

Abstract
Background
Host and parasitoid interaction is one of the most fascinating relationships of insects, which is currently receiving an increasing interest. Understanding the mechanisms evolved by the parasitoids to evade or suppress the host immune system is important for dissecting this interaction, while it was still poorly known. In order to gain insight into the immune response of Tenebrio molitor to parasitization by Scleroderma guani, the transcriptome of T. molitor pupae was sequenced with focus on immune-related gene, and the non-parasitized and parasitized T. molitor pupae were analyzed by digital gene expression (DGE) analysis with special emphasis on parasitoid-induced immune-related genes using Illumina sequencing.
Methodology/Principal Findings
In a single run, 264,698 raw reads were obtained. De novo assembly generated 71,514 unigenes with mean length of 424 bp. Of those unigenes, 37,373 (52.26%) showed similarity to the known proteins in the NCBI nr database. Via analysis of the transcriptome data in depth, 430 unigenes related to immunity were identified. DGE analysis revealed that parasitization by S. guani had considerable impacts on the transcriptome profile of T. molitor pupae, as indicated by the significant up- or down-regulation of 3,431 parasitism-responsive transcripts. The expression of a total of 74 unigenes involved in immune response of T. molitor was significantly altered after parasitization.
Conclusions/Significance
obtained T. molitor transcriptome, in addition to establishing a fundamental resource for further research on functional genomics, has allowed the discovery of a large group of immune genes that might provide a meaningful framework to better understand the immune response in this species and other beetles. The DGE profiling data provides comprehensive T. molitor immune gene expression information at the transcriptional level following parasitization, and sheds valuable light on the molecular understanding of the host-parasitoid interaction.
Introduction
Multicellular animals are continually exposed to foreign invaders such as viruses, bacteria, fungi, protozoans as well as various metazoan parasites and parasitoids. In order to defend themselves against invaders, they have evolved two effective immune systems known as acquired and innate immunity. The acquired immune system relies on the generation of random and highly diverse repertoires of antigens that allow organisms to develop an immunological memory, whereas the innate immune system uses germline encoded factors to recognize and kill foreign invaders [1], [2]. Vertebrates possess both acquired and innate immunity. Insects and other invertebrates lack the acquired immune system but have a well-developed innate immune system that successfully combats foreign invaders. The first defense of insects against invaders is the physical barriers, including the cuticle and peritrophic matrix [3]. Once the invaders overcome the physical barriers, insects will provoke a complex series of immune reactions to cope with the invaders. The immune system of insects can be divided into two categories: (i) humoral defense, including the antimicrobial peptides, reactive intermediates of oxygen or nitrogen, melanin formation and clotting; and (ii) cellular defense mainly based on haemocytes, such as phagocytosis, encapsulation, microaggregation and nodulation [4]–[6].
Parasitoids are insects that spend a significant portion of their life in the body or on the body surface of other invertebrates, mostly other insects, and whose larvae feed on and eventually kill the host [7]. During this process, oviposited eggs and developing larvae must contend with host immune responses. An amazing array of mechanisms have been evolved that enable parasitoids to effectively evade or deactivate host defenses [8]. These include introduce or secrete various virulent factors such as venom, polydnaviruses and virus like particles into host's body upon parasitization [9]–[11]. Most studies have focused on these parasitic factors and their effects on the host. Despite both their molecular identity and physiological role involving in hosts immune suppression have been investigated to various extents in many parasitoid-host systems, the transcriptional immune responses of host to parasitic wasps are still poorly understood [12]–[15]. This research trajectory has only over the last several years become a focal point of research. In addition, most of these researchers so far have only focused on the differential expression of individual or small group of host genes following parasitization. The recent development of transcriptome and digital gene expression (DGE) based on next generation deep-sequencing technology provides extensive data in much shorter time period with enormous depth and coverage to globally analyse the blueprint of host's gene expression profile under parasitization challenge.
The ant-like bethylid wasp Scleroderma guani (Hymenoptera: Bethylidae) is a generalist ectoparasitoid of wood-boring insects, which is widely dispread in China and has been firstly discovered in Guangdong and Shandong provinces in 1973 and 1975, respectively [16], [17]. In China, since its discovery, S. guani as a biocontrol agent has been widely used to control longhorned beetles, including Monochamus alternatus that is the most important insect vector of the pinewood nematode Bursaphelenchus xylophilus, the causal agent of pine wilt disease [18]. This polyphagous parasitoid can parasitize larval or pupal stage of more than 50 species of insects belonging to 22 families from three orders, including the yellow mealworm beetle, Tenebrio molitor (Coleoptera: Tenebrionidae) [19]. T. molitor pupa is one of the preferred hosts for mass rearing S. guani in the laboratory.
For the present study, we used the Illumina sequencing to explore the T. molitor immune response induced by S. guani parasitization. Firstly, we obtained and characterized the transcriptome of T. molitor pupae with special emphasis on immune related genes. Additionally, we constructed two DGE libraries and compared the gene expression profiles of non-parasitized and parasitized T. molitor pupae. A combination of transcriptome and DGE analyses revealed key differentially expressed immune genes in parasitized versus non-parasitized T. molitor pupae. The results give us a comprehensive view of global gene expression profiles of host response to parasitization, and shed light on the molecular basis of host-parasitoid interaction.
Materials and Methods
Ethics statement
Regarding the field study, no specific permits were required. The location is not privately-owned or protected in any way. The field studies did not involve endangered or protected species.
Insects and parasitization
T. molitor were mass reared under natural photo regime and temperature conditions on wheat bran. Vegetables were placed on top of the bran to provide water. S. guani strain was originally collected from the pine forestry in suburb of Kunming, China. The parasitoid had been maintained in the laboratory for over 2 years on T. molitor pupae. After emergence, adult parasitoids were fed with 20% honey solution. Parasitization by S. guani was carried out by exposing newly pupated pupae of T. molitor to female wasps. When a pupa was seen to be parasitized, it was transferred to a fresh glass petri dish, where it was allowed to develop at 25°C before harvesting for RNA extraction.
cDNA library preparation and Illumina sequencing
Total RNA was isolated from the pupae at various developmental stages separately using TRIzol® Reagent (Invitrogen) according to the manufacturer's instructions. Then, all total RNAs were pooled together. RNA integrity was confirmed using the 2100 Bioanalyzer (Agilent Technologies). The mRNA was enriched from 20 μg total RNA using oligo(dT) magnetic beads and fragmented with RNA Fragmentation Reagent. The cleaved RNA fragments were transcribed into the first-strand cDNA using random hexamer-primer, followed by second-strand cDNA synthesis. Short fragments were then purified with a QiaQuick PCR extraction kit (Qiagen, Valencia, CA, USA) and were resolved with EB buffer for end reparation and adding poly(A). After that, they were ligated to sequencing adapters. Finally, these products were purified and enriched with PCR to create the final cDNA library. The library was sequenced using Illumina HiSeq™ 2000 platform. Raw data were deposited to DDBJ database under accession DRA000603.
Transcriptome analysis
Data analysis and base calling were performed by the Illumina instrument software. After removing the sequence reads containing sequencing adapters and low quality sequence reads, the clean reads were assembled using SOAPdenovo software [20]. The resultant contigs were joined into scaffolds using paired-end joining and gap-filling. Paired-end reads are used again for gap filling of scaffolds to get sequences with least Ns and cannot be extended on either end. The final sequences were named unigenes. For assignments of predicted gene descriptions, the assembled unigenes were compared to the protein of NCBI nr databases with the BLASTX algorithm at the threshold of E-value <10−5. For functional annotation, the Gene Ontology (GO), Cluster of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genome (KEGG) annotation for each unigene was performed using the automatic annotation tool Blast2GO program [21].
DGE library preparation and sequencing
Total RNA was extracted separately from non-parasitized and parasitized T. molitor pupae at 6, 12, 24 and 48 h post-parasitization following the manufacturer's instruction, as described above. Total RNAs extracted from the two different treatments were pooled together. Next, a DGE library was prepared using an Illumina gene expression sample prep kit. Briefly, mRNA was enriched by using the oligo(dT) magnetic beads from the total RNA, and cDNAs were synthesized as described above for each sample. Bead-bound cDNA was digested with the restriction enzyme Nla III, which recognizes and cuts off the CATG sites. Those cDNA fragments with 3′ ends were purified from the magnetic beads, and Illumina adaptor 1 was added to their 5′ ends. After digestion with Mme I, which recognizes the junction of the adapter 1 and the CATG site, and makes a cut at 17 bp downstream of the NlaIII recognition site, 21-bp tags containing adaptor 2 were ligated to the 3′ ends of the tags to create a tag library. The required fragments were enriched by linear PCR amplification for 15 cycles and purified by 6% TBE PAGE gel. After denaturation, the single-chain molecules were anchored to Illumina chip, and sequenced via Illumina HiSeq™ 2000.
DGE analysis
The original image data was transferred into sequence data by base calling. The raw data were filtered to get the clean reads by removing the dirty reads with adaptors, low-quality reads (reads with unknown sequences), empty reads (no read sequence between the adaptors), and reads with only one copy number (probable sequencing error). Then, clean reads were mapped to the above transcriptome reference database using SOAPaligner/soap2 [22], allowing no more than 2 mismatches. The remainder of the clean tags were designed unambiguous clean reads. The unigene expression level was calculated by using RPKM (reads per kb per million reads) method [23]. To identify the DGEs in the two samples, a strict algorithm was developed for statistical analysis according to the method of Audic and Claverie [24]. False discovery rate (FDR) is a method to determine the threshold of P-value in multiple tests. During the analysis, “FDR ≤0.001 and the absolute value of log2Ratio ≥1” was used as the threshold to judge the significance of gene expression difference. Next, all DGEs were subjected to GO and KEGG enrichment analysis compared to the transcriptome background using hypergeometric test. The calculated P-value goes through Bonferroni Correction, taking corrected P-value ≤0.05 as a threshold. GO terms or KEGG pathways fulfilling this condition were defined as significantly enriched terms in DEGs.
Quantitative real-time PCR (qRT-PCR) validation
Total RNA was extracted as described for DGE library preparation and sequencing, and then was reverse-transcribed according to the protocol provided with the M-MLV first strand kit (Invitrogen, China). Thirteen differentially expressed genes were randomly selected to verify DGE sequencing results using three replicates. The actin gene was used as a constitutive expression control for normalization. The primers are shown in Table S1. qRT-PCR was carried out using a BIO-Rad IQ5 Real-Time PCR system (Bio-Rad) under the following conditions: 95°C for 5 min; and 40 cycles of 95°C for 10 s, 60°C for 15 s, and 72°C for 20 s, followed by melting curve generation (68°C to 95°C). EvaGreen (Bio-RAD, U.S.) was used as DNA-binding fluorescent dye according to the manufacturer's protocol.
Results and Discussion
Illumina sequencing and assembly
A total of 264,698 raw reads were generated through one pyrosequencing run (Table 1). An average read length was 90 bp, which is consistent with the Illumina sequencing capacity. The GC percentage of the reads is 44.03%, which is comparable with genome sequence of other insects. After cleaning of dirty reads and quality checks, these short reads were assembled into 389,298 contigs (Figure S1). These contigs were further assembled into 106,202 scaffolds with a mean length of 325 bp by using paired end-joining and gap-filling (Figure S2). Clustering of these scaffolds revealed 71,514 unigenes. They ranged from as small as 150 bp to as large as 12518 bp. The mean size of them was 424 bp. The lengths of the 5,078 unigenes were more than 1,000 bp (Figure 1). Most unigenes were shorter than 500 bp, and only near 25% of them were longer than 500 bp, which is similar to that obtained in previous transcriptome analyses in different insects using Illumina platform [25], [26]. This drawback might be resulted from the short length of the sequencing read. In addition, successful assembly of next generation sequencing data into relatively long sequences is affected by the coverage of individual reads [27]. The unigene could be lengthened by the increased sequence depth using normalized library with 454 platform.
Table 1. Sequence statistics of the Illumina sequencing assembly.
| Reads | Contigs | Scaffolds | Unigenes | |
| Number of sequences | 26,666,668 | 389,298 | 106,202 | 715,14 |
| Mean length (bp) | 90 | 148 | 325 | 424 |
| Total length (bp) | 2,400,000,120 | 57,799,627 | 34,470,701 | 30,319,407 |
Figure 1. Length distribution of Tenebrio molitor unigenes.
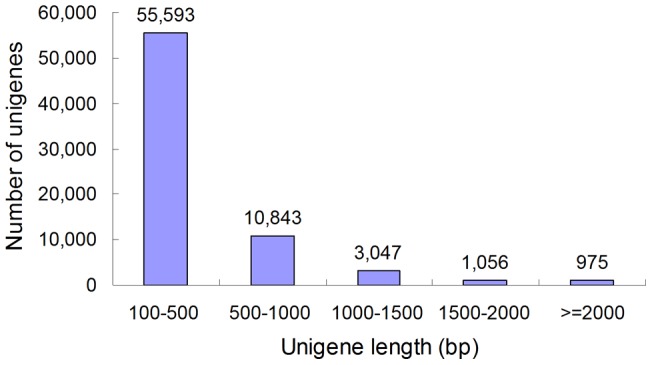
Horizontal axis represents the length of unigenes and vertical axis represents number of unigenes.
Functional annotation and classification
For BLASTX annotation, unigenes were searched against proteins of the NCBI nr database using a cut-off E-value of 10−5, just over half (52.26%) of all unigenes returned an above cut-off BLAST result. The remaining 34,141 unigenes (48.74%) could not be done and need more genetic data to annotate. As longer sequences were more likely to obtain BLAST matches in the protein databases [28], the relatively low annotated percentage is partially resulted by a high frequency of short sequences in our database. This also might be due to the transcripts derived from the cDNA of untranslated regions, chimerical sequences (assemblage errors) and nonconserved areas of proteins where homology is not detected, which could be resolved by enhancing the accuracy of the assembly and perfecting gene annotation strategies [29], [30]. Additionally, it could be estimated that a large part of the genes in T. molitor transcriptome database are with unknown functions. As shown in Figure 2A, among the annotated unigenes, 13,507 (36.15%) showed strong homology (smaller than 1.0E-45) (Table S2), whereas 8,699 (23.28%) showed poor matches with E-values between 1E-14 and 1E-5. For species distribution, most (71.58%) of the BLASTX hits are matched to the Tribolium castaneum sequences followed by Drosophila (3.35%), Apis mellifera (3,25%) and Acyrthosiphon pisum (1.99%) (Figure 2B). It is in accordance with the results of transcriptome profiling of other beetles including Tomicus yunnanensis and Dendroctonus ponderosae, of which over 60% genes were most closely related to T. castaneum [26], [30], [31]. This can be explained by the fact that T. castaneum genome is the only fully sequenced beetle genome and currently represents the vast majority of deposited coleopteran sequences in NCBI [32].
Figure 2. Homology analysis of Tenebrio molitor unigenes.
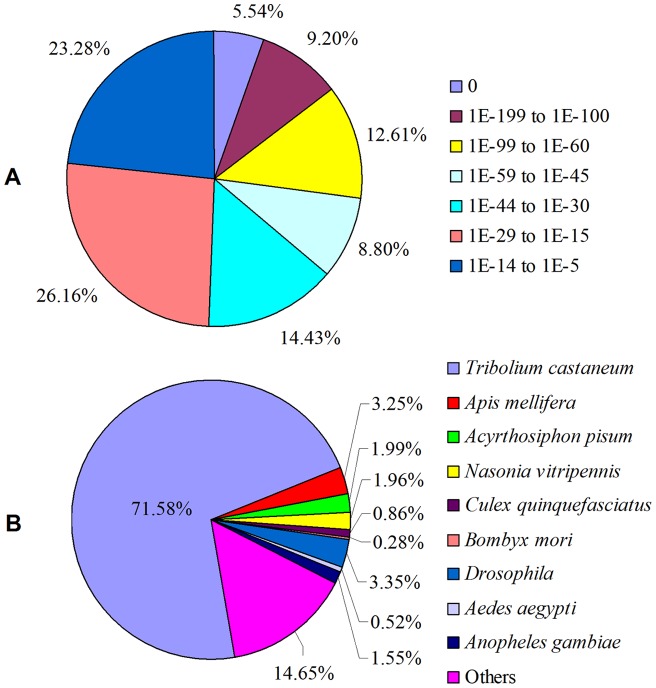
(A) E-value distribution. (B) Species distribution. All unigenes that had BLASTX annotations within the NCBI nr database with a cut-off E-value of 10−5 were analyzed. We used the first hit of each sequence for analysis.
Based on the ‘best hit’ BLASTX search, unigenes were further assigned to GO classification using Blast2GO. A total of 51,301 unigenes were annotated and assigned to GO terms (Table S3). They were classified into the three main GO categories: biological process, cellular component, and molecular function, covering with a wide range of subcategories. 23,831 (46.45%) unigenes were grouped under biological process, 17,179 (33.49%) under cellular component, and 10,291 (20.06%) under molecular function (Figure 3). Under the category of biological process, cellular process (19.63%) and metabolic process (15.55%) represented the most abundant subcategories, indicating the importance of cell cycle, generation as well as metabolic activities in this developmental stage. In the category of cellular component, cell (28.94%) and cell part (28.94%) represented the most abundant subcategories followed by organelle (17.08%) and organelle part (9.10%). The molecular function category was mostly dominated by the group of binding (45.65%) and catalytic activity (36.18%). With regard to the subcategories that took up a large proportion, similar observations for them were reported in transcriptomic studies of other insects [29], [33]. Comparing T. molitor with T. yunnanensis and D. ponderosae in the transcriptome-wide GO classification [26], [30], [31], they had high similarity with each other in the percentages of subcategory assignations for the three main categories. T. molitor sequences fall into GO categories with a roughly similar distribution to that of T. castaneum genome. It suggests that the sequencing provided a comprehensive representation of the T. molitor transcriptome, and the T. molitor sequence data do not contain notable biases towards particular categories of genes.
Figure 3. Gene ontology (GO) classification of Tenebrio molitor unigenes.
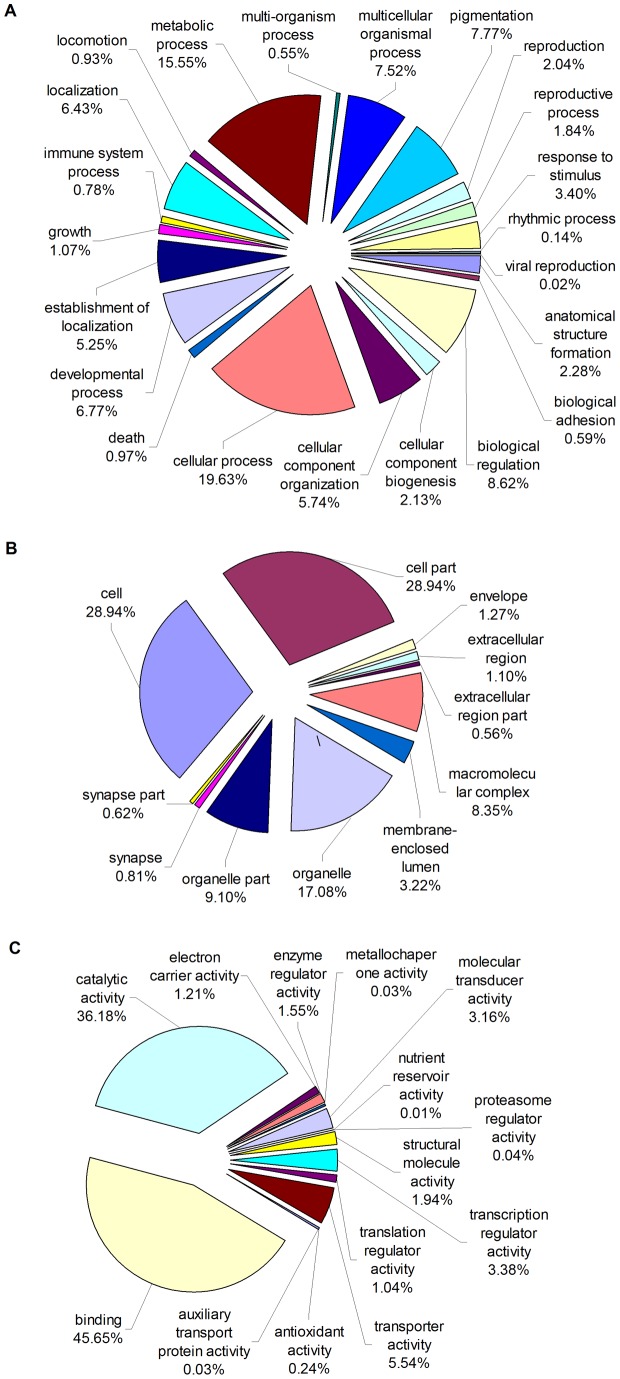
Categories for (A) biological processes, (B) cellular components, and (C) molecular processes. The data presented represent the level 2 analysis. The percentage of a specific category of genes in that main category is shown.
To further analyze the putative protein functions, the assembled unigenes were compared against COG. Altogether 13,599 functional annotations were produced, which were assigned to the appropriate COG clusters. These COG classifications were grouped into 25 functional categories (Figure 4). The largest category was general function prediction only, which contained 2,189 unigenes (16.10%). It was followed by translation, ribosomal structure and biogenesis (1,340, 9.85%), posttranslational modification, protein turnover, chaperones (1,162, 8.54%), and replication, recombination and repair (1,026, 7.54%). Only 3 unigenes (0.02%) belonged to extracellular structures, which was the smallest group. Then, nuclear structure, cell motility, and RNA processing and modification were the relatively smaller groups, containing 12, 83 and 95 unigenes, respectively.
Figure 4. Clusters of orthologous groups (COG) classification of Tenebrio molitor unigenes.
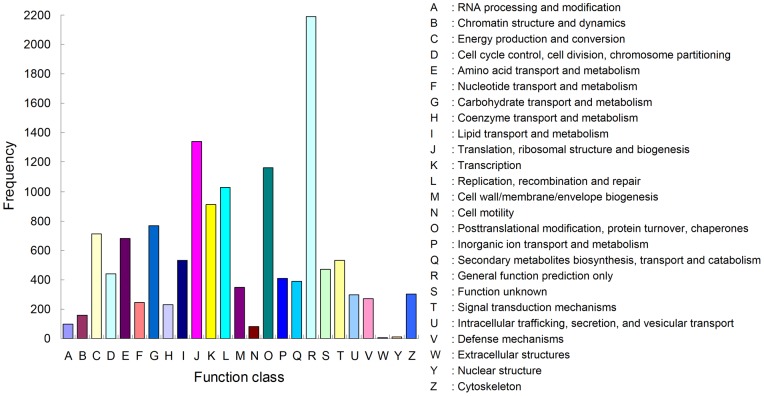
A total of 13,599 produced functional annotations were among the 25 categories.
More specifically, KEGG pathway analysis was performed on unigenes to identify the biological pathways active in T. molitor. As a result, there were 21,022 unigenes assigned to 205 KEGG pathways (Table S4). Highest number of sequences belonged to metabolic pathways (3,440, 16.36%) followed by spliceosome, pathways in cancer, purine metabolism, regulation of actin cytoskeleton, endocytosis, focal adhesion, Huntington's disease, tight junction, lysosome and so on. Except for metabolic pathways, the gene numbers and percentages of other pathways were below 1,000 and 5%, respectively.
Putative immune related genes
Well-categorized and annotated transcriptome serve as important and valuable resources for gene identification. Despite 50 of the 284 T. molitor proteins including serine proteinase (SP) and its homologue (SPH), serpin, β-1,3-glucan recognition protein (GRP)/ gram-negative binding protein (GNBP), peptidoglycan recognition protein (PGRP), prophenoloxidase (PPO), and melanization inhibitor that participate in immune responses were available in the NCBI database (prior to July 22, 2012), these molecules appear to represent only a small portion of the T. molitor immune system. In order to gain deep insight into the molecular biology of immune systems in T. molitor, the immune-relevant genes were discovered by referencing to those identified in T. castaneum genome [34]. Table 2 gives an overview of immune-related unigenes identified. 430 unigenes were noticed with significant similarity to a variety of immune system-related genes, revealing the presence of an elevated number of relevant molecules for the immune response (Table S5). This finding further demonstrates the high quality of our sequencing data. Based on molecular functions, immunity genes in insects can be assigned to three groups: effector genes encoding proteins that directly inhibit pathogen growth and survival, recognition genes that encode pathogen surveillance proteins, and signaling genes that encode proteins involved in immune-related signaling pathways [35]. Among the discovered sequences, the transcripts involved in these three different functional groups such as effector genes (e.g., antimicrobial peptide (AMP), and nimrod), recognition genes (e.g., PGRP, GRP, scavenger receptor (SR), and thiolester containing protein (TCP)), and signaling genes (e.g., Toll, relish and tab2) were identified. In T. castaneum genome, 388 immune genes have been identified [34], which are lower than those of Bombyx mori (203), D. melanogaster (268) or Anopheles gambiae (297) but are substantially higher than those of A. mellifera (121) [36]. Although the current number of immune sequences in T. molitor is more than that obtained from fully sequenced T. castaneum genome, it is noteworthy that some remainders might be absent. There was evidence of existence of few immune gene families such as spätzle, glutathione oxidase, and heme peroxidase that were not present in our transcriptome database. Furthermore, many unigenes obtained by next generation sequencing should be different fragments or even allelic or splice variants of the same gene [37]. In this study, the number of gene sequences predicted to encode immune genes would be therefore an overestimate of the actual number of genes belonging to each of the currently characterized gene families. The putative genes uncovered provide the basis for further identification of the physiological functions of candidate genes in T. molitor immune responses. However, most of these interesting genes are partial sequences. Their full-length sequences are needed to be obtained by RACE PCR for further study.
Table 2. Number of immunity-related genes in Tenebrio molitor transcriptome and Tribolium castaneum genome.
| Gene name | Gene numbers | |
| Tenebrio molitor a | Tribolium castaneum b | |
| Peptidoglycan recognition protein | 15 | 6 |
| β-1,3-Glucan recognition protein/Gram-negative binding proteins | 9 | 3 |
| C-type lectin | 24 | 16 |
| Galectin | 6 | 3 |
| Fibrinogen-like | 4 | 7 |
| Thioester-containing protein | 3 | 4 |
| Serine proteinase or its homolog | 90 | 168 |
| Serpin | 87 | 35 |
| Spätzle | 0 | 7 |
| Toll-like receptor | 2 | 9 |
| MD2-like | 0 | 8 |
| Cactus | 2 | 1 |
| Pelle | 1 | 1 |
| Myd88 | 3 | 1 |
| Tube | 1 | 1 |
| Pellino | 3 | 1 |
| Traf | 0 | 1 |
| Cactin | 7 | 1 |
| Relish | 6 | 4 |
| FADD | 0 | 1 |
| IKKb | 0 | 2 |
| IKKg | 0 | 1 |
| IMD | 0 | 1 |
| TAK | 0 | 1 |
| Caspase | 15 | 9 |
| IAP | 0 | 4 |
| Tab2 | 2 | 1 |
| Hep | 0 | 1 |
| Basket | 3 | 3 |
| Jra | 0 | 1 |
| Kay | 0 | 1 |
| DOME | 0 | 1 |
| HOP | 6 | 1 |
| STAT | 0 | 1 |
| Prophenoloxidase | 12 | 3 |
| Melanization inhibitor | 0 | 1 |
| Hexamerin | 23 | 6 |
| Catalase | 7 | 4 |
| Glutathione oxidase | 0 | 3 |
| Heme peroxidase | 0 | 11 |
| Peroxiredoxin | 10 | 6 |
| Superoxide dismutase | 8 | 4 |
| Antimicrobial peptide | 14 | 17 |
| Neuroglian/hemolin | 7 | 1 |
| Scavenger receptor | 36 | 21 |
| Nimrod | 13 | 4 |
| Draper | 11 | 1 |
Number of sequences obtained in this study that annotated with NCBI nr database.
Number of sequences from Zou et al. (2007).
DGE library sequencing
Using DGE method, the gene expression profile of T. molitor in response to parasitization by S. guani was globally analyzed. Two DGE libraries were sequenced from the pooled non-parasitized and parasitized pupae at four different time intervals post parasitization using parallel sequencing on the Illumina platform. A total of 6.07 and 6.16 million raw reads were generated, respectively (Table 3). After removing the low quality reads, 6,005,103 clean reads for the non-parasitized pupae and 6,102,041 clean reads for the parasitized pupae were obtained. About 99% of the raw reads passed the filter, reflecting the high quality of sequencing (Figure S3). The number of read entities with unique nucleotide sequences was 52,038 for non-parasitized pupae and 53416 for parasitized pupae, which was similar between each library. In addition, the distribution of the read expression over different read abundance categories demonstrated that both libraries displayed a similar pattern (Figure S4). These indicate that there was no bias in the construction of the libraries and the DGE data had good normality. Among the distinct clean reads, around 17% of them were high-expression reads with copy numbers larger than 100, with around 32% appearing below 6 times and most of them (around 50%) appearing between 6 and 100 times.
Table 3. Statistics of DGE sequencing.
| NP | P | |
| Raw data | 6,072,324 | 6,164,853 |
| Total clean reads | 6,005,103 | 6,102,041 |
| Total basepairs | 300,255,150 | 305,102,050 |
| Total mapped | 3,023,449 | 2,968,527 |
| Perfect match | 2,267,131 | 2,226,161 |
| < = 2 bp mismatch | 756,318 | 742,366 |
| Unique match | 3,022,715 | 2,967,863 |
| Multi-position match | 734 | 664 |
| Total unmapped | 2,981,654 | 3,133,514 |
NP, non-parasitization. P, parasitization.
After mapping to the above created transcriptome database, 3,023,449 and 2,968,527 total mapped reads were acquired for non-parasitized and parasitized pupae, respectively (Table 3). Of the total clean reads, 50.34% and 48.64% of distinct clean reads uniquely matched to unigenes, only 0.01% mapped to multiple unigenes, and 49.65% and 51.35% did not map to the dataset. The lower mapped ratio might result from insufficient reference transcriptome data. It also would be due to the incomplete NlaIII digestion during library preparation, many reads matching noncoding RNAs and the usage of alternative polyadenylation and/or splicing sites [38]. Additionally, incomplete T. molitor genome sequencing is the most likely reason. In order to further evaluate the unique mapping reads, we analyzed the distribution of gene coverage in each sample, which is the number of distinct clean reads that aligned to the reference genes (Figure S5). Very similar coverage patterns were observed in each library. Most gene coverage was >50% in each sample, with approximate 28% appearing between 90–100%. Fewer than 2% of the genes were covered by 0–10%.
Identification of DGEs
To gain insight into the global transcriptional changes taking place in S. guani parasitized T. molitor pupae, the DGEs were identified from the normalized DGE data by pairwise comparisons between non-parasitized and parasitized libraries. After parasitization, 3,431 mapped genes showed significant changes in expression. The up-regulated and down-regulated genes were shown in Figure 5. The number of down-regulated genes was larger than that of up-regulated. The detected fold changes (log2 ratio) of gene expression ranged from −16.48 to 17.96 (Table S6). 94.80% and 97.68% of genes in the two DGE libraries were up- or down-regulated between 1.0- and 5.0-fold. To investigate the most differentially up-and down-regulated genes, top 20 up-regulated and 20 down-regulated genes were screened out (Table S7). Only 20 genes had hits in the NCBI nr database, such as cathepsin B10 cysteine protease, reverse transcriptase-like protein, fatty acid synthase S-acetyltransferase, and cytochrome P450. Interestingly, it has been reported previously that cytochrome P450 known to be insecticide resistance/detoxification was up-regulated following parasitism, while it was the top down-regulated genes after parasitization in this study [39]. In Plutella xylostella larvae parasitized by Diadegma semiclausum, the transcripts of two cytochrome P450 genes were changed with one up-regulated and another down-regulated [40]. With respect to half of the top changed genes with no defined functions, it is consistent with the result that near 50% unigenes in the above transcriptome database remains to be annotated. However, it indicates that DGE tool has contributed greatly to identify novel genes involved in parasitoid and host interaction.
Figure 5. Changes in gene expression profile between non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae.
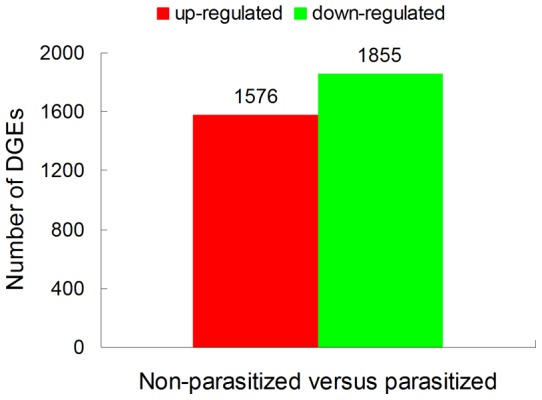
The number of up-regulated and down-regulated genes between NP and P groups is summarized here.
To facilitate the global analysis of gene expression, GO and KEGG enrichment analysis were performed by mapping each differentially expressed gene into the records of the GO and KEGG database. Of the enriched GO categories, the major subcategories were as follows: cell and cell part in the cellular component cluster; binding and catalytic activity in the molecular function cluster; and cellular process and metabolic process in the biological process cluster (Figure 6). There were 9, 15 and 14 GO terms significantly enriched in the cellular component, molecular function and biological process, respectively (Table S8). For the KEGG analysis, the major enrichment was observed in metabolic pathways, purine metabolism, Huntington's disease, vibrio cholerae infection, and MAPK signaling pathway (Table S9). Thirty five pathways were significantly enriched. Interestingly, MAPK signaling pathway, metabolic pathways and insect hormone biosynthesis were among them.
Figure 6. Selected major enriched GO terms.

Transcripts were annotated in three categories: cellular components, molecular function, and biological processes.
To validate the DGE result, qRT-PCR analysis was performed using gene-specific primers for the 13 up-regulated unigenes selected at random. The results showed that all unigenes exhibited the up-regulated expression trend in qRT-PCR analysis as the original DGE results (Figure 7). This illustrates that the results of gene expression profiling from DGE are reliable. It is worth noting that the expression levels of Unigene51387 and Unigene65859 were obviously different between qRT-PCR and DGE. This difference might be caused by a sensitivity of biases occurred between qRT-PCR and DGE.
Figure 7. Verification of differentially expressed genes by qRT-PCR.
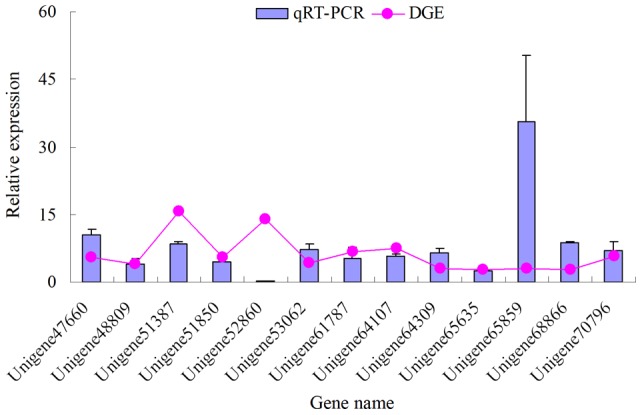
The relative expression levels of these unigenes indicate the log2 ratio of non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae by DGE and qRT-PCR, respectively. Error bars indicate standard deviations of averages from three replicates.
DGEs involved in immune response
By combining analysis of the indentified putative immune genes and DGEs, 16 out of the 47 immune gene families containing 74 unigenes were found to be respondent to parasitization (Table 4). They included AMP, C-type lectin (CTL), PGRP, GRP/GNBP, PPO, SP, serpin, superoxide dismutase (SOD), peroxiredoxin, hexamerin, cactus, pellino, cactin, caspase, and catalase. The highest changed expression levels in these genes were 6.77 fold for AMP and −13.12 fold for peroxiredoxin. There were 52 unigenes altered lower than two-fold change. As the transcriptional levels of some genes might be distinctly changed at early or late stage, the low fold change would be resulted by the pooled samples from different time intervals ranged from 6–48 h post-parasitization. Also, other immune genes whose transcription levels related to parasitization by S. guani could be revealed by detailed analysis of the samples from each time point.
Table 4. A list of Tenebrio molitor immune-related genes that were differentially transcribed after parasitization by Scleroderma guani.
| GeneID | Length | NP-RPKM | P-RPKM | Fold change | P-value | FDR |
| Antimicrobial peptide | ||||||
| Unigene48479 | 283 | 24.55 | 183.35 | 2.90 | 2.03E-09 | 5.64E-08 |
| Unigene8990 | 394 | 20.99 | 141.96 | 2.76 | 1.63E-10 | 5.32E-09 |
| Unigene12213 | 416 | 34.20 | 140.93 | 2.04 | 1.14E-13 | 6.38E-12 |
| Unigene15283 | 392 | 56.54 | 312.02 | 2.46 | 0 | 0 |
| Unigene22242 | 159 | 76.99 | 504.35 | 2.71 | 2.13E-13 | 1.10E-11 |
| Unigene61787 | 473 | 5.60 | 610.49 | 6.77 | 5.63E-06 | 8.58E-05 |
| Lysozyme | ||||||
| Unigene43694 | 251 | 358.51 | 122.16 | −1.55 | 1.59E-21 | 1.76E-19 |
| Unigene46654 | 269 | 290.24 | 71.40 | −2.02 | 1.56E-26 | 2.14E-24 |
| C-type lectin | ||||||
| Unigene11733 | 1225 | 85.34 | 34.38 | −1.31 | 1.49E-19 | 1.48E-17 |
| Unigene16572 | 921 | 4.31 | 15.00 | 1.80 | 3.98E-05 | 0.00048907 |
| Unigene25850 | 172 | 94.25 | 31.34 | −1.59 | 4.57E-05 | 0.00055274 |
| Unigene64817 | 578 | 11.45 | 34.98 | 1.61 | 3.85E-06 | 6.04E-05 |
| Unigene69689 | 1051 | 7.55 | 26.61 | 1.82 | 3.44E-09 | 9.27E-08 |
| Peptidoglycan recognition protein | ||||||
| Unigene2556 | 689 | 10.56 | 40.10 | 1.92 | 2.00E-09 | 5.56E-08 |
| Unigene46415 | 268 | 60.49 | 218.76 | 1.85 | 3.82E-14 | 2.44E-12 |
| Unigene54494 | 341 | 74.70 | 761.83 | 3.35 | 0 | 0 |
| Unigene65399 | 605 | 70.54 | 226.67 | 1.68 | 0 | 0 |
| Unigene68368 | 834 | 12.30 | 28.28 | 1.20 | 6.35E-05 | 0.00073674 |
| β-1,3-Glucan recognition protein/ Gram-negative binding proteins | ||||||
| Unigene59692 | 425 | 78.62 | 233.88 | 1.57 | 0 | 0 |
| Unigene69724 | 1058 | 49.72 | 128.34 | 1.37 | 5.28E-14 | 3.24E-12 |
| Prophenoloxidase | ||||||
| Unigene20627 | 154 | 406.02 | 109.40 | −1.89 | 7.58E-20 | 7.64E-18 |
| Unigene20889 | 155 | 192.09 | 58.69 | −1.71 | 4.84E-09 | 1.27E-07 |
| Unigene37907 | 221 | 667.64 | 167.71 | −1.99 | 8.13E-48 | 1.77E-45 |
| Unigene40274 | 232 | 1232.05 | 325.32 | −1.92 | 9.24E-87 | 2.38E-84 |
| Unigene68337 | 830 | 536.90 | 135.99 | −1.98 | 9.24E-140 | 2.49E-137 |
| Unigene11469 | 539 | 25.17 | 6.88 | −1.87 | 3.00E-05 | 0.00038056 |
| Serine protein or its homolog | ||||||
| Unigene1207 | 1292 | 10.75 | 29.21 | 1.44 | 5.38E-09 | 1.40E-07 |
| Unigene13777 | 892 | 14.46 | 32.49 | 1.17 | 1.44E-05 | 0.000199 |
| Unigene2204 | 386 | 42.00 | 13.97 | −1.59 | 4.57E-05 | 0.000553 |
| Unigene2906 | 208 | 17.50 | 79.38 | 2.18 | 1.30E-06 | 2.25E-05 |
| Unigene39230 | 227 | 148.65 | 400.77 | 1.43 | 0 | 0 |
| Unigene41772 | 240 | 64.79 | 190.93 | 1.56 | 9.09E-12 | 3.45E-10 |
| Unigene45616 | 263 | 42.77 | 98.65 | 1.21 | 2.57E-05 | 0.000332 |
| Unigene51777 | 311 | 61.70 | 222.10 | 1.85 | 7.77E-15 | 5.51E-13 |
| Unigene54421 | 340 | 12.65 | 74.33 | 2.55 | 2.93E-07 | 5.75E-06 |
| Unigene60508 | 442 | 62.87 | 190.58 | 1.60 | 0 | 0 |
| Unigene61013 | 454 | 160.31 | 414.87 | 1.37 | 3.00E-13 | 1.49E-11 |
| Unigene61795 | 474 | 25.82 | 105.92 | 2.04 | 2.13E-13 | 1.10E-11 |
| Unigene69393 | 989 | 122.76 | 58.26 | −1.08 | 8.18E-17 | 6.86E-15 |
| Unigene69732 | 1058 | 44.40 | 140.45 | 1.66 | 2.60E-13 | 1.30E-11 |
| Unigene70037 | 1131 | 551.38 | 1828.31 | 1.73 | 2.26E-12 | 9.35E-11 |
| Unigene9960 | 952 | 34.75 | 2.83 | −3.62 | 2.96E-21 | 3.24E-19 |
| Serpin | ||||||
| Unigene1138 | 230 | 1329.07 | 438.03 | −1.60 | 2.76E-72 | 6.94E-70 |
| Unigene1484 | 775 | 10.67 | 54.35 | 2.35 | 1.63E-10 | 5.32E-09 |
| Unigene16750 | 547 | 99.79 | 38.81 | −1.36 | 1.80E-11 | 6.58E-10 |
| Unigene34207 | 204 | 77.84 | 181.68 | 1.22 | 3.50E-07 | 6.78E-06 |
| Unigene44272 | 254 | 61.22 | 14.59 | −2.07 | 1.75E-06 | 2.95E-05 |
| Unigene45167 | 260 | 27.99 | 116.63 | 2.06 | 1.10E-09 | 3.18E-08 |
| Unigene54405 | 339 | 105.40 | 44.73 | −1.24 | 4.73E-07 | 8.96E-06 |
| Unigene55316 | 351 | 67.86 | 233.27 | 1.78 | 0 | 0 |
| Unigene57316 | 381 | 26.92 | 305.99 | 3.51 | 3.98E-12 | 1.58E-10 |
| Unigene59916 | 429 | 45.50 | 223.06 | 2.29 | 1.22E-14 | 8.38E-13 |
| Unigene60952 | 453 | 70.84 | 238.02 | 1.75 | 4.69E-14 | 2.91E-12 |
| Unigene60995 | 454 | 67.77 | 138.04 | 1.03 | 8.35E-09 | 2.11E-07 |
| Unigene63284 | 518 | 68.98 | 26.02 | −1.41 | 2.66E-08 | 6.26E-07 |
| Unigene69118 | 940 | 82.36 | 333.36 | 2.02 | 4.38E-13 | 2.07E-11 |
| Unigene69212 | 958 | 67.69 | 17.59 | −1.94 | 3.11E-21 | 3.39E-19 |
| Unigene69220 | 959 | 63.82 | 460.97 | 2.85 | 0 | 0 |
| Unigene70002 | 1119 | 68.00 | 29.51 | −1.20 | 5.38E-13 | 2.47E-11 |
| Superoxide dismutase | ||||||
| Unigene66938 | 701 | 11.80 | 1.44 | −3.03 | 1.88E-05 | 0.00025097 |
| Peroxiredoxin | ||||||
| Unigene14679 | 593 | 8.93 | 0.00 | −13.12 | 1.78E-05 | 0.00023959 |
| Unigene40514 | 233 | 160.44 | 368.76 | 1.20 | 2.62E-13 | 1.31E-11 |
| Hexamerin | ||||||
| Unigene16646 | 1348 | 138.42 | 297.95 | 1.11 | 8.62E-13 | 3.82E-11 |
| Unigene18014 | 150 | 760.91 | 276.29 | −1.46 | 1.03E-24 | 1.33E-22 |
| Unigene28282 | 181 | 1398.25 | 571.50 | −1.29 | 5.26E-44 | 1.08E-41 |
| Unigene30910 | 191 | 1032.32 | 365.17 | −1.50 | 4.65E-43 | 9.28E-41 |
| Unigene36412 | 214 | 1290.85 | 603.03 | −1.10 | 1.98E-37 | 3.62E-35 |
| Unigene65901 | 631 | 2635.62 | 1255.39 | −1.07 | 2.06E-207 | 5.65E-205 |
| Cactus | ||||||
| Unigene66277 | 654 | 56.66 | 126.22 | 1.16 | 2.50E-13 | 1.26E-11 |
| Unigene70354 | 1227 | 64.98 | 188.38 | 1.54 | 1.85E-13 | 9.70E-12 |
| Pellino | ||||||
| Unigene62396 | 490 | 50.64 | 102.46 | 1.02 | 3.15E-07 | 6.14E-06 |
| Cactin | ||||||
| Unigene2785 | 487 | 42.80 | 13.84 | −1.63 | 2.38E-06 | 3.91E-05 |
| Caspase | ||||||
| Unigene13475 | 850 | 17.90 | 41.23 | 1.20 | 1.01E-06 | 1.78E-05 |
| Catalase | ||||||
| Unigene42771 | 246 | 110.28 | 16.44 | −2.75 | 6.15E-14 | 3.68E-12 |
NP, non-parasitization. P, parasitization. RPKM, Reads per kb per million reads. FDR, false discovery rate. FDR ≤0.001 and the absolute value of log2 Ratio ≥1 were used in this study.
In insects, recognition of nonself is the initial process of the innate immune response. Regarding prokaryotic pathogens and fungi, it is notably known that the recognition step is mediated by a group of proteins, known as pattern recognition proteins (PRPs) such as GRP/GNBP, PGRP, and CTL [41]. GRP/GNBP was first discovered in the insect order Lepidoptera (butterflies and moths), which recognizes the β-1,3-glucans of fungal cell walls and contains a C-terminal glycosyl hydrolase 16 (GH16) domain lacking the active site of GH16 [42]. PGRP was first discovered from the hemolymph and cuticle of Bombyx mori, which specifically binds to peptidoglycan [43]. CTLs are a group of carbohydrate-binding proteins that share a common structural motif, the carbohydrate recognition domain [44]. These PRPs can specifically recognize sugars on the surface of microorganisms and cause a series of immune responses to effectively resist pathogenic invasions. In invertebrates, they are important components of cellular as well as humoral innate immune responses. They function as pathogen recognition receptors in the innate immune system by activating PPO in hemolymph, promoting the hemocyte phagocytosis, and by participating in hemocyte nodule formation and encapsulation [43], [45]–[48]. In this study, transcriptions of GRP/GNBP, PGRP, and CTL were regulated in parasitized T. molitor pupae. Previously studies also support these PRPs as candidates for the recognition function involved in the parasitoid-host interaction. Microarray experiments showed that two PGRPs of Drosophila were significantly upregulated after parasitoid attack [49]. Hyposoter didymator polydnavirus infection can down-regulate a Spodoptera frugiperda gene with high similarity with the Manduca sexta immulectin-2 [50]. Especially, it was reported that the gene lectin-24A was massively up-regulated in parasitized Drosophila larvae after parasitization at the time when the capsule was formed [51]. A similar investigation indicated that the inhibitory effect of P. puparum venom on host encapsulation is consistent with its effect in suppressing CTL expression [52]. Thus, CLT is an important immune candidate involved in the suppression of host cellular immune defense induced by the parasitoid. The other PRPs altered by parasitization might also be needed for the egg or offspring of parasitoid to avoid or evade the host immune response, which are mediated by the introduction of venomous maternal factor of parasitoid wasps.
For opposing the invasion of foreign agents, a hallmark of the holometabolous insect hosts defense is the challenge-induced synthesis and secretion of potent AMPs that accumulate in the hemolymph [53]. AMP production is an important humoral immune reaction in insects. It plays crucial roles in eliminating infections from various bacteria, fungi, viruses and protozoa by acting as pore-formers or metabolic inhibitors [54]. Eight unigenes encoding putative AMPs were found to be regulated in the parasitized T. molitor pupae (Table 4), including attacin (Unigene48479 and Unigene22242), acaloleptin (Unigene8990, Unigene12213, and Unigene15283), lysozyme (Unigene43694 and Unigene46654), and a protein that is similar to antibacterial peptide Cp1 of T. castaneum (Unigene61787). Except for lysozyme, the other AMPs were up-regulated. Similarly, few earlier investigations indicated that parasitization induces elevation of AMP transcription in the host. For instance, the parasitoid Leptopilina boulardi can trigger the transcription of the genes encoding diptericin and cecropin in Drosophila susceptible strain [55]. Nicolas et al. [56] also reported that diptericin and cecropin were either unchanged or minimally induced in Drosophila susceptible strain following attack by larval (L. boulardi and L. heterotoma) and pupal stage (Trichopria) parasitoids. In P. xylostella, gloverin, moricin, lysozyme II, and cecropin were up-regulated by D. semiclausum attack [40]. In contrast, like the lysozyme down-regulated by S. guani attack, there were a number of host AMPs that responded similarly when parasitized by parasitoid or injected with parasitoid's maternal factors. For example, transcript levels of cecropin and gloverin in eggs of M. sexta were suppressed after parasitization by Trichogramma evanescens [57]. After Pieris rapae pupae injected with P. puparum venom, ten potential antimicrobial molecules including cecropin, lysozyme, attacin, lebocin, proline-rich AMP, cysteine-rich peptide, gallerimycin and immune inducible peptide had lower transcript levels in hemocytes or fat body of the venom injected hosts[52], demonstrating that the transcriptional down regulation of these antimicrobial genes is due to the venom. Nevertheless, parasitization of Drosophila resistant strain by the corresponding parasitoids did not induce transcripts of diptericin and cecropin whilst the susceptible strain did [55], [56]. This lack of antibacterial transcript induction in the parasitized resistant strain suggests that the maternal factors injected during oviposition by the parasitoids are not strong elicitors. Also, some AMPs of lepidopteran larvae and pupae such as moricin and gloverin were found to be not inducible by bacterial challenge [58], [59], while they were appeared to be in response to parasitoid attack [40], [57]. Accordingly, we would speculate that the induction of AMP following parasitization might be different from that case of bacterial. A specific genetic factor (or highly linked factors), designated humoral response to parasitoid (hrtp) in D. melanogaster, has been found to be essential for the activation of diptericin by the parasitoid wasp [60], indicating that the expression of antibacterial gene after parasitoid infection is linked to a genetic regulation. However, it remains confusing that why the AMPs were differentially regulated in the same or different parasitoid-host systems, e.g., lysozyme in the hosts up-regulated by D. semiclausum whilst down-regulated by S. guani in this study. Furthermore, since parasitoid attack involves wounding and penetration, parasitized insects are subsequently more susceptible to opportunistic infections. It is possible that the regulation of antimicrobial peptides is associated with damage to the exoskeleton and low-level exposure to microbial factors on the surface of the host or ovipositor of the wasp [51].
In response to invaders, an essential component of the cell-mediated immediate immune response in insects is the melanization reaction observed at the site of cuticular injury or on the surface of parasites invading the hemocoel [61]. Melanization is the result of proteolytic cascade triggered by minute amounts of elicitors, which is associated with a number of vital proteins such as PPO, SP, SPH, and serpin [62]. It is well documented that parasitoid can survive despite hosts melanotic encapsulation, or destroy with no evidence of this host response [63]. SP, SPH and serpin that retain the ability to inhibit melanization have been indentified from the venom of parasitoids [15]. The inhibition is most likely due to interference with the proteolytic cascade that leads to the activation of PPO by these venom proteins. In the current study, we found that the transcripts of PPO, SP or SPH and serpin were changed after parasitism in T. molitor. In addition to transcription, these genes can be regulated at the translational level. In P. xylostella larvae parasitized by Cotesia plutellae, the expression of pxSerpin 2 gene increased as parasitism progressed, while its protein profile was reduced compared to that of the control, and continued to decrease with the progress of parasitism [64]. Genes with altered expression that are associated with the PPO system are often observed following parasitization [65]–[68]. This should result in a general decrease in PO activation and inhibition of melanization observed in parasitized hosts. Thus, the regulation of these genes upon immune challenge in parasitized host could be part of a parasitoid immune suppressive strategy. It has been reported that the transcripts of PPO in Spodoptera frugiperda were differentially affected by Hyposoter didymator Ichnovirus and Microplitis demolitor Bracovirus [69].
Reactive oxygen species (ROS) production is an immediate acute-phase oxidative defense in response to pathogen assault or cellular stress such as phagocytosis and melanotic encapsulation [70]. Due to the cytotoxicity of ROS, its production is tightly regulated by immune responsive antioxidant enzymes such as SOD, catalase, glutathione oxidase, thioredoxin reductase, and peroxidase [71]. Our analysis shows that the transcript levels of SOD, catalase, and peroxiredoxin (belonging to peroxidase) genes were altered after parasitization by S. guani. Moreover, two peroxidases, thiol peroxiredoxin and peroxiredoxin were strongly induced in the plasma of Papilio xuthus after parasitization [72]. However, the expression change may cause perturbation in normal SOD and peroxiredoxin function, which would lead to the production of parasitoid egg or offspring killing by cytotoxic ROS. Supposedly, the excessive produced ROS might be sequestered and localized to the surface of the parasitoid, thereby preventing adverse systemic reactions from occurring in the open circulatory system [48]. Given the diverse roles of ROS in immune defense, altered transcription of these genes following parasitization could be an epithelial host response to the physiological injury brought about during the process of oviposition or an alteration that adversely affects metabolism.
Hexamerins consist of six identical or similar subunits with a molecular mass in the range of 80 kDa each [73]. They belong to a protein superfamily that also comprises arthropod PPOs and hemocyanins, crustacean pseudohemocyanins, and the hexamerin receptors discovered in the diptera [74]. In insects, as hexamerins have been found at very high concentrations in the hemolymph of many insect species, they are thought to act mainly as storage proteins in non-feeding periods [75]. Among the identified immune related DGEs, six unigenes encoding hexamerin (one up-regulate and five down-regulated) were found. Similarly, the transcriptional responses of hexamerins to parasitization have been documented in several host-parasitoid systems with different regulation strategies [76]–[79]. Although distinct regulatory mechanisms at transcriptional level may exist in different host–parasitoid systems, studies so far indicate that the titer of hexamerins in the hemolymph increase in the hosts following parasitism [80]–[83]. Regardless of the critical role of hexamerins acting as storage proteins in growth and development, it has been evidenced that some hexamerins are part of the innate immune system in various arthropods, acting as pro-coagulants [84]. They also fulfill immune functions in insects. For example, it was demonstrated that the expression of genes and proteins in the honey bee was significantly changed after activation of the immune system by bacterial challenge or even after injury caused by injection of water [85], [86]. It has been speculated that arylphorin suppresses hemocyte degranulation and subsequent immune reactions that lead to the encapsulation of the parasitoid's egg [87]. The high content of aromatic amino acids in the arylphorins may enhance the cross-linking capabilities of this protein to form ideal clotting, thereby isolating parasitoid egg from host immune response or reducing host humoral immune reaction cascade [73]. Based on these hypotheses, hexamerins are probably related to some function in host protection, while much work still needs to be done to explore the mechanisms.
The innate immune system of insects relies on both humoral and cellular immune responses that are triggered by the immune challenge and mediated via activation of signaling pathways [88]. Four signal transduction pathways, Toll, Imd, JNK and JAK/STAT, are known to be present in insect immunity [89]. On the other hand, the immune repertoire genes participating in the signal transduction pathways have remained highly conserved throughout the different insect orders [36]. The expression of most of these genes is switched on following invasion of microbes, but the signaling pathways involved in anti-parasite responses are not well- understood [90]. Our results indicated that several transcripts associated with Toll and Imd pathway including cactus, pellino, cactin, and caspase were affected after S. guani attack. Similarly, even though the transcripts of most components of the different signaling pathways were found in the transcriptome database of P. xylostella, only transcription levels of proteins that showed similarity to the Toll receptor were up-regulated after parasitoid attack [40]. In addition to the components of Toll and Imd pathways, microarray-based genome-wide analyses indicated that parasitism by Asobara tabida or L. boulardi, but not L. heterotoma can induce multiple genes encoding signaling components of the JAK/STAT pathway in Drosophila [49], [51]. The data confirmed that the signaling pathways play a role in the anti-parasitoid immune response. It is likely that a deactivation of the signaling cascade might be directly linked to a suppression of host immune defense by the parasitoid.
Conclusion
In summary, the transcriptome of T. molitor pupae was sequenced with massive parallel pyrosequencing using Illumina sequencing technology, permitting the discovery of a great number of genes involved in immunity. The transcriptome profiling data sets obtained in this study make a significant contribution to existing sequence resources for the beetle. The explored immune genes provide valuable resource for future research in understanding immune system and defense mechanisms of T. molitor as well as many other Coleopteran species. Additionally, we investigated the global gene transcription profiles of T. molitor in response to parasitization by S. guani, especially with focus on the immune aspect. A large number of T. molitor genes, including immune-related gene, were differentially expressed after parasitization by S. guani. It is noteworthy that parasitization induces sets of genes that had not previously been implicated in immune function, or are not activated upon bacterial infection. This provides new insight into the host immune defense mechanisms against parasitoid, and the ways in which parasitoids overcome these mechanisms from a molecular prospective.
Supporting Information
Length distribution of Tenebrio molitor contigs. Horizontal axis represents the length of contigs and vertical axis represents number of contigs.
(TIF)
Length distribution of Tenebrio molitor scaffolds. Horizontal axis represents the length of scaffolds and vertical axis represents number of scaffolds.
(TIF)
Classification of raw reads in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Numbers in parentheses show the percentage of each type of read among the total raw reads.
(TIF)
Distribution of distinct clean reads in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Numbers in the square brackets indicate the range of copy numbers for a specific category of reads. The data in parentheses indicate the percentage of corresponding reads among the total distinct reads.
(TIF)
Distribution of gene coverage in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae.
(TIF)
Primers used in qRT-PCR for validating differentially expressed genes.
(XLS)
Top Blast hits from NCBI nr database. BLASTX against the nr protein database was used with a cutoff E-value of 10−5.
(XLS)
Gene ontology (GO) annotation of unigenes.
(XLS)
Kyoto Encyclopedia of Genes and Genome (KEGG) annotation of unigenes.
(XLS)
Immunity-related genes identified in Tenebrio molitor transcriptome.
(XLS)
Differentially expressed genes between non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Raw intensity, the total number of reads sequenced for each gene. RPKM, Reads per kb per million reads. FDR, false discovery rate. FDR ≤0.001 and the absolute value of log2Ratio ≥1 were used in this study.
(XLS)
Description of the top 20 most up- and down-regulated genes.
(XLS)
Gene Ontology (GO) enrichment analysis of differentially expressed genes.
(XLS)
Kyoto Encyclopedia of Genes and Genome (KEGG) enrichment analysis of differentially expressed genes.
(XLS)
Acknowledgments
We thank Xiao-Qing Chen for assistance in quantitative RT-PCR analysis.
Funding Statement
This work was supported by the Key Project of Chinese Ministry of Education (211171), Applied Basic Research Programs of Yunnan Province (2010CD063), and National Natural Science Foundation of China (31260449). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fearon DT (1997) Seeking wisdom in innate immunity. Nature 388: 323–324. [DOI] [PubMed] [Google Scholar]
- 2. Lavine MD, Strand MR (2002) Insect hemocytes and their role in immunity. Insect Biochem Mol Biol 32: 1295–1309. [DOI] [PubMed] [Google Scholar]
- 3. Stanley D, Miller J, Tunaz H (2009) Eicosanoid actions in insect immunity. J Innate Immun 1: 282–290. [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann JA (1995) Innate immunity of insects. Curr Opin Immunol 7: 4–10. [DOI] [PubMed] [Google Scholar]
- 5. Schmid-Hempel P (2005) Evolutionary ecology of insect immune defenses. Annu Rev Entomol 50: 529–551. [DOI] [PubMed] [Google Scholar]
- 6. Feldhaar H, Gross R (2008) Immune reactions of insects on bacterial pathogens and mutualists. Microbes Infect 10: 1082–1088. [DOI] [PubMed] [Google Scholar]
- 7.Quicke DLJ (1997) Parasitic Wasps, Chapman & Hall, London, UK.
- 8. Beckage NE, Gelman DB (2004) Wasp parasitoid disruption of host development: implications for new biologically based strategies for insect control. Annu Rev Entomol 49: 299–330. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt O, Schuchmann-Feddersen I (1989) Role of virus-like particles in parasitoid-host interaction of insects. Subcell Biochem 15: 91–119. [DOI] [PubMed] [Google Scholar]
- 10. Shelby KS, Webb BA (1999) Polydnavirus-mediated suppression of insect immunity. J Insect Physiol 45: 507–514. [DOI] [PubMed] [Google Scholar]
- 11. Moreau SJ, Guillot S (2005) Advances and prospects on biosynthesis, structures and functions of venom proteins from parasitic wasps. Insect Biochem Mol Biol 35: 1209–1223. [DOI] [PubMed] [Google Scholar]
- 12. Pennacchio F, Strand MR (2006) Evolution of developmental strategies in parasitic hymenoptera. Annu Rev Entomol 51: 233–258. [DOI] [PubMed] [Google Scholar]
- 13. Schmidt O (2006) At the core of parasitoid-host interactions. Arch Insect Biochem Physiol 61: 107–109. [DOI] [PubMed] [Google Scholar]
- 14. Kraaijeveld AR, Godfray HC (2009) Evolution of host resistance and parasitoid counter-resistance. Adv Parasitol 70: 257–280. [DOI] [PubMed] [Google Scholar]
- 15. Asgari S, Rivers DB (2011) Venom proteins from endoparasitoid wasps and their role in host-parasite interactions. Annu Rev Entomol 56: 313–335. [DOI] [PubMed] [Google Scholar]
- 16. Zhang L (1980) A preliminary study on the use of a bethylid (Scleroderma sp.) to control Semanotus bifasciatus . Sci Silvae Sin 16: 41–45. [Google Scholar]
- 17. Qin X, Gao R, Xu C, Yao D, Li Y, et al. (1982) Artificial propagation and applied research on Scleroderma spp. in Beijing. Sci Silvae Sin 18: 209–213. [Google Scholar]
- 18. Li X, Lu D, Liu X, Zhang Q, Zhou X (2011) Ultrastructural characterization of olfactory sensilla and immunolocalization of odorant binding and chemosensory proteins from an ectoparasitoid Scleroderma guani (Hymenoptera: Bethylidae). Int J Biol Sci 7: 848–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Cheng H (2000) Advances in applied research on Scleroderma spp. Chin J Biol Con 16: 166–170. [Google Scholar]
- 20. Li R, Zhu H, Ruan J, Qian W, Fang X, et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 22. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 23. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 24. Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7: 986–995. [DOI] [PubMed] [Google Scholar]
- 25. Xue J, Bao YY, Li BL, Cheng YB, Peng ZY, et al. (2010) Transcriptome analysis of the brown planthopper Nilaparvata lugens . PLoS One 5: e14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu JY, Zhao N, Yang B (2012) Global transcriptome profiling of the pine shoot beetle, Tomicus yunnanensis (Coleoptera: Scolytinae). PLoS One 7: e32291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carvalho RA, Azeredo-Espin AM, Torres TT (2010) Deep sequencing of New World screw-worm transcripts to discover genes involved in insecticide resistance. BMC Genomics 11: 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, et al. (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics 12: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bai X, Mamidala P, Rajarapu SP, Jones SC, Mittapalli O (2011) Transcriptomics of the bed bug (Cimex lectularius). PLoS One 6: e16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu JY, Zhao N, Yang B (2012) Global transcriptional analysis of olfactory genes in the head of pine shoot beetle, Tomicus yunnanensis. Comp Funct Genomics Article ID: 491748. [DOI] [PMC free article] [PubMed]
- 31. Keeling CI, Henderson H, Li M, Yuen M, Clark EL, et al. (2012) Transcriptome and full-length cDNA resources for the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major insect pest of pine forests. Insect Biochem Mol Biol 42: 525–536. [DOI] [PubMed] [Google Scholar]
- 32. Richards S, Gibbs RA, Weinstock GM, Brown SJ, Denell R, et al. (2008) The genome of the model beetle and pest Tribolium castaneum . Nature 452: 949–955. [DOI] [PubMed] [Google Scholar]
- 33. Badisco L, Huybrechts J, Simonet G, Verlinden H, Marchal E, et al. (2011) Transcriptome analysis of the desert locust central nervous system: production and annotation of a Schistocerca gregaria EST database. PLoS One 6: e17274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zou Z, Evans JD, Lu Z, Zhao P, Williams M, et al. (2007) Comparative genomic analysis of the Tribolium immune system. Genome Biol 8: R177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, et al. (2007) Dynamic evolution of the innate immune system in Drosophila . Nat Genet 39: 1461–1468. [DOI] [PubMed] [Google Scholar]
- 36. Tanaka H, Ishibashi J, Fujita K, Nakajima Y, Sagisaka A, et al. (2008) A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori . Insect Biochem Mol Biol 38: 1087–1110. [DOI] [PubMed] [Google Scholar]
- 37. Zagrobelny M, Scheibye-Alsing K, Jensen NB, Møller BL, Gorodkin J, et al. (2009) 454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides. BMC Genomics 10: 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, Jiang R, Wu H, Liu P, Xie J, et al. (2012) Next-generation sequencing-based transcriptome analysis of Cryptolaemus montrouzieri under insecticide stress reveals resistance-relevant genes in ladybirds. Genomics 100: 35–41. [DOI] [PubMed] [Google Scholar]
- 39. Takeda T, Nakamatsu Y, Tanaka T (2006) Parasitization by Cotesia plutellae enhances detoxifying enzyme activity in Plutella xylostella . Pestic Biochem Physiol 86: 15–22. [Google Scholar]
- 40. Etebari K, Palfreyman RW, Schlipalius D, Nielsen LK, Glatz RV, et al. (2011) Deep sequencing-based transcriptome analysis of Plutella xylostella larvae parasitized by Diadegma semiclausum . BMC Genomics 12: 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu XQ, Zhu YF, Ma C, Fabrick JA, Kanost MR (2002) Pattern recognition proteins in Manduca sexta plasma. Insect Biochem Mol Biol 32: 1287–1293. [DOI] [PubMed] [Google Scholar]
- 42. Hughes AL (2012) Evolution of the βGRP/GNBP/β-1,3-glucanase family of insects. Immunogenetics 64: 549–558. [DOI] [PubMed] [Google Scholar]
- 43. Dziarski R (2004) Peptidoglycan recognition proteins (PGRPs). Mol Immunol 40: 877–886. [DOI] [PubMed] [Google Scholar]
- 44. Tian YY, Liu Y, Zhao XF, Wang JX (2009) Characterization of a C-type lectin from the cotton bollworm, Helicoverpa armigera . Dev Comp Immunol 33: 772–779. [DOI] [PubMed] [Google Scholar]
- 45. Yu XQ, Kanost MR (2004) Immulectin-2, a pattern recognition receptor that stimulates hemocyte encapsulation and melanization in the tobacco hornworm, Manduca sexta . Dev Comp Immunol 28: 891–900. [DOI] [PubMed] [Google Scholar]
- 46. Ourth DD, Narra MB, Chung KT (2005) Isolation of mannose-binding C-type lectin from Heliothis virescens pupae. Biochem Biophys Res Commun 335: 1085–1089. [DOI] [PubMed] [Google Scholar]
- 47. Pal S, Wu LP (2009) Pattern recognition receptors in the fly: lessons we can learn from the Drosophila melanogaster immune system. Fly (Austin) 3: 121–129. [DOI] [PubMed] [Google Scholar]
- 48. Carton Y, Poirié M, Nappi AJ (2008) Insect immune resistance to parasitoids. Insect Sci 15: 67–87. [Google Scholar]
- 49. Schlenke TA, Morales J, Govind S, Clark AG (2007) Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster . PLoS Pathog 3: 1486–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barat-Houari M, Hilliou F, Jousset FX, Sofer L, Deleury E, et al. (2006) Gene expression profiling of Spodoptera frugiperda hemocytes and fat body using cDNA microarray reveals polydnavirus-associated variations in lepidopteran host genes transcript levels. BMC Genomics 7: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wertheim B, Kraaijeveld AR, Schuster E, Blanc E, Hopkins M, et al. (2005) Genome-wide gene expression in response to parasitoid attack in Drosophila . Genome Biol 6: R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fang Q, Wang F, Gatehouse JA, Gatehouse AM, Chen XX, et al. (2011) Venom of parasitoid, Pteromalus puparum, suppresses host, Pieris rapae, immune promotion by decreasing host C-type lectin gene expression. PLoS One 6: e26888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lemaitre B, Hoffmann J (2007) The host defense of Drosophila melanogaster . Annu Rev Immunol 25: 697–743. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Z, Zhu S (2012) Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev Comp Immunol 38: 262–274. [DOI] [PubMed] [Google Scholar]
- 55. Coustau C, Carkion Y, Nappi A, Shotkoski F, Ffrench-Constant R (1996) Differential induction of antibacterial transcripts in Drosophila susceptible and resistant to parasitism by Leptopilina boulardi . Insect Mol Biol 5: 167–172. [DOI] [PubMed] [Google Scholar]
- 56. Nicolas E, Nappi AJ, Lemaitre B (1996) Expression of antimicrobial peptide genes after infection by parasitoid wasps in Drosophila . Dev Comp Immunol 20: 175–181. [DOI] [PubMed] [Google Scholar]
- 57. Abdel-latief M, Hilker M (2008) Innate immunity: eggs of Manduca sexta are able to respond to parasitism by Trichogramma evanescens . Insect Biochem Mol Bio 38: 136–145. [DOI] [PubMed] [Google Scholar]
- 58. Hara S, Yamakawa M (1995) Moricin, a novel type of antibacterial peptide isolated from the silkworm, Bombyx mori . J Biol Chem 270: 29923–29927. [DOI] [PubMed] [Google Scholar]
- 59. Axén A, Carlsson A, Engström A, Bennich H (1997) Gloverin, an antibacterial protein from the immune hemolymph of Hyalophora pupae. Eur J Biochem 247: 614–619. [DOI] [PubMed] [Google Scholar]
- 60. Benassi V, Coustau C, Carton Y (2000) Insect immunity: a genetic factor (hrtp) is essential for antibacterial peptide expression in Drosophila after infection by parasitoid wasps. Arch Insect Biochem Physiol 43: 64–71. [DOI] [PubMed] [Google Scholar]
- 61. Christensen BM, Li J, Chen CC, Nappi AJ (2005) Melanization immune responses in mosquito vectors. Trends Parasitol 21: 192–199. [DOI] [PubMed] [Google Scholar]
- 62. Eleftherianos I, Revenis C (2011) Role and importance of phenoloxidase in insect hemostasis. J Innate Immun 3: 28–33. [DOI] [PubMed] [Google Scholar]
- 63. Nappi A, Poirié M, Carton Y (2009) The role of melanization and cytotoxic by-products in the cellular immune responses of Drosophila against parasitic wasps. Adv Parasitol 70: 99–121. [DOI] [PubMed] [Google Scholar]
- 64. Song KH, Jung MK, Eum JH, Hwang IC, Han SS (2008) Proteomic analysis of parasitized Plutella xylostella larvae plasma. J Insect Physiol 54: 1270–1280. [DOI] [PubMed] [Google Scholar]
- 65. Hartzer KL, Zhu KY, Baker JE (2005) Phenoloxidase in larvae of Plodia interpunctella (Lepidoptera: Pyralidae): molecular cloning of the proenzyme cDNA and enzyme activity in larvae paralyzed and parasitized by Habrobracon hebetor (Hymenoptera: Braconidae). Arch Insect Biochem Physiol 59: 67–79. [DOI] [PubMed] [Google Scholar]
- 66. Doucet D, Béliveau C, Dowling A, Simard J, Feng Q, et al. (2008) Prophenoloxidases 1 and 2 from the spruce budworm, Choristoneura fumiferana: molecular cloning and assessment of transcriptional regulation by a polydnavirus. Arch Insect Biochem Physiol 67: 188–201. [DOI] [PubMed] [Google Scholar]
- 67. Mahadav A, Gerling D, Gottlieb Y, Czosnek H, Ghanim M (2008) Parasitization by the wasp Eretmocerus mundus induces transcription of genes related to immune response and symbiotic bacteria proliferation in the whitefly Bemisia tabaci . BMC Genomics 9: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhu JY, Yang P, Wu GX (2011) Prophenoloxidase from Pieris rapae: gene cloning, activity, and transcription in response to venom/calyx fluid from the endoparasitoid wasp Cotesia glomerata . J Zhejiang Univ Sci B 12: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Provost B, Jouan V, Hilliou F, Delobel P, Bernardo P, et al. (2011) Lepidopteran transcriptome analysis following infection by phylogenetically unrelated polydnaviruses highlights differential and common responses. Insect Biochem Mol Biol 41: 582–591. [DOI] [PubMed] [Google Scholar]
- 70. Ribou AC, Reinhardt K (2012) Reduced metabolic rate and oxygen radicals production in stored insect sperm. Proc Biol Sci 279: 2196–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Corona M, Robinson GE (2006) Genes of the antioxidant system of the honey bee: annotation and phylogeny. Insect Mol Biol 15: 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhu JY, Ye GY, Fang Q, Hu C (2009) Proteome changes in the plasma of Papilio xuthus (Lepidoptera: Papilionidae): effect of parasitization by the endoparasitic wasp Pteromalus puparum (Hymenoptera: Pteromalidae). J Zhejiang Univ Sci B 10: 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Burmester T (1999) Evolution and function of the insect hexamerins. Eur J Entomol 96: 213–225. [Google Scholar]
- 74. Pick C, Schneuer M, Burmester T (2009) The occurrence of hemocyanin in Hexapoda. FEBS J 276: 1930–1941. [DOI] [PubMed] [Google Scholar]
- 75. Telfer WH, Kunkel JG (1991) The function and evolution of insect storage hexamers. Annu Rev Entomol 36: 205–228. [DOI] [PubMed] [Google Scholar]
- 76. Dong K, Zhang DQ, Dahlman DL (1996) Down-regulation of juvenile hormone esterase and arylphorin production in Heliothis virescens larvae parasitized by Microplitis croceipes . Arch Insect Biochem Physiol 32: 237–248. [Google Scholar]
- 77. Shelby KS, Webb BA (1994) Polydnavirus infection inhibits synthesis of an insect plasma protein, arylphorin. J Gen Virol 75: 2285–2292. [DOI] [PubMed] [Google Scholar]
- 78. Shelby KS, Webb BA (1997) Polydnavirus infection inhibits translation of specific growth-associated host proteins. Insect Biochem Mol Biol 27: 263–270. [DOI] [PubMed] [Google Scholar]
- 79. Zhu JY, Ye GY, Fang Q, Hu C, Akhtar ZR (2009) cDNA of an arylphorin-type storage protein from Pieris rapae with parasitism inducible expression by the endoparasitoid wasp Pteromalus puparum . Insect Sci 16: 227–236. [Google Scholar]
- 80. Kunkel JG, Grossniklaus-Buergin C, Sharon T, Karpells ST, Lanzrein B (1990) Arylphorin of Trichoplusia ni: Characterization and parasite-induced precocious increase in titer. Arch Insect Biochem Physiol 13: 117–125. [Google Scholar]
- 81. Coudron TA, Jones D, Jones G (1994) Premature production of late larval storage proteins in larvae of Trichoplusia ni parasitized by Euplectrus comstockii . Arch Insect Biochem Physiol 26: 97–109. [Google Scholar]
- 82. Coudron TA, Brandt SL, Raqib A (1997) Comparison of the response of Heliothis virescens to parasitism by Euplectrus comstockii and Euplectrus plathypenae . Comp Biochem Physiol B 116: 197–202. [Google Scholar]
- 83. Asgari S, Schmidt O (2004) Isolation of an imaginal disc growth factor homologue from Pieris rapae and its expression following parasitization by Cotesia rubecula . J Insect Physiol 50: 687–894. [DOI] [PubMed] [Google Scholar]
- 84. Hall M, Wang R, Van Antwerpen R, Sottrup-Jensen L, Soderhall K (1999) The crayfish plasma clotting protein: a vitellogenin-related protein responsible for clot formation in crustacean blood. Proc Natl Acad Sci USA 96: 1965–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lourenço AP, Martins JR, Bitondi MM, Simões ZL (2009) Trade-off between immune stimulation and expression of storage protein genes. Arch Insect Biochem Physiol 71: 70–87. [DOI] [PubMed] [Google Scholar]
- 86. Scharlaken B, de Graaf DC, Goossens K, Peelman LJ, Jacobs FJ (2008) Differential gene expression in the honeybee head after a bacterial challenge. Dev Comp Immunol 32: 883–889. [DOI] [PubMed] [Google Scholar]
- 87. Hayakawa Y (1994) Cellular immunisuppressive protein in the plasma of parasitized insect larvae. J Biol Chem 269: 14536–14540. [PubMed] [Google Scholar]
- 88. Marmaras VJ, Lampropoulou M (2009) Regulators and signalling in insect haemocyte immunity. Cell Signal 21: 186–195. [DOI] [PubMed] [Google Scholar]
- 89. Evans JD, Aronstein K, Chen YP, Hetru C, Imler JL, et al. (2006) Immune pathways and defence mechanisms in honey bees Apis mellifera . Insect Mol Biol 15: 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Agaisse H, Perrimon N (2004) The roles of JAK/STAT signaling in Drosophila immune responses. Immunol Rev 198: 72–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Length distribution of Tenebrio molitor contigs. Horizontal axis represents the length of contigs and vertical axis represents number of contigs.
(TIF)
Length distribution of Tenebrio molitor scaffolds. Horizontal axis represents the length of scaffolds and vertical axis represents number of scaffolds.
(TIF)
Classification of raw reads in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Numbers in parentheses show the percentage of each type of read among the total raw reads.
(TIF)
Distribution of distinct clean reads in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Numbers in the square brackets indicate the range of copy numbers for a specific category of reads. The data in parentheses indicate the percentage of corresponding reads among the total distinct reads.
(TIF)
Distribution of gene coverage in non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae.
(TIF)
Primers used in qRT-PCR for validating differentially expressed genes.
(XLS)
Top Blast hits from NCBI nr database. BLASTX against the nr protein database was used with a cutoff E-value of 10−5.
(XLS)
Gene ontology (GO) annotation of unigenes.
(XLS)
Kyoto Encyclopedia of Genes and Genome (KEGG) annotation of unigenes.
(XLS)
Immunity-related genes identified in Tenebrio molitor transcriptome.
(XLS)
Differentially expressed genes between non-parasitized (NP) and parasitized (P) Tenebrio molitor pupae. Raw intensity, the total number of reads sequenced for each gene. RPKM, Reads per kb per million reads. FDR, false discovery rate. FDR ≤0.001 and the absolute value of log2Ratio ≥1 were used in this study.
(XLS)
Description of the top 20 most up- and down-regulated genes.
(XLS)
Gene Ontology (GO) enrichment analysis of differentially expressed genes.
(XLS)
Kyoto Encyclopedia of Genes and Genome (KEGG) enrichment analysis of differentially expressed genes.
(XLS)