

Abstract
Ca2+ signaling in synaptic terminals plays a critical role in neurotransmitter release and short-term synaptic plasticity. In the present study, we examined the role of synaptic Ca2+ handling mechanisms in the synaptic terminals of mammalian rod bipolar cells, neurons that play a pivotal role in the high-sensitivity vision pathway. We found that mitochondria sequester Ca2+ under conditions of high Ca2+ load, maintaining intraterminal Ca2+ near resting levels. Indeed, the effect of the mitochondria was so powerful that the ability to clamp intraterminal Ca2+ with a somatically positioned whole cell patch pipette was compromised. The plasma membrane Ca2+-ATPase (PMCA), but not the Na+/Ca2+ exchanger (NCX) or the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), was an important regulator of resting Ca2+. Furthermore, PMCA activity, but not NCX or SERCA activity, was essential for the recovery of Ca2+ levels following depolarization-evoked Ca2+ entry. Loss of PMCA function was also associated with impaired restoration of membrane surface area following depolarization-evoked exocytosis. Given its roles in the regulation of intraterminal Ca2+ at rest and after a stimulus-evoked Ca2+ rise, the PMCA is poised to modulate luminance coding and adaptation to background illumination in the mammalian rod bipolar cell.
Keywords: bipolar cell, endocytosis, synaptic vesicle, ribbon synapse
the free intracellular Ca2+ concentration in a nerve terminal regulates key aspects of the exocytotic pathway that leads to neurotransmitter release (Heidelberger 2001b; Neher and Sakaba 2008). While an abrupt rise in intraterminal Ca2+ can trigger exocytosis and the release of neurotransmitter into the synaptic cleft, relatively modest elevations in cytosolic Ca2+ have been found to modulate synaptic output and short-term plasticity (Awatramani et al. 2005; Janz et al. 1999; Wan and Heidelberger 2011). Ca2+ regulation has been studied extensively in neuronal somata; however, less is known about Ca2+ homeostasis mechanisms in synaptic boutons.
Retinal bipolar cells are second-order glutamatergic neurons of the vertebrate retina that respond to a sustained change in illumination with a sustained change in membrane potential (Dowling 1987); this gives rise to “continuous” synaptic transmission (Heidelberger et al. 2005; Lagnado et al. 1996). In addition to triggering neurotransmitter release, the associated rise in intraterminal Ca2+ may regulate additional aspects of presynaptic function such as the replenishment rate of fusion-competent vesicles (Singer and Diamond 2006; Wan and Heidelberger 2011; Wan et al. 2008), the rate of membrane recovery after exocytosis (Heidelberger 2001a; Neves and Lagnado 1999; Oltedal and Hartveit 2010; von Gersdorff and Matthews 1994b; Wan et al. 2008), and the apparent Ca2+ sensitivity of neurotransmitter release (Wan and Heidelberger 2011). Thus, in addition to preventing Ca2+-mediated excitotoxicity, presynaptic Ca2+ clearance mechanisms might play a role in setting the gain at this synapse.
Ca2+ that enters a neuron may be buffered, sequestered, and/or extruded to maintain homeostasis. Transporters important for the regulation of intracellular Ca2+ levels include the plasma membrane Na+/Ca2+ exchangers (NCX or NCKX), P-type Ca2+-transporting ATPases such as the plasma membrane Ca2+-ATPases (PMCAs) and the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pumps, and the calcium uniporter of the mitochondria. The importance of these mechanisms differs across types of neuron and with stimulation intensity. For example, both NCXs and PMCAs have important functional roles at conventional synapses (Gleason et al. 1994, 1995; Kim et al. 2005; Komori et al. 2010; Molinaro et al. 2011), whereas the PMCAs play a dominant role in Ca2+ extrusion at primary sensory ribbon synapses (Morgans et al. 1998; Tucker and Fettiplace 1995). A functional role for SERCA pumps in axon terminals remains to be clearly established (Brini and Carafoli 2009; Carter et al. 2002; Collin et al. 2005; Empson and Knopfel 2012; Kim et al. 2003, 2005; Sasaki et al. 2005). Mitochondria provide ATP for the pumps, but at higher Ca2+ concentrations mitochondria may also sequester Ca2+, buffering cytosolic Ca2+ in the 0.5–1.0 μM range (Billups and Forsythe 2002; Nicholls 2005; Szabadkai and Duchen 2008; Zenisek and Matthews 2000).
In this study, we provide a functional examination of the Ca2+ extrusion mechanisms of the mouse rod bipolar cell, a neuron exclusive to the high-sensitivity pathway of the mammalian retina. Previous studies looking at Ca2+ handling mechanisms in the mixed-input (Mb1) bipolar cell of the goldfish retina have established a role for the PMCA in the maintenance of basal Ca2+ and in the restoration of depolarization-evoked rises in intraterminal Ca2+, but there are conflicting reports on the role of the NCX on intraterminal Ca2+ recovery (Kobayashi and Tachibana 1995; Zenisek and Matthews 2000). In the rodent retina, PMCAs have been localized to the axon terminals of the rod bipolar cell (Krizaj et al. 2002; Morgans et al. 1998), and there is functional evidence for a role for the NCX in restoration of resting Ca2+ in rod bipolar cells after 2 min of strong depolarization (Krizaj et al. 2002). Here, we performed quantitative measurements of intraterminal Ca2+ in the terminals of rod bipolar cells isolated from the retina of adult mice. These were combined with patch-clamp recording techniques and membrane capacitance measurements to study the effects of transporter blockade on intraterminal Ca2+, exocytosis, and membrane recovery. Under conditions of a large maintained Ca2+ load, mitochondrial sequestration played a dominant role in the regulation of intraterminal Ca2+. The PMCA, but not the NCX or SERCA, played a central role in maintaining resting intraterminal Ca2+ levels. The restoration of intraterminal Ca2+ levels after depolarization-evoked Ca2+ entry was also regulated by the PMCA. Although the PMCA did not influence the extent of exocytosis from the releasable pool of synaptic vesicles, inhibition of the PMCA significantly prolonged the onset of recovery of membrane surface area and produced a general slowing in the restoration of membrane surface area.
MATERIALS AND METHODS
Cell isolation and electrophysiology.
All animal procedures conformed to National Institutes of Health guidelines and were approved by the Animal Welfare Committee of the University of Texas Health Science Center at Houston. Mouse rod bipolar neurons and goldfish Mb1 bipolar cells were acutely isolated from the retinas of 2- to 6-mo-old C57BL/6 mice and 3- to 5-in. goldfish, respectively, by enzymatic digestion followed by mechanical trituration, as described previously (Heidelberger and Matthews 1992; Zhou et al. 2006). Isolated bipolar neurons were identified and selected for recording based on morphological criteria (Heidelberger and Matthews 1992; Zhou et al. 2006).
For the mouse rod bipolar cell, whole cell patch-clamp recordings were performed with a 5- to 6-MΩ pipette placed on the soma of an isolated rod bipolar cell. The standard mouse intracellular solution contained (in mM) 125 Cs-gluconate, 10 tetraethylammonium chloride (TEA-Cl), 3 MgCl2, 2 Na2ATP, 0.5 guanosine 5′-triphosphate (GTP), 0.5 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.2 bis-fura-2, and 35 HEPES, pH 7.2, 305–310 mosM. The standard external solution contained (in mM) 138 NaCl, 2.5 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 5 KCl, 0.44 KH2PO4, 0.34 Na2HPO4, 10 d-glucose, and 10 HEPES, adjusted to pH 7.2, 310–315 mosM.
For the goldfish mixed-input (Mb1) bipolar cell, whole cell recordings were made with a 5- to 7-MΩ patch placed either on the cell soma or directly on the synaptic terminal, as indicated. The internal solution with free Ca2+ set to 50 μM for Mb1 bipolar cells was prepared similarly to that described below for the mouse rod bipolar cell except that the concentration of CsCl was lowered such that the final osmolarity was 260–270 mosM, as appropriate for goldfish. The goldfish external solution contained (mM) 120 NaCl, 2.6 KCl, 1.0 MgCl2, 0.5 CaCl2, 10 HEPES, and 10 glucose, pH 7.25–7.3, 260–270 mosM.
For experiments in which the internal solution contained 50 μM free Ca2+, Cs-gluconate was replaced with Cs-Cl to avoid Ca2+ precipitation (von Gersdorff and Matthews 1994a), EGTA was replaced with a mixture of 10 mM NTA and 3 mM CaCl2, and bis-fura-2 was replaced with an equimolar mixture (0.1 mM of each) of the high- and low-affinity fluorescence Ca2+ indicator dyes bis-fura-2 and furaptra (Voets 2000). In addition, CaCl2 in the external solution was reduced from 2.5 to 0.5 mM. In experiments in which PMCA activity was inhibited by adenosine 5′-O-(3-thiotriphosphate) (ATPγS), Li4-ATPγS replaced Na2ATP in the internal solution, and free Ca2+ in the internal solution was defined by replacing 0.5 mM EGTA with a combination of 4 mM EGTA and 1.5 mM CaCl2 (Heidelberger et al. 2002). All experiments were conducted at room temperature. Holding potential (Vhold) was −70 mV for the mouse rod bipolar cell and −60 mV for the goldfish Mb1 bipolar cell. Rod bipolar cells with access resistances >35 MΩ were excluded from analysis (Zhou et al. 2006). Cells with conductances >1 nS at Vhold were excluded from analysis unless otherwise specified.
PMCA function was blocked by the addition of 1 mM sodium orthovanadate (Na3VO4) to the intracellular solution (DiPolo et al. 1979; Zenisek and Matthews 2000) or replacement of ATP in the internal solution with ATPγS, as indicated. In experiments in which cells were dialyzed with 50 μM free Ca2+ internal solution, PMCA function was blocked by orthovanadate and by the addition of 1 mM La3+ to the external solution (Carafoli 1991; Herrington et al. 1996). To remove the influence of the NCX, NaCl in the external solution was replaced with choline-chloride. In some experiments, 2 μM KB-R7943 (Tocris) was additionally added the bath to block reversal of the NCX (Hoyt et al. 1998; Iwamoto et al. 1996; Watano et al. 1996). Cells were incubated in either solution for 10 min prior to the establishment of the whole cell recording configuration. The protonophore CCCP (1 μM), used to uncouple the mitochondria, was delivered to the cell by switching the local external perfusion from that of standard external solution to standard external solution that additionally contained CCCP. In other experiments, ruthenium red, an inhibitor selective for the mitochondrial uniporter at low concentrations (Kirichok et al. 2004; Matlib et al. 1998), was added to the internal recording solution at a concentration of 5 μM. Other chemicals, such as thapsigargin (TG, 1 μM), were applied to the cells in the standard external solution for 10 min prior to the establishment of the whole cell recording configuration (Chen et al. 2003; Duman et al. 2008).
[Ca2+]i measurements.
Ca2+ measurements were ratiometrically made with a computer-controlled monochrometer-based system (ASI/T.I.L.L.Photonics, Eugene, OR) (Messler et al. 1996) that provided alternating excitation at 340 and 380 nm. The collection field for emitted fluorescence was selectively positioned over the cell soma or the terminal (Heidelberger and Matthews 1992; Zhou et al. 2006). For experiments using standard intracellular solution containing bis-fura-2, free Ca2+ concentration ([Ca2+]i) was calculated from the background-subtracted fluorescence ratios, using in vivo calibration constants obtained as described previously (Heidelberger and Matthews 1992; Wan et al. 2008). For experiments in which the intracellular solution had a calculated [Ca2+]i of 50 μM and contained a mixture of bis-fura-2 (0.1 mM) and furaptra (0.1 mM) (Voets 2000), an in vitro calibration curve was constructed to convert the background-subtracted ratio of fluorescent signals into [Ca2+]i. The following equation was fit to the measured ratio values (R) to find the five unknown parameters as well as the Ca2+ calibration curve:
Calcium calibration solutions contained (in mM) 35 HEPES, 10 TEA-Cl, 3 MgCl2, 2 Na2ATP, 0.5 GTP, 0.1 furaptra, 0.1 bis-fura-2, calcium buffers 10 EGTA (Kd = 150 nM at pH 7.2), 10 HEDTA (Kd = 3.6 μM), or 10 NTA (Kd = 134 μM) (Martell and Smith 1974; Nuccitelli et al. 1994), and sufficient CaCl2 to reach the calculated Ca2+ concentrations of (in μM) 0, 0.02, 0.36, 1.7, 12.5, 21, 52.5, 97, and 10,000. Cs-gluconate levels were adjusted to obtain the appropriate osmolarity for mouse or goldfish bipolar cells (Heidelberger and Matthews 1992; Zhou et al. 2006), and the pH was adjusted to 7.2 with CsOH. MaxChelator (http://maxchelator.stanford.edu) and the Patcher's Power Tools XOP module (Department of Membrane Biophysics, MPI-BPC, Goettingen, Germany) for IGOR Pro (WaveMetrics, Portland, OR) were used to calculate [Ca2+]i. The K2H2EGTA and Ca2+-saturated K2CaEGTA stock solutions were purchased from Molecular Probes/Life Technologies (Eugene, OR). HEDTA and Ca2+-saturated HEDTA and NTA and Ca2+-saturated NTA were prepared with a pH-metric approach (Neher 1988). The CaCl2 stock solution was purchased from Ricco Chemical (Arlington, TX). The obtained Ca2+ calibration curve was verified against a curve generated with a commercially available Ca2+ calibration kit (Molecular Probes/Life Technologies).
Data analysis.
Data analysis was performed with IGOR Pro software (WaveMetrics). Results are presented as means ± SE, where n indicates the number of cells. The resting Ca2+ concentration was defined as the average Ca2+ concentration over a 5-s period immediately before a stimulus or drug delivery. The increase in the spatially averaged Ca2+ was measured as the difference between the peak Ca2+ measured after a stimulus and resting Ca2+. To measure the rate of calcium recovery, for each record the recovery of calcium transients to baseline was described by the exponential equation Y = Y0 +A1e[−(x − x0)/τ1]. Statistical comparisons were performed with the two-tailed, unpaired Student's t-test or one-way ANOVA with Dunnett's multiple comparisons test (GraphPad InStat, GraphPad Software La Jolla, CA) as appropriate.
RESULTS
During short-term dialysis with high Ca2+, intraterminal Ca2+ is substantially lower than expected.
The free Ca2+ concentration within a nerve terminal governs key aspects of synaptic physiology, and there are multiple mechanisms that influence the intracellular Ca2+ profile. To better understand the regulation of presynaptic Ca2+ signals in a mammalian neuron, we used a combination of physiological and pharmacological approaches to examine the roles of the mitochondria, PMCA, NCX, and SERCA in Ca2+ handling in the synaptic boutons of rod bipolar neurons.
In the first set of experiments, rod bipolar neurons were acutely dissociated from the retina of adult mice (Wan et al. 2008; Zhou et al. 2006) and held under voltage-clamp control via a whole cell recording pipette placed on the cell soma. The whole cell pipette was additionally used to internally dialyze the rod bipolar cell with a Ca2+-buffered internal solution that contained 50 μM free Ca2+ (see materials and methods). The free Ca2+ concentration in either the somatic or the synaptic terminal compartment was ratiometrically calculated with a combination of fluorescent calcium indicator dyes (Voets 2000) that have on-rates comparable to the fast Ca2+ buffer BAPTA (Naraghi 1997). Two types of responses were observed. In ≈60% of the recordings, the neurons developed a large inward current at Vhold (−124 ± 21 pA, n = 23) and the intraterminal Ca2+ concentration rose quickly to ∼50 μM (68 ± 18 μM, n = 23), as did the concentration in the soma (44 ± 11 μM, n = 14). However, in ≈40% of the bipolar neurons (Fig. 1A), the current at Vhold was more typical of control cells (−28 ± 4 pA, n = 16) and the mean spatially averaged Ca2+ concentration in the synaptic terminal remained submicromolar, reaching only 200 ± 55 nM after 2 min of dialysis (n = 16), while the somatic Ca2+ concentration under identical conditions rose to 11.2 ± 2.1 μM (n = 10). Although higher than the resting Ca2+ level measured under control conditions (≈50 nM; Wan et al. 2010 and Fig. 3, this study), the Ca2+ concentration in terminals in this second group was surprisingly low given that 2 min of dialysis with a somatically positioned pipette is typically sufficient to achieve steady-state levels of fluorescent Ca2+ indicator dyes in the terminal (Wan et al. 2008, 2010).
Fig. 1.
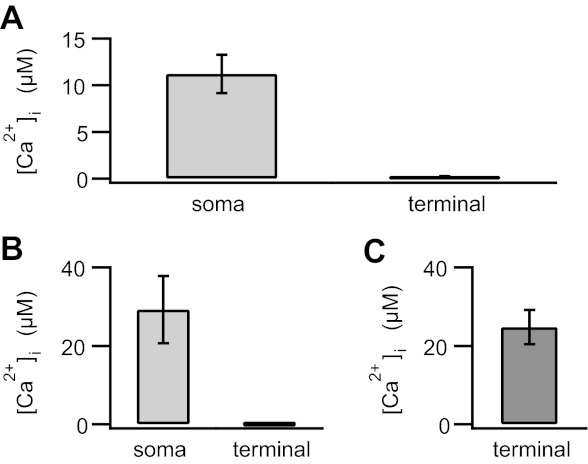
Despite 2 min of dialysis with 50 μM free Ca2+ from a somatically positioned pipette, intraterminal Ca2+ concentration remained below 1 μM. A: in isolated mouse rod bipolar cells voltage-clamped at −70 mV, dialysis with 50 μM free Ca2+ raised the average somatic Ca2+ concentration ([Ca2+]i) to greater than ≈10 μM (11.2 ± 2.1 μM; n = 10), while the intraterminal Ca2+ concentration remained submicromolar (200 ± 55 nM; n = 16). B: in isolated goldfish Mb1 bipolar cells voltage-clamped at −60 mV, dialysis with 50 μM free Ca2+ resulted in ≈30 μM Ca2+ in the soma (29.2 ± 8.5 μM; n = 4), while the intraterminal Ca2+ concentration remained submicromolar (73.6 ± 29 nM; n = 5). C: when the terminal of a Mb1 bipolar cell was directly patched with high-Ca2+ internal solution, the intraterminal Ca2+ rose to a concentration (24.7 ± 4.3 μM; n = 6) similar to that obtained in the soma with a somatically positioned pipette. Error bars represent SE.
Fig. 3.
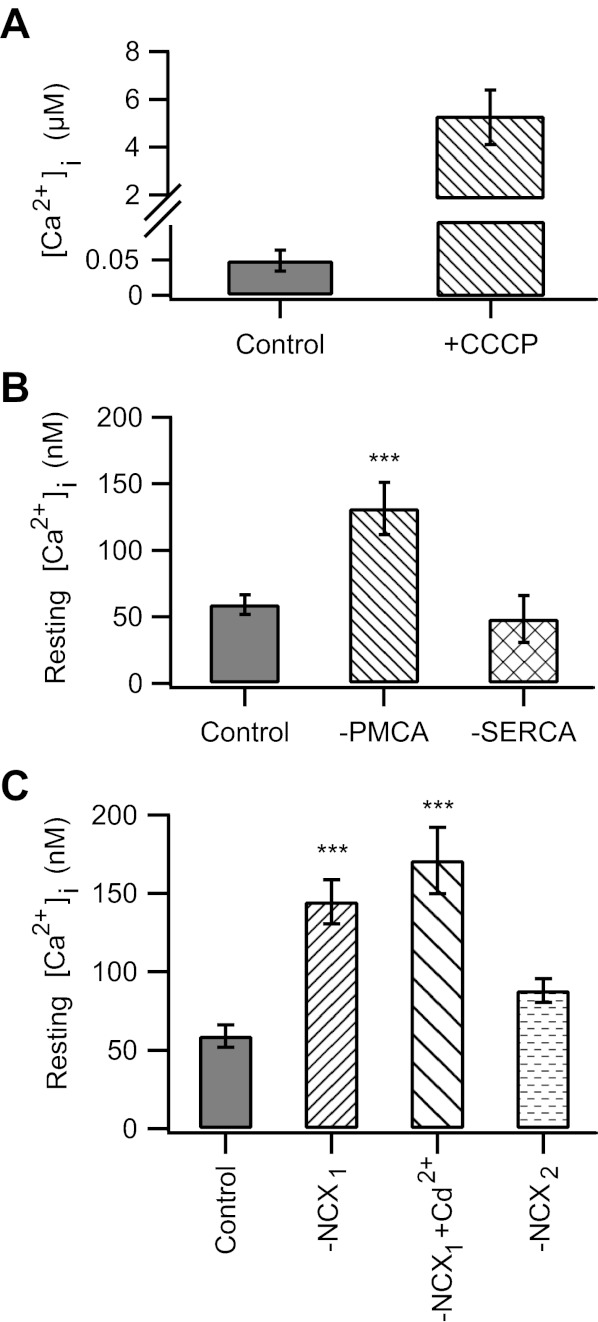
Mitochondria and PMCA, but not the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) or the Na+/Ca2+ exchanger (NCX), regulate resting intraterminal Ca2+. A: in control cells dialyzed with standard internal solution, treatment with CCCP resulted in a ≈100-fold increase in intraterminal Ca2+ (without CCCP: 49 ± 15 nM, with CCCP: 5.3 ± 1.2 μM; n = 9). B: inhibition of PMCA by orthovanadate (Na3VO4) increased the resting Ca2+ level (131 ± 20 nM; n = 14). Pretreatment for ≈10 min with thapsigargin to block SERCA had no effect (48 ± 18 nM, n = 6). C: inhibition of NCX by replacement of external NaCl with choline-Cl (NCX1) caused an increase in resting Ca2+ level [control: 59 ± 7 nM (n = 26), NCX1: 145 ± 14 nM (n = 12)]. This was not due to unclamped Ca2+ channel activity, as the addition of 200 μM Cd2+ to the bath did not reduce the increased in resting Ca2+ (NCX1 + Cd2+: 171 ± 21 nM, n = 7). However, the addition of 2 μM KB-R7943 to the 0 Na+ external solution to block the reverse mode of NCX kept resting Ca2+ near baseline levels (NCX2: 88 ± 8 nM, n = 11). For all, holding potential (Vhold) = −70 mV. ***P < 0.001.
We performed a similar set of experiments in the mixed-input (Mb1) bipolar cells of the goldfish retina, which have a shorter, stouter axon and in which direct biophysical access to the synaptic terminal can be readily achieved (Heidelberger et al. 1994; Heidelberger and Matthews 1992; Matthews 1996). After 2 min of dialysis with a somatically positioned pipette filled with internal solution containing 50 μM free Ca2+, the average somatic Ca2+ concentration was 29.2 ± 8.5 μM (n = 4); however, the intraterminal Ca2+ concentration remained near resting levels (73.6 ± 29 nM, n = 5; Fig. 1B). By contrast, when we directly patched the terminal, the mean intraterminal Ca2+ rose within 2 min to 24.7 ± 4.3 μM (Fig. 1C; n = 6). Together, these results suggest the presence of Ca2+ handling mechanisms that can outperform the pace of Ca2+ delivery from the soma to the terminal in bipolar cells, at least in the short term.
Mitochondria regulate intraterminal Ca2+ under conditions of internal dialysis with high Ca2+.
To investigate the Ca2+ handling mechanism(s) that hinder the ability to clamp intraterminal Ca2+, acutely dissociated mouse rod bipolar cells were held at −70 mV and dialyzed with internal solution containing 50 μM free Ca2+ from a somatically positioned patch pipette, and free Ca2+ was measured after 2 min of dialysis. In neurons that did not develop a large, inward current, the average intraterminal Ca2+ concentration was 200 ± 55 nM (n = 16; Fig. 2). When PMCA function was inhibited by the addition of 1 mM orthovanadate (Na3VO4) to the pipette solution and 1 mM La3+ to the bath solution (Bond and Hudgins 1980; DiPolo et al. 1979; Zenisek and Matthews 2000), the mean intraterminal Ca2+ concentration was 332 ± 92 nM (n = 3; Fig. 2). When NCX was additionally blocked by replacing NaCl in the external solution with choline-Cl, intraterminal Ca2+ rose to 760 ± 330 nM (n = 12; Fig. 2). Nevertheless, the achieved Ca2+ concentration remained tens of micromolar below the target concentration of 50 μM. The addition of the mitochondrial uncoupler CCCP to PMCA and NCX blockade triggered a dramatic rise in intraterminal Ca2+ to 33 ± 13 μM (n = 4; Fig. 2). To compare how much Ca2+ might be released from the mitochondria under more physiological conditions, cells dialyzed with standard internal solution were also treated with CCCP. The intraterminal Ca2+ concentration in the presence of CCCP in control cells was an order of magnitude lower (5.3 ± 1.2 μM, n = 9; Fig. 3A). Together, these data demonstrate a dominant role for the mitochondria in managing a large Ca2+ load in the synaptic terminal of a rod bipolar cell. In addition, they suggest sequestration of Ca2+ by the mitochondria as a mechanism.
Fig. 2.
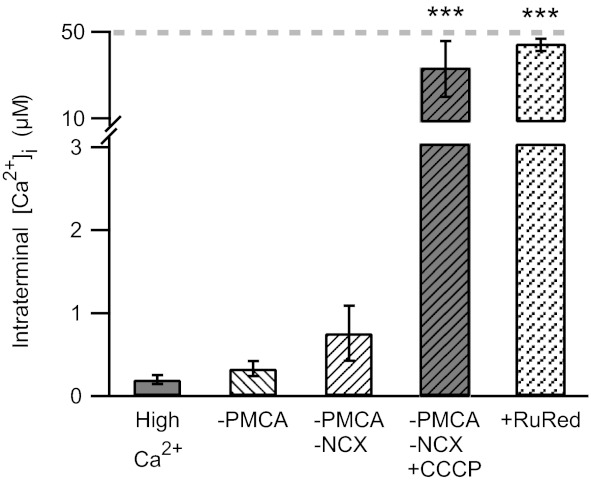
Mitochondria play a dominant role in regulating synaptic Ca2+ during internal dialysis with 50 μM free Ca2+. Addition of La3+ to the bath (1 mM) and orthovanadate (1 mM) to the internal solution to block the plasma membrane Ca2+-ATPase (PMCA) had little effect on intraterminal Ca2+ levels [high Ca2+: 200 ± 55 nM (n = 16), −PMCA: 332 ± 92 nM (n = 3)]. Replacement of Na+ in the bath by choline+, in addition to inhibition of PMCA, resulted in a rise in intraterminal Ca2+ (−PMCA −NCX: 760 ± 330 nM; n = 12). When the mitochondrial uncoupler CCCP (1 μM) was additionally applied, a large increase in intraterminal Ca2+ to 33 ± 13 μM (n = 4) was observed. When the mitochondrial Ca2+ uniporter was blocked by 5 μM ruthenium red (RuRed), the intraterminal Ca2+ concentration rose to 44 ± 3 μM (n = 6). Gray dashed line indicates the free Ca2+ concentration in the internal solution. ***P < 0.001.
To confirm a role for mitochondrial Ca2+ uptake, cells were again dialyzed with a Ca2+-buffered internal solution containing 50 μM free Ca2+. In test cells, this solution additionally contained 5 μM ruthenium red to selectively block the mitochondrial Ca2+ uniporter (Kirichok et al. 2004; Matlib et al. 1998). Within 4–5 min after the whole cell recording configuration was achieved, cells treated with ruthenium red exhibited an average intraterminal Ca2+ concentration of 44 ± 3 μM (n = 6; Fig. 2). The average holding current at this time point was −85 ± 21 pA. Using pairwise controls, we found that the intraterminal Ca2+ remained submicromolar (262 ± 8 nM, n = 2) in 40% of terminals without ruthenium red, consistent with earlier results (Fig. 1A), while in the remaining control terminals, Ca2+ rose to 39 ± 22 μM (n = 3). These results are consistent with mitochondrial sequestration of Ca2+ during dialysis with high Ca2+.
PMCA, but not NCX or SERCA, activity regulates basal intraterminal Ca2+.
In mammalian retina, PMCAs have been localized to the terminals of rod bipolar cells (Krizaj et al. 2002). These is also faint labeling for NCXs in the inner plexiform layer of the rodent retina, suggestive of the presence of NCX in some bipolar cells (Morgans et al. 1998). We therefore investigated the role of these transporters and the SERCA pumps in setting the resting Ca2+ in the synaptic terminals of rod bipolar cells. In rod bipolar cells held at −70 mV and dialyzed with standard internal recording solution, the resting Ca2+ concentration in the synaptic terminal was ≈50 nM (Fig. 3B), consistent with a previous report (Wan et al. 2010). In the presence of the PMCA inhibitor orthovanadate (Fig. 3B, −PMCA), the average resting Ca2+ in the terminals of rod bipolar cells rose to 131 ± 20 nM (n = 14). Inhibition of the NCX for 10 min by removal of external Na+, a manipulation that blocks the forward mode of NCX activity (Blaustein and Lederer 1999), also resulted in an increase in the average resting intraterminal Ca2+ (145 ± 14 nM, n = 12; Fig. 3C, −NCX1). SERCA pumps, however, did not appear to play a significant role in regulating basal Ca2+, as pretreatment with TG (1 μM) failed to significantly alter the average resting intraterminal Ca2+ (TG-treated cells: 48 ± 18 nM, n = 6; Fig. 3B, −SERCA).
The above results in Na+-free external solution indicate that the NCX is present and functional in rod bipolar cell synaptic terminals. However, it is unusual to observe an elevation of resting Ca2+ with NCX inhibition (Kobayashi and Tachibana 1995; but see Medler 2010). Therefore, we wondered whether this surprising result might be related to spontaneous activation of voltage-gated Ca2+ during the incubation period, during which time the cells are not voltage clamped. To address this possibility, cells were incubated in Na+-free external solution to which 200 μM Cd2+, a blocker of voltage-gated Ca2+ channels, was added. Under these conditions, intraterminal resting Ca2+ in Na+-free external solution was similar to that in 0 Na+ without Cd2+ [with Cd2+: 171 ± 21 nM (n = 7), without Cd2+: 145 ± 14 nM (n = 12); P > 0.05], indicating that Ca2+ entry through voltage-gated Ca2+ channels did not contribute to the rise in resting intraterminal Ca2+ (Fig. 3C). We then asked whether the reverse mode of action of the NCX might be responsible for the rise in resting intraterminal Ca2+ (Fig. 3C, −NCX2). To test this possibility, the forward mode of the NCX was inhibited by Na+-free external solution, while the reverse mode was inhibited by the NCX inhibitor KB-R7943 (2 μM) (Hoyt et al. 1998; Iwamoto et al. 1996; Watano et al. 1996). Under these conditions, intraterminal Ca2+ remained near resting values (88 ± 8 nM, n = 11; P > 0.05). These data are consistent with the interpretation that in 0 Na+ external solution the NCX runs in reverse, bringing Ca2+ into the terminals, rather than pumping it out. Thus, while functional NCX are present on rod bipolar cells, they are unlikely to regulate resting intraterminal Ca2+ provided that Na+ ions are present in the external space. Together, the data indicate a prominent role for the PMCA, but not NCX or SERCA, in the regulation of resting intraterminal Ca2+ levels.
PMCA activity is critical for restoration of intraterminal Ca2+ following a depolarization-evoked Ca2+ rise.
Next, we asked which Ca2+ handling mechanisms contribute to the recovery of a depolarization-evoked rise in intraterminal Ca2+. As shown in Fig. 4, A and B, under control conditions, a 1-s depolarization from −70 to 0 mV evoked an increase in the spatially averaged intraterminal Ca2+ that recovered to baseline within a few seconds. On average, the time constant of recovery under control conditions was 1.6 ± 0.2 s (n = 11), similar to what has been previously reported (Wan et al. 2008). As exemplified by the representative traces in Fig. 4A, pretreatment with TG to inhibit SERCA or replacement of external Na+ with choline+ to block NCX had no appreciable effect on the time constant of Ca2+ recovery [−SERCA: τ = 1.6 ± 0.4 s (n = 6); −NCX: τ = 1.8 ± 0.2 s (n = 21)]. Importantly, inhibition of PMCA either by sodium orthovanadate (Fig. 4B, −PMCA1) or by replacement of ATP in the internal solution with ATPγS (Fig. 4B, −PMCA2) significantly impeded intraterminal Ca2+ recovery. In many cases, resting Ca2+ was not restored to pretreatment values, but rather the terminals rested at a new, higher level after stimulation (i.e., Fig. 4B), precluding our ability to compare the time constants of recovery to the prestimulus baseline. Therefore, we compared the percentage of Ca2+ recovery to the prestimulus baseline at 8 s after Ca2+ channel closure. In contrast to the ≈80% recovery observed in controls or when NCX or SERCA pumps were inhibited, when PMCA was blocked recovery was <50% 8 s after membrane repolarization (Fig. 4C). Neither method of inhibiting the PMCA increased the magnitude of the Ca2+ transient (Fig. 4D), suggesting that the slowing of Ca2+ recovery could not be attributed to the saturation of clearance mechanisms caused by an enhanced evoked Ca2+ load (Wan et al. 2008). Finally, given literature reports that NCX may be important for handling large Ca2+ loads that overwhelm the capacity of the PMCA, we stimulated the cells with a 5-s depolarization. This elevated intraterminal Ca2+ above 300 nM (Fig. 4F). However, inhibition of NCX by either Na+-free external solution (NCX1) or Na+-free external solution supplemented with 2 μM KB-R7943 (NCX2) failed to slow the time course of Ca2+ recovery (Fig. 4E). In light of these data, the PMCA emerges as an important Ca2+ clearance mechanism after a depolarization-evoked rise in the presynaptic Ca2+ concentration in the mammalian rod bipolar cell.
Fig. 4.
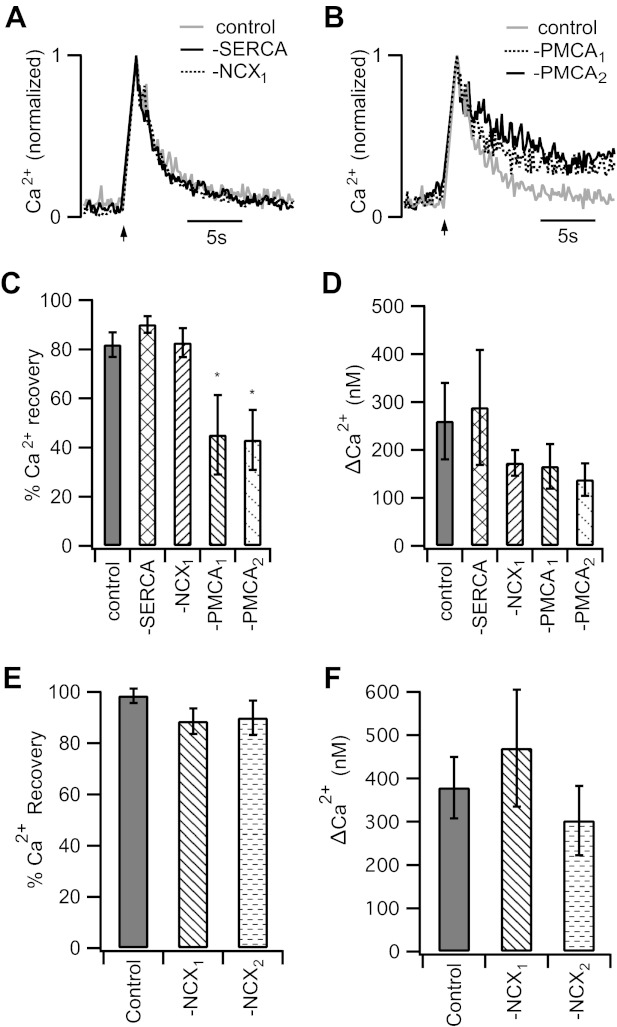
PMCA activity is critical for restoration of intraterminal Ca2+ following membrane depolarization. A and B: representative traces show the normalized Ca2+ responses evoked by a 1-s depolarization (−70 to 0 mV; arrow). The time courses of Ca2+ recovery in cells pretreated with thapsigargin to block SERCA (−SERCA) or incubated in 0 Na+ extracellular solution to block NCX (−NCX1) are similar to that of the control. However, inhibition of PMCA function resulted in failure to fully restore resting Ca2+ levels during the observation period. −PMCA1 shows the response of a cell pretreated with the PMCA blocker Na3VO4, and −PMCA2 shows the response of a cell in which adenosine 5′-O-(3-thiotriphosphate) (ATPγS) replaced ATP in the pipette solution. C: 8 s after Ca2+ calcium channel closure, restoration of intraterminal Ca2+ was 82 ± 5% complete in control rod bipolar cells (n = 11). Similar values were observed in cells in the presence of thapsigargin (90 ± 3%; n = 5) or 0 Na+ (83 ± 6%; n = 16). However, the extent of the Ca2+ recovery was significantly reduced in rod bipolar cell terminals treated with either PMCA blocker [PMCA1: 45 ± 16% (n = 8), PMCA2: 43 ± 12% (n = 8)]. D: the mean peak ΔCa2+ after a 1-s depolarization was not significantly different among the groups. E: after a 5-s depolarization, restoration of intraterminal Ca2+ was 99 ± 3% complete in control rod bipolar cells (n = 8). Similar values were observed in cells in 0 Na+ (NCX1: 89 ± 5%, n = 7) or in the presence of 0 Na+ and 2 μM KB-R7943 (NCX2: 90 ± 7%, n = 5). F: the mean peak ΔCa2+ after a 5-s depolarization was similar among the groups. Error bars represent SE. *P < 0.05.
Inhibition of PMCA activity alters time course of membrane recovery following exocytosis.
Intracellular Ca2+ concentration governs multiple aspects of synaptic vesicle dynamics (Neher and Sakaba 2008; Wan and Heidelberger 2011). In bipolar cells, both the onset of membrane retrieval following exocytosis and the time course of membrane retrieval are influenced by intraterminal Ca2+ (Rouze and Schwartz 1998; von Gersdorff and Matthews 1994b; Wan et al. 2008). Here, we studied the effect of inhibition of PMCA on membrane recovery in isolated mouse rod bipolar cells after a 1-s depolarization from −70 to 0 mV (Fig. 5). This stimulus triggers the fusion of a pool of vesicles that are available for Ca2+-dependent release (Wan et al. 2008; Zhou et al. 2006). Exocytosis and membrane recovery were monitored with membrane capacitance measurements. Inhibition of PMCA by orthovanadate had little effect on the magnitude of the evoked capacitance increase (Fig. 5A), consistent with the lack of a significant effect of PMCA inhibition on the evoked Ca2+ transient (Fig. 4D). However, after Ca2+ channel closure, the recovery of membrane capacitance to baseline was significantly prolonged when PMCAs were inhibited relative to controls. This inhibition can be seen in representative normalized capacitance records shown in Fig. 5B. To quantify this effect, we compared the percentage of recovery of the stimulus-evoked membrane increase 8 s after the depolarization ended. Results of this analysis indicate a severe defect in membrane recovery in neurons in which PMCA activity is inhibited (Fig. 5C). In addition, the brief, Ca2+-dependent delay prior to the onset of endocytosis (Wan et al. 2008) was also prolonged when PMCA activity was inhibited (Fig. 5D). These data suggest that the PMCA influences the recovery of membrane surface area via its role in regulating the intraterminal Ca2+ concentration.
Fig. 5.
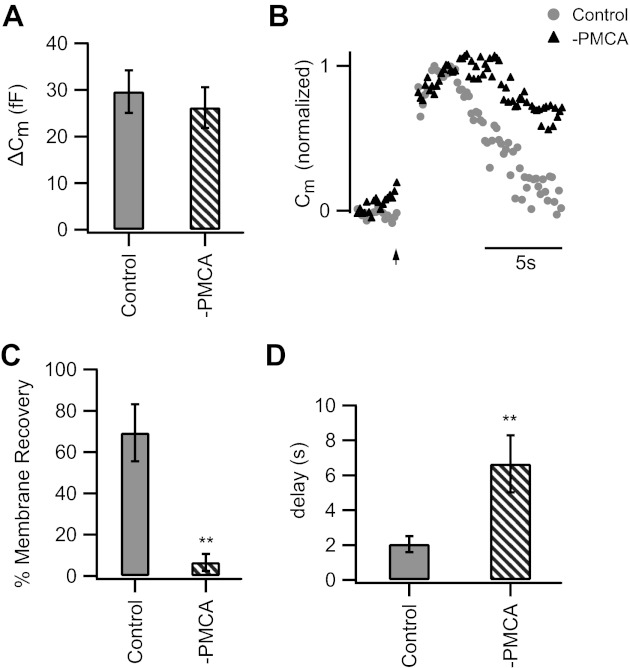
Inhibition of PMCA impedes recovery of membrane surface area following exocytosis. A: inhibition of PMCA activity had little effect on the extent of exocytosis from the releasable pool (1 s, −70 to 0 mV). Cm, membrane capacitance. B: representative normalized Cm records show the response to a 1-s depolarization from −70 to 0 mV (given at arrow). Note that while Cm returns to baseline in the control cell, membrane recovery is impaired when PMCA was inhibited by Na3VO4 (−PMCA). C: % of added membrane surface area recovered 8 s after Ca2+ channel closure was significantly reduced with PMCA inhibition [−PMCA: −7 ± 4% (n = 5), control cells: 69 ± 14% (n = 11); P < 0.01]. D: the Ca2+-dependent delay in the onset of recovery of membrane surface area was prolonged with PMCA inhibition [−PMCA: 6.7 ± 1.6 s (n = 6), control: 2 ± 0.5 s (n = 9); P < 0.01]. Error bars represent SE. **P < 0.01.
DISCUSSION
The magnitude and duration of changes in presynaptic Ca2+ are believed to be important determinants of the pattern of neurotransmitter release and synaptic signaling (Heidelberger 2001b; Neher and Sakaba 2008; Wan and Heidelberger 2011). In this study, we demonstrate a clear role for mitochondria and PMCA, but not NCX or SERCA, in the regulation of intraterminal Ca2+ in the mammalian rod bipolar cell. In addition, blockade of PMCA inhibited the recovery of membrane surface area following the depolarization-evoked fusion of the entire releasable pool of synaptic vesicles.
The role of mitochondria in neuronal Ca2+ regulation is twofold. They can both supply the ATP that is required for pump activity and sequester Ca2+ by action of the mitochondrial uniporter (Gunter and Sheu 2009; Malli and Graier 2010; Szabadkai and Duchen 2008). Typically the sequestration of Ca2+ mitochondria is observed in secretory cells when the bulk cytosolic Ca2+ rises above several hundred nanomolar and other Ca2+ handling mechanisms are saturated (Duman et al. 2008; Kim et al. 2005; von Gersdorff and Matthews 1994b). In the present study, we find that the mitochondria are surprisingly effective in managing supramicromolar Ca2+ loads imposed by the internal recording solution. In almost half of the cells, intraterminal Ca2+ remained submicromolar even though the somatic Ca2+ level was >10 μM (Fig. 1). Several lines of evidence indicate that this particular effect is due to the mitochondrial sequestration of Ca2+ rather than ATP supply. First, sufficient MgATP was provided in the internal solution to support ATPase function under normal recording conditions (i.e., Figs. 3–5). Second, it was only after mitochondrial uncoupling was added to inhibition of PMCA and NCX that the intraterminal Ca2+ concentration approached that of the internal solution; PMCA inhibition alone failed to significantly elevate intraterminal Ca2+ under these conditions. Third, uncoupling of mitochondria caused a far greater rise in intraterminal Ca2+ when cells were dialyzed with 50 μM Ca2+ solution than with control internal solution (Fig. 3A), consistent with load-dependent Ca2+ uptake and release. Finally, when the mitochondrial Ca2+ uniporter was selectively inhibited with low-dose ruthenium red (Kirichok et al. 2004; Matlib et al. 1998), intraterminal Ca2+ rose to the target level. Given that the somatic Ca2+ level was significantly elevated when rod bipolar cells were dialyzed with the high-Ca2+ internal solution, sequestration of Ca2+ by mitochondria located in the axon terminal may play a particularly important role.
While our interest was in the Ca2+ handling mechanisms that prevented us from readily clamping intraterminal Ca2+ in a cohort of rod bipolar cells, it was possible to clamp intraterminal Ca2+ in the remainder of rod bipolar cells. Why the two groups differ is not clear. Conceivably, they may relate to the two types of rod bipolar cells reported in the rodent retina. These neurons differ with respect to photoreceptor inputs and inhibitory feedback (Pang et al. 2004, 2010) and thus would be expected to experience different presynaptic Ca2+ histories. In addition, the axon lengths differ between the two subtypes of rod bipolar cells (Pang et al. 2004, 2010). This could impact the rate of Ca2+ delivery to the terminal, altering the balance between delivery and sequestration. The current observed in the high-Ca2+ group, which reversed at ECl− and was likely a Ca2+-activated Cl− conductance (not shown), developed in all rod bipolar cells once the intraterminal Ca2+ rose above a few micromolar, and therefore the presence of this current does not distinguish the two groups. It is also conceivable that in the second group of neurons the mitochondria were nonfunctional. However, this seems unlikely because application of CCCP to these cells resulted in a further rise in intraterminal Ca2+, indicating that they had been actively sequestering Ca2+. In addition, the CCCP-induced Ca2+ rise was completely reversible upon CCCP washout in three of seven cells (not shown). Further studies beyond the scope of the present study would be needed to understand the reasons for the two types of responses. Perhaps more importantly, with the increasing interest in recording from rod bipolar cells and their postsynaptic targets to study synaptic mechanisms, our results raise an important caution: the ability to define the intraterminal Ca2+ concentration by using a somatically positioned patch pipette cannot be assumed. It must be verified.
PMCAs constitute a major clearance pathway in photoreceptors and hair cells (Duncan et al. 2006; Morgans et al. 1998; Tucker and Fettiplace 1995). Similar to bipolar cells, these neurons make ribbon-style synapses, respond to changes in stimulation with graded changes in membrane potential, and experience Ca2+ influx over extended periods. We found that Ca2+ extrusion via PMCA also plays a major role in the recovery of intraterminal Ca2+ following a depolarization-evoked rise in the mouse rod bipolar cell (Fig. 4), in general agreement with previous reports from the goldfish mixed-input bipolar cell (Kobayashi and Tachibana 1995; Zenisek and Matthews 2000). In addition, PMCA function was essential for the maintenance of the resting intraterminal Ca2+ concentration in the mouse rod bipolar cell (Fig. 3). This is somewhat surprising, as PMCAs are reported to have Kd for Ca2+ in the 200–500 nM range when optimally activated and 10–30 μM at rest (Brini and Carafoli 2009). However, the mammalian rod bipolar cell expresses PMCA2 (Krizaj et al. 2002). This PMCA has an unusually high activity in the absence of activation by calmodulin, has a higher affinity for Ca2+/calmodulin than other isoforms, and can respond to Ca2+ in the tens of nanomolar range (Brini and Carafoli 2009; Brini et al. 2003; Hilfiker et al. 1994). In other neurons, PMCA2 has been found to regulate both residual Ca2+ and poststimulus Ca2+ recovery (Empson et al. 2007; Huang et al. 2010). Our data demonstrating the role of PMCA in the regulation of steady-state and poststimulus Ca2+ recovery, in combination with the localization of PMCA2 to the rod bipolar terminals (Duncan et al. 2006; Krizaj et al. 2002; Morgans et al. 1998), suggest that our results are likely to be mediated by this high-affinity, high-activity neuronal PMCA.
NCXs, along with the PMCAs, play an important modulatory role in synaptic transmission in neurons that form conventional synapses (Gleason et al. 1994, 1995; Kim et al. 2005; Molinaro et al. 2011), with loss of NCX function affecting basal and evoked Ca2+ signals, triggering asynchronous release, and altering short- and long-term synaptic plasticity (Gleason et al. 1994; Gomez-Villafuertes et al. 2005; Molinaro et al. 2011). By contrast, in primary sensory neurons that make ribbon-style synapses, the NCX plays a minor role at best, leaving the majority of extrusion to the PMCAs (Morgans et al. 1998; Tucker and Fettiplace 1995). Similarly, in the goldfish mixed-input bipolar cell, NCX does not regulate resting intraterminal Ca2+ levels (Kobayashi and Tachibana 1995), and a role for NCX in Ca2+ recovery following membrane depolarization has been only inconsistently observed (Kobayashi and Tachibana 1995; Zenisek and Matthews 2000). In general, these studies have led to the suggestion that the PMCA, not NCX, is an important Ca2+ extrusion mechanism in ribbon-style synapses.
In the mouse rod bipolar cell, functional NCXs are present on rod bipolar cells as evidenced by results shown in Figs. 2 and 3. However, we found no evidence of a role for NCX in moving Ca2+ out of the cell under resting, physiological conditions (Fig. 3). Neither could we demonstrate a role for NCX in Ca2+ clearance following a 1- to 5-s depolarization (Fig. 4) or a brief stimulus train (not shown). It may be that the role of NCX gains prominence in response to larger or more prolonged Ca2+ signals (Duman et al. 2008; Gleason et al. 1994; Krizaj et al. 2002) than explored here that exceed the capacity of the PMCA (i.e., Fig. 2). Also, if K+-dependent Na+/Ca2+ exchangers (NCKX) were additionally present on rod bipolar cells, their potential contributions to Na+/Ca2+ exchange might be underestimated in this study because of the relatively small Ca2+ rises and the composition of our internal solutions (Kim et al. 2005; Visser and Lytton 2007).
In many neurons, inhibition of SERCA pumps has failed to reveal a role for these pumps in the clearance of Ca2+ from synaptic boutons (Billups and Forsythe 2002; Chuhma and Ohmori 2002; Kim et al. 2005) or on basal Ca2+ (Sasaki et al. 2005). Similarly, in the mouse rod bipolar cell, we did not detect a role for SERCA pumps in setting the resting intraterminal Ca2+ or in Ca2+ recovery following stimulus-evoked Ca2+ influx. It may be that other protocols, with respect to either treatment (Galante and Marty 2003) or stimulation, may yet reveal a role for SERCA pumps in the mammalian rod bipolar cell synaptic terminal.
Compensatory endocytosis follows exocytosis to restore the membrane surface area to baseline values. Electrophysiological and optical studies of synaptic vesicle fusion and retrieval in goldfish mixed-input bipolar cells and hippocampal neurons have indicated that endocytosis can be inhibited by elevated cytosolic Ca2+ (Heidelberger 2001a; Leitz and Kavalali 2011; Rouze and Schwartz 1998; Neves and Lagnado 1999; von Gersdorff and Matthews 1994b). Data from mammalian rod bipolar cells obtained by membrane capacitance measurements have been similarly interpreted as being consistent with Ca2+-dependent inhibition of endocytosis (Wan et al. 2008; Oltedal and Hartveit 2010). In the mouse rod bipolar cell, PMCA inhibition had little effect on the magnitude of the depolarization-evoked exocytotic response (Fig. 5A), allowing us to study the role of the PMCA in isolation of possible confounding effects caused by differences in membrane load on the endocytic machinery. Inhibition of PMCA was associated with both a delay in the onset of membrane surface area recovery and a reduction in the amount of membrane surface area recovery measured at a given time point after Ca2+ channel closure (Fig. 5). Since capacitance measurements track the net change in membrane surface area, a slowing of the onset might reflect the inhibition of endocytosis and/or the contributions of asynchronous release. Short-lived asynchronous release has been noted in rod bipolar cells when endocytosis was blocked with a small-molecule inhibitor (Wan and Heidelberger 2011), and a small amount of asynchronous release is evident in Fig. 5B of the present study, particularly with PMCA inhibition. Thus there might be an increase in asynchronous release following PMCA inhibition that contributes to a delay in the onset of membrane recovery, in addition to inhibition of endocytosis by elevated cytoplasmic Ca2+. Both mechanisms predict that the encoding of visual information would ultimately be modulated by changes in PMCA activity.
Finally, in mammalian rod bipolar cells, the supply of vesicles to the fusion-competent pool is accelerated by elevated Ca2+, with elevations in the ≈100–150 nM range appearing to be sufficient (Singer and Diamond 2006; Wan and Heidelberger 2011; Wan et al. 2010). Our data show that the PMCA in the mouse rod bipolar cell operates within this Ca2+ range. The modulation of vesicle supply rates provides an additional means by which the PMCA, and the mitochondria that supply ATP, might regulate synaptic function. It has been suggested that a high-contrast stimulus triggers the rapid release of fusion-competent vesicles, while luminance coding is associated with maintained release (Oesch and Diamond 2011). While inhibition of PMCA activity did not influence the extent of exocytosis to a single stimulus, our data suggest that it will alter the ability to maintain release. Therefore, we propose that the PMCA, acting via Ca2+ and the Ca2+-dependent regulation of synaptic vesicle dynamics, is an important element in luminance coding.
GRANTS
This work was supported by National Eye Institute Grant EY-012128 (R. Heidelberger) and Core Grant for vision research EY-010608.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Q.-F.W. and R.H. conception and design of research; Q.-F.W. and E.N. performed experiments; Q.-F.W., E.N., and R.H. analyzed data; Q.-F.W., E.N., and R.H. interpreted results of experiments; Q.-F.W. and R.H. prepared figures; Q.-F.W. drafted manuscript; Q.-F.W., E.N., and R.H. approved final version of manuscript; R.H. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Alice Chuang for assistance with statistical analyses.
Present address of E. Nixon, Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, TX 76107.
REFERENCES
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48: 109–121, 2005 [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22: 5840–5847, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999 [DOI] [PubMed] [Google Scholar]
- Bond GH, Hudgins PM. Inhibition of red cell Ca2+-ATPase by vanadate. Biochim Biophys Acta 600: 781–790, 1980 [DOI] [PubMed] [Google Scholar]
- Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev 89: 1341–1378, 2009 [DOI] [PubMed] [Google Scholar]
- Brini M, Coletto L, Pierobon N, Kraev N, Guerini D, Carafoli E. A comparative functional analysis of plasma membrane Ca2+ pump isoforms in intact cells. J Biol Chem 278: 24500–24508, 2003 [DOI] [PubMed] [Google Scholar]
- Carafoli E. The calcium pumping ATPase of the plasma membrane. Annu Rev Physiol 53: 531–547, 1991 [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci 22: 21–28, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Koh DS, Hille B. Dynamics of calcium clearance in mouse pancreatic beta-cells. Diabetes 52: 1723–1731, 2003 [DOI] [PubMed] [Google Scholar]
- Chuhma N, Ohmori H. Role of Ca2+ in the synchronization of transmitter release at calyceal synapses in the auditory system of rat. J Neurophysiol 87: 222–228, 2002 [DOI] [PubMed] [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol 15: 275–281, 2005 [DOI] [PubMed] [Google Scholar]
- DiPolo R, Rojas HR, Beauge L. Vanadate inhibits uncoupled Ca2+ efflux but not Na+-Ca2+ exchange in squid axons. Nature 281: 229–230, 1979 [DOI] [PubMed] [Google Scholar]
- Dowling JE. The Retina: An Approachable Part of the Brain. Cambridge, MA: Belknap, 1987 [Google Scholar]
- Duman JG, Chen L, Hille B. Calcium transport mechanisms of PC12 cells. J Gen Physiol 131: 307–323, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JL, Yang H, Doan T, Silverstein RS, Murphy GJ, Nune G, Liu X, Copenhagen D, Tempel BL, Rieke F, Krizaj D. Scotopic visual signaling in the mouse retina is modulated by high-affinity plasma membrane calcium extrusion. J Neurosci 26: 7201–7211, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Garside ML, Knopfel T. Plasma membrane Ca2+ ATPase 2 contributes to short-term synapse plasticity at the parallel fiber to Purkinje neuron synapse. J Neurosci 27: 3753–3758, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Knopfel T. Functional integration of calcium regulatory mechanisms at Purkinje neuron synapses. Cerebellum 11: 640–650, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Marty A. Presynaptic ryanodine-sensitive calcium stores contribute to evoked neurotransmitter release at the basket cell-Purkinje cell synapse. J Neurosci 23: 11229–11234, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason E, Borges S, Wilson M. Control of transmitter release from retinal amacrine cells by Ca2+ influx and efflux. Neuron 13: 1109–1117, 1994 [DOI] [PubMed] [Google Scholar]
- Gleason E, Borges S, Wilson M. Electrogenic Na-Ca exchange clears Ca2+ loads from retinal amacrine cells in culture. J Neurosci 15: 3612–3621, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Villafuertes R, Torres B, Barrio J, Savignac M, Gabellini N, Rizzato F, Pintado B, Gutierrez-Adan A, Mellstrom B, Carafoli E, Naranjo JR. Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J Neurosci 25: 10822–10830, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta 1787: 1291–1308, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R. ATP is required at an early step in compensatory endocytosis in synaptic terminals. J Neurosci 21: 6467–6474, 2001a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R. Electrophysiological approaches to the study of neuronal exocytosis and synaptic vesicle dynamics. Rev Physiol Biochem Pharmacol 143: 1–80, 2001b [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371: 513–515, 1994 [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol 447: 235–256, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Sterling P, Matthews G. Roles of ATP in depletion and replenishment of the releasable pool of synaptic vesicles. J Neurophysiol 88: 98–106, 2002 [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Thoreson WB, Witkovsky P. Synaptic transmission at retinal ribbon synapses. Prog Retin Eye Res 24: 682–720, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron 16: 219–228, 1996 [DOI] [PubMed] [Google Scholar]
- Hilfiker H, Guerini D, Carafoli E. Cloning and expression of isoform 2 of the human plasma membrane Ca2+ ATPase. Functional properties of the enzyme and its splicing products. J Biol Chem 269: 26178–26183, 1994 [PubMed] [Google Scholar]
- Hoyt KR, Arden SR, Aizenman E, Reynolds IJ. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol Pharmacol 53: 742–749, 1998 [PubMed] [Google Scholar]
- Huang H, Nagaraja RY, Garside ML, Akemann W, Knopfel T, Empson RM. Contribution of plasma membrane Ca ATPase to cerebellar synapse function. World J Biol Chem 1: 95–102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem 271: 22391–22397, 1996 [DOI] [PubMed] [Google Scholar]
- Janz R, Goda Y, Geppert M, Missler M, Südhof TC. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 24: 1003–1016, 1999 [DOI] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci 25: 6057–6065, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Lee SH, Park KH, Ho WK. Distribution of K+-dependent Na+/Ca2+ exchangers in the rat supraoptic magnocellular neuron is polarized to axon terminals. J Neurosci 23: 11673–11680, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–364, 2004 [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Tachibana M. Ca2+ regulation in the presynaptic terminals of goldfish retinal bipolar cells. J Physiol 483: 79–94, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori Y, Tanaka M, Kuba M, Ishii M, Abe M, Kitamura N, Verkhratsky A, Shibuya I, Dayanithi G. Ca2+ homeostasis, Ca2+ signalling and somatodendritic vasopressin release in adult rat supraoptic nucleus neurones. Cell Calcium 48: 324–332, 2010 [DOI] [PubMed] [Google Scholar]
- Krizaj D, Demarco SJ, Johnson J, Strehler EE, Copenhagen DR. Cell-specific expression of plasma membrane calcium ATPase isoforms in retinal neurons. J Comp Neurol 451: 1–21, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado L, Gomis A, Job C. Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron 17: 957–967, 1996 [DOI] [PubMed] [Google Scholar]
- Leitz J, Kavalali ET. Ca2+ influx slows single synaptic vesicle endocytosis. J Neurosci 31: 16318–16326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malli R, Graier WF. Mitochondrial Ca2+ channels: great unknowns with important functions. FEBS Lett 584: 1942–1947, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell AE, Smith RM. (Editors). Critical Stability Constants, vol. 1 New York: Plenum, 1974 [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 273: 10223–10231, 1998 [DOI] [PubMed] [Google Scholar]
- Matthews G. Synaptic exocytosis and endocytosis: capacitance measurements. Curr Opin Neurobiol 6: 358–364, 1996 [DOI] [PubMed] [Google Scholar]
- Medler KF. Calcium signaling in taste cells: regulation required. Chem Senses 35: 753–765, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messler P, Harz H, Uhl R. Instrumentation for multiwavelengths excitation imaging. J Neurosci Methods 69: 137–147, 1996 [DOI] [PubMed] [Google Scholar]
- Molinaro P, Viggiano D, Nistico R, Sirabella R, Secondo A, Boscia F, Pannaccione A, Scorziello A, Mehdawy B, Sokolow S, Herchuelz A, Di Renzo GF, Annunziato L. Na+-Ca2+ exchanger (NCX3) knock-out mice display an impairment in hippocampal long-term potentiation and spatial learning and memory. J Neurosci 31: 7312–7321, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW, El Far O, Berntson A, Wassle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci 18: 2467–2474, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium 22: 255–268, 1997 [DOI] [PubMed] [Google Scholar]
- Neher E. The influence of intracellular calcium concentration on degranulation of dialysed mast cells from rat peritoneum. J Physiol 395: 193–214, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59: 861–872, 2008 [DOI] [PubMed] [Google Scholar]
- Neves G, Lagnado L. The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol 515: 181–202, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondria and calcium signaling. Cell Calcium 38: 311–317, 2005 [DOI] [PubMed] [Google Scholar]
- Nuccitelli R, Wilson L, Matsudaira PT. A Practical Guide to the Study of Calcium in Living Cells (vol. 40, Methods in Cell Biology) San Diego, CA: Academic, 1994 [Google Scholar]
- Oesch NW, Diamond JS. Ribbon synapses compute temporal contrast and encode luminance in retinal rod bipolar cells. Nat Neurosci 14: 1555–1561, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltedal L, Hartveit E. Transient release kinetics of rod bipolar cells revealed by capacitance measurement of exocytosis from axon terminals in rat retinal slices. J Physiol 588: 1469–1487, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Gao F, Lem J, Bramblett DE, Paul DL, Wu SM. Direct rod input to cone BCs and direct cone input to rod BCs challenge the traditional view of mammalian BC circuitry. Proc Natl Acad Sci USA 107: 395–400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Gao F, Wu SM. Light-evoked current responses in rod bipolar cells, cone depolarizing bipolar cells and AII amacrine cells in dark-adapted mouse retina. J Physiol 558: 897–912, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouze NC, Schwartz EA. Continuous and transient vesicle cycling at a ribbon synapse. J Neurosci 18: 8614–8624, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Dayanithi G, Shibuya I. Ca2+ clearance mechanisms in neurohypophysial terminals of the rat. Cell Calcium 37: 45–56, 2005 [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol 95: 3191–3198, 2006 [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23: 84–94, 2008 [DOI] [PubMed] [Google Scholar]
- Tucker T, Fettiplace R. Confocal imaging of calcium microdomains and calcium extrusion in turtle hair cells. Neuron 15: 1323–1335, 1995 [DOI] [PubMed] [Google Scholar]
- Visser F, Lytton J. K+-dependent Na+/Ca2+ exchangers: key contributors to Ca2+ signaling. Physiology (Bethesda) 22: 185–192, 2007 [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28: 537–545, 2000 [DOI] [PubMed] [Google Scholar]
- Von Gersdorff H, Matthews G. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature 367: 735–739, 1994a [DOI] [PubMed] [Google Scholar]
- Von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature 370: 652–655, 1994b [DOI] [PubMed] [Google Scholar]
- Wan QF, Heidelberger R. Synaptic release at mammalian bipolar cell terminals. Vis Neurosci 28: 109–119, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QF, Vila A, Zhou ZY, Heidelberger R. Synaptic vesicle dynamics in mouse rod bipolar cells. Vis Neurosci 25: 523–533, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QF, Zhou ZY, Thakur P, Vila A, Sherry DM, Janz R, Heidelberger R. SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron 66: 884–895, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watano T, Kimura J, Morita T, Nakanishi H. A novel antagonist, No. 7943, of the Na+/Ca2+ exchange current in guinea-pig cardiac ventricular cells. Br J Pharmacol 119: 555–563, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25: 229–237, 2000 [DOI] [PubMed] [Google Scholar]
- Zhou ZY, Wan QF, Thakur P, Heidelberger R. Capacitance measurements in the mouse rod bipolar cell identify a pool of releasable synaptic vesicles. J Neurophysiol 96: 2539–2548, 2006 [DOI] [PubMed] [Google Scholar]