

Abstract
Various pattern recognition receptors (PRRs) have been implicated in the detection of viral RNA and subsequent interferon (IFN) gene expression, including the double-stranded RNA-dependent proteinkinase R (PKR). Now, a novel role of PKR has been unveiled, as it was shown that, upon the infection with certain viruses, PKR is crucial for the integrity of newly synthesized IFN mRNA, thereby generating an optimal host antiviral immune response. There is a need of future studies to investigate additional roles of PKR in innate immunity and the molecular understanding of this novel function of PKR.
Keywords: PKR, IFN mRNA, innate immunity, virus infections
The immediate response to viral pathogens relies on the innate immune system, which becomes active long before adaptive immune responses like neutralizing antibodies and cytotoxic T lymphocytes. A key component of innate immunity is the production of type I interferons (IFN-α/β), a family of cytokines produced by many different cell types to limit viral replication as well as virus dissemination in vivo. To elicit antiviral IFN responses, mammalian cells have evolved a variety of cellular sensors, also called pattern recognition receptors (PRRs). These sensors detect nucleic acids or other conserved structural components of invading microbes and subsequently initiate signaling cascades leading to the transcriptional induction of IFN-α/β.1 Upon their production, IFN-α/β are secreted and trigger cell defense mechanisms in an autocrine or paracrine manner through the binding to a common IFN-α/β receptor (IFNAR). IFNAR signaling finally results in the transcription of hundreds of IFN-inducible genes (ISGs), involved in apoptosis and cell growth inhibition to prevent virus propagation. One of the ISGs with a key role in the resistance to viral infections is the double-stranded RNA-dependent proteinkinase R (PKR), whose multiple antiviral actions are the focus of this article.
PKR was initially discovered due to its ability to potently block the translation of viral and cellular mRNAs in IFN-treated vaccinia virus (VV)-infected cells.2 The attempt to understand the mechanism behind the inhibition of protein synthesis during VV replication led to the identification of another enzyme, 2′, 5′-oligoadenylate synthetase (2′-5′-OAS).3 PKR phosphorylates one of the key regulatory factors in protein synthesis, the eukaryotic translation initiation factor 2 α (eIF2-α). Phosphorylation of eIF2-α by PKR leads to the inhibition of host and viral mRNA translation, thereby potently suppressing virus propagation (Fig. 1).4
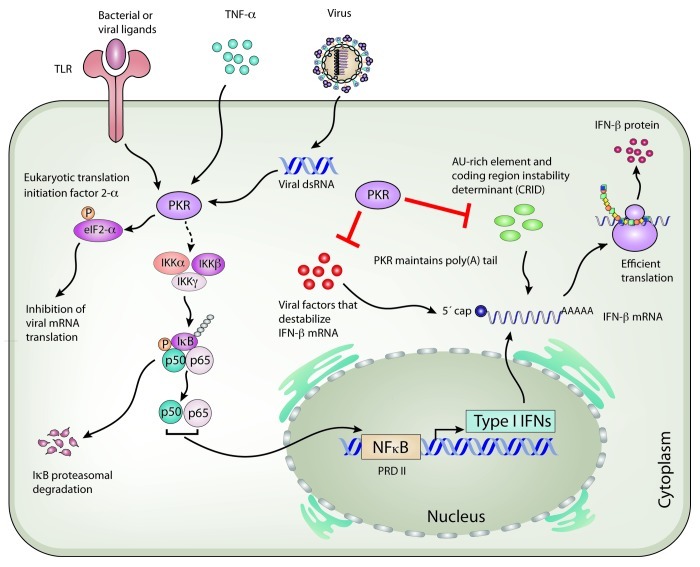
Figure 1. Role of PKR in IFN-β induction and possible regulation of IFN-β mRNA integrity. Several stimuli such as viral origin dsRNA, pathogen ligands and cytokines (e.g., TNF-α) activate the proteinkinase R (PKR). An induced PKR phosphorylates the eukaryotic translation initiation factor 2 (eIF2-α) leading to its inactivation, thereby suppressing protein synthesis. PKR also activates the IKKα/β/γ-complex, which induces the ubiquitination and proteasomal degradation of inhibitor of NFκB (IκB), resulting in the liberation of active NFκB dimers [RelA/p65 and NFκB1 or 2 (p50/p52)]. Translocation of NFκB into the nucleus leads to the transcription of several genes, including IFN-β to limit viral replication. Upon the infection of MDA5-dependent viruses such as encephalomyocarditis virus (ECMV) and Semliki Forest virus (SFV), PKR also regulates the integrity of IFN-β mRNA by establishing or maintaining its poly(A) tail. It is also possible that PKR positively regulates IFN transcript stability by interacting with destabilizing elements in the mRNA, such as AU-rich elements or coding region instability determinant (CRID). Alternatively, PKR might counteract virus-encoded factors that destabilize IFN-β mRNA to suppress the host antiviral IFN response. P indicates phosphorylation, Ub indicates ubiquitination, and dotted lines indicate indirect action of PKR.
PKR is a serine/threonine protein kinase consisting of two functionally distinct domains: an N-terminal regulatory domain for binding dsRNA (dsRBD) and a C-terminal catalytic domain, which is shared by other eIF2-α kinases.5 PKR is maintained as an inactive monomer under normal conditions and undergoes activation in response to dsRNA of viral or synthetic origin such as poly(I:C). In addition, PKR can be activated, either directly or indirectly, by various other stimuli, including oxidative stress, growth factors, cytokines and cellular proteins such as PKR-associated activator (PACT), or following the stimulation of Toll-like receptors (TLRs). PKR activation by these stimuli is believed to disrupt the autoinhibitory state of PKR, enabling its homodimerization and autophosphorylation, and thereby eIF2-α substrate binding to block viral protein synthesis.6
In addition to its well-established role as regulator of mRNA translation, PKR is also indirectly involved in a plethora of different signal transduction pathways, including the activation of c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription 1 and 3 (STAT1 and 3) and the tumor suppressor p53.6 Moreover, PKR indirectly activates the nuclear factor κB (NFκB), which has among other functions an important role in type I IFN induction (Fig. 1). The ability to detect viral dsRNA and to initiate signaling pathways that result in IFN gene expression, has suggested that PKR is a sensor for viral infections. Recently, it has been revealed that PKR functions through an eIF-2-mediated translational control mechanism to indirectly enhance IFN-β transcript levels. It was found that transient knockdown of PKR impaired IFN-β transcript induction, suggests that PKR differentially function in the signaling response leading to activation of IFN-β transcription.7 In line with this, various studies within the past 15 years found that PKR-deficient cells are defective in IFN-α/β production upon stimulation with poly(I:C) or viral infection.8-11 However, this clear picture of PKR antiviral function was complicated by the discovery of the cytosolic viral RNA sensors, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5). RIG-I and MDA5 have been shown to be the major receptor systems for the detection of viral RNA in the cytoplasm and subsequent induction of type I IFN transcription.12 Whereas RIG-I detects short dsRNA and/or ssRNA containing a 5′-triphosphate group, MDA5 is thought to sense long dsRNA. Consistent with this, in vitro as well as in vivo studies using RIG-I or MDA5 knockout mice demonstrated the critical role of RIG-I for the detection of paramyxoviruses, influenza viruses, vesicular stomatitis virus, and some flaviviruses including hepatitis C virus (HCV). In contrast, MDA5 recognizes primarily picornaviruses, including encephalomyocarditis virus (ECMV) and Theiler’s murine encephalomyelitis virus (TMEV). While our understanding of the receptor-signaling systems was substantially advanced by the identification of RIG-I and MDA5, the role of PKR and its relative contribution to antiviral IFN production remains elusive.
An important piece to this intricate puzzle of PKR function has recently been added, where it was demonstrate that PKR plays an essential role in IFN-α/β production in response to viral infections by regulating the integrity of IFN-β transcripts.13 By using PKR-deficient cells, the authors show that PKR is crucial for the IFN-α/β production in response to MDA5-dependent viruses like ECMV, TMEV and Semliki Forest virus (SFV), but not to RIG-I-dependent viruses such as Sendai virus or influenza. Surprisingly, Schulz et al. (2010) observed a striking difference between the IFN-β mRNA and protein levels produced by these cells: whereas IFN-β mRNA was highly induced in PKR-deficient cells upon ECMV or SFV infection, no or little IFN-β protein was made, supporting the idea that PKR has an important role in the post-transcriptional regulation of IFN-β production.13
In the attempt to decipher the molecular mechanism by which PKR regulates the synthesis of IFNs at a post-transcriptional level, Schulz et al. (2010) observed that the IFN-β transcripts produced in ECMV-infected PKR-deficient cells completely lacked a poly(A) tail, demonstrating that PKR is important for the integrity of IFN-β mRNA and therefore its translation into functional protein.13 Remarkably, the defect in poly(A)-tailing seen in PKR-deficient cells was specific for IFN-β transcripts since the poly(A)-tail of other mRNAs was fully intact. In addition, this effect of PKR on IFN-β mRNA stability was only observed upon the infection with MDA5-dependent viruses, but not with Sendai virus, which is detected by RIG-I. In accordance with the in vitro experiments, PKR knockout mice infected with ECMV exhibited significantly lower levels of IFN-β in the serum compared with wild-type mice, confirming the critical role of PKR in type I IFN production in vivo.13
While the findings by Schulz et al. (2010) constitute an important step to improve our understanding of PKR function, the precise molecular mechanism by which PKR regulates IFN transcript stability has yet to be elucidated. Several questions remain to be answered, including (1) what are the molecular targets of PKR for controlling IFN mRNA stability, (2) how does PKR exert its IFN mRNA-specific action and (3) why is this mechanism of PKR mRNA stabilizing function specific for cells infected with MDA5-dependent viruses? Given that the translational regulator eIF2-α is the best-characterized substrate of PKR, Schulz et al. (2010) asked whether PKR-dependent IFN-α/β production requires phosphorylation of eIF2-α.13 To address this question, mouse embryonic fibroblasts (MEFs) expressing either wild-type eIF2-α, or a mutant protein in which the critical site for phosphorylation is mutated, were either infected with ECMV or stimulated with poly(I:C), followed by measuring IFN-α or IFN-β protein in the supernatant. This showed that PKR-mediated IFN production did not require phosphorylation of eIF2-α, suggesting that PKR inhibits viral translation and promotes IFN protein synthesis through two different mechanisms. To this end, it will be interesting to determine in which cellular compartment PKR exerts its effect on IFN transcript stability. PKR has been shown to be associated to ribosomes, and several ribosomal proteins have been reported to bind to PKR.14 In addition, PKR protein expression has been described in the nuclei of human and murine cells,15 as well as in HCV-infected patients.16 Moreover, it has been shown that IFN-γ mRNA activates PKR through a pseudoknot in its 5′ untranslated region and controls its own translation yield.17 Thus, future studies directed toward correlating PKR multiple functions and its subcellular localizations will give us more insights into the molecular details of PKR antiviral activity.
The production of cytokines and in particular of IFN-β is achieved by various post-transcriptional mechanisms, including the regulation of translation initiation and mRNA decay.18 Many cellular factors have been described to control the turnover of cytokine-encoding mRNAs. These factors interact with specific elements located within the mRNA, and through multiple mechanisms, either promote or hamper transcript amount and stability. Recently, several destabilizing elements were identified in the IFN-β mRNA, including AU-rich elements (AREs) and coding region instability determinant (CRID). While the presence of these motifs promotes IFN-β mRNA deadenylation and decay, their removal abolished the shortening of the poly(A) tail, thereby increasing IFN-β transcript stability and protein expression (Fig. 1).19 The cellular factors recruited to these destabilizing elements and their detailed molecular mechanisms for controlling IFN mRNA stability have not yet been well characterized. For other cytokine-encoding transcripts it has been shown that their AREs recruit specific ARE-binding proteins (ARE-BPs), such as TIA-1 (T-cell internal antigen-1) and AUF1/heterogeneous nuclear ribonucleoprotein D (hnRNP D). Each of these ARE-BPs can individually affect the translation or decay of the mRNA, however, in most cases it is the combined effect of multiple ARE-BPs that determines the final outcome of cytokine production.18 Interestingly, the ability of ARE-BPs to bind to AREs, as well as their functional activity in mRNA translation or decay, is regulated by covalent modifications, including kinase-dependent phosphorylation and phosphatase-dependent dephosphorylation.18 It is therefore intriguing to speculate that some of the ARE-BPs implicated in IFN-β mRNA destabilization may be the target of PKR-mediated phosphorylation to modulate antiviral IFN production. Thus, determining how PKR regulates IFN protein synthesis, as well as the identification of new PKR interacting partners may improve our knowledge about the cellular machinery for controlling mRNA stability. Alternatively to the hypothesis that PKR is directly involved in IFN transcript integrity, it is also possible that PKR antagonizes viral factors promoting the instability of IFN mRNAs (Fig. 1). On the other hand, viruses have developed a myriad of ways to suppress PKR antiviral function, including sequestration of dsRNA, suppressing PKR-induced eIF2-α phosphorylation and inducing PKR degradation.20 Therefore, it would be not surprising if some viruses have also evolved strategies to specifically suppress the transcript stabilizing role of PKR.
The idea of PKR as key regulator of IFN protein synthesis is supported by other recent studies demonstrating that PKR plays a non-redundant role in the IFN response to viral infections.21-23 On the other hand, it is well-known that PKR is a potent activator of NFκB, thereby inducing IFN transcription in concerted action with the interferon-regulatory factors 3 and 7 (IRF-3/7). Specially, PKR was shown to activate the IκB kinase (IKK)-complex, which results into the degradation of the inhibitor of NFκB (IκB), leading to NF nuclear translocation (Fig. 1).24 Consistent with this, activation of NFκB and IFN induction were impaired in response to poly(I:C) treatment in PKR knockout MEFs in contrast to wild-type MEFs.14 In addition, PKR is also implicated in the activation of interferon regulatory factor 1 (IRF 1), whose expression is strongly upregulated upon viral infection, and which mainly acts as a transcriptional activator of IFN-α/β gene expression.25 It will be thus necessary to determine the relative contribution of PKR’s roles in transcriptional control and mRNA stability for establishing an efficient IFN response. In this regard, PKR may be preferentially engaged in type I IFN transcription in response to some viruses, while mainly controlling IFN protein synthesis in response to other viruses. Alternatively, it is also possible that the combined effect of both mechanisms, promoting IFN gene expression and its translation into functional protein, may account for the potent increase of type I IFN production induced by PKR, as seen in vitro and in vivo. In support of this concept are the results of Barry et al. (2009), who show that wild-type MEFs infected with SFV showed significantly higher levels of IFN transcripts and functional IFN compared with PKR knockout cells; remarkably, however, the levels of IFN protein were increased more profoundly than IFN mRNA levels,21 suggesting that the concerted action of PKR’s transcriptional as well as posttranscriptional control of IFN synthesis may be important for a successful antiviral response.
PKR was one of the first PRR discovered, and it was long believed that the function of PKR is well understood. Studies in the past few years have led to new insights into the multifaceted function of PKR and to the re-evaluation of this dogma. Especially its recently identified role as IFN transcript stabilizing factor substantially advances our understanding about PKR, and demonstrates the crucial role of this kinase for an optimal host antiviral IFN response.
Acknowledgments
The authors wish to apologize to all authors whose work was not cited due to space restrictions. This work was supported by the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas 221-2007-935). We would like to thank Michaela U. Gack for critical reading of this manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/virulence/article/23134
References
- 1.Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–83. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Metz DH, Esteban M. Interferon inhibits viral protein synthesis in L cells infected with vaccinia virus. Nature. 1972;238:385–8. doi: 10.1038/238385a0. [DOI] [PubMed] [Google Scholar]
- 3.Hovanessian AG, Kerr IM. The (2′-5′) oligoadenylate (pppA2′-5’A2′-5’A) synthetase and protein kinase(s) from interferon-treated cells. Eur J Biochem. 1979;93:515–26. doi: 10.1111/j.1432-1033.1979.tb12850.x. [DOI] [PubMed] [Google Scholar]
- 4.Williams BR. Signal integration via PKR. Sci STKE. 2001;2001:re2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 5.Feng GS, Chong K, Kumar A, Williams BR. Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc Natl Acad Sci U S A. 1992;89:5447–51. doi: 10.1073/pnas.89.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–60. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAllister CS, Taghavi N, Samuel CE. Protein Kinase PKR Amplification of Interferon β Induction Occurs through Initiation Factor eIF-2α-mediated Translational Control. J Biol Chem. 2012;287:36384–92. doi: 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–8. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 9.McAllister CS, Samuel CE. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem. 2009;284:1644–51. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith EJ, Marié I, Prakash A, García-Sastre A, Levy DE. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem. 2001;276:8951–7. doi: 10.1074/jbc.M008717200. [DOI] [PubMed] [Google Scholar]
- 11.Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schäfer R, Kumar A, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–61. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu S, Romano PR, Wek RC. Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J Biol Chem. 1997;272:14434–41. doi: 10.1074/jbc.272.22.14434. [DOI] [PubMed] [Google Scholar]
- 15.Jeffrey IW, Kadereit S, Meurs EF, Metzger T, Bachmann M, Schwemmle M, et al. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp Cell Res. 1995;218:17–27. doi: 10.1006/excr.1995.1126. [DOI] [PubMed] [Google Scholar]
- 16.MacQuillan GC, Caterina P, de Boer B, Allan JE, Platten MA, Reed WD, et al. Ultra-structural localisation of hepatocellular PKR protein using immuno-gold labelling in chronic hepatitis C virus disease. J Mol Histol. 2009;40:171–6. doi: 10.1007/s10735-009-9227-0. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–32. doi: 10.1016/S0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 18.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–9. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 19.Pasté M, Huez G, Kruys V. Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3′-untranslated region and an instability sequence in the coding region. Eur J Biochem. 2003;270:1590–7. doi: 10.1046/j.1432-1033.2003.03530.x. [DOI] [PubMed] [Google Scholar]
- 20.Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–10. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Barry G, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, Kohl A, et al. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J Gen Virol. 2009;90:1382–91. doi: 10.1099/vir.0.007336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpentier PA, Williams BR, Miller SD. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia. 2007;55:239–52. doi: 10.1002/glia.20450. [DOI] [PubMed] [Google Scholar]
- 23.Gilfoy FD, Mason PW. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:11148–58. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–90. doi: 10.1128/MCB.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 1997;16:406–16. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]