

Abstract
Vascular endothelial growth factor (VEGF)-initiated angiogenesis requires both coordinated proteolytic degradation of extracellular matrix provided by the urokinase plasminogen activator/urokinase receptor (uPA/uPAR) system and regulation of cell-migration provided by integrin–matrix interaction. Previously we have shown that stimulation of pericellular proteolysis induced by VEGF occurs via the VEGF receptor-2 leading to redistribution of uPAR to focal adhesions at the leading edge of endothelial cells. In our recent work published in Cardiovascular Research, we investigated the mechanisms underlying the uPAR-dependent modulation of VEGF-induced endothelial migration. By applying a micropatterning technique we described that VEGF stimulation results in complex formation between uPAR and α5β1-integrin on the cell surface. The subsequent internalization of this complex, important for receptor redistribution, was demonstrated by flow-cytometry and immunohistochemistry. Targeting of the interaction site between uPAR and α5β1 impairs receptor internalization and leads to the inhibition of endothelial cell migration in vitro and in an angiogenesis model in vivo. This proof-of-principle that the interface of uPAR and α5β1-integrin may represent a promising site to therapeutically target tumor angiogenesis raises hope for the development of an anti-angiogenic approach that is limited to only the mobilizing effect of VEGF to endothelial cells, and does not interfere with the inarguably positive effect of VEGF as survival factor.
Keywords: VEGF, angiogenesis, blood vessels, endothelial cell migration, integrin α5 β1, uPAR
In pathological and physiological angiogenesis, new endothelial cells penetrate avascular zones by sprouting from existing vascular vessels. For this process, vascular endothelial growth factor (VEGF) plays a critical role, because it regulates all steps required for angiogenesis, i.e., it induces endothelial cell proliferation and migration, increases vascular permeability and allows for the expression of active proteases on the cell surface.1 As a consequence, matrix molecules are degraded and new provisional extracellular matrix (ECM) is created that promotes invasion of the surrounding tissue by endothelial cells.
The urokinase-plasminogen activator receptor (uPAR) binds urokinase-plasminogen activator (uPA),2,3 which in turn localizes the activation of plasminogen to the extracellular protease, plasmin.4 Plasmin then catalyzes degradation of the ECM and also activates other proteases, which together facilitate endothelial cell invasion.
As we have shown previously,5 the above described activation of the proteolytic activity can be induced by VEGF via the VEGF receptor-2 (VEGFR-2). This process involves activation of pro-urokinase, its inactivation by plasminogen activator inhibitor-1 (PAI-1) and leads to subsequent redistribution of uPAR to focal adhesions at the leading edge of endothelial cells. It also involves internalization of uPAR via an LDL receptor-like molecule. We showed that such redistribution of uPAR was dependent on the VEGFR-2 since the VEGF isoforms VEGF165 and VEGF-E (both interacting with VEGFR-2), but not PlGF (binding only to VEGFR-1) induced internalization of uPAR via an LDL receptor-like molecule within minutes. We concluded that VEGF-induced uPAR redistribution to the leading edge of migrating endothelial cells provides them with the localized proteolytic capacity to degrade the basement membrane and invade the surrounding tissue during sprout formation.
In our recent work published in Cardiovascular Research, we studied the uPAR-dependence of endothelial cell migration and angiogenesis upon VEGF stimulation. We had previously shown that the migratory response of endothelial cells to VEGF depends on the uPAR.5 This we had observed on vitronectin—an ECM protein that is a ligand for uPAR.6 Here, we addressed our observation, that also on fibronectin, which is not a ligand for uPAR, the VEGF-induced endothelial migration still depended on uPAR. We surmised that uPAR, which has been described to be able to interact with and bind to several members of the integrin receptor family, might thereby mediate integrin redistribution in response to VEGF thus fostering migration.
In our work we demonstrate that the VEGF-signal is funneled through uPAR in order to control—besides proteolysis—a second limb of the angiogenic response, namely integrin-dependent cell migration. For this we provided several lines of evidence: (1) VEGF promoted assembly of a complex comprising uPAR and integrin α5β1; (2) The internalization of the complex into endosomes, that were also decorated with clathrin immunoreactivity, was mediated by LDLR-like protein; (3) In the absence of uPAR, VEGF failed to trigger internalization of the fibronectin receptor α5β1 and thus initiate the redistributive cycle of integrin endocytosis and exocytosis. This translated into impaired endothelial cell migration in vitro and reduced endothelial cell invasion and vessel formation in vivo (Fig. 1).
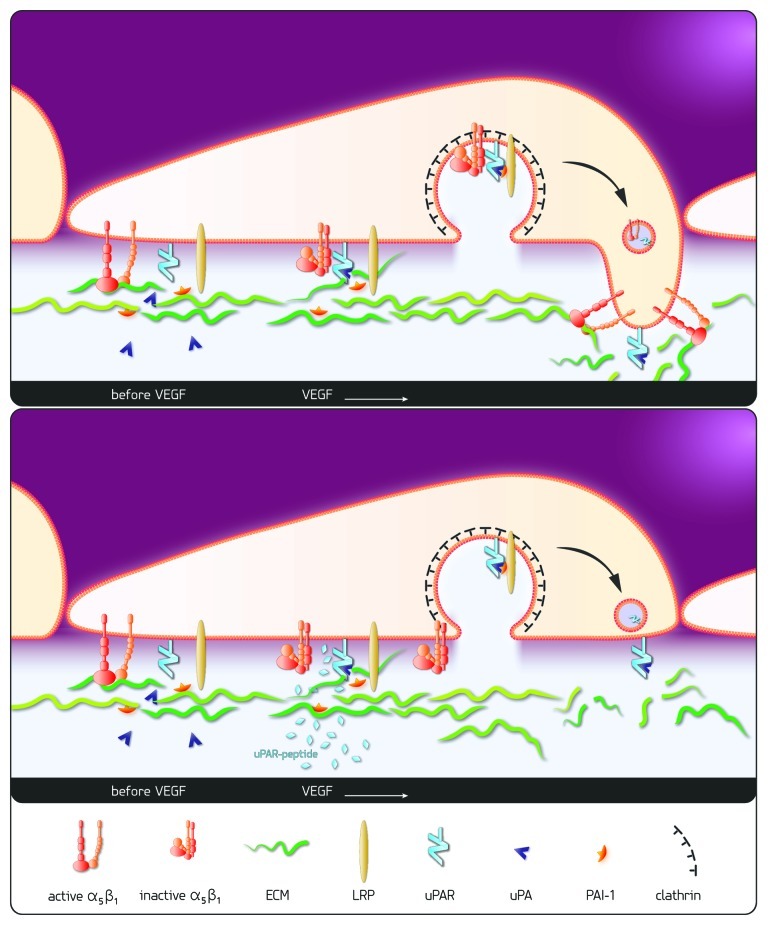
Figure 1. Scheme of proposed mechanism: VEGF leads to interaction of uPAR and the inactivated fibronectin receptor, integrin α5β1, resulting ultimately in the cointernalization of integrin α5β1 via “LRP/clathrin dependent endocytosis” and redistribution to the leading edge VEGF-stimulation of endothelial cells leads to inactivation of the fibronectin receptor integrin α5β1, to activation of urokinase (uPA) bound to its receptor (uPAR) and to matrix degradation. PAI-1, released from the degraded matrix, inactivates uPA and the resulting trimolecular complex (uPAR/uPA/PAI-1) is internalized via “LRP/clathrin dependent endocytosis” dragging along the fibronectin receptor α5β1. The redistribution of both, proteolytic as well as adhesive capacity, to the leading edge of the cell enables invasive cell migration and fosters angiogenesis. The uPAR derived peptide aa 243–251 corresponding to the proposed interaction site of uPAR and integrin α5β1 prevents the VEGF-induced interaction of the two receptors. Thus, the co-internalization and redistribution of integrin α5β1 is prevented impairing cell migration and invasion during VEGF-stimulated angiogenesis.
That integrin α5β1 and uPAR would be able to interact had been suggested by earlier experiments: in uPAR rich cells, α5β1-integrin had been recovered in the immuno-precipitate7,8 and this correlated with persistent ERK1/2 activation and enhanced tumor growth in vivo.9
In our study we employed a micropatterning approach10 that allowed us for direct visualization of VEGF-promoted formation of a complex between uPAR and α5β1-integrin on the endothelial cell surface. This method does not require any modification of the interacting molecules (e.g., by attaching fluorescent moieties) or any change in their stoichiometry (e.g., by heterologous expression). The fact that integrin internalization is precluded in this experimental setup did not only facilitate quantitative assessment of uPAR recruitment but also afforded the unequivocal demonstration that the interaction occurred indeed at the cell surface. This new approach does not differentiate though, whether there is a direct interaction between uPAR and integrins or whether additional proteins—other than LDLR-like proteins—must be recruited to stabilize and internalize the complex. In detergent solution, in any case, purified uPAR and purified α5β1-integrin can directly interact. Furthermore, that functional association of uPAR with integrin α5β1 can indeed be mediated by a specific site of uPAR, was shown by the use of a 9-mer peptide derived from a sequence in domain III (residues 240–248) which was able to bind purified α5β1-integrin. Substitution of a single amino acid (S245A) in this peptide, or in full-length soluble uPAR, impaired binding of the purified integrin.11
Our observations in the micropatterning experiment suggest that the interaction of uPAR and integrin α5β1 in the cell membrane is subject to regulation that is brought about by a conformational change. This increases the mutual affinity of the partners or drives their association by recruiting an additional molecule into the complex. Our earlier results suggest that the integrin is inactivated by VEGF-induced inside-out signaling via a PI3-kinase dependent mechanism12 before interacting with uPAR. This is underscored by the fact that in our micropatterning experiment the activating antibody 12G10 to α5β113 inhibited the VEGF-induced recruitment of uPAR.
In our publication in Cardiovascular Research, we furthermore report that VEGF triggered in endothelial cells endocytosis of another integrin, α3β1. Surprisingly however, we did not detect any direct interaction of integrin α3β1 with uPAR in our micropatterning experiment, though this method is, in our opinion, among the most sensitive methods to record interactions in the native cell membrane.10 Thus, it seems likely that, in endothelial cells, VEGF indeed does not promote complex formation between integrin α3β1 and uPAR. This our conclusion is also supported by our observation that VEGF165-triggered endocytosis of α3β1-integrin was not impaired by the receptor-associated protein (RAP), which blocks LDLR like protein mediated internalization of uPAR.
While previous reports documented direct binding of purified integrin α3β1 to uPAR,7,14 the discrepancy between these and our findings could most likely be accounted for by cell type-dependent differences. In fact, uPAR can recruit various integrins in a cell type-dependent manner.11,14,15 An alternative explanation would be delocalization of integrin α3β1by the tetraspanin CD9 upon VEGF stimulation previously described.16
The findings presented in our recent study strongly suggest that absence of integrin α5β1 or targeting of the interaction sites between uPAR and α5β1 might lead to the inhibition of endothelial cell migration and angiogenesis.
In fact, blockage of integrin α5β1 by an antibody was recently shown to inhibit angiogenesis in a murine tumor model, where human rhabdomyosarcoma cells were xenografted into immuno-deficient mice.17 Accordingly, integrin α5β1 is currently being targeted by using the pertinent chimeric antibody in phase II clinical trials in advanced human cancer.18
The binding sites for integrin α5β1 to uPAR are thought to reside on domain-3 of uPAR and point mutation of S245 or H249 resulted in inhibition of the association of uPAR and integrin α5β.11,19 We employed a peptide comprising the residues 243–251 of uPAR (TASMCQHAH) containing both, S245 and H249. This peptide efficiently blocked VEGF-induced internalization of the fibronectin receptor in vitro and angiogenesis in vivo.
The interaction site between uPAR and integrin α5β1 fulfils several criteria of a candidate drug binding site: (1) it is readily accessible, because it is on the extracellular surface thus obviating the cell membrane as a permeation barrier; (2) it allows for discrimination, because it can be specifically targeted; (3) there is no major toxicity that can be a priori anticipated. Furthermore, targeting only VEGF-induced integrin redistribution—without affecting VEGF-mediated maintenance of the endothelium is expected to reduce the adverse side effects of a broad anti-VEGF therapy and might even foster the normalization of the tumor vasculature. Our results therefore provide a proof-of-principle that the interface of uPAR and α5β1-integrin may represent a site to therapeutically target tumor vasculature.
Acknowledgments
Supported by funds of the Oesterreichische Nationalbank (Anniversary Fund, project number: 13204).
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/22124
References
- 1.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Stoppelli MP, Corti A, Soffientini A, Cassani G, Blasi F, Assoian RK. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc Natl Acad Sci U S A. 1985;82:4939–43. doi: 10.1073/pnas.82.15.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassalli JD, Baccino D, Belin D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J Cell Biol. 1985;100:86–92. doi: 10.1083/jcb.100.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephens RW, Pöllänen J, Tapiovaara H, Leung KC, Sim PS, Salonen EM, et al. Activation of pro-urokinase and plasminogen on human sarcoma cells: a proteolytic system with surface-bound reactants. J Cell Biol. 1989;108:1987–95. doi: 10.1083/jcb.108.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prager GW, Breuss JM, Steurer S, Olcaydu D, Mihaly J, Brunner PM, et al. Vascular endothelial growth factor receptor-2-induced initial endothelial cell migration depends on the presence of the urokinase receptor. Circ Res. 2004;94:1562–70. doi: 10.1161/01.RES.0000131498.36194.6b. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269:32380–8. [PubMed] [Google Scholar]
- 7.Wei Y, Eble JA, Wang Z, Kreidberg JA, Chapman HA. Urokinase receptors promote beta1 integrin function through interactions with integrin alpha3beta1. Mol Biol Cell. 2001;12:2975–86. doi: 10.1091/mbc.12.10.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei Y, Czekay RP, Robillard L, Kugler MC, Zhang F, Kim KK, et al. Regulation of alpha5beta1 integrin conformation and function by urokinase receptor binding. J Cell Biol. 2005;168:501–11. doi: 10.1083/jcb.200404112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarzenbacher M, Kaltenbrunner M, Brameshuber M, Hesch C, Paster W, Weghuber J, et al. Micropatterning for quantitative analysis of protein-protein interactions in living cells. Nat Methods. 2008;5:1053–60. doi: 10.1038/nmeth.1268. [DOI] [PubMed] [Google Scholar]
- 11.Chaurasia P, Aguirre-Ghiso JA, Liang OD, Gardsvoll H, Ploug M, Ossowski L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J Biol Chem. 2006;281:14852–63. doi: 10.1074/jbc.M512311200. [DOI] [PubMed] [Google Scholar]
- 12.Prager GW, Breuss JM, Steurer S, Mihaly J, Binder BR. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood. 2004;103:955–62. doi: 10.1182/blood-2003-07-2214. [DOI] [PubMed] [Google Scholar]
- 13.Mould AP, Garratt AN, Askari JA, Akiyama SK, Humphries MJ. Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the beta 1 subunit. FEBS Lett. 1995;363:118–22. doi: 10.1016/0014-5793(95)00301-O. [DOI] [PubMed] [Google Scholar]
- 14.Mazzieri R, D’Alessio S, Kenmoe RK, Ossowski L, Blasi F. An uncleavable uPAR mutant allows dissection of signaling pathways in uPA-dependent cell migration. Mol Biol Cell. 2006;17:367–78. doi: 10.1091/mbc.E05-07-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohuslav J, Horejsí V, Hansmann C, Stöckl J, Weidle UH, Majdic O, et al. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med. 1995;181:1381–90. doi: 10.1084/jem.181.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamisasanuki T, Tokushige S, Terasaki H, Khai NC, Wang Y, Sakamoto T, et al. Targeting CD9 produces stimulus-independent antiangiogenic effects predominantly in activated endothelial cells during angiogenesis: a novel antiangiogenic therapy. Biochem Biophys Res Commun. 2011;413:128–35. doi: 10.1016/j.bbrc.2011.08.068. [DOI] [PubMed] [Google Scholar]
- 17.Bhaskar V, Zhang D, Fox M, Seto P, Wong MH, Wales PE, et al. A function blocking anti-mouse integrin alpha5beta1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J Transl Med. 2007;5:61. doi: 10.1186/1479-5876-5-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell-McGuinn KM, Matthews CM, Ho SN, Barve M, Gilbert L, Penson RT, et al. A phase II, single-arm study of the anti-α5β1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol Oncol. 2011;121:273–9. doi: 10.1016/j.ygyno.2010.12.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Y, Tang CH, Kim Y, Robillard L, Zhang F, Kugler MC, et al. Urokinase receptors are required for alpha 5 beta 1 integrin-mediated signaling in tumor cells. J Biol Chem. 2007;282:3929–39. doi: 10.1074/jbc.M607989200. [DOI] [PubMed] [Google Scholar]