

Abstract
Partial hearing loss often results in enlarged representations of the remaining hearing frequency range in primary auditory cortex (AI). Recent studies have implicated certain types of synaptic plasticity in AI map reorganization in response to transient and long-term hearing loss. How changes in neuronal excitability and morphology contribute to cortical map reorganization is less clear. In the present study, we exposed adult rats to a 4-kHz tone at 123 dB, which resulted in increased thresholds over their entire hearing range. The threshold shift gradually recovered in the lower-frequency, but not the higher-frequency, range. As reported previously, two distinct zones were observed 10 days after the noise exposure, an enlarged lower-characteristic frequency (CF) zone displaying normal threshold and enhanced cortical responses and a higher-CF zone showing higher threshold and a disorganized tonotopic map. Membrane excitability of layer II/III pyramidal neurons increased only in the higher-CF, but not the lower-CF, zone. In addition, dendritic morphology and spine density of the pyramidal neurons were altered in the higher-CF zone only. These results indicate that membrane excitability and neuronal morphology are altered by long-term, but not transient, threshold shift. They also suggest that these changes may contribute to tinnitus but are unlikely to be involved in map expansion in the lower-CF zone.
Keywords: homeostatic plasticity, sensory map, intrinsic property, hearing lesions
hearing loss causes molecular, cellular, morphological, and functional changes at multiple levels of the auditory pathway (Dong et al. 2009, 2010; Engineer et al. 2011; Kraus et al. 2011; Milbrandt et al. 2000; Norena and Eggermont 2005; Popescu and Polley 2010; Sanes and Bao 2009; Schwaber et al. 1993; Seki and Eggermont 2003; Syka 2002; Vale and Sanes 2002; Wang et al. 2002, 2009; Willott et al. 1993). In the auditory cortex, for example, hearing loss modulates the expression of genes involved in neural transmitter metabolism, encoding synaptic receptors and ion channels, or required for synaptic and morphological plasticity and alters synaptic transmission (Kotak et al. 2005, 2008; Sarro et al. 2008; Scholl and Wehr 2008; Sun et al. 2008; Tan et al. 2007; Xu et al. 2007). These molecular and cellular events are hypothesized to underlie cortical map reorganization (Dorrn et al. 2010; Feldman 2009; Froemke et al. 2007) and pathological consequences of hearing loss such as hyperacusis and tinnitus (Brozoski et al. 2007; Eggermont 2006; Eggermont and Roberts 2004; Engineer et al. 2011; Yang et al. 2011). However, correlating the documented molecular and cellular changes with cortical reorganization and the associated functional consequences has been difficult because the results were often obtained in separate experiments and under different experimental conditions. Recent studies indicate that different forms of synaptic and sensory map plasticity may be induced under different procedures, for example, by transient versus long-term hearing loss (Yang et al. 2011).
In addition to changing synaptic transmission, hearing loss also increases intrinsic excitability and alters the morphology of cortical neurons (Bogart et al. 2011; Bose et al. 2010; Kotak et al. 2005; Rao et al. 2010). For example, bilateral cochlear removal in juvenile gerbils results in increased resting membrane potential, larger input resistance, and more sustained firing in primary auditory cortex (AI) layer II/III pyramidal neurons. Noise-induced deafness in juvenile rats also reduces the length of apical dendrites and spine density on basal and apical dendrites of AI pyramidal neurons (Bose et al. 2010). Interestingly, exposure in an acoustically enriched environment produced opposite effects—increased basal dendritic length and spine density (Bose et al. 2010). Although these results strongly suggest that morphological changes are involved in sensory cortical plasticity, their specific roles in hearing loss-induced map reorganization and behavioral pathology are still unclear.
In the present study, we examined intrinsic excitability and the morphology of pyramidal neurons in layers II/III of AI after noise-induced hearing loss (NIHL). As shown previously (Yang et al. 2011), NIHL results in two distinct zones in AI: the lower-characteristic frequency (CF) zone, which shows a transient threshold increase followed by long-term threshold decrease and response enhancement, and the zone previously representing higher frequencies, which shows a long-lasting threshold increase. Increased intrinsic excitability and altered neuronal morphology were observed in the higher-CF area with long-lasting threshold increase but not in the lower-CF area with transient threshold increase. Thus these types of structural and functional changes are unlikely to be involved in the map expansion and response enhancement observed in the lower-CF area. Instead, they may contribute to noise exposure-induced tinnitus.
MATERIALS AND METHODS
Noise exposure and auditory brain stem response recording.
Adult female rats (Sprague-Dawley, Charles River, Germantown, WI) weighing 250–300 g were used in all experiments. All experimental procedures were reviewed and approved by the UC Berkeley Animal Care and Use Committee. NIHL was performed by placing animals into a small wire mesh cage located in a sound attenuation chamber where a continuous pure tone of 4 kHz was played at 123 dB SPL through a loudspeaker for 7 h. The sound level was calibrated with a condenser microphone (¼ in., model 4135, Brüel and Kjær) before every NIHL procedure.
Hearing thresholds were assessed by auditory brain stem responses (ABRs) recorded before and after noise exposure. Animals were anesthetized with ketamine and xylazine (90 mg/kg and 5 mg/kg, respectively, ip) and were maintained on a heating pad. Tone pips (3-ms full-cycle sine waves at 1, 2, 4, 8, 16, and 32 kHz at 5-dB intensity steps from 70 to 0 dB) were delivered to the left ear at a rate of 19 times per second through a calibrated earphone (Stax) using BioSigRP software on a Tucker Davis Technology Sys3 recording rig (Alachua, FL). ABR signals were recorded with three electrodes subcutaneously inserted behind ipsilateral (−) and contralateral ears (ground) and at the vertex of the skull (+). The sound level that activated a minimal discernible response was defined as the auditory threshold. Thresholds were compared between before and after NIHL at each tested frequency to calculate threshold shift.
Surgical procedures and acute auditory mapping.
Three groups of rats (naive, 1 day after NIHL, 10 days after NIHL; 4 animals in each group) were mapped. Animals were anesthetized with pentobarbital (50 mg/kg) or both ketamine (90 mg/kg) and xylazine (5 mg/kg) throughout surgery and recording. Respiratory rate, heart rate, and corneal and hindpaw withdrawal reflexes were monitored to ensure that a surgical plane of anesthesia was uniformly maintained throughout the recording procedure. Lactated Ringer solutions were administered subcutaneously periodically throughout the experiment to ensure adequate hydration of the animals. The animal's body temperature was maintained at 37°C with a rectal probe and a homeothermic blanket (Harvard Apparatus). A cisternal drain was introduced to minimize brain edema. The temporal muscle, cranium, and dura overlying the auditory cortex were removed, and the exposed region was covered with silicone oil for electrophysiological recording. Multiunits were evenly sampled from the primary auditory cortex at a depth of ∼450 μm. Their responses to 25-ms tone pips of 51 frequencies (1 to 32 kHz, 0.1-octave spacing) and 8 sound pressure levels (0–70 dB SPL, 10-dB steps) were recorded to reconstruct the frequency-intensity receptive field (RF). Data analysis was done off-line with custom MATLAB programs. Briefly, response onset, peak, and offset latencies were determined with a peristimulus time histogram of the spikes and a two-standard deviation threshold. Spikes in the response window were used to reconstruct the RF, which were smoothed and thresholded at 25% of the maximum response magnitude. The frequency-intensity response area so defined matches well with the response area defined by experienced observers. Response threshold, tuning bandwidth, size of RF, and mean response magnitude (spikes per tone) were calculated from this response area.
Slice physiology.
Brain slices were prepared from a total of 19 animals (9 naive and 10 NIHL). After brain isolation, the cortical region was sectioned along the transverse plane into 300-μm slices with a vibrating microtome (Leica, VT1200). Lower- and higher-CF areas of AI were located by landmarks of adjacent hippocampal and thalamic structures as described previously (Yang et al. 2011). Two slices were collected from lower- and higher-CF areas of AI each from each hemisphere. They were immersed in oxygenated (95% O2-5% CO2) external solution (in mM: 140 NaCl, 4 KCl, 1.4 MgCl2, 10 d-glucose, 25 NaHCO3, 2.4 CaCl2, pH 7.4) in a tissue chamber for at least 1 h at room temperature before electrophysiological recording; the chamber was continuously perfused with external solution at a rate of 2 ml/min. A fixed-stage microscope (Olympus, BX50WI) equipped with differential interference contrast optics and a ×63 water-immersion objective was used to visualize individual neurons in the auditory cortex. We recorded from pyramidal neurons in layers II/III, 200–500 μm below pia and 2.3–2.7 mm dorsal to the dorsal end of the rhinal fissure. All recorded pyramidal neurons displayed a regular firing pattern in response to current steps.
Patch electrodes were fabricated from 1.5-mm-diameter borosilicate glass micropipettes and had impedance of 3–5 MΩ when back-filled with the internal solution (in mM: 117 K-gluconate, 13 KCl, 1.0 MgCl2·6H2O, 0.07 CaCl2·2H2O, 0.1 EGTA, 10 HEPES, 3 ATP-Mg, 0.3 GTP-Na, pH 7.3 and 290 mosM). The initial serial resistance typically ranged from 15 to 23 MΩ and remained stable during the recording session. Serial resistances were also continuously monitored with a brief voltage pulse. Recordings were accepted when cells had a serial resistance of 15–25 MΩ and <20% fluctuation during the recording session and a resting membrane potential of at least −68 mV (membrane potentials over −68 mV were rare significant outliers). All recordings were performed at room temperature (∼22.5°C) with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA).
A total of 87 layer II/III pyramidal neurons were recorded (43 from naive animals and 44 from animals 10 days after NIHL). Data were collected and analyzed with pCLAMP software (Molecular Devices).
Biocytin staining.
After full characterization of biophysical properties, all recorded cells were filled with biocytin and stained for morphological examination. Biocytin-filled cells were processed with a standard 3,3′-diaminobenzidine (DAB) staining protocol. After electrical recording and fixation, slices were rinsed in phosphate buffer (PB) and subsequently treated with hydrogen peroxide and permeabilized in a solution of 2% Triton X-100 for 1 h. The slices were then incubated in PB containing 1% avidin-biotinylated horseradish peroxidase complex (ABC, Vector Laboratories) for 2 h at 4°C. Excess ABC was removed by four rinses in PB. Slices were developed with 0.1% DAB and then embedded in Permount (Sigma). The cells displayed typical morphological features such as spiny apical and basal dendrites, pyramidal somas, and axonal projections to deep layers.
Dendrite and spine morphology.
Cells were digitally reconstructed in three dimensions with Neurolucida software at ×20 and ×40 magnification. Dendritic length and branch patterns were analyzed with applications within NeuroExplorer (Microbrightfield). All of the cells included in the analysis were chosen with the same criteria of recording loci for whole cell patch recording. Only cells in which the filling was optimal in both apical and basal dendrites, having no major truncation of dendrite branches during the slicing procedure, were used for anatomical analysis. For spine count, light microscope and high-magnification oil-immersion objectives (×100, 1.2 NA, Olympus) were used to count the number of dendritic spines. For each neuron, counting was performed on second principal apical dendrites (i.e., the split principal apical dendrites) and basal dendrites. Spines were counted only if they had both visible neck and head. All measurements and analyses were performed by experienced experimenters who were “blind” to the groups of the examined neurons. There were no differences in the spine numbers from the same segments of dendrites when counted by different experimenters.
Statistics.
Statistical differences were determined by ANOVA (Tukey) test unless otherwise stated; the significance level was set at 0.05. Data are presented as means ± SE.
RESULTS
NIHL results in two areas with transient and long-lasting threshold shift.
Hearing loss was induced by exposing the animals to a 4-kHz tone at 123 dB for 7 h (Yang et al. 2011). Significant hearing threshold shift in ABR was observed at all frequencies measured 1 day after NIHL. After 10 days, the threshold had completely recovered at the lower frequencies of 1 and 2 kHz (1 kHz: F1,22 =0.04, P = 0.83; 2 kHz: F1,22 =0.01, P = 0.91; Fig. 1A). By contrast, the threshold remained significantly elevated for the higher frequencies of 4, 8, 16, and 32 kHz (P < 0.001). Similar results were observed in responses of AI neurons. One day after NIHL, we found responses to high-intensity and lower-frequency (∼1 kHz) tones over an area that was larger than that of naive animals (Fig. 1C; at 1–2 kHz, threshold increase, F2,74 =8.96, P = 0.003; Fig. 1B, i and ii, area expansion, F1,6 =5.99, P = 0.05). In naive animals, high-intensity, low-frequency tones mainly activate lower-CF neurons in a smaller area in caudal AI but not higher-CF neurons in rostral AI (see Fig. 1D of Yang et al. 2011). Ten days later, response thresholds recovered for frequencies up to 2 kHz in animals with NIHL compared with naive animals (Fig. 1C; threshold × frequency ANOVA interaction, F4,295 =20.4, P < 0.001). The area representing these lower frequencies was enlarged in animals with NIHL (F1,6 =18.3, P = 0.0052). By contrast, the threshold increase for frequencies higher than 2 kHz persisted at 10 days (Fig. 1C). Neurons responded to those frequencies with higher threshold (ANOVA, main effect, F1,295 = 136.4, P < 0.001). Tonotopic organization of the frequency representations was also disrupted (Fig. 1Biii). These results indicate that NIHL results in two areas, one with transient and the other with long-lasting threshold increase. Although the animals with NIHL did not show increased thresholds in the lower frequency range, they could still have impaired cochlear output function (Kujawa and Liberman 2009).
Fig. 1.
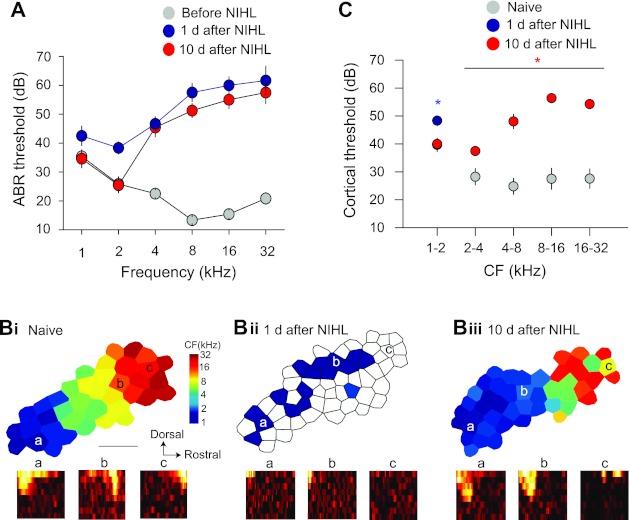
Map reorganization caused by transient and long-term hearing loss. A: noise exposure-induced transient and long-term threshold shift of the auditory brain stem responses (ABRs). ABRs were recorded in the same animals before and after noise-induced hearing loss (NIHL). B: cortical maps before (i) and 1 day (ii) and 10 days (iii) after NIHL. Bottom: receptive fields (RFs). The locations of the RFs are marked in the maps. Horizontal axis of the RF depicts frequencies from 1 to 32 kHz; vertical axis depicts intensity from 0 to 70 dB SPL. CF, characteristic frequency. C: response threshold of auditory cortical neurons. One day after NIHL, only lower-frequency responses were observed. Ten days after NIHL, neurons showed lower response threshold at lower frequencies. *P < 0.05.
It should be noted that the expansion of lower-frequency representations after NIHL (Fig. 1B) was possibly caused by two processes. First, the threshold increase in the higher frequency range would lower the CFs of some neurons that were previously tuned to the frequencies in the hearing loss range, resulting in apparent increase in the number of lower-CF neurons and enlarged lower-CF representations. This passive process is simply a result of NIHL and does not involve cortical plasticity. The second process is an increase in responses to lower-frequency sounds after higher-frequency hearing loss, which has been documented in a previous report in which the same NIHL protocol was used (see Fig. 1D of Yang et al. 2011). Such an active process likely involves cortical plasticity (Yang et al. 2011).
NIHL increase response latencies of cortical neurons.
Analysis of latencies of AI neurons indicates that both onset and peak latencies were increased by NIHL. Figure 2, A and B, show response latencies as functions of neuronal thresholds. Both naive and NIHL animals had neurons in the threshold range from 10 to 60 dB, which permitted statistical tests. A group × threshold two-way ANOVA indicated that both onset and peak latencies were significantly increased by NIHL (onset: main effect, F1,289 = 8.01, P < 0.005, no interaction; peak: main effect, F1,289 = 13.11, P < 0.0003, interaction, F5,289 = 5.79, P < 0.0001). Post hoc t-test with correction for multiple comparisons indicated that peak latencies were different between the two groups for neurons with thresholds from 20 to 50 dB (Fig. 2, A and B). The differences in onset latencies between naive and NIHL animals ranged from 2.16 to 3.94 ms across the four threshold bins (20–50 dB). The differences in peak latencies were much greater. For example, neurons in naive animals that had a 20-dB threshold had mean peak latency of 18.97 ± 0.78 ms, whereas mean peak latency of 20-dB-threshold neurons in NIHL animals was 40.00 ± 2.27 ms. Because lower-CF neurons tended to have longer latencies (linear regression: F1,309 = 19.86, P < 0.0001), the increase in latencies may be partly due to NIHL-induced increase in lower-CF neurons (blue dots in Fig. 2A; also see Fig. 1B).
Fig. 2.
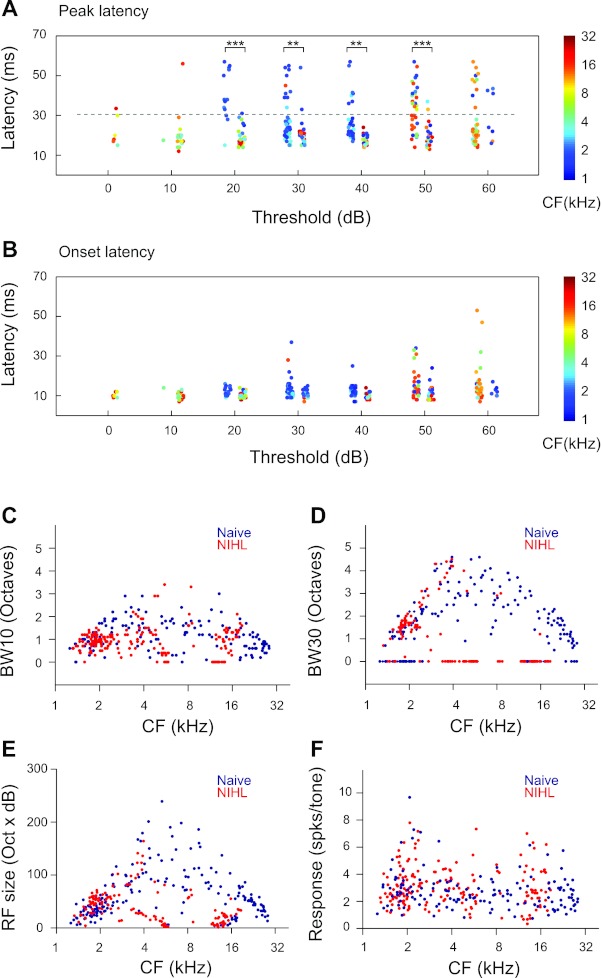
NIHL alters cortical neuron response properties. A: peak latencies of neurons in NIHL animals (left columns) and naive animals (right columns) as functions of the threshold of the neurons. Random jitters were added to the thresholds to facilitate visualization. Colors of the dots code for the neuronal CFs. B: onset latencies of the same neurons as in A. C: bandwidth at 10 dB above threshold (BW10) as functions of neuronal CFs. BW10 was not measured for neurons with a threshold higher than 20 dB. Those neurons were assigned a BW10 value of 0. D: bandwidth at 30 dB above threshold (BW30). BW30 was not measured for neurons with a threshold higher than 40 dB. Those neurons were assigned a BW10 value of 0. E: RF sizes as functions of neuronal CFs. The RF size was calculated as the number of frequency-intensity combinations in the range of 1–30 kHz at 0.1-octave steps and 0–70 dB at 10-dB steps. F: mean response magnitude, which is measured as the mean responses per tone for all the tones in the response area of the neuron. Unpaired t-tests: **P < 0.005, ***P < 0.0001.
Effects of NIHL on response properties of cortical neurons.
We characterized tuning bandwidth at 10 and 30 dB above the response threshold, referred to as BW10 and BW30, respectively. BW10 was reduced in NIHL animals compared with naive animals (F1,295 = 15.39, P < 0.0001; Fig. 2C), and the differences depended on CFs (group × CF interactions, F1,295 = 11.69, P = 0.0007). BW30 was also different between naive and NIHL animals (group, F1,196 = 32.60, P < 0.0001; group × CF, F1,196 =21.61, P < 0.0001). However, the difference appears to be due to an increase in bandwidth in the lower-CF (<4 kHz) neurons of the NIHL group, i.e., bandwidths of the NIHL group tended to track the upper bound of the bandwidth range of the naive group. In the above analysis, multiunits with thresholds higher than 40 dB were not included, because they did not allow measurement of BW30. They were arbitrarily assigned a bandwidth of 0 for plotting (Fig. 2D).
We also compared the RF sizes of cortical neurons (i.e., size of response areas in the RF) and found significant group × CF interaction (F1,316 = 9.72, P = 0.002; Fig. 2E) but no overall group differences. This is likely due to the fact that NIHL increased RF sizes in lower-CF neurons (<4 kHz; group effect, F1,160 =12.12, P = 0.0006) but reduced RF sizes in higher-CF neurons (≥4 kHz; group effect, F1,152 =35.93, P < 0.0001). Although NIHL altered response threshold, latency, tuning bandwidth, and receptive sizes, the mean response magnitude was not changed (F1,316 =42,400, P = 0.9984; Fig. 2F).
The finding of increased RF sizes in lower-CF neurons is consistent with increased tuning bandwidth in lower-CF neurons (Fig. 2D). Overall, the effects of NIHL on response threshold, latency, tuning bandwidth, and receptive size tended to be different for higher-CF versus lower-CF neurons.
Long-lasting but not transient NIHL increases intrinsic membrane excitability.
Previous studies have implicated the potentiation of excitatory synapses in the expansion of lower-frequency representations after NIHL and the depression of inhibitory synapses in the origin of hearing loss-induced tinnitus (Yang et al. 2011). To determine how intrinsic excitability may contribute to those processes, we performed whole cell clamp recording in AI pyramidal neurons in layers II/III and examined the resting membrane potential, input resistance, and action potential thresholds. In the lower-CF area, where NIHL induced map expansion, resting membrane potential, input resistance, and voltage and current thresholds were not significantly altered by the NIHL (resting membrane potential: naive −76.7 ± 0.4 mV, NIHL −76.4 ± 0.4 mV, F1,41 = 0.12, P = 0.72; input resistance: naive 78.2 ± 7.1 MΩ, NIHL 96.0 ± 6.5 MΩ, F1,41 = 3.37, P = 0.07; voltage threshold: naive −39.7 ± 0.8 mV, NIHL −40.6 ± 0.9 mV, F1,41 = 0.48, P = 0.49; current threshold: naive 262 ± 18 pA, NIHL 227 ± 14 pA, F1,41 = 2.07, P = 0.15; Fig. 3, B and C). In the higher-CF area, input resistance was significantly increased and current threshold was reduced. Resting membrane potential and voltage threshold were not changed (input resistance: naive 73.5 ± 3.7 MΩ, NIHL 107.9 ± 12.1 MΩ, F1,42 = 4.42, P = 0.04; current threshold: naive 288 ± 17 pA, NIHL 224 ± 23.7 pA, F1,42 = 5.85, P = 0.02; resting membrane potential: naive −77.4 ± 0.4 mV, NIHL −76.9 ± 0.3 mV, F1,42 = 1.14, P = 0.34; voltage threshold: naive −42.2 ± 0.7 mV, NIHL −41.5 ± 0.9 mV, F1,42 = 0.32, P = 0.57; Fig. 3, B and C). We also measured the number of spikes activated by a range of current amplitudes. While NIHL enhanced excitability in higher-CF neurons over the entire range of current amplitudes, there was no consistent change of intrinsic excitability in the lower-CF area (2-factor repeated-measures ANOVA group effect: higher-CF area F1,34 = 4.483, P = 0.042; lower-CF area F1,39 = 0.948, P = 0.336; Fig. 3D). These results demonstrate that hearing loss can induce homeostatic increase of intrinsic excitability in the hearing loss area of AI.
Fig. 3.
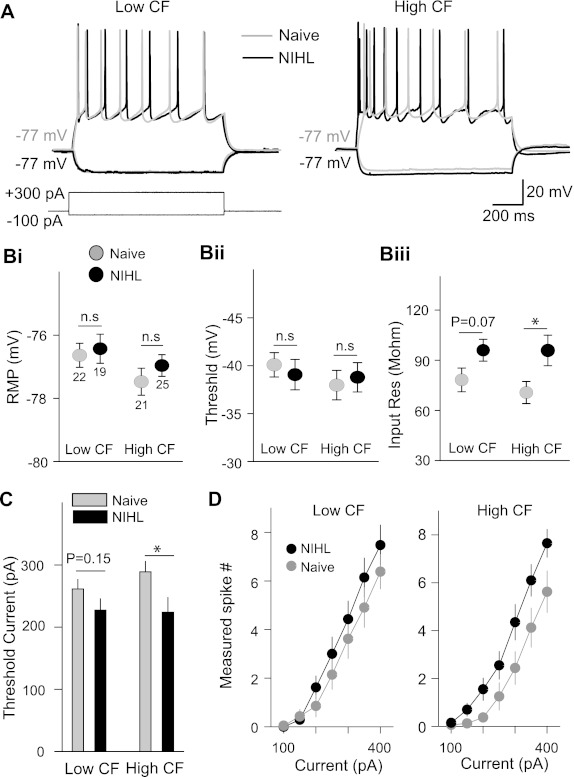
Long-term hearing loss increases intrinsic excitability of AI pyramidal neurons. A: voltage responses to current injection recorded from layer II/III pyramidal neurons of the auditory cortex. B: membrane properties of AI pyramidal neurons: resting membrane potential (RMP; i), response threshold (ii), and input resistance (iii). Only input resistance in the higher-CF area significantly increased. C: threshold of minimal current injected to activate spikes in the pyramidal neurons. D: number of spikes as a function of current amplitude. The number of spikes increased in the higher-CF area, but not the lower-CF area, of the NIHL animals (see text for statistical tests). *P < 0.05; n.s., not significant. Numbers in Bi indicates numbers of cells tested.
Long-lasting but not transient NIHL causes dendrite and spine reorganization.
After characterization of intrinsic properties, pyramidal neurons were filled with biocytin, stained, and analyzed (Fig. 4A). First, we measured dendrite length and branch number. In the lower-CF area, there was no difference in either apical or basal dendrite lengths between naive and NIHL animals (apical dendrite length: F1,34 = 0.21, P = 0.64; apical dendrite branch number: F1,34 = 1.6, P = 0.20; basal dendrite length: F1,34 = 0.190, P = 0.665; basal dendrite branch number: F1,34 = 0.193, P = 0.662; Fig. 4B). In the higher-CF area, we observed a redistribution of dendrite arborization in apical and basal dendrite compartments after the NIHL. The apical dendrite branch numbers were significantly increased, whereas basal dendrite length was decreased (apical dendrite length: naive 1,328 ± 77 μm, NIHL 1,574 ± 101 μm, F1,40 = 3.8, P = 0.05; apical dendrite branch number: naive 13.6 ± 0.8, NIHL 16.7 ± 0.8, F1,40 = 6.1, P = 0.02; basal dendrite length: naive 733 ± 65 μm, NIHL 547 ± 39 μm, F1,40 = 5.08, P = 0.02; basal dendrite branch number: naive 10.8 ± 1.1, NIHL 9.1 ± 0.7, F1,40 = 1.58, P = 0.21; Fig. 4B). However, the overall dendritic length was unaltered in both lower- and higher-CF areas (lower: F1,34 = 1.08, P = 0.30; higher: F1,40 = 0.08, P = 0.76; Fig. 4Dii).
Fig. 4.
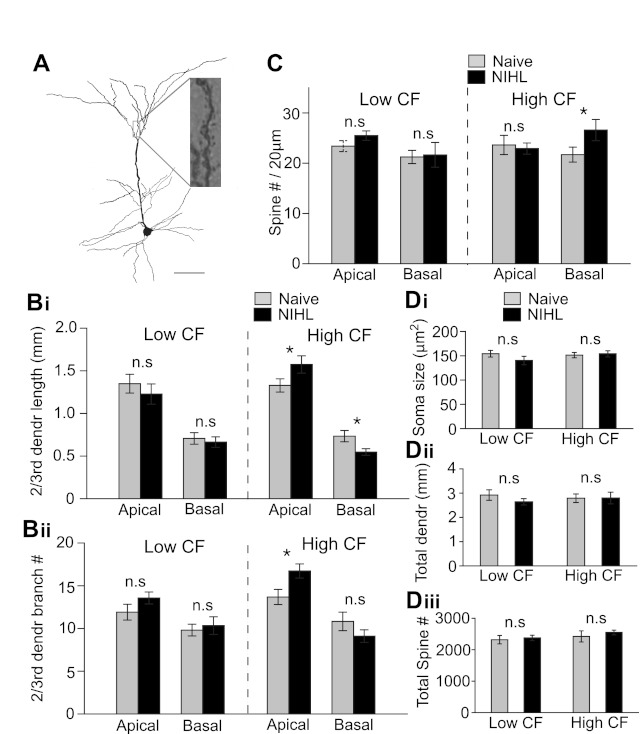
Long-term hearing loss alters dendritic morphology and spine density. A: an example neuron filled with biocytin to visualize dendrites and spines. Scale bar, 100 μm. B: length (i) and number of branches (ii) of 2nd- and 3rd-order dendrites. Basal dendritic length decreased in the higher-CF area after NIHL. Number of branches of apical dendrites increased in the higher-CF area. C: spine density increased on the basal dendrites in the higher-CF area of NIHL animals. D: soma size (i), total dendritic length (ii), and total number of spines (iii) were not changed by NIHL. *P < 0.05; n.s., not significant.
It was somewhat unexpected that dendritic morphology was unchanged in the lower-CF area, since increases in miniature excitatory postsynaptic current (mEPSC) frequency and amplitude were observed in the same area after NIHL (Yang et al. 2011). We further counted dendritic spine density of the pyramidal neurons. Significant changes were only observed in basal dendrites in the higher-CF area (higher-CF spine number: naive 1.09 ± 0.07/μm, NIHL 1.33 ± 0.09/μm, F1,13 = 4.3, P < 0.05; Fig. 4C). After considering dendritic lengths, we found no significant changes in total number of spines (lower: F1,13 = 0.02, P = 0.88; higher: F1,13 = 0.27, P = 0.60; Fig. 4Diii). There was no significant difference in soma sizes between naive and NIHL groups (lower: F1,34 = 0.91, P = 0.34; higher: F1,40 = 0.04, P = 0.83; Fig. 4Di). These morphological results indicate that transient and long-lasting NIHL in adulthood can differentially modulate dendrite geometry and spine distribution.
DISCUSSION
Hearing loss following cochlear ablation, noise exposure, and genetically determined processes has been shown to cause diverse effects at multiple stages in the central auditory pathway, including seemingly opposite effects in different experiments (Dong et al. 2009, 2010; Engineer et al. 2011; Kraus et al. 2011; Milbrandt et al. 2000; Norena and Eggermont 2005; Popescu and Polley 2010; Sanes and Bao 2009; Schwaber et al. 1993; Seki and Eggermont 2003; Syka 2002; Vale and Sanes 2002; Wang et al. 2002, 2009; Willott et al. 1993). For example, both increases and decreases of excitatory and inhibitory synaptic transmission have been reported (Kotak et al. 2005; Scholl and Wehr 2008; Yang et al. 2011). In addition, different effects on dendritic morphology and spine density were observed in noise-induced versus genetically induced hearing loss (Bogart et al. 2011; Bose et al. 2010). It is likely that different hearing lesion procedures used in those studies resulted in different degrees and durations of inner ear pathology and associated structural and functional changes (Kujawa and Liberman 2009). To better understand the relationship between structure and function, we need to quantify each of the NIHL-induced changes under the same experimental conditions. In the present study, we examined intrinsic excitability and dendritic morphology of the layer II/III pyramidal neurons, in order to correlate them with cortical map reorganization and perceptual consequences following the same NIHL procedure (Yang et al. 2011). NIHL differentially affected the lower- and higher-CF regions. In neurons that represented higher frequencies, the threshold shifts were long-lasting, and cortical neurons became unresponsive to moderate-intensity tones after NIHL. Tonotopy was also disrupted in the higher-CF region of the cortical map. We observed an increase in intrinsic membrane excitability and diverse changes in dendritic morphology. These changes may contribute to tinnitus perception, which has been hypothesized to originate in neurons representing hearing loss frequencies (Yang et al. 2011). The lower-CF area expanded significantly after NIHL but showed no changes in intrinsic membrane excitability and dendritic morphology. Thus the observed changes in intrinsic membrane excitability and neuronal morphology are unlikely to be involved in expansion of lower-frequency map representations. Since we only examined these properties in layer II/III pyramidal neurons, we cannot exclude the possibility that cortical sensory map reorganization involves intrinsic excitability or neuronal morphology changes in other types of neurons or in other cortical layers. It should be noted that our in vitro experiments examined pyramidal neurons in layers II/III, where robust synaptic, intrinsic morphological and sensory map plasticity have been observed (Diamond et al. 1994; Hickmott 2005; Lendvai et al. 2000; Maravall et al. 2004; Petersen et al. 2004), whereas the in vivo cortical responses were recorded presumably from layers III/IV, where experience-dependent cortical map reorganization is often reported (Yang et al. 2011). Thus the observed cellular and morphological changes might underlie map reorganizations occurring in more superficial layers, which were not examined here.
In the higher-CF neurons, we observed a redistribution of pyramidal dendrites—shrinkage of basal and expansion of apical dendrites. The reduction of basal dendritic length is likely due to the loss of sensory inputs via the thalamocortical synapses primarily located on the those dendrites. The expanded apical dendrites and unaltered spine density would result in more spines on apical dendrites, which may represent a compensatory increase of corticocortical connectivity. However, the total length of dendrites and number of spines of individual pyramidal neurons were not changed by hearing loss in either the lower- or higher-CF areas (Fig. 4). The discrepancy between our results and the previously reported decrease of apical dendritic length and unchanged basal dendritic length after NIHL (Bose et al. 2010) is likely due to differences in the ages of animals (>90 days in our experiments and <30 days in the previous report) and the severity of the NIHL.
The altered intrinsic properties and morphology of cortical pyramidal neurons are likely to be separate responses to NIHL. The increases in input resistance and neuronal excitability observed in the present study are global effects. By contrast, redistributions of dendrites and spines require polarized apical and basal cues, such as imbalanced thalamocortical versus corticocortical inputs. In addition, because the total dendritic length and the number of spines did not change, their redistributions are unlikely to change global neuronal properties such as cell surface area and total synaptic input, factors that could influence input resistance and neuronal excitability. Instead, the observed increase in input resistance and neuronal excitability may be due to altered expression of voltage-gated ion channels or hearing loss-induced reduction in tonic inhibitory currents in layer II/III pyramidal neurons (Yang et al. 2011).
In the present study, we observed changes in neuronal morphology and increased intrinsic excitability in layer II/III pyramidal neurons in the higher-CF area but not the lower-CF area. Together with the previously reported decrease of cortical inhibitory synaptic transmission in the higher-CF area, our results indicate that multiple mechanisms may be involved in homeostatic increase of neuronal activity after hearing loss. These mechanisms may contribute to hearing loss-induced sensory pathology such as hyperacusis and tinnitus (Sun et al. 2012; Yang et al. 2011).
GRANTS
This work was supported by the American Tinnitus Association and by National Institute on Deafness and Other Communicative Disorders Grant DC-009259.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.Y. and S.B. conception and design of research; S.Y. and W.S. performed experiments; S.Y. and W.S. analyzed data; S.Y. interpreted results of experiments; S.Y. prepared figures; S.Y. and S.B. drafted manuscript; S.Y. and S.B. edited and revised manuscript; S.Y., W.S., and S.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Chris Xu for help with morphological analysis and Robert Gibboni for comments on the manuscript.
REFERENCES
- Bogart LJ, Levy AD, Gladstone M, Allen PD, Zettel M, Ison JR, Luebke AE, Majewska AK. Loss of prestin does not alter the development of auditory cortical dendritic spines. Neural Plast 2011: 305621, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose M, Munoz-Llancao P, Roychowdhury S, Nichols JA, Jakkamsetti V, Porter B, Byrapureddy R, Salgado H, Kilgard MP, Aboitiz F, Dagnino-Subiabre A, Atzori M. Effect of the environment on the dendritic morphology of the rat auditory cortex. Synapse 64: 97–110, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozoski TJ, Spires TJ, Bauer CA. Vigabatrin, a GABA transaminase inhibitor, reversibly eliminates tinnitus in an animal model. J Assoc Res Otolaryngol 8: 105–118, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ME, Huang W, Ebner FF. Laminar comparison of somatosensory cortical plasticity. Science 265: 1885–1888, 1994 [DOI] [PubMed] [Google Scholar]
- Dong S, Mulders WH, Rodger J, Robertson D. Changes in neuronal activity and gene expression in guinea-pig auditory brainstem after unilateral partial hearing loss. Neuroscience 159: 1164–1174, 2009 [DOI] [PubMed] [Google Scholar]
- Dong S, Mulders WH, Rodger J, Woo S, Robertson D. Acoustic trauma evokes hyperactivity and changes in gene expression in guinea-pig auditory brainstem. Eur J Neurosci 31: 1616–1628, 2010 [DOI] [PubMed] [Google Scholar]
- Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC. Developmental sensory experience balances cortical excitation and inhibition. Nature 465: 932–936, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont JJ. Cortical tonotopic map reorganization and its implications for treatment of tinnitus. Acta Otolaryngol Suppl (Stockh) 556: 9–12, 2006 [DOI] [PubMed] [Google Scholar]
- Eggermont JJ, Roberts LE. The neuroscience of tinnitus. Trends Neurosci 27: 676–682, 2004 [DOI] [PubMed] [Google Scholar]
- Engineer ND, Riley JR, Seale JD, Vrana WA, Shetake JA, Sudanagunta SP, Borland MS, Kilgard MP. Reversing pathological neural activity using targeted plasticity. Nature 470: 101–104, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci 32: 33–55, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Merzenich MM, Schreiner CE. A synaptic memory trace for cortical receptive field plasticity. Nature 450: 425–429, 2007 [DOI] [PubMed] [Google Scholar]
- Hickmott PW. Changes in intrinsic properties of pyramidal neurons in adult rat S1 during cortical reorganization. J Neurophysiol 94: 501–511, 2005 [DOI] [PubMed] [Google Scholar]
- Kotak VC, Fujisawa S, Lee FA, Karthikeyan O, Aoki C, Sanes DH. Hearing loss raises excitability in the auditory cortex. J Neurosci 25: 3908–3918, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Takesian AE, Sanes DH. Hearing loss prevents the maturation of GABAergic transmission in the auditory cortex. Cereb Cortex 18: 2098–2108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus KS, Ding D, Jiang H, Lobarinas E, Sun W, Salvi RJ. Relationship between noise-induced hearing-loss, persistent tinnitus and growth-associated protein-43 expression in the rat cochlear nucleus: does synaptic plasticity in ventral cochlear nucleus suppress tinnitus? Neuroscience 194: 309–325, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci 29: 14077–14085, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404: 876–881, 2000 [DOI] [PubMed] [Google Scholar]
- Maravall M, Koh IY, Lindquist WB, Svoboda K. Experience-dependent changes in basal dendritic branching of layer 2/3 pyramidal neurons during a critical for developmental plasticity in rat barrel cortex. Cereb Cortex 14: 655–664, 2004 [DOI] [PubMed] [Google Scholar]
- Milbrandt JC, Holder TM, Wilson MC, Salvi RJ, Caspary DM. GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hear Res 147: 251–260, 2000 [DOI] [PubMed] [Google Scholar]
- Norena AJ, Eggermont JJ. Enriched acoustic environment after noise trauma reduces hearing loss and prevents cortical map reorganization. J Neurosci 25: 699–705, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, Brecht M, Hahn TT, Sakmann B. Synaptic changes in layer 2/3 underlying map plasticity of developing barrel cortex. Science 304: 739–742, 2004 [DOI] [PubMed] [Google Scholar]
- Popescu MV, Polley DB. Monaural deprivation disrupts development of binaural selectivity in auditory midbrain and cortex. Neuron 65: 718–731, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao D, Basura GJ, Roche J, Daniels S, Mancilla JG, Manis PB. Hearing loss alters serotonergic modulation of intrinsic excitability in auditory cortex. J Neurophysiol 104: 2693–2703, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Bao S. Tuning up the developing auditory CNS. Curr Opin Neurobiol 19: 188–199, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarro EC, Kotak VC, Sanes DH, Aoki C. Hearing loss alters the subcellular distribution of presynaptic GAD and postsynaptic GABAA receptors in the auditory cortex. Cereb Cortex 18: 2855–2867, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl B, Wehr M. Disruption of balanced cortical excitation and inhibition by acoustic trauma. J Neurophysiol 100: 646–656, 2008 [DOI] [PubMed] [Google Scholar]
- Schwaber MK, Garraghty PE, Kaas JH. Neuroplasticity of the adult primate auditory cortex following cochlear hearing loss. Am J Otol 14: 252–258, 1993 [PubMed] [Google Scholar]
- Seki S, Eggermont JJ. Changes in spontaneous firing rate and neural synchrony in cat primary auditory cortex after localized tone-induced hearing loss. Hear Res 180: 28–38, 2003 [DOI] [PubMed] [Google Scholar]
- Sun W, Deng A, Jayaram A, Gibson B. Noise exposure enhances auditory cortex responses related to hyperacusis behavior. Brain Res. (February 9, 2012). doi:10.1016/j.brainres.2012.02.008 [DOI] [PubMed] [Google Scholar]
- Sun W, Zhang L, Lu J, Yang G, Laundrie E, Salvi R. Noise exposure-induced enhancement of auditory cortex response and changes in gene expression. Neuroscience 156: 374–380, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J. Plastic changes in the central auditory system after hearing loss, restoration of function, and during learning. Physiol Rev 82: 601–636, 2002 [DOI] [PubMed] [Google Scholar]
- Tan J, Ruttiger L, Panford-Walsh R, Singer W, Schulze H, Kilian SB, Hadjab S, Zimmermann U, Kopschall I, Rohbock K, Knipper M. Tinnitus behavior and hearing function correlate with the reciprocal expression patterns of BDNF and Arg3.1/arc in auditory neurons following acoustic trauma. Neuroscience 145: 715–726, 2007 [DOI] [PubMed] [Google Scholar]
- Vale C, Sanes DH. The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur J Neurosci 16: 2394–2404, 2002 [DOI] [PubMed] [Google Scholar]
- Wang H, Turner JG, Ling L, Parrish JL, Hughes LF, Caspary DM. Age-related changes in glycine receptor subunit composition and binding in dorsal cochlear nucleus. Neuroscience 160: 227–239, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ding D, Salvi RJ. Functional reorganization in chinchilla inferior colliculus associated with chronic and acute cochlear damage. Hear Res 168: 238–249, 2002 [DOI] [PubMed] [Google Scholar]
- Willott JF, Aitkin LM, McFadden SL. Plasticity of auditory cortex associated with sensorineural hearing loss in adult C57BL/6J mice. J Comp Neurol 329: 402–411, 1993 [DOI] [PubMed] [Google Scholar]
- Xu H, Kotak VC, Sanes DH. Conductive hearing loss disrupts synaptic and spike adaptation in developing auditory cortex. J Neurosci 27: 9417–9426, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Weiner BD, Zhang LS, Cho SJ, Bao S. Homeostatic plasticity drives tinnitus perception in an animal model. Proc Natl Acad Sci USA 108: 14974–14979, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]