic acid synthase (cyclopropane synthase) |
C57BL/6 mice (iv)† |
Failed to persist in the spleens |
ND |
27 |
| |
C57BL/6 mice (iv) |
Failed to persist in organs Increased animal survival |
Yes |
||||
| |
C57BL/6 mice (lda) |
Reduced CFUs in lung |
Yes |
28 |
|||
| |
mymA operon |
Rv3083 to Rv3089 |
Propable Monooxygenase (Hydroxylase) |
Activated J774 macrophages and guinea pigs (sc) |
Reduced CFUs |
ND |
32 |
| |
- |
Rv2869c |
Membrane bound metalloprotease |
C57BL/6 mice (lda) |
Reduced CFUs in lung |
Yes |
33 |
| |
treS |
Rv0126 |
Trehalose synthase |
C57BL/6 mice (iv) |
Reduced CFUs in lung Increased animal survival |
ND |
34 |
|
Synthesis of complex lipids |
pks15 pks1 |
Rv2946c Rv2947c |
Probable polyketide synthases |
C57BL/6J mice (in) and MAM MH-S |
Reduced CFUs |
ND |
36 |
|
PDIM |
B6D2 F1 mice (lda) |
Increased animal survival |
ND |
37 |
|||
| |
Rabbits (intracisternally) |
Reduced CFUs in cerebrospinal fluid and organs |
ND |
38 |
|||
| |
pks10 |
Rv1660 |
Possible chalcone synthase |
C57BL/6J mice (in) and MAM MH-S |
Reduced CFUs |
ND |
36 |
| |
pks12 |
Rv2048c |
Probable polyketide synthase |
C57BL/6J mice (in) and MAM MH-S |
Reduced CFUs |
ND |
39 |
| |
fadD26 |
Rv2930 |
Fatty-acid-Coa synthase |
C57BL/6 mice (iv) |
Reduced CFUs in lung |
ND |
41 |
| |
|
|
|
BALB/c mice (iv) |
Reduced CFUs in lung |
ND |
42 |
| |
|
|
|
BALB/c mice (in) |
Reduced CFUs in organs |
ND |
44 |
| |
fadD28 |
Rv2941 |
Fatty-acid-Coa synthase |
C57BL/6 mice (iv) |
Reduced CFUs in lung |
ND |
41 |
| |
mmpL7 |
Rv294 |
Conserved transmembrane transport protein |
C57BL/6 mice (iv) |
Reduced CFUs in lung |
ND |
41 |
| |
drrC |
Rv2938 |
Probable daunorubicin-dim-integral membrane ABC-transporter |
BALB/c mice (iv) |
Reduced CFUs in lung |
ND |
42 |
| |
C57BL/6 mice (lda.) |
Reduced CFUs in organs. Increased animal survival |
ND |
45 |
|||
| |
pks5 |
Rv1527c |
Probable polyketide synthase |
BALB/c mice (lda) |
Reduced CFUs in organs |
ND |
46 |
| |
pks7 |
Rv1661 |
Probable polyketide synthase |
BALB/c mice (lda) |
Reduced CFUs in organs |
ND |
46 |
|
SL |
mmpL8 |
Rv3823c |
Probable conserved integral membrane transport protein |
C57BL/6 mice (iv) |
Reduced CFUs in organs |
ND |
21 |
|
Others genes related in lipid synthesis |
fadD33 |
Rv1345 |
Possible polyketide synthase |
C57BL/6 and B6D2/F1 mice (lda) |
Increased animal survival |
ND |
45 and 54 |
| BALB/c mice (iv) |
Reduced CFUs in liver |
Yes |
55 |
||||
| |
icl1 |
Rv0467 |
Isocitrate lyases |
C57BL/6 and BALB/c mice (iv). Activated MBMDM |
Failed to persist. Increased animal survival Reduced lung pathology |
Yes |
57 |
| |
icl1 and icl2 |
Rv0467 and Rv1915-Rv1916 |
Isocitrate lyases |
C57BL/6; IFN-γ −/− and TNF-R1−/− mice (iv). Non-activated and activated BMDM and HBMDM |
Reduced CFUs, increased animal survival reduced lung pathology |
Yes |
58 |
| |
plcA plcB plcC plcD |
Rv2351c Rv2350c Rv2349c Rv1755c |
Probable phospholipase C |
BALB/c mice (lda) |
Reduced CFUs in organs |
Yes |
60 |
|
Catabolism of cholesterol |
choD |
Rv3409c |
Putative cholesterol oxidase |
C57BL/6 (iv) and mouse peritoneal macrophages |
Reduced CFUs |
Yes |
64 |
| |
hsaC |
Rv3568c |
3,4-DHSA dioxygenase |
SCID mice (iv) |
Increase survival |
Yes |
65 |
| Guinea pig (a) |
Modestly reduced CFUs and granulome formation in lungs. |
Yes |
|||||
|
igr operon: cyp125 |
Rv3545c |
Putative cytochrome P450 |
C57BL/6 mice (a) |
Reduced CFUs |
ND |
67 |
|
|
fadE28/29 |
Rv3544c-Rv3543c |
Acyl coenzyme A dehydrogenases |
|||||
|
- |
Rv3542c-Rv3541c |
CHPs |
|||||
|
ltp2 |
Rv3540c |
Probable lipid carrier protein |
|||||
|
Cell Envelope Proteins |
erp |
Rv3810 |
Exported repetitive protein |
MBMDM and BALB/c mice (iv)‡ |
Reduced CFUs in macrophages and organs |
Yes |
82 |
|
Cell wall proteins |
fbpA |
Rv3804 |
Fibronectin binding protein, mycolyltransferese |
BALB/c mice (ip)* |
Reduced CFUs in organs. |
Yes |
85 |
| THP-1 and J774 macrophages |
Reduced CFUs. |
ND |
31 |
||||
| |
mce1 |
Rv0166 to Rv0174 |
Mammalian cell entry proteins. Possible lipids ABC-transporters. |
BALB/c mice (it) (mce1, 2 and 3 mutants) |
Reduced CFUs in organs, reduced tissue pathology and increased animal survival |
ND |
95 |
| |
mce2 |
Rv0586-0594 |
|
C57BL/6 mice (lda) |
Reduced CFUs and gross lesion in lung. Increased animal survival |
ND |
96 |
| |
mce3 |
Rv1964 to Rv1971 |
Mammalian cell entry proteins. Possible lipids ABC-transporters. |
C57BL/6 mice (lda) (mce3, 4 mutants) |
Reduced CFUs in organs and less tissue pathology in lung. Increased survival |
ND |
97 |
| |
mce4 |
Rv3501c to Rv3494c |
Cholesterol transporter |
||||
| |
ompATb |
Rv0899 |
Pore-forming protein |
THP1 and MBMDM BALB/c mice (lda) |
Reduced CFUs in macrophagues and organs |
ND |
107 |
| |
hbhA |
Rv0475 |
Heparin binding hemaglutinin protein (Adhesine) |
A549 pneumocytes. BALB/c mice (lda)‡ |
Reduced adhesion and CFUs in pneumocytes and reduced CFUs in spleen |
Yes |
111 |
| |
pstA1 |
Rv0930 |
Inorganic phosphate-ABC transporter |
Resting and activated MBMDM |
Reduced CFUs |
Partial |
99 |
| |
phoT |
Rv0820 |
Resting and activated MBMDM and C57BL/6J mice (iv) |
Reduced CFUs in macrophages and lung |
Yes |
||
| |
caeA |
Rv2224c |
Carboxylesterase for esters of 3 to 7 carbon atoms |
BALB/c mice (lda) |
Reduced CFUs and gross pathology in organs |
Yes |
113 |
| BALB/c mice (iv) |
Increased survival and weight |
Yes |
|||||
| C57BL/6 mice (lda) |
Moderated reduction in CFUs |
Yes |
114 |
||||
| |
kefB |
Rv3236c |
K+/H+ antiporter, affecting ROS production |
J774 macrophages† |
Reduced phagosome ROS production |
ND |
116 |
| |
oppABCD |
Rv3666c to Rv3663c |
Oligopeptide ABC-transporter |
BALB/c mice (lda) |
Reduced CFUs in organs in the chronic infection. Increased survival |
No |
118 |
| |
ctaC |
Rv2200c |
Citocrome C oxidase unit II |
MBMDM |
Reduced CFUs |
ND |
119 |
|
Lipoproteins |
lppX |
Rv2945c |
Carrier of DIM and antigen |
BALB/c mice (lda) |
Reduced CFUs in lung |
ND |
127 |
| |
lpqH |
Rv3763 |
Antigen Apoptogenic |
C57BL/6 mice (lda) |
Reduced CFUs in lung |
Yes |
138 |
| |
lprG |
Rv1411c |
Cell wall assembly TLR2 agonist |
BALB/c mice (ip)* |
Reduced bacterial load in spleens |
Yesa |
147 |
| |
BALB/c mice (it)* |
Reduced CFUs in lung |
Yesa |
145 |
|||
| |
lprG-p55 |
Rv1411c-Rv1410c |
Antibiotic efflux pump (P55) |
J774macrophages* |
Reduced CFUs |
Yesa |
|
| |
pstS-1 |
Rv0934 |
Inorganic phosphate transport. Antigen and apoptogenic |
BALB/c and C57BL/6 mice (iv) |
Reduced CFUs in organs |
ND |
157 |
| |
Mouse peritoneal macrophages |
Reduced multiplication |
ND |
|
|||
| |
lpqY |
Rv1235 |
ABC-transporter (Recycling system of threalose) |
SCID mice (BALB/c mice background) (lda) |
Increased survival |
Yes |
158 |
| |
C57BL/6 mice (lda) |
Reduced UFCs in organs. |
Yes |
||||
| |
modA |
Rv1857 |
Molybdenum ABC transporter |
CBA/J mice (lda) |
Reduced CFUs in ungsand slight increased |
ND |
42 |
| |
BALB/c mice (iv) |
Reduced CFUs in lung |
ND |
||||
|
Secretion system |
esxA |
Rv3875 |
Esx-1 component or substrate (C or S) |
Guinea pigs (sc)* |
Reduced CFUs in spleen |
ND |
170 |
| |
RD1 |
Rv3868 to Rv3875 and Rv3877 |
Esx-1 C or S |
THP-1 macrophage |
Reduced CFUs |
No |
175 |
| |
C57BL/6 mice (a) |
Reduced CFUs in organs Total survival |
No |
||||
| |
esxB |
Rv3874 |
Esx-1 C or S |
THP-1 macrophage |
Reduced CFUs |
Yes |
187 |
| |
espH |
Rv3867 |
Esx-1 C or S |
BMDM |
Reduced CFUs |
NU |
186 |
| |
C57BL/6 mice (lda) |
Reduced CFUs in organs |
Partial |
||||
| |
espG1 |
Rv3865 |
Esx-1 C or S |
BMDM |
Reduced CFUs |
NU |
186 |
| |
C57BL/6 mice (lda) |
Reduced CFUs in organs Total survival |
Partial |
||||
| |
espA |
Rv3614 |
Esx-1 C or S |
C57BL/6 mice (iv) |
Reduced CFUs in organs Increased animal survival |
Yes |
181 |
| |
espC |
Rv3615 |
Esx-1 C or S |
BMDM |
Reduced CFUs |
Yes |
182 |
| |
eccCd |
Rv3877 |
Esx-1 C or S |
BMDM |
Reduced CFUs |
Yes |
182 |
| |
espR |
Rv3849 |
Esx-1 C or S |
C57BL/6 mice (iv) |
Reduced CFUs in lung |
NU |
185 |
| |
mycP1 |
Rv3883 |
Esx-5 C or S |
MBMDM |
Reduced CFUs |
Yes |
191 |
| |
|
|
BALB/c mice (lda) |
Reduced CFUs in lung |
Yes |
||
| |
eccD5 |
Rv1795 |
Esx-5 C or S |
MBMDM |
Reduced CFUs |
Yes |
202 |
| |
ppe25 to pe19 |
Rv1787 to Rv1791 |
Signal peptidase for lipoproteins |
MBMDM |
Reduced CFUs |
Yes |
202 |
| |
lspA |
Rv1539 |
Preprotein translocase ATPase |
J774 macrophages |
Reduced CFUs |
NU |
123 |
| |
CBA/J mice |
Reduced CFUs in lung |
Yes |
||||
| |
secA2 |
Rv1821 |
Accesory SecA protein |
C57BL/6 mice (iv) |
Reduced CFUs in organs Increased animal survival |
Partial |
206 |
| |
C57BL/6 -SCID mice (iv) |
Increased animal survival |
ND |
||||
| |
C57BL/6 mice (lda) |
Increased animal survival Reduced CFUs in organs |
ND |
272 |
|||
| |
secA2 |
Rv1821 |
Accesory SecA protein |
BMDM from C57BL/6 mice, NOS2−/− and pho−/− mice (lda) |
Reduced CFUs |
ND |
272 |
|
Proteins Inhibiting Antimicrobial Effectors of the Macrophage |
acr1 (hspX) |
Rv2031c |
Dormancy-associated protein |
MBMDM /THP-1 macrophages |
Reduced CFUs |
ND |
211 |
| |
acr2 |
Rv0251c |
Alpha-crystallin (Acr) family of molecular chaperones |
C57BL/6 mice (iv) |
Reduced weight loss |
Partial |
217 |
|
Oxidative and nitrosative stresses |
- |
Rv2136c |
Likely involved in the synthesis of peptidoglycan |
C57BL/6 mice (lda) |
Reduced CFUs in organs and reduced gross pathology in lung |
ND |
114 |
| |
ponA2 |
Rv3682 |
Probable transglycosylase and transpeptidase |
C57BL/6 mice (lda) |
Moderated reduction in CFUs |
ND |
114 |
| |
ahpC |
Rv2428 |
Alkyl ydroperoxide reductase c |
Guinea pigs (sc) (Antisense)* |
Reduced tissue pathology and CFUs |
ND |
221 |
| |
J774 macrophages |
Reduced CFUs |
ND |
225 |
|||
| |
sodC |
Rv0432 |
Superoxide dismutase (SOD) protein |
Activated murine peritoneal C57BL/6 mice and iNOS−/− macrophages |
Reduced CFUs |
Yes |
230 |
| |
mel2 |
Rv1936 to Rv1941 |
Bioluminescence-related proteins |
Activated J774, MBMDM from infected mice and human PBMC-derived macrophages |
Reduced CFUs |
Yes |
232 |
| |
C57BL/6 and Phox−/− and iNOS−/− mice (lda) |
Moderated reduction of CFUs in organs |
Yes |
||||
| |
katG |
Rv1908c |
Catalase-peroxidase enzyme |
BALB/c mice (iv) |
Reduced CFUs and increased survival |
Yes |
235 |
| |
Guinea pigs (im) |
Reduced CFUs in spleen and reduced number of lesion in tissues |
Yes |
||||
| |
C57BL/6 mice (iv) |
Reduced CFUs |
Yes |
237 |
|||
| |
NOS2−/− mice (iv) |
Reduced CFUs |
ND |
||||
| |
BMDM from C57BL/6 and NOS2−/− mice |
Reduced CFUs |
ND |
||||
| |
Activated BMDM NOS2−/− |
Reduced CFUs |
ND |
||||
| |
Guinea pigs* |
Moderated reduction in CFUs |
Yes |
236 |
|||
| |
BALB/c mice and MHC class II-knockout mice (ip) |
Reduced baterial load in organs |
ND |
234 |
|||
| |
tpX |
Rv1932 |
Thiol peroxidase |
Resting and activated BMDM from BALB/c and C57BL/6 mice |
Reduced CFUs |
Yes |
240 |
| BALB/c mice (iv) |
Reduced CFUs and increased animal survival |
Yes |
|||||
|
Phagosome arresting |
ndk |
Rv2445c |
Nucleoside diphosphate kinase |
RAW 264.7 macrophages (Antisense)† |
Reduced CFUs |
ND |
244 |
|
ptpA |
Rv2234 |
Low-molecular weight tyrosine phosphatase |
THP-1 macrophage |
Reduced CFUs |
Yes |
247 |
|
|
pe_pgrs30 |
Rv1651c |
Member of the PE family |
BALB/c mice (lda) |
Reduced CFUs |
Yes |
249 |
|
| BALB/c mice (lda) |
Reduced tissue damage |
ND |
|||||
| J774.1 and THP-1 macrophages |
Reduced CFUs |
Yes |
|||||
|
Inhibition of apoptosis |
nuoG |
Rv3151 |
Subunit of the type I NADH dehydrogenase, NADH-1 |
BALB/c mice (iv) |
Increased animal survival and reduced CFUs in organs |
Yes |
268 |
| SCID mice (iv) |
Increased animal survival |
Yes |
|||||
|
pknE |
Rv1743 |
Serine/threonine kinase E |
THP-1 macrophages |
Reduced bacterial survival |
Yes |
275 |
|
| - |
Rv3654c-Rv3655c |
CHP |
U937 apoptotic macrophages |
Reduced CFUs |
ND |
276 |
|
|
Protein Kinases |
pknD |
Rv0931c |
Protein kinase D |
Brain microvascular endothelial cells (HBMEC) |
Impaired invasion |
Yes |
311 |
| BALB/c mice (iv) |
Reduced CFUs in brain |
ND |
|||||
| BALB/c mice (iv) |
Increased animal survival |
ND |
|||||
|
pknG |
Rv0410c |
Protein kinase G |
BALB/c mice (iv) |
Reduced CFUs in organs |
ND |
312 |
|
| SCID mice (iv) |
Increased animal survival |
ND |
|||||
|
Proteases |
mycP1 |
Rv3883c |
Subtilisin-like serine protease. |
MBMDM /BALB/c (ld a) |
Reduced CFUs in organs |
Yes |
191 |
|
Serine proteases |
htrA2 (pepD) |
Rv0983 |
HtrA-like serine protease and chaperone. |
BALB/c, SCID and C57BL/6 mice(iv) |
Increased animal survival and less tissue pathology |
Yes |
328 |
| |
- |
Rv3671c |
Serine protease |
C57BL/6 mice (lda) |
Reduced CFUs in organs and reduced gross pathology in lung |
Yes |
335 |
|
ATP-dependent proteases |
clgR |
Rv2745c |
Transcriptional regulator |
MBMDM |
Reduced CFUs in organs |
Yes |
334 |
|
Metalloproteases |
zmp1 |
Rv0198c |
Zn2+ Metalloprotease |
J774and RAW264.7 macrophages BALB/c mice (lda) |
Reduced CFUs in organs |
Yes |
340 |
| THP1 macrophage |
No differences in CFU’s |
No |
341 |
||||
| SCID mice (iv) |
Reduced survival |
No |
|||||
| C57BL/6 mice (lda) |
Increased CFU’s in organs |
No |
|||||
|
rip1 |
Rv2869c |
S2P class of metalloproteases |
C57BL/6 mice (a) |
Reduced CFUs in organ and reduced tissue pathology |
Yes |
342 |
|
|
Proteasome-associated proteins |
pafA |
Rv2097c |
Mycobacterial proteasomal ATPase |
C57BL/6 and iNOS−/− mice Resting BMDM from both |
Reduced CFUs in organs and pathology |
ND |
354 |
|
mpa |
Rv2115c |
Mycobacterial proteasomal ATPase |
MBMDM BALB/c mice (lda) |
Reduced CFUs in organs and less tissue pathology. Increased animal survival |
Yes |
349 |
|
|
Metals-Transporter Proteins |
mbtB |
Rv2383c |
Iron ABC Transporter |
Macrophage-like cell line THP-1. |
Reduced CFUs and retarded for ability to grow in this cell line. |
ND |
352 |
|
Metal importers |
irtAB |
Rv1348-Rv1349 |
Iron-dependent regulatory protein. Repressor of mtb and mtb-2 loci. |
THP-1 macrophages. C57B/6 mice (lda) |
Reduced CFUs in macrophages and lung |
Yes |
354 |
|
ideR |
Rv2711 |
|
- |
Essential |
- |
356 |
|
|
mgtC |
Rv1811 |
Mg2+ transport P-type ATPase. Mg2+ uptake |
HBMDM. BALB/c mice (iv) |
Reduced CFUs in macrophages and organs |
Yes |
357 |
|
|
Metal exporters |
ctpC |
Rv3270 |
Zn2+ efflux transporter P-type ATPase |
Human macrophages |
Reduced CFUs |
Yes |
358 |
|
ctpV |
Rv0969 |
Cu2+ efflux transporter P-type ATPase |
BALB/c mice (lda) |
Increased survival and lower tissue damage in lung |
Yes |
359 |
|
| Guinea pigs (lda) |
Reduced CFUs and lower tissue damage in lung |
Yes |
|||||
|
Gene Expression Regulators |
phoPR |
Rv0757 to Rv0758 |
TCS |
THP1 macrophages |
Reduced CFUs |
Yes |
190 |
| C57BL/6 MICE (lda) |
Reduced CFUs in lung |
||||||
|
Two component system (TCS) |
phoP |
Rv0757 |
Transcriptional regulator |
MBMDM |
Reduced CFUs |
Yes |
362 and 364 |
| BALB/c mice (iv) |
Reduced CFUs |
NU |
|||||
| |
SCID mice (lda/iv) |
Improved survival |
Yes |
363 |
|||
| |
aprABC |
Rv2396abc |
Expressed in acidic medium dependent on PhoP |
Resting and activated C57BL/6 BMDM |
Defects in intracellular replication |
Yes |
366 |
| |
senX3-regX3 |
Rv0490-Rv0491 |
TCS |
THP-1 macrophages |
Reduced CFUs |
ND |
367 |
| |
|
|
|
DBA mice |
Moderate reduced CFUs in lung |
|
|
| |
senX3 |
Rv0490 |
Sensor |
BALB/c mice (iv) |
Reduced CFUs in lung |
Yes |
370 |
|
regX3 |
Rv0491 |
Transcriptional Regulator |
BALB/c mice (iv) |
Reduced CFUs in lung |
Yes |
||
|
dosRS dosT |
Rv3133c-Rv3132c Rv2027c |
TCS |
C57BL/6 MICE (lda) |
Reduced lung pathology. Increased CFUs |
Yes |
375 |
|
|
dosR (devR) |
Rv3133c |
Transcriptional Regulator |
C57BL/6 mice (lda) |
Moderate reduction CFUs in lung |
ND |
376 |
|
| Guinea pigs (lda) |
Reduction CFUs in lung |
No |
|||||
| Rabbit (lda) |
Moderate reduction CFUs in lung |
ND |
|||||
| Guinea pigs (sc) |
Reduced lung pathology |
NU |
378 |
||||
|
mprAB |
Rv0981-Rv0982 |
TCS |
BALB/c mice (iv) |
Reduced CFUs in lung latent stage |
ND |
385 |
|
|
Sigma factors |
sigA |
Rv2703 |
Sigma factor A |
C57BL/6 mice (lda) (Antisense) |
Reduced CFUs in lung |
ND |
390 |
| MonoMac6 cells (Antisense) |
Reduced CFUs |
ND |
|||||
|
sigC |
Rv2069 |
Sigma factor C |
DBA/2 mice (lda) |
Modest reduction of CFUs in lung, increased animal survival and reduced lung pathology |
Yes |
394 |
|
| SCID mice (lda) |
Modest reduction of CFUs in lung and increased animal survival |
Yes |
|||||
| Guinea pigs (lda) |
Reduced lung pathology |
Yes |
|||||
| DBA/2 mice (lda/iv) |
Increased survival and reduced lung pathology |
Yes |
395 |
||||
| Guines pigs (ida) |
Reduced lung and spleen pathology |
ND |
396 |
||||
|
sigD |
Rv3414c |
Sigma factor D |
C3H mice (iv) |
Increased animal survival |
Partial |
397 |
|
| BALB/c mice (iv) |
Increased animal survival |
Partial |
398 |
||||
|
sigE |
Rv1221 |
Sigma factor E |
SCID mice |
Increased animal survival |
No |
403 |
|
| BALB/c mice (iv) |
Reduced CFUs in organs |
Yes |
|
||||
| C3H/HeJ mice (a) |
Increased animal survival |
Partial |
402 |
||||
| Unactivated THP-1 and J774 or activated J774 macrophages |
Reduced CFUs |
Yes |
404 |
||||
|
sigF |
Rv3286c |
Sigma factor F |
BALB/c mice (iv) |
Increased animal survival |
ND |
413 |
|
| |
|
|
|
BALB/c mice (iv) |
Reduced CFUs and pathology in tissues |
ND |
414 |
| |
|
|
|
Guinea pigs (ida) |
Moderated reduction in organ pathology |
ND |
396 |
| |
sigG |
Rv0182c |
Sigma factor G |
J774macrophages |
Moderated reduction in CFUs |
Partial |
416 |
| |
sigH |
Rv3223c |
Sigma factor H |
C3H and C57BL/6 mice |
Reduced pathology in tissues |
Yes |
424 |
| |
|
|
C3H:He mice |
Increased animal survival |
Yes |
|
|
| |
|
|
BMDM from rhesus monkey |
Reduced CFUs at late time post infection |
Yes |
422 |
|
|
sigL |
Rv0735 |
Sigma factor L |
BALB/c mice (iv) |
Increased animal survival |
Yes |
426 |
|
|
Other transcriptional regulators |
- |
Rv0485 |
Member of the nagc/xylr repressor family |
BALB/c mice (iv) |
Reduced lung pathology |
Yes |
437 |
| |
|
|
BALB/c mice (iv) |
Modestly increased survival |
ND |
||
| |
|
|
SCID (iv) |
Modestly increased survival |
ND |
||
|
- |
Rv1931c |
AraC transcriptional regulator |
BALB/c mice (iv) and BMDM from BALB/c mice |
Reduced CFUs |
Yes |
441 |
|
|
hspR |
Rv0353 |
Transcriptional repressor |
C57BL/6 mice (iv) |
Reduced CFUs in organs |
ND |
442 |
|
|
whiB3 |
Rv3416 |
Whib-like regulator family |
C57BL/6 mice (iv)* |
Increased animal survival |
ND |
393 |
|
|
mosR |
Rv0348 |
Mycobacterial operons of survival regulator |
BALB/c mice (lda) |
Reduced CFUs |
No |
449 |
|
|
virS |
Rv3082c |
AraC family of transcriptional regulator |
BALB/c mice (lda) |
Increased animal survival |
Partial |
450 |
|
|
phoY2 |
Rv0821c |
Probable phosphate-transport system transcriptional regulator |
Activated J774 macrophage. Guinea pig (sc) |
Reduced CFUs |
Yes |
453 |
|
|
Proteins of Unknown Function |
pe_pgrs33 |
Rv1818c |
PE_PGRS family protein |
BALB/c mice (iv) |
Reduced CFUs |
Yes |
194 |
|
The PE/PPE families |
pe_pgrs51 |
Rv3367 |
PE_PGRS family |
J774 and BMDM macrophages† |
Reduced CFUs |
Yes |
100 |
| |
ppe46 |
Rv3018 |
PPE family |
BALB/c mice (iv) |
Moderate reduced CFUs in lung |
ND |
42 |
| |
- |
Rv1099c |
CHP |
C57BL/6J mice (iv) |
Reduced CFUs in organs |
ND |
100 |
|
Others proteins with unknown function |
- |
Rv0573c |
CHP |
C57BL/6J mice (iv) |
Reduced CFUs in organs |
ND |
100 |
|
- |
Rv0204c |
Integral membrane protein |
BALB/c mice (iv) |
Moderate reduced CFUs in lung |
ND |
42 |
|
|
- |
Rv2452c |
Hypothetical proteins |
BALB/c mice (iv) |
Moderate reduced CFUs in lung |
ND |
42 |
|
|
- |
Rv1290c |
CHP |
CB-17/Icr SCID mice (iv) |
Markedly increased surival |
ND |
460 |
|
|
- |
Rv1891 |
CHP |
CB-17/Icr SCID mice (iv) |
Moderate increased survival |
ND |
460 |
|
|
- |
Rv3404c |
CHP |
CB-17/Icr SCID mice (iv) |
Moderate increased survival |
ND |
460 |
|
|
- |
Rv1503c to Rv1507c |
CHP |
BALB/c mice (lda) |
Markely reduced CFUs in organs |
Yes |
461 |
|
|
- |
Rv0199 |
Conserved membrane protein |
MBMDM |
Reduced CFUs |
Yes |
119 |
|
| |
mmpl4 |
Rv0450c |
Conserved membrane transport protein |
C57BL/6 and (C57BL/6 x DBA2)F1 mice (a) |
Reduced CFUs in organs and increased life survival |
No |
45 |
|
- |
Rv2136c |
Conserved transmembrane protein |
C57BL/6 mice (lda) |
Markedly reduced CFUs in organs. Decreased gross pathology in lung |
No |
114 |
|
| Other Virulence Factors |
RD2 |
Rv1979c to Rv1982 |
Region of Difference |
C57BL/6 mice (lda) |
Reduced CFUs in lung and tissue pathology |
Yes |
467 |
| RAW macrophages |
Reduced CFUs at late times |
Yes |
|||||
|
acg |
Rv2032 |
Uncertain |
BALB/c mice (iv) |
Reduced CFUs |
Yes |
472 |
|
| SCID mice (iv) |
Increased animal survival |
Yes |
|||||
| BMDM from BALB/c mice |
Reduced CFUs |
Yes |
|||||
|
pckA |
Rv0211 |
Phosphoenolpyruvate carboxykinase |
BMDM from BALB/c mice† |
Reduced CFUs |
ND |
473 |
|
| |
|
|
BALB/c mice (iv)† |
Reduced CFUs in spleen |
ND |
||
|
ptpB |
Rv0153c |
Tyrosine phosphatase |
Guinea pigs |
Reduced CFUs in spleen |
Yes |
475 |
|
| |
|
|
Activated J774macrophages |
Reduced CFUs |
ND |
|
|
| hsp22.5 | Rv0990c | Novel heat shock protein | BALB/c mice (a) | Reduced CFUs in organs and increased animal survival | Yes | 476 | |
The mutant used was made in M. bovis; †in M. bovis BCG or ‡in both M. tuberculosis and M. bovis BCG. Route of infection: lda, low dosis aerosol; a, aerosol; it: intratracheal; iv, intravenous; ip, intraperitoneal; im, intramuscular; sc, subcutaneous. Complementation: ND, a complemented strain was not reported by the authors; No, a complemented strait was done but the phenotype was not restored; Yes, a complemented strait was done and the phenotype restored; Yesa, the phenotype was restored by the insertion of the entire operon; Partial, a complemented strait was done and the phenotype partially restored; NU: a complemented strait was done but not used by the authors. Abbreviations: CHP, conserved hypothetical proteins; MBMDM, murine bone marrow-derived macrophages; MAM, murine alveolar macrophage; HBMDM, human blood monocyte-derived macrophages.
Lipid and Fatty Acid Metabolism
M. tuberculosis is unique among bacterial pathogens in that it displays a wide array of complex lipids and lipoglycans on its cell surface.21 These exclusive cell wall lipids are known to play an important role in pathogenesis; therefore, the genes responsible for their biosynthesis, degradation and transport are potential virulence factors that offer new targets for drug design. This section is dedicated to proteins involved in the metabolism and transport of lipids that have been shown to influence mycobacterial pathogenesis and virulence.
Overview of mycolic acids biosynthesis
In order to help understanding the information on mycolic acids related to virulence, we will provide a brief overview of their biosynthetic pathways as shown below.
Mycobacteria are de facto Gram-positive bacteria; however, we can simplify the M. tuberculosis cell wall structure describing it as comparable to that of a Gram-negative bacterium. The first macromolecular layer after the peptidoglycan is composed of an heteropolysaccharide composed of arabinan and galactan (thus designated arabinogalactan) to which very long chain α-alkyl β-hydroxy fatty acids (mycolic acids) are esterified. Importantly these mycolic acids are similar in length but different in structure, having either cyclopropanations (cis or trans) or keto or methoxy groups, creating a number of sub-families. It is also important that besides their structural role, covalently attached to the arabinogalactan, these mycolic acids are also esterified to glycerol and trehalose; in the latter case, trehalose can contain one or two molecules of mycolic acids forming trehalose dimycolates (TDM) and trehalose monomycolates (TMM). Both compounds are present in the cell wall envelope interacting with other complex lipids and lypoglycans as it will be described below. The synthesis of mycolic acids was one of the first of several big surprises, since mycobacteria specialized in the synthesis of fatty acids to make different products. However, mycobacteria keep the essence of the chemistry and use comparable enzymes to the ones used by most of the other bacterial genera. So, while most of the prokaryotes use the fatty acid synthetase (FAS) system to produce fatty acids in the C14-C18 range, mycobacteria use it to make these long chains (up to 86–95 carbon atoms in length) hypothetically starting from a medium length fatty acid.14 The next surprise was the source of the latter, which is made by a FAS system homologous to eukaryotic systems.22 In these systems FASI contains all the catalytic domains in one polypeptide, whereas in the bacterial system (and the mycobacterial mycolic acid synthesis system), known as FASII, are several enzymes sequentially functioning.
Mycolic acid synthesis apparatus: its relation to virulence
The cytoplasmic synthesis of fatty acids is coordinated with a set of steps in charge of leading them to their final destination out of the cell. If any of the assembly and exporting steps is somehow affected, this will cause an effect in their proper localization.
Different areas of research on tuberculosis have focused the interest on synthesis and export of mycolic acids; in the first place, because several anti-mycobacterial drugs such as isoniazid, ethionamide, isoxyl and thiacetazone affect FASII enzymes23,24 (Belardinelli, personal communication); in the second place, different strategies, mainly based on signature tagged mutagenesis (STM) and specific gene deletion, unmistakably have shown that affecting mycolic acids synthesis or their structure caused alterations in virulence.
Recently Bhatt et al. constructed an M. tuberculosis mutant defective in one of the two β-keto acyl synthetases of the FASII system encoded by the kasB gene (Fig. 1).25 Although the gene was clearly not essential (as opposed to the essential kasA gene), this deletion resulted in loss of acid-fast staining, alteration of the colony morphology and abolition of classic serpentine growth (traditionally known as “cording” owing to its rope-like form under the microscope). Biochemical analyses revealed that the ΔkasB strain produced mycolates with slightly shorter chain lengths, compared with the parental strain. In addition to the distinct phenotypes of this mutant strain, the most remarkable effect of kasB disruption was its ability to persist in infected C57BL/6 immuno-competent mice for up to 600 d without causing disease or mortality. These results imply that the kasB gene is involved in the pathogenesis of M. tuberculosis and that the ΔkasB strain could be used as a model to study latent infections.25 Moreover, the extensive identity and similarity between kasA and kasB (both present in all the mycobacterial species) lead to asking why both genes have been conserved through evolution and how kasB expression and/or KasB activity is regulated in order to give biological meaning and relevance to the synthesis of mycolic acids that are just 2–4 carbons shorter than usual.
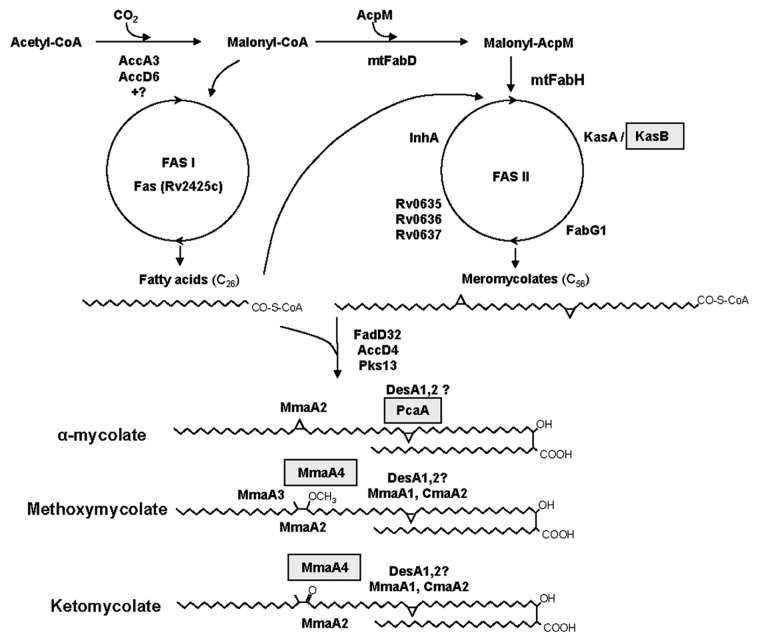
Figure 1. Pathway leading to the biosynthesis of the three types of mycolic acids in M. tuberculosis. The genes highlighted in gray were described as virulence factors. KasB, β-keto acyl synthetase; PcaA, Mycolic acid synthase A (cyclopropane synthase); MmaA4, Methoxy mycolic acid synthase 4.
Another topic in which important advances have been achieved over the last ten years is the mycolic acid methyl transferases (MAMTs), S-adenosyl methionine dependent enzymes which function is to introduce methyl groups, further modified by their conversion to cyclopropane rings, methoxy or keto groups (Fig. 1). As a whole, expression of these enzymes leads to the synthesis of a variety of sub-families for which the studies described below have found amazing and specific roles in spite of very subtle structural differences. Dubnau et al. previously demonstrated that an M. tuberculosis mutant with an inactivated hma (also known as cmaA and more recently as mmaA4) gene displayed a profound alteration in its envelope permeability as well as the loss of oxygenated mycolic acids. This mutant, when tested in a mouse (C57BL/6 mice) model of infection by aerosolization, showed an attenuated phenotype, suggesting that the oxygenated mycolic acids are important in the process of infection.26 Importantly this publication clearly pointed to specific roles of each MAMTs in the biogenesis of mycolic acids and their role in virulence. Continuing this line of research, Glickman et al. demonstrated that the deletion of pcaA, whose product catalyzes the proximal cyclopropanation of α-mycolates (Fig. 1), led to loss of cording. They also found that this mutant, despite its normal initial replication, failed to persist and kill infected C57BL/6 mice.27 These results suggest that the site-specific cyclopropane modification of mycolic acids could be an important determinant of the interaction between M. tuberculosis and the host. Since this modification of mycolic acids is absent in non-pathogenic mycobacteria, the phenotypes of the ΔpcaA strain suggest that the cyclopropyl modification system evolved to mediate principal virulence functions such as interaction with host innate immune receptors. To test this hypothesis, these authors performed further research focused on TDM, a glycolipid that contains cyclopropane modified mycolic acids. In order to explore the role of the ΔpcaA phenotype in the recognition by the innate host immune, this group set out to examine in greater detail the behavior of the ΔpcaA mutant during the early stages of infection in the lungs. They infected C57BL/6 mice by aerosol inoculation with the parental or the ΔpcaA mutant strains and determined bacterial titers at weekly intervals. Examination at earlier time points showed a drastic initial delay in the growth of the ΔpcaA mutant. However, bacterial titers equalized four weeks after infection. The early growth defect of the mutant could be reversed in the complemented strain, demonstrating that the pcaA gene was responsible for the observed phenotype. These results indicate that the pcaA gene is a temporally restricted determinant of bacterial growth after infection. In addition, the authors showed that purified TDM isolated from the cyclopropane-deficient pcaA mutant was hypo-inflammatory for macrophages and induced less severe granulomatous inflammation in mice, demonstrating that the structure of this diffusible glycolipid is critical to its pro-inflammatory activity. Hence, these results imply that the cyclopropyl modification of mycolates on TDM not only modify innate immune recognition but also have a profound effect on the function of these lipids as important virulence factors of the bacteria.28 Moreover, it demonstrated in an unquestionable way through several very elegant experiments that by subtle structural modifications and different locations (esterified in the cell wall skeleton or loosely attached to the outer membrane) mycolic acids are fundamental not only to preserve cell wall structure and functionality but also to modulate the interaction with the host immune system. This area of research has been recently reinforced by the analysis of the essentiality of each MAMT and the deletion of the seven MAMTs present in M. tuberculosis29 demonstrating that none of them is per se essential and that all of them may be deleted although a certain sequence in the deletion has to be followed to allow cellular adjustment of cell permeability and thus survival. This technically challenging work (severely limited by the availability of antibiotic resistance markers) generated a viable strain uncapable of making oxygenated or cyclopropanated mycolic acids. This strain showed massive changes in cell appearance such as total loss of acid fastness and susceptibility to detergents. Interestingly, it also displayed a severe attenuation during the first or second week of infection after aerosol infection of mice, depending on whether cyclopropanating enzymes or all the MAMTs were lost. At the same time, the strain generated a marked hyperinflammatory response from the host. This highly valuable work shows the immunomodulatory role of the mycolic acid modifications in the interaction with the host immune system, underscoring the importance of this group of enzymes as molecular targets. It also clearly shows that a novel strategy—targeting non-essential genes that alter the bacilli virulence—is possible.
The mentioned studies on mycolic acid synthesis correlated with others performed on their mechanism(s) of transport and assembly in the mycobacterial cell envelope. In this regard, a gene designated FbpA (part of the antigen 85 complex, Ag85A) has been studied. Belisle et al. identified members of this complex as enzymes responsible for the transfer of mycolic acids to α-α′-trehalose to form α-α′-TMM and α-α′-TDM.30 In a later study, the fbpA gene was disrupted in M. tuberculosis H37Rv and it was found that it plays a role in the pathogenesis of these bacteria.31 The phenotype of this mutant will be further discussed (see the section on Cell wall proteins).
Another M. tuberculosis system involved in mycolic acid export that has been found to be important for the cell envelope of the bacteria is the mymA operon. To further investigate the function of the genes within this operon, Singh et al. disrupted them in M. tuberculosis and analyzed the phenotypes of the obtained mutant strains. The biochemical characterization of the virS (the AraC/XylS repressor of the mym operon) mutant and the mymA mutant showed that both strains displayed reduced contents and altered composition of mycolic acids, along with accumulation of saturated C24 and C26 fatty acids as compared with the wild-type strain. They also found that mutation of these genes impaired the ability of M. tuberculosis to survive in activated murine J774 macrophages, but not in resting macrophages, suggesting the importance of the mymA operon in protecting the bacterium under unfavorable conditions. Infection of guinea pigs with the mutant and wild-type strains resulted in reduced spleen bacillary loads of the mutant strains as compared with the wild type in animals at 20 weeks post-infection. However, the bacillary load in lungs of animals infected with the mutants was comparable to that one of animals infected with the parental strain, suggesting that the mymA operon is a virulence factor specifically required for growth of M. tuberculosis in the spleens of guinea pigs at later stages of the disease32 (see the section on “Other Transcriptional Regulators”).
Finally, a last member of this group of virulence factors is the product of the gene Rv2869c, a member of the M50 class of membrane-bound zinc metalloproteases; its disruption translated into various alterations in mycolic acid biosynthesis and phosphatidylinositol mannoside (PIM) composition. These data are consistent with a model in which the Rv2869c protease participates in multiple lipid biosynthetic pathways possible through cleavage of membrane bound transcriptional regulators.33 The constructed mutation resulted in a strain defective for initial replication in the lungs of C57BL/6 mice after aerosol infection and also severely defective for persistence, indicating that although this gene product is not necessary for viability, it is related to virulence.
Even though the synthesis of mycolic acid per se provided a large number of enzymes to be study as virulence factors, the fact that these fatty acids are also acting as diffusible factors in pathogenesis as TDM and TMM led to investigate the pathways involved in the synthesis of trehalose, the major free sugar in the cytoplasm of mycobacteria. In M. tuberculosis there are three putative pathways for trehalose synthesis, which is catalyzed by OtsAB, TreS and TreYZ. In an effort to assess their relative contribution to mycobacterial biology, Murphy et al. disrupted five genes from the three pathways: otsA, otsB1, otsB2, treS and treY. The deletion of the otsA gene resulted in marked growth defects of M. tuberculosis in vitro and in C57BL/6 mice. Of the otsB homologs present in the genome of M. tuberculosis, only otsB2 has a functional role in the pathway and is strictly essential for growth. Inactivation of the TreYZ pathway (ΔtreY), which can generate trehalose from α-1,4-linked-glucose polymers, had no effect on the growth of M. tuberculosis both in vitro and in vivo. The deletion of the treS gene altered the late stages of pathogenesis of M. tuberculosis in the mouse model, significantly increasing the time to death in chronic infection. The results showed that the OtsAB pathway, which generates trehalose from glucose and glucose-6-phosphate, is the dominant pathway required for M. tuberculosis growth. However, since treS is the only gene whose deletion resulted in defective growth in vivo but not in vitro, this is the only gene of the three pathways that can be considered a virulence factor of the MTBC according to the criteria taken in this review.34
Synthesis of complex lipids: an overview of their pathways
Although mycolic acids are a remarkable feature shared by all mycobacterial species, M. tuberculosis is also characterized by a plethora of complex lipids and glycolipids present in its cell envelope. These lipids and glycolipids are loosely associated to the cell envelope, and thus they can be diffusible factors to modulate the host’s immune response or can act as infection stage signals for the pathogen. Cell wall lipids of M. tuberculosis containing multiple methyl-branched fatty acids play important roles in pathogenesis and thus offer targets for new anti-mycobacterial drugs. In M. tuberculosis, lipids esterified with multiple methyl-branched fatty acyl substituents include sulfolipids (SL), di- and tri-acylated trehaloses (DAT and TAT), poly-acyltrehaloses (PAT) and phthiocerol dimycocerosates (PDIM). The PDIM structure consists on a long-chain β-diol - phthiocerol - esterified by one type of such long-chain multiple methyl-branched fatty acids called mycocerosic acids. PDIMs constitute major virulence factors of M. tuberculosis, in particular during the early step of infection when bacilli encounter their host macrophages. However, although the chemical nature of most of these lipids and glycolipids have been known for some time, only the advent of M. tuberculosis H37Rv genome sequence shed some light on their biosynthetic pathways. The exploration of the genome sequence revealed the presence of a large number of genes involved in fatty acid synthesis/modification.22 Intriguingly, it contains a high number of genes with homology to fatty acyl CoA synthases and dehydrogenases as well as a set of genes with homology to polyketide synthases. The latter are multifunctional enzymes that, in other Actinomycetes such as Streptomyces, participate in the synthesis of secondary metabolites like antibiotics, using as building blocks acetate and propionate units among others. Thus, it was tempting to speculate that those pks gene products (along with adjacent or neighboring Acyl-CoA synthases and dehydrogenases) could be involved in the synthesis of complex lipids such as mycocerosic acid, sulfolipids, DAT and PATs. In this regard, a seminal work by Kolattukudy’s group proposed the hypothetical products of Pks based on the nature of their catalylitic domains.35
Biosynthesis apparatus of complex lipids: its relation to virulence
So far, two different approaches have produced information on Pks and accompanying lipid biosynthetic genes. In some cases, the gene of interest was disrupted and the effects of the disruption on the lipid synthesis were assessed. In others, mutants with attenuated virulence were selected using STM.
One of the earliest complex lipids and fatty acids chemically and biochemically characterized is mycocerosic acid, a multiple methyl-branched fatty acid. Seven hypothetical mycocerosic acid synthase (mas)-like genes (msl) have been identified in the genome of M. tuberculosis. One of them, msl7, was disrupted in M. tuberculosis H37Rv by replacement of an internal segment with a hygromycin resistance gene. This mutant could produce mycocerosic acids but not pthiocerol dimycocerosic acid (PDIM), a molecule biosynthetically derived from the latter. Based on that, the authors suggest that Msl7 is required for phthiocerol biosynthesis. When the virulence of the mutant was tested infecting intra-nasally C57BL/6J mice, it showed an attenuated phenotype, supporting the hypothesis that PDIMs are important virulence factors.36
In spite of these results, Reed et al. found that an M. tuberculosis mutant in the pks1/15(msl7) gene was deficient in the production of phenolic glycolipids (PGLs), but not in the synthesis of PDIM. PGLs contain a lipid core composed of phenolphthiocerol esterified by two chains of multiple methyl-branched fatty acids (phthioceranic acids or mycocerosic acids) and a variable carbohydrate moiety that, according to the mycobacterial species, is composed of one to four O-methylated deoxysugars. PGLs are produced by M. leprae, M. kansasii, M. bovis, a few slow-growing mycobacteria and only some strains of M. tuberculosis. The work performed by Reed et al. established that the production of PGL in M. tuberculosis is associated to the hyper virulent phenotype displayed by a subset of M. tuberculosis isolates belonging to the W-Beijing family. These strains showed a “hyper-lethal” behavior in murine infection models. Thus, disruption of pks1/15 resulted in a strain attenuated in its ability to kill mice following aerosol infection. However, this phenotype was not associated with a defect in multiplication or persistence within the lung or spleen, underlying the exquisite complexity of M. tuberculosis pathogenic mechanisms. Thus, the absence of PGLs results in the loss of the hyper virulent phenotype without affecting bacterial load during disease.37 Extending these results, Tsenova et al. tested the role of PGL-tb in a rabbit model of tuberculous meningitis to correlate the severity of disease caused by the M. tuberculosis clinical isolates CDC1551 and HN878 or W4, two members of the W-Beijing family strains. Compared with the infection produced by CDC1551, central nervous system (CNS) infection with HN878 or W4 resulted in higher bacillary loads in the cerebrospinal fluid and brain, increased dissemination of bacilli to other organs, persistent levels of tumor necrosis factor-α (TNF-α), higher leukocytosis and more severe clinical manifestations. These authors showed that the disruption of the pks1/15 gene in HN878 lead to reduced virulence in the rabbit model of infection. Thereby, they concluded that the pathogenic process is associated with the production of PGLs by HN878.38 Altogether these results certainly demonstrate the role of pks1/15 gene in the synthesis of PGLs and thus in the virulence of pathogenic mycobacteria, whether or not this gene is implicated in PDIMs metabolism of M. tuberculosis needs further investigation.
Another pks gene whose function was studied is pks10, a chalcone synthase-like gene. A Δpks10 mutant strain displayed the same phenotypes as the pks1/15 mutant regarding PDIM production and virulence attenuation in C57BL/6J mice, thus it was concluded that Pks10 is also involved in phthiocerol biosynthesis.36 This scenario becomes more complex and puzzling because a mutant with a deletion in pks12, the largest open reading frame in the genome of M. tuberculosis H37Rv, was deficient in the synthesis of PDIMs. However, the synthesis of mycocerosic acids was unaffected by this mutation and thus Pks12 is probably another Pks required for the production of phthiocerol. The growth of this mutant was attenuated in mouse alveolar macrophage and in C57BL/6J mice infected by the intranasal route. Hence, the expression of pks12 is probably involved in pathogenesis.39
As expected, STM yielded valuable information in connection with the role of pathways leading to the synthesis of PDIM. Two independent STM experiments showed an attenuated phenotype of FadD26 mutants, a fatty-acid-CoA synthase involved in the biosynthesis of these complex lipids. In one of these experiments, done with M. tuberculosis H37Rv, the transposon inserted in the promoter region of fadD26 and affected the expression of the downstream ppsA-ppsE operon (Rv2931 to 2935) encoding a polyketide synthase required for phthiocerol biosythesis,40 thus making the strain deficient in PDIM production. The deletion of the identified genes led to a reduced bacillary load recovered from lungs of intravenously infected mice (C57BL/6) during the initial phase of the infection. In contrast, CFUs recovered from liver and spleen appeared to be unaffected by the mutations. Thus, it was suggested that PDIM would be a virulence factor specifically required for growth of M. tuberculosis in the lungs of infected mice.41 No complementation studies were performed in this experiment; therefore, it is still unknown whether the observed phenotypes of the mutant are solely due to a polar effect on the downstream pps operon or reflect a role for FadD26 in these processes. Almost simultaneously, a second independent STM experiment42 in M. tuberculosis strain 103 yielded two strains with insertions in the fadD26 gene and in its upstream region. These mutants showed reduced in vivo growth when BALB/c mice were infected intravenously. These strains produced little to no PDIMs. Complementation of fadD26::Tn produced only 5% of the PDIM level found in the wild type, suggesting that polar effects may have contributed to their corresponding phenotypes.43
In contrast to the results described by Cox et al.,41 Rousseau et al.44 found that CFUs of fadD26::Tn M. tuberculosis 103 recovered from lungs and spleens of intra-nasally infected BALB/c mice were reduced as compared with those of the parental strain, raising doubts about the role of PDIM in tissue-specific replication. The reason for the discrepancy between both experiments is not clear but could be a consequence of the differences in the used protocols.
During the STM experimental work, Cox et al. also isolated a Tn5370 insertion in fadD28, another fatty-acid-CoA synthase. Its disruption translated in a PDIM production deficiency.41 As a consequence of these results, Camacho et al. searched for a strain mutant in the fadD28 gene by screening an insertional mutant library they had previously constructed. This new fadD28 mutant was also defective in PDIM biosynthesis and its complemented strain had only 15% the level of PDIM produced by the parental stain, which suggests that other genes apart from fadD28 are responsible for the observed phenotype.
Although the role of fadD28 in virulence was only evaluated by a high throughput technique and it was not individually tested, its involvement in the production of the pathogenesis associated-PDIM lipid family implies that FadD28 is very likely required for mycobacteria virulence. Nevertheless, further investigation is needed to demonstrate that this gene is indeed a virulence factor.
Along with the identification of the genes involved in the biosynthesis of PDIM, STM experiments brought up the identity of genes whose products were involved in the transport of this important complex lipid. One of such genes encoded for MmpL7, a member of the MmpL protein family that is located within the DIM locus. Insertion mutants in this gene were capable of synthesizing PDIMs but failed to localize them on the cell surface, confirming the role of MmpL7 in the translocation of these lipids across the plasma membrane. Consistent with these results, Domenech et al. showed that MmpL7 is required for normal growth of M. tuberculosis H37Rv in aerosol-infected mice.45 Another component of the secretion of PDIM is DrrC, which is a member of an ATP-binding cassette (ABC) transporter and works along with MmpL7; thus defects in any of them lead to PDIM accumulation. DrrC was identified in one of the STM transposon searches as being important for M. tuberculosis virulence.42 Complementation of the DrrC mutant with a copy of the wild-type gene led to full restoration of PDIM production and translocation, demonstrating that the DrrABC transporter, like MmpL7, is essential for PDIM translocation.43 Since the virulence of DrrC was only evaluated by a high throughput technique, further research is required to assert its role in M. tuberculosis pathogenesis. Another protein required for the translocation of PDIMs to the outer membrane of M. tuberculosis is the lipoprotein Lppx. A mutant in the gene encoding this protein was identified as being highly attenuated in the STM experiment performed by Camacho et al.42 Lpp× will be extensively described below in the section referred to lipoproteins.
A study performed by Rousseau et al.46 led to the construction and analysis of a Δpks5 M. tuberculosis mutant. Disruption of this gene, which encodes a mas-like polyketide enzyme, showed no difference in cell envelope lipid composition although it displayed severe growth defects in a mouse model of infection (BALB/c mice). Simultaneously, these researchers also disrupted pks7, another mas-like polyketide gene. In contrast with the results obtained for pks5, the M. tuberculosis Δpks7 strain was deficient in the production of PDIMs. As expected, the growth of this mutant in BALB/c mice infected via the respiratory route was severely affected.46 Thus, a large body of evidence supports the role of PDIM as a virulence factor through the identification of several genes involved in its synthesis. This molecule displays its role in virulence mainly in the early step of infection, when bacilli encounter their host macrophages. Although available information pointed at a mechanism that modulate immune response at the macrophage level, the mechanisms by which the bacilli modultate it are still unknown. A recent study by Astarie-Dequeker et al. in which they used a ΔppsE M. tuberculosis strain reported that PDIM participates in the receptor-dependent phagocytosis of the tubercle bacilli, as well as in the prevention of phagosomal acidification.47 This study demonstrated that this effect was mediated by insertion of PDIM in the host membrane, affecting lipid organization and increasing the efficiency of receptor-mediated phagocytosis of bacilli. These results will clearly help understanding the molecular events through which complex lipids interact with infected host cells, modulating their response to the pathogen’s advantage.
Apart from PDIMs, another predominant cell wall lipid is sulpholipid-1 (SL-1), a sulphated glycolipid that has been studied for over 50 years rendering controversial results. Goren et al. showed that SL prevented phagosome-lysosome fusion in cultured macrophages, produced toxic effects on mytochondria by blocking oxidative phosphorylation and suppressing the production of reactive oxygen.48,49 Only recently genetic manipulation and the analysis techniques available allowed a detailed characterization of this molecule,50,51 which is thought to mediate host-pathogen interactions during infection. However, a direct involvement of SL-1 in mycobacterial virulence has not yet been established. There are two independent studies that show that MmpL8, a member of a membrane protein family potentially involved in lipid transport in M. tuberculosis, is required for SL-1 production. Both studies show that the mutation of the gene encoding MmpL8 in M. tuberculosis leads to the accumulation of an SL-1 precursor, indicating that MmpL8 is necessary for an intermediate step in the SL-1 biosynthesis pathway. This precursor, called SL1278, was found accumulate inside the cell, whereas SL-1 was present on the cell surface. These results suggest that the transport and biogenesis of SL-1 are coupled. Both works also showed that mmpL8 mutants are attenuated for growth in C57Bl/6 mice. However, SL-1 per se is not required for establishing infection, since pks2 mutants that are defective in SL-1 biosynthesis have no obvious in vivo growth defect.52,53 These results suggest that either MmpL8 transports other molecules that are implicated in virulence (other than SL-1) or that the accumulation of SL1278 prevents bacterial growth during infection.21,45,54
Other virulence genes involved in fatty acid/lipid metabolism
Although we have offered in the precedent sections an overview of the synthesis of mycolic acids and complex lipids and their roles in the virulence of M. tuberculosis, several other pathways are also of importance in the success of this pathogen. Below, we summarize what is currently known about those pathways, and what lies ahead.
FadD33
FadD33 is an acyl-CoA synthase whose gene shows much higher expression in the virulent strain M. tuberculosis H37Rv as compared with the avirulent H37Ra strain. Thus, Rindi et al. set out to investigate the potential pathogenic role of this protein. In a first approach, the authors complemented M. tuberculosis H37Ra to restore gene expression and studied if this condition conferred any growth advantage to this strain in an infection model of BALB/c mice. They found that, although the growth of the attenuated strain H37Ra was impaired in liver, complementation of this strain with fadD33 restored bacterial replication in this organ. In a second approach, the fadD33 gene of M. tuberculosis H37Rv was disrupted and the virulence of the generated mutant was evaluated by mouse infection. Again, the absence of the FadD33 protein affected the growth of M. tuberculosis in liver but not in lungs or spleen, suggesting that fadD33 plays a role in M. tuberculosis virulence by supporting tissue-specific replication.55
Icl1 and Icl2
As an intracellular pathogen, M. tuberculosis has to rely on carbon sources obtained from the host’s cells to survive. Those metabolic adaptations are important as possible points of intervention through the design of novel drugs. Importantly, the fact that M. tuberculosis primarily uses fatty acids instead of carbohydrates during infection is known since the mid 1950s, as reported by Bloch and Segal.56 Fatty acids may be used by their catabolism through β-oxidation generating acetyl-CoA, which can be incorporated into the Krebs cycle using an anaplerotic cycle, the glyoxylate cycle. Isocitrate lyase (Icl) is an enzyme that converts isocitrate to succinate in the glyoxylate cycle. This allows bacteria to grow on acetate or fatty acids as sole carbon sources, since the glyoxylate cycle provides a source of carbon that can be further metabolized. In M. tuberculosis the glyoxylate cycle apparently comprises a single gene encoding malate synthase and two genes encoding Icl. The smaller icl gene (icl1) encodes an enzyme closely related to Icls in other eubacteria, while the larger gene (icl2) encodes a protein more homologous to eukaryotic Icls. McKinney et al. have mutated icl1 gene of M. tuberculosis and studied the contribution of this gene to the in vivo metabolism of these bacteria infecting C57BL/6 mice. The mutant initially grew normally in mice, but from the second week onwards the Δicl mutant was eliminated progressively from the lungs and extra-pulmonary organs. In addition, the mutant exhibited wild-type growth in IFN-γ knockout mice and in inactivated macrophages but was killed more rapidly than the parental strain when these macrophages were activated.57 Extending these results, Muñoz-Elías and McKinney showed that both prokaryotic- and eukaryotic-like isoforms of the Icl are jointly required for fatty acid catabolism and pathogenesis in M. tuberculosis. While mutation of icl1 or icl2 had little effect on bacterial growth in a mouse model of infection (C57BL/6 mice) and in both murine BMDM and human blood derived macrophages, the deletion of both genes resulted in complete impairment of intracellular replication and rapid elimination from the lungs.58 Additional evidence indicating the importance of Icl includes the observation that icl mRNA levels increase in lungs of M. tuberculosis-infected C57BL/6 mice as the infection progresses.59 These data establish a link between the requirement of Icl and the immune status of the host, suggesting that the in vivo metabolism of M. tuberculosis is profoundly influenced by the host response to infection. Thus, the identification and characterization of M. tuberculosis glyoxylate cycle not only helped to finally confirm Bloch and Segall’s experiments, but also generated at the same time a great deal of information that can be translated into drug design taking advantage of the absence of this cycle in humans. Obviously, more research in this area is warranted considering the proven role of the M. tuberculosis Icl enzymes during infection.
PlcA, PlcB, PlcC and PlcD
The M. tuberculosis genome has four open reading frames (ORFs) which encode phospholipase C-type enzymes: plcA, plcB, plcC and plcD. To study the contribution of these genes to the pathogenesis of M. tuberculosis, Raynaud et al. constructed four single mutants of M. tuberculosis, each inactivated in one of the plc genes, a triple plcABC mutant and a quadruple plcABCD mutant. Phospholipase C activity was determined in cell extracts of these strains and it was found that all individual mutants had lower enzyme activities compared with the wild-type M. tuberculosis 103. Although RT-PCR analysis of the plc genes transcripts showed that the expression of these genes was strongly upregulated during the first 24 h of macrophage infection, the triple and quadruple plc mutants of M. tuberculosis grew normally in these cells. However, the growth kinetics of the triple and quadruple mutants in BALB/c mice revealed that both strains were attenuated in the late phase of the infection, suggesting a role of plc in the virulence of M. tuberculosis.60
Catabolism of cholesterol
As mentioned above, in the intracellular environment, M. tuberculosis adapts its metabolism, shifting from one carbohydrate-based to one fatty acid-based.58 It has been demonstrated that M. tuberculosis uses cholesterol as energy source and for the biosynthesis of the virulence-associated lipid PDIM.61 In addition, an increasing number of reports indicated that M. tuberculosis metabolizes cholesterol during host infections and that degradation of this sterol contributes to the survival of M. tuberculosis in the host.61,62 However, while the mutation of genes associated with cholesterol catabolism choD, hsaC and mce4 operon attenuated the virulence of M. tuberculosis, mutation of hsd (encoding a 3β-hydroxysteroid dehydrogenase) did not reduce M. tuberculosis growth inside macrophages or guinea pigs but it was found that this gene is required for bacterial growth on cholesterol as a sole carbon source.63 Therefore, it is plausible to speculate that the in vivo attenuation observed for the above mentioned mutants was due to the toxicity of accumulated metabolites of M. tuberculosis cholesterol pathway rather than impairment in the utilization of cholesterol as carbon source. In this regard, several studies focused on choD, which encodes a putative cholesterol oxidase. This enzyme catalyzes the oxidation and isomerization of cholesterol to cholestenone (4-cholesten-3-one), which is an initial step in the cholesterol degradation process. The lack of choD from M. tuberculosis resulted in an impaired replication in C57BL/6 mouse lungs and spleens as compared with the wild-type and complemented strains. Infection of mouse peritoneal macrophages also showed a replication defect in the mutant strain.64 Whether the inability of the mutant to use intracellular cholesterol is the reason of the reduced virulence of choD mutant needs further investigation.
In the same line of research, HsaC, another enzyme linked to cholesterol catabolism, has been recently studied. hsaC encodes a putative extradiol dioxygenase that catalyzes the cleavage of catechols and their analogs. Yam et al. have reported that M. tuberculosis produces catechols such as DHSA [3,4-dihydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione] through the metabolism of cholesterol.65 In that study, the authors demonstrated that HsaC is a key enzyme in the cholesterol catabolic pathway. Importantly, an M. tuberculosis mutant deleted in the hsaC gene completely failed to grow on cholesterol and killed SCID mice in more time than the wild-type and complemented strains. Moreover, bacterial loads of lungs from guinea pigs infected with the mutant strain were modestly reduced as compared with those of the animals infected with the wild-type and complemented strains. This attenuated phenotype in lungs correlated with reduced granulome formation in this organ.
Finally, another player identified in the metabolism of cholesterol by M. tuberculosis is the igr operon, which contains genes for a putative cytochrome P450 (cyp125), two acyl coenzyme A dehydrogenases (fadE28/29), two conserved hypothetical proteins (Rv3541c, Rv3542c), and a probable lipid carrier protein (ltp2). Based on the homology with a Rhodococcus cholesterol-catabolic locus and the genetic interaction with genes encoding the cholesterol transporter Mce4,66 it has been proposed that the igr operon encodes enzymes of cholesterol catabolism. Consistent with this proposed role for igr operon, Chang et al. have demonstrated that the lack of this locus inhibits the growth of M. tuberculosis in the presence of cholesterol and that this growth inhibition was not due to inability of the bacteria to use the steroid as carbon source but to accumulation of toxic intermediate generated in the initial steps of its degradation.67 A M. tuberculosis deleted in igr operon replicated less in C57BL/6 mouse lungs and could not colonized the spleens, as compared with the wild-type H37Rv strain. Remarkably, the deletion of the mce4 operon from this mutant strain rescued this in vivo attenuated phenotype, indicating that in the absence of cholesterol uptake the M. tuberculosis igr mutant restored its full virulence.67 Based on their finding, the authors proposed that the in vivo attenuation of igr mutant was most likely due to its inability to metabolize cholesterol fully.
Thus, all the information currently available reinforces the concept of the importance of cholesterol metabolism for a successful mycobacterial infection, although the relative contribution of the partial degradation of this compound as carbon source seems to be of lesser importance than the total metabolism to avoid accumulation of toxic intermediates. As cholesterol is a major component of human cell membranes and its concentration there modulates membrane fluidity and engulfment processes, it is clear that this area of research will be quite active in the near future. Moreover, cholesterol is a precursor of steroideal sex hormones, which (among other functions) control immune response. Recent studies have reported that the changes of these hormones correlate to the severity of TB in humans68; thus it is not farfetched to assume that we are just starting to learn about a new way by which M. tuberculosis became a successful pathogen adapted to humans.
In conclusion, as it was mentioned above, M. tuberculosis is exclusive among bacterial pathogens in that it displays a large array of complex lipids on its cell envelope. Indeed, the lipid content of the cell envelope of mycobacteria may represent up to 40% of the cell dry mass.69 This exclusive feature reflects the large proportion of the mycobacterial genome devoted to lipid and fatty acid metabolism: about 250 genes are involved in lipid metabolism in M. tuberculosis, vs. only 50 such genes in E. coli, which has a similar genome size.70 Many of these lipids are documented to be important virulence factors of the bacteria and the mechanisms by which they play relevant roles during infection are diverse. Some lipids present in the mycobacterial cell envelope constitute key ligands for the host cell receptors, allowing molecular docking between the host phagocytes and the mycobacteria, leading to cell invasion. Once inside phagocytic cells, M. tuberculosis avoids lysosomal fusion and acidification, residing in an immature phagosome.71,72 It was found that some mycobacterial lipids such as TDMs, Man-LAM and PDIMs47,73,74 play important roles in intracellular trafficking and the vacuole maturation arrest by M. tuberculosis. In addition, some mycobacterial lipids modify host cell signaling, affect the secretion of cytokines necessary for protection, participate in the inflammation process during tuberculous infection, or are recognized as antigens by the adaptive immune system. Apart from being involved in these mechanisms of host-cell interactions, some lipids also influence virulence because they display a structural function as being part of the mycobacterial cell wall, which presents a notably low permeability to nutrients and antibacterial drugs. This feature slows down the growth of the bacteria and makes disease caused by pathogenic species difficult to treat. As a consequence of these mechanisms of action, many mycobacterial lipids, and therefore the genes encoding proteins involved in their metabolism, constitute very important virulence factors of the pathogenic species. However, some lipids, such as mycolic acids and PIMs, present only a few genes involved in their metabolism whose mutation affect the pathogenesis of the bacteria. The reason of this feature is that they are essential for mycobacterial viability and thus the variations in their structure have to be minimal. On the other hand, there are other lipids, such as PDIMs, which are not required for viability but display an important function during the infection process. As a consequence, there are no clinical isolates lacking PDIMs, but it was reported that M. tuberculosis H37Rv is highly prone to losing the ability to synthesize PDIMs during extended periods of in vitro culture.75 Due to this feature there is a large list of genes involved in PDIMs metabolism that influence the pathogenesis of the bacteria when they are mutated. Finally, there is a third group of lipids, such as PGLs and SLs, which are not required for virulence per se, but influence the development or clinical signs of the disease. This is the explanation for the different proportions of genes involved in the biosynthetic pathways of the diverse lipids described to date as virulence factors. However, further research is needed to identify new genes important in the disease process, since many proteins described as virulence factors by high throughput techniques were not yet individually tested.
Cell Envelope Proteins
As mentioned above, the cell wall, an intricate structure of complex lipids and proteins, is the hallmark of mycobacteria. Its “core” is comprised of peptidoglycan covalently bound to a linear galactofuran, in turn attached to several strands of a highly branched arabinofuran joined to mycolic acids that are very long chain α-alkyl β-hydroxy fatty acids. These lipids are perpendicularly oriented to the plane of the membrane and provide a special barrier responsible for many of the physiological and disease-inducing aspects of mycobacteria. Intercalated within this “core” are the PDIMs, TDM, SLs and PIMs.50 In turn, phosphatidyl-myoinositol mannosides, lipomannan (LM) and lipoarabinomanan (LAM) are anchored to the plasma membrane and extend to the exterior of the cell wall. The most external layer of mycobacteria is the capsule, a structure that has been recognized and described later than other cell envelope components.76 The capsule contains polysaccharides and minor amounts of inner lipids. There are a number of proteins embedded in this matrix whose function is related to the synthesis and maintenance of the cell wall and that are also responsible for the adhesion, infection, transport of solutes (porins) and survival of mycobacteria in the host cells (Fig. 2).
Figure 2. Schematic representation of cell envelope proteins embedded in the cell wall of the of M. tuberculosis complex. Proteins like Erp, Fbp, HbpA and the porin OmpATb are exposing to the surface. The ABC transporters: Pst-1, Pst-3, Opp and the Mce proteins consist in the substrate binding proteins (SBP), the permeases (MSD) and the nucleotide binding protein (NBD). The two component systems: PhoR-P, DosS/T-R and MprAB consist in the sensor kinase (SK) that senses the stimuli and the response regulator (RR), which induce the gene transcription. AG, arabinogalactan; PG, peptidoglycan; CM, cytoplasmic membrane; TMM, TDM, trehalose mono and di mycolate; DAT, PAT, di and poli acyl trehalose; PGL, phenolic glycolipid; PDIM, phthiocerol dimycocerosate; SL, sulfolipid.
In this section, we review (1) proteins that are either localized entirely in the cell wall or with a trans-membrane domain exposed toward it, (2) lipoproteins and (3) secretion systems.
Cell wall proteins
Proteomic studies of the mycobacteria cell wall have identified more than five hundred proteins in this cellular structure, including secreted cell wall proteins and lipoproteins.77,78 Most of these putative cell wall proteins are involved in cell wall processes and intermediary metabolism. Among these identified putative cell wall proteins, more than 5% have been classified in the Virulence and Detoxification category and about 15% in the Lipid Metabolism category. Cell wall proteins include the outer membrane proteins (OMPs) that are likely localized in the recently discovered mycobacterial outer membrane bilayer.79,80 By combining the use of an algorithm entirely based on physical principles to predict outer membrane proteins (OMPs) of M. tuberculosis with biological knowledge, Song et al.81 have predicted 144 proteins as OMPs of M. tuberculosis. These OMPs may participate in the uptake of hydrophobic compounds across outer membranes, efflux processes and energy-dependent uptake of nutrients. They also may play a role in the attachment and invasion of host cell and the degradation of host’s structures. Importantly, some of these OMPs would comprise the unknown outer membrane component of the inner membrane transport systems, already identified in MTBC.
The identification and characterization of cell wall and secreted proteins is critical to the understanding of bacterial survival and immune modulation in the host. In this section we describe mycobacterial cell wall proteins, including outer membrane and cell-associated secreted proteins that were demonstrated to play a role in the interaction of MTBC species with their host.
Erp
Erp (exported repetitive protein) is a cell-wall-associated surface protein, usually secreted into the culture medium with a molecular mass of 36-kDa, therefore, it is known as P36 protein82-84 (Fig. 2). Erp proteins have a modular organization consisting of three domains: two well-conserved N- and C-terminal domains, and a central domain containing several Pro-Gly-Leu-Thr-Ser (PGLTS) repeats which vary in number, with four repeats in M. leprae and 24 in M. xenopi. This variability of the central domain has shown to be related to virulence.84 Although the function of Erp still remains unknown, the contribution to virulence has been well demonstrated. Berthet et al.82 have shown that M. tuberculosis and M. bovis BGC erp mutants were impaired for multiplication in cultured macrophages and in BALB/c mice compared with the wild type. The re-introduction of erp into the mutants restored their ability to multiply, suggesting that Erp contributes to the virulence of M. tuberculosis. Similarly, Bigi et al. have observed the same phenotype in an M. bovis erp mutant infecting BALC/c mice: the CFU counts in spleen and lung were markedly reduced compared with the wild-type or complemented strain.85 Additionally, an M. tuberculosis erp mutant complemented with the erp from M. leprae, which has a reduction in the number of PGLTS repeats in the central domain, correlated well with an increase in the multiplication and damage of mycobacteria in the lung of mice.84 This result suggests that the contribution of Erp to virulence is directly related with the extension and variability of its central PGLTS repeated domain. Besides, the Erp family is not restricted to pathogenic mycobacteria and is also present in the saprophytic ones, which suggests a more physiological role for Erp. The erp gene is located between glf and csp, which encode proteins involved in lipopolysaccharide biosynthesis. Given that this genomic organization is well conserved in many mycobacterial species, it is tempting to hypothesize that Erp could have a possible role in cell wall biosynthesis.86
Fbp
Fbp (fibronectin binding protein) is a complex of three proteins: FbpA, FbpB and FbpC2, and their name derives from their capabilities to bind fibronectin (FN). This complex is commonly known as antigen 85 (Ag85) and includes Ag85A (FbpA), Ag85B (FbpB) and Ag85C (FbpC2). These proteins are encoded by the fbpA, fbpB and fbpC2 genes, which are located in different genomic regions: Rv3804, Rv1886c and Rv0129c, respectively. A fourth gene fbpD, or fbpC1, related to Ag85 is annotated in the M. tuberculosis genome. However, FbpC1 did not show an in vitro mycolyl transferase activity, a signature of this complex.87 The Fbp complex is the major secreted protein constituent of mycobacterial cell culture and it is also found in association with the bacterial surface.78,88,89 This protein complex plays an essential role in the pathogenesis of tuberculosis. Its ability to bind FN promotes the adhesion of mycobacteria to the mucosal surface, thus facilitating its entry into the host cells.89 However, its main contribution to the virulence of M. tuberculosis is likely due to its physiological role in the synthesis of cell wall lipids.
Belisle et al.30 have shown that FbpA, FbpB, and FbpC have mycolyltransferase activity, required for maintaining the integrity of the mycobacterial cell envelope. All three proteins catalyze the transfer of mycolates to trehalose, leading to the formation of α,α′-trehalose monomycolate (TMM) and α,α′-trehalose dimycolate (TDM) (Fig. 2). While FbpA and FbpC2 have similar specific activities, the FbpB specific activity values decreased by up to 80% compared with FbpC2. These proteins conserve the carboxyl esterase consensus sequence (G-X-S-X-G) and have a catalytic triad formed by Ser, Asp/Glu and His, in which the Ser is the active site.30 Studies on the crystal structure of FbpC2 have revealed a hydrophobic pocket and tunnel extending into the protein structure as the probable trehalose monomycolate binding site, and a large region of conserved residues in the surface of the proteins a putative site for the interaction with FN.90
The role of Fpb proteins in the virulence of mycobacteria has been evaluated in individual mutant strains. While an M. tuberculosis H37Rv fbpA mutant exhibited a marked decrease in growth in monocyte-like human THP-1 and murine J774 macrophage cell lines compared with the parental strain, an H37Rv fbpB mutant grew at the same rate than the wild type in those macrophage models of infection.31 This could be a direct consequence of the catalytic efficiency of each protein. In an additional study, an M. tuberculosis Mt103 mutant in fbpC2 gene showed 40% less mycolates in the cell wall as compared with the parental strains, although changes in the types of mycolates esterifying arabinogalactan or in the composition of non-covalently linked mycolates were not observed. Despite this profound modification in its cell wall, the fbpC mutant showed a similar intracellular replication and survival in mouse bone marrow macrophages as compared with the wild type.91 Altogether, these results suggest that FbpA, but not FbpB or FbpC, would be essential in the virulence of M. tuberculosis.
Mce
The Mce proteins are a large group of secreted or surface-exposed proteins organized in large operons. Their name Mce, mammalian cell entry, is due to the first function described for the Mce proteins: it has been shown that Mce1 conferred mycobacteria the ability to enter into mammalian cells and survive inside the macrophage.92 These operons comprise eight genes each and are organized in identical manner: two yrbE genes (A and B) followed by six mce genes (A, B, C, D, E and F).22 M. tuberculosis contains four mce loci: mce1, mce2, mce3 and mce4, whereas M. bovis lacks the mce3 locus.93 Mce proteins are homologous to ATP-binding cassette transporters (ABC-transporters), presenting a typical gene arrangement of importers in which YrbEs are homologous to permeases and have a region within the penultimate cytoplasmic loop that may serve as the site of interaction to ATPases, whereas Mces are homologous to substrate-binding proteins94 (Fig. 2). The contribution of each Mce to virulence has been demonstrated from a set of M. tuberculosis knockout mutants. When mce1, mce2 and mce3 mutants were used to infect BALB/c mice by an intratracheal route, they showed an attenuated phenotype compared with the wild type.95 The same behavior was observed when mutants for the mce2, mce3 and mce4 loci were used to infect C57BL/6 mice after aerosol infection.96,97 However, Δmce1 M. tuberculosis strain showed a hyper virulent phenotype as compared with parental or complemented strains98 in mice infected by a systemic or intraperitoneal95 route with this mutant. This indicates that in the absence of the Mce1 M. tuberculosis could trigger an active infection; therefore, a specific control of the mce1 expression would be necessary to keep the mycobacteria proliferation in check and allow the establishment of a latent infection in the host.98 Contrary to this, some high-throughput techniques have shown that the mce genes, mainly mce1, are required for mycobacterial survival in macrophages or mice models of infection.99-101 Perhaps the importance of Mce proteins in MTBC virulence lies in their hypothetical role as transporters and, therefore, their absence would prevent these mycobacteria from importing essential but yet unidentified compounds. The first Mce operon with a demonstrated transport function was Mce4,61 which is involved in cholesterol import in M. tuberculosis. Importantly, cholesterol is essential for maintaining a chronic infection in the host as it has been described in the cholesterol metabolism section. Later on, Santangelo et al. demonstrated that the transcriptional regulator of mce3 operon, Mce3R, not only controls the expression of the mce3 operon but also of the Rv1933c-Rv1935c and Rv1936-Rv1941 operons, which encode proteins that are predicted to be involved in lipid metabolism and redox reactions.102,103 Additionally, a mutant deleted in the fadD5 gene, which is located within the mce1 operon, shows a decreased growth in minimal medium containing mycolic acids as a carbon source.104 This suggests that the Mce1 proteins also may be related to the recycling of these lipids. Furthermore, a mutant in the mce2 operon shows an increased accumulation of sulpholipids.105 Thus, although a clear role for mce operons has been only firmly established for mce4, it is reasonable in light of the results of mce1, mce2 and mce3 to postulate that these operons have evolved to fulfil specific roles, most likely related to lipid metabolism, modulating pathogenicity through changes in M. tuberculosis lipid pathways.
OmpATb
OmpATb is a pore-forming protein (porin) that belongs to the OmpA family of outer membrane proteins. Its β-sheet/β-barrel structure enables it to form pores with a diameter of 1.4 and 1.8 nm, facilitating the passage of small hydrophilic molecules such as arabinose, glucose, sucrose and serine to the cytoplasm106 (Fig. 2). Like other members of the OmpA family, this porin plays a role in pathogenicity. An M. tuberculosis mutant in the ompATb gene showed a significantly reduced multiplication in macrophages compared with the wild type, and its growth in lungs and spleen of BALB/c mice was markedly reduced when compared with the levels of the wild type.107 The transcription of the ompATb gene is highly increased at low pH suggesting an activity of this porin in acidic conditions. As expected, the growth of ompATb mutant at low pH is affected as well as the uptake of some water-soluble metabolites such as serine.107 These observations suggest that OmpATb is a pore-forming protein that functions under acidic conditions. Therefore, in the environment prevailing within the phagosome, OmpATb allows the bacteria to continue acquiring molecules and, therefore, makes its survival possible.
HbhA
HbhA (heparin-binding hemagglutinin) is the major adhesin exposed at the surface of the cell (Fig. 2). This protein binds sulphated glycoconjugates like heparin, promoting the attachment of the mycobacteria to the epithelial cells and fibroblasts, but not to macrophage-like cells. In addition, the protein promotes the agglutination of rabbit erythrocytes and also induces mycobacterial aggregation,108 which gives the bacteria the ability to form a primary biofilm. HbhA is a 28-kDa protein with three functional domains: a large N-terminal domain of 81 amino acids consistent with an α-helical coiled-coil region, which promotes bacteria-bacteria interaction, a trans-membrane domain of 18 amino acids residing near the N-terminus, which is the anchoring to the cell wall, and a C-terminal domain with a Lys-Pro-Ala-rich repeat that mediates binding to proteoglycans.109,110 In addition, the relevance of HbhA in virulence has also been demonstrated in an in vivo model of infection. The disruption of the M. tuberculosis M103 or M. bovis BCG hbhA gene markedly affects mycobacterial interactions with A549 pneumocytes but not with murine J774 macrophage cells. Also, when the mutants are inoculated into BALB/c mice, they are severely impaired in spleen colonization, but not in lung colonization compared with the parental or complemented strains. These results indicate that HbhA is required for extrapulmonary dissemination, and that interactions with non-phagocytic cells have an important role in the pathogenesis of tuberculosis.111 Interestingly, HbhA is immunogenic in humans during mycobacterial infections108; additionally, coating wild-type mycobacteria with anti-HbhA antibodies also impaired dissemination in a mouse model of infection; which suggests that the antibody responses to HbhA may be playing an important role to immune protection against tuberculosis.111
PstA1 and PhoT
PstA1 and PhoT (encoded by pstA1 and phoT, respectively) are proteins involved in the transport of inorganic phosphate. Whereas pstA1 is clustered together with pstC2 and phoS2 (also called pstS3) in the same operon, phoT is located elsewhere in the genome. PhoS2 is the substrate-binding protein, PstC2 and PstA1 are the permeases containing the membrane spanning domain (MSD) and PhoT has homology to the nucleotide-binding domain protein, responsible for the energy coupling to the transport system99,112 (Fig. 2). M. tuberculosis contains three ABC phosphate transporters: PstS-1 (described below with the 38-kDa protein), PstS-2 and PstS-3. All of them are involved in phosphate import during starvation, a condition prevailing inside the phagosome. Rengarajan et al.99 showed that the PstS-3 transporter plays a role in virulence, although Sassetti and Rubin had previosuly shown that it was not essential for mycobacterial survival in mice.100 The M. tuberculosis transposon mutants in the pstA1 or in the phoT genes were more sensitive to grow in a phosphate-limited culture compared with the wild type; however, they grew at the same levels in a high phosphate concentration cultures.99 Mutants grew poorly in resting and activated macrophages compared with the wild type, although the attenuated phenotype could be partially complemented by overexpression of each wild-type gene. In addition, the phoT mutant has shown a reduced growth in mouse lungs but not in spleen or liver,99 compared with the wild-type or complemented strains. These results clearly indicate that PstS-3 is required for phosphate uptake and survival within macrophages.99
CaeA
CaeA, also known as Rv2224c, is a carboxylesterase located in the cell surface. The protein is an esterase/lipase, preferentially hydrolyzing ester bonds of substrates with intermediate carbon chain length, about 3 to 7 carbon atoms.113 The enzyme is a 54-kDa monomer with the active-site consensus sequences (G-X-S-X-G), where the replacement of Ser215, Asp453 and His477 by Ala completely abolishes the esterase activity, suggesting that these residues form the catalytic triad with Ser215 as the active site residue. It serine active site is characteristic of α/β hydrolase-fold family members, namely proteases, esterases, and lipases. In a former study it has been shown that CaeA is required for full virulence of M. tuberculosis in mice.113 A deletion mutant has been used to infect BALB/c mouse, showing reduced CFU load in lungs and spleen and diminished lung pathology as compared with the wild-type or to complemented strains.113 Similar results have been reported with an M. tuberculosis Rv2224c insertional mutant when used to infect C57BL/6 8 mice.114 In addition, these authors have shown that this mutant is hyper-susceptible to acid or oxidative stress (see section on “Proteins Inhibiting Antimicrobial Responses of the Macrophage”). Lately, Rengarajan et al. also demonstrated that Rv2224c is critical for M. tuberculosis virulence in vivo. In C57BL/6 infected mice, the mutant survived significantly longer and caused reduced lung pathology than the wild type. In primary macrophages at early stages of infection, both wild-type and mutant strains grew equally well, but the mutant failed to continue growing, probably because of its inability to modulate the innate immune control of infection. Interestingly, these authors have reported that Rv2224c is needed to the release of GroEL2 from the cell wall of M. tuberculosis, infering a protease activity for this protein. GroEL2 is a highly expressed, immunodominant stress-induced protein that is present as both a full-length and smaller N-terminally processed form.115 The role of Rv2224c in the virulence of M. tuberculosis is unquestionably, possibly due to its role as lipase113. Although it has been observed that some proteases may also have a lipase activity, whether or not GroEL2 is a direct substrate for Rv2224c, remains unclear and awaits further biochemical characterization.
KefB
The potassium efflux system KefB functions via potassium/proton antiport. Bacteria inside phagosomes can release potassium from their cytoplasm via KefB and uptake proton from the phagosomal lumen, increasing luminal pH. This latter interferes with the phagosomal maturation and hence with the elimination of the bacteria. An M. tuberculosis kefB mutant generated by transposon insertion localized mostly in acidified phagosomes,101 suggesting that the role of KefB is to avoid the phagosomal acidification induced in macrophages in response to bacterial infection. However, paradoxically, the opposite has been observed in an M. bovis BCG mutant in the kefB gene (BCGΔkef). The BCGΔkef mutant exhibits an increased intracellular survival phenotype in resting and activated murine J774 macrophages compared with the wild-type BCG Pasteur.116 Despite the absence of KefB, this mutant retains the capability to inhibit phagosome acidification. In addition, the mutant is able to inhibit the induction of antimicrobial mechanisms from the macrophages, such as ROS. The production of ROS by macrophages infected with BCGΔkef decreased in relation with those infected with wild-type BCG, suggesting that reduction of the macrophage oxidative burst could be the cause of the increased intracellular survival of BCGΔkef.116 Therefore, the production of ROS in the mycobacterial phagosome, paradoxically, would depend on the bacterial potassium transporter KefB, whose activity is altering the ionic contents of the phagosome and promoting the production of ROS. So, the role of KefB in the virulence of M. tuberculosis is still controversial and requires further studies.
Opp-Dpp ABC transporter
Two predicted operons encoding permeases involved in the uptake of small peptides are annotated in the M. tuberculosis H37Rv genome based on homology to other described transporters: OppABCD (oligopeptide permease, Rv1283c-Rv1280c) and DppABCD (dipeptide permease, Rv3666c-Rv3663c). Different approaches have been used to elucidate their function and role in bacterial survival. Based on the results from Green et al.117 and Flores-Valdez et al.,118 it is now clear that the Rv3666c-Rv3663c locus encodes an oligopeptide transport system, whereas the Rv1283c-Rv1280c locus encodes a dipeptide system. As a consequence, special attention should be paid to the nomenclature in bibliography. Two insertional mutants in the ATP-binding component of both operons (which renders the transporter not functional because the ATP hydrolysis is impaired) have shown different results. BCG Rv1281c (formerly oppD) transposon-insertion mutant in inactivated murine macrophages failed to show an affected survival,117 whereas an Rv3663c (formerly dppD) mutant strain, generated by allelic exchange in H37Rv, showed an affected survival at the initial phase of infection in mice.100 Along the same line of evidence, in a microarray-based screening of a BCG transposon library in macrophage culture, dppC disruption has shown among the most attenuating mutations of that study.101 Moreover, Flores-Valdez et al.118 obtained an opp (Rv3666c-Rv3662c) M. tuberculosis knockout strain and assessed its virulence in BALB/c mice. This opp mutant did not show differential virulence phenotype in mice during the active phase of infection. However, bacterial load in lungs and spleen in the chronic phase of infection was diminished when compared with the wild-type strain. In addition, the survival time of the opp mutant in infected animals was higher than in the wild type, but the reintroduction of a copy of opp into the mutant strain failed to restore the wild-type phenotype. These authors have proposed that Opp incorporates oligopeptides that modulate intracellular signaling pathways, fauvoring the survival of M. tuberculosis inside cells. Indeed, Opp was required to modulate the expression of several genes like fasI, desA3, icl, fadE13 and PE13 among others, most of them encoding surface-exposed molecules such as mycolic acids, PDIMs as well as PE-family proteins. An interesting hypothesis is that these permeases are involved in the import of peptides or lipopeptides to signal the need to adequate/remodel cell wall envelope components. A comparable system is the basis of the natural uptake of DNA (competence) displayed by Bacillus subtilis, which uses formation and import of oligopeptides to signal metabolic stages. Regarding the important role of lipid molecules in the mycobacterial physiology and in the molecular mechanisms of pathogenesis displayed by M. tuberculosis, more studies are undoubtedly warranted.
CtaC
CtaC is the subunit II of the cytochrome c oxidase important for growth under aerobic conditions. An in silico analysis showed the cytochrome oxidase domain is on the extracytoplasmic face of the membrane.119 CtaC has been predicted to be essential in M. tuberculosis H37Rv. An ctaC::Tn_blaTEM-1 mutant, which expresses a version of CtaC truncated in the final 13 amino acids of the C-terminus, was able to grow in vitro, but showed a rough and spread out colony phenotype compared with the wild-type strain. This mutant displayed a growth defect in macrophages, suggesting that CtaC has a role in virulence. However, no experiment has been done to rule out the possibility that the attenuated phenotype of the ctaC mutant was due to a polar effect on the downstream ctaF gene, which has been predicted to encode for an additional subunit of the cytochrome c protein.119
Lipoproteins
Lipoproteins (Lpps) constitute a major component of the cell envelope in mycobacteria. Bioinformatics studies have predicted that the M. tuberculosis genome potentially encodes 48 to 99 lipoproteins (depending on the different algorithms used) representing 1.2 to 2.5% of the proteome.120,121 However, only a few experimental reports have found evidence of acylation. Lipoproteins in MTBC are predicted to be associated to diverse cellular functions, including transport, cell wall metabolism, cell adhesion, signaling and protein degradation and thus some lipoproteins will play a significant role in virulence. The association of Lpps to virulence was clearly demonstrated in 2004. The recent discovery of novel cell envelope associated potential Lpps through Triton X114 phase separation coupled to mass spectrometry122 emphasizes that there is much to be explored in this protein family. Different evidence indicates that Lpps could be directly or indirectly involved in virulence and hence data from recent research are integrated herein this section.
The first gene to be described in this section is LspA, the lipoprotein signal peptidase responsible for removal of the signal peptide following trans-acetylation and translocation. The peptidase cleavage generates the mature form of the lipoprotein that is anchored in the membrane.121 Sander et al. disrupted lspA by allelic replacement in M. tuberculosis. The obtained lspA mutant, impaired in lipoprotein synthesis, resulted markedly attenuated in the relatively resistant mouse strain BALB/c and the more susceptible strain CBA/J to infection with M. tuberculosis.123 Early in the 1990s, Young and Garbe had identified four lipoprotein antigens in M. tuberculosis through detergent phase separation and metabolic labeling.124 Since then, others Lpps have been identified or predicted in silico.120,121
LppX
LppX was primary described as a mycobacterial protein antigen (22-kDa) common to M. leprae and the MTBC.125 The protein is secreted and is also highly expressed in the bacterial cell wall and membrane compartment but has not been found in the cytosol.126 STM strategy has established the link of this Lpp to virulence. The disruption of lppX had no effect on in vitro growth, but the mutant obtained by the disruption of this gene showed an attenuated phenotype in BALB/c mice.127 This result was concordant with that obtained in a previous study in C57BL/6J mice.100 Chromosomal localization of lppX in a region of genes involved in PDIM metabolism has led to a functional characterization of LppX related to this complex lipid. A biochemical approach through lipid fractionation coupled to MALDI-TOF has shown that the wild type and its isogenic lppX mutant produced the same types and amount of PDIM molecules. However, while 36% of the PDIM synthesized by the wild type was found in the culture filtrate, no PDIM was detected in the culture filtrate of the mutant; it remained associated to the cell wall and cytosol plus plasma membrane fractions. In addition to these results, a structural biology study has provided strong support regarding a possible role of this protein as a PDIM carrier transport from the outlet leaflet of the plasma membrane to the outer membrane.127 In this way, the possible contribution to virulence could be explained through the role of PDIM as explained above.
LpqH (19-kDa lipoprotein antigen)
LpqH has been first described as a 19-kDa lipoprotein antigen of M. tuberculosis.124 A recent study has described this Lpp as a putative glycoprotein.128 The M. tuberculosis 19-kDa glycolipoprotein can inhibit MHC-II antigen processing and presentation in macrophages.129 This inhibition occurs by blocking gamma interferon (IFN-γ) signaling through a Toll-like receptor-2 dependent (TLR-2) mechanism. The 19-kDa lipoprotein, as well as synthetic lipopeptides, induces dendritic cell (DC) maturation. The lipid moiety of the lipopeptide has been found to be essential for induction of DC maturation.130 In addition to its powerful immunomodulatory properties, exposure of neutrophils to the M. tuberculosis 19-kDa lipoprotein promotes neutrophil priming and activation.131 Exposure of macrophages to this protein may also induce apoptosis, allowing escape and dissemination of the bacilli.132 Recently, it has been shown that the 19-kDa lipoprotein activates autophagy in human monocytes and, thus, could exert antimycobacterial activity through the induction of this mechanism.133 Besides, the 19-kDa Ag of M. tuberculosis has been described as a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria.134 These properties reinforce the pleiotropic effects of this molecule at different levels of the infection process. Even more contradictory results have been published: for instance, while a report has shown that despite inducing an Ag-specific Th1 response, overexpression or addition of the 19-kDa Ag has a detrimental effect on protective efficacy of different mycobacterial vaccines such as M. vaccae or BCG.28,135 Other authors, however, have reported that neither overexpression of the 19-kDa Ag, nor deletion of the endogenous 19-kDa Lpp gene altere the ability of BCG to protect against M. tuberculosis challenge in a mouse model.136 Interestingly, M. tuberculosis 12646 and Sl strains that do not express the 19-kDa Lpp in vitro and in vivo, exhibit lower virulence than the transformant-producing native 19-kDa glycolipoprotein, as assessed by bacterial loads and lesions in infected organs.137 Later on, a high-throughput mutagenesis screen showed that disruption of the 19-kDa Lpp gene is associated with moderately reduced bacterial load in mice.100 More recently, an M. tuberculosis H37Rv ΔlpqH mutant displayed high attenuation in C57BL/6 mice; a phenotype that was lost upon complementation of the mutant.138
LprG (27-kDa lipoprotein Ag, P27)
LprG is a secreted surface-expressed lipoprotein antigen, which together with 19-kDa Lpp Ag is one of the most extensively studied lipoproteins (Fig. 2). It has been shown that it is also a glycoprotein128 and described as an important antigen of M. bovis and M. tuberculosis139,140 but not exclusive of the MTBC. In spite of the Th1 immune response in BALB/c mice induced by this antigen, an adverse effect on the protection afforded by BCG has been observed.140 Moreover, co-administration of LprG with M. tuberculosis aggravates the infection,141 suggesting that the 27-kDa Lpp plays a role in M. tuberculosis infection by inducing increased suppression of the immune response. The gene that encodes LprG constitutes an operon together with Rv1410c gene,142 usually known as p55. The p27-p55 (Rv1411c-Rv1410c) operon, also called lprG-p55, is a bicistronic operon conserved across several non-pathogenic and pathogenic mycobacterial species.143 It has been suggested that Rv1410c codes for the P55 protein, an antibiotic efflux pump, since overexpression of M. tuberculosis P55 in M. smegmatis confers resistance to streptomycin and aminoglycosides.143 However, a later study in which M. tuberculosis P55 was overexpressed in M. smegmatis failed to demonstrate this phenotype.144 In spite of this, evidence suggests that P55 clearly functions as a transporter. This is also supported by the homology of P55 to other major facilitator superfamily pumps and because it is a target for MDR (multidrug resistance) inhibitors such as reserpine and verapamil.143,144 Sequence homology between LprG and LppX may also suggest a common function, as mentioned above for LppX, related to lipid transport. Several studies have shown that cell wall composition is altered in mycobacterium mutants defective on lprG and p55.144,145 Thus, LprG and P55 are required for the translocation of cell wall components or their precursors. Knockout of lprG or its operon resulted in attenuated growth and survival in mice and macrophages.100,146,147
In addition, two studies have provided evidence of a cooperative effect between LprG and P55. In the first study, the altered phenotypes of an M. smegmatis strain lacking both p55 and lprG could be complemented by a plasmid encoding both genes, but none of the genes alone could restore the pheotype.144 Moreoverer, the second study has shown that complementatin of M. bovis mutant (lacking the operon) with either lprG or p55 failed to fully complement the wild-type phenotype.146 These studies suggest the importance of both components on virulence.
Recently, it has been proposed that LprG serves as a carrier to facilitate assembly or trafficking of glycolipids to the mycobacterial cell wall, in this way contributing to virulence.148 Additionally, LprG has been recognized as a ligand for the dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) receptor in M. bovis BCG. DC-SIGN has an important role in mediating adherence of mycobacteria species, including M. tuberculosis and M. bovis BCG, to human dendritic cells and macrophages. DC-SIGN is a C-type lectin receptor, and interactions with mycobacterial cells are believed to occur via mannosylated structures on the mycobacterial surface.149 This study provides new insights on the role of LprG on host-pathogen adhesion. The mechanisms through which this gene/operon could play a key role on virulence may be explained by its contribution to M. tuberculosis cell wall assembly and/or by its contribution to the delivery of glycolipids to TLR2.
PstS-1 (38-kDa Ag; phoS or phoS1)
PstS-1 belongs to the ABC phosphate transporters family (Fig. 2), homologous to the Pst system in E. coli. PstS-1, the phosphate binding subunit, is encoded by a gene surrounded by pstB, pstC-1 and pstA-2 within a potential operon (pstB, pstS-1, pstC-1 and pstA-2).150 This protein has been described as one of the first glycoproteins reported in M. tuberculosis.151 It has also been described as an immunodominant antigen in TB patients.152 Later on, through detergent phase separation and metabolic labeling, it was classified as a glycolipoprotein.124 It has been shown that this 38-kDa glycolipoprotein possesses structural motifs to interact with both TLR2 and Toll-like receptor 4 (TLR4). In addition, the intact protein moieties of the 38-kDa Ag would be responsible for the induction of the pro-inflammatory cytokines, TNF-α and IL-6 induction in human primary monocytes. Purified 38-kDa glycolipoprotein induced the activation of ERK1/2 and p38 MAPK via TLR2 and TLR4, and the subsequent TNF-α and IL-6 in human primary monocytes. This line of evidence suggests that this antigen could play a role in early inflammatory responses during mycobacterial infection.153
Further investigation has been focused on this antigen as evidence indicated its possible role in phosphate metabolism.154 In immunolocalization experiments, gold-particles labeling 38-kDa Ag were located mainly in the wall and on the outer surface of the bacilli. This location is in agreement with the possible role of P38 in phosphate metabolism. It is also consistent with previous studies suggesting that P38 is a secretion antigen.155 Additionally, this protein is overexpressed under phosphate starvation.155,156 pstS1 and pstS2 knockout strains showed a significantly reduced multiplication rate within mouse peritoneal macrophages and were attenuated in an in vivo infection model.157 This study has also provided important evidence on a possible role of Pst-1 and Pst-2 in phosphate uptake from media with low phosphate concentration contributing to the intracellular survival of M. tuberculosis.
LpqY
LpqY is a component of a putative sugar ABC transport of M. tuberculosis and lpqY forms an operon with sugC, sugB and sugA. Different studies based on TraSH have identified lpqY as a gene required for growth in mice and in macrophages.99,100 This could mean that sugars (not only host lipids) could be important for infection. A study based on this ABC transporter has provided new insights into the role of sugar transporters in pathogenesis. It has been shown that this importer plays a role in recycling of extracellular trehalose, released from trehalose-containing molecules synthesized by the bacillus.158 SCID mice infected via the aerosol route with the ΔlpqY or ΔsugC mutants survived significantly longer than mice infected with the wild-type or the complemented mutant strains. In C57BL/6 mice, trehalose transporter mutants showed a severe growth defect in lungs of infected animals during the acute phase of infection, but failed to persist during the chronic phase, albeit at a strongly reduced bacillary organ burden.158 Genetic complementation of the mutants fully restored virulence in this model. This finding highlights the crucial importance of the LpqY-SugA-SugB-SugC transporter-mediated uptake of trehalose in the virulence of M. tuberculosis.
ModA
ModA (molybdate-binding lipoprotein) is encoded in the modABC-operon of the ABC-type transport system for molybdenum.22 Further characterization of the attenuated mutants from a STM library screened in mice has shown that the interruption of this putative gene confers moderate attenuation.42 This result has implicated molybdenum uptake in virulence. In the majority of molybdenum-containing enzymes, the metal is coordinated to the dithiolene group of molybdopterin (MPT) to form molybdenum cofactor (MoCo), with the exception of nitrogenases, in which molybdenum is coordinated in an iron-molybdenum cofactor.159 Deficient transport of molybdenum may, therefore, impact MoCo biosynthesis and lead to a function deficiency of several molybdenum-dependent enzymes essential for the survival of the bacteria.159 However, involvement of these enzymes in virulence might be further investigated.
Other Lpps
Wide genome screening of M. tuberculosis H37Rv mutants has allowed the identification of additional Lpps encoding genes specifically required for mycobacterial survival in vivo100 or in vitro.99 Several Lpps have been identified in both studies: lpqT, lpqY, lprG (described elsewhere), lpqZ and lprK. On the other hand, lppX (described elsewhere) and lprN are exclusively required for in vivo survival,100 whereas lppP and lprO have been predicted to be required for growth in macrophages.99 Additional studies are needed to fully characterize the function of these Lpps and their implication in virulence.
The cell envelope represents an interphase considered relevant in the host-pathogen interaction and several studies have been performed to reveal potential drug targets and vaccine candidates. Knowledge of cell wall proteins and other proteins, together with their biological function, are critical to understand how mycobacterium survives and modulates the host immune response. The main virulence factors described in this section are proteins involved in transport of metabolites (hydrophilic molecules, inorganic phosphate, peptides or lipids) or in lipid and glicolipid synthesis. In relation to the latter function, the degradation and reconstruction of the cell wall keep its architecture intact which is necessary for virulence and survival of mycobacteria in the host. Several lines of evidence point out to the Ag85 complex as a remarkable multitask protein component, with adhesive properties and related to process of cell wall synthesis. As mentioned above, the crystal structure of Ag85C has revealed several potential targets to design preventive or therapeutic strategies as the location of the trehalose monomycolate binding site and the region for fibronectin interaction.90 Additionally, several novel tuberculosis vaccines currently in clinical trials involve the use of these antigens: Ag85B fused to other antigens160,161 or Ag85A expressed on modified vaccinia ankara virus.162 Nowadays, this Ag complex remains as a good candidate to control the infection and attention on its use has not declined.
Lpps are part of the uncommon cell envelope of mycobacteria, and its acylation allows them to anchor and/or sort to the cell surface. Their main function is structural but they may also be involved in synthesis and/or transport of important components of the cell envelope. Besides its clearly structural role, Lpps could act as stimulatory and inhibitory ligands of TLRs allowing modulation of the immune response to mycobacteria. This has been addressed and confirmed by targeting of these genes through mutagenesis experiments used to reveal their relevance in host-pathogen interaction. The export of a variety of potential virulence factors through membrane vesicles (MV)163,164 could be considered a process directly involved with the cell envelope. This mechanism has also been described in mycobacteria and can serve to deliver virulence factors to other compartments of the eukaryotic cell or to their incorporation into host cell–derived exosomes.164,165 Despite that several different molecules are present in M. tuberculosis complex-MVs, Lpps—such as 19-kDa Lpp, LprG, and LppX—constitute a large fraction of them (20% of total proteins), underlying their importance in the interplay with the host immune system.165 A detrimental effect of MVs for the host during M. tuberculosis infection has been also observed, since mice pretreated with pathogenic-MVs developed a more acute inflammation and higher bacterial loads in the lungs compared with control and nonpathogenic MVs-treated mice. Moreover, in the same study, preexposure to MVs also increased dissemination of bacilli from the lung to other lymphoid organs.165
These observations highlight the intriguing mechanisms involved in immune response modulation, in which Lpps appears as relevant players. Most of the Lpps characterized as virulence factors by mutagenesis have also been described as antigenic, some of them with potential diagnostic value. Surprisingly, no deletion of the MPT83 antigen (or its counterpart, MPB83 in M. bovis), also described as a glycolipoprotein,166 has been reported yet; however, it has been successfully evaluated as part of a DNA and protein vaccine in a mice model of tuberculosis.167,168
Secretion systems
Protein secretion is a very important mechanism in bacterial functioning and operation in their natural surrounding environment for adaptation and survival. Moreover, these systems are also essential for interacting with host cells by exporting toxins/signal proteins that allow different bacterial species to cause pathology. Even more, secretion systems have evolved, duplicating and diverging to carry on specific functions; some of them are involved in pathogenicity, leading to the description of a large number of such systems. Several gene clusters encoding proteins that are secreted into the environment via specific pathways have been identified in mycobacteria; therefore, they are considered important for mycobacterial pathogenesis.
Recent evidence shows that mycobacteria have developed a novel and specialized secretion system for the transport of extracellular proteins across their hydrophobic and highly impermeable, cell wall. Abdallah et al. have discussed this novel secretion pathway and considered variants that are present in various Gram-positive bacteria. Since the composition of this secretion system is unique and of general importance, these authors have proposed that, in line with the accepted nomenclature, it should be called type seven secretion system (T7SS).169 M. tuberculosis contains a total of five T7SS, also called ESX, that show similarity in gene content and gene order. These systems contain genes conserved in four of the five systems named as ESX conserved components (Ecc) and also genes coding for proteins defined as ESX-1 secretion-associated proteins (Esp). Strikingly, at least four types of secretion systems are encoded in the M. tuberculosis H37Rv genome, but only secretion systems type II and VII are involved in virulence.
Two members of T7SS, ESX-1 and ESX-5, have been shown to be involved in virulence. Both affect the cell-to-cell migration of pathogenic mycobacteria.
The ESX-1 secretion system
A 9.5 kb fragment absent in BCG encodes for several genes of the T7SS in bacteria. ESAT6 (6-kDa early secretory antigenic target, also known as ESXA) and CFP10 (10-kDa culture filtrate protein, also known as ESXB) are two relevant proteins associated to virulence in MTBC that are secreted by T7SS. The specialized T7SS for ESXA and ESXB is called ESAT6 secretion system 1 (ESX-1). ESAT6 and CFP10 are needed for full virulence of MTBC species.170 Importantly, the genes coding for T7SS are absent in M. microtti and in the BCG vaccine. ESAT6 and CFP10 genes are located in a segment called Region of Difference 1 (RD1).171Figure 3A shows the genomic region where ESX-1 is placed.
Figure 3. Schematic representation of the ESX-1 secretion system. (A) Schematic organization of the M. tuberculosis genomic region containing the RD1 genes. (B) Model. The abbreviation ecc stands for esx conserved component, whereas esp stands for ESX-1 secretion-associated proteins. The topology of the different proteins in the cytoplasmic membrane is shown and refers to the ESX-1 cluster based on predictions made using the MEMSAT3 algorithm. Note that the channel drawn in the outer membrane of this model refers to a hypothetical pore, whose existence has not been experimentally demonstrated. Reproduced from Bitter et al.176
ESAT6 and CFP10 are small proteins, around 9 and 10 kDa respectively, with very high self-affinity and appear as a heterodimer in culture supernatants. Both proteins are part of a large protein family in mycobacteria and some Gram-positive bacteria. This family is characterized by a WXG signature located in the central part of the around 100 aa size protein.172 ESAT6 and CFP10 have been described as dominant antigens recognized by T-cells in natural infection in humans and bovines and in experimentally infected animals.173,174
By means of genetic manipulations, the segment corresponding to RD1 have been deleted in wild-type M. tuberculosis171 or re-inserted in BCG. The M. tuberculosis mutant in RD1 was attenuated in comparison to its parental strain but more virulent than BCG,175 which has other genomic regions deleted (in addition to RD1). This RD1 M. tuberculosis mutant has been useful for complementation studies that allowed the identification of genes in ESX-1 locus required for the full virulence of M. tuberculosis. The BCG strain complemented with RD1 gained virulence, measured as persistence and growth in mice, and secretion of ESAT6 and CFP10.171
The different nomenclatures of ESX-1 are shown in Table 2. ESX-1 is conserved in many other mycobacteria. For instance, in M. smegmatis ESX-1 encodes for genes involved in transfer of genetic material by conjugation. It is also present in M. leprae but absent in M. avium complex species. In the complemented BCG, it has been observed that only the use of a full RD1 DNA fragment, contrasting to smaller fragments, restored secretion of ESAT6 and CFP10 and increased virulence. This was the first indication that RD1 encodes for a secretion system.
Table 2. Nomenclature of proteins involved in the ESX-1 type seven secretion system in M. tuberculosis176.
| Rv number | Other usual name | New nomenclature |
|---|---|---|
| Rv3614 |
Snm 10 |
EspA |
| Rv3615 |
Snm 9 |
EspC |
| Rv3616 |
|
EspD |
| Rv3849 |
|
EspR |
| Rv3864 |
|
EspF |
| Rv3865 |
|
EspG1 |
| Rv3866 |
Snm 5 |
EspG |
| Rv3867 |
|
EspH |
| Rv3868 |
|
EccA |
| Rv3869 |
Snm 6 |
EccB |
| Rv3870 |
|
EccCa |
| Rv3871 |
|
EccCb |
| Rv3872 |
PE-35 |
PE-35 |
| Rv3873 |
PPE-68 |
PPE-68 |
| Rv3874 |
CFP10 |
ESXB |
| Rv3875 |
ESAT6 |
ESXA |
| Rv3876 |
Snm 3 |
EspI |
| Rv3877 |
Snm 4 |
EccCd |
| Rv3878 |
Tb-27 |
EspJ |
| Rv3879 |
|
EspK |
| Rv3880 |
|
EspL |
| Rv3881 |
|
EspB |
| Rv3882 |
|
EccE |
| Rv3883 | MycP1, Snm 8 |
M. tuberculosis uses the ESX-1 secretion system to deliver virulence proteins during infection of host cells (Fig. 3B). Furthermore, the finding that the C-terminal signal sequence of CFP10 is sufficient to secrete yeast fusion proteins via the ESX-1 system supports that CFP10 is a secreted protein.177 Sequence analysis of the ESX-1 proteins shows that EccA has an ATPase signature. The proteins EccB, EccCa and EccDd exhibit 1, 3 or 11 predicted transmembrane domains, respectively, and they probably are the secretion apparatus. CFP10 has a C-terminal signal sequence, recognized by EccCb, that itself interacts with the membrane protein EccCa. A point mutation in this signal sequence abolished the binding of CFP10 to EccCb and prevented secretion of ESAT6 and CFP10.177Mycobacterium marinum has a T7SS very similar to that of M. tuberculosis, and its polar localization in this bacillus has been demonstrated by confocal and electronic microscopy.178 Similar results have been obtained in M. smegmatis.179
EspB is secreted through ESX-1 and, after its secretion, is cleaved yielding a 50-kDa protein from the original 61-kDa. The EspB C-terminus is dispensable for its own secretion, as the expression of a truncated form of EspB in an espB transposon mutant led to a normal secretion of EspB. In M. marinum, however, the C-terminus is essential for the interaction of EspB with ESAT6, maintenance of intracellular levels of ESAT6 and secretion of ESAT6 and CFP10, indicating that cleavage of EspB could have a regulatory function in ESX-1 secretion.180
In addition to ESAT6, CFP10 and EspB, three other substrates of the ESX-1 system have been described: EspA, EspB, EspC and EspR.180-185 An unusual feature that distinguishes ESX-1 from other systems is that the secretion of all substrates is mutually dependent. For example, secretion of EspA is blocked in an ESAT6 mutant, and vice versa.181
In turn, EspF and EspG1 are virulence factors that are not part of the ESX-1 system, because their disruption did not impact the secretion and T cell recognition of ESAT6/CFP10 but still caused severe attenuation in BMDM, although no attenuation was observed in another cell types, such as pneumocytes.
In aerosol-infected C57BL/6 mice, 3- and 4-log reductions in bacillary load were observed in lungs for espG1 and espF mutants, respectively186.
The mode of action of ESAT6 in eukariotyc cells has not been precisely defined. The most frequently reported observations are the lysis of cells and/or membranes. Guinn et al. have reported that ESAT6 deletion mutants of M. tuberculosis are capable to multiply within THP-1 macrophage cells lines but fail to spread to uninfected macrophages. Moreover, ESAT6 destabilizes and lyses liposomes, whereas CFP10 lacks these characteristics.187 Thus, individual RD1-region genes are required for export of ESAT6/CFP10 and for virulence of M. tuberculosis.
Moreover, the fact that certain single amino acid changes in ESAT6 fail to prevent secretion of the modified ESAT6 molecules, but cause attenuation of the recombinant M. tuberculosis strains argues in favor of ESAT6 being a secreted effector molecule.
It has been recently described that translocation of M. tuberculosis from the phagosome into the host cell cytoplasm at later stages of infection is facilitated by ESAT6 and CFP10.188 While at days four to seven after infection with M. tuberculosis more than one-third of the non-apoptotic human dendritic cells contained translocated bacteria, this effect was not observed when BCG or M. tuberculosis ESX-1 transposon mutants were used for infection.188 These observations suggest that the ESX-1 system might provide M. tuberculosis with the required tools for escaping from the phagosomal compartment of professional phagocytic cells and/or releasing ESAT6 proteins to the cytoplasm, where they gain access to the class I-processing machinery contained in the proteasome. Such events would explain the recruitment and activation of CD8+ T cells found in the lungs of mice aerosol infected with M. tuberculosis. These are strong indications that ESX-1 secreted proteins reach the eukaryotic cytoplasm. In addition, Pathak et al. have demonstrated that ESAT6 prevents antigen-presenting cell function by inhibition of TLR signaling pathways, which in turn reduces IL-12 production by THP1 macrophages and inhibits macrophage apoptosis signals as well.189
A transcription factor named EspR, encoded by Rv3849, binds to the espACD operon promoter and is then secreted from M. tuberculosis by the ESX-1 system.185 This operon is under positive regulation control of EspR but its activation is turned off when EspR is secreted via the ESX-1 system, leading to downregulation of espACD transcription.
Transcriptomic analyses of M. tuberculosis have also shown that PhoP regulates the expression of the espACD operon, which are significantly less expressed in the phoP inactivated strain.190 This and other results remark the influence of PhoP-PhoR regulation on ESX-1 activity.
In addition, serine protease MycP1 post-transcriptionally regulates the ESX-1 secretion activity, being required for substrate secretion through ESX-1 as demonstrated by the deletion of mycP-1191. The mutagenic inactivation of MycP1 proteolytic activity increases secretion of ESX-1 substrates and attenuates the virulence of M. tuberculosis in chronic infection models191 (see section on “Serine proteases”). Therefore, MycP1 contributes to the fine-tuning of ESAT6 and CFP10 secretion, balancing the virulence and immunogenic properties of these proteins, which is essential for successful maintenance of long-term M. tuberculosis infection.
In conclusion, ESX-1 is a fascinating secretion system with many unique regulatory and functional characteristics. It is also one of the best-understood examples of virulence factors in M. tuberculosis complex. Upon deletion of RD1 or esxAB M. tuberculosis almost completely lost its virulence in animal models. However, it fails to fulfill all the requirements for a molecular virulence factor as proposed by Falkow,192 as orthologous genes are present in some non-pathogenic mycobacteria (such as M. smegmatis).193 This fact suggests that ESX-1 has evolved to be adapted to life inside eukaryotic cells. Recent results indicate that ESX-1 allows M. tuberculosis to escape from the phagosome. Importantly, there are still many proteins essential for ESX-1 function whose precise functional role or topological location still needs to be determined. In addition, ESX-5 allows the secretion of many PE and PPE proteins, in example PE_PGRS33. Disruption of the PE_PGRS33 encoding gene causes attenuation of M. tuberculosis.194 The exact role of PE/PPE proteins on virulence, however, remains largely unknown (see section on “Proteins of Unknown Function”).
The operon espACD (Rv3616c-Rv3614c)
EspA is a protein secreted by ESX-1 whose gene is outside of the RD1 region and, therefore, is present in BCG. EspC and EspD show significant homology to Rv3865 and Rv3867, respectively.181 Although its small size, EspC is secreted by ESX-1; the secretion of EspD, unlike EspA and ESXA, does not exclusively require the ESX-1 system. Evidence for stabilization of cellular levels of EspA and EspC by EspD has been presented, and depletion of EspD results in loss of ESXA secretion. Site-directed mutagenesis of espD reveals that its role in the maintenance of cellular levels of EspA in M. tuberculosis is distinct from its facilitation of ESXA secretion. The polycistronic nature of espA, C and D was evident when the virulence and ESX-1 function of an M. tuberculosis mutant in espC was only complemented with the entire espA-espC-espD gene cluster.182 In spite of this genetic relatedness, there are strong functional dissimilarities among proteins encoded in the espACD operon. EspA and EspC secretion require EspD, but EspD secretion does not require EspA, EspC or ESX-1.195 EspC is a potent antigen in both active and latent TB infection. T-cell responses to EspC were highly specific (93%) for M. tuberculosis infection. The immunodominance of EspC, equivalent to that of ESAT6 and CFP10, and its high antigenic specificity make this protein a promissory TB vaccine candidate and a potential T-cell-antigen.196
M. tuberculosis EspC and EspA mutants have shown a highly reduced capability to grow in BMDM.182 In addition, an M. tuberculosis H37Rv mutant in espA is also highly attenuated in lungs of C57BL/6 infected mice in terms of both bacterial load and pulmonary damage.181 The deletion of the entire espACD operon leads to a large reduction of M. tuberculosis H37Rv load in SCID mice.197 It is still not clear if the impact of EspA, EspC and EspD on the virulence of M. tuberculosis is only due to their role in ESXA and ESXB secretion or if these proteins exert a direct interaction with the host. Regarding the versatility of this pathogen, this cannot be discarded and should be addressed.
The ESX-5 secretion system
MTBC species contain a large number of genes that encode for an M. tuberculosis unique family of proteins whose N-termini contain the characteristic motifs Pro-Glu (PE) or Pro-Pro-Glu (PPE). A subgroup of the PE proteins contains polymorphic GC-rich sequences (PGRS). The function of most of these proteins remains unknown and speculative. Abdallah et al. have shown that PE_PGRS proteins from M. marinum are secreted by components of the ESX-5 (Rv1782-Rv1798) system, which is different from ESX-1, but also belongs to T7SS.198 These observations, which now need to be addressed and confirmed in M. tuberculosis, have opened new perspectives on the function of these highly abundant proteins.199 In contrast to ESX-1, the effector proteins secreted by ESX-5 are not required for the translocation of M. tuberculosis or M. marinum to the cytosol of host cells. These results reveal distinct roles for two different type VII secretion systems during infection and shed light on how virulent mycobacteria manipulate the host cell in various ways to replicate and spread.200 It has been reported that ESX-5 is essential for PE_PGRS33 export.201 The PE domain of PE_PGRS33 (Rv1818c) is crucial for its surface localization, and it has been demonstrated that a PE domain lacking its first 30 amino acids loses its function. However, single amino acid substitutions in two extremely well-conserved regions within the N-terminal domain in all PE proteins had some effect on the stability of PE_PGRS33, but not on its localization.
Recently, Bottai et al.202 have knocked out the ESX-5 locus in M. tuberculosis H37Rv. Whereas the M. tuberculosis mutant in Rv1794 displayed no obvious phenotype, the other four mutants (eccA5, eccD5, ESXM genes and the ppe25-pe19 region) showed defects in secretion of the ESX-5-encoded ESXN and PPE41; the latter is a representative member of the large PPE protein family. Mutants in eccD5 and ppe25-pe19 are attenuated both in BMDM and in the SCID mouse infection model. These findings indicate an essential role of ESX-5 for transport of PPE proteins, cell wall integrity and full virulence of tuberculosis.
Type II secretion system
In bacteria, lipoproteins are exported via type II secretion systems or general Sec secretory pathway; however, a signal peptidase (the lipoprotein signal peptidase LspA) different from the canonical signal peptidase of SecII pathway cleaves the signal peptide of lipoproteins exported by this secretory pathway. The genome of M. tuberculosis potentially encodes for more than 90 lipoproteins (see above). When the M. tuberculosis lspA gene was inactivated by homologous recombination, lipoproteins were synthesized but not processed; although this gene is dispensable for in vitro growth, its deletion markedly affectes the growth of mycobacteria in murine J774 macrophages and severely attenuates their virulence in BALB/c mice123 Interestingly, Rampini et al. have observed that the loss of LspA results in attenuation without afecting phagosome maturation arrest (a central point for M. tuberculosis survival in phagocytes) showing a low degree of co-localization with lysosomal-associated membrane protein 1 (LAMP1) and with acidic compartments.203
In conclusion, the genome of M. tuberculosis encodes a very large number of potential lipoproteins, largely contributing to cell envelope structure. It is surprising that M. tuberculosis is viable in vitro without properly modified lipoproteins while mutant lacking the signal peptidase LspA cannot survive in vivo. This property, together with the fact that lipoproteins are well-known TLR ligands that may influence the intracellular fate of the bacteria, highlights the role of lipoproteins in vivo. It has been observed that the genes for lipoprotein synthesis and modification are dispensable for in vitro growth of Gram-positive bacteria but are essential for Gram-negative bacteria. However, the essentiality of LspA seems to depend on the mycobacterial specie since the gene encoding this protein could not be mutated in BCG (Sander P, personal communication). Also, the first enzyme in lipoprotein synthesis, prolipoprotein diacylglyceryl transferase, is also essential in M. tuberculosis but not in M. smegmatis.204
Accessory SecA2 export system
Mycobacteria have two SecA proteins, SecA1 and accesory SecA2.205 SecA2 is a nonessential preprotein translocase ATPase. Contrasting with the canonical preprotein translocase ATPase SecA1, SecA2 exports a limited number of proteins. SecA2 is essential for the full virulence of M. tuberculosis, suggesting that one or more of the proteins it exports are important in pathogenesis.206
SecA1 and SecA2 have independent functions, since SecA2 cannot compensate for the lethal SecA1 deletion. Similarly, overexpression of SecA1 fails to rescue the phenotypes of a secA2 deletion mutant. The analysis of proteins exported into the culture media (culture filtrates) by an M. tuberculosis secA2 mutant has revealed that among the very few proteins absent exclusively in the mutant strain was the superoxide dismutase (SodA), which is an oxygen radical detoxifying enzyme. Therefore, this finding indicates that SodA depends on SecA2 for export despite the absence of a signal peptide.207 Another antioxidant enzyme, KatG, also lacks a signal peptide and is dependent on SecA2 for export. A more detailed description of the role of both SecA2 and SodA in the virulence of MTBC species is given below in this review (see section on “Inhibition of apoptosis”).
Proteins Inhibiting Antimicrobial Responses of the Macrophage
During infection, macrophages ingest and destroy pathogens, recruit other cells of the immune system, and present antigens from the microbe to cells of the adaptive immune system. However, pathogenic mycobacteria have evolved mechanisms to counteract the macrophage microbicidal ability, some of those mechanisms are: (1) increasing in the resistance to host toxic compounds, (2) arrest of the normal progression of the phagosome and (3) avoidance of the induction of apoptosis. In this section, we discuss the advance in knowledge of these survival mycobacterial strategies and described those virulence proteins involved in these process and the genes encoded by them.
Oxidative and nitrosative stresses
Resistance to macrophage-mediated killing is critical to the virulence of M. tuberculosis. Upon phagocytosis of bacteria, host cells produce reactive oxygen species (ROS) and reactive nitrogen species (RNS) with potential bactericidal activity. These reactive oxygen/nitrogen intermediates (ROI/RNI) react with a wide variety of molecules, including nucleic acids, proteins, lipids and carbohydrates. Most of intracellular pathogens, including mycobacterial species, have developed a range of defense strategies to protect themselves against the damaging effects of these agents. However, for pathogenic mycobacterial species these strategies seem to be different to those of other intracellular bacteria. For instance, the classic defense mechanism OxyR is dysfunctional in all members of the MTBC.208 Yet, other mechanisms or processes compensate for the lack of a functional OxyR. In fact, several proteins directly involved in these resistance responses have been identified and characterized in mycobacteria. In addition, the finding that several genes likely involved in cell envelope processes are also important in M. tuberculosis resistance to oxidative and nitrosative stresses114 suggests that the highly impermeable mycobacterial cell wall acts as a barrier for ROI and RNI. The present section focuses on proteins that, either directly or indirectly, are necessary for the MTBC species to counteract oxidative and nitrosative stresses, and that have also been shown to be relevant for the virulence of pathogenic mycobacterial species. These proteins and their mechanisms are schematized in Figure 4.
Figure 4. Antimicrobial properties of the macrophage. (A) Oxidative and nitrosative stresses, (B) phagosome arresting, (C) apoptosis and cross presentation. Proteins involved in the inhibition of antimicrobial mechanisms are indicated. *Proteins likely involved in cell wall associated resistance to oxidative burst.
Acr Family
There are two members of the α-crystallin (Acr) family of molecular chaperones in M. tuberculosis:
Acr1, also called 16-kDa α-crystallin or HspX, is part of the DosR regulon, a genetic program of M. tuberculosis induced by conditions that inhibit aerobic respiration and prevent bacillus replication that regulates the expression of a large number of dormancy-associated proteins.209 HspX is a dominant protein present in old, stationary phase cultures, but undetectable during logarithmic growth of M. tuberculosis. acr1 transcription is induced by exposure to hypoxia or nitric oxide in the context of the dormancy program of M. tuberculosis210 and during the course of in vitro infection of macrophages.211 The role of HspX in M. tuberculosis virulence has been tested in vitro and in vivo, but the results are not conclusive. While Yuan et al. have reported that a Δacr mutant in M. tuberculosis is significantly impaired for growth in both murine BMDM and THP-1 cells,211 Hu et al. have shown just the opposite: another Δacr mutant in M. tuberculosis exhibits increased growth following infection of BALB/c mice and in both resting and activated BMDM.212 Consistent with the latter, overexpression of HspX results in reduced growth of BCG within mouse lungs and liver.213 Based on these findings, it has been proposed that HspX plays an active role in slowing the growth of M. tuberculosis.212
Acr2 is the other Acr family member. Its expression is induced by heat shock,214 oxidative stress215 and uptake by both human monocytes and monocyte-derived macrophages.216 In addition, Acr2 is highly expressed during both acute and chronic infection in a mouse model. The deletion of the acr2 gene fails to impair the growth of M. tuberculosis either in murine BMDM or in mouse organs.217 However, a delay in the disease progression has been observed over a prolonged mice infection with an M. tuberculosis mutant in acr2, evidenced by a reduced weight loss and diminished size of lung lesions in animals infected with the mutant strain as compared with those infected with the parental strain.217
Altogether, these findings indicate that both α-crystallins, HspX and Acr2, contribute somehow to the infection with M. tuberculosis. Infections of macrophages that produce limited oxidative and nitrosative bursts with acr1 and acr2 mutants are necessary to determine whether there is a connexion between the role of HspX and Acr2 as stress responders and their virulence properties.
Rv2136c, Rv2224c and PonA2
M.tuberculosis mutants with transposon insertions in Rv2136c, Rv2224c and ponA2 displayed a hypersensitive phenotype to low pH, antibiotics, sodium dodecyl sulfate, heat shock and reactive oxygen and nitrogen intermediates. These transposon mutant strains have also shown a variable level of attenuation when assayed C57BL/6 mice. While the M. tuberculosis H37Rv mutant in Rv2136c gene shows a remarkable impairment in its persistence in mouse lungs and spleen and markedly reduced gross pathology in lungs,114 the mutant in Rv2224c and ponA2 shows attenuated phenotypes but not as strong as that detected in mutant ΔRv2136c. Although the function of Rv2136c gene is at present unknown, the close homology to an E. coli enzyme involved in the synthesis of peptidoglycan suggests an equivalent function in M. tuberculosis. PonA2 is predicted to have transglycosylase and transpeptidase activities, and thus likely to be also involved in peptidoglycan biosynthesis. Finally, Rv2224c encodes a putative carboxylesterase with lipase and proteinase activities (see sections on “Cell wall proteins” and “ATP-dependent proteases”). These predicted functions for Rv2136c, Rv2224c and PonA2 suggest that these proteins participate in cell wall processes, highlighting the importance of M. tuberculosis cell wall in protection against diverse stresses.
AhpC
AhpC is an alkyl hydroperoxide reductase C, an enzyme that reduces organic peroxides, similar to a family of bacterial and eukaryotic antioxidant proteins that directly reduces peroxides and peroxynitrites. Many lines of evidence have shown that AhpC is involved in the response to oxidative stress in mycobacterial species: in M. tuberculosis, as in most mycobacterial species, ahpC is located adjacent to oxyR, a central regulator of the peroxydative and nitrosative stress response that is dysfunctional in the MTBC species. The AhpC contribution to the peroxydative homeostasis of M. tuberculosis has been demonstrated in isoniazid resistance of M. tuberculosis isolates that overexpress AhpC in the absence of catalase-peroxidase KatG (see below).218 Therefore, M. tuberculosis compensates the lack of KatG catalase-peroxidase activity overexpressing AhpC.219 Although AhpC is unlikely to confer resistance to rifampicin, its expression is upregulated by this antibiotic.220 Overexpression of AhpC in an M. bovis katG mutant strain has conferred resistance to both hydrogen peroxide and the organic peroxide cumene hydroperoxide.221 AhpC crystal has been resolved222 and its peroxynitrite reductase activity has been demonstrated in vitro.223,224 In addition, an M. tuberculosis mutant in ahpC showed higher susceptibility to peroxynitrite, but not to nitric oxide, than the wild-type strain.225 Importantly, this mutant strain also showed decreased survival in unstimulated macrophages, but the effect was no longer detectable upon IFNγ activation,225 suggesting that AhpC has an essential role in the resistance mechanisms to host oxidative agents only very early during the infection. Consistent with this latter finding, the knockdown of ahpC in M. bovis attenuates the virulence of the bacteria in a guinea pig model.221 In conclusion, these findings clearly demonstrate that AhpC plays a role in the resistance of M. tuberculosis to oxidative stress; however, the contribution of this protein to bacterial virulence needs further investigation.
SodC
M. tuberculosis has two genes encoding superoxide dismutase (SOD) proteins, sodA and sodC. The function of Sod enzymes is the detoxification of ROS by conversion of O2- into molecular oxygen and hydrogen peroxide.226 SodC is a copper-containing SOD outer-membrane lipoprotein227,228 involved in the protection of M. tuberculosis against the extracellular superoxide generated by host cells. Consistently, sodC transcription is upregulated upon macrophage infection.229 Moreover, an M. tuberculosis mutant in sodC has shown very high susceptibility to superoxide generated externally, as compared with the wild-type strain.230 This mutant strain also showed decreased survival in IFN-γ-activated murine peritoneal macrophages and, importantly, the effect was no longer detectable in unstimulated macrophages or in activated macrophages from respiratory burst-deficient mice.230 Nonetheless, a mutant of M. bovis BCG in sodC did not show impaired growth in both activated and resting mouse BMDM.231 One possible explanation for these apparently contradictory results could be the type of macrophages used in both studies. It has been proposed that murine-activated peritoneal macrophages produce higher amounts of ROS in response to infection than BMDM.230 Therefore, the lack of attenuation of the mutant strain in BMDM could be due to the absence of an effective oxidative burst. However, the fact that different genetic backgrounds were used to generate both sodC mutant strains cannot be overlooked. Paradoxically, an M. tuberculosis H37Rv mutant in sodC was virulent in guinea pig.231 These controversial findings highlight the need for further research in different in vivo models to clearly establish SodC role in M. tuberculosis virulence.
Mel2
Mel2 is encoded by mel2, a member of a new class of bioluminescence-related genes present in numerous non-bioluminescent pathogens and symbionts. In a recent study by Cirillo et al. it has been shown that in M. tuberculosis Mel2 confers protection against ROI in vitro.232 In this study, an M. tuberculosis strain deleted in mel2 gene was less persistent in both the lungs and spleen of C57BL/6 mice, with reduced pathology in these organs, as compared with its parental strain. The replication of the mutant strain was also significantly impaired in a murine macrophage cell line, in human peripheral blood monocyte-derived macrophages and in activated BMDM obtained from the wild-type C57BL/6 and oxidative burst defective (phox−/− and iNOS−/−) mouse strains. These attenuated phenotypes for the mutant strain in macrophages defective in the generation of either ROS or RNS provided evidence that mel2 plays a role in resistance to both ROS and RNS in activated macrophages. However, when evaluated iNOS−/− mice, the mel2 mutant mantained an attenuated phenotype as compared with the wild type, while in phox−/− mice the replication of the mutant in organs was similar to the wild type. These observations suggest that the primary mechanism by which Mel2 affects pathogenesis is through its ability to confer resistance to ROS.
KatG
KatG is the only enzyme with catalase-peroxidase activity that degrades H2O2 and organic peroxides in M. tuberculosis. KatG activates the anti-tuberculose drug isoniazid (INH), converting it to several reactive species that inhibits a mycolic acid biosynthetic enzyme;233 thus, almost of all the described INH resistant mutants bear mutations in this gene. In two independent studies, INH-resistant M. tuberculosis H37Rv strains that showed no KatG activity were attenuated in both mice (BALB/c and MHC class II-knockout mice) and guinea pigs, as determined by bacterial counts in organs or animal survival.234,235 In one of these reports, both the catalase activity and the virulence were restored by re-introduction of a wild-type copy of katG.235 Similarly, an INH-resistant and catalase negative M. bovis strain was attenuated in guinea pigs as compared with the same strain complemented with a functional copy of katG.236 Finally, an M. tuberculosis Erdman strain with a katG deletion was attenuated in wild-type C57Bl/6 mice and iNOS−/− mice, but not in gp91phox−/− mice lacking the gp91 subunit of NADPH oxidase.237 Similar results have been reported in resting and activated BMDM obtained from the same mouse strains and in activated iNOS−/− macrophages, therefore, suggesting that the main role of KatG in the virulence of M. tuberculosis is to catabolise the peroxydes generated by the phagocyte NADPH oxidase.237 Altogether, these results indicate that the catalase-peroxidase activity exerted by KatG is higly important for the virulence of M. tuberculosis.
TpX
TpX is a thiol peroxidase enzyme that catalizes the reduction of hydroperoxides and peroxynitrite in M. tuberculosis. These enzymatic activities have been demonstrated in mycobacteria both in vitro using purified recombinant protein238,239 and also in vivo by comparative analysis of an M. tuberculosis mutant in tpx gene and its parental strain.240 In this latter study, Hu et al. have reported that the M. tuberculosis tpx mutant is more susceptible to oxidative and nitrosative stress, showing reduced peroxidase activity.240 Importantly, when tested in mice, the tpx mutant strain was less lethal and persistent than the parental strain. In macrophages, the tpx mutant strain showed impaired replication in both activating and resting macrophages, as compared with the wild-type strain. In contrast, the attenuated phenotype was not observed in the macrophages iNOS−/−, which produce a limited oxidative burst.240 Undoubtedly, the study mentioned above indicates that TpX is essential for protecting M. tuberculosis against the RNS and ROS produced by the host cells during tuberculosis infection.
Enzymes that directly participate in the RNI and ROI detoxification mechanisms such as AhpC, SodC, KatG and TpX are essential for the full virulence of the MTBC species. However, virulence proteins with non-conserved detoxification activities have also shown to confer to the bacteria protection against oxidative and nitrosative stresses, indicating that MTBC contain additional antioxidant mechanisms to respond to the hostile environment in the phagosome and phagolysosome.
Some of these non-classical oxidative and nitrosative stress response virulence proteins, such as Rv2136c, Rv224c and PonA2, are likely implicated in cell wall integrity, suggesting that the mycobacterial lipid-rich cell envelope could act as an effective barrier against the entry of RNI and ROI. Other proteins of this class are the two members of α-crystallin family, HspX and Acr2, molecular chaperones primarily described as proteins expressed in multiple stress conditions. Although the role of these mycobacterial chaperones in the in vivo essential antioxidant mechanism is still unclear, the current knowledgment of their eukarytic counterpart may allow to make some conjectures about their function: it has been described that the eukaryotic heat shock protein 90 (Hsp 90) and α-crystallin protect some peptidase activities of the active form 20 S proteasome against oxidative inactivation.241 In turn, authors of several studies have reported that the proteasome plays a crucial role in the selective recognition and degradation of oxidized proteins.242 Therefore, a possible role of HspX and Acr2 in nitrosative and oxidative stresses may be through their interaction with the mycobacterial proteasome. In connection with the latter, the transcriptional regulator ClgR positively regulates the expression of Acr2 together with a number of proteases, thus, supporting a role of Acrs in protein homeostasis (see section on “Proteases”).
Phagosome arresting
A phagosome is a cellular compartment in which pathogenic microorganisms can be killed and digested. Phagosomes fuse with lysosomes in their maturation process, forming phagolysosomes. Some intracellular bacterial pathogens reside inside phagosomes and either divide or grow inside of the formed phagolysosome or escape into the cytoplasm before the phagosome fuses with the lysosome. Many mycobacteria manipulate the host macrophage to prevent nitrous acid-containing lysosomes from fusing with phagosomes and creating mature phagolysosomes. Therefore, one of the main mechanisms elicited by intracellular mycobacteria to survive and replicate inside the host cells is to arrest the normal process of phagosome maturation, which enables bacterial survival in a non-acidified intracellular compartment. The identification of mycobacterial components that interfere with maturation of the phagosomal compartment after ingestion has become a central issue of investigation in tuberculosis.
Based on microarray-based screening of a transposon library, Stewart et al. have found that ppe10, Rv3707c, cut2 and glyA1, among other genes, were relevant for M. bovis BCG to arrest phagosome acidification following uptake by macrophages. cut2 encodes a protein member of a family of serine esterases and glyA1 encodes a protein member of a family of serine hydroxymethyltransferases. ppe10 encodes a member of PPE family and Rv3707c encodes a conserved hypothetical protein (CHP). Transposon mutants in these genes have shown reduced replication inside macrophages as compared with the parental BCG strain, being these attenuations most pronounced at later point times.101 These findings highlight the importance of avoiding phagosomal acidification for the intracellular survival and persistence of pathogenic mycobacteria.
Other mycobacterial proteins involved in arresting macrophage phagosomal maturation with a clear role in the virulence of pathogenic Mycobacterium species are Ndk, PtpA and PE_PGRS30.
These proteins and their mechanisms are schematized in Figure 4.
Ndk
ndk encodes a nucleoside diphosphate kinase with ATP and GTP binding activity as well as hydrolysis activity, demonstrated in vitro.243 It has also been shown that this protein is autophosphorylated and secreted into the culture medium by M. tuberculosis, and that purified Ndk in combination with ATP produces cytotoxicity in macrophages.243 Recently, Sun et al. have shown that recombinant Ndk dephosphorylates the cellular Rab7-GTP and Rab5-GTP in a cell-free biochemical assay, resulting in the deactivation of these enzymes. Moreover, the authors have found evidence suggesting that Ndk inhibits the recruitment of Rab5 and Rab7 effectors to phagosomes (early endosome antigen 1-EEA1 and Rab7-interacting lysosomal protein, respectively). Based on those results, it has been proposed that, within the phagosome, Ndk released from pathogenic mycobacteria might have access to the cytosolic face of the phagosomal membrane so as to interact with and inhibit effectors of phagosome maturation. Consistently, infection of macrophages with a ndk knock-down M. bovis BCG (Pasteur 1173P2) strain resulted in increased fusion of its phagosome with lysosomes along with significantly decreased survival of the mutant.244
PtpA
PtpA is a low-molecular weight tyrosine phosphatase245 reported to dephosphorylate VPS33B, a host protein involved in the regulation of membrane fusion within the endocytic pathway. This phosphatase activity resulted in arrested phagosome maturation by M. tuberculosis. Later on, it has been found that PtpA binds to subunit H of the macrophage vacuolar-H+-ATPase machinery, a multisubunit protein complex in the phagosome membrane that drives luminal acidification. This interaction and dephosphorylation of VPS33B are both required for PtpA inhibition of macrophage phagosome-lysosome fusion and phagosome acidification.246 Consistent with this proposed role for PtpA in arresting phagosomal maduration, an M. tuberculosis H37Rv mutant in ptpA has shown impaired replication in the human THP-1 macrophage cell line.247 However, the knockout of ptpA in M. tuberculosis Erman strain showed no apparent variation in the bacterial replication in mouse organs compared with the wild type.248 The two different infection models used in these studies could explain these opposite findings.
PE_PGRS30
PE_PGRS30 is a member of the PE_PGRS family. In a very recent study by Iantomasi et al. it was shown that PE_PGRS30 is essential for the full virulence of M. tuberculosis in both mice and macrophage models. These authors found that the elimination of PE_PGRS30 from M. tuberculosis H37Rv results in an attenuated strain in terms of replication in mouse organs and lung pathology. The mutant strain showed impaired replication in mouse lungs during the chronic steps of infection and this phenotype was correlated with reduced tissue damage in lungs of infected BALB/c mice. When assessed in human THP-1 and murine J774 macrophages, the mutant also showed reduced replication in both cell lines as compared with that of the wild-type strain. Most importantly, this protein seems to have a role in the phagosomal arresting strategies developed by M. tuberculosis after macrophage uptake, since the mutant strain showed higher colocalization with a lysosomal marker. This finding strongly suggests that the inability of the mutant strain to inhibit phagosome-lysosome fusion is the reason of the attenuated phenotype observed in the in vivo and ex vivo models. In conclusion, the study of Iantomasi et al. has characterized one of the first PE_PGRS proteins with a certain role in the virulence of M. tuberculosis.249
Likely, the most efficient strategy used by MTBC to counteract the macrophage antimicrobial actions is to subvert the normal progression of the phagosomal compartment and prevent it from maturing into an active phagolysosome. This modulation of the intracellular endosomal trafficking allows the bacteria to stay in a non-acidic hospitable niche suitable for replication and helps to avoid its immunological detection. Although the process of phagosomal arresting induced by M. tuberculosis is not fully understood, some aspects of this process are coming into sight. The endosomal tethering molecule EEA1 is a specific Rab5 effector that plays an essential role in phagosomal maturation.250 EEA1 associates with phophatidylinositol 3-phophate (PI3P) in endosomal membranes via its PI3P binding FYVE domain, and it has been demonstrated that this association is essential for phagosome maturation since inhibition of PI3P production arrests the process. M. tuberculosis prevents EEA1 recruitment to phagosomes in infected macrophages, thus precluding phagolysosome formation.251 Besides, M. tuberculosis inhibits Ca2+ signaling, which together with calmodulin are necessary for recruitment of PI3PK responsible for the production of PIP3 in the endosomal membranes. Therefore, one possible mechanism by which M. tuberculosis prevents EEA1 recruitment to phagosomes is through inhibition of Ca2+ efflux.252 However, a reduced level of Ca2+ in macrophages infected with M. tuberculosis is not sufficient to completely explain the phagosome maturation blocking in these infected cells, suggesting that others steps of this phagosomal maturation pathway are targeted by this pathogen. Vergne et al. found that M. tuberculosis culture supernatant proteins desphosphorylate PIP3P in vitro and that this PIP3P phophatase activity prevented the in vitro phagosome-lysosome fusion. The authors identified the secreted acid phophatase M (SapM) as one of the culture supernatant proteins responsible of this PIP3P dephosphorylation.252 However, further studies have shown that a mutant in sapM gene of M. bovis BCG fails to prevent phagosome arresting, therefore, suggesting that other phosphatases could compensate the lack of SapM from BCG.253 In addition to PIP3P, other proteins (Rab7, Rab5 and the vacuolar protein VPS33B) have been also proposed as potential target of phosphorylation/desphosphorylation mediated by M. tuberculosis. In this regards, the mycobacterial kinase Ndk has been demonstrated to phosphorylate Rab7 and Rab5 in vitro.244 Besides, PtpA contributes to the inhibition of phagosome acidification induced by Mycobacterium spp and this PtpA action is dependent on its binding to the H subunit of the vacuolar ATPase and its phosphatase activity on VPS33B.246 It has been proposed a model in which PtpA inhibits the vacuolar ATPase trafficking to the mycobacterial phagosome. In this model, PtpA is secreted by Mycobacterium into the host cytosol, binds to subunit H of the vacuolar ATPase complex, disrupting the interaction between the two protein complexes and localizing itself near to VPS33B. PtpA then dephosphorylates and inactivates VPS33B; consequently, the normal progression of membrane fusions is blocked.246
Proteins involved in the biosynthesis of cell wall lipids, such as PhoP254 and Ag85A,255 also have a role in the phagosome arresting exerted by MTBC. Most likely, these proteins are not direct effectors of phagosome trafficking, instead they participate in the synthesis of compounds that are actually implicated in this cellular process. For instance, the synthesis of cell wall TDM and the SL is regulated by the two-component system PhoP/PhoR and these lipids have been described as implicated in blocking phagosome/lysosome fusion.256
The M. tuberculosis SecA2 system, a specialized protein export system, is also required for phagosome maturation arrest,257 suggesting that there are effectors of phagosome maturation that are exported into the host environment by this system. However, one important question that remains unclear is how mycobacterial secreted proteins gain access to the host cytoplasm and thereby to their endosomal trafficking pathway targets. The bacterial escape from endosomal compartment to the cytoplasm has been demonstrated in the infection of macrophages with Mycobacterium marinum258; however, for MTBC this point is still matter of discussion.259
Inhibition of apoptosis
The programmed cell death or apoptosis is one of the major mechanisms of the innate immune response elicited by eukaryotic organisms against pathogens. In this process, the host controls the infection at the expense of killing infected cells, but favoring efficient cytotoxic T cell priming via the detour pathway of antigen cross-presentation.260 Accordingly, viral, protozoan and bacterial pathogens have developed anti-apoptotic capacities to counteract this host microcidal activity. It has been extensively reported in the literature that M. tuberculosis induces apoptosis upon infection of host cells.261-263 However, the magnitude of the apoptotic response varies depending on the MTBC strain that infects the cell. In addition, a negative correlation between the bacterial virulence and its capacity to induce apoptosis has been found.261,264 Thus, M. tuberculosis infection mainly results in necrosis, whereas attenuated mutant infections primarily induce apoptosis.265-267 Recent studies have reported the identification and characterization of several anti-apoptotic M. tuberculosis genes, specifically nuoG, katG, sodA/secA2, pknE and Rv3654c/Rv3655c. It is not surprising that most of these genes play roles in the bacterial redox homeostasis, since phagosomal ROS, which are generated after M. tuberculosis infection, trigger the induction of apoptosis.266
In this section, we describe those anti-apoptotic factors that in turn have been shown to be essential for the full virulence of M. tuberculosis species (Fig. 4).
NuoG
NuoG is one of the 14 subunits of the type I NADH dehydrogenase, NADH-1, and the gene that encodes it was first described by Velmurugan et al. as an anti-apoptotic gene in a gain-of-function screening for anti-apoptotic M. tuberculosis genes performed in M. smegmatis. Its function was then confirmed using an M. tuberculosis strain deleted in nuoG.268 The mutant induced apoptosis in human THP-1 cells and cultured primary mouse macrophages, while the wild-type strain did not. Importantly, both BALB/c and SCID mice inoculated with the mutant strain survived longer than those inoculated with the wild-type strain. Furthermore, in the lungs of BALB/c mice, the bacterial load of the mutant was smaller than that of the wild-type strain.268 Thus, these findings are in line with previous evidence indicating that there is a positive correlation between virulence and inhibition of apoptosis. Recently, the same research group demonstrated that NuoG is involved in inhibiting an extrinsic TNF-α-dependent apoptosis pathway.269 Furthermore, they have found that the pro-apoptotic phenotype of the nuoG mutant is ROS-dependent, since in murine BMDM derivative and primary human alveolar macrophages apoptosis was abolished in the presence of both ROS scavengers and in the absence of a functional cellular NADPH oxidase system (NOX2).269 In addition, it has been recently reported that NuoG also suppresses neutrophil apoptosis and that the lack of nuoG in M. tuberculosis accelerates CD4 T cell priming, suggesting that inhibiting neutrophil apoptosis delays adaptive immunity in tuberculosis.270
SecA2/SodA
secA2 encodes a preprotein translocase ATPase. SecA2 is a putative new type of secretion pathway that translocates superoxide dismutase A (SodA) and some others proteins to the culture supernatant (see section 3, Inhibition of apoptosis).206,271 In two different studies, the secA2 deletion in M. tuberculosis H37Rv displayed an attenuation of the bacterial replication in organs of immunocompetent C57BL/6 mice.206,272 One of these studies has also shown longer survival of both C57BL/6 and SCID mice infected with the mutant strain as compared with animals infected with the wild-type strain.206 In addition, the mutant was attenuated in resting BMDM from both wild-type and phox−/− (deficient in oxidative burst) mice, but not in activated wild-type macrophages.272 These results indicate that SecA2 contributes to intracellular growth even in the absence of an oxidative burst.
In another study, Hinchey et al. have demonstrated that SecA2 prevents apoptosis in BMDM since an M. tuberculosis mutant in secA2 has induced higher macrophages apoptosis than its parental strain.273 Based on the activity of different caspases (2, 8 and 9) in the mutant and wild-type strains, these authors have suggested that the lack of SecA2 induces the initiation of apoptosis trought both the extrinsic and intrinsic caspase-dependent pathways. Importantly, the pro-apoptotic phenotype of macrophages infected with the mutant in secA2 was reverted with the introduction of SecA2-dependently secreted sodA gene, indicating that secretion of SodA is likely to be the major SecA2-dependent process involved in the inhibition of host cell apoptosis.273 Consistently, it has been reported that the mutation of sodA in M. tuberculosis H37Rv conferred high susceptibility to hydrogen peroxide and attenuation in a mice model (see the section on “Oxidative and nitrosative stresses”). In addition, the mutant in sodA induced higher apoptosis in mouse lungs than the wild type.274
Finally, it has been recently reported that SecA2 is required for phagosome maturation arrest.257 In this study, it has been shown that shortly after infection, phagosomes containing an M. tuberculosis mutant in secA2 are more acidified and show a higher association with markers of late endosomes than phagosomes containing the wild-type strain. These results suggest that the phagosome maturation arrest defect of the mutant is responsible for the intracellular growth defect. Altogether, these results indicate that SecA2 secretion system, most likely through SodA, inhibits extrinsic and intrinsic apoptosis pathways induced upon M. tuberculosis macrophages infection, in a mechanism probably independent of oxidative burst. The in vivo attenuation detected in secA2 M. tuberculosis mutants are likely to be due to the capacity of SecA2 to avoid apoptosis and antigen-specific CD8+ cross-presentation273 as well as the capacity of altering the intracellular trafficking in favor of the bacteria.
PknE
pknE encodes the serine/threonine kinase E, PknE. Jayakumar et al.’s study has shown that the deletion of pknE from M. tuberculosis results in increase in nitric oxide-mediated apoptosis in human THP-1 macrophages and decrease in production of pro-inflammatory cytokines, TNF-α and IL-6.275 In vitro assays of the same research have also shown that the mutant is more resistant to nitric oxide donors than the wild-type strain. Altogether, PknE may inhibit apoptosis by eliminating reactive nitrogen species that would be indispensable for the TNF-α-mediated induction of apoptosis. The only assessment of the essentiality of pknE for M. tuberculosis virulence, done in human THP-1 cells, has shown that a mutant in pknE displays a moderate impairment in the intracellular replication at late points of infection.275 In this regard, further in vivo studies are needed to establish the role of oxide-mediated apoptosis induced by PknE in the virulence of M. tuberculosis.
Rv3654c and Rv3655c
The role of Rv3660c-Rv3654c operon in apoptosis inhibition was initially demonstrated after screening for the lack of ability to inhibit macrophage apoptosis of an M. tuberculosis H37Rv transposon library.276 This study has also revealed that the proteins encoded in this operon affect mainly the extrinsic apoptosis pathway, since significant suppression of caspase-8 activation (part of extrinsic apoptosis cascade) was observed in macrophages infected with the wild-type strain, but not in those infected with the mutant strain. The virulence of this mutant strain has been evaluated in both apoptotic (detached macrophages) and non-apoptotic macrophages (detached macrophages) and non-apoptotic macrophages (attached macrophages), showing that the mutation impairs the replication of M. tuberculosis in apoptotic macrophages, but not in those non-apoptotic.276 These finding suggest that the products of Rv3660c-Rv3654c operon are somehow important for the virulence of M. tuberculosis.
Further experiments using macrophages transfected with each gene of the Rv3660c-Rv3654c operon indicate that Rv3654c and Rv3655c are the proteins responsible for the extrinsic inhibition of apoptotic macrophage response.276 Despite these important advances, more research is necessary to confirm the role of Rv3654c and Rv3655c in the virulence of M. tuberculosis and to define the precise role of these proteins in the manipulation of cell apoptosis responses.
Apoptosis is an innate defense mechanism by which the host eliminates the niche for bacterial growth. In addition, increasing numbers of publication have shown that apoptosis of infected macrophages produces vesicles containing bacterial antigens that can be engulfed by dendritic cells to prime antigen-specific T cells.277 Therefore, apoptosis would promote the induction of the adaptive immune response apart from its role in innate immunity.
There are alternative pathways by which the apoptosis is triggered, including the apoptosis induced by granzyme B, the lysosomal pathway,278 and the extrinsic and intrinsic apoptotic pathways. In the extrinsic pathway, the binding of TNF-α and FasL ligands to their receptors triggers apoptosis.266 In the intrinsic apoptotic pathway the mitochondria releases cytochrome c and other factors from the mitochondrial intermembrane space that promote apoptosis.
The generation of ROS in phagosome containing bacteria by the action of NOX2, is one of the mechanisms by which infected macrophages induce TNF-α-mediated apoptosis.
The concept that virulent mycobacteria modulate the cell death program inhibiting apoptosis and favoring necrosis is gaining acceptance in the research community. It has been proposed that M. tuberculosis affects different cell signaling pathways to inhibit macrophage apoptosis. One of the anti-apoptotic strategies used by mycobaceria is to control the production of ROS.266 In this section we describe two mycobacterial proteins involved in this ROS dependant-anti-apoptotic mechanism: NuoG subunit of the type I NADH and SecA2, a protein required for the secretion of the superoxide dismutase SodA, among other proteins. On the other hand, the mycobacterial PknE inhibits macrophages TNF-α-mediated apoptosis through elimination of RNS, indicating that RNS also participate in the cellular death signaling. Finally, the precise mechanism by which proteins encoded in Rv3600-3654c operon inhibit the TNF-α-mediated apoptosis remains to be deciphered.
Other models describing how M. tuberculosis inhibits apoptosis favoring cell necrosis have been proposed: Lee et al.279 have demonstrated that after reaching a high intracellular load, virulent bacilli trigger a necrotic mode of macrophage cell death, releasing them to infect new host cells. In addition, M. tuberculosis can modify the surface of infected macrophages preventing completion of the apoptotic envelope and favoring a necrotic cell death outcome.280 Moreover, it has been shown that M. tuberculosis infection prevent macrophage cell membrane repair, which is important for the induction of apoptosis, predisposing the induction of cellular necrosis.281 The study of Chen et al. provides a model by which M. tuberculosis manipulates the apoptosis/necrosis cellular balance. Their study showed that virulent M. tuberculosis negatively modulates the production of prostaglandin E2 (PGE2) in infected macrophages.282 PGE2 induces plasma membrane repair and prevent mitochondrial damage, protecting infected macrophages against necrosis. They also reported that virulent mycobacteria induce the production of LXA4 lipoxin, which is generated by 5- and 15-lipoxygenases (5- and 15-LO)282 and that necrosis is positively regulated by LXA4 through inhibition of PGE2 synthesis. Based on these findings, together with the fact that 5 LO−/− mice are significantly more resistant to tuberculosis infections,282 Chen et al. have proposed that lipoxin production is involved in macrophage necrosis and in inhibition of the apoptosis induced by virulent mycobacteria. However, to completely elucidate these cell death signaling in the context of tuberculosis infections, it is necessary to identify the bacterial anti-apoptotic factors involved in these pathways and to understand the mechanism by which these factors affect the apoptotic/necrotic pathways.
Protein Kinases
Reversible protein phosphorylation is one of the principal signal-transduction pathways by which both eukaryotic and prokaryotic cells regulate the metabolism in response to external stimuli. In bacteria, signal transduction events are performed by two-component regulatory systems and by specific protein kinases and protein phosphatases.
M. tuberculosis genome encodes 11 eukaryotic-like serine-threonine protein kinases (PknA to PknL, except for PknC). All of these pkn genes have been shown to encode functional serine-threonine kinases and some of them have assigned roles in the modulation of different cellular events such as environmental adaptation, differentiation and cell division. These kinase proteins are mainly localized in membrane cell and cell wall of M. tuberculosis, but PknG is predominantly found soluble in cytoplasm.283 Only four of the 11 M. tuberculosis kinases (PknA, PknB, PknG and PknL) are conserved in M. leprae,284 as well as in Corynebacterium glutamicum, a more distantly related actinomycete.285 PknA, PknB and PknG have been also predicted to be essential in M. tuberculosis.286,287 As the massive genome decay shown by M. leprae suggests that only essential genes (coding for functional proteins) have been left unmutated; therefore, the essentiality of pknL in M. tuberculosis should be analyzed.
PknA and PknB are encoded in the same operon as genes involved in cell wall synthesis. Consistently, increasing evidence indicates that PknA and PknB play important roles in determining cell shape, morphology, and cell division.287-289 In addition, enzymes involved in M. tuberculosis peptidoglycan and mycolic acid biosynthesis are substrates of PknA and PknB,290-293 supporting the idea that these protein kinases modulate cell morphology and division in response to environmental cues through regulation of specific pathways involved in cell wall skeleton synthesis. Similarly, PknF is directly or indirectly involved in the regulation of cell growth and septum formation in M. tuberculosis as well as in glucose transport294 and also in the regulation of the mycolic acid biosynthesis through phosphorylation of the β-ketoacyl-acyl carrier protein synthase III (FabH).293 It has been reported that PknF phosphorylates Rv1747, an ABC transporter, and this protein modification seems to be relevant for the replication of M. tuberculosis inside macrophages. However, a mutant deleted in pknF replicates normally in macrophages, indicating that other kinases compensate the lack of PknF-mediated phosphorylation in Rv1747 protein.295 PknI and PknK also seem to have a potential role in growth regulation of M. tuberculosis: The lack of pknI results in an increased growth of this bacterium within macrophages and a hypervirulence phenotype in severe combined immunodeficiency mice.296 Similarly, the absence of pknK displays an increased replication rate both in vitro and in immunocompetent mice297 compared with its parental strain. It has been proposed that VirS, a regulator of mono-oxygenase (mymA) operon, and four other proteins encoded by the mymA operon are potential substrates for PknK.298 In addition, pknK deletion from M. tuberculosis resulted in altered colony morphology and in increased resistance to acidic pH, hypoxia, oxidative and stationary-phase stresses in vitro.297
In M. tuberculosis, PknH phosphorylates InhA, a key enzyme of the fatty-acid synthase II system involved in mycolic acid biosynthesis,291 and EmbR, a putative transcriptional regulator of the embCAB operon.299 The embCAB operon encodes arabinosyltransferases involved in the biosynthesis of arabinogalactan and lipoarabinomannans.300-302 Studying the putative substrate of PknH, Chao et al. have recently demonstrated a case of convergence of the two major signaling systems in M. tuberculosis: the two-component systems and serine-threonine protein kinases. These authors have found that PknH phosphorylates DosR, which in turns regulates the transcription of hypoxia and NO-inducible dormancy (DosR) regulon.303 Importantly, pknH deletion from M. tuberculosis induces hypervirulent phenotype in BALB/c mice in terms of bacterial load in mouse organs.304 Therefore, similarly to PknI and PknK, PknH seems to mediate the growth rate of M. tuberculosis, but unlike these Pkns, mutation in pknH has shown an unaltered in vitro and intracellular growth of M. tuberculosis. Evenmore, the pknH mutant replicated less in non-stimulated human macrophage cell line than the wild-type and complemented strains.304 This result may indicate that PknH controls the intracellular growth of M. tuberculosis through a signaling pathway that requires activation with external stimuli. In fact, it has been demonstrated that in the absence of PknH, M. tuberculosis is more resistant to nitric oxide in vitro; thus, PknH probably senses free radicals produced in response to activation of the host cells that contribute to its survival.304 Altogether, these results suggest that some protein kinases mediate the signaling that slow down the growth of intracellular M. tuberculosis. Uncontrolled replication inside the host cell may be disadvantageous if it contributes to rapid death of the host.
Regarding the function of PknJ and PknL, recent studies suggest that the glycolytic enzyme pyruvate kinase A (mtPykA)305 and Rv2175c, a DNA binding protein,306 are potential substrates of PknJ and PknL, respectively. Interestingly, pknJ is only conserved in MTBC genomes,307 suggesting a role of PknJ in the adaptation of MTBC species to the intracellular life.
The M. tuberculosis infection process involves cross-talk signals between the host and the bacterium; which results in the reprogramming of cell events in both organims. Therefore, it is expected that protein kinases, as key components of the signal transduction pathways of M. tuberculosis, play key roles in the signaling network that allow M. tuberculosis to survive in the aggressive microenvironment of the host. Supporting this hypothesis, three members of the serine-threonine kinase family, PknD, PKnE and PknG, have been shown to be required for the survival and persistence of M. tuberculosis inside hosts (see below).
PknD
It has been proposed that PknD phosphorylates MmpL7,308 a transporter of the RND family essential for M. tuberculosis virulence45 (see “Lipids and Fatty Acid Metabolism”) as well as Rv0516c.309 Some evidence have suggested that Rv0516c is an anti-sigma factor antagonist (or anti-anti sigma factor) that regulates the expression of sigma factor SigF of M. tuberculosis in response to stress signals.310 Greenstein et al. have found that Rv0516c is phosphorylated by PknD in a Thr residue.309 Based on these finding, together with the fact that SigF is essential for the full virulence of M. tuberculosis, it is plausible to speculate that PknD transduces environmental signals by controlling expression of specific groups of genes that are relevant for adaptation to the environment during infections. Consistently, it has been demonstrated that PknD is essential for invasion of mice brain endothelia by M. tuberculosis. An M. tuberculosis CDC1551transposon mutant in pknD has shown impaired invasion and survival in brain microvascular endothelial cells, but not in activated murine J774 macrophages, epithelial A549 cells, and umbilical vein endothelia. In addition, this mutant strain replicated less in brain than the wild type when inoculated in BALB/c mice,311 indicating that PknD may be a key factor required for central nervous system tuberculosis.
PknE
PknE seems to play a role in the virulence of M. tuberculosis, since it contributes to the persistence of M. tuberculosis in human macrophages cell line via an anti-apoptotic mechanism (see the section on “Inhibition of apoptosis”).
PknG
The pknG gene is in a putative operon containing glnH, a gene encoding a protein potentially involved in glutamine uptake. Nevertheless, whether or not PknG is implicated in the regulation of glutamate metabolism of M. tuberculosis is controversial. While Cowley et al. have found that mutation of pknG alters the level of glutamate/glutamine in M. tuberculosis,312 in another study Nguyen et al. have reported that an M. bovis BCG mutant in pknG showed no differences either in the intracellular level of glutamine or in the uptake of glutamine.313
It has been reported that PknG is unique in the fact that it undergoes autophosphorylation on Thr residues located at the N-terminus. Although this autophosphorylation seems unrelated to the regulation of its kinase activity, it is essential for the prevention of M. bovis BCG trafficking to lysosomes and for the bacterial survival in murine BMDM.314
An M. tuberculosis H37Rv mutant strain in pknG gene was attenuated in lungs, spleen and liver of immunocompetent BALB/c mice, but not in those of CD-1 mice. This mutant also caused delayed mortality in SCID mice. Surprisingly, pknG mutation has been shown to produce some defects in the in vitro growth of M. tuberculosis,312 but not in that of M. bovis BCG.313 Therefore, further confirmative analysis shoud be performed to consider PknG as a virulence factor.
Pkns together with two-component systems and sigma factors are the constituents of the mycobacterial machine that regulates the adaptive gene expression in response to external stimuli. Increasing evidence suggest a crosstalk among all components of these regulatory network. For instance, it has been demonstrated that PknH phophorylates DosR, a member of a two-component system, and that various sigma factor regulators are substrates of Pkns.309
Although the signaling pathways and endogenous substrates remain to be clearly established, the functional roles have been defined for most of the Pkns. PknA, PknB, PknF, PKnI and PknK mediate the signaling that slows down the in vitro growth of M. tuberculosis. Moreover, PknI seems to play a role in the intracellular growth regulation of M. tuberculosis, suggesting that the uncontrolled replication inside the host cell may be disadvantageous if it contributes to a rapid death of the host. Similarly, the lack of PknH or K significantly increases the replication of M. tuberculosis in immune competent mice. However, whether these Pkns regulate the mycobacterial growth in animal models is still uncertain. This is mainly because of the difficulties to accurately determine the rate of bacterial replication in organs, as an increase in bacterial counts might be due to an increment of the growth rate but also to any adaptive advantage in the hostile intracellular environment. In addition, potential roles in central carbon metabolism and in cell wall biosynthesis have been assigned to PknG, F, J, L and PknA, B, H, D, F, respectively.
As mentioned above, only three Pkns have been demonstrated to be implicated in different aspects of M. tuberculosis infection. PksD is essential for M. tuberculosis brain endothelia invasion, and PknE participates in the anti-apoptotic mechanisms displayed by M. tuberculosis to avoid a host efficient immune response. Furthermore, PknE is involved in the resistance to nitrosative stress.315 However, the in vivo role of PknG is less precise. The rest of the Pkns either seem to downregulate the in vivo replication (PknH, I, K and L), are dispensable for intracellular growth (PknF), or are essential for the basic metabolism, such as PknB and probably PknA.286
Proteases
Proteases play crucial roles in cellular homeostasis by controlling proteins involved in transcription, regulation, metabolism and virulence. Microbial pathogens frequently utilize extracellular proteases as virulence factors that can play different roles in tissue destruction or modulation of the immune response by inactivation of host defense molecules such as immunoglobulins and complement components. They can also activate key regulatory proteins or peptides, acquire nutrients by hydrolyzing host’s proteins and process signaling molecules that regulate gene expression.13
The genome of M. tuberculosis H37Rv encodes over 100 proteases. Some are potential secreted proteins22 and 38 of them are conserved among M. leprae, M. bovis, M. avium paratuberculosis and M. tuberculosis.316 Despite this, very little is known about the biology of these enzymes in these organisms.
Serine proteases
Serine proteases are enzymes that cleave peptide bonds in proteins, in which serine serves as the nucleophilic amino acid at the enzyme's active site. There is a family of five subtilisin-like serine proteases, the mycosins (mycosin-1 to -5), which share a high degree of similarity and are constitutively expressed in M. tuberculosis H37Rv. Bacterial subtilases are typically secreted and they degrade non-specific proteins to provide bacteria with readily importable peptides.317
mycP1, mycP3 and mycP5 homologs have been detected in the genome of M. leprae and mycP2 to 5 in M. avium. Only mycP3 has been detected in the avirulent M. smegmatis so it appears that the multiplicity of the mycP genes may occur only in virulent mycobacteria.318mycP1 encodes a protein that localizes to the cell wall/membrane fraction and is not expressed in the attenuated M. bovis strain BCG, although the gene is present in the genome. The expression of mycP1 also occurs in intracellular bacteria and seems to be upregulated during growth in macrophages and may be processed intracellularly.319
MycP1
Ohol et al. have recently reported that MycP1 controls ESX-1 protein export by cleaving EspB, one of the substrates of the ESX-1 secretion system.191 As it was mentioned in a previous section (see “Secretion systems”), ESX-1 secretion is required for early replication and full virulence in macrophages and mice.320 Other substrates of the ESX-1 system are ESAT6 and CFP10, both essential for virulence and highly immunogenic.321-324 Thus, it may be essential for M. tuberculosis to tightly regulate the amount of ESAT6 being exported in order to maintain an optimal balance between virulence and immunogenicity. EspB is probably secreted as a full-length protein into the periplasm, promoting secretion of the ESX-1 substrates, but its proteolysis by MycP1 serves to turn off secretion. EspB is proteolyzed at least at three sites, two of which require MycP1, while the third one could be hydrolyzed by one of the four homologous of MycP1.191
The inability of ΔmycP1 mutant bacteria to secrete ESAT6 leads to severe attenuation in the murine BMDM and BALB/c mouse model. While bacteria expressing a mutated version of MycP1 in the catalytic site grew to similar bacterial loads as wild type, they were attenuated in survival assays and histopathological analysis compared with wild-type M. tuberculosis. In this strain, the loss of MycP1 protease activity led to the constitutive secretion of ESAT6 and promoted ESAT6 antigen presentation by antigen-presenting cells during chronic infection, probably priming T cells to generate a stronger immune response against M. tuberculosis.191
HtrA
The HtrA (high-temperature requirement A) family of oligomeric serine proteases (S1, chymotrypsin family) is conserved from prokaryotes to humans. The structure of the protease comprises a serine protease domain and one or more C-terminal PDZ or protein-protein interaction domains, which regulate their protease activities by binding to regions of unfolded proteins in the periplasm. These proteins often possess the activities of chaperones and/or proteases.325 In bacteria, HtrA-like family members have been shown to participate in stress response networks, including those regulated through sigma factors and two-component systems326 and they are involved in a variety of biological functions and pathogenicity.327
M. tuberculosis contains three HtrA proteases22 with a moderate homology among themselves (32–40% identity).328 HtrA1 (or DegP) has been predicted to be essential in M. tuberculosis.286,328,329 HtrA2 (or PepD) is involved in virulence,328 while HtrA3 (or PepA) is not (the ΔhtrA3 deletion had no effect on the mean survival time of mice or any other detectable phenotype).328M. tuberculosis HtrA1 forms integral part of the membrane,330 while the other two proteases may be exported, as they are detected in the culture filtrate.329,331,332
HtrA2 or PepD
HtrA2 (or PepD) encodes an HtrA-like serine protease and is thought to process proteins that have been altered following exposure of M. tuberculosis to extra-cytoplasmic stress. HtrA2 functions in vitro both as a protease and as a chaperone, and it is required for aspects of M. tuberculosis virulence in vivo. HtrA2 undergoes autolytic processing which might be involved in modulating its activity. C57BL/6 mice infected with a ΔhtrA2 M. tuberculosis mutant exhibit increased time to death, and less tissue pathology than animals infected with wild-type or complemented strains.328 However, loss of htrA2 (ΔpepD) neither alters the ability of M. tuberculosis to resist SDS-mediated killing nor it affects the growth and/or survival characteristics of M. tuberculosis either within peripheral human blood monocyte-derived macrophages or within resting or activated murine J774 macrophages in vitro. Rather, the deletion of htrA2 in M. tuberculosis upregulates the expression of sigE and other stress-responsive determinants, which may compensate for the loss of this protein. In turn, the expression of hstrA2 is indirectly regulated by the extracytoplasmic function (ECF) sigma factor SigE326 and directly regulated by MprAB, the stress-responsive two-component signal transduction system enconded in operon mprAB. Remarkably, htrA2 is part of this operon. Based on the similarities with the heat shock regulon CpxAR and RpoE systems in E. coli, White et al. have proposed a model for M. tuberculosis, in which MprAB and SigE systems sense and process stress resulting from the accumulation of unfolded or misfolded protein substrates and regulate the expression of chaperones/proteases like HtrA2. One identified target of HtrA2/PepD protease is the 35-kDa antigen of M. tuberculosis (Rv2744c), a member of the PspA family of proteins. These proteins participate in the phage shock response and likely in other multiple functions. The Rv2744c gene lies in an operon with clgR,333 a transcription factor that regulates its own expression and those of several other genes in M. tuberculosis, including proteases and chaperones.334 Therefore, one possible role of HtrA2 is to regulate the activity of proteins involved in manteining cell wall homeostasis.333 clgR is further reviewed below in this section.
Rv3671c
Rv3671c is a membrane-associated protein,335 predicted to be a serine protease with conserved aspartate, histidine and serine active site residues and four transmembrane domains, two β-barrel subdomains and six antiparallel β-strands. It protects M. tuberculosis from acidification and oxidative stress probably by degrading proteins unfolded owing to acid and oxidative stresses. Alternately, it could be essential for maintenance of the cell wall integrity, remodelling ion channels, proton pumps, or membrane lipids and thus, maintaining the internal pH.336
An M. tuberculosis transposon mutant disrupted in the Rv3671c gene, was sensitive to acid and failed to maintain intracellular pH homeostasis both in vitro and in activated BMDM. The mutant was also hypersensitive to the cell wall-damaging detergent SDS and to the lipophilic antibiotics erythromycin and rifampin, suggesting that it has some defect in cell wall function. Growth of the mutant was also severely attenuated in C57BL/6 mice and it induced markedly less pulmonary pathology than wild-type bacteria and complemented strains.335
ATP-dependent proteases
Self-compartmentalized or chambered proteases are common in most bacteria.337 M. tuberculosis encodes two ClpP (caseinolytic protease) homologs, ClpP1 and ClpP2, in a single operon, as well as the associated ATPase chaperones ClpC and ClpX (Rv2457c, Rv2667).316 ClpP1 and ClpP2 are required for growth in vitro and in a mouse model of infection.338
The clp gene regulator (clgR) expression is highly increased in stress conditions such as heat shock339 and during macrophage infection.215 ClgR activates the transcription of four protease systems (ClpP1/C, ClpP2/C, PtrB and HtrA-like protease Rv1043c) and three chaperones (Acr2, ClpB and the chaperonin Rv3269). ClgR-regulated transcriptional activation of these systems is essential for M. tuberculosis to replicate in murine BMDM. M. tuberculosis lacking ClgR is deficient in the ability to control phagosome pH. This attenuation could be explained if the rapid upregulation of the ClgR-dependent proteases and chaperones is necessary for protein homeostasis during exposure to the macrophage antimicrobial mechanisms, such as the oxidative burst. Regulation of proteases is vital to avoid the uncoordinated destruction of cellular proteins; this occurs at the level of substrate selection and also by controlling the expression of proteases via transcriptional regulators.334
Metalloproteases
Metalloproteases are a subfamily of proteases that use metals, mostly zinc, for their catalytic activities and are involved in virulence, cell wall processes and intermediary metabolism. There are three putative Zn-dependent metalloproteases in the genome of M. tuberculosis. We will only refer to two of them (Zmp1 and Rip1) because the third metalloprotease gene in M. tuberculosis, Rv1977, coding for a putative iminopeptidase that probably acts as a Zn-dependent enzyme with chaperone function, is deleted in M. bovis, so it is unlikely to be involved in virulence.
Zmp1
Master et al. have demonstrated that the zmp1 (Rv0198c) gene is required for the survival of M. tuberculosis and BCG in murine J774 and RAW264.7 macrophages and for the full virulence of M. tuberculosis in a model of tuberculosis in C57BL/6 mice. It is also essential for prevention of inflammasome activation (a specialized inflammatory caspase activating protein complex, and component of the innate immune system) and IL-1β production. The generation of active IL-1β, a potent pro-inflammatory cytokine from its pro-protein precursor, is a tightly regulated process dependent on the inflammasome and caspase-1. Its production results in a vigorous host response to the pathogen and, when induced, it is an effective anti-tuberculosis agent. M. tuberculosis inhibits this mechanism through the function of Zmp1.340
In a more recent study, Muttucumaru et al. have reported that Zmp1 is not required for survival of M. tuberculosis within THP1 macrophage-like human cell line, but the mutant in zmp1 resulted hyper-virulent in SCID and C57BL/6 mice. The absence of Zmp1 led to changes in the expression of other genes in the same pathway or in compensatory pathways. Yet, the mechanism by which the deletion of zmp1 leads to changes in gene expression is not known, but it is likely that the absence of this protease and its proteolytic activity results in the lack of the cleavage of a substrate signal for one regulatory protein, which in turn modulates the expression of other genes.341 The discrepancies between these two studies are not well understood, but it could be due to the nature of the different M. tuberculosis mutants.
Rip1
Rip1 is a major virulence determinant of M. tuberculosis through its role in regulating cell envelope composition growth and persistence in vivo.342 ECF sigma factors, alternative sigma factors that direct RNA polymerase to specific promoters (see “Sigma factors”), are often held inactive by trans-membrane anti-sigma factors, which are degraded by proteolysis in response to extracellular stimuli. Rip1 participates in signaling across the cell envelope through proteolysis of three anti-sigma factor substrates RskA, RslA and RsmA (anti-SigK, anti-SigL and anti-SigM, negative regulators of Sigma K, L and M respectively).343
Proteasome-associated proteins
The proteasome accessory or associated factors, Mpa and PafA, are important for defense against RNI and for virulence of M. tuberculosis in mouse. mpa (Rv2115c) encodes an ATPase with an AAA-protein family signature (AAA stands for ATPases associated with diverse cellular activities) homologous to that found in proteasome regulatory caps in eukaryotes344 and is probably involved in substrate recognition, unfolding and translocation into the proteasome core.345,346 In addition, the proximity of Rv2115c to putative proteosomal genes also indicates that this gene could be associated to proteosomal functions in M. tuberculosis.22 Darwin et al. have demonstrated that the products of Rv2115c (Mpa) and Rv2097c (PafA) confer M. tuberculosis protection against RNI, since mutants of M. tuberculosis H37Rv in any of these genes are highly susceptible to nitrite at low pH. Rv2097c encodes a hypothetical protein that was also potentially associated to proteosomal functions.345 The biochemical activity of PafA remains to be discovered, but it seems that PafA has a role similar to that of Mpa in protein degradation since mpa and pafA mutants were similarly sensitive to NO in vitro, and had the same attenuated phenotype in wild-type mice.347 When tested in iNOS−/− mice, which do not produce NO in their macrophages, the mpa and pafA mutants were not as attenuated as in wild-type mice, supporting the hypothesis that these genes are required for resistance of M. tuberculosis to NO in vivo. However, both mutants not only showed impaired growth in resting primary macrophages from wild-type mice but also in those from iNOS−/− mice, suggesting that mpa and pafA are involved in the response to more macrophage-associated stresses than those dependent on iNOS.345
As mentioned above, it has been hypothesized that both Mpa and PafA proteins are involved in proteasome functions to protect the bacilli against nitrosative stress.345 It remains to be determined how the proteasome protects against NO and other stresses in the host. A plausible hypothesis is that Mpa, PafA and the proteasome dispose of NO-damaged proteins that are toxic to the cell. Another possibility is that the proteasome directly or indirectly regulates antioxidant or virulence gene expression.345,348 A role in bacterial growth both in vivo and in vitro has also been assigned to Mpa.349 An M. tuberculosis Erman mutant in mpa showed an impaired growth in standard culture medium and in BALB/c mice.349 Surprisingly, this gene showed to be not required for in vitro growth of M. tuberculosis H37Rv.345 Therefore, it is still a matter of discussion whether or not mpa can be considered a virulence gene of M. tuberculosis and what is the precise role of both, Mpa and PafA proteins in the mycobacterial proteasome.
Metal Transporter Proteins
Metals such as iron, magnesium, cobalt, cupper, manganese and zinc are essential for survival of prokaryotes and eukaryotes either in the environment or within the cell. Metals are part of prosthetic groups or are co-factors of many enzymes. Microorganisms need traces of these micronutrients and in excess these metals could be toxic. Consequently, microoganisms have evolved many strategies in order to import metals into their cytoplasm or pump them out to the extracellular medium. Some of the proteins detailed in this section have shown to be essential for Mycobacterium growth in some in vitro extraordinary conditions, such as limiting metal condition. Although these proteins, according to our definition, are not actual virulence factors, they have been included in this review because they provide the bacteria with significant advantages for adaptation and survival in host cells.
Metal importers
MbtB, IrtAB and IdeR
MbtB, IrtAB and IdeR are proteins involved in iron acquisition. Iron is an essential cofactor, required in the heme of cytochromes and hemeproteins. Iron is also cofactor of proteins involved in amino acid and pyrimidine biogenesis, enzymes involved in the tricarboxylic acid cycle and in DNA synthesis.350 In the restrictive environment of nature or of the mammalian macrophage, the iron is 1,000 times less concentrated than that required by the bacterium and it is also in an insoluble state. This situation has led pathogenic and non-pathogenic bacteria to evolve efficient iron-acquisition systems.
Siderophores are the most important iron-chelating compounds synthetised by microorganisms, being mycobactin and carboxymycobactin the major ones in Mycobacterium. These compounds are biosynthesized through the action of proteins encoded by the mbt cluster, which includes the genes mbtA to mbtJ. Proteins encoded by these genes are: MbtA, predicted as a salicyl-AMP lipase/salicyl-S-aroyl carrier protein domain synthetase; MbtB, MbtE and MbtF predicted as non-ribosomal peptide synthetases; MbtC and MbtD predicted as polyketyde synthases; MbtG a predicted lysine-N-oxygenase; MbtJ a putative esterase/acetyl hydrolase; MbtI required for salicylic acid biosynthesis and MbtH of unknown function.351 The mutation of any mbt gene disrupts the synthesis of these siderophores, which, in turn, unables the bacterium to acquire the metal from the medium. Therefore, the bacteria fail to survive in the host cell. As expected, an M. tuberculosis H37Rv mutant, in which the mbtB gene was replaced by recombination with a hygromycin-resistance cassette, was restricted for growth in iron-limited media but grew normally in iron-replete media. In addition, the biosynthesis of all siderophores-like molecules derived from salicylic acid was interrupted in the mutant, a defect that impaired its growth in macrophage-like THP-1 cells as compared with the wild type.351 These results clearly suggest that siderophore production is required for M. tuberculosis virulence.
An alternatively mbt-2 cluster of genes involved in iron uptake is present in the M. tuberculosis genome. It includes two operons containing the mbtK to mbtN genes and the irtA and irtB genes, encoding for the IrtA and IrtB proteins which have the typical domains of an ABC transporter.352 IrtA is different to the common ABC transporters; it has the transmembrane domain fused to a cytoplasm substrate-binding domain (SBD), which is essential for iron acquisition. IrtB only harbours the permease and ATPase domains.353 The inactivation of the irtAB system in M. tuberculosis H37Rv, by a two-step recombination, results in a mutant with a decrease ability to survive in iron-deficient conditions and shows reduced ability to use iron from the Fe-carboxymycobactin.354 These results indicate that IrtAB is a transporter of the Fe3+-siderophore complex. It has been proposed that IrtAB transports the Fe3+-siderophore complex toward the cytoplasm and, once there, the SBD domain of IrtA, functioning as a reductase, reduces the iron and releases it from the complex for its assimilation into metalloproteins.355 The relevance of IrtAB in the virulence of M. tuberculosis has been demonstrated in an irtAB mutant, which showed a reduced ability to replicate in THP-1 human macrophages and in the lungs of C57B/6 mice compared with the parental strain.354
The transcription of the mbt and mbt-2 cluster of genes is negatively regulated by IdeR. IdeR is an iron-dependent regulatory protein essential in M. tuberculosis that functions as a repressor. In an abundant iron condition, IdeR is found complexed with Fe3+, and this complex binds to the promoter regions of mbt and mbt-2 clusters preventing their transcription. On the contrary, in a depleted iron condition, there is not sufficient iron to form the Fe3+-IdeR complex, and IdeR releases the promoter leading to the transcription of the genes of this cluster which, as was described ealier, leads to the synthesis of proteins essential for the acquisition and incorporation of iron to the bacteria.356
MgtC
MgtC is a transmembrane P-type ATPase protein involved in Mg2+ uptake. Magnesium is essential as a cofactor for enzymes binding to phosphate and it is important in DNA and RNA synthesis. However, Mg2+ is found in low concentration inside macrophages. Therefore, the uptake of this metal is needed for the survival of the bacteria inside the host. The inactivation of mgtC in M. tuberculosis Erdman resulted in a mutant that grew slower than the parental or complemented strains in media with limiting magnesium (20 μM), but it grew equally to the parental strain in medium with high magnesium. In addition, in a medium containing limiting Mg2+ at slightly acidic medium (but not at neutral pH), the growth of the mutant was more affected than that of the parental or complemented strains, suggesting that in the environment of the phagosome, that is, in a limiting and acidic environment, MgtC would be important in Mg2+ uptake. As expected, mgtC mutant was attenuated for virulence in cultured human macrophages and impaired for growth in the lungs and spleens of BALB/c mice compared with the parental or complemented strains.357 This suggests that the ability to acquire magnesium is essential for virulence in pathogens that proliferate within macrophage phagosomes.
Metal exporters
CtpC
CtpC is a Zn2+ efflux P-type ATPase that functions as an exporter. Even though Zn2+ is essential for bacterium growth, an increased concentration results poisonous to the bacteria. M. tuberculosis contains a set of 11 ctp genes, ctpA to J and ctpV, encoding for proteins predicted as probable cation transporters P-type ATPases, which unlike ABC transporters have auto-hydrolytic ATP activity required for exporting metals. It has been reported that macrophages may make use of heavy metal poisoning as mechanisms of antimicrobial immunity. Interestingly, a burst of free zinc inside macrophages and intraphagosomal zinc accumulation was observed a few hours post mycobacterium infection.358 In this condition of high Zn2+ concentrations, M. tuberculosis induces high level transcription of ctpC and the upstream Rv3269 gene but also, at a lower level, of the ctpG and ctpJ genes, suggesting a role of CtpC in Zn2+ detoxification. In addition, an M. tuberculosis GC1237 mutant in ctpC gene, generated by allelic exchange, has shown higher levels (as much as three-times higher) of zinc retention within the mycobacterial cytoplasm compared with the wild-type strain. This mutant has also shown impaired intracellular growth in human macrophages.358 Taken together, these results suggest that the P1-type ATPases neutralize the toxic effects of zinc in macrophages by pumping the metal outside the mycobacteria.
CtpV
CtpV is a Cu2+ efflux transporter P-type ATPase required by M. tuberculosis to maintain resistance to copper toxicity. Copper is a required micronutrient but, similarly to Zn2+, is toxic at excess concentrations. A ctpV mutant, generated in M. tuberculosis H37Rv, has an increased copper sensitivity relative to wild-type or complemented strains when grown under toxic copper conditions (500 mM CuCl2). Also, the mutant shows higher levels of intracellular copper than the wild type, suggesting that CtpV is necessary to maintain copper homeostasis.359 These authors have also demonstrated that CtpV has a role in host infection: when guinea pigs were infected with an M. tuberculosis H37Rv ctpV mutant, they showed significantly lower CFUs counts in lungs as compared with the wild-type or complemented strains. Additionally, the tissue damage and granulomatous responses were less severe in lungs infected with the ctpV mutant.359 These results suggest a connection between bacterial copper response mediated by CtpV and the virulence of M. tuberculosis, supporting the hypothesis that copper response could be important for intracellular pathogens.
It is clear that the metal transporter proteins, either importers or exporters, confer mycobacteria a significant adaptive advantage allowing them to survive in metal-limited environments, as inside the phagosome or the host, or to tolerate environment with high metal concentration that would otherwise be toxic and lead to bacterium death. That is why these proteins have been conserved throughout the evolution from a saprophytic to an intracellular life into the host and why these proteins are relevant to the virulence of many bacteria. However, it is important to mention that based on the cirteria here established to define a virulence factor, the proteins described in this section are not true virulence factors because their absence affect the in vitro growth of bacteria.
Gene Expression Regulators
M. tuberculosis is able to establish lifelong infections in individuals within granulomatous lesions that are formed following a productive immune response. Adaptation to this highly dynamic environment is thought to be mediated primarily through transcriptional reprogramming initiated in response to recognition of stimuli, including low-oxygen tension, nutrient depletion, reactive oxygen and nitrogen species, altered pH, toxic lipid moieties, cell wall/cell membrane-perturbing agents and other environmental cues.
To survive to the continued exposure to these potentially adverse factors, M. tuberculosis encodes a variety of regulatory factors, including 11 complete two-component systems (TCSs) and several orphan response regulators (RRs) and sensor kinases (SKs).
Two-component systems
Two-component regulatory signal transduction systems are important elements of the adaptive response of the tubercle bacillus, among other prokaryotes, to a variety of environmental stimuli. They typically consist of a membrane-bound histidine kinase (sensor kinase) that senses a specific environmental stimulus and a corresponding response regulator, phosphorilated by the sensor kinase that mediates the cellular response, mostly through differential expression of target genes (Fig. 2).
PhoP-PhoR
So far, 11 paired two-component systems have been described. Specifically, the two-component system PhoP-PhoR is the one whose disruption has been shown to most dramatically affect the ability of M. tuberculosis to replicate in cellular and animal models.
The phoP-phoR operon (conserved in most mycobacteria) encodes PhoR, a histidine protein kinase (sensor of stimuli), and PhoP, a transcriptional regulator that receives a phosphate from PhoR.
First thought to be implicated in phophate metabolism and transport, the two-component system PhoP-PhoR is involved in diverse aspects of metabolic physiology, lipid metabolism regulation and respiration. PhoP-PhoR is likely to sense magnesium. It increases triacylated mannose-capped lipoarabinomannans (ManLAM) acyl forms. Monoacylated ManLAM, which predominates in phoP-phoR mutants, fails to inhibit the IL-12 production in human dendritic cells. Many genes involved in lipid metabolism seem to be regulated by PhoP-PhoR: pks3 (polyketide β-ketoacyl synthase), pks5 (polyketide synthase), papA3 (polyketide synthase associated protein), fadD26 (a fatty acid-coenzyme A ligase), lipF (an esterase/lipase), fbpA (secreting type Ag85A FbpA), mmpL10 and mmpL8 (transport proteins belonging to the RND superfamily). Some M. tuberculosis lipid components such as DAT, PAT and SL are also diminished in the phoP mutants.360 In the attenuated M. tuberculosis H37Ra strain, a single nucleotide mutation within a DNA binding domain of PhoP can abolish the binding between PhoP and its own promoter.361 M. tuberculosis H37Rv ΔphoP shares many features with M. tuberculosis H37Ra, such as low content of DAT, PAT and SL, and show significant overlap in their transcription expression profile. This is strong evidence of the critical role played by PhoP-PhoR in metabolic control with a considerable impact on the virulence of M. tuberculosis. Moreover, PhoP regulates genes related to ESX-1 secretion system required for virulence and ESAT6 secretion. PhoP modulates the expression of both EspB (secreted ESX-1 substrate protein B) and EspR (transcriptional regulatory protein).362 As a result, M. tuberculosis H37Rv ΔphoP and M. tuberculosis H37Ra synthesize, but are incapable of secreting, both ESAT6 and CFP10.
Importantly, phoP and phoP-phoR mutants of M. tuberculosis are attenuated for growth in various cultured or primary cell types, including murine BMDM, murine alveolar macrophages, murine J774 macrophage cells, human THP-1 cells, and SCID and BALB/c mice and guinea pigs. These ex vivo and in vivo assessments of the role of PhoP-PhoR to M. tuberculosis virulence have been reviewed by Bretl et al.365
Abramovitch et al. have suggested that an specific subset of the phoP regulon is controlled by aprABC operon,366 since similar altered expression profiles were detected in M. tuberculosis mutants for either phoP-phoR or aprABC locus. The authors have demonstrated that the aprABC operon, which is unique to the MTBC, is expressed in acidic medium in vitro and in macrophages in a manner dependent on PhoP-PhoR. Moreover, this study showed that the deletion of aprABC operon from M. tuberculosis CDC1551 caused defects in intracellular replication in both resting and activated C57BL/6 BMDM, but also affected the mycobacterial in vitro growth, showing the mutant aggregation in liquid media and reduced colony size.366 These authors propose a model where PhoP-PhoR senses the acidic pH of the phagosome and induces aprABC expression to fine-tune processes unique for intracellular adaptation of M. tuberculosis.
SenX3-RegX3
This TCS is involved in phosphate sensing and is homologous to the master aerobic regulator ArcB-ArcA of E. coli.
In M. tuberculosis, RegX3 both positively and negatively regulates a large and functionally diverse regulon comprised of 100 genes. Several of these genes encode hypothetical proteins, while others are involved in important physiological activities, including energy metabolism, cell envelope maintenance, and regulatory functions.367
RegX3 is an activator of the phoA gene that encodes the alkaline phosphatase, PhoA. In turn, PhoA activates the expression of ptsS belonging to the phosphate transport system PstSCAB. RegX3 also upregulates the expression of another phosphate transport system, PhnDCE. Phosphate limitation restricts M. tuberculosis growth in a concentration-dependent manner. Three other genes, ald, encoding alanine dehydrogenase, cyd, encoding a subunit of the cytochrome D ubiquinol oxidase, and gltA1, encoding a citrate synthase, also are regulated by SenX3-RegX3 system.368
In the intergenic region of senX3 and regx3 genes, there is a mycobacterial interspersed repetitive unit (MIRU) element, precisely the first one to be identified.369 While regX3 appears to be essential in M. smegmatis, this gene is dispensable for the in vitro growth of M. tuberculosis.367
senX3 and regX3 are necessary for M. tuberculosis full virulence,367 as an M. tuberculosis H37Rv senX3-regX3 mutant is attenuated for growth in the human THP-1 macrophage cell line and murine BMDM (115), similar to what is observed in the lungs and spleens of SCID and DBA mice after infection with a this mutant.367 Also, individual senX3 and regX3 mutants were attenuated in BALB/c mice.370
DosR/S/T (DevR/S/T)
Together with PhoP-PhoR, DosR/S/T is the most studied TCS system from the MTBC. This TCS system is formed by one response regulator (DosR), which is activated in response to hypoxia and nitric oxide, and by either DosS or DosT, both histidine kinases. This TCS was initially termed DevR/S/T by the original discoverers. DosR refers to “dormancy survival regulator” and this denomination has been at present adopted by most researchers. Boon and Dick have observed that DosR is responsible for the dormancy stage of M. tuberculosis, and that upon mutation of DosR the bacteria failed to enter in dormancy and died in a Wayne culture system of hypoxia.371 It has been observed that exposure of M. tuberculosis mutants lacking DosR to low oxygen tension induces the expression of more than 100 genes, from which 48 are under the control of DosR whose C-terminal segment recognizes a 18–20 bp palindromic sequence upstream of the regulated genes.372 Both DosS and DosT autophosphorylate and transfer the phosphate group to DosR. DosS and DosT are indispensable for the induction of the Dos regulon in a dormancy model in vitro. DosS is expressed during hypoxia and DosT is constitutively expressed. However, once expressed DosS can be replaced by DosT. DosS and DosT possess GAF domains, a receptor of cyclic GMP and a β-type heme group that senses CO and NO molecules,373 which are a cell signal of hypoxia and a marker of bactericide activity by macrophages respectively. Heme oxygenase-1 produces carbon monoxide in the macrophage, inducing the M. tuberculosis Dos regulon.
DosT is inactive when bound to O2. However, during hypoxia, CO and NO may displace O2 in DosT, restoring its active form. O2 can oxidize the heme group and inhibit DosS. The oxidative form Fe3+-DosS does not autophosphorylate, while the reduce Fe2+-DosS does.373 On the other hand, in hypoxia, FAD and FMN reduce the heme group from DosS further supporting the concept that DosS is a redox sensor.
The emerging and highly pathogenic M. tuberculosis Beijing strain normally overexpresses the Dos regulon during the exponential growth phase, and many isolates of Beijing lineage possess a frameshift mutation in DosT. The molecular explanation for this difference has yet to be found.374
In spite of the central role of the DosR/T/S system in M. tuberculosis sensing of oxygen tension and redox state, its involment in virulence is not clear, yielding contrasting results. For example, the persistence of an M. tuberculosis H37Rv mutant in ΔdosR seemed to be unaltered in C57BL/6 mice; this mutant, however, produced less lung pathology.375 Other authors have shown that in C57BL/6 and BALB/c mice, rabbit and guinea pig models, a ΔdosR-S mutant exhibited a slight growth defect and induced lung pathology generation when compared with the wild-type M. tuberculosis.376,377 Malhotra et al. have observed that guinea pigs infected with a devR mutant showed a significant decrease in gross lesions in lung, liver and spleen; and a three log lower bacterial burden in spleen compared with guinea pigs infected with the parental strain.378 Contrary to this, a devR deleted of M. tuberculosis showed hypervirulence in an SCID mouse model. ΔdevR also grew more rapidly in the acute stage of infection in immunocompetent DBA mice.379
MprA/MprB
Named after mycobacterial persistence regulator, MprB is the sensor kinase and MprA is the response regulator in this two-component system. MprB not only phosphorilates MprA but also acts as its phosphatase, regulating in this way its own activity.380
The genes coding for this TCS are part of an in vivo-expressed genomic island in which a set of 20 genes are activated only during BALB/c mice infection and not in SCID mice or in vitro. The region is called an in vivo-expressed genomic island (iVEGI).381
The use of MprA and B deletion mutants has led to the identification of around 200 genes that are regulated by the pair. Positive regulation is observed in mprA, pepD and moaB2, as well as in acr2 (hspX) (a member of the DosR regulon).382-384 This TCS is part of a highly complex regulatory network and, as a consequence, it is difficult to define the mprAB regulon. For example, sigE and sigB are regulated by MprAB system that responds to membrane damaging and stressing agents such as detergents and alkaline pH. SigE regulates ppk1 that generates polyphosphates, which are in turn phosphate donors for MprB. Also, MprA and SigE regulate pepD that cleaves Rv2774 protein, implicated in cell wall antibiotic susceptibility. Mutants in mprA or in both mprAB genes are hypervirulent in human macrophages derived from peripheral blood monocytes but attenuated in C57BL/6 mice,385 indicating that this complex regulatory system is required for full virulence. Moreover, mprAB orthologous genes are required for Rhodococcus equi virulence.386
The authors of the whole genome sequence of M. tuberculosis22 have noted that it encodes for 11 putative complete TCS and a few isolated kinase and regulatory genes. This number is clearly lower than in B. subtilis and E. coli, whose genomes encode more than 30 genes for two-component regulatory systems.22 Some of these TCS were not included in this review because mutants in these systems have not been constructed or obtained. Among them, there is Rv0600c-Rv0601c-tcrA,387 which is composed of two SKs (Rv0600c and Rv0601c) and one RR, but the signals sensed by the SKs have not yet been identified. Similarly, mutants in MtrA-MtrB have not been obtained, although it has been described that the overexpression of mtrA causes attenuation in mice.365 Some others may be essential for M. tuberculosis growth, as no mutants could be obtained. In our knowledge, no articles using alternative gene inactivation approaches such as conditional lethal or gene silencing have been published in order to solve the question of the role of these TCS in virulence. In other cases, the mutation of a TCS has no a clear impact in virulence. One example is narL-Rv0845, enoding a TCS similar to NarQ-NarL from E. coli that regulates genes expression in response to nitrate concentrations. The deletion of genes encoding this TCS produced no alteration of M. tuberculosis virulence in mice.379 The mutation of TCS prrA-prrB diminishes the growth in macrophages only at initial stages and has no impact on mice infection.365 Other mutants like those of kdpD-kdpE and tcrX (Rv3765c)-tcrY (Rv3764c) have been tested only in SCID mice. Moreover, mutation in trcR (Rv1033c)-trcS (Rv1032c) has produced a hypervirulence effect.
PhoP/PhoR and DosR/T/S are the most studied TCSs in M. tuberculosis. Even though PhoP-PhoR have been studied extensively regarding its role in the virulence of M. tuberculosis, the input signals and the molecular aspects of sensing need more research. For example, it has been postulated that PhoP senses Mg2+,190 but other authors failed to reproduce this finding (C. Martin, University of Zaragoza, Spain, personal communication). Regarding the role of DosRTS in virulence, the results are inconclusive or even contradictory. Despite the current understanding of the signals sensed, a central repiratory gas, such as CO2, has not been tested as a heme ligand for these sensors.
Sigma factors
Bacteria adapt to changes in the environment and life-style mainly using RNA polymerase (RNAP) holoenzymes with different promoter specificities. The holoenzyme comprises a core RNAP with five subunits and a dissociable subunit called sigma factor (σ). Sigma factors contain many of the promoter recognition determinants that provide promoter specificity to the RNAP holoenzyme. Therefore, the association of a variety of sigma factors with the core RNAP allows the transcription of genes required for the different environmental conditions that the bacteria encounter during its cycle of life.
M. tuberculosis encodes 13 sigma factors (σA, σB, σC, σD, σE, σF, σG, σH, σI, σJ, σK, σL and σM), of which σA is the main sigma factor and the other are alternative sigma factors. Along with the alternative sigma factors, ten (σC, σD, σE, σG, σH, σI, σJ, σK, σL and σM) are members of the so-called ECF subfamily, comprising sigma factors that respond to signals from the extra-cytoplasmic environment.388
This section summarizes current knowledge regarding mycobacterial sigma factors that have been demonstrated to be essential for the full virulence of M. tuberculosis.
σA (SigA)
SigA, also known as RpoV,389 is the main sigma factor that regulates housekeeping genes and its expression may be induced in response to stress, but increasing evidence indicates that SigA also regulates the expression of virulence genes.389,390 The first evidence of this role was the finding that attenuation of a defined M. bovis mutant strain, assessed in a guinea pig model, was restored when a wild-type copy of sigA genes was introduced in this strain by electroporation. Further analysis of the sigA sequence of the M. bovis attenuated strain has shown that this mutant has a single point mutation at position 522, which caused an arginine → histidine change.389 The mutation is located in a region highly conserved among major sigma factors and their homologs, with the characteristic helix-turn-helix motif known as to probably interact with promoters.391 However, the same M. bovis sigA mutant did not show attenuation in Australian brushtail possum,392 supporting the idea that mycobacteria employ different mechanisms to replicate and survive in its hosts.
In M. tuberculosis, overexpression of sigA improves the bacterial replication in mononuclear phagocytes and in C57BL/6 mice compared with control strain.390 Knockdown of sigA in two M. tuberculosis strains negatively affected the bacterial replication inside a human macrophage cell line and in lungs of C57BL/6 mice, and elicited a minimal local inflammatory response.390 Moreover, it has been reported that SigA interacts with WhiB3, a putative transcription regulator and that the inactivation of whiB3 conferred an attenuated phenotype to a wild-type M. bovis, thus, suggesting that the SigA-WhiB3 interaction was the cause of the observed attenuation.393 However, inactivation of whiB3 in M. tuberculosis resulted in partial attenuation of its virulence,393 implying that the genetic background may define the relevance of particular genes in the virulence of pathoghenic mycobacteria.
σC (SigC)
SigC is important for pathogenesis and survival within granulomas in low-dose aerosol guinea pig infection model: sigC-mutant produced fewer and smaller lung and spleen granulomas as compared with the parental M. tuberculosis H37Rv.394 In an M. tuberculosis CDC1551 background, the mutation of sigC significantly impaired the replication of the bacilli in lungs of infected guinea pigs,394 and it dramatically reduced its lethality in DBA2 and SCID mice.394,395 However, there are reported discrepancies in the impact of sigC in the bacterial replication in immunocompetent DBA2 mice organs. While one study395 showed that the mutant replicates at the same rate as the parental strain in mice organs, another one394 reported that the infection remained between 1 to 1.5 log10 units lower than the replication of sigC-mutant three weeks post infection. In the latter study, equivalent rates of replication were detected in lung of SCID mice infected with the wild-type and mutant strains.394 Although the role of sigC in the replication of M. tuberculosis in mice needs more investigation, the role of sigC in the immunopathology induced by M. tuberculosis in animal models, in terms of both mortality and histopathologic progression of pulmonary disease, is undoubted.
In this regards, it has been shown that lack of sigC produces reduced levels of pro-inflammatory cytokines TNF-α, IL-1β, IL-6 and IFN-γ in the lungs of both SIDC and DBA2 mice and reduced level of neutrophiles, suggesting that the attenuated phenotype of the sigC mutant is associated with its inability to trigger a strong early immune response. Consistently, an M. tuberculosis H37Rv mutant in sigC did not produced necrotic granulomas in lungs and spleen of guinea pigs.396
A complete genomic microarray study has demonstrated that SigC modulates the expression of several key virulence-associated factors including a crystallin homolog hspX, a two-component sensor kinase senX3 and a two-component response regulator mtrAr.395
Altogether, these results indicate that SigC is an important regulator of virulence. Additionally, sigC toghether with sigE are the only ECF sigma factors encoded by the reduced M. leprae genome, therefore, supporting the role of sigC in the virulence of pathogenic mycobacteria.284 Moreover, sigC is only present in the genome of pathogenic mycobacteria.
σD (SigD)
The role of SigD in the virulence of M. tuberculosis has been evaluated in H37Rv and CDC1551. Mutants of sigD in both M. tuberculosis strains are able to replicate and persist in the lungs and spleen of immunocompetent mice at similar rates as those of the parental strain. However, the survival of mice infected with either of the two mutant strains is moderately improved as compared with that of the parental strain. In addition, no such longer survival is detected in immunocompromised CB17-SIDC mice infected with the mutant strain.397 Immunopathology associated to sigD is found in lungs of BALB/c mice,398 but not in those of C3H mice.397
In regards to the genes whose expression is regulated by SigD, two independent global transcriptional studies have defined different subsets of SigD-targeted genes. While the analysis of Raman et al. has identified a gene encoding the autocrine growth factor RpfC and a gene of unknown function, Rv1815, as directly being regulated by this sigma factor during exponential growth phase, Calamita et al. have reported that SigD governs the expression of a small set of ribosomal genes expressed in stationary phase during in vitro growth. In conclusion, evidence obtained so far indicates that sigD may play a role in the tuberculosis disease, but further investigation is needed to clearly establish the action of SigD in regards to the mechanism of virulence of M. tuberculosis, for instance: which genes are regulated by SigD, and in which in vivo situation these genes are expressed.
σE (SigE)
The ECF σE is one of the major regulators involved in the mycobacterial stress responses326,399,400 and it is also upregulated upon macrophage infection.229,400,401 Moreover, the stress-responsive two-component system MprAB directly regulates the expression of sigE, as well as the expression of sigB, another stress-responsive sigma factor gene in M. tuberculosis.382
A sigE mutant strain of M. tuberculosis has been independently constructed and characterized by two laboratories in the CDC1551 and H37Rv strains.402,403 The disruption of sigE in both strains resulted in delayed lethality when the strains were used to infect both immunocompetent and immunocompromised mice. However, while the replication of the mutant in H37Rv was impaired in BALB/c mouse lungs, as compared with its parental strain,403 this attenuated phenotype was not detected in C3H/HeJ mice infected with the CDC1551 mutant strain.402 This apparent discrepancy on the ability of sigE mutant to replicate and persist in mouse lungs could be explained in part by the differences in the genomic background of both the bacterial and mice strains used in these studies. In addition, the mutation of sigE impaired the growth of H37Rv in both human and mice macrophages404 and in human dendritic cells,405 suggesting that this sigma factor controls genes directly related to the intracellular survival of the bacterium. These findings are also consistent with the proposed role of sigE in the response of M. tuberculosis to oxidative stress.404
Most of the genes regulated by σE are involved in the maintenance of M. tuberculosis cell envelope integrity and lipid metabolism.406 Importantly, it has been recently reported that σE is a global regulator of the central metabolism genes of the methylcitrate cycle,407 a metabolic pathway for the assimilation of propionyl-CoA produced during catabolism of lipids.408 Because M. tuberculosis activates the lipid catabolism during the host infection, the involvement of sigE in this metabolic pathway also supports its role in the mycobacterial virulence. Moreover, the global transcriptional profile of M. tuberculosis infected macrophages has demonstrated that σE induces the expression of host genes involved in pro-inflammatory response,406 which could explain the alteration in the granuloma structure observed in mouse lungs infected with an M. tuberculosis mutant in sigE.409
σF (SigF)
It has been shown that σF is upregulated upon nutrient depletion of M. tuberculosis cultures410 and during infection of cultured human macrophages.401 Based on these and other evidence,411 σF has been defined as a stress response alternative sigma factor of Mycobacterium species. However, the viability of a mutant of sigF in M. tuberculosis CDC1551 strain was unaltered either under prolonged nutrient starvation conditions or upon macrophage infection,412 questioning the role of σF in the responses to stress conditions, at least in this mycobacterial species.
Although the mutant of sigF (mentioned above) produced a lethal infection of mice, it was less virulent than its parental strain in a time to death analysis.413 In a long-term virulence assay the mutant strain replicated in the lung and spleen of immunocompetent mice at moderate lesser rates than the parental strain.414 Moreover, at a later stage of the disease, the mutant in the CDC1551 strain showed smaller and fewer lesions and less inflammation in the lungs and spleen.414 This view is consistent with another study in which a sigF mutant of M. tuberculosis H37Rv produced diffused granulomas lacking necrosis in guinea pig lungs.396
σG (SigG)
sigG is significantly upregulated within human macrophages.229,415 It has been proposed that SigG is also involved in the SOS response of M. tuberculosis,416 but recent evidence suggests the opposite.417 Consistently with an essential role of sigG during intracellular growth of M. tuberculosis, an M. tuberculosis deleted in sigG gene displayed impaired survival in a macrophage infection model.416 Despite these findings, the relevance of sigG for the infection of M. tuberculosis in animal models remains to be demonstrated.
σH (SigH)
σH is a key regulator of the response to oxidative, nitrosative, and heat stresses in M. tuberculosis and other mycobacterial species.418-420 Microarray analysis has shown that σH regulates the expression of other sigma factors, several heat shock proteins, detoxification enzymes, virulence factors and protein processing, among others proteins.421 This sigma factor is significantly upregulated in human macrophages401 and in its absence M. tuberculosis showed impaired replication in monkey macrophages at late stage.422 In this last macrophage model, a mutant M. tuberculosis strain in the sigH gene induced the expression of numerous inducible and homeostatic β-chemokines and several apoptotic markers, suggesting a role of σH as a modulator of innate immune responses directed against M. tuberculosis.422 Moreover, an M. tuberculosis mutant deleted in sigH did not produce acute tuberculosis when tested in a non-human primate model of acute tuberculosis.423 In resistant C57BL/6 mice an M. tuberculosis CDC1551 sigH mutant showed significantly reduced lethality, comparing to the wild-type strain, but high bacterial counts in lung and spleen. In addition, the mutant produced less tissue pathology in lungs than the parental strain. In susceptible C3H mice, the mutant again showed diminished immunopathology, and the survival of mice infected with the mutant was significantly recovered as compared with that of animal infected with the wild-type strain.424 Altogether, these findings suggest that σH modulates the immune response elicited against M. tuberculosis.
σL (SigL)
σL regulates the expression of proteins involved in lipid metabolism and cell envelope, such as polyketide synthase, lipid transporters, enzymes of lipid biogenesis, etc., among other mycobacterial genes.425,426
The relevance of sigL in M. tuberculosis virulence has been addressed in two independent studies. Hahn et al. have shown that a sigL mutant of M. tuberculosis produced less lethality in BALB/c mice than its parental strain.426 On the other hand, Dainese et al. have performed a similar analysis using two different mouse strains: the resistant C57BL/6 and the susceptible DBA/2 mice strains. In these two animal models, the mutant strain replicated and persisted in organs at a similar rate to that of the parental strain. Consistent with the finding of Hahn et al., a significant extension of the survival time was reported for DBA/2 mice infected with the sigL mutant strain, comparing to that of mice infected with the parental strain.425 Therefore, the lack of sigL in M. tuberculosis results in an immunopathology defect (Imp phenotype) of virulence in mice as it has been reported for mutants in many other sigma factors.
While sigma factors B, I, J and M have shown so far to be dispensable for in vivo growth of MTBC species, mutants in the sigma factors A, C, D, E, F, G, H, K and L were attenuated at least in one infection model, showing differences in their attenuated virulence phenotypes. When tested in either mice or guinea pigs, mutants in sigma factors F, H, L and likely D have revealed an Imp phenotype, in which high tissue bacterial counts were observed but the tissue pathology and lethality were reduced. σC and σE are, to our knowledge, the only examples of a sigma factor with a role in the immunopathology of tuberculosis as well as bacterial replication in mouse organs. However, it is important to take in consideration that attenuated virulence phenotypes are sometime restricted to the animal model used. For instance, it has been recently reported that a mutant in sigH was not only severely attenuated for lethality and immunopathology but also for bacterial burden, when assayed in non-human primates.423 Little information about the role of σK and σG in the virulence of pathogenic mycobacteria is still available. While a mutant in sigG has only been addressed in a macrophage infection model, the unique evidence of σK as a virulence factor was obtained from a signature-tagged mutagenesis experiment; in which sigK gene showed to be part of a locus lost in an attenuated M. bovis strain.427
As expected, in MTBC species, virulence sigma factors regulate the expression of genes essential for a successful infection. For instance, σC regulates the expression of several key virulence-associated genes, such as hspX, senX3 and mtrA395; σD controls the expression of the Rv0169, a member of the virulence mce1 operon397 ; σE governs the expression of esat-6, sodA fbpB (ag85B), fbpC and icl;404 σF is involved in the regulation of biosynthesis and structure cell envelope and lipid metabolism, among other processes;414 the mutation of sigG in M. tuberculosis have been demonstrated to affect the expression of sigH and sigD, showing the complex interplay of sigma factors in M. tuberculosis. Similarly, under diamine stress, σH regulates the expression of several sigma factors, together with many virulence genes with a wide variety of functions, such as: mce1 operon, pirG, regulatory virulence genes, cfp10 and sodA, among others.413 Finally, the regulation of the virulence genes pks10 and pks7, both involved in fatty acid metabolism, by σL, has been demonstrated in two independent studies.425,426 In addition, it has been reported that the in vivo dispensable sigma factors M and J govern the expression of four esat-6 homologs and alternative H2O2 resistance pathway, respectively.428,429 Moreover, transcriptional analysis of an M. tuberculosis sigB mutant strain revealed regulation of the virulence gene katG, among others.430 Altogether, this evidence suggest that almost all of the sigma factors control the expression of potential virulence genes of MTBC, irrespectively their role in the bacterial virulence.
It is remarkable that, with the exception of σC, sigma factors are conserved in non-pathogenic mycobacterial species, suggesting that the regulatory systems are conserved across the Mycobacterium genus, whereas the regulon under their control varies across species.
Other transcriptional regulators
The genome of M. tuberculosis encodes more than one hundred putative transcriptional regulators (http://genolist.pasteur.fr/TubercuList/). This extensive platform of regulatory genes together with those encoding two-component systems and sigma factors would indicate that there is a fine-tuning at the level of protein expression defining the success of pathogenic mycobacteria in infection, colonization and persistence inside of hosts. The regulatory genes are classified in families: AraC, TetR, MarR, GntR, LuxR, AcrR, ArsR, LysR, AsnC and CRP/FNR among others, based on the presence of conserved domains; and depending on their effects in the transcription, they are repressors, activators or both. Members of all these subfamilies are present in the M. tuberculosis genome, but, surprisingly, few have been mutated; thus, the impact of these mutations in general physiology and virulence needs further studying.
Some regulatory genes have been mutated in pathogenic mycobacteria, such as mce3R,102 araC,431 mabR,102,432 furB and furA433,434 and a subset of them have been assessed in infection models. For instance, it has been demonstrated that the lack of Mce1R in M. tuberculosis increases the virulence of the bacteria in a mice model,435,436 likely due to overexpression of mce1 operon. Other regulatory mutants, most of them mutants in activators, are attenuated either in animal models, macrophages or both. Below, we describe the main features of this kind of attenuated regulatory mutants.
Rv0485
Rv0485 is a highly conserved gene in mycobacteria and other closely related species, which encodes a putative transcriptional regulator and a member of the NagC/XylR repressor family.22,437 The disruption of Rv0485 in M. tuberculosis leads to a reduced expression of the pe13 (Rv1195), ppe18 (Rv1196) and Rv2626c, as well as overexpression of a putative operon Rv2391 to Rv2394. Members of the PE and PPE gene families seems to be involved in different aspects of mycobacterial pathogenesis, for example, in granuloma and macrophage persistence438 acid resistance, vacuole acidification439 and induction of apoptosis and pro-inflammatory cytokine secretion.440 The Rv0485 mutant strain replicates in organs of both immunocompetent and immunocompromise (SCID) BALB/c mice at a similar rate as the wild type, but with milder lung pathology, and the survival of animals inoculated with the mutant is modestly longer than that of those inoculated with the wild type. These findings suggest that disruption of Rv0485 alters the immunomodulatory characteristics of the mutant strain rather than impacting on the growth or strain clearance.437 Since the mutation alters the expression profile of M. tuberculosis in both senses (up- and down-regulations) and the relationship among these Rv0485-regulated genes is not obvious, it is difficult to speculate about the role of this regulon in the virulence of M. tuberculosis; thus, more research is necessary to clarify the function of Rv0485.
Rv1931c
Rv1931c encodes a putative AraC transcriptional regulator. A region of Rv1931c gene has been deleted from M. tuberculosis strain 1424 genome and the resultant mutant has been tested in mice and in macrophages. The mutant strain exhibited reduced replication in mouse BMDM compared with the parental and complemented strains.441 The replication of the mutant was also impaired in lungs and spleen of mice, but the attenuated phenotype of the mutant was more consistent during chronic infection. Although there is no available information on the genes regulated by Rv1931c, altogether, these results suggest that this protein is directly or indirectly implicated in the expression of virulence factors in M. tuberculosis.
HspR
hspR (Rv0353) encodes the transcriptional repressor HspR, a member of the MerR family. In the M. tuberculosis genome, hspR is the fourth gene in an operon also comprising hsp70, grpE and dnaJ, all of them encoding heat shock proteins.
It has been experimentally demonstrated, by proteomic analysis, that HspR represses the expression of Hsp70, ClpB, GrpE and DnaJ by binding to a consensus upstream sequence known as HAIR (HspR-associated inverted repeats).442 The transcriptional analysis of an M. tuberculosis hspR deletion strain indicated that HspR controls also the transcription of Rv0251c-Rv0249c operon, which encodes Acr2, a member of the low-molecular-mass α-crystallin family.339 However, it has been later reported that the transcription of acr2 is also regulated by MprA.383 Importantly, the transcriptional regulation exerted by HspR resulted relevant for the persistence of M. tuberculosis in mice. The M. tuberculosis mutant in hspR showed reduced survival and less histological damage in C57BL/6 mouse organs than the parental strain during the chronic phase of infection. However, no complementation studies have been performed to confirm the role of HspR in these observed attenuated phenotypes. On the other hand, when assayed in BMDM, the mutant was as virulent as the wild-type strain. The authors have proposed an immune-mediated attenuation on the hspR mutant owing to overexpression of Hsp70. This is supported with their finding that an M. bovis BCG mutant in hspR induced higher production of IFNγ in splenocytes of infected mice as compared with its parental strain.214 Therefore, overexpression of Hsp70, and very likely of the other HspR-regulated proteins, seems to favor the host immune response against pathogenic mycobacteria. This hypothesis is consistent with the antigenic properties of heat-shock proteins.443,444
WhiB3
WhiB3 is one of the seven members of the WhiB-like regulator family of M. tuberculosis. The role of WhiB3 in the virulence of pathogenic Mycobacteium species has been extensively addressed. Steyn et al. have found that this regulator binds to the main sigma factor of M. tuberculosis, SigA,393 suggesting a concerted transcriptional control between both transcriptional regulators. Singh et al. have reported that WhiB3 senses redox signals through its four Fe-S cluster.445 The same authors further suggested that WhiB3 responds to the reductive stress generated by host lipid catabolism by controlling the expression of pks2, pks3 and fbpA genes, which are involved in the synthesis of the methyl-branched lipids PAT, DAT, SL-1 and TMM/TDM.446 Consistently, an M. tuberculosis mutant in whiB3 showed alteration in the production of these methyl-branched lipids, as well as PDIMs and triacylglycerol (TAG), both in vitro under defined oxidizing and reducing condition and inside macrophages. Therefore, the authors have proposed that under intracellular oxidative or reductive stress, M. tuberculosis modulates the anabolism of diverse polyketides to maintain the redox homeostasis. WhiB3 would participate in this process by channeling toxic reducing equivalents, produced as a result of host lipid catabolism, into bacterial lipid anabolism. The absence of whiB3 in M. tuberculosis induced the production of pro- and anti-inflammatory cytokines by macrophages, without modifying its intracellular replication. It is most probable that this modulation of macrophage innate immune response is due to the lack of WhiB3-mediated regulation of complex lipids that M. tuberculosis uses to suppress the host immune responses. Therefore, it is tempting to speculate that WhiB3 contributes to the virulence of pathogenic mycobacteria by indirect modulation of the host immune response.446
Remarkably, the mutation of whiB3 in M. bovis significantly impaired the bacterial replication in spleen of guinea pigs, and this attenuated phenotype is reverted in the complemented strain.393 Taking into consideration that some methyl-branched lipids are not produced by M. bovis, it is plausible that the lipid regulation mediated by WhiB3 has a more essential role in M. bovis for modulating the host immune response in its favor.
In another study, Banaiee et al. have demonstrated that the lack of WhiB3 in M. tuberculosis H37Rv did not alter the replication of the bacteria in C57BL/6 mouse or guinea pig organs, but extended the survival of infected imunocompentent mice as compared with the wild-type strain.447 However, the time to death curve of immunodeficient IFN-γR−/− mice infected with an M. tuberculosis whiB3 mutant was equivalent to that of mice infected with the parental strain. Although these results appear to be contradictory, it is important to take into consideration the differential immunological features of the two models used in these studies.
MosR (Rv0348)
MosR (named derived from regulator of mycobacterial operons of survival) regulates the transcription of several operons and regulons, either by repression or activation, including those involved in mammalian cell entry (mce1), hypoxia (tgs1) and starvation. In fact, by means of global transcriptional examination, it has been found that MosR indirectly regulates the transcription of 163 genes. Consistent with the high number of MosR-regulated genes, the analysis of M. tuberculosis transcriptional regulatory network indicates that MosR is the most connected hub of this network.448
Disruption of the mosR gene from the genome of M. tuberculosis H37Rv impairs the replication of the bacteria in lungs of BALB/c mice, but this attenuated phenotype is more evident after 30 weeks post-infection, suggesting a critical role for mosR in the chronic phase of tuberculosis. In addition, the mutant strain is less lethal to BALB/c mice as compared with the wild-type strain. The reintroduction of a wild-type copy of mosR into the mutant produces a partial complementation in vivo.449 Although the heterologous expression of M. tuberculosis mosR gene in M. smegmatis would indicate that MosR participates in anaerobic responses,449 the precise role of this global regulator in the virulence of pathogenic mycobacteria is still unclear.
VirS
VirS of M. tuberculosis belongs to the AraC family of transcriptional regulators (AraC/Xyls) and controls the transcription of the mymA operon, which is encoded divergently to virS.450 In turn, VirS is phosphorylated by the serine/threonin kinase PknK, and this phosphorylation increases the affinity for the mymA promoter.298 M. tuberculosis mutants in either virS or mymA operon exhibited reduced contents and altered composition of mycolic acids together with the accumulation of saturated fatty acids as compared with the parental strain. These mutants were more susceptible to major anti-tubercular drugs at acidic pH and also showed increased sensitivity to detergent and to acidic stress than the parental strain.
The disruption of virS resulted in an impaired replication of M. tuberculosis in spleen of guinea pigs at chronic phase of infection, but not in lungs as compared with the complemented and wild-type strains. Moreover, this reduced virulence for virS mutant strain was also detected upon infection of activated JJ74 murine macrophage cell line but not in resting macrophages, indicating a role of VirS in the response to oxidative burst. Importantly, the mutant in the mymA operon showed the same attenuated phenotype observed for virS mutant.32 The role of the mymA operon in the intracellular replication and persistence of M. tuberculosis has been further studied in many in vitro models. For instance, an M. tuberculosis mutant deleted in mymA operon failed to grow in human THP-1 cells, pneumocyte cells (A549) and murine J774 macrophage cells451 as compared with the complemented and wild-type strains.
Altogether, these results indicate that the expression of the mymA operon, activated by VirS, is required for maintaining the appropriate mycolic acid composition and bacterial wall permeability. Each of these postulated biologic roles is consistent with the attenuation observed for virS and mymA mutants.
PhoY2
phoY2 encodes a probable phosphate-transport system transcriptional regulatory protein. However, the role of PhoY2 as a transcriptional regulator has not been assessed yet. It has been demonstrated that its E. coli homolog, PhoU, is a global repressor for cellular metabolism involved in the generation of persistent bacteria.452 Remarkably, the disruption of phoY2 in M. tuberculosis H37Rv increased its susceptibility to rifampicin and pyrazinamide. Moreover, the mutant was less capable to survive and persist in mouse lungs and spleens than the complemented and parental strains.453 These observed phenotypes for the phoY2 mutant suggest that PhoY2 may have a role in the generation of persistent M. tuberculosis. However, further studies are needed for deciphering the function of this putative regulator, as well as for identifying its target genes.
Proteins of Unknown Function
The advent of the genomic era has allowed sequencing the genome of 12 species of Mycobacterium. This, added to the studies in the transcriptomic, proteomic, metabolomic and bioinformatic fields have allowed to know or to infer the biological function of many ORFs. Despite this, the physiological function of many proteins related to Mycobacterium virulence remains unknown.
Below, we describe MTBC proteins of unknown function, in which their involvement in bacterial virulence has been demonstrated.
The PE/PPE families
The PE/PPE families have been found only in mycobacteria and consist of two large unrelated families of acidic and glycine-rich proteins whose genes are clustered.22 About 10% of the coding capacity of M. tuberculosis H37Rv genome consists of pe and ppe genes. Many of these genes are located upstream or within the ESX operons. The PE name derives from the signature motif Pro-Glu, at the residues 8 and 9 on the N-terminus, whereas the PPE represents the Pro-Pro-Glu motif. Most of these proteins are localized on the cell surface and/or are secreted, and they induce a strong immune response in the host. The N-terminus of these proteins is well conserved and is linked to a variable C-terminus, which is thought to be a source of antigenic and genetic variations.454-456 The high level of polymorphism among members of these protein families suggests a role in the mechanism of host immune-response evasion, even though a wide variety of functions have also been assigned to them.457 However, whether or not these PE/PPE proteins modulate the host’s immune system in favor of the bacilli is still uncertain, since only few PE/PPE proteins have shown to be certainly relevant for the virulence of MTBC. Some of these virulence associated-PE/PPE proteins are encoded in the ESX-1 locus, which is detailed in the section on Secretion Systems; the PE_PGRS30 protein, which belongs to the PE family is detailed in “Phagosome arresting” and the rest are described below.
PE_PGRS33 is a cell surface protein encoded by the Rv1818c gene that promotes mycobacterial aggregation.458,459 This aggregative phenotype was observed in a BCG mutant for the Rv1818c gene, which was unable to form typical large cell aggregates when grown in liquid media but rather showed a dispersed growth phenotype, suggesting a role of this protein in cell surface structure. The role of this protein in mycobacteria virulence was studied in a macrophage model. Murine J774 macrophage cells were infected with the BCG mutant and its replication was significantly reduced as compared with the parental and complemented strains.194 Similarly, PE_PGRS51, encoded by Rv3367, and PPE46, encoded by Rv3018, have shown to be essential for full replication of M. tuberculosis mutants in mouse organs. M. tuberculosis H37Rv mutant in the Rv3367 gene replicated less than its wild-type strain in lung and spleen of C57BL/6J mice100 and an M. tuberculosis strain MT103 mutant in Rv3018 have a reduced multiplication rate in the lung of BALB/c mice compared with wild type.42 Although the precise mechanism by which these virulence associated PE/PPE proteins are involved in the survival and replication of MTBC in their host is still uncertain. The findings here described highlight the role of these exclusive proteins in the virulence of mycobacteria.
Other proteins with unknown functions
By means of TraSH technique, it has been demonstrated that Rv1099c and Rv0573c, two proteins predicted as conserved hypothetical proteins (CHP), are required for M. tuberculosis survival during infection in a mouse model of tuberculosis.100 Individual mutants in each gene were constructed and used to infect C57BL/6J mice. In fact, mutant growth in lung and spleen did indeed show a significant defect compared with that of the parental strain, indicating that these proteins have a role in virulence but that their mechanisms are still unknown.100
The Rv0204c and Rv2452c proteins are predicted as an integral membrane protein and a hypothetical protein, respectively. None of them show any sequence similarity to entries in the protein database. Mutant strains in both genes generated in M. tuberculosis strain MT103 background have shown reduced multiplication in the lung of BALB/c mice compared with wild type.42
The Rv1290c, Rv1891 and Rv3404c proteins are described as CHP. CB-17/Icr SCID mice infected with M. tuberculosis H37Rv mutants for Rv1290c, Rv1891 or Rv3404c presented a highly significant increase in their survival time as compared with mice infected with the parental strain.460 For instance, the survival time of the mice infected with the mutant in the Rv1290c gene increased from a median of 25 to 62 d.460 This strong in vivo attenuation for this mutant clearly shows that Rv1290c has an important function in determining the extent of the virulence of M. tuberculosis.
The Rv1503c to Rv1506c proteins, encoded by the Rv1503c to Rv1506c operon, are also predicted as CHP. M. tuberculosis transposon mutants in the Rv1503c or Rv1506c genes fail to transcribe the entire Rv1503c to Rv1506c operon. These mutants grew poorly inside host macrophages and their ability to infect BALB/c mice was strongly impaired, since the CFU counts from spleen and lungs were markedly reduced as compared with the parental. The attenuated phenotype was restored to wild-type levels in a complemented strain carrying the entire operon.461 Interestingly, the absence of the Rv1503c to Rv1506c proteins in the mutants lead to a noticeable overproduction of tetracylated sulphoglycolipids (Ac4SGL), and to lower production of 2,3-di-O-acyltrehaloses as compared with the parental or complemented strains.461 This imbalance could indicate that these proteins play a role in glycolipid metabolism and this may account, at least in part, for the attenuated phenotype observed in mutants.
Rv0199 is a probable conserved membrane protein with homology to some Mce-associated proteins.101 It has been predicted to have a transmembrane domain at the N-terminus, the majority of which is located on the extra-cytoplasmic side of the membrane.119 The Rv0199 gene is located in the virulence-associated membrane proteins (VAMP) region, together with 10 genes that encode for VAMP.101 While the function of Rv0199 is still unknown, its role in the mycobacteria virulence has been already demonstrated in a TraSH analysis performed in a mouse model of infection.100 Furthermore, an M. tuberculosis mutant by a transposon insertion in the Rv0199 gene was defective for growth in macrophages as compared with the wild type, and this intracellular growth defect was complemented by addition of a plasmid expressing Rv0199.119
MmpL4 is predicted as a probable conserved transmembrane transport protein. It belongs to the MmpL family of proteins (see “Lipid and Fatty Acid Metabolism”) but, unlike them, a role in fatty acid transport has not been yet reported. An M. tuberculosis H37Rv mutant in mmpL4 used to infect C57/BL6 mice was significantly impaired for growth as compared with the parental strain.45 The mutant strain is so attenuated that the same phenotype was observed in a more susceptible mice model of tuberculosis, a (C57BL/6xDBA2) F1 hybrid mouse race. Although the mutation in the mmpL4 strain has not been complemented, the expression of the genes around mmpL4 has been evaluated by an RT-PCR experiment demonstrating that the insertion of the hyg cassette did not cause a polar effect.45 Thus, the attenuated phenotype is likely due to the absence of MmpL4.
Finally, Rv2136c is predicted as a possible conserved transmembrane protein of unknown function with a possible role in the virulence of M. tuberculosis. An M. tuberculosis transposon mutant in Rv2136c was hypersensitive to acid pH, SDS, heat shock, reactive oxygen and nitrogen intermediates and to lipophilic (rifampicin or ethanbutol) or non-lipophilic (isoniazid) antibiotics.114 The mutant was unable to maintain a neutral cytoplasmic pH in activated macrophages and its growth and survival in C57BL/6 mice was severely attenuated in lungs and spleen as compared with the wild type.114 These results suggest an important role for this protein in the virulence of the bacteria. However, the lack of complementation of the mutant phenotype with either the single Rv2136c gene or the putative operon (Rv2133 to Rv2137c) could indicate a possible alternative mutation as the cause of the defects observed.
In conclusion, just over half (52%) of the open reading frames in the M. tuberculosis genome have been assigned a biological function, while the rest of the ORFs are annotated as hypothetical proteins of unknown function.462 The PE/PPE family of proteins are the most relevant in this group; despite that their specific function remains unknown, they have been linked with the virulence of mycobacteria as a source for antigenic biodiversity and as modulators of the host immune response.454,456 However, almost half of the ORFs in the mycobacteria genome remain as uncharacterized proteins, and most of these proteins have no homology with other and in many cases are unique and play a specific role in the organism. Therefore, our understanding on the pathogenicity and virulence of mycobacteria is greatly reduced. Moreover, in the current era of informatics, the in silico analyses are essential and the use of tools such as BLAST, PFAM, COG, among others, have allowed us to predict the function of proteins described as yet hypothetical based on the degree of homology they share with those whose function have been determined by biochemical and/or molecular studies. Therefore, it has been possible to predict the function of 12% of the hypothetical proteins in the M. tuberculosis genome, with a confidence greater than 75%.463,464 Our challenge now is to integrate the large amount of data resulting from the sequencing of genomes with the annotation of biological function for each ORF and, thus, be able to build the puzzle which involves the understanding of the biology of the organisms in order to have the proper artillery to control the development of pathogenic bacteria.
Other Virulence Proteins
Regions of differences (RD)
Only RD1 and RD2 have been demonstrated to be essential for the full virulence of the MTBC species among the already known region of differences. RD1 has been already described in the previous section.
RD2 is abstent in some BCG sub-strains. The deletion in BCG goes from Rv1978 to Rv1988 and comprises 12 genes. In M. pinnipedi there is a deletion of 1.94 kb involving Rv1978 and Rv1979c. In addition, some M. microtti strains have a deletion going from yrb3A (Rv1964) to Rv1979c.465 Little is known about the biological function of the proteins encoded in this region apart from the fact that many of the proteins encoded here have been shown to be antigenic. A putative amino acid efflux pump gene, lysE (Rv1986), from M. tuberculosis, together with the divergently transcribed putative lysR-type regulator gene (Rv1985c), is encoded in RD2.466 Rv1987 is a putative cellulose-targeting protein. Rv1982c is predicted to encode a toxin-antitoxin of the VapBC family; this family of proteins has been shown to inhibit translation through RNase activity.185 RD2 was disrupted in M. tuberculosis H37Rv to test whether its loss might contribute to the attenuation of BCG.467 The deletion of RD2 did not affect in vitro growth; in contrast, the mutant manifested a decrease in pulmonary and splenic bacterial burdens and reduced pathology in C57BL/6 mice at early time points. This attenuated phenotype was complemented by reintroducing the genes Rv1979c to Rv1982 (including mpt64), but not Rv1985c to Rv1986. Rv1983 and Rv1984 have been naturally deleted in a clinical strain of M. tuberculosis, demonstrating that these genes are dispensable for full virulence in humans.468 In RAW264.7 macrophages, the mutant H37RvΔRD2 has showen a decreased proliferation and impaired modulation of the host innate immune response; both observed phenotypes were complemented with Rv1979c to Rv1982 genes.467
The RD2-containing BCG Russia, BCG Pasteur (which has a natural deletion of RD2) and a BCG Pasteur strain complemented with RD2 genes Rv1979c-Rv1982 have been compared through various in vitro and in vivo assays for immunogenicity and protection. In a mouse vaccine-challenge model, the presence of RD2 displayed no effect on pulmonary TB, as measured by M. tuberculosis burden and degree of histopathology, until 12 weeks post-infection. RD2 deletion was, however, associated with decreased dissemination of M. tuberculosis to the spleen. The data demonstrate that the loss of RD2 resulted in decreased immunogenicity but did not affect protection against pulmonary TB, indicating dissociation between these phenotypes commonly associated with BCG vaccination.469 Rv1980 encodes for Mpt64, a well-known antigen and exported protein containing an N-terminal signal sequence whose function is presently unknown. Surprisingly, no attempts to mutate mpt64 gene to assess the impact on virulence have been published so far.
Acg
In M. tuberculosis, acg transcription is controlled by the TCS DosR-DosS in response to hypoxia and nitric oxide conditions within macrophages and mice.470 Acg had been first proposed as a member of a superfamily of classical nitroreductases.471 However, Hu et al. have shown that the deletion of acg from M. tuberculosis does not affect bacterium growth and survival in acidic, nitric oxide and hydrogen peroxide environments in vitro.472 Besides, this mutant was attenuated in macrophages and in acute and persistent murine infection models.472 The replication of the acg mutant strain was significantly impaired in both resting and activated BMDM. In addition, BALB/c mice infected with the mutant strain showed significant lower bacillary load in organs and longer survival than its parental strain and less cytokine production in lungs (TNF-α, IL6 and IL-1b at later times). Although the authors of this study have not addressed the impact of the acg mutation in the in vitro culture of M. tuberculosis, it is plausible to speculate that this mutation has only affected the in vivo growth of the bacilli, since it has been possible to obtain the mutant in standard conditions in vitro growth.
PckA
pckA encodes a phosphoenolpyruvate carboxykinase that catalyzes the reversible decarboxylation and phosphorylation of oxaloacetate (OAA) to form phosphoenolpyruvate (PEP) or the reverse reaction. It has been demonstrated that the expression of pckA is upregulated in the presence of either palmitate or acetate. The pckA disruption in M. bovis BCG results in attenuation of the obtained mutant strain in macrophages and mice. The mutant strain replicates less in both spleen of BALBc mice and resting murine BMDM than its parental strain.473 The authors have postulated that the attenuation observed shortly after infection with the mutant in pckA is due to a reduced capacity to respond to the changing environment within the macrophage, possibly when OAA is needed to fulfil an anaplerotic role in the tricarboxylic acid cycle.
PtpB
Reversible phosphorylation of proteins by protein kinases and phosphatases is a major mechanism of signal transduction events that regulate several cellular processes. The M. tuberculosis genome encodes two tyrosine phosphatase, ptpA (Rv2234) and ptpB (Rv0153c),474 but, surprisingly, it apparently does not encode any tyrosine kinase. Both tyrosine phosphatases have been shown to be important for the virulence of M. tuberculosis. It has been reported that PtpA is relevant for the arrestment of phagosomal maturation by M. tuberculosis (see “Phagosome arresting”), and thus, to survival inside macrophages. However, this phosphatase seems to be nonessential for bacterial in vivo replication. On the other hand, an M. tuberculosis mutant in ptpB was less persistent in guinea pig spleen than wild-type and complemented strains. Moreover, animals infected with this mutant showed a reduction in the percentage of granuloma in liver and lung and a highly lymphocytic granuloma when compared with the wild-type and complemented strains. The authors of this study have suggested that the histological phenotype observed in guinea pigs infected with the ptpB mutant is consistent with an infection controlled by the host immune response. The mutant has also displayed a moderated attenuation in IFNγ activated murine J774 macrophages,475 but not in resting macrophages. These findings highlight the role of tyrosine phosphatase in the pathogenesis of M. tuberculosis.
Hsp22.5
The study of Talaat et al. describes an M. tuberculosis genomic region containing about 20 genes highly expressed in BALB/c mice. Among these, there is an in vivo upregulated gene, Rv0990c.381 As the expression of Rv0990c has been also shown to be upregulated at high temperature339 and under treatment with H2O2 and SDS, this protein (named as Hsp22.5) has been proposed as a novel heat shock protein. The deletion of Rv0990c from M. tuberculosis H37Rv genome affects the replication of the bacteria in lungs of BALB/c mice during chronic phase of the disease. However, this attenuation is not detected in acute infection of guinea pigs. In addition, the survival of mice infected with the mutant strains is longer than that of the wild type. The introduction of a wild-type copy of Rv0990c in the mutant strain partially complements the lethal phenotype of M. tuberculosis.476 These results, together with the findings that the mutation of hspX and acr2 genes results in alteration of the M. tuberculosis virulence, highlight the role of heat shock proteins during the infection of pathogenic mycobacteria. Although the function of Hsp22.5 is still unclear, expression profile studies have shown that the absence of this protein in M. tuberculosis affects the expression of a large number of genes, among them several heat shock proteins.476
Concluding Remarks
Global searches have allowed the identification of more than a hundred potential virulence genes in pathogenic mycobacteria. A pioneering work in this field was that of Camacho et al., who used STM to identify M. tuberculosis mutants exhibiting an attenuated phenotype in large pools of mutants.42 Remarkably, most of the genes identified in that study have been implicated in lipid metabolism. Later on, the outstanding work of Sassetti and Rubin100 allowed the definition of 194 genes as specifically required for mycobacterial growth in mice by using the TraSH methodology. Again, lipid metabolic genes as well as those involved in the transport or metabolism of inorganic ions and carbohydrates were prominently represented among the genes required for in vivo growth. Although data from these high-throughput screenings have been considered in this review, the main focus is mainly on those genes whose role in virulence has been individually demonstrated. Remarkable, more than 20% of these loci have also been identified as virulence factors using the global virulence analysis of Sassetti and Rubin.100
The virulence factors described in this review are mainly involved in the interaction of MTBC species with the host macrophages: One set of these virulence factors is implicated in the adaptation of the bacilli to the limited nutritional condition of the macrophages and includes proteins required for the uptake of nutrients and ions as well as for the switching of carbon metabolism that occurs when mycobacteria reside inside host cells. Another set comprises proteins that participate in the mechanisms triggered by mycobacteria to counteract the microbicidal host cell responses, such as: (1) arresting the normal progression of the phagosome and increasing the resistance to host toxic compounds (cell wall barrier and specific effectors), (2) escaping from the intracellular compartment and (3) avoiding the development of localized, productive immune responses.
An additional category of virulence factors is that encompassing proteins with a role in the modulation of host immune responses. Among them, those implicated in the inhibition of inflammatory responses and apoptosis are highly represented. Mutant strains in genes encoding these virulence proteins have been shown to induce poor tissue damage and, in some cases, wild type levels of bacteria replication in organs.
The thorough description of M. tuberculosis virulence factors summarized in this review is expected to contribute to a better understanding of the mechanisms involved in the interaction of pathogenic mycobacteria with their hosts. This information is, thus, essential for the development of new treatments and vaccines that should help prevent or control this pandemic. However, it is important to take into consideration the words of Smith, “TB will be completely eradicated only when poverty and unequal development are ended throughout the world.”7
Acknowledgments
The authors are grateful to Dr. Rodolfo Biekofsky for critical reading of the manuscript. This study was supported by grant NIH/NIAID 1R01AI083084-03. M.P.S., A.A.C. and F.B. are CONICET fellows. H.R.M. is a member of the Research Council, University of Rosario (CIUNR).
Glossary
Abbreviations:
- ABC-transporter
ATP-binding cassette transporter
- BMDM
bone marrow-derived macrophages
- CFU
colony-forming unit
- DAT
di-acylated trehalose
- DC
dendritic cell
- ECF
extracytoplasmic function
- IFN-γ
gamma interferon
- LAM
lipoarabinomannane
- Lpp
lipoprotein
- MAMTs
mycolic acid methyl transferases
- MHC-II
major histocompatibility complex class II
- MTBC
Mycobacterium tuberculosis complex
- ORF
open reading frame
- PAT
poly-acylated trehalose
- PDIM
phthiocerol dimycocerosate
- PGL
phenolic glycolipid
- p-HBADs
p-hydroxybenzoic acid derivatives
- PIM
phosphatidylinositol mannoside
- RD
region of difference
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SL
sulfolipid
- STM
signature-tagged mutagenesis
- TAG
triacylglycerides
- TAT
tri-acylated trehalose
- TB
tuberculosis
- TCS
two-component system
- TLR2
Toll-like receptor 2
- TDM
trehalose di-mycolates
- TMM
trehalose mono-mycolates
- TNF-α
tumor necrosis factor-alpha
- TraSH
transposon site hybridization
- T7SS
type VII secretion system
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/virulence/article/22329
References
- 1.Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niemann S, Richter E, Rüsch-Gerdes S. Differentiation among members of the Mycobacterium tuberculosis complex by molecular and biochemical features: evidence for two pyrazinamide-susceptible subtypes of M. bovis. J Clin Microbiol. 2000;38:152–7. doi: 10.1128/jcm.38.1.152-157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cousins DV, Bastida R, Cataldi A, Quse V, Redrobe S, Dow S, et al. Tuberculosis in seals caused by a novel member of the Mycobacterium tuberculosis complex: Mycobacterium pinnipedii sp. nov. Int J Syst Evol Microbiol. 2003;53:1305–14. doi: 10.1099/ijs.0.02401-0. [DOI] [PubMed] [Google Scholar]
- 4.Aranaz A, Cousins D, Mateos A, Domínguez L. Elevation of Mycobacterium tuberculosis subsp. caprae Aranaz et al. 1999 to species rank as Mycobacterium caprae comb. nov., sp. nov. Int J Syst Evol Microbiol. 2003;53:1785–9. doi: 10.1099/ijs.0.02532-0. [DOI] [PubMed] [Google Scholar]
- 5.Meena LS, Rajni Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. 2010;277:2416–27. doi: 10.1111/j.1742-4658.2010.07666.x. [DOI] [PubMed] [Google Scholar]
- 6.Saunders BM, Frank AA, Orme IM. Granuloma formation is required to contain bacillus growth and delay mortality in mice chronically infected with Mycobacterium tuberculosis. Immunology. 1999;98:324–8. doi: 10.1046/j.1365-2567.1999.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. 2003;16:463–96. doi: 10.1128/CMR.16.3.463-496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins DM. Virulence factors of Mycobacterium bovis. Tuberculosis (Edinb) 2001;81:97–102. doi: 10.1054/tube.2000.0255. [DOI] [PubMed] [Google Scholar]
- 9.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–8. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenreich W, Dandekar T, Heesemann J, Goebel W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol. 2010;8:401–12. doi: 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- 11.Cardona PJ, Ruiz-Manzano J. On the nature of Mycobacterium tuberculosis-latent bacilli. Eur Respir J. 2004;24:1044–51. doi: 10.1183/09031936.04.00072604. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y. Persistent and dormant tubercle bacilli and latent tuberculosis. Front Biosci. 2004;9:1136–56. doi: 10.2741/1291. [DOI] [PubMed] [Google Scholar]
- 13.Zhao QJ, Xie JP. Mycobacterium tuberculosis proteases and implications for new antibiotics against tuberculosis. Crit Rev Eukaryot Gene Expr. 2011;21:347–61. doi: 10.1615/CritRevEukarGeneExpr.v21.i4.50. [DOI] [PubMed] [Google Scholar]
- 14.Barry CE., 3rd Interpreting cell wall ‘virulence factors’ of Mycobacterium tuberculosis. Trends Microbiol. 2001;9:237–41. doi: 10.1016/S0966-842X(01)02018-2. [DOI] [PubMed] [Google Scholar]
- 15.Guenin-Macé L, Siméone R, Demangel C. Lipids of pathogenic Mycobacteria: contributions to virulence and host immune suppression. Transbound Emerg Dis. 2009;56:255–68. doi: 10.1111/j.1865-1682.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- 16.Hotter GS, Collins DM. Mycobacterium bovis lipids: virulence and vaccines. Vet Microbiol. 2011;151:91–8. doi: 10.1016/j.vetmic.2011.02.030. [DOI] [PubMed] [Google Scholar]
- 17.Mehrotra J, Bishai WR. Regulation of virulence genes in Mycobacterium tuberculosis. Int J Med Microbiol. 2001;291:171–82. doi: 10.1078/1438-4221-00113. [DOI] [PubMed] [Google Scholar]
- 18.Sachdeva P, Misra R, Tyagi AK, Singh Y. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. FEBS J. 2010;277:605–26. doi: 10.1111/j.1742-4658.2009.07479.x. [DOI] [PubMed] [Google Scholar]
- 19.Brodin P, Rosenkrands I, Andersen P, Cole ST, Brosch R. ESAT-6 proteins: protective antigens and virulence factors? Trends Microbiol. 2004;12:500–8. doi: 10.1016/j.tim.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Simeone R, Bottai D, Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Converse SE, Mougous JD, Leavell MD, Leary JA, Bertozzi CR, Cox JS. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc Natl Acad Sci U S A. 2003;100:6121–6. doi: 10.1073/pnas.1030024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 23.Quémard A, Lanéelle G, Lacave C. Mycolic acid synthesis: a target for ethionamide in mycobacteria? Antimicrob Agents Chemother. 1992;36:1316–21. doi: 10.1128/AAC.36.6.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilchèze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, et al. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J Bacteriol. 2000;182:4059–67. doi: 10.1128/JB.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, et al. Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci U S A. 2007;104:5157–62. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubnau E, Chan J, Raynaud C, Mohan VP, Lanéelle MA, Yu K, et al. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol Microbiol. 2000;36:630–7. doi: 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- 27.Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–27. doi: 10.1016/S1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 28.Rao V, Fujiwara N, Porcelli SA, Glickman MS. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. 2005;201:535–43. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkan D, Hedhli D, Yan HG, Huygen K, Glickman MS. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperinflammatory in mice. Infect Immun. 2012;80:1958–68. doi: 10.1128/IAI.00021-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–2. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 31.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun. 2000;68:767–78. doi: 10.1128/IAI.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh A, Gupta R, Vishwakarma RA, Narayanan PR, Paramasivan CN, Ramanathan VD, et al. Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J Bacteriol. 2005;187:4173–86. doi: 10.1128/JB.187.12.4173-4186.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makinoshima H, Glickman MS. Regulation of Mycobacterium tuberculosis cell envelope composition and virulence by intramembrane proteolysis. Nature. 2005;436:406–9. doi: 10.1038/nature03713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy HN, Stewart GR, Mischenko VV, Apt AS, Harris R, McAlister MS, et al. The OtsAB pathway is essential for trehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2005;280:14524–9. doi: 10.1074/jbc.M414232200. [DOI] [PubMed] [Google Scholar]
- 35.Kolattukudy PE, Fernandes ND, Azad AK, Fitzmaurice AM, Sirakova TD. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol Microbiol. 1997;24:263–70. doi: 10.1046/j.1365-2958.1997.3361705.x. [DOI] [PubMed] [Google Scholar]
- 36.Sirakova TD, Dubey VS, Cynamon MH, Kolattukudy PE. Attenuation of Mycobacterium tuberculosis by disruption of a mas-like gene or a chalcone synthase-like gene, which causes deficiency in dimycocerosyl phthiocerol synthesis. J Bacteriol. 2003;185:2999–3008. doi: 10.1128/JB.185.10.2999-3008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–7. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 38.Tsenova L, Ellison E, Harbacheuski R, Moreira AL, Kurepina N, Reed MB, et al. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis. 2005;192:98–106. doi: 10.1086/430614. [DOI] [PubMed] [Google Scholar]
- 39.Sirakova TD, Dubey VS, Kim HJ, Cynamon MH, Kolattukudy PE. The largest open reading frame (pks12) in the Mycobacterium tuberculosis genome is involved in pathogenesis and dimycocerosyl phthiocerol synthesis. Infect Immun. 2003;71:3794–801. doi: 10.1128/IAI.71.7.3794-3801.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azad AK, Sirakova TD, Fernandes ND, Kolattukudy PE. Gene knockout reveals a novel gene cluster for the synthesis of a class of cell wall lipids unique to pathogenic mycobacteria. J Biol Chem. 1997;272:16741–5. doi: 10.1074/jbc.272.27.16741. [DOI] [PubMed] [Google Scholar]
- 41.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 42.Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34:257–67. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 43.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, et al. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001;276:19845–54. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 44.Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Avé P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. 2004;6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 45.Domenech P, Reed MB, Barry CE., 3rd Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. 2005;73:3492–501. doi: 10.1128/IAI.73.6.3492-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rousseau C, Sirakova TD, Dubey VS, Bordat Y, Kolattukudy PE, Gicquel B, et al. Virulence attenuation of two Mas-like polyketide synthase mutants of Mycobacterium tuberculosis. Microbiology. 2003;149:1837–47. doi: 10.1099/mic.0.26278-0. [DOI] [PubMed] [Google Scholar]
- 47.Astarie-Dequeker C, Le Guyader L, Malaga W, Seaphanh FK, Chalut C, Lopez A, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 2009;5:e1000289. doi: 10.1371/journal.ppat.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goren MB, D’Arcy Hart P, Young MR, Armstrong JA. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1976;73:2510–4. doi: 10.1073/pnas.73.7.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Goren MB, Holzer TJ, Andersen BR. Effect of Mycobacterium tuberculosis-derived sulfolipid I on human phagocytic cells. Infect Immun. 1988;56:2876–83. doi: 10.1128/iai.56.11.2876-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mougous JD, Petzold CJ, Senaratne RH, Lee DH, Akey DL, Lin FL, et al. Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nat Struct Mol Biol. 2004;11:721–9. doi: 10.1038/nsmb802. [DOI] [PubMed] [Google Scholar]
- 51.Seeliger JC, Holsclaw CM, Schelle MW, Botyanszki Z, Gilmore SA, Tully SE, et al. Elucidation and chemical modulation of sulfolipid-1 biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2012;287:7990–8000. doi: 10.1074/jbc.M111.315473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sirakova TD, Thirumala AK, Dubey VS, Sprecher H, Kolattukudy PE. The Mycobacterium tuberculosis pks2 gene encodes the synthase for the hepta- and octamethyl-branched fatty acids required for sulfolipid synthesis. J Biol Chem. 2001;276:16833–9. doi: 10.1074/jbc.M011468200. [DOI] [PubMed] [Google Scholar]
- 53.Rousseau C, Turner OC, Rush E, Bordat Y, Sirakova TD, Kolattukudy PE, et al. Sulfolipid deficiency does not affect the virulence of Mycobacterium tuberculosis H37Rv in mice and guinea pigs. Infect Immun. 2003;71:4684–90. doi: 10.1128/IAI.71.8.4684-4690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Domenech P, Reed MB, Dowd CS, Manca C, Kaplan G, Barry CE., 3rd The role of MmpL8 in sulfatide biogenesis and virulence of Mycobacterium tuberculosis. J Biol Chem. 2004;279:21257–65. doi: 10.1074/jbc.M400324200. [DOI] [PubMed] [Google Scholar]
- 55.Rindi L, Fattorini L, Bonanni D, Iona E, Freer G, Tan D, et al. Involvement of the fadD33 gene in the growth of Mycobacterium tuberculosis in the liver of BALB/c mice. Microbiology. 2002;148:3873–80. doi: 10.1099/00221287-148-12-3873. [DOI] [PubMed] [Google Scholar]
- 56.Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72:132–41. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKinney JD, Höner zu Bentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–8. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 58.Muñoz-Elías EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–44. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc Natl Acad Sci U S A. 2003;100:241–6. doi: 10.1073/pnas.0136863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raynaud C, Guilhot C, Rauzier J, Bordat Y, Pelicic V, Manganelli R, et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol Microbiol. 2002;45:203–17. doi: 10.1046/j.1365-2958.2002.03009.x. [DOI] [PubMed] [Google Scholar]
- 61.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–80. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miner MD, Chang JC, Pandey AK, Sassetti CM, Sherman DR. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol. 2009;47:407–11. [PubMed] [Google Scholar]
- 63.Yang X, Gao J, Smith I, Dubnau E, Sampson NS. Cholesterol is not an essential source of nutrition for Mycobacterium tuberculosis during infection. J Bacteriol. 2011;193:1473–6. doi: 10.1128/JB.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brzostek A, Dziadek B, Rumijowska-Galewicz A, Pawelczyk J, Dziadek J. Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2007;275:106–12. doi: 10.1111/j.1574-6968.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- 65.Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A. 2006;103:11760–5. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, et al. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–9. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bottasso O, Bay ML, Besedovsky H, del Rey A. Immunoendocrine alterations during human tuberculosis as an integrated view of disease pathology. Neuroimmunomodulation. 2009;16:68–77. doi: 10.1159/000180261. [DOI] [PubMed] [Google Scholar]
- 69.Goren MB, Brennan PJ. Mycobacterial lipids: chemistry and biologic activities. In G P Youmans (ed), Tuberculosis W B Saunders Company, Philadelphia, PA 1979:63-193. [Google Scholar]
- 70.Neyrolles O, Guilhot C. Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis (Edinb) 2011;91:187–95. doi: 10.1016/j.tube.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 71.Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med. 1971;134:713–40. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–77. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 73.Axelrod S, Oschkinat H, Enders J, Schlegel B, Brinkmann V, Kaufmann SH, et al. Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell Microbiol. 2008;10:1530–45. doi: 10.1111/j.1462-5822.2008.01147.x. [DOI] [PubMed] [Google Scholar]
- 74.Welin A, Winberg ME, Abdalla H, Särndahl E, Rasmusson B, Stendahl O, et al. Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun. 2008;76:2882–7. doi: 10.1128/IAI.01549-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Domenech P, Reed MB. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology. 2009;155:3532–43. doi: 10.1099/mic.0.029199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daffé M, Etienne G. The capsule of Mycobacterium tuberculosis and its implications for pathogenicity. Tuber Lung Dis. 1999;79:153–69. doi: 10.1054/tuld.1998.0200. [DOI] [PubMed] [Google Scholar]
- 77.Mawuenyega KG, Forst CV, Dobos KM, Belisle JT, Chen J, Bradbury EM, et al. Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling. Mol Biol Cell. 2005;16:396–404. doi: 10.1091/mbc.E04-04-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolfe LM, Mahaffey SB, Kruh NA, Dobos KM. Proteomic definition of the cell wall of Mycobacterium tuberculosis. J Proteome Res. 2010;9:5816–26. doi: 10.1021/pr1005873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci U S A. 2008;105:3963–7. doi: 10.1073/pnas.0709530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zuber B, Chami M, Houssin C, Dubochet J, Griffiths G, Daffé M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol. 2008;190:5672–80. doi: 10.1128/JB.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song H, Sandie R, Wang Y, Andrade-Navarro MA, Niederweis M. Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2008;88:526–44. doi: 10.1016/j.tube.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berthet FX, Lagranderie M, Gounon P, Laurent-Winter C, Ensergueix D, Chavarot P, et al. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science. 1998;282:759–62. doi: 10.1126/science.282.5389.759. [DOI] [PubMed] [Google Scholar]
- 83.Bigi F, Alito A, Fisanotti JC, Romano MI, Cataldi A. Characterization of a novel Mycobacterium bovis secreted antigen containing PGLTS repeats. Infect Immun. 1995;63:2581–6. doi: 10.1128/iai.63.7.2581-2586.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Mendonça-Lima L, Bordat Y, Pivert E, Recchi C, Neyrolles O, Maitournam A, et al. The allele encoding the mycobacterial Erp protein affects lung disease in mice. Cell Microbiol. 2003;5:65–73. doi: 10.1046/j.1462-5822.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 85.Bigi F, Gioffré A, Klepp L, Santangelo MP, Velicovsky CA, Giambartolomei GH, et al. Mutation in the P36 gene of Mycobacterium bovis provokes attenuation of the bacillus in a mouse model. Tuberculosis (Edinb) 2005;85:221–6. doi: 10.1016/j.tube.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 86.de Mendonça-Lima L, Picardeau M, Raynaud C, Rauzier J, de la Salmonière YO, Barker L, et al. Erp, an extracellular protein family specific to mycobacteria. Microbiology. 2001;147:2315–20. doi: 10.1099/00221287-147-8-2315. [DOI] [PubMed] [Google Scholar]
- 87.Kremer L, Maughan WN, Wilson RA, Dover LG, Besra GS. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett Appl Microbiol. 2002;34:233–7. doi: 10.1046/j.1472-765x.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- 88.Horwitz MA, Lee BW, Dillon BJ, Harth G. Protective immunity against tuberculosis induced by vaccination with major extracellular proteins of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1995;92:1530–4. doi: 10.1073/pnas.92.5.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiker HG, Harboe M. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol Rev. 1992;56:648–61. doi: 10.1128/mr.56.4.648-661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ronning DR, Klabunde T, Besra GS, Vissa VD, Belisle JT, Sacchettini JC. Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat Struct Biol. 2000;7:141–6. doi: 10.1038/72413. [DOI] [PubMed] [Google Scholar]
- 91.Jackson M, Raynaud C, Lanéelle MA, Guilhot C, Laurent-Winter C, Ensergueix D, et al. Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol Microbiol. 1999;31:1573–87. doi: 10.1046/j.1365-2958.1999.01310.x. [DOI] [PubMed] [Google Scholar]
- 92.Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science. 1993;261:1454–7. doi: 10.1126/science.8367727. [DOI] [PubMed] [Google Scholar]
- 93.Zumárraga M, Bigi F, Alito A, Romano MI, Cataldi A. A 12.7 kb fragment of the Mycobacterium tuberculosis genome is not present in Mycobacterium bovis. Microbiology. 1999;145:893–7. doi: 10.1099/13500872-145-4-893. [DOI] [PubMed] [Google Scholar]
- 94.Casali N, Riley LW. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics. 2007;8:60. doi: 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gioffré A, Infante E, Aguilar D, Santangelo MP, Klepp L, Amadio A, et al. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 2005;7:325–34. doi: 10.1016/j.micinf.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 96.Marjanovic O, Miyata T, Goodridge A, Kendall LV, Riley LW. Mce2 operon mutant strain of Mycobacterium tuberculosis is attenuated in C57BL/6 mice. Tuberculosis (Edinb) 2010;90:50–6. doi: 10.1016/j.tube.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, et al. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J Med Microbiol. 2008;57:164–70. doi: 10.1099/jmm.0.47454-0. [DOI] [PubMed] [Google Scholar]
- 98.Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, et al. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci U S A. 2003;100:15918–23. doi: 10.1073/pnas.2433882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102:8327–32. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stewart GR, Patel J, Robertson BD, Rae A, Young DB. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 2005;1:269–78. doi: 10.1371/journal.ppat.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de la Paz Santangelo M, Klepp L, Nuñez-García J, Blanco FC, Soria M, García-Pelayo MC, et al. Mce3R, a TetR-type transcriptional repressor, controls the expression of a regulon involved in lipid metabolism in Mycobacterium tuberculosis. Microbiology. 2009;155:2245–55. doi: 10.1099/mic.0.027086-0. [DOI] [PubMed] [Google Scholar]
- 103.Santangelo MP, Blanco FC, Bianco MV, Klepp LI, Zabal O, Cataldi AA, et al. Study of the role of Mce3R on the transcription of mce genes of Mycobacterium tuberculosis. BMC Microbiol. 2008;8:38. doi: 10.1186/1471-2180-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dunphy KY, Senaratne RH, Masuzawa M, Kendall LV, Riley LW. Attenuation of Mycobacterium tuberculosis functionally disrupted in a fatty acyl-coenzyme A synthetase gene fadD5. J Infect Dis. 2010;201:1232–9. doi: 10.1086/651452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marjanovic O, Iavarone AT, Riley LW. Sulfolipid accumulation in Mycobacterium tuberculosis disrupted in the mce2 operon. J Microbiol. 2011;49:441–7. doi: 10.1007/s12275-011-0435-4. [DOI] [PubMed] [Google Scholar]
- 106.Senaratne RH, Mobasheri H, Papavinasasundaram KG, Jenner P, Lea EJ, Draper P. Expression of a gene for a porin-like protein of the OmpA family from Mycobacterium tuberculosis H37Rv. J Bacteriol. 1998;180:3541–7. doi: 10.1128/jb.180.14.3541-3547.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Raynaud C, Papavinasasundaram KG, Speight RA, Springer B, Sander P, Böttger EC, et al. The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol Microbiol. 2002;46:191–201. doi: 10.1046/j.1365-2958.2002.03152.x. [DOI] [PubMed] [Google Scholar]
- 108.Menozzi FD, Rouse JH, Alavi M, Laude-Sharp M, Muller J, Bischoff R, et al. Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med. 1996;184:993–1001. doi: 10.1084/jem.184.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Delogu G, Brennan MJ. Functional domains present in the mycobacterial hemagglutinin, HBHA. J Bacteriol. 1999;181:7464–9. doi: 10.1128/jb.181.24.7464-7469.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Menozzi FD, Bischoff R, Fort E, Brennan MJ, Locht C. Molecular characterization of the mycobacterial heparin-binding hemagglutinin, a mycobacterial adhesin. Proc Natl Acad Sci U S A. 1998;95:12625–30. doi: 10.1073/pnas.95.21.12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pethe K, Alonso S, Biet F, Delogu G, Brennan MJ, Locht C, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature. 2001;412:190–4. doi: 10.1038/35084083. [DOI] [PubMed] [Google Scholar]
- 112.Braibant M, Gilot P, Content J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2000;24:449–67. doi: 10.1111/j.1574-6976.2000.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 113.Lun S, Bishai WR. Characterization of a novel cell wall-anchored protein with carboxylesterase activity required for virulence in Mycobacterium tuberculosis. J Biol Chem. 2007;282:18348–56. doi: 10.1074/jbc.M700035200. [DOI] [PubMed] [Google Scholar]
- 114.Vandal OH, Roberts JA, Odaira T, Schnappinger D, Nathan CF, Ehrt S. Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J Bacteriol. 2009;191:625–31. doi: 10.1128/JB.00932-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rengarajan J, Murphy E, Park A, Krone CL, Hett EC, Bloom BR, et al. Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc Natl Acad Sci U S A. 2008;105:264–9. doi: 10.1073/pnas.0710601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Butler RE, Cihlarova V, Stewart GR. Effective generation of reactive oxygen species in the mycobacterial phagosome requires K+ efflux from the bacterium. Cell Microbiol. 2010;12:1186–93. doi: 10.1111/j.1462-5822.2010.01463.x. [DOI] [PubMed] [Google Scholar]
- 117.Green RM, Seth A, Connell ND. A peptide permease mutant of Mycobacterium bovis BCG resistant to the toxic peptides glutathione and S-nitrosoglutathione. Infect Immun. 2000;68:429–36. doi: 10.1128/IAI.68.2.429-436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Flores-Valdez MA, Morris RP, Laval F, Daffé M, Schoolnik GK. Mycobacterium tuberculosis modulates its cell surface via an oligopeptide permease (Opp) transport system. FASEB J. 2009;23:4091–104. doi: 10.1096/fj.09-132407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McCann JR, McDonough JA, Sullivan JT, Feltcher ME, Braunstein M. Genome-wide identification of Mycobacterium tuberculosis exported proteins with roles in intracellular growth. J Bacteriol. 2011;193:854–61. doi: 10.1128/JB.01271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sutcliffe IC, Harrington DJ. Lipoproteins of Mycobacterium tuberculosis: an abundant and functionally diverse class of cell envelope components. FEMS Microbiol Rev. 2004;28:645–59. doi: 10.1016/j.femsre.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 121.Rezwan M, Grau T, Tschumi A, Sander P. Lipoprotein synthesis in mycobacteria. Microbiology. 2007;153:652–8. doi: 10.1099/mic.0.2006/000216-0. [DOI] [PubMed] [Google Scholar]
- 122.Målen H, Pathak S, Søfteland T, de Souza GA, Wiker HG. Definition of novel cell envelope associated proteins in Triton X-114 extracts of Mycobacterium tuberculosis H37Rv. BMC Microbiol. 2010;10:132. doi: 10.1186/1471-2180-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sander P, Rezwan M, Walker B, Rampini SK, Kroppenstedt RM, Ehlers S, et al. Lipoprotein processing is required for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;52:1543–52. doi: 10.1111/j.1365-2958.2004.04041.x. [DOI] [PubMed] [Google Scholar]
- 124.Young DB, Garbe TR. Lipoprotein antigens of Mycobacterium tuberculosis. Res Microbiol. 1991;142:55–65. doi: 10.1016/0923-2508(91)90097-T. [DOI] [PubMed] [Google Scholar]
- 125.Oftung F, Wiker HG, Deggerdal A, Mustafa AS. A novel mycobacterial antigen relevant to cellular immunity belongs to a family of secreted lipoproteins. Scand J Immunol. 1997;46:445–51. doi: 10.1046/j.1365-3083.1997.d01-150.x. [DOI] [PubMed] [Google Scholar]
- 126.Lefèvre P, Denis O, De Wit L, Tanghe A, Vandenbussche P, Content J, et al. Cloning of the gene encoding a 22-kilodalton cell surface antigen of Mycobacterium bovis BCG and analysis of its potential for DNA vaccination against tuberculosis. Infect Immun. 2000;68:1040–7. doi: 10.1128/IAI.68.3.1040-1047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sulzenbacher G, Canaan S, Bordat Y, Neyrolles O, Stadthagen G, Roig-Zamboni V, et al. LppX is a lipoprotein required for the translocation of phthiocerol dimycocerosates to the surface of Mycobacterium tuberculosis. EMBO J. 2006;25:1436–44. doi: 10.1038/sj.emboj.7601048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.González-Zamorano M, Mendoza-Herńndez G, Xolalpa W, Parada C, Vallecillo AJ, Bigi F, et al. Mycobacterium tuberculosis glycoproteomics based on ConA-lectin affinity capture of mannosylated proteins. J Proteome Res. 2009;8:721–33. doi: 10.1021/pr800756a. [DOI] [PubMed] [Google Scholar]
- 129.Tobian AA, Potter NS, Ramachandra L, Pai RK, Convery M, Boom WH, et al. Alternate class I MHC antigen processing is inhibited by Toll-like receptor signaling pathogen-associated molecular patterns: Mycobacterium tuberculosis 19-kDa lipoprotein, CpG DNA, and lipopolysaccharide. J Immunol. 2003;171:1413–22. doi: 10.4049/jimmunol.171.3.1413. [DOI] [PubMed] [Google Scholar]
- 130.Hertz CJ, Kiertscher SM, Godowski PJ, Bouis DA, Norgard MV, Roth MD, et al. Microbial lipopeptides stimulate dendritic cell maturation via Toll-like receptor 2. J Immunol. 2001;166:2444–50. doi: 10.4049/jimmunol.166.4.2444. [DOI] [PubMed] [Google Scholar]
- 131.Neufert C, Pai RK, Noss EH, Berger M, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein promotes neutrophil activation. J Immunol. 2001;167:1542–9. doi: 10.4049/jimmunol.167.3.1542. [DOI] [PubMed] [Google Scholar]
- 132.López M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003;170:2409–16. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 133.Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, et al. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010;12:1648–65. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Diaz-Silvestre H, Espinosa-Cueto P, Sanchez-Gonzalez A, Esparza-Ceron MA, Pereira-Suarez AL, Bernal-Fernandez G, et al. The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb Pathog. 2005;39:97–107. doi: 10.1016/j.micpath.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 135.Abou-Zeid C, Gares MP, Inwald J, Janssen R, Zhang Y, Young DB, et al. Induction of a type 1 immune response to a recombinant antigen from Mycobacterium tuberculosis expressed in Mycobacterium vaccae. Infect Immun. 1997;65:1856–62. doi: 10.1128/iai.65.5.1856-1862.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yeremeev VV, Stewart GR, Neyrolles O, Skrabal K, Avdienko VG, Apt AS, et al. Deletion of the 19kDa antigen does not alter the protective efficacy of BCG. Tuber Lung Dis. 2000;80:243–7. doi: 10.1054/tuld.2000.0252. [DOI] [PubMed] [Google Scholar]
- 137.Lathigra R, Zhang Y, Hill M, Garcia MJ, Jackett PS, Ivanyi J. Lack of production of the 19-kDa glycolipoprotein in certain strains of Mycobacterium tuberculosis. Res Microbiol. 1996;147:237–49. doi: 10.1016/0923-2508(96)81384-2. [DOI] [PubMed] [Google Scholar]
- 138.Henao-Tamayo M, Junqueira-Kipnis AP, Ordway D, Gonzales-Juarrero M, Stewart GR, Young DB, et al. A mutant of Mycobacterium tuberculosis lacking the 19-kDa lipoprotein Rv3763 is highly attenuated in vivo but retains potent vaccinogenic properties. Vaccine. 2007;25:7153–9. doi: 10.1016/j.vaccine.2007.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bigi F, Espitia C, Alito A, Zumarraga M, Romano MI, Cravero S, et al. A novel 27 kDa lipoprotein antigen from Mycobacterium bovis. Microbiology. 1997;143:3599–605. doi: 10.1099/00221287-143-11-3599. [DOI] [PubMed] [Google Scholar]
- 140.Hovav AH, Mullerad J, Davidovitch L, Fishman Y, Bigi F, Cataldi A, et al. The Mycobacterium tuberculosis recombinant 27-kilodalton lipoprotein induces a strong Th1-type immune response deleterious to protection. Infect Immun. 2003;71:3146–54. doi: 10.1128/IAI.71.6.3146-3154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hovav AH, Mullerad J, Maly A, Davidovitch L, Fishman Y, Bercovier H. Aggravated infection in mice co-administered with Mycobacterium tuberculosis and the 27-kDa lipoprotein. Microbes Infect. 2006;8:1750–7. doi: 10.1016/j.micinf.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 142.Bigi F, Alito A, Romano MI, Zumarraga M, Caimi K, Cataldi A. The gene encoding P27 lipoprotein and a putative antibiotic-resistance gene form an operon in Mycobacterium tuberculosis and Mycobacterium bovis. Microbiology. 2000;146:1011–8. doi: 10.1099/00221287-146-4-1011. [DOI] [PubMed] [Google Scholar]
- 143.Silva PE, Bigi F, Santangelo MP, Romano MI, Martín C, Cataldi A, et al. Characterization of P55, a multidrug efflux pump in Mycobacterium bovis and Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2001;45:800–4. doi: 10.1128/AAC.45.3.800-804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Farrow MF, Rubin EJ. Function of a mycobacterial major facilitator superfamily pump requires a membrane-associated lipoprotein. J Bacteriol. 2008;190:1783–91. doi: 10.1128/JB.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bianco MV, Blanco FC, Imperiale B, Forrellad MA, Rocha RV, Klepp LI, et al. Role of P27 -P55 operon from Mycobacterium tuberculosis in the resistance to toxic compounds. BMC Infect Dis. 2011;11:195. doi: 10.1186/1471-2334-11-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bianco MV, Blanco FC, Forrellad MA, Aguilar D, Campos E, Klepp LI, et al. Knockout mutation of p27-p55 operon severely reduces replication of Mycobacterium bovis in a macrophagic cell line and survival in a mouse model of infection. Virulence. 2011;2:233–7. doi: 10.4161/viru.2.3.15888. [DOI] [PubMed] [Google Scholar]
- 147.Bigi F, Gioffré A, Klepp L, Santangelo MP, Alito A, Caimi K, et al. The knockout of the lprG-Rv1410 operon produces strong attenuation of Mycobacterium tuberculosis. Microbes Infect. 2004;6:182–7. doi: 10.1016/j.micinf.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 148.Drage MG, Tsai HC, Pecora ND, Cheng TY, Arida AR, Shukla S, et al. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol. 2010;17:1088–95. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carroll MV, Sim RB, Bigi F, Jäkel A, Antrobus R, Mitchell DA. Identification of four novel DC-SIGN ligands on Mycobacterium bovis BCG. Protein Cell. 2010;1:859–70. doi: 10.1007/s13238-010-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Andersen AB, Hansen EB. Structure and mapping of antigenic domains of protein antigen b, a 38,000-molecular-weight protein of Mycobacterium tuberculosis. Infect Immun. 1989;57:2481–8. doi: 10.1128/iai.57.8.2481-2488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Espitia C, Mancilla R. Identification, isolation and partial characterization of Mycobacterium tuberculosis glycoprotein antigens. Clin Exp Immunol. 1989;77:378–83. [PMC free article] [PubMed] [Google Scholar]
- 152.Espitia C, Cervera I, González R, Mancilla R. A 38-kD Mycobacterium tuberculosis antigen associated with infection. Its isolation and serologic evaluation. Clin Exp Immunol. 1989;77:373–7. [PMC free article] [PubMed] [Google Scholar]
- 153.Jung SB, Yang CS, Lee JS, Shin AR, Jung SS, Son JW, et al. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun. 2006;74:2686–96. doi: 10.1128/IAI.74.5.2686-2696.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Andersen AB, Ljungqvist L, Olsen M. Evidence that protein antigen b of Mycobacterium tuberculosis is involved in phosphate metabolism. J Gen Microbiol. 1990;136:477–80. doi: 10.1099/00221287-136-3-477. [DOI] [PubMed] [Google Scholar]
- 155.Espitia C, Elinos M, Herńndez-Pando R, Mancilla R. Phosphate starvation enhances expression of the immunodominant 38-kilodalton protein antigen of Mycobacterium tuberculosis: demonstration by immunogold electron microscopy. Infect Immun. 1992;60:2998–3001. doi: 10.1128/iai.60.7.2998-3001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lefèvre P, Braibant M, de Wit L, Kalai M, Röeper D, Grötzinger J, et al. Three different putative phosphate transport receptors are encoded by the Mycobacterium tuberculosis genome and are present at the surface of Mycobacterium bovis BCG. J Bacteriol. 1997;179:2900–6. doi: 10.1128/jb.179.9.2900-2906.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Peirs P, Lefèvre P, Boarbi S, Wang XM, Denis O, Braibant M, et al. Mycobacterium tuberculosis with disruption in genes encoding the phosphate binding proteins PstS1 and PstS2 is deficient in phosphate uptake and demonstrates reduced in vivo virulence. Infect Immun. 2005;73:1898–902. doi: 10.1128/IAI.73.3.1898-1902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR., Jr Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2010;107:21761–6. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Williams MJ, Kana BD, Mizrahi V. Functional analysis of molybdopterin biosynthesis in mycobacteria identifies a fused molybdopterin synthase in Mycobacterium tuberculosis. J Bacteriol. 2011;193:98–106. doi: 10.1128/JB.00774-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Langermans JA, Doherty TM, Vervenne RA, van der Laan T, Lyashchenko K, Greenwald R, et al. Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine. 2005;23:2740–50. doi: 10.1016/j.vaccine.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 161.Skeiky YA, Dietrich J, Lasco TM, Stagliano K, Dheenadhayalan V, Goetz MA, et al. Non-clinical efficacy and safety of HyVac4:IC31 vaccine administered in a BCG prime-boost regimen. Vaccine. 2010;28:1084–93. doi: 10.1016/j.vaccine.2009.10.114. [DOI] [PubMed] [Google Scholar]
- 162.McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, Huygen K, et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med. 2004;10:1240–4. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 163.Ellis TN, Kuehn MJ. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev. 2010;74:81–94. doi: 10.1128/MMBR.00031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Marsollier L, Brodin P, Jackson M, Korduláková J, Tafelmeyer P, Carbonnelle E, et al. Impact of Mycobacterium ulcerans biofilm on transmissibility to ecological niches and Buruli ulcer pathogenesis. PLoS Pathog. 2007;3:e62. doi: 10.1371/journal.ppat.0030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Prados-Rosales R, Baena A, Martinez LR, Luque-Garcia J, Kalscheuer R, Veeraraghavan U, et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J Clin Invest. 2011;121:1471–83. doi: 10.1172/JCI44261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Harboe M, Wiker HG, Ulvund G, Lund-Pedersen B, Andersen AB, Hewinson RG, et al. MPB70 and MPB83 as indicators of protein localization in mycobacterial cells. Infect Immun. 1998;66:289–96. doi: 10.1128/iai.66.1.289-296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chambers MA, Vordermeier H, Whelan A, Commander N, Tascon R, Lowrie D, et al. Vaccination of mice and cattle with plasmid DNA encoding the Mycobacterium bovis antigen MPB83. Clin Infect Dis. 2000;30(Suppl 3):S283–7. doi: 10.1086/313875. [DOI] [PubMed] [Google Scholar]
- 168.Kao FF, Mahmuda S, Pinto R, Triccas JA, West NP, Britton WJ. The secreted lipoprotein, MPT83, of Mycobacterium tuberculosis is recognized during human tuberculosis and stimulates protective immunity in mice. PLoS One. 2012;7:e34991. doi: 10.1371/journal.pone.0034991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, et al. Type VII secretion--mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–91. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 170.Wards BJ, de Lisle GW, Collins DM. An esat6 knockout mutant of Mycobacterium bovis produced by homologous recombination will contribute to the development of a live tuberculosis vaccine. Tuber Lung Dis. 2000;80:185–9. doi: 10.1054/tuld.2000.0244. [DOI] [PubMed] [Google Scholar]
- 171.Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A, et al. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med. 2003;9:533–9. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- 172.Pallen MJ. The ESAT-6/WXG100 superfamily -- and a new Gram-positive secretion system? Trends Microbiol. 2002;10:209–12. doi: 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 173.Ravn P, Demissie A, Eguale T, Wondwosson H, Lein D, Amoudy HA, et al. Human T cell responses to the ESAT-6 antigen from Mycobacterium tuberculosis. J Infect Dis. 1999;179:637–45. doi: 10.1086/314640. [DOI] [PubMed] [Google Scholar]
- 174.Aagaard C, Govaerts M, Meikle V, Gutiérrez-Pabello JA, McNair J, Andersen P, et al. Detection of bovine tuberculosis in herds with different disease prevalence and influence of paratuberculosis infection on PPDB and ESAT-6/CFP10 specificity. Prev Vet Med. 2010;96:161–9. doi: 10.1016/j.prevetmed.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 175.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, et al. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guérin attenuation. J Infect Dis. 2003;187:117–23. doi: 10.1086/345862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bitter W, Houben EN, Bottai D, Brodin P, Brown EJ, Cox JS, et al. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 2009;5:e1000507. doi: 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Champion PA, Stanley SA, Champion MM, Brown EJ, Cox JS. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science. 2006;313:1632–6. doi: 10.1126/science.1131167. [DOI] [PubMed] [Google Scholar]
- 178.Carlsson F, Joshi SA, Rangell L, Brown EJ. Polar localization of virulence-related Esx-1 secretion in mycobacteria. PLoS Pathog. 2009;5:e1000285. doi: 10.1371/journal.ppat.1000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wirth SE, Krywy JA, Aldridge BB, Fortune SM, Fernandez-Suarez M, Gray TA, et al. Polar assembly and scaffolding proteins of the virulence-associated ESX-1 secretory apparatus in mycobacteria. Mol Microbiol. 2012;83:654–64. doi: 10.1111/j.1365-2958.2011.07958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, Cox JS, et al. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007;3:e105. doi: 10.1371/journal.ppat.0030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, et al. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A. 2005;102:10676–81. doi: 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.MacGurn JA, Raghavan S, Stanley SA, Cox JS. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol. 2005;57:1653–63. doi: 10.1111/j.1365-2958.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- 183.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol. 2004;53:1677–93. doi: 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- 184.Xu J, Laine O, Masciocchi M, Manoranjan J, Smith J, Du SJ, et al. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol Microbiol. 2007;66:787–800. doi: 10.1111/j.1365-2958.2007.05959.x. [DOI] [PubMed] [Google Scholar]
- 185.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature. 2008;454:717–21. doi: 10.1038/nature07219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bottai D, Majlessi L, Simeone R, Frigui W, Laurent C, Lenormand P, et al. ESAT-6 secretion-independent impact of ESX-1 genes espF and espG1 on virulence of Mycobacterium tuberculosis. J Infect Dis. 2011;203:1155–64. doi: 10.1093/infdis/jiq089. [DOI] [PubMed] [Google Scholar]
- 187.Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51:359–70. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–98. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 189.Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, et al. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol. 2007;8:610–8. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 190.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60:312–30. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 191.Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, Cox JS. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe. 2010;7:210–20. doi: 10.1016/j.chom.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–6. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- 193.Coros A, Callahan B, Battaglioli E, Derbyshire KM. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol Microbiol. 2008;69:794–808. doi: 10.1111/j.1365-2958.2008.06299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Brennan MJ, Delogu G, Chen Y, Bardarov S, Kriakov J, Alavi M, et al. Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect Immun. 2001;69:7326–33. doi: 10.1128/IAI.69.12.7326-7333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen JM, Boy-Röttger S, Dhar N, Sweeney N, Buxton RS, Pojer F, et al. EspD is critical for the virulence-mediating ESX-1 secretion system in Mycobacterium tuberculosis. J Bacteriol. 2012;194:884–93. doi: 10.1128/JB.06417-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Millington KA, Fortune SM, Low J, Garces A, Hingley-Wilson SM, Wickremasinghe M, et al. Rv3615c is a highly immunodominant RD1 (Region of Difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A. 2011;108:5730–5. doi: 10.1073/pnas.1015153108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, et al. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog. 2010;6:e1000957. doi: 10.1371/journal.ppat.1000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jiménez C, et al. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. 2009;73:329–40. doi: 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- 199.Bottai D, Brosch R. Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol Microbiol. 2009;73:325–8. doi: 10.1111/j.1365-2958.2009.06784.x. [DOI] [PubMed] [Google Scholar]
- 200.Abdallah AM, Bestebroer J, Savage ND, de Punder K, van Zon M, Wilson L, et al. Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J Immunol. 2011;187:4744–53. doi: 10.4049/jimmunol.1101457. [DOI] [PubMed] [Google Scholar]
- 201.Cascioferro A, Daleke MH, Ventura M, Donà V, Delogu G, Palù G, et al. Functional dissection of the PE domain responsible for translocation of PE_PGRS33 across the mycobacterial cell wall. PLoS One. 2011;6:e27713. doi: 10.1371/journal.pone.0027713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol. 2012;83:1195–209. doi: 10.1111/j.1365-2958.2012.08001.x. [DOI] [PubMed] [Google Scholar]
- 203.Rampini SK, Selchow P, Keller C, Ehlers S, Böttger EC, Sander P. LspA inactivation in Mycobacterium tuberculosis results in attenuation without affecting phagosome maturation arrest. Microbiology. 2008;154:2991–3001. doi: 10.1099/mic.0.2008/018895-0. [DOI] [PubMed] [Google Scholar]
- 204.Tschumi A, Grau T, Albrecht D, Rezwan M, Antelmann H, Sander P. Functional analyses of mycobacterial lipoprotein diacylglyceryl transferase and comparative secretome analysis of a mycobacterial lgt mutant. J Bacteriol. 2012;194:3938–49. doi: 10.1128/JB.00127-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Braunstein M, Brown AM, Kurtz S, Jacobs WR., Jr Two nonredundant SecA homologues function in mycobacteria. J Bacteriol. 2001;183:6979–90. doi: 10.1128/JB.183.24.6979-6990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Braunstein M, Espinosa BJ, Chan J, Belisle JT, Jacobs WR., Jr. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol Microbiol. 2003;48:453–64. doi: 10.1046/j.1365-2958.2003.03438.x. [DOI] [PubMed] [Google Scholar]
- 207.Ligon LS, Hayden JD, Braunstein M. The ins and outs of Mycobacterium tuberculosis protein export. Tuberculosis (Edinb) 2012;92:121–32. doi: 10.1016/j.tube.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zahrt TC, Deretic V. Reactive nitrogen and oxygen intermediates and bacterial defenses: unusual adaptations in Mycobacterium tuberculosis. Antioxid Redox Signal. 2002;4:141–59. doi: 10.1089/152308602753625924. [DOI] [PubMed] [Google Scholar]
- 209.Kendall SL, Movahedzadeh F, Rison SC, Wernisch L, Parish T, Duncan K, et al. The Mycobacterium tuberculosis dosRS two-component system is induced by multiple stresses. Tuberculosis (Edinb) 2004;84:247–55. doi: 10.1016/j.tube.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 210.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198:705–13. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yuan Y, Crane DD, Simpson RM, Zhu YQ, Hickey MJ, Sherman DR, et al. The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc Natl Acad Sci U S A. 1998;95:9578–83. doi: 10.1073/pnas.95.16.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hu Y, Movahedzadeh F, Stoker NG, Coates AR. Deletion of the Mycobacterium tuberculosis alpha-crystallin-like hspX gene causes increased bacterial growth in vivo. Infect Immun. 2006;74:861–8. doi: 10.1128/IAI.74.2.861-868.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Spratt JM, Britton WJ, Triccas JA. In vivo persistence and protective efficacy of the bacille Calmette Guerin vaccine overexpressing the HspX latency antigen. Bioeng Bugs. 2010;1:61–5. doi: 10.4161/bbug.1.1.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, et al. The heat shock response of Mycobacterium tuberculosis: linking gene expression, immunology and pathogenesis. Comp Funct Genomics. 2002;3:348–51. doi: 10.1002/cfg.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wilkinson KA, Stewart GR, Newton SM, Vordermeier HM, Wain JR, Murphy HN, et al. Infection biology of a novel alpha-crystallin of Mycobacterium tuberculosis: Acr2. J Immunol. 2005;174:4237–43. doi: 10.4049/jimmunol.174.7.4237. [DOI] [PubMed] [Google Scholar]
- 217.Stewart GR, Newton SM, Wilkinson KA, Humphreys IR, Murphy HN, Robertson BD, et al. The stress-responsive chaperone alpha-crystallin 2 is required for pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2005;55:1127–37. doi: 10.1111/j.1365-2958.2004.04450.x. [DOI] [PubMed] [Google Scholar]
- 218.Silva MS, Senna SG, Ribeiro MO, Valim AR, Telles MA, Kritski A, et al. Mutations in katG, inhA, and ahpC genes of Brazilian isoniazid-resistant isolates of Mycobacterium tuberculosis. J Clin Microbiol. 2003;41:4471–4. doi: 10.1128/JCM.41.9.4471-4474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sherman DR, Mdluli K, Hickey MJ, Barry CE, 3rd, Stover CK. AhpC, oxidative stress and drug resistance in Mycobacterium tuberculosis. Biofactors. 1999;10:211–7. doi: 10.1002/biof.5520100219. [DOI] [PubMed] [Google Scholar]
- 220.Wilson M, DeRisi J, Kristensen HH, Imboden P, Rane S, Brown PO, et al. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci U S A. 1999;96:12833–8. doi: 10.1073/pnas.96.22.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wilson T, de Lisle GW, Marcinkeviciene JA, Blanchard JS, Collins DM. Antisense RNA to ahpC, an oxidative stress defence gene involved in isoniazid resistance, indicates that AhpC of Mycobacterium bovis has virulence properties. Microbiology. 1998;144:2687–95. doi: 10.1099/00221287-144-10-2687. [DOI] [PubMed] [Google Scholar]
- 222.Guimarães BG, Souchon H, Honoré N, Saint-Joanis B, Brosch R, Shepard W, et al. Structure and mechanism of the alkyl hydroperoxidase AhpC, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J Biol Chem. 2005;280:25735–42. doi: 10.1074/jbc.M503076200. [DOI] [PubMed] [Google Scholar]
- 223.Chauhan R, Mande SC. Characterization of the Mycobacterium tuberculosis H37Rv alkyl hydroperoxidase AhpC points to the importance of ionic interactions in oligomerization and activity. Biochem J. 2001;354:209–15. doi: 10.1042/0264-6021:3540209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hillas PJ, del Alba FS, Oyarzabal J, Wilks A, Ortiz De Montellano PR. The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J Biol Chem. 2000;275:18801–9. doi: 10.1074/jbc.M001001200. [DOI] [PubMed] [Google Scholar]
- 225.Master SS, Springer B, Sander P, Boettger EC, Deretic V, Timmins GS. Oxidative stress response genes in Mycobacterium tuberculosis: role of ahpC in resistance to peroxynitrite and stage-specific survival in macrophages. Microbiology. 2002;148:3139–44. doi: 10.1099/00221287-148-10-3139. [DOI] [PubMed] [Google Scholar]
- 226.Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 227.D’orazio M, Folcarelli S, Mariani F, Colizzi V, Rotilio G, Battistoni A. Lipid modification of the Cu,Zn superoxide dismutase from Mycobacterium tuberculosis. Biochem J. 2001;359:17–22. doi: 10.1042/0264-6021:3590017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Spagnolo L, Törö I, D’Orazio M, O’Neill P, Pedersen JZ, Carugo O, et al. Unique features of the sodC-encoded superoxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking zinc in the active site. J Biol Chem. 2004;279:33447–55. doi: 10.1074/jbc.M404699200. [DOI] [PubMed] [Google Scholar]
- 229.Volpe E, Cappelli G, Grassi M, Martino A, Serafino A, Colizzi V, et al. Gene expression profiling of human macrophages at late time of infection with Mycobacterium tuberculosis. Immunology. 2006;118:449–60. doi: 10.1111/j.1365-2567.2006.02378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun. 2001;69:4980–7. doi: 10.1128/IAI.69.8.4980-4987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dussurget O, Stewart G, Neyrolles O, Pescher P, Young D, Marchal G. Role of Mycobacterium tuberculosis copper-zinc superoxide dismutase. Infect Immun. 2001;69:529–33. doi: 10.1128/IAI.69.1.529-533.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Cirillo SL, Subbian S, Chen B, Weisbrod TR, Jacobs WR, Jr., Cirillo JD. Protection of Mycobacterium tuberculosis from reactive oxygen species conferred by the mel2 locus impacts persistence and dissemination. Infect Immun. 2009;77:2557–67. doi: 10.1128/IAI.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Timmins GS, Deretic V. Mechanisms of action of isoniazid. Mol Microbiol. 2006;62:1220–7. doi: 10.1111/j.1365-2958.2006.05467.x. [DOI] [PubMed] [Google Scholar]
- 234.Heym B, Stavropoulos E, Honoré N, Domenech P, Saint-Joanis B, Wilson TM, et al. Effects of overexpression of the alkyl hydroperoxide reductase AhpC on the virulence and isoniazid resistance of Mycobacterium tuberculosis. Infect Immun. 1997;65:1395–401. doi: 10.1128/iai.65.4.1395-1401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Li Z, Kelley C, Collins F, Rouse D, Morris S. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J Infect Dis. 1998;177:1030–5. doi: 10.1086/515254. [DOI] [PubMed] [Google Scholar]
- 236.Wilson TM, de Lisle GW, Collins DM. Effect of inhA and katG on isoniazid resistance and virulence of Mycobacterium bovis. Mol Microbiol. 1995;15:1009–15. doi: 10.1111/j.1365-2958.1995.tb02276.x. [DOI] [PubMed] [Google Scholar]
- 237.Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol. 2004;52:1291–302. doi: 10.1111/j.1365-2958.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 238.Jaeger T, Budde H, Flohé L, Menge U, Singh M, Trujillo M, et al. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch Biochem Biophys. 2004;423:182–91. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 239.Rho BS, Hung LW, Holton JM, Vigil D, Kim SI, Park MS, et al. Functional and structural characterization of a thiol peroxidase from Mycobacterium tuberculosis. J Mol Biol. 2006;361:850–63. doi: 10.1016/j.jmb.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 240.Hu Y, Coates AR. Acute and persistent Mycobacterium tuberculosis infections depend on the thiol peroxidase TpX. PLoS One. 2009;4:e5150. doi: 10.1371/journal.pone.0005150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Conconi M, Petropoulos I, Emod I, Turlin E, Biville F, Friguet B. Protection from oxidative inactivation of the 20S proteasome by heat-shock protein 90. Biochem J. 1998;333:407–15. doi: 10.1042/bj3330407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteom. 2011;10:R110 006924. doi: 10.1074/mcp.M110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chopra P, Singh A, Koul A, Ramachandran S, Drlica K, Tyagi AK, et al. Cytotoxic activity of nucleoside diphosphate kinase secreted from Mycobacterium tuberculosis. Eur J Biochem. 2003;270:625–34. doi: 10.1046/j.1432-1033.2003.03402.x. [DOI] [PubMed] [Google Scholar]
- 244.Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One. 2010;5:e8769. doi: 10.1371/journal.pone.0008769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cowley SC, Babakaiff R, Av-Gay Y. Expression and localization of the Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Res Microbiol. 2002;153:233–41. doi: 10.1016/S0923-2508(02)01309-8. [DOI] [PubMed] [Google Scholar]
- 246.Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108:19371–6. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe. 2008;3:316–22. doi: 10.1016/j.chom.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 248.Grundner C, Cox JS, Alber T. Protein tyrosine phosphatase PtpA is not required for Mycobacterium tuberculosis growth in mice. FEMS Microbiol Lett. 2008;287:181–4. doi: 10.1111/j.1574-6968.2008.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Iantomasi R, Sali M, Cascioferro A, Palucci I, Zumbo A, Soldini S, et al. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol. 2012;14:356–67. doi: 10.1111/j.1462-5822.2011.01721.x. [DOI] [PubMed] [Google Scholar]
- 250.Mills IG, Urbé S, Clague MJ. Relationships between EEA1 binding partners and their role in endosome fusion. J Cell Sci. 2001;114:1959–65. doi: 10.1242/jcs.114.10.1959. [DOI] [PubMed] [Google Scholar]
- 251.Vergne I, Chua J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: selective targeting of PI3P-dependent membrane trafficking. Traffic. 2003;4:600–6. doi: 10.1034/j.1600-0854.2003.00120.x. [DOI] [PubMed] [Google Scholar]
- 252.Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102:4033–8. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Festjens N, Bogaert P, Batni A, Houthuys E, Plets E, Vanderschaeghe D, et al. Disruption of the SapM locus in Mycobacterium bovis BCG improves its protective efficacy as a vaccine against M. tuberculosis. EMBO Mol Med. 2011;3:222–34. doi: 10.1002/emmm.201000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ferrer NL, Gomez AB, Neyrolles O, Gicquel B, Martin C. Interactions of attenuated Mycobacterium tuberculosis phoP mutant with human macrophages. PLoS One. 2010;5:e12978. doi: 10.1371/journal.pone.0012978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Katti MK, Dai G, Armitige LY, Rivera Marrero C, Daniel S, Singh CR, et al. The Delta fbpA mutant derived from Mycobacterium tuberculosis H37Rv has an enhanced susceptibility to intracellular antimicrobial oxidative mechanisms, undergoes limited phagosome maturation and activates macrophages and dendritic cells. Cell Microbiol. 2008;10:1286–303. doi: 10.1111/j.1462-5822.2008.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Rohde K, Yates RM, Purdy GE, Russell DG. Mycobacterium tuberculosis and the environment within the phagosome. Immunol Rev. 2007;219:37–54. doi: 10.1111/j.1600-065X.2007.00547.x. [DOI] [PubMed] [Google Scholar]
- 257.Sullivan JT, Young EF, McCann JR, Braunstein M. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect Immun. 2012;80:996–1006. doi: 10.1128/IAI.05987-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Stamm LM, Morisaki JH, Gao LY, Jeng RL, McDonald KL, Roth R, et al. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med. 2003;198:1361–8. doi: 10.1084/jem.20031072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Harriff MJ, Purdy GE, Lewinsohn DM. Escape from the Phagosome: The Explanation for MHC-I Processing of Mycobacterial Antigens? Front Immunol. 2012;3:40. doi: 10.3389/fimmu.2012.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–46. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 261.Danelishvili L, McGarvey J, Li YJ, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003;5:649–60. doi: 10.1046/j.1462-5822.2003.00312.x. [DOI] [PubMed] [Google Scholar]
- 262.Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 2007;9:1547–55. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 263.Rojas M, Barrera LF, Puzo G, Garcia LF. Differential induction of apoptosis by virulent Mycobacterium tuberculosis in resistant and susceptible murine macrophages: role of nitric oxide and mycobacterial products. J Immunol. 1997;159:1352–61. [PubMed] [Google Scholar]
- 264.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–20. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 265.Briken V, Miller JL. Living on the edge: inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 2008;3:415–22. doi: 10.2217/17460913.3.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011;4:279–87. doi: 10.1038/mi.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol. 1998;161:2636–41. [PubMed] [Google Scholar]
- 268.Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Miller JL, Velmurugan K, Cowan MJ, Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 2010;6:e1000864. doi: 10.1371/journal.ppat.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 2012;11:81–90. doi: 10.1016/j.chom.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Gibbons HS, Wolschendorf F, Abshire M, Niederweis M, Braunstein M. Identification of two Mycobacterium smegmatis lipoproteins exported by a SecA2-dependent pathway. J Bacteriol. 2007;189:5090–100. doi: 10.1128/JB.00163-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kurtz S, McKinnon KP, Runge MS, Ting JP, Braunstein M. The SecA2 secretion factor of Mycobacterium tuberculosis promotes growth in macrophages and inhibits the host immune response. Infect Immun. 2006;74:6855–64. doi: 10.1128/IAI.01022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest. 2007;117:2279–88. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeStefano MS, Tham KT, et al. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2001;164:2213–9. doi: 10.1164/ajrccm.164.12.2106093. [DOI] [PubMed] [Google Scholar]
- 275.Jayakumar D, Jacobs WR, Jr., Narayanan S. Protein kinase E of Mycobacterium tuberculosis has a role in the nitric oxide stress response and apoptosis in a human macrophage model of infection. Cell Microbiol. 2008;10:365–74. doi: 10.1111/j.1462-5822.2007.01049.x. [DOI] [PubMed] [Google Scholar]
- 276.Danelishvili L, Yamazaki Y, Selker J, Bermudez LE. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS One. 2010;5:e10474. doi: 10.1371/journal.pone.0010474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Behar SM, Martin CJ, Nunes-Alves C, Divangahi M, Remold HG. Lipids, apoptosis, and cross-presentation: links in the chain of host defense against Mycobacterium tuberculosis. Microbes Infect. 2011;13:749–56. doi: 10.1016/j.micinf.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 279.Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J. 2009;50:1–11. doi: 10.3349/ymj.2009.50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. 2008;9:1189–97. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Koul A, Choidas A, Tyagi AK, Drlica K, Singh Y, Ullrich A. Serine/threonine protein kinases PknF and PknG of Mycobacterium tuberculosis: characterization and localization. Microbiology. 2001;147:2307–14. doi: 10.1099/00221287-147-8-2307. [DOI] [PubMed] [Google Scholar]
- 284.Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–11. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 285.Greenstein AE, Grundner C, Echols N, Gay LM, Lombana TN, Miecskowski CA, et al. Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J Mol Microbiol Biotechnol. 2005;9:167–81. doi: 10.1159/000089645. [DOI] [PubMed] [Google Scholar]
- 286.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 287.Fernandez P, Saint-Joanis B, Barilone N, Jackson M, Gicquel B, Cole ST, et al. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol. 2006;188:7778–84. doi: 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Dasgupta A, Datta P, Kundu M, Basu J. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology. 2006;152:493–504. doi: 10.1099/mic.0.28630-0. [DOI] [PubMed] [Google Scholar]
- 289.Kang JL, Lee HW, Kim HJ, Lee HS, Castranova V, Lim CM, et al. Inhibition of SRC tyrosine kinases suppresses activation of nuclear factor-kappaB, and serine and tyrosine phosphorylation of IkappaB-alpha in lipopolysaccharide-stimulated raw 264.7 macrophages. J Toxicol Environ Health A. 2005;68:1643–62. doi: 10.1080/15287390500192114. [DOI] [PubMed] [Google Scholar]
- 290.Gupta M, Sajid A, Arora G, Tandon V, Singh Y. Forkhead-associated domain-containing protein Rv0019c and polyketide-associated protein PapA5, from substrates of serine/threonine protein kinase PknB to interacting proteins of Mycobacterium tuberculosis. J Biol Chem. 2009;284:34723–34. doi: 10.1074/jbc.M109.058834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Khan S, Nagarajan SN, Parikh A, Samantaray S, Singh A, Kumar D, et al. Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J Biol Chem. 2010;285:37860–71. doi: 10.1074/jbc.M110.143131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Thakur M, Chakraborti PK. Ability of PknA, a mycobacterial eukaryotic-type serine/threonine kinase, to transphosphorylate MurD, a ligase involved in the process of peptidoglycan biosynthesis. Biochem J. 2008;415:27–33. doi: 10.1042/BJ20080234. [DOI] [PubMed] [Google Scholar]
- 293.Veyron-Churlet R, Zanella-Cléon I, Cohen-Gonsaud M, Molle V, Kremer L. Phosphorylation of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J Biol Chem. 2010;285:12714–25. doi: 10.1074/jbc.M110.105189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Deol P, Vohra R, Saini AK, Singh A, Chandra H, Chopra P, et al. Role of Mycobacterium tuberculosis Ser/Thr kinase PknF: implications in glucose transport and cell division. J Bacteriol. 2005;187:3415–20. doi: 10.1128/JB.187.10.3415-3420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Curry JM, Whalan R, Hunt DM, Gohil K, Strom M, Rickman L, et al. An ABC transporter containing a forkhead-associated domain interacts with a serine-threonine protein kinase and is required for growth of Mycobacterium tuberculosis in mice. Infect Immun. 2005;73:4471–7. doi: 10.1128/IAI.73.8.4471-4477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Gopalaswamy R, Narayanan S, Chen B, Jacobs WR, Av-Gay Y. The serine/threonine protein kinase PknI controls the growth of Mycobacterium tuberculosis upon infection. FEMS Microbiol Lett. 2009;295:23–9. doi: 10.1111/j.1574-6968.2009.01570.x. [DOI] [PubMed] [Google Scholar]
- 297.Malhotra V, Arteaga-Cortés LT, Clay G, Clark-Curtiss JE. Mycobacterium tuberculosis protein kinase K confers survival advantage during early infection in mice and regulates growth in culture and during persistent infection: implications for immune modulation. Microbiology. 2010;156:2829–41. doi: 10.1099/mic.0.040675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Kumar P, Kumar D, Parikh A, Rananaware D, Gupta M, Singh Y, et al. The Mycobacterium tuberculosis protein kinase K modulates activation of transcription from the promoter of mycobacterial monooxygenase operon through phosphorylation of the transcriptional regulator VirS. J Biol Chem. 2009;284:11090–9. doi: 10.1074/jbc.M808705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sharma K, Gupta M, Krupa A, Srinivasan N, Singh Y. EmbR, a regulatory protein with ATPase activity, is a substrate of multiple serine/threonine kinases and phosphatase in Mycobacterium tuberculosis. FEBS J. 2006;273:2711–21. doi: 10.1111/j.1742-4658.2006.05289.x. [DOI] [PubMed] [Google Scholar]
- 300.Belanger AE, Besra GS, Ford ME, Mikusová K, Belisle JT, Brennan PJ, et al. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A. 1996;93:11919–24. doi: 10.1073/pnas.93.21.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Amin AG, Goude R, Shi L, Zhang J, Chatterjee D, Parish T. EmbA is an essential arabinosyltransferase in Mycobacterium tuberculosis. Microbiology. 2008;154:240–8. doi: 10.1099/mic.0.2007/012153-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Goude R, Amin AG, Chatterjee D, Parish T. The critical role of embC in Mycobacterium tuberculosis. J Bacteriol. 2008;190:4335–41. doi: 10.1128/JB.01825-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Chao JD, Papavinasasundaram KG, Zheng X, Chávez-Steenbock A, Wang X, Lee GQ, et al. Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J Biol Chem. 2010;285:29239–46. doi: 10.1074/jbc.M110.132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Papavinasasundaram KG, Chan B, Chung JH, Colston MJ, Davis EO, Av-Gay Y. Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J Bacteriol. 2005;187:5751–60. doi: 10.1128/JB.187.16.5751-5760.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Arora G, Sajid A, Gupta M, Bhaduri A, Kumar P, Basu-Modak S, et al. Understanding the role of PknJ in Mycobacterium tuberculosis: biochemical characterization and identification of novel substrate pyruvate kinase A. PLoS One. 2010;5:e10772. doi: 10.1371/journal.pone.0010772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Cohen-Gonsaud M, Barthe P, Canova MJ, Stagier-Simon C, Kremer L, Roumestand C, et al. The Mycobacterium tuberculosis Ser/Thr kinase substrate Rv2175c is a DNA-binding protein regulated by phosphorylation. J Biol Chem. 2009;284:19290–300. doi: 10.1074/jbc.M109.019653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Narayan A, Sachdeva P, Sharma K, Saini AK, Tyagi AK, Singh Y. Serine threonine protein kinases of mycobacterial genus: phylogeny to function. Physiol Genomics. 2007;29:66–75. doi: 10.1152/physiolgenomics.00221.2006. [DOI] [PubMed] [Google Scholar]
- 308.Pérez J, Garcia R, Bach H, de Waard JH, Jacobs WR, Jr., Av-Gay Y, et al. Mycobacterium tuberculosis transporter MmpL7 is a potential substrate for kinase PknD. Biochem Biophys Res Commun. 2006;348:6–12. doi: 10.1016/j.bbrc.2006.06.164. [DOI] [PubMed] [Google Scholar]
- 309.Greenstein AE, MacGurn JA, Baer CE, Falick AM, Cox JS, Alber T. M. tuberculosis Ser/Thr protein kinase D phosphorylates an anti-anti-sigma factor homolog. PLoS Pathog. 2007;3:e49. doi: 10.1371/journal.ppat.0030049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Parida BK, Douglas T, Nino C, Dhandayuthapani S. Interactions of anti-sigma factor antagonists of Mycobacterium tuberculosis in the yeast two-hybrid system. Tuberculosis (Edinb) 2005;85:347–55. doi: 10.1016/j.tube.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 311.Be NA, Bishai WR, Jain SK. Role of Mycobacterium tuberculosis pknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol. 2012;12:7. doi: 10.1186/1471-2180-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Cowley S, Ko M, Pick N, Chow R, Downing KJ, Gordhan BG, et al. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol. 2004;52:1691–702. doi: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 313.Nguyen L, Walburger A, Houben E, Koul A, Muller S, Morbitzer M, et al. Role of protein kinase G in growth and glutamine metabolism of Mycobacterium bovis BCG. J Bacteriol. 2005;187:5852–6. doi: 10.1128/JB.187.16.5852-5856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Scherr N, Müller P, Perisa D, Combaluzier B, Jenö P, Pieters J. Survival of pathogenic mycobacteria in macrophages is mediated through autophosphorylation of protein kinase G. J Bacteriol. 2009;191:4546–54. doi: 10.1128/JB.00245-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kumar D, Narayanan S. pknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect Genet Evol. 2012;12:737–47. doi: 10.1016/j.meegid.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 316.Ribeiro-Guimarães ML, Pessolani MC. Comparative genomics of mycobacterial proteases. Microb Pathog. 2007;43:173–8. doi: 10.1016/j.micpath.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 317.Gupta R, Beg QK, Lorenz P. Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol. 2002;59:15–32. doi: 10.1007/s00253-002-0975-y. [DOI] [PubMed] [Google Scholar]
- 318.Brown GD, Dave JA, Gey van Pittius NC, Stevens L, Ehlers MR, Beyers AD. The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene. 2000;254:147–55. doi: 10.1016/S0378-1119(00)00277-8. [DOI] [PubMed] [Google Scholar]
- 319.Dave JA, Gey van Pittius NC, Beyers AD, Ehlers MR, Brown GD. Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol. 2002;2:30. doi: 10.1186/1471-2180-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A. 2003;100:13001–6. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Brandt L, Elhay M, Rosenkrands I, Lindblad EB, Andersen P. ESAT-6 subunit vaccination against Mycobacterium tuberculosis. Infect Immun. 2000;68:791–5. doi: 10.1128/IAI.68.2.791-795.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Colangeli R, Spencer JS, Bifani P, Williams A, Lyashchenko K, Keen MA, et al. MTSA-10, the product of the Rv3874 gene of Mycobacterium tuberculosis, elicits tuberculosis-specific, delayed-type hypersensitivity in guinea pigs. Infect Immun. 2000;68:990–3. doi: 10.1128/IAI.68.2.990-993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Coler RN, Campos-Neto A, Ovendale P, Day FH, Fling SP, Zhu L, et al. Vaccination with the T cell antigen Mtb 8.4 protects against challenge with Mycobacterium tuberculosis. J Immunol. 2001;166:6227–35. doi: 10.4049/jimmunol.166.10.6227. [DOI] [PubMed] [Google Scholar]
- 324.Dietrich J, Andersen C, Rappuoli R, Doherty TM, Jensen CG, Andersen P. Mucosal administration of Ag85B-ESAT-6 protects against infection with Mycobacterium tuberculosis and boosts prior bacillus Calmette-Guerin immunity. J Immunol. 2006;177:6353–60. doi: 10.4049/jimmunol.177.9.6353. [DOI] [PubMed] [Google Scholar]
- 325.Kim DY, Kim KK. Structure and function of HtrA family proteins, the key players in protein quality control. J Biochem Mol Biol. 2005;38:266–74. doi: 10.5483/BMBRep.2005.38.3.266. [DOI] [PubMed] [Google Scholar]
- 326.White MJ, He H, Penoske RM, Twining SS, Zahrt TC. PepD participates in the mycobacterial stress response mediated through MprAB and SigE. J Bacteriol. 2010;192:1498–510. doi: 10.1128/JB.01167-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Singh N, Kuppili RR, Bose K. The structural basis of mode of activation and functional diversity: a case study with HtrA family of serine proteases. Arch Biochem Biophys. 2011;516:85–96. doi: 10.1016/j.abb.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 328.Mohamedmohaideen NN, Palaninathan SK, Morin PM, Williams BJ, Braunstein M, Tichy SE, et al. Structure and function of the virulence-associated high-temperature requirement A of Mycobacterium tuberculosis. Biochemistry. 2008;47:6092–102. doi: 10.1021/bi701929m. [DOI] [PubMed] [Google Scholar]
- 329.Braunstein M, Griffin TJ IV, Kriakov JI, Friedman ST, Grindley ND, Jacobs WR., Jr Identification of genes encoding exported Mycobacterium tuberculosis proteins using a Tn552’phoA in vitro transposition system. J Bacteriol. 2000;182:2732–40. doi: 10.1128/JB.182.10.2732-2740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Xiong Y, Chalmers MJ, Gao FP, Cross TA, Marshall AG. Identification of Mycobacterium tuberculosis H37Rv integral membrane proteins by one-dimensional gel electrophoresis and liquid chromatography electrospray ionization tandem mass spectrometry. J Proteome Res. 2005;4:855–61. doi: 10.1021/pr0500049. [DOI] [PubMed] [Google Scholar]
- 331.Downing KJ, McAdam RA, Mizrahi V. Staphylococcus aureus nuclease is a useful secretion reporter for mycobacteria. Gene. 1999;239:293–9. doi: 10.1016/S0378-1119(99)00408-4. [DOI] [PubMed] [Google Scholar]
- 332.Skeiky YA, Lodes MJ, Guderian JA, Mohamath R, Bement T, Alderson MR, et al. Cloning, expression, and immunological evaluation of two putative secreted serine protease antigens of Mycobacterium tuberculosis. Infect Immun. 1999;67:3998–4007. doi: 10.1128/iai.67.8.3998-4007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.White MJ, Savaryn JP, Bretl DJ, He H, Penoske RM, Terhune SS, et al. The HtrA-like serine protease PepD interacts with and modulates the Mycobacterium tuberculosis 35-kDa antigen outer envelope protein. PLoS One. 2011;6:e18175. doi: 10.1371/journal.pone.0018175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Estorninho M, Smith H, Thole J, Harders-Westerveen J, Kierzek A, Butler RE, et al. ClgR regulation of chaperone and protease systems is essential for Mycobacterium tuberculosis parasitism of the macrophage. Microbiology. 2010;156:3445–55. doi: 10.1099/mic.0.042275-0. [DOI] [PubMed] [Google Scholar]
- 335.Vandal OH, Pierini LM, Schnappinger D, Nathan CF, Ehrt S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat Med. 2008;14:849–54. doi: 10.1038/nm.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Biswas T, Small J, Vandal O, Odaira T, Deng H, Ehrt S, et al. Structural insight into serine protease Rv3671c that Protects M. tuberculosis from oxidative and acidic stress. Structure. 2010;18:1353–63. doi: 10.1016/j.str.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.De Mot R, Nagy I, Walz J, Baumeister W. Proteasomes and other self-compartmentalizing proteases in prokaryotes. Trends Microbiol. 1999;7:88–92. doi: 10.1016/S0966-842X(98)01432-2. [DOI] [PubMed] [Google Scholar]
- 338.Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, et al. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 2012;8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology. 2002;148:3129–38. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 340.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–32. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Muttucumaru DG, Smith DA, McMinn EJ, Reese V, Coler RN, Parish T. Mycobacterium tuberculosis Rv0198c, a putative matrix metalloprotease is involved in pathogenicity. Tuberculosis (Edinb) 2011;91:111–6. doi: 10.1016/j.tube.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 342.Makinoshima H, Glickman MS. Site-2 proteases in prokaryotes: regulated intramembrane proteolysis expands to microbial pathogenesis. Microbes Infect. 2006;8:1882–8. doi: 10.1016/j.micinf.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 343.Sklar JG, Makinoshima H, Schneider JS, Glickman MS. M. tuberculosis intramembrane protease Rip1 controls transcription through three anti-sigma factor substrates. Mol Microbiol. 2010;77:605–17. doi: 10.1111/j.1365-2958.2010.07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–58. doi: 10.1016/S1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 345.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–6. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 346.Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55:561–71. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- 347.Festa RA, Pearce MJ, Darwin KH. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J Bacteriol. 2007;189:3044–50. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Butler SM, Festa RA, Pearce MJ, Darwin KH. Self-compartmentalized bacterial proteases and pathogenesis. Mol Microbiol. 2006;60:553–62. doi: 10.1111/j.1365-2958.2006.05128.x. [DOI] [PubMed] [Google Scholar]
- 349.Lamichhane G, Raghunand TR, Morrison NE, Woolwine SC, Tyagi S, Kandavelou K, et al. Deletion of a Mycobacterium tuberculosis proteasomal ATPase homologue gene produces a slow-growing strain that persists in host tissues. J Infect Dis. 2006;194:1233–40. doi: 10.1086/508288. [DOI] [PubMed] [Google Scholar]
- 350.Quadri LEN. Iron uptake in Mycobacteria. In: Daffé, M and Reyrat, JM, ed(s). The Mycobacterial Cell Envelope. Washington: ASM Press, 2008:167-184. [Google Scholar]
- 351.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci U S A. 2000;97:1252–7. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.De Voss JJ, Rutter K, Schroeder BG, Barry CE., 3rd Iron acquisition and metabolism by mycobacteria. J Bacteriol. 1999;181:4443–51. doi: 10.1128/jb.181.15.4443-4451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Farhana A, Kumar S, Rathore SS, Ghosh PC, Ehtesham NZ, Tyagi AK, et al. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS One. 2008;3:e2087. doi: 10.1371/journal.pone.0002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Rodriguez GM, Smith I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol. 2006;188:424–30. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ryndak MB, Wang S, Smith I, Rodriguez GM. The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J Bacteriol. 2010;192:861–9. doi: 10.1128/JB.00223-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. ideR, An essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002;70:3371–81. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Buchmeier N, Blanc-Potard A, Ehrt S, Piddington D, Riley L, Groisman EA. A parallel intraphagosomal survival strategy shared by mycobacterium tuberculosis and Salmonella enterica. Mol Microbiol. 2000;35:1375–82. doi: 10.1046/j.1365-2958.2000.01797.x. [DOI] [PubMed] [Google Scholar]
- 358.Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. 2011;10:248–59. doi: 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol Microbiol. 2010;77:1096–110. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Gonzalo-Asensio J, Mostowy S, Harders-Westerveen J, Huygen K, Herńndez-Pando R, Thole J, et al. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One. 2008;3:e3496. doi: 10.1371/journal.pone.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Lee JS, Krause R, Schreiber J, Mollenkopf HJ, Kowall J, Stein R, et al. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe. 2008;3:97–103. doi: 10.1016/j.chom.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 362.Li AH, Waddell SJ, Hinds J, Malloff CA, Bains M, Hancock RE, et al. Contrasting transcriptional responses of a virulent and an attenuated strain of Mycobacterium tuberculosis infecting macrophages. PLoS One. 2010;5:e11066. doi: 10.1371/journal.pone.0011066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Martin C, Williams A, Hernandez-Pando R, Cardona PJ, Gormley E, Bordat Y, et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine. 2006;24:3408–19. doi: 10.1016/j.vaccine.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 364.Pérez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martín C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. 2001;41:179–87. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 365.Bretl DJ, Demetriadou C, Zahrt TC. Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol Mol Biol Rev. 2011;75:566–82. doi: 10.1128/MMBR.05004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Abramovitch RB, Rohde KH, Hsu FF, Russell DG. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol. 2011;80:678–94. doi: 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Parish T, Smith DA, Roberts G, Betts J, Stoker NG. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology. 2003;149:1423–35. doi: 10.1099/mic.0.26245-0. [DOI] [PubMed] [Google Scholar]
- 368.Roberts G, Vadrevu IS, Madiraju MV, Parish T. Control of CydB and GltA1 expression by the SenX3 RegX3 two component regulatory system of Mycobacterium tuberculosis. PLoS One. 2011;6:e21090. doi: 10.1371/journal.pone.0021090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Supply P, Magdalena J, Himpens S, Locht C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997;26:991–1003. doi: 10.1046/j.1365-2958.1997.6361999.x. [DOI] [PubMed] [Google Scholar]
- 370.Rickman L, Saldanha JW, Hunt DM, Hoar DN, Colston MJ, Millar JB, et al. A two-component signal transduction system with a PAS domain-containing sensor is required for virulence of Mycobacterium tuberculosis in mice. Biochem Biophys Res Commun. 2004;314:259–67. doi: 10.1016/j.bbrc.2003.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Boon C, Dick T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J Bacteriol. 2002;184:6760–7. doi: 10.1128/JB.184.24.6760-6767.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc Natl Acad Sci U S A. 2001;98:7534–9. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Cho HY, Cho HJ, Kim YM, Oh JI, Kang BS. Structural insight into the heme-based redox sensing by DosS from Mycobacterium tuberculosis. J Biol Chem. 2009;284:13057–67. doi: 10.1074/jbc.M808905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Fallow A, Domenech P, Reed MB. Strains of the East Asian (W/Beijing) lineage of Mycobacterium tuberculosis are DosS/DosT-DosR two-component regulatory system natural mutants. J Bacteriol. 2010;192:2228–38. doi: 10.1128/JB.01597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Bartek IL, Rutherford R, Gruppo V, Morton RA, Morris RP, Klein MR, et al. The DosR regulon of M. tuberculosis and antibacterial tolerance. Tuberculosis (Edinb) 2009;89:310–6. doi: 10.1016/j.tube.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Converse PJ, Karakousis PC, Klinkenberg LG, Kesavan AK, Ly LH, Allen SS, et al. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect Immun. 2009;77:1230–7. doi: 10.1128/IAI.01117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Rustad TR, Harrell MI, Liao R, Sherman DR. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One. 2008;3:e1502. doi: 10.1371/journal.pone.0001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Malhotra V, Sharma D, Ramanathan VD, Shakila H, Saini DK, Chakravorty S, et al. Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2004;231:237–45. doi: 10.1016/S0378-1097(04)00002-3. [DOI] [PubMed] [Google Scholar]
- 379.Parish T, Smith DA, Kendall S, Casali N, Bancroft GJ, Stoker NG. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect Immun. 2003;71:1134–40. doi: 10.1128/IAI.71.3.1134-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Zahrt TC, Wozniak C, Jones D, Trevett A. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect Immun. 2003;71:6962–70. doi: 10.1128/IAI.71.12.6962-6970.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Talaat AM, Lyons R, Howard ST, Johnston SA. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci U S A. 2004;101:4602–7. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.He H, Hovey R, Kane J, Singh V, Zahrt TC. MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J Bacteriol. 2006;188:2134–43. doi: 10.1128/JB.188.6.2134-2143.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Pang X, Howard ST. Regulation of the alpha-crystallin gene acr2 by the MprAB two-component system of Mycobacterium tuberculosis. J Bacteriol. 2007;189:6213–21. doi: 10.1128/JB.00492-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Pang X, Vu P, Byrd TF, Ghanny S, Soteropoulos P, Mukamolova GV, et al. Evidence for complex interactions of stress-associated regulons in an mprAB deletion mutant of Mycobacterium tuberculosis. Microbiology. 2007;153:1229–42. doi: 10.1099/mic.0.29281-0. [DOI] [PubMed] [Google Scholar]
- 385.Zahrt TC, Deretic V. Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc Natl Acad Sci U S A. 2001;98:12706–11. doi: 10.1073/pnas.221272198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.MacArthur I, Parreira VR, Lepp D, Mutharia LM, Vazquez-Boland JA, Prescott JF. The sensor kinase MprB is required for Rhodococcus equi virulence. Vet Microbiol. 2011;147:133–41. doi: 10.1016/j.vetmic.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 387.Shrivastava R, Ghosh AK, Das AK. Intra- and intermolecular domain interactions among novel two-component system proteins coded by Rv0600c, Rv0601c and Rv0602c of Mycobacterium tuberculosis. Microbiology. 2009;155:772–9. doi: 10.1099/mic.0.019059-0. [DOI] [PubMed] [Google Scholar]
- 388.Bashyam MD, Hasnain SE. The extracytoplasmic function sigma factors: role in bacterial pathogenesis. Infect Genet Evol. 2004;4:301–8. doi: 10.1016/j.meegid.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 389.Collins DM, Kawakami RP, de Lisle GW, Pascopella L, Bloom BR, Jacobs WR., Jr. Mutation of the principal sigma factor causes loss of virulence in a strain of the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 1995;92:8036–40. doi: 10.1073/pnas.92.17.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Wu S, Howard ST, Lakey DL, Kipnis A, Samten B, Safi H, et al. The principal sigma factor sigA mediates enhanced growth of Mycobacterium tuberculosis in vivo. Mol Microbiol. 2004;51:1551–62. doi: 10.1111/j.1365-2958.2003.03922.x. [DOI] [PubMed] [Google Scholar]
- 391.Lonetto M, Gribskov M, Gross CA. The sigma 70 family: sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–9. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Collins DM, Kawakami RP, Buddle BM, Wards BJ, de Lisle GW. Different susceptibility of two animal species infected with isogenic mutants of Mycobacterium bovis identifies phoT as having roles in tuberculosis virulence and phosphate transport. Microbiology. 2003;149:3203–12. doi: 10.1099/mic.0.26469-0. [DOI] [PubMed] [Google Scholar]
- 393.Steyn AJ, Collins DM, Hondalus MK, Jacobs WR, Jr., Kawakami RP, Bloom BR. Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc Natl Acad Sci U S A. 2002;99:3147–52. doi: 10.1073/pnas.052705399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Abdul-Majid KB, Ly LH, Converse PJ, Geiman DE, McMurray DN, Bishai WR. Altered cellular infiltration and cytokine levels during early Mycobacterium tuberculosis sigC mutant infection are associated with late-stage disease attenuation and milder immunopathology in mice. BMC Microbiol. 2008;8:151. doi: 10.1186/1471-2180-8-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Sun R, Converse PJ, Ko C, Tyagi S, Morrison NE, Bishai WR. Mycobacterium tuberculosis ECF sigma factor sigC is required for lethality in mice and for the conditional expression of a defined gene set. Mol Microbiol. 2004;52:25–38. doi: 10.1111/j.1365-2958.2003.03958.x. [DOI] [PubMed] [Google Scholar]
- 396.Karls RK, Guarner J, McMurray DN, Birkness KA, Quinn FD. Examination of Mycobacterium tuberculosis sigma factor mutants using low-dose aerosol infection of guinea pigs suggests a role for SigC in pathogenesis. Microbiology. 2006;152:1591–600. doi: 10.1099/mic.0.28591-0. [DOI] [PubMed] [Google Scholar]
- 397.Calamita H, Ko C, Tyagi S, Yoshimatsu T, Morrison NE, Bishai WR. The Mycobacterium tuberculosis SigD sigma factor controls the expression of ribosome-associated gene products in stationary phase and is required for full virulence. Cell Microbiol. 2005;7:233–44. doi: 10.1111/j.1462-5822.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- 398.Raman S, Hazra R, Dascher CC, Husson RN. Transcription regulation by the Mycobacterium tuberculosis alternative sigma factor SigD and its role in virulence. J Bacteriol. 2004;186:6605–16. doi: 10.1128/JB.186.19.6605-6616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Wu QL, Kong D, Lam K, Husson RN. A mycobacterial extracytoplasmic function sigma factor involved in survival following stress. J Bacteriol. 1997;179:2922–9. doi: 10.1128/jb.179.9.2922-2929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Jensen-Cain DM, Quinn FD. Differential expression of sigE by Mycobacterium tuberculosis during intracellular growth. Microb Pathog. 2001;30:271–8. doi: 10.1006/mpat.2001.0431. [DOI] [PubMed] [Google Scholar]
- 401.Graham JE, Clark-Curtiss JE. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS) Proc Natl Acad Sci U S A. 1999;96:11554–9. doi: 10.1073/pnas.96.20.11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Ando M, Yoshimatsu T, Ko C, Converse PJ, Bishai WR. Deletion of Mycobacterium tuberculosis sigma factor E results in delayed time to death with bacterial persistence in the lungs of aerosol-infected mice. Infect Immun. 2003;71:7170–2. doi: 10.1128/IAI.71.12.7170-7172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Manganelli R, Fattorini L, Tan D, Iona E, Orefici G, Altavilla G, et al. The extra cytoplasmic function sigma factor sigma(E) is essential for Mycobacterium tuberculosis virulence in mice. Infect Immun. 2004;72:3038–41. doi: 10.1128/IAI.72.5.3038-3041.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol Microbiol. 2001;41:423–37. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- 405.Giacomini E, Sotolongo A, Iona E, Severa M, Remoli ME, Gafa V, et al. Infection of human dendritic cells with a Mycobacterium tuberculosis sigE mutant stimulates production of high levels of interleukin-10 but low levels of CXCL10: impact on the T-cell response. Infect Immun. 2006;74:3296–304. doi: 10.1128/IAI.01687-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Fontán PA, Aris V, Alvarez ME, Ghanny S, Cheng J, Soteropoulos P, et al. Mycobacterium tuberculosis sigma factor E regulon modulates the host inflammatory response. J Infect Dis. 2008;198:877–85. doi: 10.1086/591098. [DOI] [PubMed] [Google Scholar]
- 407.Datta P, Shi L, Bibi N, Balázsi G, Gennaro ML. Regulation of central metabolism genes of Mycobacterium tuberculosis by parallel feed-forward loops controlled by sigma factor E (σ(E)) J Bacteriol. 2011;193:1154–60. doi: 10.1128/JB.00459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–22. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- 409.Manganelli R, Provvedi R, Rodrigue S, Beaucher J, Gaudreau L, Smith I. Sigma factors and global gene regulation in Mycobacterium tuberculosis. J Bacteriol. 2004;186:895–902. doi: 10.1128/JB.186.4.895-902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–31. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 411.DeMaio J, Zhang Y, Ko C, Young DB, Bishai WR. A stationary-phase stress-response sigma factor from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1996;93:2790–4. doi: 10.1073/pnas.93.7.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Williams EP, Lee JH, Bishai WR, Colantuoni C, Karakousis PC. Mycobacterium tuberculosis SigF regulates genes encoding cell wall-associated proteins and directly regulates the transcriptional regulatory gene phoY1. J Bacteriol. 2007;189:4234–42. doi: 10.1128/JB.00201-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Chen P, Ruiz RE, Li Q, Silver RF, Bishai WR. Construction and characterization of a Mycobacterium tuberculosis mutant lacking the alternate sigma factor gene, sigF. Infect Immun. 2000;68:5575–80. doi: 10.1128/IAI.68.10.5575-5580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Geiman DE, Kaushal D, Ko C, Tyagi S, Manabe YC, Schroeder BG, et al. Attenuation of late-stage disease in mice infected by the Mycobacterium tuberculosis mutant lacking the SigF alternate sigma factor and identification of SigF-dependent genes by microarray analysis. Infect Immun. 2004;72:1733–45. doi: 10.1128/IAI.72.3.1733-1745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Cappelli G, Volpe E, Grassi M, Liseo B, Colizzi V, Mariani F. Profiling of Mycobacterium tuberculosis gene expression during human macrophage infection: upregulation of the alternative sigma factor G, a group of transcriptional regulators, and proteins with unknown function. Res Microbiol. 2006;157:445–55. doi: 10.1016/j.resmic.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 416.Lee JH, Geiman DE, Bishai WR. Role of stress response sigma factor SigG in Mycobacterium tuberculosis. J Bacteriol. 2008;190:1128–33. doi: 10.1128/JB.00511-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Smollett KL, Dawson LF, Davis EO. SigG does not control gene expression in response to DNA damage in Mycobacterium tuberculosis H37Rv. J Bacteriol. 2011;193:1007–11. doi: 10.1128/JB.01241-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Manganelli R, Dubnau E, Tyagi S, Kramer FR, Smith I. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol. 1999;31:715–24. doi: 10.1046/j.1365-2958.1999.01212.x. [DOI] [PubMed] [Google Scholar]
- 419.Raman S, Song T, Puyang X, Bardarov S, Jacobs WR, Jr., Husson RN. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J Bacteriol. 2001;183:6119–25. doi: 10.1128/JB.183.20.6119-6125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Park ST, Kang CM, Husson RN. Regulation of the SigH stress response regulon by an essential protein kinase in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2008;105:13105–10. doi: 10.1073/pnas.0801143105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Mehra S, Kaushal D. Functional genomics reveals extended roles of the Mycobacterium tuberculosis stress response factor sigmaH. J Bacteriol. 2009;191:3965–80. doi: 10.1128/JB.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Dutta NK, Mehra S, Martinez AN, Alvarez X, Renner NA, Morici LA, et al. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS One. 2012;7:e28958. doi: 10.1371/journal.pone.0028958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Mehra S, Golden NA, Stuckey K, Didier PJ, Doyle LA, Russell-Lodrigue KE, et al. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis. 2012;205:1203–13. doi: 10.1093/infdis/jis102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Kaushal D, Schroeder BG, Tyagi S, Yoshimatsu T, Scott C, Ko C, et al. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc Natl Acad Sci U S A. 2002;99:8330–5. doi: 10.1073/pnas.102055799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Dainese E, Rodrigue S, Delogu G, Provvedi R, Laflamme L, Brzezinski R, et al. Posttranslational regulation of Mycobacterium tuberculosis extracytoplasmic-function sigma factor sigma L and roles in virulence and in global regulation of gene expression. Infect Immun. 2006;74:2457–61. doi: 10.1128/IAI.74.4.2457-2461.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Hahn MY, Raman S, Anaya M, Husson RN. The Mycobacterium tuberculosis extracytoplasmic-function sigma factor SigL regulates polyketide synthases and secreted or membrane proteins and is required for virulence. J Bacteriol. 2005;187:7062–71. doi: 10.1128/JB.187.20.7062-7071.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Collins DM, Skou B, White S, Bassett S, Collins L, For R, et al. Generation of attenuated Mycobacterium bovis strains by signature-tagged mutagenesis for discovery of novel vaccine candidates. Infect Immun. 2005;73:2379–86. doi: 10.1128/IAI.73.4.2379-2386.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Agarwal N, Woolwine SC, Tyagi S, Bishai WR. Characterization of the Mycobacterium tuberculosis sigma factor SigM by assessment of virulence and identification of SigM-dependent genes. Infect Immun. 2007;75:452–61. doi: 10.1128/IAI.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Hu Y, Kendall S, Stoker NG, Coates AR. The Mycobacterium tuberculosis sigJ gene controls sensitivity of the bacterium to hydrogen peroxide. FEMS Microbiol Lett. 2004;237:415–23. doi: 10.1016/j.femsle.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 430.Fontán PA, Voskuil MI, Gomez M, Tan D, Pardini M, Manganelli R, et al. The Mycobacterium tuberculosis sigma factor sigmaB is required for full response to cell envelope stress and hypoxia in vitro, but it is dispensable for in vivo growth. J Bacteriol. 2009;191:5628–33. doi: 10.1128/JB.00510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Recchi C, Sclavi B, Rauzier J, Gicquel B, Reyrat JM. Mycobacterium tuberculosis Rv1395 is a class III transcriptional regulator of the AraC family involved in cytochrome P450 regulation. J Biol Chem. 2003;278:33763–73. doi: 10.1074/jbc.M305963200. [DOI] [PubMed] [Google Scholar]
- 432.Salzman V, Mondino S, Sala C, Cole ST, Gago G, Gramajo H. Transcriptional regulation of lipid homeostasis in mycobacteria. Mol Microbiol. 2010;78:64–77. doi: 10.1111/j.1365-2958.2010.07313.x. [DOI] [PubMed] [Google Scholar]
- 433.Maciag A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, et al. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol. 2007;189:730–40. doi: 10.1128/JB.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Zahrt TC, Song J, Siple J, Deretic V. Mycobacterial FurA is a negative regulator of catalase-peroxidase gene katG. Mol Microbiol. 2001;39:1174–85. doi: 10.1111/j.1365-2958.2001.02321.x. [DOI] [PubMed] [Google Scholar]
- 435.Cheigh CI, Senaratne R, Uchida Y, Casali N, Kendall LV, Riley LW. Posttreatment reactivation of tuberculosis in mice caused by Mycobacterium tuberculosis disrupted in mce1R. J Infect Dis. 2010;202:752–9. doi: 10.1086/655224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Uchida Y, Casali N, White A, Morici L, Kendall LV, Riley LW. Accelerated immunopathological response of mice infected with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional regulator. Cell Microbiol. 2007;9:1275–83. doi: 10.1111/j.1462-5822.2006.00870.x. [DOI] [PubMed] [Google Scholar]
- 437.Goldstone RM, Goonesekera SD, Bloom BR, Sampson SL. The transcriptional regulator Rv0485 modulates the expression of a pe and ppe gene pair and is required for Mycobacterium tuberculosis virulence. Infect Immun. 2009;77:4654–67. doi: 10.1128/IAI.01495-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Ramakrishnan L, Federspiel NA, Falkow S. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science. 2000;288:1436–9. doi: 10.1126/science.288.5470.1436. [DOI] [PubMed] [Google Scholar]
- 439.Li Y, Miltner E, Wu M, Petrofsky M, Bermudez LE. A Mycobacterium avium PPE gene is associated with the ability of the bacterium to grow in macrophages and virulence in mice. Cell Microbiol. 2005;7:539–48. doi: 10.1111/j.1462-5822.2004.00484.x. [DOI] [PubMed] [Google Scholar]
- 440.Basu S, Pathak SK, Banerjee A, Pathak S, Bhattacharyya A, Yang Z, et al. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J Biol Chem. 2007;282:1039–50. doi: 10.1074/jbc.M604379200. [DOI] [PubMed] [Google Scholar]
- 441.Frota CC, Papavinasasundaram KG, Davis EO, Colston MJ. The AraC family transcriptional regulator Rv1931c plays a role in the virulence of Mycobacterium tuberculosis. Infect Immun. 2004;72:5483–6. doi: 10.1128/IAI.72.9.5483-5486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Stewart GR, Snewin VA, Walzl G, Hussell T, Tormay P, O’Gaora P, et al. Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat Med. 2001;7:732–7. doi: 10.1038/89113. [DOI] [PubMed] [Google Scholar]
- 443.Uto T, Tsujimura K, Uchijima M, Seto S, Nagata T, Suda T, et al. A novel vaccine strategy to induce mycobacterial antigen-specific Th1 responses by utilizing the C-terminal domain of heat shock protein 70. FEMS Immunol Med Microbiol. 2011;61:189–96. doi: 10.1111/j.1574-695X.2010.00762.x. [DOI] [PubMed] [Google Scholar]
- 444.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–8. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 445.Singh A, Guidry L, Narasimhulu KV, Mai D, Trombley J, Redding KE, et al. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci U S A. 2007;104:11562–7. doi: 10.1073/pnas.0700490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, et al. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 2009;5:e1000545. doi: 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Banaiee N, Jacobs WR, Jr., Ernst JD. Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung and macrophages. Infect Immun. 2006;74:6449–57. doi: 10.1128/IAI.00190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Sanz J, Navarro J, Arbués A, Martín C, Marijún PC, Moreno Y. The transcriptional regulatory network of Mycobacterium tuberculosis. PLoS One. 2011;6:e22178. doi: 10.1371/journal.pone.0022178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Abomoelak B, Hoye EA, Chi J, Marcus SA, Laval F, Bannantine JP, et al. mosR, a novel transcriptional regulator of hypoxia and virulence in Mycobacterium tuberculosis. J Bacteriol. 2009;191:5941–52. doi: 10.1128/JB.00778-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Singh A, Jain S, Gupta S, Das T, Tyagi AK. mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol Lett. 2003;227:53–63. doi: 10.1016/S0378-1097(03)00648-7. [DOI] [PubMed] [Google Scholar]
- 451.Cheruvu M, Plikaytis BB, Shinnick TM. The acid-induced operon Rv3083-Rv3089 is required for growth of Mycobacterium tuberculosis in macrophages. Tuberculosis (Edinb) 2007;87:12–20. doi: 10.1016/j.tube.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 452.Li Y, Zhang Y. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob Agents Chemother. 2007;51:2092–9. doi: 10.1128/AAC.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Shi W, Zhang Y. PhoY2 but not PhoY1 is the PhoU homologue involved in persisters in Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:1237–42. doi: 10.1093/jac/dkq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Brennan MJ, Delogu G. The PE multigene family: a ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002;10:246–9. doi: 10.1016/S0966-842X(02)02335-1. [DOI] [PubMed] [Google Scholar]
- 455.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie. 2012;94:110–6. doi: 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 456.Sampson SL. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol. 2011;2011:497203. doi: 10.1155/2011/497203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Mukhopadhyay S, Balaji KN. The PE and PPE proteins of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2011;91:441–7. doi: 10.1016/j.tube.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 458.Delogu G, Brennan MJ. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect Immun. 2001;69:5606–11. doi: 10.1128/IAI.69.9.5606-5611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Cascioferro A, Delogu G, Colone M, Sali M, Stringaro A, Arancia G, et al. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol Microbiol. 2007;66:1536–47. doi: 10.1111/j.1365-2958.2007.06023.x. [DOI] [PubMed] [Google Scholar]
- 460.McAdam RA, Quan S, Smith DA, Bardarov S, Betts JC, Cook FC, et al. Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology. 2002;148:2975–86. doi: 10.1099/00221287-148-10-2975. [DOI] [PubMed] [Google Scholar]
- 461.Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, et al. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. 2010;6:e1001100. doi: 10.1371/journal.ppat.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Camus JC, Pryor MJ, Médigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148:2967–73. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 463.Anandakumar S, Shanmughavel P. Computational Annotation for Hypothetical Proteins of Mycobacterium tuberculosis. J Comput Sci Syst Biol. 2008;1:050–62. doi: 10.4172/jcsb.1000004. [DOI] [Google Scholar]
- 464.Mazandu GK, Mulder NJ. Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins. Int J Mol Sci. 2012;13:7283–302. doi: 10.3390/ijms13067283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Bigi F, Garcia-Pelayo MC, Nuñez-García J, Peralta A, Caimi KC, Golby P, et al. Identification of genetic markers for Mycobacterium pinnipedii through genome analysis. FEMS Microbiol Lett. 2005;248:147–52. doi: 10.1016/j.femsle.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 466.Blokpoel MC, O’Toole R, Smeulders MJ, Williams HD. Development and application of unstable GFP variants to kinetic studies of mycobacterial gene expression. J Microbiol Methods. 2003;54:203–11. doi: 10.1016/S0167-7012(03)00044-7. [DOI] [PubMed] [Google Scholar]
- 467.Kozak RA, Alexander DC, Liao R, Sherman DR, Behr MA. Region of difference 2 contributes to virulence of Mycobacterium tuberculosis. Infect Immun. 2011;79:59–66. doi: 10.1128/IAI.00824-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, et al. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci U S A. 2004;101:4865–70. doi: 10.1073/pnas.0305634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Cockle PJ, Gordon SV, Lalvani A, Buddle BM, Hewinson RG, Vordermeier HM. Identification of novel Mycobacterium tuberculosis antigens with potential as diagnostic reagents or subunit vaccine candidates by comparative genomics. Infect Immun. 2002;70:6996–7003. doi: 10.1128/IAI.70.12.6996-7003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol. 2003;48:833–43. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Purkayastha A, McCue LA, McDonough KA. Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr aene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect Immun. 2002;70:1518–29. doi: 10.1128/IAI.70.3.1518-1529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Hu Y, Coates AR. Mycobacterium tuberculosis acg gene is required for growth and virulence in vivo. PLoS One. 2011;6:e20958. doi: 10.1371/journal.pone.0020958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Liu K, Yu J, Russell DG. pckA-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology. 2003;149:1829–35. doi: 10.1099/mic.0.26234-0. [DOI] [PubMed] [Google Scholar]
- 474.Koul A, Choidas A, Treder M, Tyagi AK, Drlica K, Singh Y, et al. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J Bacteriol. 2000;182:5425–32. doi: 10.1128/JB.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Singh R, Singh A, Tyagi AK. Deciphering the genes involved in pathogenesis of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2005;85:325–35. doi: 10.1016/j.tube.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 476.Abomoelak B, Marcus SA, Ward SK, Karakousis PC, Steinberg H, Talaat AM. Characterization of a novel heat shock protein (Hsp22.5) involved in the pathogenesis of Mycobacterium tuberculosis. J Bacteriol. 2011;193:3497–505. doi: 10.1128/JB.01536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]