

Abstract
CaV2.1 Ca2+ channels have a dominant and specific role in initiating fast synaptic transmission at central excitatory synapses, through a close association between release sites and calcium sensors. Familial hemiplegic migraine type 1 (FHM-1) is an autosomal-dominant subtype of migraine with aura, caused by missense mutations in the CACNA1A gene that encodes the α1A pore-forming subunit of CaV2.1 channel. We used knock-in (KI) transgenic mice harboring the FHM-1 mutation R192Q to study the consequences of this mutation in neurotransmission at the giant synapse of the auditory system formed by the presynaptic calyx of Held terminal and the postsynaptic neurons of the medial nucleus of the trapezoid body (MNTB). Although synaptic transmission seems unaffected by low-frequency stimulation in physiological Ca2+ concentration, we observed that with low Ca2+ concentrations (<1 mM) excitatory postsynaptic currents (EPSCs) showed increased amplitudes in R192Q KI mice compared with wild type (WT), meaning significant differences in the nonlinear calcium dependence of nerve-evoked transmitter release. In addition, when EPSCs were evoked by broadened presynaptic action potentials (achieved by inhibition of K+ channels) via Cav2.1-triggered exocytosis, R192Q KI mice exhibited further enhancement of EPSC amplitude and charge compared with WT mice. Repetitive stimulation of afferent axons to the MNTB at different frequencies caused short-term depression of EPSCs that recovered significantly faster in R192Q KI mice than in WT mice. Faster recovery in R192Q KI mice was prevented by the calcium chelator EGTA-AM, pointing to enlarged residual calcium as a key factor in accelerating the replenishment of synaptic vesicles.
Keywords: R192Q knock-in mice, familial hemiplegic migraine, CaV2.1 channels, excitatory postsynaptic currents, short-term synaptic plasticity
transmitter release at central synapses is triggered by Ca2+ influx through voltage-gated Ca2+ channels (VGCCs). Transmitter release is mediated by multiple Ca2+ channel subtypes during early development but on maturation increasingly relies on Cav2.1 (P/Q-type) Ca2+ channels, as was shown at the neuromuscular junction (Rosato Siri et al. 2002) and the calyx of Held (Fedchyshyn and Wang 2005). Familial hemiplegic migraine type 1 (FHM-1) is a Mendelian subtype of “migraine with aura” caused by missense mutations in the CACNA1A gene that encodes the α1A pore-forming subunit of Cav2.1 Ca2+ channels. Typical migraine attacks in FHM-1 patients are associated with transient hemiparesis, but apart from this, they are identical to those of the common forms of “migraine with aura” (Pietrobon 2005; Pietrobon and Striessnig 2003). In addition, more than two-thirds of patients with FHM-1 have attacks of “normal typical migraine” as well. Interestingly, in several families, FHM CACNA1A mutations were also found in family members who had only “normal” nonparetic migraine but no FHM. This suggests that gene mutations for FHM may also be responsible for the common forms of migraine. Therefore, FHM-1 is a promising model to study the pathogenic mechanisms of common forms of migraine (Ferrari et al. 2008). A knock-in (KI) migraine mouse model carrying the human FHM-1 R192Q mutation has been generated (van den Maagdenberg et al. 2004), which allows the mutant channels to be studied in their native neuronal environment and at their endogenous level of expression. KI mice exhibit several gain-of-function effects (Tottene et al. 2009) and an increased propensity for cortical spreading depression (CSD), the likely underlying mechanism of the migraine aura (Ayata 2009; Haerter et al. 2005). Tottene et al. (2009) have shown increased probability of glutamate release at the excitatory synapse between cortical pyramidal cell (PC) and fast-spiking (FS) interneurons from FHM-1 mice. Intriguingly, neurotransmission at the FS interneuron-PC inhibitory synapse appeared unaltered, despite being mediated by P/Q-type channels (i.e., carrying the FHM-1 mutation). This imbalance of cortical excitation and inhibition was associated with increased susceptibility for CSD in the KI mice, but the underlying mechanism changing synaptic strength in the R192Q mutation is not fully understood. In a recent paper (González Inchauspe et al. 2010), we showed that presynaptic KI CaV2.1 channels activate at more negative membrane potentials than wild-type (WT) channels. This hyperpolarized activation led to a higher inward Ca2+ influx when Ca2+ currents were evoked by long-duration action potentials (APs) (such as PC APs) but not when Ca2+ currents were elicited by short-duration APs (like the calyx of Held or interneuron APs). We postulated that the shorter AP duration of the FS interneuron may allow the unaltered inhibitory neurotransmission observed by Tottene et al. (2009) in the R192Q KI mouse.
Here we analyzed how R192Q presynaptic CaV2.1 channels altered neurotransmission at the axosomatic glutamatergic synapse formed by the presynaptic calyx of Held terminal and the postsynaptic neurons of the medial nucleus of the trapezoid body (MNTB). R192Q KI mice show enlarged AMPA receptor-mediated excitatory postsynaptic currents (EPSCs) when evoked by broadened presynaptic APs and a faster recovery of EPSCs after short-term depression (STD) following high-frequency synaptic activity.
MATERIALS AND METHODS
Preparation of brain stem slices.
Generation of the R192Q KI mouse strain was described previously (van den Maagdenberg et al. 2004). All experiments were carried out according to national guidelines and were approved by local Ethical Committees.
Mice were killed by decapitation at P12–P16, and the brain was removed and placed into an ice-cold low-sodium artificial cerebrospinal fluid (aCSF). The brain stem was mounted in the Peltier chamber of an Integraslice 7550PSDS vibrating microslicer (Campden Instruments). Transverse slices of 300-μm thickness were cut and transferred to an incubation chamber containing normal aCSF with low calcium (0.1 mM CaCl2 and 2.9 mM MgCl2) at 37°C for 1 h. Normal aCSF contained (mM) 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 0.5 ascorbic acid, 3 myo-inositol, 2 sodium pyruvate, 1 MgCl2, and 2 CaCl2. Low-sodium aCSF was as above, but NaCl was replaced by 250 mM sucrose and MgCl2 and CaCl2 concentrations were 2.9 mM and 0.1 mM, respectively. The pH was 7.3 when gassed with 95% O2-5% CO2.
Electrophysiology.
Slices were transferred to an experimental chamber perfused with normal aCSF at 25°C. Neurons were visualized with Nomarski optics on a BX50WI microscope (Olympus, Japan), with a ×40/0.90 NA water immersion objective lens (LUMPlane FI, Olympus). Whole cell voltage-clamp recordings were made with patch pipettes pulled from thin-walled borosilicate glass (GC150F-15, Harvard Apparatus). Electrodes had resistances of 2.9–3.2 MΩ when filled with internal solution of the following composition (mM): 110 CsCl, 40 HEPES, 10 TEA-Cl, 12 Na2phosphocreatine, 0.5 EGTA, 2 MgATP, 0.5 LiGTP, and 1 MgCl2. pH was adjusted to 7.3 with CsOH. To block Na+ currents and avoid postsynaptic action potentials, 10 mM N-(2,6-diethylphenylcarbamoylmethyl)triethylammonium chloride (QX-314) was added to the pipette solution. Patch-clamp recordings were made with a Multiclamp 700B amplifier (Axon CNS, Molecular Devices), a Digidata 1440A (Axon CNS, Molecular Devices), and pCLAMP 9.0 software. Data were sampled at 50 kHz and filtered at 6 kHz (low-pass Bessel). Whole cell membrane capacitances (15–25 pF) and series resistances (4–15 MΩ) were registered from the amplifier after compensation of the transient generated by a 10-ms voltage step and compensated by 40–60%.
EPSCs were evoked by stimulating the globular bushy cell axons in the trapezoid body at the midline with a bipolar platinum stimulating electrode placed in the midline and applying square pulses (0.1 ms and 4–10 V) through an isolated stimulator (model DS2A, Digitimer). To isolate EPSCs, strychnine (1 μM) was added to the external solution to block inhibitory glycinergic synaptic responses.
Presynaptic APs were recorded in whole cell configuration under current-clamp mode with a patch solution containing (mM) 110 K-gluconate, 30 KCl, 10 HEPES, 10 Na2phosphocreatine, 0.2 EGTA, 2 MgATP, 0.5 LiGTP, and 1 MgCl2. APs were elicited by injecting depolarizing step current pulses of 0.5–1 nA over 0.1 ms.
Data analysis was done with Clampfit 10.0 (Molecular Devices), Sigma Plot 10.0, SigmaStat 3.5, and Excel 2003 (Microsoft) software. Average data are expressed and plotted as means ± SE. Statistical significance was determined by Student's t-test or one-way repeated-measures analysis of variance (ANOVA) plus Student-Newman-Keuls post hoc test.
RESULTS
Glutamate release and EPSCs at the calyx of Held synapse.
EPSCs at the calyx of Held synapse from WT and R192Q mice show synchronous release, display an all-or-none behavior, and have amplitudes (when above threshold) independent of the stimulus intensity. EPSCs were abolished by ω-agatoxin IVA (200 nM), indicating that only CaV2.1 channels were mediating Ca2+ influx responsible for exocytosis in both WT and R192Q mice. EPSCs recorded from the soma of an MNTB neuron under voltage-clamp conditions at a holding potential of −70 mV with 2 mM external Ca2+ concentration ([Ca2+]o) and 1 mM [Mg2+]o show no statistically significantly different mean amplitudes between R192Q KI (10.7 ± 0.5 nA, n = 65) and WT (10.6 ± 0.6 nA, n = 46, Student's t-test, P = 0.42; González Inchauspe et al. 2010) mice. Further studies of the calcium dependence of neurotransmitter release were made with [Mg2+]o of 2 mM. (representative traces in Fig. 1A). We observed that the relation between EPSC amplitudes and [Ca2+]o is nonlinear and can be approximated by
where r is an estimative measure of the cooperative binding of intracellular Ca2+ to the sensor at a release site. Plotted in logarithmic scales, the power relation between EPSC amplitudes and [Ca2+]o is linear for [Ca2+]o up to 1 mM (Fig. 1B). In this range, the calcium dependence of nerve-evoked EPSCs is significantly different between the genotypes (P < 0.01, nonparametric ANOVA, Kruskal-Wallis test). There is an increase of transmitter release in R192Q KI synapses at low [Ca2+]o.
Fig. 1.
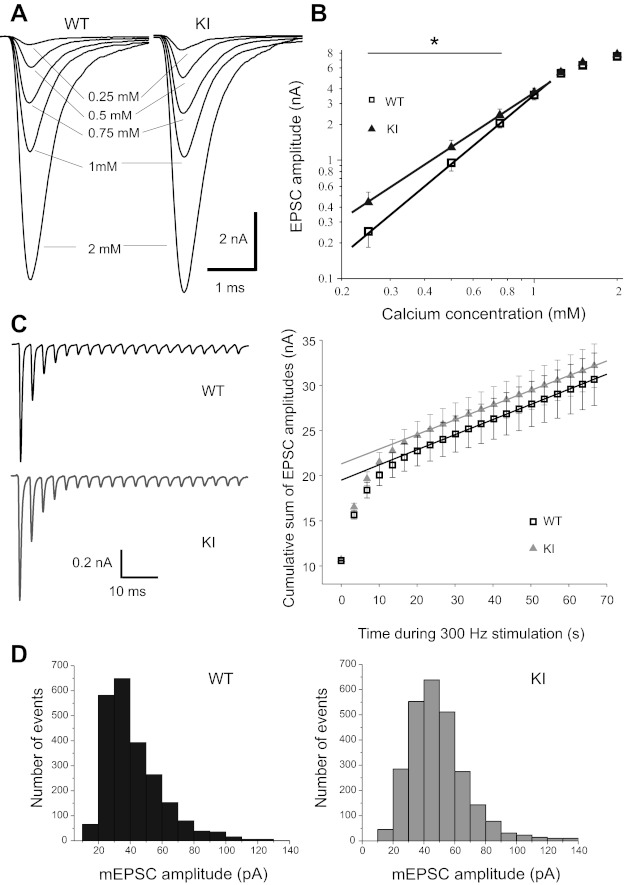
Transmitter release is increased at low external Ca2+ concentration ([Ca2+]o) in R192Q knock-in (KI) mice, while release probability and quantal content remain unchanged at 2 mM [Ca2+]o. A: representative excitatory postsynaptic currents (EPSCs) evoked in medial nucleus of the trapezoid body (MNTB) neurons from wild-type (WT, black) and R192Q KI (gray) mice during whole cell voltage-clamp recording conditions at a holding potential of −70 mV, with [Ca2+]o of 0.25, 0.5, 0.75, 1, and 2 mM in the extracellular aCSF. Transmitter release is increased in KI mice at low [Ca2+]o, while at 2 mM [Ca2+]o there are no differences in mean EPSC amplitudes between R192Q KI (7.8 ± 0.5 nA, n = 8) and WT (7.5 ± 0.4 nA, n = 9; Student's t-test, P = 0.38) mice. [Mg2+]o was kept constant at 2 mM throughout experiments. B: double logarithmic plots showing relationship between EPSC amplitudes and [Ca2+]o for WT and KI mice. In the [Ca2+]o range of 0.25–1 mM the fit to a linear function indicates that the EPSC amplitudes are proportional to the [Ca2+]o raised to a power of 1.5 ± 0.2 for R192Q KI (n = 8) and 1.9 ± 0.3 for WT (n = 9). In this range, the calcium dependence of nerve-evoked EPSCs is significantly different between the genotypes (P < 0.01, nonparametric ANOVA, Kruskal-Wallis test). C, left: recordings of EPSCs showing short-term depression (STD) during 300-Hz stimulation in WT and R192Q KI mice. Right: mean cumulative summation of EPSC amplitudes during 300-Hz stimulation in WT and R192Q KI mice plotted as a function of time. The time 0 intercept of the lineal fitting of data in the steady-state region for times > 25 ms gives an estimate of the readily releasable pool size (N) multiplied by the mean quantal amplitude (q). The Nq values obtained for WT and KI are 20 ± 1 nA and 21 ± 1 nA, respectively. The release probability (pr), calculated as the ratio between the mean amplitude of the 1st EPSC in the train and the Nq values, is not altered in KI mice (pr = 0.50 ± 0.02, n = 9) vs. WT mice (pr = 0.54 ± 0.02, n = 13; P = 0.2, Student's t-test). D: histogram showing probability distribution of miniature (m)EPSC amplitudes. At normal [Ca2+]o (2 mM) and [Mg2+]o (1 mM), mean mEPSC amplitudes are 48 ± 3 pA in R192Q KI (n = 26) and 45 ± 3 pA in WT (n = 25) mice, while frequencies are 2.8 ± 0.5 Hz and 2.4 ± 0.4 Hz, respectively.
We next calculated the probability of vesicle release by estimating the fraction of the readily releasable pool released by a single AP. We used an approach based on repetitive stimulation that induced STD of EPSC amplitudes. Assuming that depression is largely caused by a transient decrease in the number of readily releasable quanta, it is possible to estimate the pool size by calculating the cumulative summation of EPSC amplitudes for time intervals that are short with respect to the time required for recovery from depression (Schneggenburger et al. 1999). We measured the cumulative sum of EPSC amplitudes during a train of 20 stimuli at 300 Hz in WT and R192Q KI mice. Data points in a range between 25 ms and 70 ms were fitted by linear regression and back-extrapolated to time 0 (Fig. 1C). This estimation effectively takes into account the cumulative EPSC amplitudes reached within the first six stimuli, corresponding to a time interval of ∼20 ms, before depression reaches its steady-state level. It assumes that recovery from depression is negligible for the time interval of ∼20 ms used for the calculation. The validity of this assumption can be assessed in the analysis of time recovery from STD described in Fig. 4G. The zero time intersect gives an estimate of the size of the readily releasable pool of synaptic vesicles (N) multiplied by the mean quantal amplitude (q). The release probability can be estimated by dividing the mean amplitude of the first EPSC in the train by the Nq value. Although this calculation may be overestimated because Ca2+ influx during the first stimulus is lower than during the last pulses and Ca2+ concentration rises during stimulation, comparison between KI and WT mice indicates that the release probability is not altered by the mutation (P = 0.2, Student's t-test) at [Ca2+] of 2 mM. Furthermore, the amplitudes of spontaneous events [miniature (m)EPSCs], which give an estimate of the quantal size, are similar (48 ± 3 pA, n = 26 for KI and 45 ± 3 pA, n = 25 for WT). There are also no significant differences in the frequencies of mEPSCs: 2.8 ± 0.5 Hz and 2.4 ± 0.4 Hz for KI and WT respectively, as shown in Fig. 1D. All these results are in agreement with the similarity in the amplitudes of presynaptic calcium currents evoked by physiological APs in WT and R192Q KI mice (González Inchauspe et al. 2010). However, we have previously observed (González Inchauspe et al. 2010) that the time course of the APs triggering synaptic transmission was a key factor for the expression of a synaptic gain of function in the FHM-1 R192Q KI mouse. Indeed, we have shown that presynaptic Ca2+ currents (IpCa) evoked at the presynaptic terminals of the calyx of Held by AP waveforms of long duration (similar to pyramidal cell APs) had larger amplitudes in R192Q compared with WT mice, while no differences were observed when these IpCa were evoked by the short-duration APs of the calyx of Held itself. To further examine the underlying mechanism, we decided to broaden presynaptic APs by partially blocking K+ channels. The addition of TEA (1 mM) alone or in combination with 4-AP (0.1 mM) to the external solution slowed down the kinetics of APs recorded from the calyx of Held presynaptic nerve terminals (Kim et al. 2010; Wang and Kaczmarek 1998) as shown in Fig. 2A for WT mice. Mean rise times (calculated as time from 10% to 90% of peak amplitude), half-widths, and 90–10% decay times in control conditions and in the presence of TEA and 4-AP are compared in Fig. 2B. No significant differences were observed in the effect of TEA and 4-AP on APs from KI mice (data not shown) compared with WT mice.
Fig. 4.
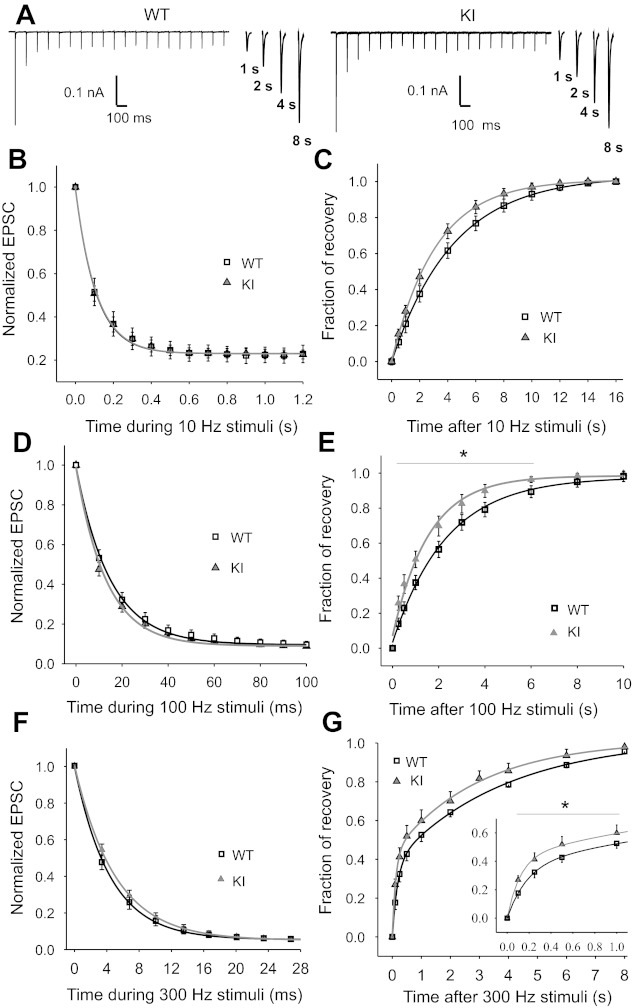
Recovery of EPSCs from STD following conditioning trains of stimuli at 10, 100, and 300 Hz is faster in R192Q KI mice. A: traces show depression of EPSC amplitudes during 2-s stimulation at 10 Hz (conditioning train) and recovery at different time delays after the conditioning train. Kynurenic acid (1 mM) was added to the extracellular solution to reduce postsynaptic AMPA receptor saturation. B: time course of depression of EPSC amplitudes (normalized to the 1st evoked EPSC) during 10-Hz stimulation. Data are fitted to a single-exponential decay function, with decay time constants τ = 108 ± 3 ms for R192Q KI (n = 16) and τ = 110 ± 3 ms for WT (n = 10, P = 0.2, Student's t-test). Magnitude of depression is 23 ± 1% and 23 ± 2% of the first pulse for R192Q KI and WT, respectively (P = 0.15, Student's t-test). C: time course of recovery from synaptic depression, measured by eliciting a single test EPSC at different increasing time intervals following the 10-Hz conditioning train. The fraction of recovery is calculated as (Itest − Iss)/(I1 − Iss), where I1 and Iss are the amplitudes of the first and last EPSCs in the train and Itest is the amplitude of the test EPSC. Data are fitted to an exponential decay function, with time constants τ = 3.24 ± 0.16 s (n = 10) and τ = 4.23 ± 0.25 s (n = 10) for R192Q KI and WT, respectively (P = 0.003, Student's t-test). D: mean time course of depression of normalized EPSC amplitudes during 100-Hz stimulation. The amplitudes of the EPSCs at the end of the stimulus are 9.0 ± 0.4% and 9.6 ± 0.3% of the first pulse for R192Q KI (n = 27) and WT (n = 15, P = 0.25, Student's t-test), respectively. Data are fitted with a single-exponential decay function, with time constants τ = 13.1 ± 0.5 ms for R192Q KI and τ = 14.8 ± 0.4 ms for WT (P = 0.18, Student's t-test). E: time course of recovery from STD after 100-Hz synaptic stimulation, fitted to an exponential decay function, with recovery time constants τ = 1.6 ± 0.1 s (n = 14) for R192Q KI and τ = 2.4 ± 0.1 s (n = 15) for WT (*P < 0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison). F: average time course of depression of normalized EPSC amplitudes during 0.07-s stimulation at 300 Hz. The amplitudes of the EPSCs at the end of the stimulus are 5.0 ± 0.4% and 5.4 ± 0.6% of the first pulse for R192Q KI (n = 9) and WT (n = 13, P = 0.43, Student's t-test), respectively. Data are fitted with a single-exponential decay function, with time constants τ = 5.0 ± 0.3 ms for R192Q KI and τ = 4.4 ± 0.3 ms for WT (P = 0.03, Student's t-test). G: recovery after the 300-Hz stimulation follows a double-exponential time course with the following time constants and percentages of contribution of fast and slow components: τfast = 130 ± 10 ms (45 ± 3%), τslow = 3.6 ± 0.5 s (55%) for R192Q KI mice (n = 9) and τfast = 180 ± 20 ms (37 ± 2%), τslow = 3.9 ± 0.2 s (63%) for WT mice (n = 13). Recovery in the range 0.1–1.0 s (inset) is significantly faster in KI than in WT mice (*P = 0.001, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc test).
Fig. 2.
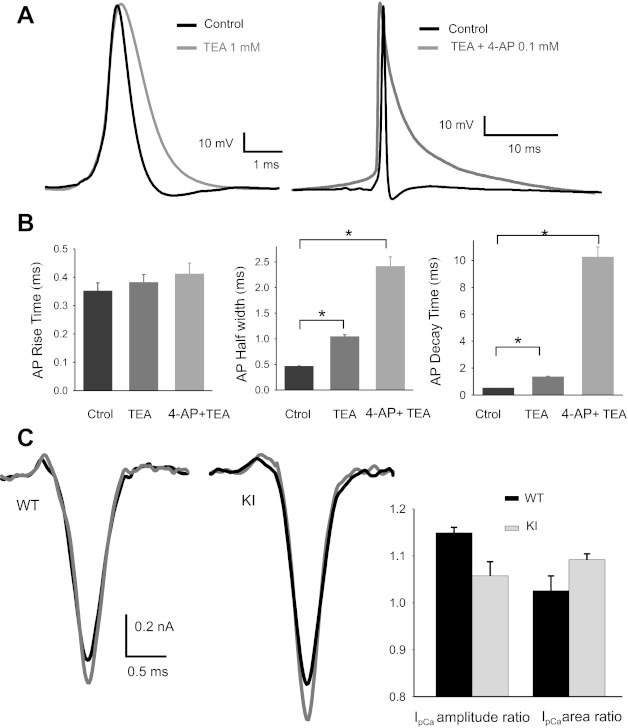
TEA and 4-AP broaden presynaptic action potentials (APs) without significant effect on presynaptic Ca2+ currents (IpCa). A: voltage-dependent potassium channel blockers slow presynaptic AP decay during whole cell current-clamp recordings at the calyx of Held of WT mice. B: in control conditions, mean rise times (10%-90%), half-widths, and decay times (90–10%) of AP from WT calyces of Held (n = 4) were 0.35 ± 0.03 ms, 0.45 ± 0.02 ms, and 0.47 ± 0.04 ms, respectively. After addition of 1 mM of TEA the same values increased to 0.38 ± 0.03, 1.03 ± 0.05 ms, and 1.3 ± 0.1 ms. With the additional application of 4-AP (100 μM), mean rise times, half-widths, and decay times became 0.41 ± 0.04 ms, 2.4 ± 0.2 ms, and 10.2 ± 0.8 ms. Values from KI mice were not statistically different from those measured in WT mice. *Significant differences (P < 0.001, Student's t-test). C: effect of 4-AP on IpCa. Left: sample traces of IpCa in control conditions (black) and after addition of 100 μM 4-AP (gray) for WT and KI mice. Right: no significant differences were observed in the ratio of IpCa amplitudes (or in the ratio of IpCa areas) with and without 4-AP in WT (n = 4) as well as in KI mice (n = 4) (P > 0.05, Student's t-test).
Wu et al. (2009) demonstrated that, in dissociated neurons from the rat dorsal root ganglion, superior cervical ganglion, and hippocampus, 4-AP and several of its analogs have a potentiating effect on high-voltage-activated Ca2+ channels themselves, independent of KV channels. Therefore, we first examined the effect of this K+ channel blocker on the IpCa at the calyx of Held. Control experiments using calyx of Held AP templates to depolarize the nerve terminal showed a minor effect of 4-AP (0.1 mM) on the IpCa as shown in traces of Fig. 2C, left. There is an increase in amplitude and consequently in area, but with no statistical differences between WT and R192Q KI mice (Fig. 2C, right). On the other hand, the kinetics of IpCa is not altered by 4-AP.
Figure 3A shows representative EPSCs in control conditions and after the sequential addition of TEA and 4-AP for WT (Fig. 3A, left) and R192Q KI (Fig. 3A, right) mice at Ca2+ and Mg2+ concentrations of 0.75 mM and 2 mM, respectively (i.e., to avoid saturating vesicle release after TEA and 4-AP bath application). When transmitter release was evoked in the presence of TEA and 4-AP, and thus by longer-duration APs, EPSCs from KI mice revealed a significant increase in amplitude and area compared with WT mice. Figure 3B summarizes the mean EPSC areas recorded from WT and KI mice in control conditions and after slices were incubated with TEA (Student's t-test, P = 0.002 between WT and KI) and 4-AP (Student's t-test, P = 0.003 between WT and KI). Figure 3C shows the ratios between EPSC areas evoked with TEA in the external solution and in control conditions, as well as the ratio of EPSC areas with TEA plus 4-AP with respect to control for WT and R192Q KI mice. These ratios indicate that the increments in EPSC amplitudes and areas are significantly greater in KI than WT neurons when AP durations are increased by TEA (P = 0.017, Student's t-test) or by TEA plus 4-AP (P = 0.018, Student's t-test).
Fig. 3.
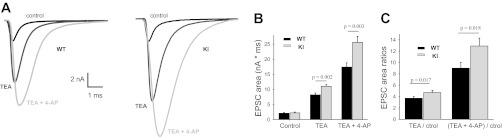
Increasing presynaptic AP duration reveals a gain of function in KI mice. A: EPSCs in WT and KI MNTB neurons in an external solution with 0.75 mM [Ca2+]o and 2 mM [Mg2+]o in control conditions (black), in the presence of 1 mM TEA (dark gray), and with TEA + 4-AP (0.1 mM, light gray). B: bar graphs show EPSC integrals (areas, representing charge of EPSCs) in control conditions and after addition of TEA and TEA + 4-AP in the external solution for WT (black, n = 12) and R192Q KI (gray, n = 14) mice. Control values: 2.06 ± 0.35 nA·ms (WT) and 2.21 ± 0.27 nA·ms (KI). With TEA: 8.2 ± 0.6 nA·ms (WT) vs. 11.0 ± 0.6 nA·ms (KI, P = 0.002, Student's t-test). With TEA plus 4-AP: 17.4 ± 1.4 nA·ms (WT) vs. 25.6 ± 2.0 nA·ms (KI, P = 0.003, Student's t-test). C: ratios of EPSC integrals (blocker/control) in the presence of the potassium blockers, relative to control conditions, for WT (black) and R192Q KI (gray) mice. With TEA: 3.7 ± 0.3 (WT) vs. 4.7 ± 0.4 (KI, P = 0.017, Student's t-test). With TEA + 4-AP: 9 ± 1 (WT) vs. 12.9 ± 1.4 (KI, P = 0.018, Student's t-test).
Short-term plasticity in synaptic transmission of R192Q KI mice.
Repetitive stimulation of the calyx of Held-MNTB synapse causes STD of EPSCs: postsynaptic current amplitudes decrease during the train of stimuli until they reach steady-state amplitudes. The efficacy of synaptic transmission during repetitive stimulation is also determined by the rate of recovery from STD, owing to the replenishment of the readily releasable pool of synaptic vesicles, which is dynamically regulated by Ca2+ influx through VGCCs in an activity-dependent manner (Wang and Kaczmarek 1998; Zucker and Regehr 2002).
We studied the time course of STD and recovery for different stimulus frequencies at the calyx of Held-MNTB synapse from WT and R192Q KI mice. EPSC amplitudes during 20-pulse trains at 10 Hz (see representative traces in Fig. 4A), 100 Hz, and 300 Hz depress after a single-exponential time course with similar time constants at the calyx of Held synapses from both WT and R192Q KI mice. The magnitude of depression at the end of the train of stimuli was similar between WT and KI mice (see Fig. 4B for 10 Hz, Fig. 4D for 100 Hz, and Fig. 4F for 300 Hz). The time course of recovery from synaptic depression was studied by eliciting a single test EPSC at varying time intervals (from 0.1 s to 14 s) after the conditioning train. The fraction of recovery was calculated as follows:
where I1 and Iss are the amplitudes of the first and last EPSCs in the train and Itest is the amplitude of the test EPSC.
The fractional recovery from STD as a function of time following the 10- or 100-Hz conditioning train of stimuli was fit by a single exponential (Fig. 4C for 10 Hz and Fig. 4E for 100 Hz). The rate of recovery is significantly increased in R192Q KI compared with WT synapses at both frequencies. Recovery from STD after 300-Hz stimuli follows a double-exponential time course (Fig. 4G) with a fast and a slow time constant. R192Q KI synapses show an enhanced recovery with a significant difference in the time constant and percentage of participation of the fast component compared with WT mice (P = 0.001, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison).
Increased calcium buffering affects fast recovery of EPSCs from STD in R192Q KI mice.
To assess the role of residual Ca2+ on the recovery kinetics of EPSCs after STD, we studied the effect of a slow Ca2+ buffer in presynaptic calyces. Slices were preincubated for 75 min with the tetra-acetoxymethyl ester form of EGTA (EGTA-AM, 0.2 mM). The recovery of EPSC amplitudes after a 0.2-s train at 100 Hz was unaltered in WT mice compared with control conditions (slices with no EGTA-AM), as shown in Fig. 5A (P > 0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison). In contrast, EGTA-AM increased the time constant of recovery from τ = 1.7 ± 0.1 s (control) to τ = 3.2 ± 0.3 s (EGTA-AM) in R192Q KI mice, as shown in Fig. 5B. The difference in recovery time course between WT and R192Q KI mice under slow intracellular Ca2+ buffering was no longer significant (P > 0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison), as shown in Fig. 5C.
Fig. 5.
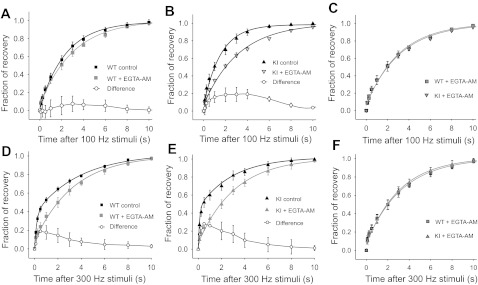
Residual calcium is involved in the acceleration of synaptic recovery after STD at the calyx of Held in R192Q KI mice. Slices were incubated for 75 min with EGTA-AM (0.2 mM) to increase the buffering of Ca2+ in presynaptic terminals prior to whole cell recording. A and B: time course of recovery from STD following a 0.2-s train at 100 Hz after loading presynaptic terminals with EGTA-AM in WT (A) and R192Q KI (B) mice, superimposed with the corresponding data in control conditions (without EGTA-AM). The difference between the data in the presence and absence of EGTA-AM is also shown. The mean recovery time constant in the presence of EGTA-AM (τ = 3.1 ± 0.4 s, n = 7) for WT mice is similar to that in control conditions (P > 0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison), but for KI mice EGTA-AM significantly slows the rate of recovery (τ = 3.2 ± 0.5 s, n = 9) compared with control (P < 0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc comparison). C: comparison of the recovery time course from STD after a 0.2-s conditioning train at 100 Hz in the presence of EGTA-AM for WT and R192Q KI mice. EGTA-AM abolished the differences in the rate of recovery observed in control conditions between WT and KI (P >0.05, 1-way repeated-measures ANOVA, Student-Newman-Keuls post hoc). D and E: EGTA-AM further decreases the rate of recovery after STD at 300-Hz frequency stimulation in synapses from both WT (D) and R192Q KI (E) mice. In control conditions (with no EGTA-AM), recovery after a 20-pulse train at 300 Hz follows a double-exponential time course (parameters in Fig. 4G legend). The fast component is eliminated in the presence of EGTA-AM, and recovery follows a single-exponential time course with similar time constants for R192Q KI (3.2 ± 0.3 s, n = 9) and WT (3.3 ± 0.3 s, n = 7) mice. The difference between data with and without EGTA-AM is also plotted. F: comparison of the time course of recovery from STD after a 0.07-s train at 300 Hz in the presence of EGTA-AM for WT and R192Q KI mice. EGTA-AM abolished the faster recovery observed in R192Q mice compared with WT in control conditions (P > 0.05, 1-way ANOVA RM, Student-Newman-Keuls post hoc test).
EGTA-AM has a more potent effect on the fast recovery after STD following higher-frequency stimulations. After a 0.07-s train of stimuli at 300 Hz, recovery from STD in control conditions (with no EGTA-AM) showed a fast (τfast) and a slow (τslow) component (see double-exponential fits shown in Fig. 4G). When experiments were performed in slices that had been incubated with EGTA-AM, the fastest recovery component was eliminated, and recovery followed a single-exponential time course, as shown in Fig. 5, D (WT) and E (KI R192Q). Similar results have been reported previously by Wang and Kaczmarek (1998). The recovery after 300-Hz stimulation under the effect of EGTA-AM did not show any significant difference between WT and R192Q KI mice (P > 0.05, 1-way repeated measures ANOVA, Student-Newman-Keuls post hoc comparison), as shown in Fig. 5F.
DISCUSSION
KI mice carrying the FHM-1 mutation R192Q in the Cacna1a gene that encodes the α1A-subunit of voltage-gated Cav2.1 (P/Q-type) Ca2+ channels have been used to evaluate the functional consequences of this mutation on synaptic transmission at the calyx of Held-MNTB synapse, where transmitter release is exclusively triggered by Cav2.1 channels.
The mutation in the CaV2.1 channels linked to FHM-1 affects the biophysical properties of IpCa at the calyx of Held synapse, allowing them to activate at more hyperpolarizing potentials. Despite this, synapses from WT and R192Q KI mice have similar EPSC amplitudes, release probability (Fig. 1C), and spontaneous release (Fig. 1D) when [Ca2+]o = 2 mM. This is consistent with the observed similarity in IpCa evoked by APs (González Inchauspe et al. 2010). However, the most significant impact of the FHM R192Q mutation is revealed when exocytosis is triggered by longer-duration APs. When the half-width and decay times of presynaptic APs were increased by inhibiting K+ channels (which normally contribute to membrane potential repolarization of the AP), the evoked EPSCs in R192Q KI mice showed enhanced current amplitudes and charge transfer compared with WT mice. This gain of function is correlated to the increase of IpCa amplitudes and charge observed at KI calyx of Held terminals when induced by APs of long duration (half with ≥1 ms; González Inchauspe et al. 2010).
Furthermore, CaV2.1 channels with the R192Q mutation affect neurotransmitter release at low Ca2+ concentrations, as indicated by the dependence of EPSC amplitudes on [Ca2+]o when <1 mM (Fig. 1B). The broadening of APs at low Ca2+ concentration will contribute to a certain extent to raising Ca2+ influx through presynaptic terminals (and consequently to larger EPSCs) in R192Q KI compared with WT mice, as previously demonstrated (González Inchauspe et al. 2010) and ratified here with the results in Fig. 3. The effect on AP shape of lowering [Ca2+]o from 2 mM to 0.5 mM showed an increase of 11 ± 1% in the half-width and 16 ± 3% in the decay time, while the amplitude decreased by 6 ± 1% (n = 6; Student's t-test, P < 0.001). Endogenous Ca2+ buffering and Ca2+ extrusion in R192Q KI mice would also enhance transmitter release, particularly in low [Ca2+]o, since these mechanisms would be less saturated than in normal extracellular calcium. In fact, the in vivo conditions are better approximated in slices by lowering the extracellular Ca2+ concentration from 2 to 1.2 mM (Borst 2010). Finally, following Frankenhaeuser and Hodgkin's theory of surface potential contribution from divalent ions (“charge shielding”; Frankenhaeuser and Hodgkin 1957), a reduction in calcium concentration might shift the voltage dependence of Ca2+ and K+ channel gating to negative voltages. Shifts have been described for activation of Ca2+ channels, Na+ channels, delayed-rectifier K+ channels, and Ih. Although the absolute shift might be similar in normal and mutated channels, the consequences for the increase of AP-evoked EPSC amplitudes would be more pronounced in KI mice because the mutated Ca2+ channels are already shifted toward hyperpolarized potentials (González-Inchauspe et al. 2010). Thus, in low [Ca2+]o, Ca2+ channel activation will be closer to the resting potential, and on opening the driving force will be greater for KI than for WT mice.
We show that synapses driven by short-duration APs (e.g., such as those at the calyx of Held and cortical interneurons) are less affected by the mutation-induced hyperpolarizing shift in Ca2+ channel activation than those driven by longer-duration APs (e.g., pyramidal neuron APs). These results provide a plausible explanation for the diverse consequences of FHM-1 mutations observed in inhibitory (lack of effect) versus excitatory (gain of function) cortical synapses (Tottene et al. 2009), although further experiments are needed to confirm this hypothesis in cortical circuitry. The underlying mechanism influencing synaptic strength by the R192Q mutation might be related to the variability in calcium currents induced by the different shapes of APs.
We also observed that FHM-1 mutation increases the rate of recovery from STD across a range of frequencies (10, 100, and 300 Hz). STD reflects depletion of the release-ready vesicle pool (Schneggenburger et al. 1999; von Gersdorff and Borst 2002; Wang and Kaczmarek 1998; Wong et al. 2003; Wu and Borst 1999; Zucker and Regehr 2002). However, other mechanisms might be involved, such as receptor desensitization (Neher and Sakaba 2001; Schneggenburger et al. 2002; Taschenberger et al. 2002; Wong et al. 2003), calcium channel inactivation (Forsythe et al. 1998; Muller et al. 2008; Xu and Wu 2005), and calcium channel inhibition by presynaptic metabotropic glutamate receptors (Takahashi et al. 1996; Von Gersdorff et al. 1997) or AMPA receptor activation (Neher and Sakaba 2001; Scheuss et al. 2002; Takago et al. 2005). The rate of recovery from STD is an important feature in determining synaptic efficacy, particularly in burst-firing neurons (Birtoli and Ulrich 2004; Ma et al. 2012; Xiang et al. 2002). It is mainly dependent on the recycling rate of vesicles into the readily releasable pool (Wang and Kaczmarek 1998; Zucker and Regehr 2002), which is strongly influenced by preceding synaptic activity. There is good evidence that cytosolic Ca2+ plays multiple roles in regulating the vesicle cycle. For instance, previous reports have shown that increased presynaptic Ca2+ buffering slows the recovery from synaptic depression (Dittman and Regehr 1998; Stevens and Wesseling 1998; Wang and Kaczmarek 1998). Exogenous mobile Ca2+ buffers (such as EGTA) freely diffuse throughout the cytoplasm and close to the site of Ca2+ influx. Here we have shown that presynaptic terminals experimentally manipulated to have higher Ca2+ buffering (i.e., after EGTA-AM incubation) exhibited slower recovery from synaptic depression at high stimulation frequencies, consistent with recycling being calcium dependent. After 100-Hz STD, EGTA-AM did not significantly change the rate of recovery in WT mice (Fig. 5A), but the decrease was significant in R192Q KI mice (Fig. 5B). On the other hand, at 300 Hz the fast component of the refilling was eliminated in both WT and R192Q KI animals (Fig. 5, D and E, respectively) by EGTA-AM. Thus at high conditioning frequencies (100–300 Hz), EGTA-AM abolished the faster recovery observed in R192Q KI compared with WT mice (Fig. 5, C and F), consistent with the idea that increased residual Ca2+ may be responsible for accelerating the recovery process following STD in synapses from R192Q KI mice. The rise in residual calcium is a consequence of the mutated CaV2.1 channels that activate at more hyperpolarized potentials (González Inchauspe et al. 2010), together with a faster (smaller time constant) voltage-dependent activation of Ca2+ currents (unpublished observations) at the calyx of Held of R192Q KI mice. Because activation and deactivation of calcium channels are strongly voltage dependent, AP-evoked calcium entry can have a steep dependence on AP half-width and waveform. This is especially significant in presynaptic terminals, where modest changes in AP waveform dramatically modify calcium entry and transmitter release. Cumulative inactivation of potassium channels can broaden AP half-width in response to increased firing frequency. Such frequency-dependent broadening of the AP is prominent in hippocampal CA1 pyramidal neurons (Shao et al. 1999) and pyramidal-like projection neurons in the lateral amygdala (Faber and Sah 2003), and elsewhere, due to inactivation of BK channels. Inactivation of KV4-mediated IA currents also contributes to spike broadening in pyramidal neuron somata (Kim et al. 2005). A particularly dramatic example of frequency-dependent spike broadening occurs at mossy fiber boutons in the hippocampus, probably mediated by inactivation of KV1 family channels (Geiger and Jonas 2000). The open probability and open time of the mutated CaV2.1 calcium channels might also contribute to an increased Ca2+ influx that would lead to a faster recovery from STD in the R192Q KI mice. Studies of FHM mutation R192Q CaV2.1 channels in heterologous expression systems revealed changes in single-channel biophysical properties, exhibiting increased channel open probability and unitary channel Ca2+ influx (as measured by the product of single-channel current and open probability) over a wide range of voltages, mainly due to the shift to more negative voltages of channel activation (Catterall et al. 2008; Hans et al. 1999; Kraus et al. 1998; Tottene et al. 2002). The IpCa evoked by the 100-Hz AP template previously recorded in current-clamp conditions (at a resting potential of −64 ± 1 mV) showed an amplitude and charge increase due to short-term facilitation in both WT and KI mice; there was no significant difference between the genotypes. Such changes would not be easy to detect because of the difficulty of achieving perfect voltage control during voltage-clamp recordings (Williams and Mitchell 2008).
On the other hand, the discrepancy between the lack of change in EPSC amplitudes and release probability and the faster recovery after STD in KI mice may be explained by the differences in the reliance of transmitter release and recovery from STD on presynaptic internal calcium concentration ([Ca2+]i). Transmitter release is steeply Ca2+ dependent with an onset > 2 μM [Ca2+]i (Bollmann et al. 2000; Schneggenburger and Neher 2000), whereas Ca2+-dependent vesicle recruitment is efficiently augmented by residual [Ca2+]i at lower concentrations of ∼0.5–2 μM. Therefore, an increase in resting Ca2+ by accumulation after high-frequency stimulation in presynaptic terminals from KI compared with WT mice could affect the recovery phase but would be insufficient close to the release site, to influence release probability.
van den Maagdenberg et al. (2004) showed that gain of function of CaV2.1 channels, resulting from the lower threshold of channel activation and increased channel open probability, leads to a lower threshold for induction of CSD, an increased velocity of propagation, and a longer duration of the CSD in the R192Q KI mouse in vivo. The dependence of CSD on the open probability of CaV2.1 channels can be explained by the prominent role of these channels in triggering glutamate release from cortical synapses (Pietrobon 2005; Pietrobon and Striessnig 2003). It is very likely that the acceleration in the rate of recovery could cause important changes in network activity leading to nervous dysfunction. The faster recovery of vesicle recycling during high-frequency transmission might contribute to the increased excitability in FHM-1 pathology. It is worth mentioning that the faster recovery in the R192Q KI model is opposite to the effect observed in knockout mice with ablated CaV2.1 channels (α1A−/−). Synapses from α1A−/− mice show a slower kinetics of recovery after STD compared with WT mice, as a consequence of the reduced Ca2+ influx and transmitter release at the presynaptic terminals (González Inchauspe et al. 2007).
While an established model that explains migraine attacks is still lacking, a favored hypothesis considers that the abnormal balance of cortical excitation-inhibition and the resulting persistent state of hyperexcitability of neurons in the cerebral cortex may be associated with the increased susceptibility for CSD, which is believed to initiate the attacks of migraine with aura (Ayata 2009; Haerter et al. 2005; Zhang et al. 2010, 2011).
The mutations in the three genes for FHM (CACNA1A, ATP1A2 and SCN1A) are the only established molecular links to migraine (Pietrobon 2010). The identification and analysis of gene mutations in FHM revealed a major role for disturbance of ion transport in this disorder, which could lead to altered synaptic function (van den Maagdenberg et al. 2007). The specific dominant and efficient role of CaV2.1 channels in controlling fast neurotransmitter release from central excitatory synapses suggests that the human and mouse CaV2.1 channelopathies and their episodic neurological symptoms, ranging from migraine to absence epilepsy and ataxia, might primarily be synaptic diseases. The different disorders probably arise from disruption of the finely tuned balance between excitation and inhibition in neuronal circuits of specific brain regions: the cortex in the case of migraine, the thalamus in the case of absence epilepsy, and the cerebellum in the case of ataxia.
GRANTS
This work was supported by the following grants: UBACYT X-223, PICT BID 1728 OC.AR.PICT 2005 N 32113, FONCYT (ANPCyT) PICT BID 1728 OC.AR.PICT 2006 N 199 (Argentina), and Wellcome Trust no. 084636 (UK) (to O. D. Uchitel) and FONCYT (ANPCyT) PICT 2007-1009, PICT 2008-2019, and PIDRI-PRH 2007 (to F. J. Urbano).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.G.I. and M.N.D.G. performed experiments; C.G.I. and M.N.D.G. analyzed data; C.G.I., M.N.D.G., and O.D.U. interpreted results of experiments; C.G.I. prepared figures; C.G.I. drafted manuscript; C.G.I., F.J.U., and O.D.U. edited and revised manuscript; C.G.I., F.J.U., M.N.D.G., M.D.F., A.M.v.d.M., I.F., and O.D.U. approved final version of manuscript; F.J.U., I.F., and O.D.U. conception and design of research.
ACKNOWLEDGMENTS
We thank María Eugenia Martin for her invaluable technical assistance and Paula Felman for administrative assistance. F. J. Urbano is a 2011 fellow of the John Simon Guggenheim Memorial Foundation.
REFERENCES
- Ayata C. Spreading depression: from serendipity to targeted therapy in migraine prophylaxis. Cephalalgia 29: 1095–1114, 2009 [DOI] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci 24: 4935–4940, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289: 953–957, 2000 [DOI] [PubMed] [Google Scholar]
- Borst JG. The low synaptic release probability in vivo. Trends Neurosci 33: 259–266, 2010 [DOI] [PubMed] [Google Scholar]
- Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci 28: 11768–11777, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng L, Maeno-Hikichi Y, Lai M, Chang S, Chen G, Zhang JF. Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell 115: 37–48, 2003 [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci 18: 6147–6162, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Ca2+-activated K+ (BK) channel inactivation contributes to spike broadening during repetitive firing in the rat lateral amygdala. J Physiol 552: 483–497, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang L. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci 25: 4131–4140, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MD, van den Maagdenberg AM, Frants RR, Goadsby PJ. Migraine as a cerebral ionopathy with impaired central sensory processing. In: Molecular Neurology, edited by Waxman SG. Oxford, UK: Elsevier, 2008 [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 20: 797–807, 1998 [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol 137: 218–244, 1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron 28: 927–939, 2000 [DOI] [PubMed] [Google Scholar]
- González Inchauspe C, Urbano FJ, Di Guilmi MN, Forsythe ID, Ferrari MD, van den Maagdenberg AM, Uchitel OD. Gain of function in FHM-1 Cav2.1 knock-in mice is related to the shape of the action potential. J Neurophysiol 104: 291–299, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González Inchauspe C, Forsythe ID, Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol 584: 835–851, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerter K, Ayata C, Moskowitz MA. Cortical spreading depression: a model for understanding migraine biology and future drug targets. Headache Currents 2: 97–103, 2005 [Google Scholar]
- Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon DJ. Functional consequences of mutations in the human α1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci 19: 1610–1619, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kushmerick C, von Gersdorff H. Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J Neurosci 30: 15479–15490, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol 569: 41–57, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change α1A Ca2+ channel kinetics. J Biol Chem 273: 5586–5590, 1998 [DOI] [PubMed] [Google Scholar]
- Ma Y, Hu H, Agmon A. Short-term plasticity of unitary inhibitory-to-inhibitory synapses depends on the presynaptic interneuron subtype. J Neurosci 32: 983- 988, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Felmy F, Schneggenburger R. A limited contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release at the rat calyx of Held. J Physiol 586: 5503–5520, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of Held. J Neurosci 21: 444–461, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci 4: 386–398, 2003 [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Migraine: new molecular mechanisms. Neuroscientist 11: 373–386, 2005 [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Cav2.1 channelopathies. Pflügers Arch 460: 375–393, 2010 [DOI] [PubMed] [Google Scholar]
- Rosato Siri MD, Piriz J, Giugovaz Tropper B, Uchitel OD. Differential Ca2+-dependence of transmitter release mediated by P/Q- and N-type calcium channels at neonatal rat neuromuscular junctions. Eur J Neurosci 15: 1874–1880, 2002 [DOI] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci 22: 728–739, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23: 399–409, 1999 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406: 889–893, 2000 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci 25: 206–212, 2002 [DOI] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol 521: 135–146, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron 21: 415–424, 1998 [DOI] [PubMed] [Google Scholar]
- Takago H, Nakamura Y, Takahashi T. G protein-dependent presynaptic inhibition mediated by AMPA receptors at the calyx of Held. Proc Natl Acad Sci USA 102: 7368–7373, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe I, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science 274: 594–597, 1996 [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leao RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36: 1127–1143, 2002 [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AM, Ferrari MD, Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Cav2.1 knockin migraine mice. Neuron 61: 762–773, 2009 [DOI] [PubMed] [Google Scholar]
- Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci USA 99: 13284–13289, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41: 701–710, 2004 [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Haan J, Terwindt GM, Ferrari MD. Migraine: gene mutations and functional consequences. Curr Opin Neurol 20: 299–305, 2007 [DOI] [PubMed] [Google Scholar]
- Virmani T, Han W, Liu X, Sudhof TC, Kavalali ET. Synaptotagmin 7 splice variants differentially regulate synaptic vesicle recycling. EMBO J 22: 5347–5357, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JGG. Short-term plasticity at the calyx of Held. Nat Rev Neurosci 3: 53–64, 2002 [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci 17: 8137–8146, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Poser C, Zhang JZ, Mineo C, Ding W, Ying Y, Sudhof TC, Anderson RG. Synaptotagmin regulation of coated pit assembly. J Biol Chem 275: 30916–30924, 2000 [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394: 384–388, 1998 [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11: 790–798, 2008 [DOI] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci 23: 4868–4877, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron 23: 821–832, 1999 [DOI] [PubMed] [Google Scholar]
- Wu Z, Li D, Chen S, Pan H. Aminopyridines potentiate synaptic and neuromuscular transmission by targeting the voltage-activated calcium channel β subunit. J Biol Chem 284: 36453–36461, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Synaptic inhibition of pyramidal cells evoked by different interneuronal subtypes in layer V of rat visual cortex. J Neurophysiol 88: 740–750, 2002 [DOI] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron 46: 633–645, 2005 [DOI] [PubMed] [Google Scholar]
- Zhang XC, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30: 8807–8814, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69: 855–865, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405, 2002 [DOI] [PubMed] [Google Scholar]