

Abstract
Laminin 332 is an essential component of the dermal-epidermal junction, a highly specialized basement membrane zone that attaches the epidermis to the dermis and thereby provides skin integrity and resistance to external mechanical forces. Mutations in the LAMA3, LAMB3 and LAMC2 genes that encode the three constituent polypeptide chains, α3, β3 and γ2, abrogate or perturb the functions of laminin 332. The phenotypic consequences are diminished dermal-epidermal adhesion and, as clinical symptoms, skin fragility and mechanically induced blistering. The disorder is designated as junctional epidermolysis bullosa (JEB). This article delineates the signs and symptoms of the different forms of JEB, the mutational spectrum, genotype-phenotype correlations as well as perspectives for future molecular therapies.
Keywords: skin fragility, blistering, hemidesmosome, granulation tissue, basement membrane
Introduction
In the skin laminin 332 is an essential component of the dermal-epidermal basement membrane.1 Its loss leads to the severe skin fragility disorder junctional epidermolysis bullosa Herlitz type, which restricts the life expectancy to few months or years.2,3 The dermal-epidermal junction zone contains a highly specialized basement membrane suprastructure, which provides the skin integrity and resistance against external mechanical forces.4 This suprastructure consists of a planar core, formed by the laminin 332- and the collagen IV-networks, which are superimposed and welded together by perlecan.5 Functional domains of a number of other proteins—keratinocyte cell surface and extracellular matrix molecules—are inserted into this network core, and highly specific protein-protein interactions6 secure the adhesion of the skin layers.
Laminin 332 can be regarded as a supramolecular bridge between the basal keratinocytes of the epidermis and the underlying dermis. Through specific interactions with integrin α6β4 in the hemidesmosomes and α3β1 in the focal adhesions it links the surface of basal keratinocytes with the dermal-epidermal basement membrane. On the dermal side, through specific binding to collagen VII,7 it links the anchoring fibrils to the basement membrane. The anchoring fibrils extend into the dermis and bind to dermal fibrils, thus securing the adhesion of the dermal-epidermal basement membrane to the dermal extracellular matrix. Apart from the structural role as an adhesion protein, laminin 332 also modulates cell behavior by transmitting information into keratinocytes via α6β4 and α3β1 integrins (“outside-in signaling”).4 The laminin 332-mediated cell signaling is believed to play a pivotal role in epidermal adhesion, cell survival, migration and regeneration (Rousselle et al., this issue).
Mutations in the LAMA3, LAMB3 and LAMC2 genes, which encode the α3, β3 and γ2 chains of laminin 332,8 abrogate or perturb the functions of this laminin. The consequences are diminished dermal-epidermal adhesion and mechanically induced skin blistering and fragility as clinical symptoms in both humans and animals. The disorder is collectively called junctional epidermolysis bullosa.9 In the following, its signs and symptoms, the mutational spectrum and genotype-phenotype correlations are delineated.
Junctional Epidermolysis Bullosa (JEB)
JEB is a clinically and genetically heterogeneous group of skin fragility disorders. It is characterized by mechanically induced skin and mucosal blistering and by chronic wounds. Based on clinical characteristics, the group can be divided into two main forms: (1) JEB-Herlitz (JEB-H), in which extreme fragility of the skin and mucous membranes usually leads to death within the first years of life and (2) milder forms collectively called JEB-other (also designated as non-Herlitz). JEB-H is caused by loss-of-function mutations in LAMA3, LAMB3 and LAMC2, leading to complete loss of laminin 332. JEB-other is associated with mutations in the above three genes or in COL17A1, the gene coding for collagen XVII, a binding ligand of laminin 332. Rare cases of JEB are associated with integrin α6β4 deficiency and result in JEB with pyloric atresia. In this review, we will focus on the clinical and molecular findings in JEB and mutations in the laminin 332 genes.
JEB Herlitz
JEB-H is a mostly lethal skin disorder. Absence of laminin 332 causes tissue separation along the lamina lucida of the dermal-epidermal basement membrane, i.e., the intact epidermis separates into the blister roof and the basement membrane proper remains on the blister floor. The clinical hallmark is profound skin fragility upon slightest trauma. Affected individuals present with mucocutaneous blistering already at birth, and with time the blistering involves most of the body surface (Fig. 1A). Exuberant granulation tissue, typically around nose and mouth, the buttocks and around the nail folds, is an almost pathognomonic sign of the disease.2 The mucous membranes are affected in all patients, and hoarseness and difficulty to eat are common. Loss of fluids and protein render the affected infants susceptible to infections. The patients suffer from extreme pain.10,11 The long-term consequences include failure to thrive, anemia, dyspnoea, pneumonia or sepsis, which account for the high mortality within the two first years of life.12
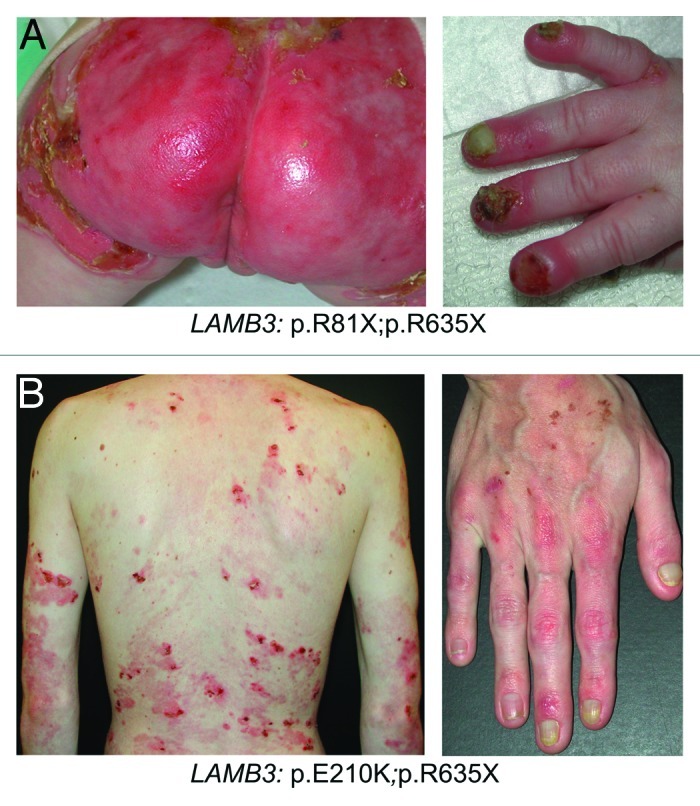
Figure 1. Clinical features of JEB-H and JEB-other. (A) Extensive blisters and erosions on the gluteal area and pronounced nail involvement in an infant with JEB-H and LAMB3 null mutations. (B) Blisters and crusts on the trunk and hand of a 25-y-old patient with JEB-other who is compound heterozygous for a nonsense and a missense mutation in the LAMB3 gene. Note also the partial nail dystrophy.
JEB-other
The term JEB-other was coined for a heterogeneous group of mild and moderate JEB phenotypes. Characteristically, laminin 332 is present in the skin, at least to some extent. Clinically, the patients exhibit mechanically induced, localized or widespread, skin blistering followed by slight atrophy and hypopigmentation at sites of healed blisters (Fig. 1B). Mucous membranes can also be affected, but not as severely as in JEB-H. Other manifestations include different degrees of hair loss, enamel defects13 and dystrophy or loss of nails. Granulation tissue and chronic wounds, or involvement of the cornea,14 larynx15 or urinary tract16 can occur. These phenotypes result from different mutation constellations in the genes encoding laminin 332. Clinically very similar phenotypes can be caused by mutations of collagen XVII,17 which is a ligand of laminin 332.
Laryngo-onycho-cutaneous (LOC) syndrome
LOC syndrome is a subtype of JEB caused by specific mutations in the LAMA3 gene.18 Thus far, the disorder is confined to patients from the Punjabi Muslim population and characterized by formation of extensive granulation tissue in skin areas of repeated trauma and in the laryngeal and conjunctival mucosa. The onset is at birth with a hoarse cry; later chronic wounds and granulation tissue develop. The ocular lesions can lead to blindness;19 the larynx involvement may require tracheostomy.18
Molecular Diagnosis of JEB
Since the molecular basis of JEB is heterogeneous, yet well understood, immunofluorescence mapping of a skin biopsy specimen is the first diagnostic tool. This technique utilizes antibodies to specific components of the dermal-epidermal junction zone to map their location in blistered skin. The constellation of immunofluorescence signals of keratins in the blister roof and collagen IV on the blister floor demonstrates tissue separation along the lamina lucida of the basement membrane, typical for JEB. This approach can also reveal the candidate gene, if the staining pattern of a particular protein is altered (Fig. 2). Negative signals with antibodies to all three laminin 332 chains (e.g., BM165 to the laminin α3 chain,20-24 6F12 to the laminin β3 chain23,25 and GB3 to the laminin γ2 chain26) indicate the diagnosis of JEB-H, and a severe prognosis. In JEB-other laminin 332 staining may appear attenuated. In contrast, loss of the collagen XVII signal suggests that JEB-other is caused by mutations in the COL17A1 gene. However, because of the intimate functional interaction between these molecules, the staining pattern of both binding partners may be altered. Identification of the candidate gene facilitates mutation analysis, which is required for precise diagnosis, prognostication and prenatal diagnosis.
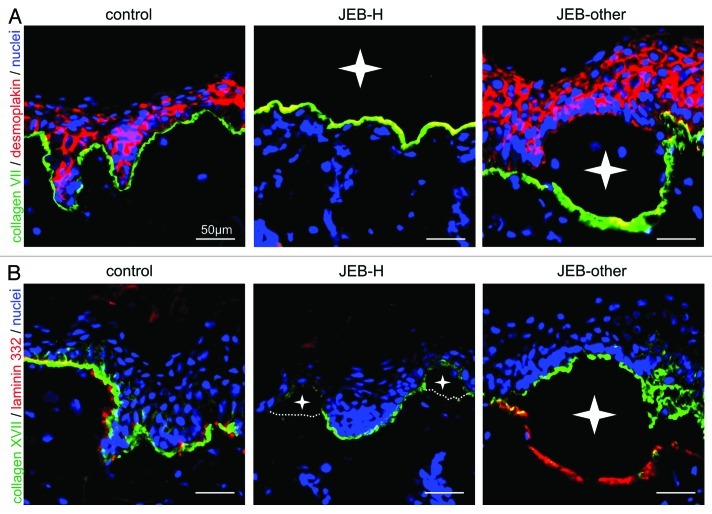
Figure 2. Immunofluorescence mapping of JEB-H and JEB-other skin. (A) Junctional blister formation in JEB skin is demonstrated by a double staining of collagen VII (green) and desmoplakin (red). Nuclei are stained with DAPI (blue). Left panel: Normal control. Middle panel: JEB-H. Right panel: JEB-other. Note that dermal-epidermal detachment leads to complete loss of the epidermis in JEB-H skin and only collagen VII staining is seen on the blister floor. In JEB-other collagen VII is found on blister floor and desmoplakin on blister roof. (B) A double staining of collagen XVII (green) and laminin 332 (red) in control, JEB-H and JEB-other skin. Nuclei are stained with DAPI (blue). In both JEB-H and JEB-other, collagen XVII is found in the blister roof. The expression of laminin 332 is completely lost in JEB-H, whereas the signal is well visible in JEB-other. Blister cavities are depicted with white stars. For all panels, bar = 50 μm.
In some centers transmission electron microscopy is used to diagnose EB. In JEB it demonstrates the blistering level along the lamina lucida of the basement membrane and abnormalities of the hemidesmosomes, which are typically absent, rudimentary or reduced in number, depending on the underlying molecular defect.
Mutational Spectrum
In laminin 332-deficient JEB, mutations are harbored by one of the three genes, LAMA3, LAMB3 and LAMC2. In about 80% of the cases, LAMB3 is affected and, therefore, this is the first gene to be screened.27 LAMB3 on 1q32 contains 23 exons and generates a transcript spanning 4,033 bp, which are translated into a polypeptide of 1,172 amino acids. So far, 87 different mutations (26 nonsense, 10 missense, 16 splicing, 23 small deletions, 8 small insertions, 1 indel, 1 gross deletion and 2 gross insertions/duplications) have been reported (HGMD Professional 2012.1). The majority of these are predicted to lead to premature termination codons, mRNA decay and synthesis of no protein or to truncated unstable polypeptides. By far the most frequent mutation is p.R635X, a mutational hot spot, which accounts for 45–63% of all mutated LAMB3 alleles in JEB-H.2,28 Several other recurrent or population-specific mutations have also been reported including, c.957ins77, p.Q243X and p.R42X.27,29-31 The milder phenotypes in patients with JEB-other often result from a combination of pathogenic variants in the laminin 332 genes, involving compound heterozygosity for a nonsense or frameshift mutation with a missense or splice-site mutation and allowing some expression of at least partially functional laminin 33232,33 (Fig. 3).

Figure 3. Missense mutations reported in patients with JEB-other. The schematic representation of the laminin 332 molecule is used to demonstrate the positions of the mutated amino acids (according to HGMD Professional 2012.2). These are predicted to allow expression of full-length, mutated laminin 332 molecules, which are functionally perturbed.
Prognostication of the consequences of the mutations often requires mRNA and protein studies. For example, an exceptional spontaneously ameliorating course of JEB was associated with the out-of-frame deletion c.1587_1588delAG, p.G530MfsX5 in compound heterozygosity with p.R635X.34 Although mRNA analysis performed shortly after birth demonstrated mRNA decay, with advancing age, the mRNA carrying the deletion generated a new internally shortened β-3 transcript through illegitimate splicing. The truncated polypeptide lacking the N-terminal part of the rod domain II was at least in part functional assuring dermal-epidermal adhesion.34
Besides LAMB3, mutations have been described in LAMA3, and in LAMC2, in about 10% of patients, each.27 LAMA3, located on 18q11.2, contains 76 exons and has five alternative transcripts. In the skin, the LAMA3A transcript comprises exons 39–76 (5,175 bp open reading frame, encoding 1,724 amino acids), and LAMA3B spans exons 1–38 and 40–76 (10,505 bp open reading frame and 3,333 amino acids).35 So far, 37 LAMA3 mutations have been reported (six missense mutations, 12 nonsense mutations, six splice site mutations and 13 small insertions and deletions; HGMD Professional 2012.1). The mutation c.151insG in exon 39, the only exon specific to the LAMA3A isoform, leads to loss of the laminin α3a polypeptide alone, and to clinical features of LOC syndrome.18 LAMC2 contains 23 exons, generates a transcript of 5,147 bp and codes for 1,193 amino acids. Thirty-five LAMC2 mutations have been described so far (13 nonsense mutations, five splice site mutations, 12 small deletions, one small insertion, three indel and one gross insertion/duplication) and are all predicted to lead to premature termination codons.
Unusual Phenotype-Genotype Correlations
JEB with laminin 332 mutations leading to premature termination codons and spontaneous amelioration with age
In very rare cases of JEB, an intriguing amelioration of the severe JEB-H phenotype developed with advancing age (own unpublished observations).32,34,36 The patients were born with severe skin fragility and both immunofluorescence staining of the skin and the mutations were suggestive of JEB-H, i.e., laminin 332 was absent and the mutation analyses disclosed homozygous nonsense or frameshift mutations in LAMA3, LAMB3 or LAMC2 genes. However, unexpectedly, with advancing age the phenotype grew milder, and laminin 332 protein appeared in the skin, allowing the patients to survive and grow normally. The molecular mechanisms of such changes encompassed alternative splicing that led to skipping of the exon carrying the null mutation32,34 or spontaneous read-through of the null allele.36 At protein level, the result was synthesis of mutated or truncated, partially functional laminin 332 polypeptides. These observations imply activation of cryptic splice-sites, which allow in-frame splicing-out of the mutated exon. Alternatively, activation of read-through mechanisms could take place after birth, suggesting age-dependent regulation of laminin 332 expression.
Digenic JEB
A unique case of a patient with severe, non-lethal JEB and mutations in COL17A1 and LAMB3 has been reported in the literature.37 Collagen XVII was negative and laminin 332 attenuated in the skin of a patient, who carried two COL17A1 null mutations and the recurrent LAMB3 mutation p.R635X. The clinical features included severe skin blistering with minimally affected mucous membranes and paronychia-like nails. The mother of the patient is a double heterozygote for a null mutation in LAMB3 and COL17A1. She shows that a reduction of about 50% of the expression of both laminin 332 and collagen XVII is tolerable, if the other hemidesmosomal components are normal.
Revertant mosaicism
Revertant mosaicsm is a phenomenon occurring in an individual with a disease-causing germline mutation, wherein a subpopulation of cells re-acquires the wild-type phenotype through a naturally occurring recombination, back or second-site mutation.38 This so called “natural gene therapy” was first reported in 1997 in a patient with JEB and mutations in COL17A1.39 Since then it has been described in patients with different EB subtypes,40-43 including two patients with JEB-other due to LAMB3 mutations.44 In contrast to the hypopigmented and atrophic JEB-other skin, the healthy-appearing skin spots were normally pigmented and not atrophic. Seven skin areas were investigated and in each a different second-site mutation was found, that corrected the germline mutation in one allele,44 resulting in expression of functional laminin 332. Interestingly, revertant mosaicism has been reported in all patients with JEB and COL17A1 mutations in a Dutch patient cohort.40 Investigation of more patients with revertant mosaicism and mutations in the laminin 332 genes is required in order to draw conclusions about the occurrence of this phenomenon in JEB.
Animal Models
A number of animal models for JEB exist; some of them caused by naturally occurring mutations in the responsible genes, others developed after genetic modification. These models have provided important insights into the disease mechanisms and are useful for testing novel therapeutic approaches.45,46 All three laminin 332 chains (α3, β3 and γ2) have been experimentally mutated in mice, which show extensive blistering at birth and, thus, recapitulate the human disease.47 The first laminin 332 knock out mouse model was a spontaneously mutated mouse with a pathogenic lamb3 genetic variant. Soon after birth, the mouse shows pronounced blistering of the entire skin surface and often large denuded areas. The nails, tongue and pharynx are also involved. The affected mice die within 24 h, usually without feeding.48
The α3 knockout mice die within the first days of life due to severe blistering.49 They have smaller stomachs and weigh less than their wild-type littermates. Further disease signs encompass abnormalities in ameloblast differentiation, and abnormal glomerulogenesis with perturbed maturation of glomerular endothelial cells and mesangial cells.50
A mouse model for deficiency of the laminin γ2 chain results from a frameshift deletion of lamc2 exon 8.51 The mice show varying levels of blistering, mostly on paws and legs. The mice grow smaller and weaker and die within 5 d due to failure to thrive.51 Despite morphologically aberrant tracheal hemidesmosomes, the lung branching morphogenesis and epithelial differentiation were not perturbed in these mice.52
Naturally occurring mutations in the laminin 332 genes have been reported in several animals, e.g., horses,53,54 sheep,55 rats,56 dogs57 and cats.58 The disease manifestations, both severe and mild forms, are very similar to human JEB. After elucidation of the underlying disease mechanisms these animals may become useful for testing experimental therapies.
Perspectives for Molecular Therapies
Currently, there is no cure for patients with JEB, and the therapeutic options are restricted to symptomatic care. Considering the high morbidity and mortality in JEB-H, a precise diagnosis is a prerequisite for the best management of the patients and their families. Although several invasive treatments have been used to treat the severe clinical features and symptoms of the patients, studies have shown that they do not prolong the life span significantly.59,60 Patients with JEB-other have a normal life span, and their management aims at alleviating symptoms and promoting wound healing.
The tremendous progress in understanding the genetic basis and the molecular disease mechanisms in JEB facilitates development of molecular therapy approaches.61-64 A successful gene therapy experiment was conducted in 2005 in an adult patient with JEB-other and LAMB3 mutations.65 An epidermal cell population enriched in stem cells of this patient was transduced with a retroviral vector expressing LAMB3 cDNA and used to prepare genetically corrected epidermal grafts. These were then transplanted onto surgically prepared areas on the patient's legs. Laminin 332 was properly synthesized in the grafts, resulting in normal levels of functional laminin 332 and firmly adherent epidermis that remained stable for the follow up period of three and a half years.65,66 This study offers a proof of principle for an ex vivo gene therapy for EB. However, concerns over the risks associated with the use of retroviral vectors have led to a more stringent regulatory environment, and “safer” therapeutic options are currently pursued.66
An approach that would circumvent the use of vectors takes advantage of spontaneous gene repair, so called revertant mosaicism (see above). Since 2007, when the first patients with JEB-other due to LAMB3 mutations and healthy skin spots were reported, revertant mosaicism has been found in all subtypes of EB40-43,67 and, indeed, it seems to be more common than anticipated. The therapeutic relevance for EB is intriguing, but currently hampered by the experience that the revertant cells are harder to cultivate than expected.68 After establishing optimal isolation and growth conditions for revertant keratinocytes, the possibility of applying cell therapy with the patient’s own naturally corrected keratinocytes will come closer. The advantages of a therapy using revertant keratinocytes are the natural correction of the mutation and the lack of immune response to therapeutic cells or proteins.
Acknowledgments
The work of the authors is supported by the Network Epidermolysis bullosa grant from the Federal Ministry for Education and Research (BMBF), the German Research Foundation DFG (SFB 850/B6), the Excellence Initiative of the German Federal Governments and the Freiburg Institute for Advanced Studies FRIAS, School of Life Sciences—LifeNet and the E-RARE grants “Kindlernet” and “RevertantEB” to C.H. and L.B.-T.
Glossary
Abbreviations:
- DAPI
4',6-diamidino-2-phenylindole stain
- EB
epidermolysis bullosa
- JEB
junctional epidermolysis bullosa
- JEB-H
junctional epidermolysis bullosa-Herlitz
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/22418
References
- 1.Sugawara K, Tsuruta D, Ishii M, Jones JC, Kobayashi H. Laminin-332 and -511 in skin. Exp Dermatol. 2008;17:473–80. doi: 10.1111/j.1600-0625.2008.00721.x. [DOI] [PubMed] [Google Scholar]
- 2.Laimer M, Lanschuetzer CM, Diem A, Bauer JW. Herlitz junctional epidermolysis bullosa. Dermatol Clin. 2010;28:55–60. doi: 10.1016/j.det.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Has C, Bruckner-Tuderman L. [Epidermolysis bullosa : Diagnosis and therapy] Hautarzt. 2011;62:82–90. doi: 10.1007/s00105-010-2049-x. [DOI] [PubMed] [Google Scholar]
- 4.Van Agtmael T, Bruckner-Tuderman L. Basement membranes and human disease. Cell Tissue Res. 2010;339:167–88. doi: 10.1007/s00441-009-0866-y. [DOI] [PubMed] [Google Scholar]
- 5.Behrens DT, Villone D, Koch M, Brunner G, Sorokin L, Robenek H, et al. The epidermal basement membrane is a composite of separate laminin- or collagen IV-containing networks connected by aggregated perlecan, but not by nidogens. J Biol Chem. 2012;287:18700–9. doi: 10.1074/jbc.M111.336073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aumailley M, El Khal A, Knöss N, Tunggal L. Laminin 5 processing and its integration into the ECM. Matrix Biol. 2003;22:49–54. doi: 10.1016/S0945-053X(03)00013-1. [DOI] [PubMed] [Google Scholar]
- 7.Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, Burgeson RE. Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol. 1997;138:719–28. doi: 10.1083/jcb.138.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–32. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Chiang LY, Poole K, Oliveira BE, Duarte N, Sierra YA, Bruckner-Tuderman L, et al. Laminin-332 coordinates mechanotransduction and growth cone bifurcation in sensory neurons. Nat Neurosci. 2011;14:993–1000. doi: 10.1038/nn.2873. [DOI] [PubMed] [Google Scholar]
- 11.Fine JD, Johnson LB, Weiner M, Suchindran C. Assessment of mobility, activities and pain in different subtypes of epidermolysis bullosa. Clin Exp Dermatol. 2004;29:122–7. doi: 10.1111/j.1365-2230.2004.01428.x. [DOI] [PubMed] [Google Scholar]
- 12.Yuen WY, Lemmink HH, van Dijk-Bos KK, Sinke RJ, Jonkman MF. Herlitz junctional epidermolysis bullosa: diagnostic features, mutational profile, incidence and population carrier frequency in the Netherlands. Br J Dermatol. 2011;165:1314–22. doi: 10.1111/j.1365-2133.2011.10553.x. [DOI] [PubMed] [Google Scholar]
- 13.Bruckner-Tuderman L. [Skin and teeth] J Dtsch Dermatol Ges. 2005;3:327–8. doi: 10.1111/j.1610-0387.2005.05716.x. [DOI] [PubMed] [Google Scholar]
- 14.Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, et al. Eye involvement in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry. Am J Ophthalmol. 2004;138:254–62. doi: 10.1016/j.ajo.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 15.Fine JD, Johnson LB, Weiner M, Suchindran C. Tracheolaryngeal complications of inherited epidermolysis bullosa: cumulative experience of the national epidermolysis bullosa registry. Laryngoscope. 2007;117:1652–60. doi: 10.1097/MLG.0b013e318093ed8e. [DOI] [PubMed] [Google Scholar]
- 16.Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, et al. Genitourinary complications of inherited epidermolysis bullosa: experience of the national epidermylosis bullosa registry and review of the literature. J Urol. 2004;172:2040–4. doi: 10.1097/01.ju.0000143200.86683.2c. [DOI] [PubMed] [Google Scholar]
- 17.Kiritsi D, Kern JS, Schumann H, Kohlhase J, Has C, Bruckner-Tuderman L. Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J Med Genet. 2011;48:450–7. doi: 10.1136/jmg.2010.086751. [DOI] [PubMed] [Google Scholar]
- 18.McLean WH, Irvine AD, Hamill KJ, Whittock NV, Coleman-Campbell CM, Mellerio JE, et al. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum Mol Genet. 2003;12:2395–409. doi: 10.1093/hmg/ddg234. [DOI] [PubMed] [Google Scholar]
- 19.Kadyan A, Aralikatti A, Shah S, Jewell R, Paul L, Darling J, et al. Laryngo-onycho-cutaneous syndrome. Ophthalmology. 2010;117:1056–e2, e2. doi: 10.1016/j.ophtha.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Rousselle P, Lunstrum GP, Keene DR, Burgeson RE. Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol. 1991;114:567–76. doi: 10.1083/jcb.114.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rousselle P, Aumailley M. Kalinin is more efficient than laminin in promoting adhesion of primary keratinocytes and some other epithelial cells and has a different requirement for integrin receptors. J Cell Biol. 1994;125:205–14. doi: 10.1083/jcb.125.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Champliaud MF, Lunstrum GP, Rousselle P, Nishiyama T, Keene DR, Burgeson RE. Human amnion contains a novel laminin variant, laminin 7, which like laminin 6, covalently associates with laminin 5 to promote stable epithelial-stromal attachment. J Cell Biol. 1996;132:1189–98. doi: 10.1083/jcb.132.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan JR, McGrath JA, Pulkkinen L, Kon A, Burgeson RE, Ortonne JP, et al. Immunohistochemical analysis of the skin in junctional epidermolysis bullosa using laminin 5 chain specific antibodies is of limited value in predicting the underlying gene mutation. Br J Dermatol. 1997;136:817–22. doi: 10.1111/j.1365-2133.1997.tb03918.x. [DOI] [PubMed] [Google Scholar]
- 24.McMillan JR, Akiyama M, Shimizu H. Ultrastructural orientation of laminin 5 in the epidermal basement membrane: an updated model for basement membrane organization. J Histochem Cytochem. 2003;51:1299–306. doi: 10.1177/002215540305101007. [DOI] [PubMed] [Google Scholar]
- 25.Marinkovich MP, Lunstrum GP, Keene DR, Burgeson RE. The dermal-epidermal junction of human skin contains a novel laminin variant. J Cell Biol. 1992;119:695–703. doi: 10.1083/jcb.119.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui C, Nelson CF, Hernandez GT, Herron GS, Bauer EA, Hoeffler WK. Gamma 2 chain of laminin-5 is recognized by monoclonal antibody GB3. J Invest Dermatol. 1995;105:648–52. doi: 10.1111/1523-1747.ep12324108. [DOI] [PubMed] [Google Scholar]
- 27.Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006;43:641–52. doi: 10.1136/jmg.2005.039685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulkkinen L, Meneguzzi G, McGrath JA, Xu Y, Blanchet-Bardon C, Ortonne JP, et al. Predominance of the recurrent mutation R635X in the LAMB3 gene in European patients with Herlitz junctional epidermolysis bullosa has implications for mutation detection strategy. J Invest Dermatol. 1997;109:232–7. doi: 10.1111/1523-1747.ep12319752. [DOI] [PubMed] [Google Scholar]
- 29.Mühle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L, Meneguzzi G, Schneider H. Novel and recurrent mutations in the laminin-5 genes causing lethal junctional epidermolysis bullosa: molecular basis and clinical course of Herlitz disease. Hum Genet. 2005;116:33–42. doi: 10.1007/s00439-004-1210-y. [DOI] [PubMed] [Google Scholar]
- 30.Castori M, Floriddia G, De Luca N, Pascucci M, Ghirri P, Boccaletti V, et al. Herlitz junctional epidermolysis bullosa: laminin-5 mutational profile and carrier frequency in the Italian population. Br J Dermatol. 2008;158:38–44. doi: 10.1111/j.1365-2133.2007.08208.x. [DOI] [PubMed] [Google Scholar]
- 31.Kon A, Pulkkinen L, Hara M, Tamai K, Tagami H, Hashimoto I, et al. Laminin 5 genes and Herlitz junctional epidermolysis bullosa: novel mutations and polymorphisms in the LAMB3 and LAMC2 genes. Mutations in brief no. 190. Online. Hum Mutat. 1998;12:288. doi: 10.1002/(SICI)1098-1004(1998)12:4<280::AID-HUMU11>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 32.Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, et al. Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet. 2002;110:41–51. doi: 10.1007/s00439-001-0630-1. [DOI] [PubMed] [Google Scholar]
- 33.Nakano A, Lestringant GG, Paperna T, Bergman R, Gershoni R, Frossard P, et al. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. J Am Acad Dermatol. 2002;46:510–6. doi: 10.1067/mjd.2002.119673. [DOI] [PubMed] [Google Scholar]
- 34.Gache Y, Allegra M, Bodemer C, Pisani-Spadafora A, de Prost Y, Ortonne JP, et al. Genetic bases of severe junctional epidermolysis bullosa presenting spontaneous amelioration with aging. Hum Mol Genet. 2001;10:2453–61. doi: 10.1093/hmg/10.21.2453. [DOI] [PubMed] [Google Scholar]
- 35.Hamill KJ, Paller AS, Jones JC. Adhesion and migration, the diverse functions of the laminin alpha3 subunit. Dermatol Clin. 2010;28:79–87. doi: 10.1016/j.det.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pacho F, Zambruno G, Calabresi V, Kiritsi D, Schneider H. Efficiency of translation termination in humans is highly dependent upon nucleotides in the neighbourhood of a (premature) termination codon. J Med Genet. 2011;48:640–4. doi: 10.1136/jmg.2011.089615. [DOI] [PubMed] [Google Scholar]
- 37.Floeth M, Bruckner-Tuderman L. Digenic junctional epidermolysis bullosa: mutations in COL17A1 and LAMB3 genes. Am J Hum Genet. 1999;65:1530–7. doi: 10.1086/302672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–8. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonkman MF, Scheffer H, Stulp R, Pas HH, Nijenhuis M, Heeres K, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88:543–51. doi: 10.1016/S0092-8674(00)81894-2. [DOI] [PubMed] [Google Scholar]
- 40.Pasmooij AM, Nijenhuis M, Brander R, Jonkman MF. Natural gene therapy may occur in all patients with generalized non-Herlitz junctional epidermolysis bullosa with COL17A1 mutations. J Invest Dermatol. 2012;132:1374–83. doi: 10.1038/jid.2011.477. [DOI] [PubMed] [Google Scholar]
- 41.Kiritsi D, He Y, Pasmooij AM, Onder M, Happle R, Jonkman MF, et al. Revertant mosaicism in a human skin fragility disorder results from slipped mispairing and mitotic recombination. J Clin Invest. 2012;122:1742–6. doi: 10.1172/JCI61976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almaani N, Nagy N, Liu L, Dopping-Hepenstal PJ, Lai-Cheong JE, Clements SE, et al. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130:1937–40. doi: 10.1038/jid.2010.64. [DOI] [PubMed] [Google Scholar]
- 43.Schuilenga-Hut PH, Scheffer H, Pas HH, Nijenhuis M, Buys CH, Jonkman MF. Partial revertant mosaicism of keratin 14 in a patient with recessive epidermolysis bullosa simplex. J Invest Dermatol. 2002;118:626–30. doi: 10.1046/j.1523-1747.2002.01715.x. [DOI] [PubMed] [Google Scholar]
- 44.Pasmooij AM, Pas HH, Bolling MC, Jonkman MF. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J Clin Invest. 2007;117:1240–8. doi: 10.1172/JCI30465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bruckner-Tuderman L, McGrath JA, Robinson EC, Uitto J. Animal models of epidermolysis bullosa: update 2010. J Invest Dermatol. 2010;130:1485–8. doi: 10.1038/jid.2010.75. [DOI] [PubMed] [Google Scholar]
- 46.Jiang QJ, Uitto J. Animal models of epidermolysis bullosa--targets for gene therapy. J Invest Dermatol. 2005;124:xi–xiii. doi: 10.1111/j.0022-202X.2005.23652.x. [DOI] [PubMed] [Google Scholar]
- 47.Durbeej M. Laminins. Cell Tissue Res. 2010;339:259–68. doi: 10.1007/s00441-009-0838-2. [DOI] [PubMed] [Google Scholar]
- 48.Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ. IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm Genome. 1997;8:673–81. doi: 10.1007/s003359900535. [DOI] [PubMed] [Google Scholar]
- 49.Ryan MC, Lee K, Miyashita Y, Carter WG. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J Cell Biol. 1999;145:1309–23. doi: 10.1083/jcb.145.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abrass CK, Berfield AK, Ryan MC, Carter WG, Hansen KM. Abnormal development of glomerular endothelial and mesangial cells in mice with targeted disruption of the lama3 gene. Kidney Int. 2006;70:1062–71. doi: 10.1038/sj.ki.5001706. [DOI] [PubMed] [Google Scholar]
- 51.Meng X, Klement JF, Leperi DA, Birk DE, Sasaki T, Timpl R, et al. Targeted inactivation of murine laminin gamma2-chain gene recapitulates human junctional epidermolysis bullosa. J Invest Dermatol. 2003;121:720–31. doi: 10.1046/j.1523-1747.2003.12515.x. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen NM, Pulkkinen L, Schlueter JA, Meneguzzi G, Uitto J, Senior RM. Lung development in laminin gamma2 deficiency: abnormal tracheal hemidesmosomes with normal branching morphogenesis and epithelial differentiation. Respir Res. 2006;7:28. doi: 10.1186/1465-9921-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graves KT, Henney PJ, Ennis RB. Partial deletion of the LAMA3 gene is responsible for hereditary junctional epidermolysis bullosa in the American Saddlebred Horse. Anim Genet. 2009;40:35–41. doi: 10.1111/j.1365-2052.2008.01795.x. [DOI] [PubMed] [Google Scholar]
- 54.Lieto LD, Swerczek TW, Cothran EG. Equine epitheliogenesis imperfecta in two american saddlebred foals is a lamina lucida defect. Vet Pathol. 2002;39:576–80. doi: 10.1354/vp.39-5-576. [DOI] [PubMed] [Google Scholar]
- 55.Mömke S, Kerkmann A, Wöhlke A, Ostmeier M, Hewicker-Trautwein M, Ganter M, et al. International Sheep Consortium A frameshift mutation within LAMC2 is responsible for Herlitz type junctional epidermolysis bullosa (HJEB) in black headed mutton sheep. PLoS One. 2011;6:e18943. doi: 10.1371/journal.pone.0018943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brenneman KA, Olivry T, Dorman DC. Rudimentary hemidesmosome formation in congenital generalized junctional epidermolysis bullosa in the Sprague-Dawley rat. Vet Pathol. 2000;37:336–9. doi: 10.1354/vp.37-4-336. [DOI] [PubMed] [Google Scholar]
- 57.Capt A, Spirito F, Guaguere E, Spadafora A, Ortonne JP, Meneguzzi G. Inherited junctional epidermolysis bullosa in the German Pointer: establishment of a large animal model. J Invest Dermatol. 2005;124:530–5. doi: 10.1111/j.0022-202X.2004.23584.x. [DOI] [PubMed] [Google Scholar]
- 58.Alhaidari Z, Olivry T, Spadafora A, Thomas RC, Perrin C, Meneguzzi G, et al. Junctional epidermolysis bullosa in two domestic shorthair kittens. Vet Dermatol. 2005;16:69–73. doi: 10.1111/j.1365-3164.2005.00420.x. [DOI] [PubMed] [Google Scholar]
- 59.Yuen WY, Duipmans JC, Molenbuur B, Herpertz I, Mandema JM, Jonkman MF. Long-term follow-up of patients with Herlitz-type junctional epidermolysis bullosa. Br J Dermatol. 2012;167:374–82. doi: 10.1111/j.1365-2133.2012.10997.x. [DOI] [PubMed] [Google Scholar]
- 60.Kho YC, Rhodes LM, Robertson SJ, Su J, Varigos G, Robertson I, et al. Epidemiology of epidermolysis bullosa in the antipodes: the Australasian Epidermolysis Bullosa Registry with a focus on Herlitz junctional epidermolysis bullosa. Arch Dermatol. 2010;146:635–40. doi: 10.1001/archdermatol.2010.109. [DOI] [PubMed] [Google Scholar]
- 61.Uitto J, McGrath JA, Rodeck U, Bruckner-Tuderman L, Robinson EC. Progress in epidermolysis bullosa research: toward treatment and cure. J Invest Dermatol. 2010;130:1778–84. doi: 10.1038/jid.2010.90. [DOI] [PubMed] [Google Scholar]
- 62.Uitto J, Christiano AM, McLean WH, McGrath JA. Novel molecular therapies for heritable skin disorders. J Invest Dermatol. 2012;132:820–8. doi: 10.1038/jid.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salas-Alanis JC, Cepeda-Valdes R, Mellerio JE, Christiano AM, Uitto J. Progress in epidermolysis bullosa: summary of a workshop in CILAD-2010*. Int J Dermatol. 2012;51:682–7. doi: 10.1111/j.1365-4632.2011.05130.x. [DOI] [PubMed] [Google Scholar]
- 64.Aumailley M, Has C, Tunggal L, Bruckner-Tuderman L. Molecular basis of inherited skin-blistering disorders, and therapeutic implications. Expert Rev Mol Med. 2006;8:1–21. doi: 10.1017/S1462399406000123. [DOI] [PubMed] [Google Scholar]
- 65.Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 66.De Luca M, Pellegrini G, Mavilio F. Gene therapy of inherited skin adhesion disorders: a critical overview. Br J Dermatol. 2009;161:19–24. doi: 10.1111/j.1365-2133.2009.09243.x. [DOI] [PubMed] [Google Scholar]
- 67.Jonkman MF, Pasmooij AM. Realm of revertant mosaicism expanding. J Invest Dermatol. 2012;132:514–6. doi: 10.1038/jid.2011.445. [DOI] [PubMed] [Google Scholar]
- 68.Gostynski A, Deviaene FC, Pasmooij AM, Pas HH, Jonkman MF. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br J Dermatol. 2009;161:444–7. doi: 10.1111/j.1365-2133.2009.09118.x. [DOI] [PubMed] [Google Scholar]