

Abstract
Exome sequencing has identified the causes of several Mendelian diseases, although it has rarely been used in a clinical setting to diagnose the genetic cause of an idiopathic disorder in a single patient. We performed exome sequencing on a pedigree with several members affected with attention deficit/hyperactivity disorder (ADHD), in an effort to identify candidate variants predisposing to this complex disease. While we did identify some rare variants that might predispose to ADHD, we have not yet proven the causality for any of them. However, over the course of the study, one subject was discovered to have idiopathic hemolytic anemia (IHA), which was suspected to be genetic in origin. Analysis of this subject’s exome readily identified two rare non-synonymous mutations in PKLR gene as the most likely cause of the IHA, although these two mutations had not been documented before in a single individual. We further confirmed the deficiency by functional biochemical testing, consistent with a diagnosis of red blood cell pyruvate kinase deficiency. Our study implies that exome and genome sequencing will certainly reveal additional rare variation causative for even well-studied classical Mendelian diseases, while also revealing variants that might play a role in complex diseases. Furthermore, our study has clinical and ethical implications for exome and genome sequencing in a research setting; how to handle unrelated findings of clinical significance, in the context of originally planned complex disease research, remains a largely uncharted area for clinicians and researchers.
Background
The advent of the capture of most known exons in the genome (the exome), followed by high-throughput sequencing, has already led to the identification of the causes of many Mendelian disorders (Bilguvar et al., 2010; Choi et al., 2009; Erlich et al., 2011; Johnston et al., 2010; Ng et al., 2010a; Ng et al., 2010b; Pierce et al., 2010), including from just a single individual (Haack et al., 2010). As the cost of exome (and even whole genome) sequencing decreases, it is becoming increasingly feasible to use exome sequencing in the diagnostic realm for Mendelian diseases with already identified genetic mutations, rather than only to investigate the genetic basis of unknown Mendelian disorders. Indeed, one of the pioneering studies examining the clinical utility of whole-exome sequencing demonstrated how an unexpected genetic diagnosis of congenital chloride diarrhea in a patient suspected with Bartter syndrome can be made from sequencing data (Choi et al., 2009). Furthermore, exome sequencing may also be an important tool for investigating complex traits. However, such studies, whether family-based or case series-based, are only just now being performed to our knowledge. Therefore, it remains an open question how much exome sequencing can advance our understanding of genetics of complex traits, particularly when sample sizes and/or pedigrees are relatively small.
To begin to address this issue, we focused on a complex illness, Attention Deficit/Hyperactivity Disorder (ADHD, OMIM: #143465). ADHD is a common disorder affecting more than 1 in 20 children in the U.S., with as many as 50% remaining symptomatic into adulthood, along with much heterogeneity in the presentation of the illness (CDC, 2010; Petersen et al., 2009). Genetic factors are thought to play a large role in the etiology of the disorder, but studies thus far remain inconclusive (Mick et al., 2010; Neale et al., 2010a; 2010b). We have hypothesized that rare, family-specific genetic variants may account for some of the “missing heritability” of ADHD, and we have presented evidence for the involvement of mGluR5 in one family (Elia et al., 2010). This is consistent with recent studies which showed that an excess of deleterious rare low-frequency nonsynonymous mutations reside in the human population (Li et al., 2010). Given the multifactorial nature of complex diseases, especially neuropsychiatric diseases, one way to reduce the complexity of searching for disease genes is to focus on rare variants with potentially higher penetrance, identified from clearly familial cases (Cirulli and Goldstein, 2010). Familial segregation of ADHD in a small family such as this one suggested to us that some major-effect genetic variants could be present and we set out to determine whether exome sequencing could shed light on the genetic basis of ADHD in this one family. Although we did identify several such rare variants, we have not yet proven that the identified variants cause the ADHD (alone or in combination), and we remark on the reasons for why this has been difficult. Our study also led to the unrelated characterization of the genetic basis of a case of idiopathic hemolytic anemia (IHA), which ended up being a clearly Mendelian trait (Pyruvate Kinase Deficiency of Red Cells, OMIM #266200). This study highlights the coming wave of unrelated findings and the resolution of “idiopathic” diseases with whole exome and whole genome sequencing.
Materials and Methods
Sample Collection
The samples used in our study all came from the same pedigree ascertained in clinics at the University of Utah. The collection and genomic analysis of the DNA were approved by the Institutional Review Board at the University of Utah, and written informed consent was obtained from all study participants. Blood samples were collected and genomic DNA extracted using alkaline lysis and ethanol precipitation (Gentra Puregene, Qiagen Corp., USA). DNA was quality-checked on agarose gels and quantified according to standard protocols.
Phenotype Analysis
The clinical records of patient 84060 were reviewed with his permission, allowing for collection of data related to his hemolytic anemia. ADHD Research scales and assessments included: Parents Rating Scale (PRS) (Ward et al., 1993), Wender Utah Rating Scale (WURS) (Ward et al., 1993), Wender-Reimherr Adult Attention Deficit Disorder Scale (WRAADDS) (Rosler et al., 2010b), Conners Adult ADHD Rating Scale - Investigator Version (CAARS-Inv) (Rosler et al., 2010a), Clinical Global Impression - Improvement (CGI-I), Clinical Global Impression - Severity (CGI-S), Wisconsin Personality Disorders Inventory-IV (Smith et al., 2003), Personality Disorder Questionnaire (Hyler et al., 1992), Iowa Personality Disorder Screen (Langbehn et al., 1999), CNS Vital Signs (CNSVS) (Gualtieri and Johnson, 2006), Weissman Social Adjustment Scale - Self Report (SAS-SR) (Weissman et al., 2001), Sheehan Disability Scale (Leon et al., 1997), Childhood Symptom Scale - Self Report Form (ChSS-SRF) (Murphy and Adler, 2004), and Disruptive Behavior Rating Scale - Retrospective Adult Form (DBRS-RAF) (Friedman-Weieneth et al., 2009).
Exome Capture and Sequencing
Exome capture for the three males was carried out in January 2010 using the commercially available Agilent SureSelect Human All Exon v1 in solution method as per the manufacturer guidelines (Agilent). This method is designed to target all human exons, regions totaling approximately 38 Mb, in a single tube. The DNA from the unaffected mother was obtained at a later date, allowing us to use the newly released SureSelect Human All Exon v.2 Kit, which targets approximately 44 Mb, covering 98.2% of the CCDS database. For both captures, the pure and high molecular weight genomic DNA samples were randomly fragmented at BGI-Shenzhen by Covaris, resulting in DNA fragments with a base pair peak of 150 to 200 bp. Adaptors were then ligated to both ends of the resulting fragments. The adaptor-ligated templates were purified by the Agencourt AMPure SPRI beads and fragments with insert size of approximately 250bp being excised. Extracted DNA was amplified by ligation-mediated PCR (LM-PCR), purified, and hybridized to the Agilent SureSelect Library for enrichment. Hybridized fragments were bound to the streptavidin beads whereas non-hybridized fragments were washed out after a 24-hour hybridization. Captured LM-PCR products were subjected to Agilent 2100 Bioanalyzer to estimate the magnitude of enrichment. Paired end sequencing was performed using the Illumina Genome Analyzer IIx platform with read lengths of 76 base pairs, providing at least 20× average coverage at the targeted region. The unaffected mother was sequenced with read lengths of 90 base pairs due to technological advancements during the course of the study, at an average coverage of 30× at the targeted region. Raw image files were processed by Illumina Pipeline v1.6 for base-calling with default parameters. FASTQ files were produced from the pipeline for downstream sequence data analysis.
We identified variants with four pipelines.
Method I: SOAP-based pipeline
The first pipeline was performed at BGI-Shenzhen. Sequence reads were aligned to human reference genome builds hg19 using SOAPaligner 2.20 (Li et al., 2008) with a maximum of two mismatches. The consensus genotypes in target regions (based on the SureSelect Human All Exon Kit) were called by SOAPsnp version 1.03 (Li et al., 2009). Single nucleotide variant (SNV) results were filtered as followed: Base quality ≥20, depth from 4–200, copy number estimate <2, and distance between two adjacent SNVs no less than 5. Insertions/deletions (indels) <5 base pairs were detected using SOAPindel. The resulting variant genotypes relative to the human reference assembly were identified.
Method II: GATK-based pipeline
BWA (Li and Durbin, 2009) version 0.5.8 was used to align the sequencing reads, with default parameters, to the human reference genome sequence build 37 (hg19), downloaded from the 1000 Genomes Project website (1000 Genomes Consortium, 2010). BWA implements a backward search with Burrows-Wheeler Transform to efficiently align short reads with perfect identity against references. Alignments were converted from SAM format to sorted, indexed BAM files with SamTools (Evjenth et al., 2009). The Picard tool (http://sourceforge.net/projects/picard/) was used to remove invalid alignments and remove duplicate reads from the BAM files. The BAM files were re-aligned with the GATK IndelRealigner too (McKenna et al., 2010). Genotypes were called by the GATK UnifiedGenotyper and the IndelGenotyperV2. Based on the recommendations from the authors of GATK (see http://www.broadinstitute.org/gsa/wiki/index.php/Best_Practice_Variant_Detection_with_the_GATK_v2), we removed variant calls based on having any of the following criteria: 1) SNVs with-in clusters (3 SNVs within 10 bp of each other); (2) more than four reads with mapping quality of zero (MQ0) and more than 10% of reads with mapping quality of zero; (3) strand bias (SB) higher than or equal to −1.0; (4) SNV quality score less than 30; (5) quality-by-depth (QD) score less than 5.0; (6) largest Contiguous Homopolymer Run of Variant Allele (HRun) more than 5; or (7) SNVs around a potential indel. Finally, we removed all variants with depth coverage less than 6.
Method III: SNVer pipeline
Similar to Method II, BWA-SW was used to align the sequencing reads to hg19 with default parameters; duplicates were removed by picard and local re-alignment was done by GATK. Instead of using the variant call method provided by GATK, we used SNVer (http://snver.sourceforge.net/) for detecting SNVs in each sample (Wei et al., 2011). We considered the mapped short reads with mapping quality >20, and counted only bases with base quality >30. Because of the cleaning step by picard and GATK, SNVer assumed a small sequencing/mapping error rate of 0.01 for the cleaned mapped short reads in making variant calls. SNVer sets the number of haploids to 2 for analysis of individual samples. We set the variant allele frequency threshold >0 for detecting both rare and common SNVs. SNVer provides multiplicity control, and we performed Bonferroni correction and controlled the family-wise error rate at 0.05 level. After calling SNVs in each sample, we selected those SNVs which are shared by the three patients but absent in the healthy control (mother). Then, the SNVs called were extracted and annotated by ANNOVAR (Wang et al., 2010b) for further analysis.
Method IV: GNUMAP-based pipeline
SNVs were verified using GNUMAP (Clement et al., 2009). GNUMAP utilizes position-weight matrices and a modified Needleman-Wunsch algorithm to probabilistically align reads to multiple locations on a reference genome. Alignment was performed for the four subjects using human genome version hg19. GNUMAP’s SNP-caller outputs coverage by base at each locus in the genome and uses a p-value statistic to call single nucleotide polymorphisms (SNPs). USeq’s Alleler software package (Nix et al., 2008) (http://useq.sourceforge.net/) was used to isolate non-synonymous variants. SamTools was used to visualize read pile-ups for manual verification and analysis of these variants.
SNP arrays and CNV analysis
All DNA samples were genotyped on the Illumina Human610-Quad version 1 SNP arrays (Illumina, San Diego, CA) with ~610,000 markers (including ~20,000 non-polymorphic markers) at the Center for Applied Genomics, Children’s Hospital of Philadelphia. Total genomic DNA extracted from whole blood was used in the experiments. Standard data normalization procedures and canonical genotype clustering files provided by Illumina were used to process the genotyping signals. Copy number variants (CNVs) were detected using PennCNV (Wang et al., 2007) and genomic wave adjustment (Diskin et al., 2008), with default parameters. Concordance between SNPs from the arrays and SNPs from exome sequencing was determined by calculating the percentage of variants from exome sequencing also with the same genotype derived from the SNP arrays.
Functional annotation of genetic variants
Variants were functionally annotated using the ANNOVAR software (Wang et al., 2010b). The variants reduction pipeline was also applied to identify a list of prioritized variants, but without filtering out variants in segmental duplication regions or conserved regions.
Validation
SNVs were validated using standard Sanger sequencing or Sequenom iPlex platform (http://www.sequenom.com/iplex).
Enzyme biochemical assays
The preparation of red cell hemolysates (to remove leucocytes and thrombocytes) and the measurements of enzymatic activities of pyruvate kinase, hexokinase, and glucose-6-phosphate dehydrogenase were performed according to standard methods (Beutler, 1984).
Results
Ascertainment of patients
The pedigree collected in our study is shown in Figure 1, with a mother, father, and four children. The father and two sons in this pedigree have ADHD, combined hyperactive and inattentive subtype, meeting DSM-IV-Text Revision and the Utah criteria for ADHD (Rosler et al., 2008), whereas the mother was evaluated and was determined to be unaffected. The status of the two sisters and more distantly related relatives is unknown, as they refused to participate in the research. The father and two sons also participated in an adult ADHD clinical trial in the Psychiatric Research Clinic at the University of Utah (Marchant et al., 2011). The clinical trial was a double-blind placebo-controlled crossover study of methylphenidate transdermal system (MTS) for adult ADHD. It was noted by the psychiatrists (G.J.L., F.R., and R.R.) that all three subjects were very similar in presentation, with all three being moderately to severely impaired with their ADHD symptoms (with a score of 4 or greater on the CGI-Severity Scale for ADHD at both Screening and Baseline visits), and all improving with active medication but not placebo. Supplementary Table 1 (see the attached patient information and supplementary tables and figures) shows ADHD assessment rating scores during the clinical trial at the following assessments: 1) baseline, 2) after treatment with active medication, and 3) after treatment with placebo.
Figure 1.
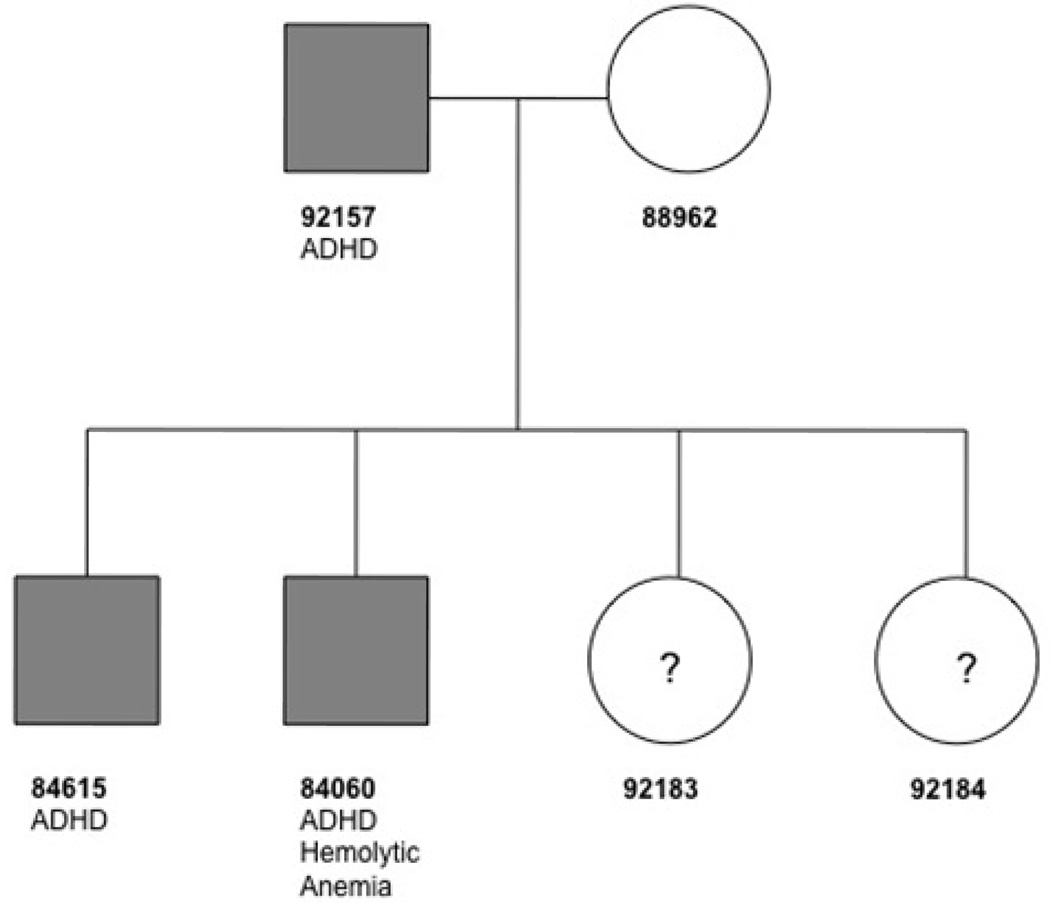
The pedigree structure is shown, with corresponding ID numbers. The three subjects in the pedigree affected with ADHD are shaded. Only 84060 has the idiopathic hemolytic anemia. The mother, father, and two sons were sequenced. The two sisters in the family declined to participate in the study, thus their phenotype status is unknown and marked as ‘?.’
Copy number variant analysis
Given that some prior studies have found large CNVs associated with ADHD (Elia et al., 2010), we initially screened for such CNVs in the family we studied. Using the data from the high-density SNV arrays, we performed CNV analysis to identify CNVs shared by all three ADHD cases. Four deletions were shared among the three individuals (Supplementary Table 5), but all of these CNVs appear to be common in the population based on Database for Genomic Variants, and thus likely not associated with ADHD or idiopathic anemia in this pedigree.
Identification of variants
In an effort to understand the genetic basis of the ADHD exhibited in this pedigree, we initially used exome capture (with Agilent SureSelect v.1) and sequenced the DNA of the index patient (84060), his father (92157), and his brother (84615) (Figure 1). To evaluate the sensitivity of exome sequencing to different data analytical strategies, we applied four computational pipelines to identifying genetic variants from the exome sequencing data (see Materials and Methods). Comparative analysis of the results from four pipelines may teach us about the sensitivity of data interpretation to the utilization of analytical approaches, highlighting the computational challenges in utilizing exome sequencing in clinical diagnosis. Approximately 20,000 single nucleotide variants (SNVs) were detected in each of the males being sequenced at a mean average coverage of ~20×, using four different computational pipelines for variant detection (Table 1, Figure 2, and Supplementary Figures 1 and 2). Given that our initial analyses revealed the desirability of filtering against an unaffected relative, we sequenced the mother’s exome at a later time, using a newer version of the exon capture (Agilent SureSelect v.2) and with a higher mean sequencing coverage of ~30×. We focused for the analysis presented here on the ~38 MB target region common to both platforms (Supplementary Tables 2–3). Given the experimental design, we benefited from the fact that we could check the relatives to verify calls, thus effectively tripling at a minimum the number of sequencing reads. This was particularly important in areas of relatively low coverage, given that the mean sequencing coverage ranged from 20–30×, which resulted in 10 or more reads in 67–75% of the target bases in each of the 4 individuals, but which was improved overall by having the sequence from the relatives. For example, after lowering the coverage threshold, there were 28,125,119 base pairs covered with six or more reads among all the three affected males. We also checked the accuracy of the exome sequencing by comparing to the Illumina Human610 SNP arrays with ~590,000 SNP markers and ~20,000 copy number markers. The concordance rate between the SNP results from the arrays and the exome sequencing variant calls was above 99.8% for all samples (Supplementary Table 4). We also verified by a combination of Sanger sequencing or Sequenom genotyping 19 of the SNVs identified by the SOAP pipeline as shared in the two brothers and father (Supplementary Figures 3–26). All 19 of these SNVs validated, demonstrating a very low rate of false positives for SNVs. On the other hand, the tested version of SOAPindel was not reliable for indel-calling, as 10 out of 13 called microindels were not validated with Sequenom genotyping. We therefore switched to the GATK pipeline for indels, as it has been shown to be more reliable in this regard (Depristo et al., 2011).
Table 1.
Summary of Single Nucleotide Variants (SNVs) for Exome Capture Samples.
| ExomeCapture | 84060 (child 1) | 84615 (child 2) | 92157 (father) | 88962 (mother) |
|---|---|---|---|---|
| Sequencing platform | GA IIX | GAIIX | GAIIX | HiSeq 2000 |
| Reads property | 76bp PE | 76bp PE | 76bp PE | 90bp PE |
| Number of SNVs (Method 1: SOAP) | 19825 | 19270 | 20430 | 22294 |
| Ti/Tv ratio | 2.8 | 2.7 | 2.9 | 2.8 |
| Number of SNVs+indels (Method 2: BWA+GATK) | 19655+947 | 18892+955 | 20100+916 | 21572+513 |
| Ti/Tv ratio | 2.9 | 2.9 | 3.0 | 2.9 |
| Number of SNVs (Method 3: Shrimp2+SNVer) | 16063 | 16704 | 18253 | 23917 |
| Ti/Tv ratio | 2.7 | 2.6 | 2.7 | 2.4 |
Note: We have not yet analyzed the mother’s exome with the 4th method (GNUMAP), so we have omitted this method from the table.
Figure 2.
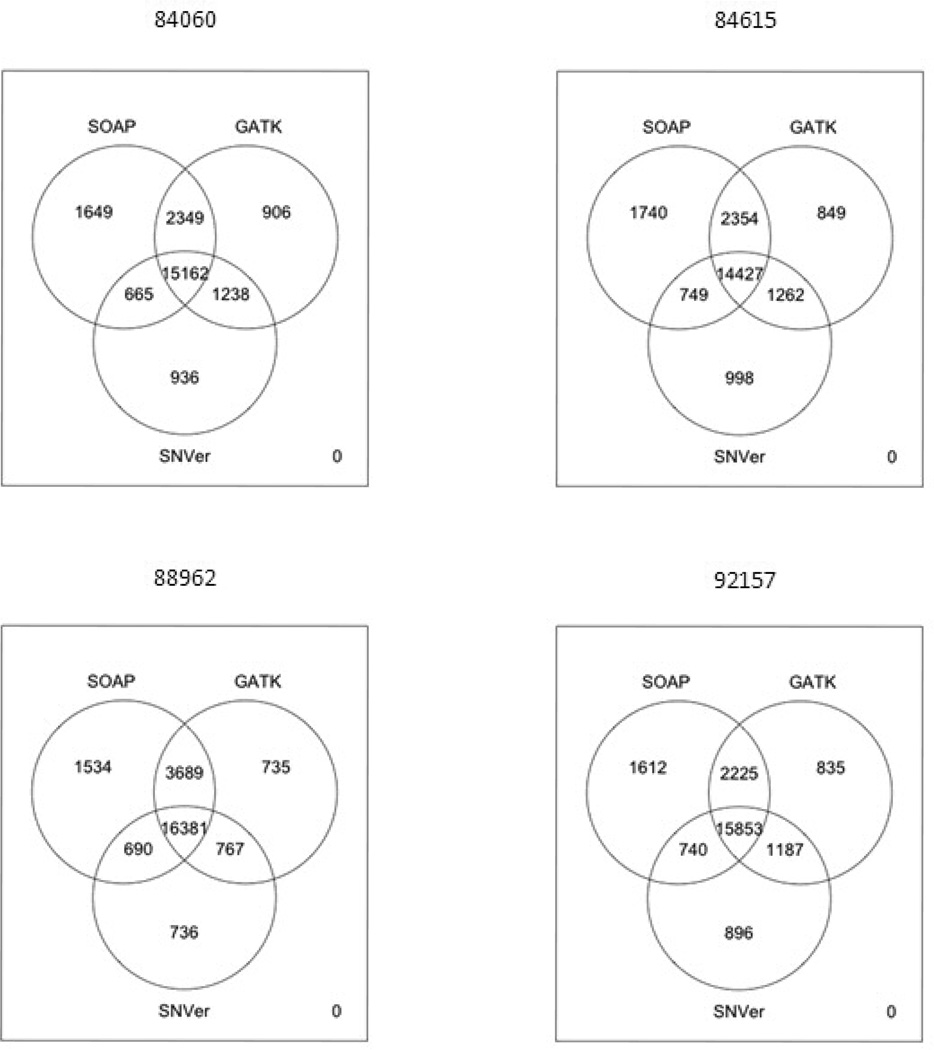
Overlap of variant calls from three computational pipelines (SOAP, GATK, and SNVer). Although PKLR loss-of-function mutations are identified by each pipeline, the amount of overlap of variant calls is modest, leading to potential concerns about clinical diagnosis using low-coverage exome data.
Analysis for ADHD
We made the assumption that any causative common variants with large effect sizes would have already been found in genome-wide association studies (GWAS). Therefore, we generated a list of rare [minor allele frequency (MAF) <1% in the 1000 Genomes project] candidate variants shared in the father and two sons that were not found in the mother (see Figure 3). It is important to note that the three different pipelines used to analyze all four exomes produced different sets of variant calls, although the vast majority of such variants (n=1,426) were found by all three pipelines (Figure 3). We also assessed the frequency of these variants in ~6,000 exomes already sequenced in other projects at BGI and Baylor (Supplementary Table 6). Nonsynonymous rare variants in ATP7B, CSTF2T, ALDH1L1, and METTL3 appeared to be the most plausible candidates for an association with ADHD in this pedigree, as these genes have been shown to be brain-expressed (at the level of RNA and/or protein) and several are implicated in possible neuropsychiatric conditions (Barley et al., 2009; Cocco et al., 2009; Elleuch et al., 2010; Kurian et al., 2011; Shankarling et al., 2009). However, to our knowledge, exome sequencing has not yet unambiguously identified many (if any) diseases variants shown to be definitely causative by themselves for complex diseases. After sequencing two parents with two sons, with the three males of the family sharing a well-phenotyped and similar version of a complex disease, we can obtain a prioritized list of potential candidate variants, but we have no certainty that any one of them can cause ADHD in the family, either by themselves or in aggregate. As it is well known among psychiatrists that there is extreme heterogeneity of ADHD in terms of its presentation, we decided to sequence this family due to the marked similarity in phenotype in the three males and what appeared to be an enhanced enrichment of ADHD in this family, based on reports from family members regarding other members of the family. We have unfortunately not been able to expand this pedigree further, as several members of the family have refused to participate in the genetics research. When we chose this family for sequencing, we did not anticipate the difficulty that we would encounter with recruiting other members of the family. Therefore, given the relatively small size of the family and the refusal of other family members to come in for an assessment, we were unable to define exactly the inheritance pattern in this family, although it seemed very reasonable to start with the assumption that variants causing the ADHD would be shared between the father and two sons.
Figure 3.
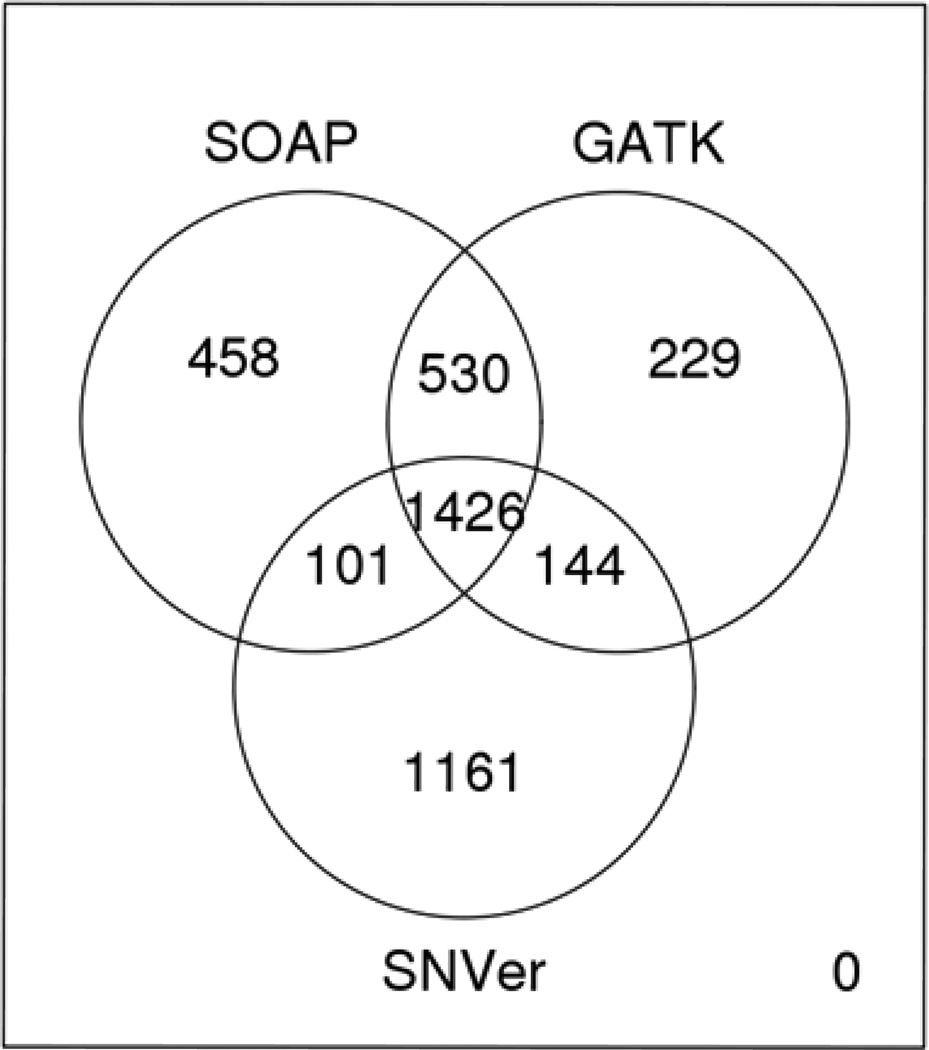
Intersection of variants. We show here the variants identified by the three main pipelines as being present in the three males with ADHD, but not present in the unaffected mother.
Genetic basis for the idiopathic hemolytic anemia
Immediately after we obtained the first set of exome sequencing data, one of the patients (patient 84060, a 28-year old Caucasian male) revealed that he had previously been diagnosed with idiopathic hemolytic anemia. After further reviewing his medical history, we recognized a long record of intermittent jaundice since childhood, and he had always been chronically anemic, with hemoglobin (Hgb) levels ranging from 10–12 g/dL (normal range 13–16). As a youth, he had been informed that he had idiopathic hemolytic anemia, but no genetic test had ever been performed to the best of his knowledge. He had visited a hematologist, who had ordered several clinical tests, but he was unaware of the results from those tests. We therefore decided to test the clinical utility of exome sequencing in solving the idiopathic hemolytic anemia in this patient. Further details of his clinical course are presented in Supplementary Information (Patient Presentation).
We began by analyzing the non-synonymous and frame-shift insertions/deletions in this individual, as such mutations are much more likely to affect protein function. There were ~6,000 such SNVs in 84060 (Table 1). A series of procedures (Figure 4) was implemented in the ANNOVAR pipeline for the purpose of reducing the number of candidate variants. Unlike previous studies on recessive diseases, we did not utilize dbSNP filtering, because causal variants for well-studied diseases [such as those that cause hearing loss or Crohn’s disease (Wang et al., 2010a)] are likely already catalogued in dbSNP. We used an MAF>1% filter for the 1000 Genomes Project for a similar reason (see Figure 4). After these filtering steps, we began with the assumption of either an autosomal recessive (compound heterozygotes or homozygotes) or X-linked mode of inheritance. Under the autosomal model, we required at least two mutations from different parents in the same gene, given that the idiopathic hemolytic anemia was not observed in either of his parents, or his brother or sisters.
Figure 4.
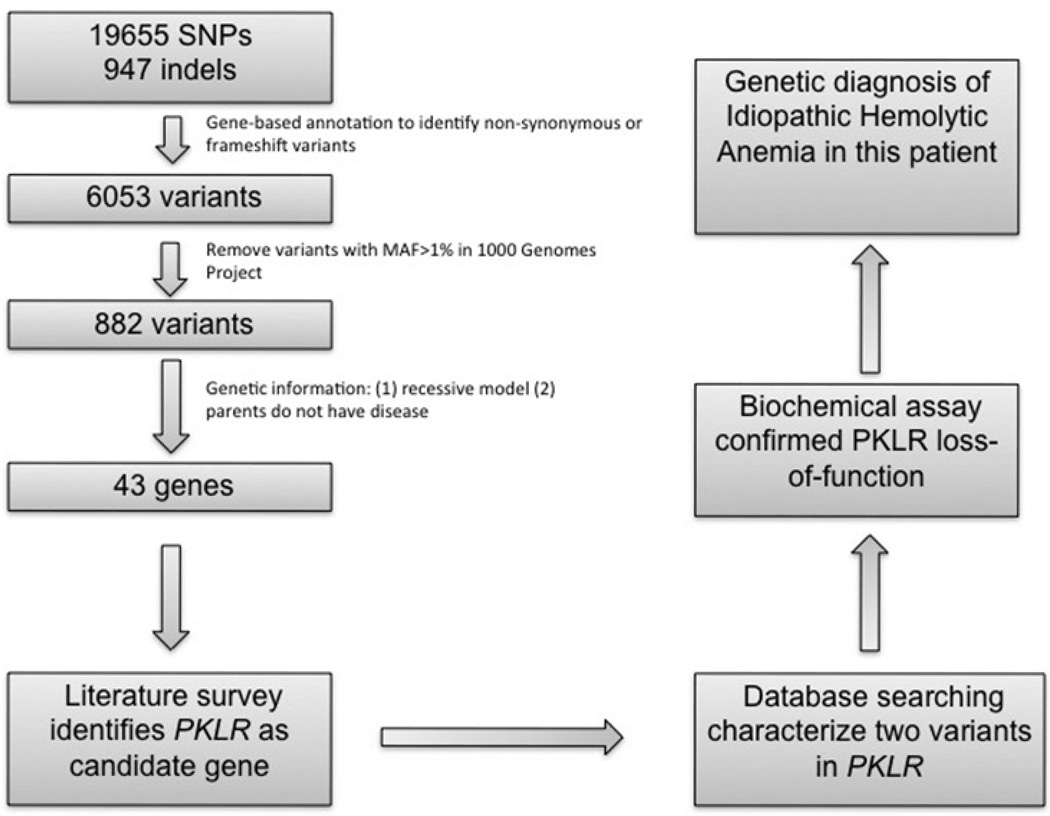
Illustration of the analysis method on IHA. We used a progressive filtering of variants from the GATK pipeline with variant calls aligned to hg19, leading to the genetic diagnosis of IHA.
The X-linked inheritance model did not reveal any mutations of functional significance. Using the autosomal recessive model we generated a list of variants in 43 genes (Supplementary Table 7). Non-immune mediated hereditary hemolytic anemia with unaltered red blood cell (RBC) morphology is mainly caused by mutations in genes involved in red cell metabolism (glycolysis, hexose monophosphate shunt, and the purine salvage pathway) (van Solinge, 2010)). A thorough literature search for the 43 candidate genes revealed evidence supporting the mutations in PKLR (pyruvate kinase, liver, and RBC isoform 2) as being well known to cause hemolytic anemia. Patient 84060 is a compound heterozygote for two very rare mutations in PKLR. These two mutations are c.1022G>C substitution in exon 8, encoding a Gly341Ala amino acid change, and a c.1706G>A substitution in exon 12, encoding an Arg569Gln amino acid change (Figures 5–6). The protein encoded by this gene is pyruvate kinase (PK), which catalyzes the transphosphorylation of phosphoenolpyruvate into pyruvate and ATP. PK acts at a rate-limiting step of glycolysis. Defects in this enzyme due to gene mutations are known to cause chronic hereditary nonspherocytic hemolytic anemia (CNSHA or HNSHA, OMIM ID #266200) (van Wijk and van Solinge, 2005; Zanella et al., 2007). From the list of 43 candidates, the patient was also homozygous for a mutation in ULK1 (NM_003565:exon23: c.A2446G:p.T816A). The ULK1 serine threonine kinase is known to be a critical regulator of mitochondrial and ribosomal clearance during the final stages of erythroid maturation (Kundu et al., 2008), but there is no evidence that mutation in this gene can result in anemia. None of the other genes had any known connection to hemolytic anemia.
Figure 5.
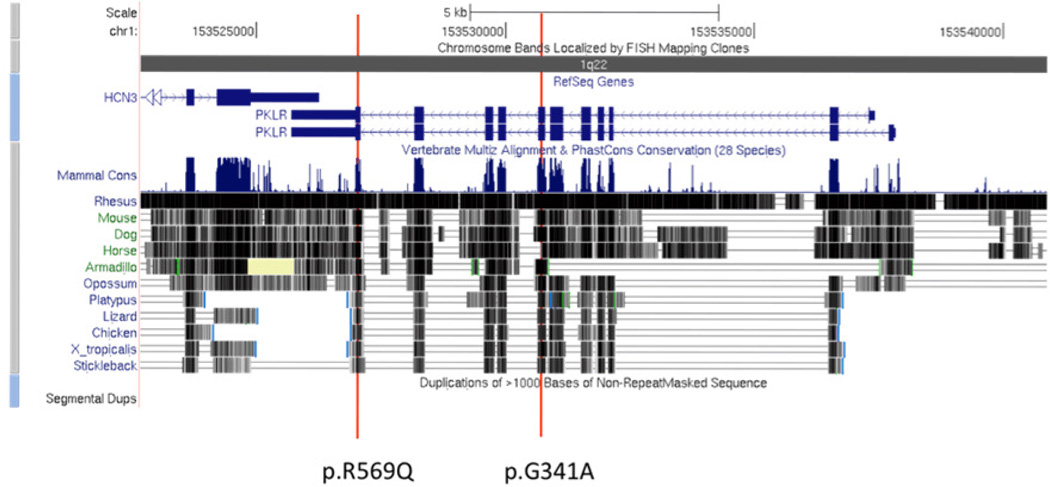
Genome browser shot of PKLR and the location of the two causal mutations. Each of the two mutations sits within an evolutionarily conserved region, and has been reported once in patients affected with PKLR deficiency.
Figure 6.

Illustration of protein domains in PKLU affected by the two mutations. Using the Protein Data Bank (PDB), we identified the protein domains in which the mutations were found to see how the mutations might affect functionality. We found that both mutations (indicated with red vertical lines in Figure 5) are near recorded binding sites (as indicated by the ‘Site Record’ entry). Arg569Gln is 5 amino acids from the PK allosteric regulator binding site for phosphoglyceric acid (PGA A 580) and Gly341Ala is 2 amino acids from the binding site for folate binding protein (FBP A 581). Additionally we note that a known SNP exists at amino acid 569.
The c.1706G>A exon 12 p.Arg569Gln mutation in PKLR was present only in the mother’s exome, whereas the c.1022G>C exon 8 p.Gly341Ala mutation was found only in the father’s exome, thus confirming an autosomal recessive mode of inheritance. Both mutations were confirmed by Sanger sequencing in the respective family members (Supplementary Figures 27–33). Neither mutation was found in 50 control Caucasian subjects sequenced in other projects; however, the c.1706G>A exon 12 p.Arg569Gln mutation was found rarely in the following datasets: 1) listed in dbSNP (version 130), 2) appeared in the 1000 Genomes project with a frequency of 0.1%, 3) seen in one out of 40 whole-genomes from the Complete Genomics Diversity Panel (http://www.completegenomics.com/sequence-data/download-data/), and 4) was seen six times in 5,680 exomes sequenced for other projects at BGI. On the other hand, the c.1022G>C exon 8 p.Gly341Ala mutation was completely unique, not being found in any of the above datasets. No other deleterious mutations in PKLR were found in patient 84060. These two mutations have been submitted and updated on the PKLR mutation database http://www.pklrmutationdatabase.com/.
After we had determined that the patient might be anemic due to PK deficiency, we obtained his permission to contact his hematologist and obtain his medical records. His records revealed a very recent medical work-up for the idiopathic hemolytic anemia, with the hematologist having concluded from a large panel of tests that the patient had pyruvate kinase deficiency, based on an abnormally low red blood cell pyruvate kinase enzyme activity. Further clinical evidence in favor of PK deficiency is the improved clinical outlook of the patient as a result of the splenectomy, which is frequently observed in patients with hemolytic anemia due to PK deficiency (Baronciani and Beutler, 1995). This clinical improvement was accompanied by an increased number of reticulocytes of 11.8% (presplenectomy: 3.3%), also typically seen in PK-deficiency (Mentzer et al., 1971).
Biochemical confirmation of a diagnosis of PK deficiency
Despite the clinical results, we decided to test more systematically for the effect of these mutations on pyruvate kinase enzymatic activity with the blood of 84060 and an age-matched Caucasian control, as these two mutations in PKLR had been reported 1–2 times in the literature in association with PK deficiency (Baronciani and Beutler, 1995; Fermo et al., 2005; van Wijk et al., 2009) but never in this particular combination (Table 2). Red blood cells of the patient showed lowered PK activity. In addition, the increased activities of hexokinase (HK) and glucose-6-phosphate dehydrogenase (G6PD), two other red blood cell age-dependent enzymes, were indicative of the presence of a red blood cell population of relatively young age. Therefore, since the activity of PK is strongly dependent on the age of the red blood cell, with the youngest cells showing the highest activity (Rijksen et al., 1990), PK activity should be considered to be reduced even more when taking into account the reticulocytosis in subject 84060 (11.8%).
Table 2.
Biochemical Assays of Enzyme Activities in the Patient Affected with Idiopathic Hemolytic Anemia Confirmed PKLR Deficiency.
| Patient 84060 | Control | Reference Values | |
|---|---|---|---|
| PK (U/gHb) | 3.3 L | 8.6 | 6.1 – 12.3 |
| HK (U/gHb) | 3.2 H | 1.1 | 0.8 – 1.5 |
| G6PD (U/gHb) | 15.8 H | 9.2 | 6.4 – 10.5 |
Abbreviations: PK, pyruvate kinase; HK, hexokinase; G6PD, glucose-6-phosphate dehydrogenase.
PK is a key enzyme in glycolysis. The sustenance of the red blood cell entirely depends on this pathway for the generation of metabolic energy (ATP). Both mutations, p.G341A and p.R569Q, are located at different subunit interfaces of the tetrameric enzyme, and the lowered activity of the enzyme leads ultimately to premature removal of the red blood cell from the circulation. Residues of the subunit interface are considered to be crucial for the enzyme’s response upon binding of its allosteric activator fructose-1,6-bisphosphate (Valentini et al., 2002). Both mutations were predicted to be deleterious by several complementary nsSNV scoring algorithms, including SIFT (Ng and Henikoff, 2003), PolyPhen version 2, LRT, PhyloP, and MutationTaster, using scores obtained from the dbNSFP database (Liu et al., 2011) (Table 3). Gly341 is a highly conserved residue at the interface between two A domains of opposing subunits in the tetrameric enzyme (A/A’ subunit interface). This is a conserved region of the protein involved in binding of phosphoenolpyruvate and ADP/ATP, and divalent cation binding (Munoz and Ponce, 2003). This mutation has been described only once in a PK-deficient Caucasian patient from the USA (Baronciani and Beutler, 1995). This latter patient carried the common p.R486W variant in trans and had received multiple blood transfusions, so the level of his or her PK deficiency could never be accurately measured. Rather, the authors deduced that this mutation was likely to have a negative effect on PK activity by conducting red cell PK assays on the patient’s mother and sister, although the actual data were not reported. Arg569 is a solvent exposed residue in the C/C’ subunit interface, located 5 residues from the C-terminal end of red blood cell PK. The arginine side chain is directed away from the surface and does not make specific hydrogen bonds or charge interactions. Introduction of a glutamine side chain at this position could cause unfavorable interactions with residues of the same or opposite subunit. However, there are also several putative conformations into which glutamine can mold that are predicted to affect its neighboring residues only to a minimal extent. We and others have previously described a patient carrying the R569Q change (Fermo et al., 2005; van Wijk et al., 2009). In one study the second mutation was never detected (Fermo et al., 2005) whereas in the other study two related patients (brother and sister) inherited the p.569Q variant together with a p.L374P variant. Both of these patients were PK-deficient although clinically mildly affected (van Wijk et al., 2009).
Table 3.
Bioinformatics Prediction on the Functional Impact of Two PKLR Mutations.
| Mutation | SIFT | PolyPhen 2 | PhyloP | LRT | MutationTaster |
|---|---|---|---|---|---|
| R569Q | 0.03 | 0.84 | 0.97 | D | D |
| G341A | 0 | 0.889 | 1 | D | D |
Note: A mutation is regarded as deleterious if the SIFT<0.05, or PolyPhen>0.85, or PhyloP>0.95, or MutationTaster/LRT prediction as “D” (deleterious).
Altogether, our results indicate that it is highly likely that the two identified changes in PKLR, in this particular combination, represent the main cause for the idiopathic hemolytic anemia as observed in this patient.
Discussion
Although this research study was initially begun as a method to discover a genetic cause for ADHD, during the course of the study we determined an unrelated explanation for this patient’s idiopathic anemia. We do not specifically refer to this as an “incidental finding,” because we did search for the cause of the anemia, once we knew that the research subject had this illness as well. Rather, we call this simply an unrelated finding. As a research test, our exome sequencing did not meet the standards necessary for a clinical test as required by Clinical Laboratory Improvement Amendments (CLIA). Therefore, we did not use our research results clinically to inform the research subject of his carrier status for two autosomal recessive mutations in PKLR. Instead, after consultation with our Institutional Review Board, we conveyed the results to the patient’s hematologist, so that the clinician could determine whether further CLIA-certified genetic testing might be warranted. It is important to note that one of the mutations was found with a low frequency (~0.1%) in the 1000 Genomes Project and dbSNP(v130), so these databases should not be used simply as a filter for any recessive mutations, given that heterozygote carriers of some mutations are likely to be found in these databases. In addition, a very rare mutation might only have manifestations in the context of compound heterozygosity with another modifying mutation, such as seen here with the low frequency (0.1%) mutation in PKLR on the other allele.
We acknowledge that any hematologist could have (and did) diagnose this patient with pyruvate kinase deficiency, based on a biochemical test. Importantly however, such results should be interpreted with care (Wijk et al., 2004) and, ideally, be confirmed by DNA analysis of PKLR. It was only in the research setting that we uncovered the fact that this is the only person currently known to carry both the p.G341A and R569Q changes, thus allowing us to conclude that this combination of mutations leads to impaired PK activity in such a way that the red blood cell life span is compromised, which is supported by the biochemical enzymatic assay results. As exome (and eventually whole genome) sequencing enters the clinic, there will be much opportunity to map and document human genetic variation, as illustrated here and in another recent paper related to hemoglobinopathies (Giardine et al., 2011).
One lesson from our analysis is that sequencing just four members of a family is likely insufficient to prove causality of any particular variant in a complex disease such as ADHD, particularly when the affected members of the family are closely related, such as the case here. A further problem is that some variants in neuropsychiatric illness might only have 50% penetrance (Mitchell and Porteous, 2011). Strategies looking for de novo mutations in sporadic cases of neuropsychiatric illness might prove more successful, although proving causation in a de novo case, rather than just association, will still be quite difficult. A very recent report in 20 autism de novo trios suggested a possible association with candidate variants in four probands out of the twenty trios, along with other possible variants for additional cases, but the study did not prove that any of the variants cause the illness (either by themselves or in aggregate with other unreported variants in the genome) (O’Roak et al., 2011). Also, given the possible oligogenic nature of autism, supported in part by recent copy number variant studies (Sanders et al., 2011), sequencing only a relatively small number of autism de novo trios (or even a large number of single cases) will not be able to prove causality of any discovered alleles, in light of penetrance issues and environmental influences. Rather, to provide more compelling evidence for causality, it is critically important to show segregation of the variants with phenotype in multiple family members in large pedigrees living in the same geographic region, including from affected distantly related individuals (such as cousins), so as to increase the power of detection and to help sort out the rate of penetrance. Given the extreme heterogeneity of ADHD and other neuropsychiatric conditions, we surmise that “intersection filtering” of the exomes (or even whole genomes) of completely unrelated individuals will be less successful, unlike the situation with rare Mendelian syndromes with monogenic causes and extremely high penetrance (Ng et al., 2010a; Rope et al., 2011), particularly given ongoing debates in regards to polygenic versus oligogenic modes of inheritance for these illnesses. We therefore believe it is critically important to control for environmental and ethnic/population stratification differences by focusing on large pedigrees living in the same geographic region.
We filtered some candidate rare variants against exome sequencing databases at BGI and Baylor to confirm whether they are truly rare variants. We did this because, despite our efforts to filter candidate variants by dbSNP and the 1000 Genomes Project, a large fraction of candidate variants were found in other exomes to be relatively common and are thus unlikely to be disease causing. This analysis suggests that the current catalog of less common variants in the 1000 Genomes Project is not comprehensive enough, perhaps due to the relatively low sequencing coverage and relatively small sample size. To identify genes for complex diseases from small families, given the large number of candidate variants, the analytical strategy depends heavily on a well-annotated catalog of rare variants, preferably collected from well-phenotyped research subjects. Recent studies suggest that there are 144,000 new variants per genome, after the first 15 individuals have been sequenced (Pelak et al., 2010). Considering the total number of variants from the 1000 Genomes Project (~30 million variants), it is clear that a catalog of rare/less common variants is far from complete, so pooling variant calls from multiple genome centers to produce a catalog of rare variants (MAF<0.1% or 0.5%) will help to take advantage of whole exome or whole genome sequencing data.
There are several limitations to our study. One of them is the need for additional sequencing to achieve a higher depth of coverage in the pedigree, as we found that exome sequencing at an average coverage of 20–30× only resulted in 10 or more reads per base pair in ~67–75% of the target region, whereas the emerging consensus at the large sequencing centers appears to be that it is better to aim for 20 or more reads per base pair in >80% of the target region.. Obviously, we plan to sequence in more depth and in more pedigrees and also to look for the identified rare variants in case-control studies, but this will require more time and funding to accomplish. Also, in the rapidly moving field of DNA sequencing, we have been grappling with the decision of whether it is worth the cost to sequence more of the exomes, or just to switch to whole genomes, given that we cannot predict a priori that the causative variants for this complex disorder will reside entirely in the exons. We have not conducted a formal cost-benefit analysis of our study, so we can only state here that if the cost of sequencing continues to plummet (and if capturing costs stay largely unchanged), it is likely that whole genome sequencing will soon become competitive with exome sequencing and even with the cost of currently marketed single gene mutational assays, which can sometimes range into thousands of US dollars. Also, another limitation of exome capture in general and thus also for our study is precisely the fact that not everything is efficiently captured, necessitating much more sequencing and expense to obtain enough data on low-capture regions as we have shown herein; this is not a problem with whole genome sequencing given that there is no capture step. It therefore seems likely that whole genome sequencing will replace exome sequencing and single gene diagnostic tests for Mendelian diseases at some point in the near future, assuming that the clinical standards for this can be established.
Whether genome sequencing can deliver any clinical benefits for complex traits is still largely unknown. The nature of these complex diseases, especially neuropsychiatric diseases, makes it possible that a single major-effect gene will not explain disease phenotypes in most subjects, with this point being highlighted at least theoretically in studies of complex phenotypes in model organisms (Buckler et al., 2009; Ehrenreich et al., 2010). It is therefore still an open question how much of the inheritance of neuropsychiatric disease will be explained by single rare variants in the context of an oligogenic model. Appropriate identification of multiple candidate variants, which together might lead to disease phenotypes in a potential epistatic and/or stochastic manner (incomplete penetrance) (i.e., a polygenic model), would certainly be more challenging in comparison to our recent effort to identify a single-gene cause for a new rare Mendelian syndrome, which we have tentatively named Ogden Syndrome, in honor of where that family resides (Rope et al., 2011).
For now, our results are meant to highlight the difficulties that our group (outside of a large sequencing center) has encountered thus far with trying to use exome sequencing in one small family to figure out the genetic basis of a complex neuropsychiatric illness. We anticipate that we and others will move to whole genome sequencing as the costs continue to plummet, given that it is obvious from the ENCODE (Birney et al., 2007; ENCODE Project Consortium et al., 2011) and other projects that there are a large number of non-coding variants, i.e., regions outside the exome, which could play a role in the pathogenesis of complex diseases. Besides the potential need to perform whole-genome sequencing, novel bioinformatics methods that can help functionally interpret genetic variants are of paramount importance to truly utilize next-generation sequencing in clinical practice for complex traits. In this regard, we and our collaborators have recently introduced a probabilistic search tool (VAAST) for identifying disease-causing variants in personal genome sequences (Rope et al., 2011; Yandell et al., 2011), and we are applying this and other new tools to the analysis of this and other datasets for rare Mendelian and complex disorders.
Supplementary Material
Acknowledgments
We thank Matthew Bainbridge and Richard Gibbs at Baylor for access to unpublished exome data. We also thank David A. Nix and the University of Utah Bioinformatics Shared Resource. This work was funded by University of Utah Department of Psychiatry funds to G.J.L. and R.R., along with funds from F.R. J.X. is supported by NIH/NHGRI K99HG005846. B.M. and M.Y. were supported by NHGRI 1RC2HG005619. K.W. and H.H. were funded by a Pilot/Methodological Study Award from NIH/NCRR Grant UL1 RR025774.
Footnotes
Author Contributions: The first two authors (G.J.L. and T.J.) should be regarded as joint first authors. G.J.L., K.W., T.J., W.W., P.M.B., J.X., L.T., M.C., Y.L., P.Z., Y.L., B.M., Z.W., and W.E.J. analyzed the exome and SNP genotyping data. T.J. and colleagues at BGI performed all Sanger sequencing and Sequenom genotyping. J.T.G. and K.W. performed CNV analysis. J.E., F.R., R.R., and G.J.L. contributed phenotyping data. R.V.W. and W.W.v.S. performed biochemical assays of PK and contributed expertise in the PK field. G.J.L. and K.W. conceived of the project, supervised all data analysis, and wrote the manuscript. All authors provided comments on the manuscript.
Disclosure
The authors do not have any competing financial conflicts of interest to report.
Contributor Information
Gholson J. Lyon, Department of Psychiatry, University of Utah, Salt Lake City, Utah 84132, USA and NYU Child Study Center, New York University, New York, New York 10016, USA.
Tao Jiang, BGI-Shenzhen, Shenzhen, 518083, China.
Richard Van Wijk, Clinical Chemistry and Haematology, University Medical Center Utrecht, Utrecht, 3583 CX, Netherlands.
Wei Wang, Department of Computer Science, New Jersey Institute of Technology, Newark, New Jersey 07102, USA.
Paul Mark Bodily, Brigham Young University, Provo, Utah, USA.
Jinchuan Xing, Eccles Institute of Human Genetics, Department of Human Genetics, University of Utah, Salt Lake City, Utah 84112, USA.
Lifeng Tian, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA.
Reid J. Robison, Utah Foundation for Biomedical Research, Salt Lake City, Utah 84107, USA.
Mark Clement, Brigham Young University, Provo, Utah, USA.
Yang Lin, BGI-Shenzhen, Shenzhen, 518083, China.
Peng Zhang, BGI-Shenzhen, Shenzhen, 518083, China.
Ying Liu, BGI-Shenzhen, Shenzhen, 518083, China.
Barry Moore, Eccles Institute of Human Genetics, Department of Human Genetics, University of Utah, Salt Lake City, Utah 84112, USA.
Joseph T. Glessner, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA.
Josephine Elia, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA.
Fred Reimherr, Department of Psychiatry, University of Utah, Salt Lake City, Utah 84132, USA.
Wouter W. van Solinge, Clinical Chemistry and Haematology, University Medical Center Utrecht, Utrecht, 3583 CX, Netherlands.
Mark Yandell, Eccles Institute of Human Genetics, Department of Human Genetics, University of Utah, Salt Lake City, Utah 84112, USA.
Hakon Hakonarson, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA and Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104, USA.
Jun Wang, BGI-Shenzhen, Shenzhen, 518083, China.
William Evan Johnson, Brigham Young University, Provo, Utah, USA.
Zhi Wei, Department of Computer Science, New Jersey Institute of Technology, Newark, New Jersey 07102, USA.
Kai Wang, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA and Zilkha Neurogenetic Institute, Department of Psychiatry and Preventive Medicine, University of Southern California, Los Angeles, California 90089, USA.
References
- 1000 Genomes Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barley K, Dracheva S, Byne W. Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr Res. 2009;112(1–3):54–64. doi: 10.1016/j.schres.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Baronciani L, Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J Clin Invest. 1995;95(4):1702–1709. doi: 10.1172/JCI117846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler E. Red Cell Metabolism: A Manual of Biochemical Methods. Orlando, Florida, USA: Grune & Stratton; 1984. [Google Scholar]
- Bilguvar K, Ozturk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoglu D, Tuysuz B, Caglayan AO, Gokben S, Kaymakcalan H, Barak T, Bakircioglu M, Yasuno K, Ho W, Sanders S, Zhu Y, Yilmaz S, Dincer A, Johnson MH, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467(7312):207–210. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroon DE, Larsson S, Lepak NK, Li H, Mitchell SE, Pressoir G, Peiffer JA, et al. The genetic architecture of maize flowering time. Science. 2009;325(5941):714–718. doi: 10.1126/science.1174276. [DOI] [PubMed] [Google Scholar]
- CDC. Increasing prevalence of parent-reported attention-deficit/hyperactivity disorder among children — United States 2003 and 2007. MMWR Morb Mortal Wkly Rep. 2010;59(44):1439–1443. [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- Clement NL, Snell Q, Clement MJ, Hollenhorst PC, Purwar J, Graves BJ, Cairns BR, Johnson WE. The GNUMAP algorithm: unbiased probabilistic mapping of oligonucleotides from next-generation sequencing. Bioinformatics. 2009;26(1):38–45. doi: 10.1093/bioinformatics/btp614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco GA, Loudianos G, Pes GM, Tolu F, Lepori MB, Barrocu M, Sechi GP. “Acquired” hepatocerebral degeneration in a patient heterozygote carrier for a novel mutation in ATP7B gene. Mov Disord. 2009;24(11):1706–1708. doi: 10.1002/mds.22659. [DOI] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, Mckenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36(19):e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464(7291):1039–1042. doi: 10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, D’Arcy M, Deberardinis R, Frackelton E, Kim C, Lantieri F, Muganga BM, Wang L, Takeda T, Rappaport EF, Grant SF, Berrettini W, Devoto M, Shaikh TH, Hakonarson H, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2010;15(6):637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleuch N, Feki I, Turki E, Miladi MI, Boukhris A, Damak M, Mhiri C, Chappuis E, Woimant F. A novel mutation in ATP7B gene associated with severe neurological impairment in Wilson’s disease. Rev Neurol (Paris) 2010;166(5):550–552. doi: 10.1016/j.neurol.2009.10.008. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Myers RM, Stamatoyannopoulos J, Snyder M, Dunham I, Hardison RC, Bernstein BE, Gingeras TR, Kent WJ, Birney E, Wold B, Crawford GE. A User’s Guide to the Encyclopedia of DNA Elements (ENCODE) PLoS Biol. 2011;9(4):e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y, Edvardson S, Hodges E, Zenvirt S, Thekkat P, Shaag A, Dor T, Hannon GJ, Elpeleg O. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21(5):658–664. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth R, Hole K, Karlsen OA, Ziegler M, Arnesen T, Lillehaug JR. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J Biol Chem. 2009;284(45):31122–31129. doi: 10.1074/jbc.M109.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermo E, Bianchi P, Chiarelli LR, Cotton F, Vercellati C, Writzl K, Baker K, Hann I, Rodwell R, Valentini G, Zanella A. Red cell pyruvate kinase deficiency: 17 new mutations of the PK-LR gene. Br J Haematol. 2005;129(6):839–846. doi: 10.1111/j.1365-2141.2005.05520.x. [DOI] [PubMed] [Google Scholar]
- Friedman-Weieneth JL, Doctoroff GL, Harvey EA, Goldstein LH. The Disruptive Behavior Rating Scale-Parent Version (DBRS-PV): Factor analytic structure and validity among young preschool children. J Atten Disord. 2009;13(1):42–55. doi: 10.1177/1087054708322991. [DOI] [PubMed] [Google Scholar]
- Giardine B, Borg J, Higgs DR, Peterson KR, Philipsen S, Maglott D, Singleton BK, Anstee DJ, Basak AN, Clark B, Costa FC, Faustino P, Fedosyuk H, Felice AE, Francina A, Galanello R, Gallivan MV, Georgitsi M, Gibbons RJ, Giordano PC, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat Genet. 2011;43(4):295–301. doi: 10.1038/ng.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualtieri CT, Johnson LG. Reliability and validity of a computerized neurocognitive test battery, CNS Vital Signs. Arch Clin Neuropsychol. 2006;21(7):623–643. doi: 10.1016/j.acn.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, Rolinski B, Iuso A, Biskup S, Schmidt T, Mewes HW, Wittig I, Meitinger T, Zeviani M, Prokisch H. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42(12):1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- Hyler SE, Skodol AE, Oldham JM, Kellman HD, Doidge N. Validity of the Personality Diagnostic Questionnaire-Revised: a replication in an outpatient sample. Compr Psychiatry. 1992;33(2):73–77. doi: 10.1016/0010-440x(92)90001-7. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Chong K, Mullikin JC, Biesecker LG. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86(5):743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian SM, Le-Niculescu H, Patel SD, Bertram D, Davis J, Dike C, Yehyawi N, Lysaker P, Dustin J, Caligiuri M, Lohr J, Lahiri DK, Nurnberger JI, Jr, Faraone SV, Geyer MA, Tsuang MT, Schork NJ, Salomon DR, Niculescu AB. Identification of blood biomarkers for psychosis using convergent functional genomics. Mol Psychiatry. 2011;16(1):37–58. doi: 10.1038/mp.2009.117. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Pfohl BM, Reynolds S, Clark LA, Battaglia M, Bellodi L, Cadoret R, Grove W, Pilkonis P, Links P. The Iowa Personality Disorder Screen: development and preliminary validation of a brief screening interview. J Pers Disord. 1999;13(1):75–89. doi: 10.1521/pedi.1999.13.1.75. [DOI] [PubMed] [Google Scholar]
- Leon AC, Olfson M, Portera L, Farber L, Sheehan DV. Assessing psychiatric impairment in primary care with the Sheehan Disability Scale. Int J Psychiatry Med. 1997;27(2):93–105. doi: 10.2190/T8EM-C8YH-373N-1UWD. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, Jin X, Li Q, Liu J, Liu X, Sparso T, Tang M, Wu H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42(11):969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human non-synonymous SNPs and their functional predictions. Human Mutation. 2011 Apr 21; doi: 10.1002/humu.21517. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant BK, Reimherr FW, Robison RJ, Olsen JL, Kondo DG. Methylphenidate transdermal system in adult ADHD and impact on emotional and oppositional symptoms. J Atten Disord. 2011;15(4):295–304. doi: 10.1177/1087054710365986. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, Depristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzer WC, Jr, Baehner RL, Schmidt-Schonbein H, Robinson SH, Nathan DG. Selective reticulocyte destruction in erythrocyte pyruvate kinase deficiency. J Clin Invest. 1971;50(3):688–699. doi: 10.1172/JCI106539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick E, Todorov A, Smalley S, Hu X, Loo S, Todd RD, Biederman J, Byrne D, Dechairo B, Guiney A, Mccracken J, Mcgough J, Nelson SF, Reiersen AM, Wilens TE, Wozniak J, Neale BM, Faraone SV. Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49(9):898–905. doi: 10.1016/j.jaac.2010.02.014. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, Porteous DJ. Rethinking the genetic architecture of schizophrenia. Psychol Med. 2011;41(1):19–32. doi: 10.1017/S003329171000070X. [DOI] [PubMed] [Google Scholar]
- Munoz ME, Ponce E. Pyruvate kinase: current status of regulatory and functional properties. Comp Biochem Physiol B Biochem Mol Biol. 2003;135(2):197–218. doi: 10.1016/s1096-4959(03)00081-2. [DOI] [PubMed] [Google Scholar]
- Murphy KR, Adler LA. Assessing attention-deficit/hyperactivity disorder in adults: focus on rating scales. J Clin Psychiatry 65 Suppl. 2004;65(Suppl 3):12–17. [PubMed] [Google Scholar]
- Neale BM, Medland S, Ripke S, Anney RJ, Asherson P, Buitelaar J, Franke B, Gill M, Kent L, Holmans P, Middleton F, Thapar A, Lesch KP, Faraone SV, Daly M, Nguyen TT, Schafer H, Steinhausen HC, Reif A, Renner TJ, et al. Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010a;49(9):906–920. doi: 10.1016/j.jaac.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP, Faraone SV, Nguyen TT, Schafer H, Holmans P, Daly M, Steinhausen HC, Freitag C, Reif A, Renner TJ, Romanos M, Romanos J, Walitza S, Warnke A, Meyer J, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010b;49(9):884–897. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010a;42(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010b;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Shianna KV, Ge D, Maia JM, Zhu M, Smith JP, Cirulli ET, Fellay J, Dickson SP, Gumbs CE, Heinzen EL, Need AC, Ruzzo EK, Singh A, Campbell CR, Hong LK, Lornsen KA, Mckenzie AM, Sobreira NL, Hoover-Fong JE, et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6(9) doi: 10.1371/journal.pgen.1001111. pii:e1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Kube DA, Whitaker TM, Graff JC, Palmer FB. Prevalence of developmental and behavioral disorders in a pediatric hospital. Pediatrics. 2009;123(3):e490–e495. doi: 10.1542/peds.2008-2750. [DOI] [PubMed] [Google Scholar]
- Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, Opitz JM, Levy-Lahad E, Klevit RE, King MC. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet. 2010;87(2):282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijksen G, Veerman AJ, Schipper-Kester GP, Staal GE. Diagnosis of pyruvate kinase deficiency in a transfusion-dependent patient with severe hemolytic anemia. Am J Hematol. 1990;35(3):187–193. doi: 10.1002/ajh.2830350309. [DOI] [PubMed] [Google Scholar]
- Rope AF, Wang K, Evjenth R, Xing J, Johnston JJ, Swensen JJ, Johnson WE, Moore B, Huff CD, Bird LM, Carey JC, Opitz JM, Stevens CA, Schank C, Fain HD, Robison R, Dalley B, Chin S, South ST, Pysher TJ, et al. The use of VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Gen. 2011 Jun 22; doi: 10.1016/j.ajhg.2011.05.017. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosler M, Retz W, Fischer R, Ose C, Alm B, Deckert J, Philipsen A, Herpertz S, Ammer R. Twenty-four-week treatment with extended release methylphenidate improves emotional symptoms in adult ADHD. World J Biol Psychiatry. 2010a;11(5):709–718. doi: 10.3109/15622971003624197. [DOI] [PubMed] [Google Scholar]
- Rosler M, Retz W, Retz-Junginger P, Stieglitz RD, Kessler H, Reimherr F, Wender PH. Attention deficit hyperactivity disorder in adults. Benchmarking diagnosis using the Wender-Reimherr adult rating scale. Nervenarzt. 2008;79(3):320–327. doi: 10.1007/s00115-007-2375-0. [DOI] [PubMed] [Google Scholar]
- Rosler M, Retz W, Stieglitz RD. Psychopathological rating scales as efficacy parameters in adult ADHD treatment investigations - benchmarking instruments for international multicentre trials. Pharmacopsychiatry. 2010b;43(3):92–98. doi: 10.1055/s-0029-1242819. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Davis Wright NR, Dhodapkar RM, Dicola M, Dilullo NM, et al. Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron. 2011;70(5):863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarling GS, Coates PW, Dass B, Macdonald CC. A family of splice variants of CstF-64 expressed in vertebrate nervous systems. BMC Mol Biol. 2009;10:22. doi: 10.1186/1471-2199-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TL, Klein MH, Benjamin LS. Validation of the Wisconsin Personality Disorders Inventory-IV with the SCID-II. J Pers Disord. 2003;17(3):173–187. doi: 10.1521/pedi.17.3.173.22150. [DOI] [PubMed] [Google Scholar]
- Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, Wang C, Bianchi P, Zanella A, Mattevi A. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002;277(26):23807–23814. doi: 10.1074/jbc.M202107200. [DOI] [PubMed] [Google Scholar]
- Van Solinge WW, Van Wijk R. Disorders of red cells resulting from enzyme abnormalities. In: Lichtman MA, Beutler E, Kaushansky KJ, Kipps TJ, Seligsohn U, Prchal JT, editors. Williams Hematology. 8th ed. New York, New York, USA: McGraw-Hill; 2010. pp. 647–674. [Google Scholar]
- Van Wijk R, Huizinga EG, Van Wesel AC, Van Oirschot BA, Hadders MA, Van Solinge WW. Fifteen novel mutations in PKLR associated with pyruvate kinase (PK) deficiency: structural implications of amino acid substitutions in PK. Hum Mutat. 2009;30(3):446–453. doi: 10.1002/humu.20915. [DOI] [PubMed] [Google Scholar]
- Van Wijk R, Van Solinge WW. The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. 2005;106(13):4034–4042. doi: 10.1182/blood-2005-04-1622. [DOI] [PubMed] [Google Scholar]
- Wang K, Bucan M, Grant SF, Schellenberg G, Hakonarson H. Strategies for genetic studies of complex diseases. Cell. 2010a;142(3):351–353. doi: 10.1016/j.cell.2010.07.025. author reply 353–355. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010b;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MF, Wender PH, Reimherr FW. The Wender Utah Rating Scale: an aid in the retrospective diagnosis of childhood attention deficit hyperactivity disorder. Am J Psychiatry. 1993;150(6):885–890. doi: 10.1176/ajp.150.6.885. [DOI] [PubMed] [Google Scholar]
- Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr599. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman MM, Olfson M, Gameroff MJ, Feder A, Fuentes M. A comparison of three scales for assessing social functioning in primary care. Am J Psychiatry. 2001;158(3):460–466. doi: 10.1176/appi.ajp.158.3.460. [DOI] [PubMed] [Google Scholar]
- Wijk R, Van Wesel AC, Thomas AA, Rijksen G, Van Solinge WW. Ex vivo analysis of aberrant splicing induced by two donor site mutations in PKLR of a patient with severe pyruvate kinase deficiency. Br J Haematol. 2004;125(2):253–263. doi: 10.1111/j.1365-2141.2004.04895.x. [DOI] [PubMed] [Google Scholar]
- Yandell M, Huff CD, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011 Jun 22; doi: 10.1101/gr.123158.111. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21(4):217–231. doi: 10.1016/j.blre.2007.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.