

Abstract
Activity of neurons in the dorsal motor nucleus of the vagus nerve (DMV) is closely regulated by synaptic input, and regulation of that input by glutamate receptors on presynaptic terminals has been proposed. Presynaptic N-methyl-d-aspartic acid (NMDA) receptors have been identified in a number of brain regions and act to modulate neurotransmitter release, but functional presynaptic NMDA receptors have not been adequately studied in the DMV. This study identified the presence and physiological function of presynaptic NMDA receptors on synaptic input to DMV neurons. Whole-cell patch-clamp recordings from DMV neurons in acute slices from mice revealed prevalent miniature excitatory postsynaptic currents, which were significantly increased in frequency, but not amplitude, by application of NMDA. Antagonism of NMDA receptors with dl-2-amino-5-phosphonopentanoic acid (100 μM) resulted in a decrease in miniature excitatory postsynaptic current frequency and an increase in the paired pulse ratio of responses following afferent stimulation. No consistent effects of presynaptic NMDA receptor modulation were observed on GABAergic inputs. These results suggest that presynaptic NMDA receptors are present in the dorsal vagal complex and function to facilitate the release of glutamate, preferentially onto DMV neurons tonically, with little effect on GABA release. This type of presynaptic modulation represents a potentially novel form of glutamate regulation in the DMV, which may function to regulate glutamate-induced activity of central parasympathetic circuits.
Keywords: autonomic, glutamate, parasympathetic, plasticity, presynaptic modulation
the dorsal vagal complex (DVC) in the caudal brain stem regulates central parasympathetic reflexive and integrative functions, including those of the digestive system. Viscerosensory inputs and centrally originating afferents form a synapse in the nucleus of the solitary tract (NTS), where information is integrated and transmitted to other autonomic centers, including the dorsal motor nucleus of the vagus nerve (DMV). The axons of DMV motorneurons project to postganglionic neurons controlling peripheral viscera (Browning and Travagli 2011; Kalia and Mesulam 1980a, 1980b; Travagli 2007; Travagli et al. 2006).
Glutamate is the principal excitatory neurotransmitter in the DMV, activating both N-methyl-d-aspartic acid (NMDA) and non-NMDA ionotropic receptors (Travagli et al. 1991). On activation by glutamate, NMDA receptors contribute to membrane depolarization and Ca2+-dependent intracellular signaling by increasing the conductance of Na+ and Ca2+ (Kandel et al. 2000). Activation of NMDA receptors by glutamate is also voltage dependent, due to voltage-dependent blockade of the channel by Mg2+ (Mayer et al. 1984). NMDA receptors are generally thought to exist as heterotetramers composed of an obligatory NR1 subunit, in addition to a combination of NR2A-D and/or NR3A-B receptor subunits (Chatterton et al. 2002; Paoletti and Neyton 2007). While NMDA receptors typically contribute to postsynaptic conductances, their role as presynaptic receptors that modulate neurotransmitter release from synaptic terminals is receiving increased attention (Corlew et al. 2008; Engelman and MacDermott 2004; Ma and Hargreaves 2000; MacDermott et al. 1999). Similar to postsynaptic receptors, various subunit combinations have been identified at preterminal receptors (Bidoret et al. 2009; Woodhall et al. 2001; Zhang et al. 2009). Their functional significance as facilitatory autoreceptors or heteroreceptors has been established in several brain regions, increasing glutamate or GABA release when activated (Berretta and Jones 1996; Duguid and Smart 2004). Presynaptic glutamate receptors, possibly including NMDA receptors, contribute to glutamatergic heterosynaptic facilitation of GABA release onto DMV neurons (Derbenev et al. 2006), but the subtype of glutamate receptor mediating this enhanced release is not known. Metabotropic glutamate receptors have been identified previously to contribute to modulation of both excitatory and inhibitory neurotransmission in the DMV (Browning and Travagli 2007). These findings are consistent with the hypothesis that glutamate released in the DMV can regulate synaptic transmission by acting at receptors on afferent terminals. Presynaptic NMDA receptors, and their potential role in contributing to facilitation of fast excitatory and/or inhibitory neurotransmission, have not been adequately studied in the DVC. Synaptic input to DMV neurons is altered in disease states affecting visceral function, underscoring the importance of understanding synaptic control of these neurons (Zsombok et al. 2011). This study aimed to identify the presence of presynaptic NMDA receptors in the DMV and their functional relevance as modulators of excitatory and/or inhibitory neurotransmission.
METHODS
Animals.
Male CD-1 mice (Harlan, Indianapolis, IN), 4–10 wk of age, were housed under a standard 12:12-h light-dark cycle, with food and water provided without restriction. All animals were treated and cared for in accordance with National Institutes of Health guidelines, and all procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
Brain stem slice preparation.
Whole-cell patch-clamp recordings were made using brain stem slices prepared from mice, as described previously (Gao et al. 2009; Glatzer et al. 2007; Zsombok et al. 2011). Mice were deeply anesthetized by halothane inhalation to effect and then decapitated. The brain was removed and blocked on an ice-cold stand, and the brain stem was glued to a sectioning stage. Transverse (i.e., coronal) brain stem slices (300 μm) from ∼300 μm rostral to area postrema (AP) to the caudal edge of AP containing the DVC were made in cold (0–2°C), oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid (ACSF) using a vibrating microtome (Vibratome Series 1000; Technical Products, St. Louis, MO). The ACSF contained the following (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl2, 1.4 NaH2PO4, 26 NaHCO3, 11 glucose (pH 7.2–7.4; osmolality 290–315 mosmol/kg). Slices were then incubated for ≥1 h in warm (32–35°C) oxygenated ACSF. For recording, a single brain slice was transferred to a chamber mounted on a fixed stage under an upright microscope (model BX51WI; Olympus, Melville, NY), where it was continually superfused with warmed (30–33°C) oxygenated ACSF. Tetrodotoxin (TTX; 1–2 μM; Tocris Bioscience, Ellisville, MO) was bath applied to record action potential-independent (i.e., miniature) excitatory and inhibitory postsynaptic currents (i.e., mEPSCs and mIPSCs). For specific experiments, dl-2-amino-5-phosphonopentanoic acid (AP-5; 100 μM), NMDA (15 μM), Ro 25–6981 (1 μM), ZnCl2 (5 μM), or picrotoxin (PTX; 100 μM; Sigma, Ellisville, MO) were added to ACSF.
Electrophysiological recording.
Whole-cell voltage-clamp recordings were obtained in the DMV using recording pipettes pulled from borosilicate glass (open tip resistance of 3–5 MΩ; King Precision Glass, Claremont, CA). The pipette solution for most recordings contained the following (in mM): 130–140 Cs-gluconate, 10 HEPES, 1 NaCl, 1 CaCl2, 3 CsOH, 5 EGTA, and 2 Mg2+-ATP. In addition, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801; 1 mM) was added to the pipette solution to block postsynaptic NMDA receptors in the recorded cell where indicated. Pipettes were back-filled with ACSF before addition of MK-801-containing pipette solution. Inadvertent ejection of MK-801 during acquisition of patch-clamp recordings was a potential concern, as unappreciated leakage might result in inconsistent blockade of receptors, especially during antagonist application experiments, so MK-801 was added to the recording pipette for agonist application experiments only. Intracellular Cs+ was used as the primary cation carrier in voltage-clamp recordings to block K+ currents, including GABAB receptor-mediated currents in the recorded neuron. Neurons in the DMV were targeted for recording under a ×40 water-immersion objective (numerical aperture = 0.8) with infrared-differential interference contrast optics, as previously described (Gao et al. 2009; Glatzer et al. 2007; Zsombok et al. 2011). Electrophysiological signals were obtained using a Multiclamp 700B amplifier (Molecular Devices, Union City, CA), low-pass filtered at 2 or 3 kHz, digitized, and recorded onto a computer (Digidata 1440A, Molecular Devices) using pClamp 10.2 or 10.3 software (Molecular Devices). Seal resistance was typically 2–5 GΩ, and series resistance, measured from brief voltage steps applied through the recording pipette (5 mV, 5 ms), was typically <25 MΩ (mean = 13.03 ± 0.86 MΩ) and monitored periodically during the recording. Recordings were discarded if series resistance changed by >20% over the course of the experiment.
Electrical stimulation.
Electrical stimulation was performed using a platinum-iridium concentric bipolar electrode (125 μm diameter, FHC, Bowdoinham, ME) placed in the medial NTS (Browning and Travagli 2009; Glatzer et al. 2007). A minimum of 15 paired current pulses (2–100 μA; 300 μs; A.M.P.I., Jerusalem, Israel) at interpulse intervals of 30 ms and a cycle rate of 0.1 Hz were administered to the NTS, and responses in DMV neurons voltage-clamped at −80 mV were recorded and analyzed for current amplitudes before, during, and after the application of AP-5.
Data analysis.
Spontaneous and miniature postsynaptic currents (PSCs) were analyzed with MiniAnalysis (Synaptosoft) to measure the peak amplitude, frequency, and decay time constant. Spontaneous or miniature synaptic events (minimum 65; typically 2-min continuous recording) were measured within a recording under each condition. Evoked responses were analyzed using pClamp. Intra-assay analysis of drug-evoked changes in spontaneous and miniature PSC frequency and amplitude within a recording were assessed using a Kolmogorov-Smirnov test. Pooled results of single comparisons of drug effects (i.e., before and after a single drug treatment) were analyzed using a paired, two-tailed t-test. For electrical stimulation, peak amplitudes from a minimum of 15 evoked responses were averaged and measured for each treatment using pClamp programs and analyzed using a paired, two-tailed Student's t-test. Paired-pulse response ratios, taken as the ratio of the amplitude of the second to the first excitatory PSC (EPSC) response to afferent stimulation, were analyzed using the nonparametric Wilcoxon signed-rank test. Statistical significance for all measures was set at P < 0.05. Statistical measurements were performed with Microsoft Excel (Microsoft, Redmond, WA) or Prism (GraphPad Software, La Jolla, CA). Numbers were expressed as means ± SE.
RESULTS
Blocking NMDA receptors decreased tonic excitatory input to DMV neurons.
Electrophysiological recordings were made in voltage-clamp mode in the presence of PTX (100 μM) and TTX (1–2 μM) using Cs+ intracellularly to block K+ currents. At a membrane potential of −80 mV, postsynaptic NMDA receptors in DMV neurons are largely under voltage-dependent Mg2+ blockade, reducing any contribution to EPSCs of NMDA receptor activation. By also blocking fast inhibitory PSCs (IPSCs) with PTX, action potential propagation with TTX, and K+ channels with intracellular Cs+, the effect of the NMDA receptor antagonist AP-5 was targeted to effects on AMPA-mediated mEPSCs. Bath application of AP-5 (100 μM) resulted in an average 31.1 ± 5.7% decrease in mEPSC frequency (n = 9), from a baseline frequency of 12.4 ± 3.6 events/s to 8.3 ± 2.5 events/s in AP-5 (P < 0.05; Fig. 1). Application of AP-5 did not result in a significant change in amplitude (19.8 ± 1.8 pA baseline; 19.2 ± 1.5 pA AP-5; P = 0.21) or decay time constant (1.6 ± 0.1 ms baseline; 1.6 ± 0.9 ms AP-5; P = 0.43). The decrease in mEPSC frequency in response to AP-5, in the absence of a change in amplitude or decay time constant, suggested that AP-5 inhibited glutamate release by acting at receptors located on presynaptic terminals.
Fig. 1.
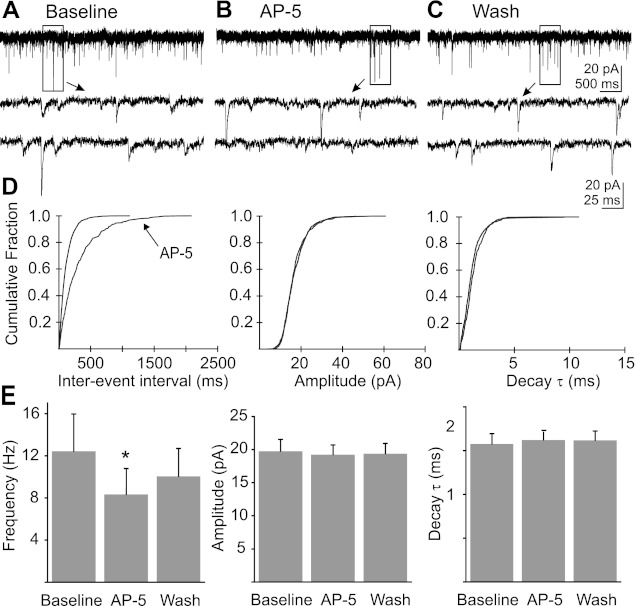
Blockade of N-methyl-d-aspartic acid (NMDA) receptors decreased miniature excitatory postsynaptic current (mEPSC) frequency without changing amplitude or decay time constant (τ). A: representative recording of mEPSCs in a dorsal motor nucleus of the vagus nerve (DMV) neuron. B: recording of mEPSCs from the same neuron during the application of dl-2-amino-5-phosphonopentanoic acid (AP-5; 100 μM; 10-min application). C: recording from the same neuron during wash to control artificial cerebrospinal fluid (ACSF) (i.e., no AP-5) indicates a partial reversal of the AP-5 effect. Boxed areas in A–C are shown expanded temporally below each trace. D: cumulative probability plots of mEPSC frequency, amplitude, and decay τ for the neuron represented in A–C. E: mean mEPSC frequency, amplitude, and decay τ before and during the application of AP-5 (n = 9; *significant difference from control; P < 0.05).
In cortical neurons, NMDA receptors located on presynaptic terminals have been reported to contain NR2B subunits (Woodhall et al. 2001; Yang et al. 2006). To test the hypothesis that NR2B-containing receptors modulated glutamate release in the DMV, the NR2B antagonist Ro 25–6981 (1 μM) was applied under conditions identical to those used for AP-5 experiments (Fig. 2). No change was observed in frequency (14.5 ± 2.9 events/s baseline; 16.8 ± 3.7 events/s Ro 25–6981; P = 0.10; n = 11), amplitude (19.5 ± 1.8 pA baseline; 17.4 ± 2.3 pA Ro 25–698; P = 0.28), or decay time constant (1.8 ms baseline; 2.0 ms Ro 25–6981; P = 0.06). Since NR2A subunit-containing NMDA receptors have also been identified at presynaptic terminals (Bidoret et al. 2009), we tested for the presence of this subunit using ZnCl2, applied at a concentration that antagonizes NR2A/NR2B, but not NR2C/NR2D subunit-containing receptors (Paoletti et al. 2009). Application of ZnCl2 (5 μM) resulted in a significant decrease in mEPSC frequency from 15.5 ± 2.9 events/s to 11.6 ± 2.2 events/s in the presence of ZnCl2 (n = 8; P < 0.05). No change in amplitude (19.6 ± 1.6 pA baseline; 20.0 ± 2.3 pA ZnCl2; P = 0.48) or decay time constant (1.7 ± 0.2 ms baseline; 1.8 ± 0.2 ms ZnCl2; P = 0.45) was observed.
Fig. 2.
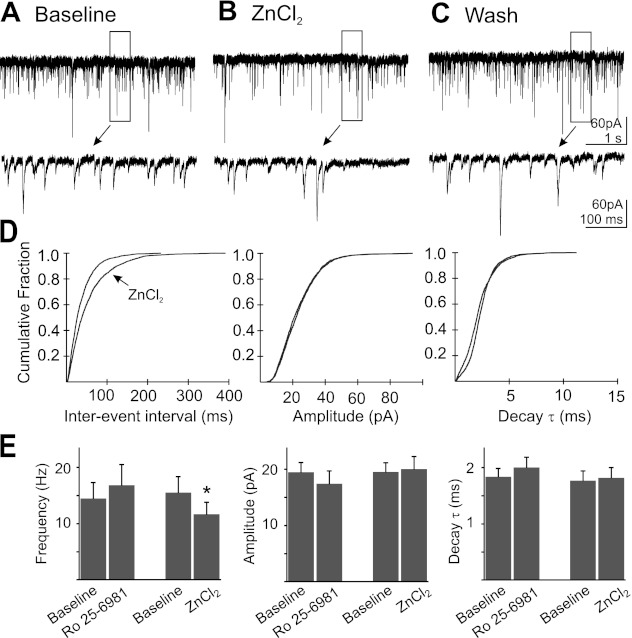
Application of Ro 25–6981 (n = 11) did not change frequency, amplitude, or decay τ of mEPSC, while ZnCl2 (n = 8) application decreased mEPSC frequency without changing amplitude or decay τ. A: representative recording of mEPSCs in a DMV neuron. B: recording of mEPSCs from the same neuron during the application of ZnCl2 (5 μM; 15-min application). C: recording from the same neuron during wash to control ACSF. Boxed areas in A–C are shown expanded temporally below each trace. D: cumulative probability plots of mEPSC frequency, amplitude, and decay τ for the neuron represented in A–C. E: mean mEPSC frequency, amplitude, and decay τ before and during the application of ZnCl2 (n = 8) or Ro 25–6981 (1 μM) (*significant difference from control; P < 0.05).
NMDA facilitates glutamatergic release onto DMV neurons.
When bath applying the receptor agonist NMDA (15 μM), MK-801 was included in the intracellular solution to permeate and block postsynaptic NMDA receptors from within the cell (Berretta and Jones 1996); neurons were first depolarized to 0 mV for 5 min to facilitate blockade of the receptor pore (Berretta and Jones 1996). These experiments were additionally conducted at a holding potential of −80 mV in the presence of PTX and TTX with intracellular Cs+. Consistent with the MK-801 blockade of postsynaptic NMDA receptors, application of NMDA (15 μM) did not induce a significant whole cell current (−3.0 ± 5.1 pA in response to NMDA), nor was mEPSC amplitude (20.0 ± 2.0 pA baseline; 20.1 ± 2.1 pA NMDA; P = 0.87) or decay time constant (1.8 ± 0.2 ms baseline; 1.7 ± 0.2 ms NMDA; P = 0.70) altered. However, NMDA application resulted in a significant increase in mEPSC frequency (27 ± 4.7%), from 10.8 ± 2.9 events/s before and 14.4 ± 3.3 events/s during NMDA application (n = 6; P < 0.05; Fig. 3). The increase in mEPSC frequency in absence of a change in whole cell current or mEPSC amplitude or decay time constant implied that NMDA augmented glutamate release by acting at presynaptic terminals.
Fig. 3.
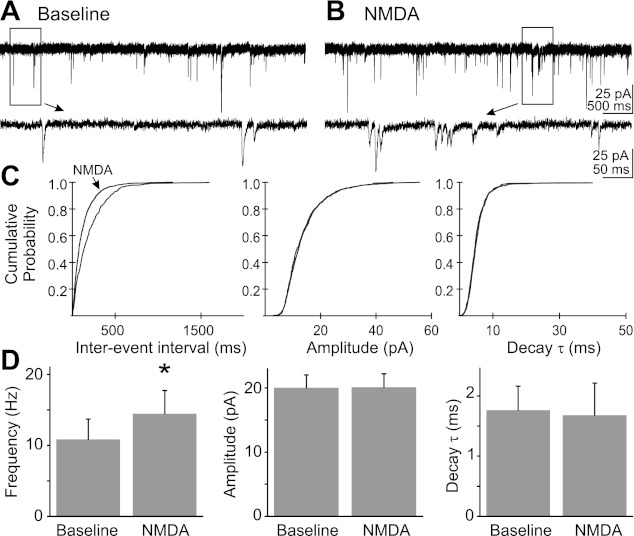
NMDA application increased mEPSC frequency without changing amplitude or decay τ. A: representative recording of mEPSCs in a DMV cell in control ACSF. B: recording of mEPSCs during the application of NMDA (10 μM). Boxed areas in A and B are shown expanded below each trace. C: cumulative probability plots of mEPSC frequency, amplitude, and decay τ for the cell represented in A and B. D: average mEPSC frequency, amplitude, and decay τ before and during the application of NMDA (n = 6; *significant difference from control; P < 0.05). Pipette solution contained MK-801 (1 mM).
Electrical stimulation.
Paired electrical stimuli (30-ms interstimulus interval) were delivered to the NTS at 0.1 Hz, and resulting paired evoked EPSC (eEPSC) amplitudes were recorded in DMV neurons. Paired stimulation of the NTS resulted in eEPSCs that typically displayed prominent paired pulse depression of the second response. The average amplitude of the first eEPSC was significantly decreased following application of AP-5 (90 ± 10 pA baseline; 51 ± 6 pA AP-5; P < 0.05), without a significant change in the amplitude of the second response (61 ± 6 pA baseline; 53 ± 5 pA AP-5; P = 0.29; Fig. 4). This greater relative effect on the first EPSC amplitude resulted in a decrease in paired-pulse depression, which was reflected in an increase in the paired-pulse ratio (PPr; 0.7 ± 0.1 baseline and 1.2 ± 0.3 in the presence of AP-5; n = 9; P < 0.05). The change in PPr induced by AP-5 was consistent with activity at receptors located on presynaptic terminals and suggested a reduction in the ongoing activity of endogenously activated NMDA receptors.
Fig. 4.
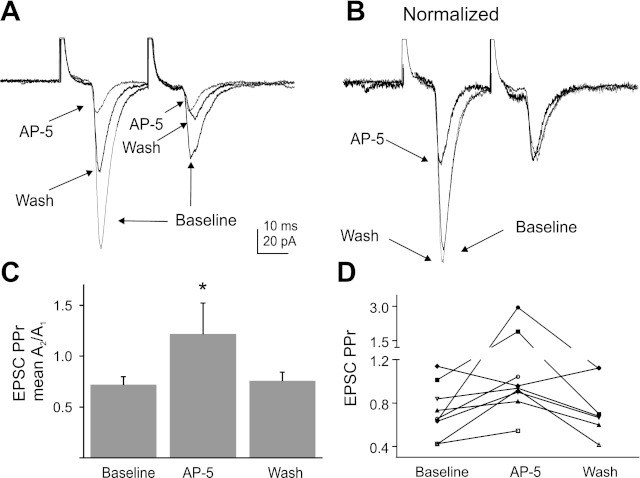
NMDA receptor blockade induced a decrease in the amplitude of excitatory postsynaptic currents (EPSCs) evoked after nucleus of the solitary tract (NTS) stimulation and an increase in paired-pulse ratio (PPr). A: paired electrical stimulation of the NTS in control ACSF, AP-5 (100 μM), and 15 min after removal of the antagonist indicated in a decrease in evoked EPSC amplitude during drug application. Averages of 15 responses are shown for each condition. B: averaged evoked EPSCs (normalized to the amplitude of the second response in A) before, during, and after the application of AP-5 indicated the antagonist induced a greater decrease in the first response amplitude than the second. C: mean PPr for control, AP-5, and wash indicates a significant increase in PPr (*significant difference from control; P < 0.05; n = 9). D: the PPr of individual cells shows the increase in PPr by AP-5 application in 8 out of 9 cells tested.
NMDA receptor modulation did not affect IPSCs.
Activation of ionotropic glutamate receptors enhances GABA release from synaptic terminals in the DMV (Derbenev et al. 2006). The hypothesis that presynaptic NMDA receptor activation increases GABA release was tested by recording mIPSCs in voltage-clamp mode at a holding potential of 0 mV in the presence of TTX and intracellular Cs+. The mean mIPSC frequency (1.0 ± 0.3 events/s baseline; 1.0 ± 0.3 events/s NMDA; P = 0.94), amplitude (26.1 ± 1.7 pA baseline; 25.2 ± 2.7 pA NMDA; P = 0.95), and decay time constant (7.0 ± 0.6 ms baseline; 7.3 ± 0.6 ms NMDA; P = 0.50) were not changed following the application of NMDA (15 μM; n = 7; Fig. 5). Likewise, blockade of NMDA receptors with AP-5 (100 μM; n = 7) did not alter mIPSC frequency (2.9 ± 0.8 events/s baseline; 2.2 ± 0.8 events/s AP-5; P = 0.922), amplitude (30.6 ± 2.7 pA baseline; 31.6 ± 3.2 pA AP-5; P = 0.97), or decay time constant (8.6 ± 0.7 ms baseline; 8.5 ± 0.6 ms AP-5; P = 0.94; Fig. 6). These results suggested that activation or blockade of NMDA receptors on GABA terminals did not consistently alter synaptic GABA release onto DMV neurons.
Fig. 5.
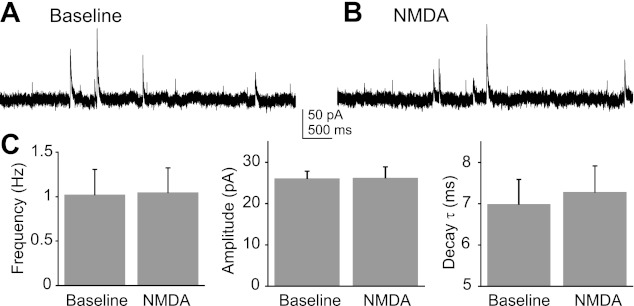
Effect of NMDA application on miniature inhibitory postsynaptic currents (mIPSCs) was not significant. A: representative baseline recording of mIPSCs. B: recoding of mIPSCs in the presence of NMDA (15 μM). C: average frequency, amplitude, and decay τ of mIPSCs in response to NMDA indicated no significant mean changes (P > 0.05; n = 7). Pipette solution contained MK-801 (1 mM).
Fig. 6.
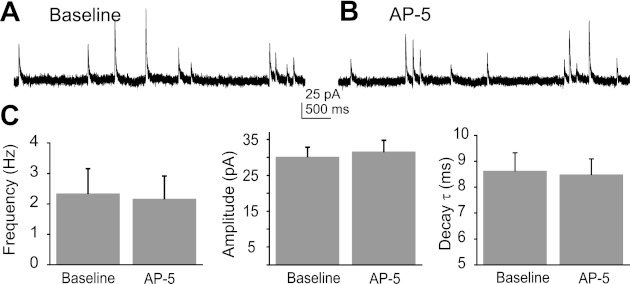
NMDA receptor blockade was without effect on mIPSCs. A: representative baseline recording of mIPSCs. B: representative recordings of mIPSCs in the presence of AP-5 (100 μM). C: average frequency, amplitude, and decay τ of mIPSCs in response to AP-5 indicated no significant mean changes (P > 0.05; n = 7).
DISCUSSION
A goal of this study was to provide evidence supporting the presence and function of NMDA receptors on presynaptic terminals contacting DMV neurons in the DVC. In other regions of the brain, including the hippocampus and the neocortex, presynaptic NMDA receptors have been found to function as autoreceptors to facilitate the release of glutamate (Berretta and Jones 1996; Brasier and Feldman 2008), participating in increasing excitatory synaptic tone (Woodhall et al. 2001). Perhaps more typically, presynaptic NMDA receptors located on GABAergic terminals have been found to function as heteroreceptors to enhance the release of GABA (Duguid and Smart 2004; Mathew and Hablitz 2011; Sjostrom et al. 2003). Given these disparate functions of presynaptic NMDA receptors and the existing evidence for glutamatergic modulation of GABA release in the DVC (Browning and Travagli 2007; Derbenev et al. 2006), this study was further aimed at identifying their putative function on synaptic activity in the DVC. Effects of NMDA receptor activation and antagonism on mEPSCs and paired evoked responses to afferent stimulation were consistent with the presence of NMDA receptors on glutamate terminals in the DMV.
A change in the frequency of action potential-independent (i.e., miniature) synaptic currents, in the absence of a change in the amplitude or decay time constant, suggests an NMDA receptor-mediated modulation of neurotransmitter release from presynaptic terminals. The decrease in mEPSC frequency in response to AP-5 or Zn2+, in the absence of a change in mEPSC decay time constant or amplitude, suggests that presynaptic NMDA receptors are activated by ambient glutamate and facilitate excitatory input to DMV neurons tonically. ZnCl2 application at a concentration that likely antagonized NR2A and NR2B subunit-containing receptors (Paoletti and Neyton 2007) resulted in a decrease in mEPSC frequency similar to that of AP-5, while the selective NR2B subunit-containing receptor antagonist Ro 25–6981 failed to have an effect on parameters measured. While it remains possible that other subunits are also present, these data suggest that preterminal NMDA receptors most likely contain NR2A, but not NR2B, subunits. Furthermore, the increase in mEPSC frequency in response to applied NMDA implies that the receptors are not fully saturated by ambient glutamate levels in the slice preparation.
Blockade of NMDA receptors can reduce the fidelity of synaptic currents evoked subsequent to stimulation of the NTS, and the change in PPr further suggests that this occurs subsequent to modulation of NMDA receptors located on presynaptic terminals. The paired-pulse depression observed here and previously (Browning and Travagli 2009; Browning and Travagli 2003; Glatzer and Smith 2005; Williams et al. 2007) suggests a synaptic connection with relatively high release probability, with the second response being of smaller amplitude due to depletion of the readily releasable primed pool during the first activated release (Debanne et al. 1996). A change in the PPr is often interpreted to indicate preterminal modulation of neurotransmitter release probability, with an increase in the ratio implicating a decrease in the probability of release (Debanne et al. 1996; Dobrunz et al. 1997; Dobrunz and Stevens 1997). Modulation of the amplitude of the first eEPSC by NMDA receptor blockade suggests that presynaptic NMDA receptors modulate terminal Ca2+ influx tonically. The absence of change in the second amplitude may imply that successive stimuli activate additional Ca2+-permeable receptors, allowing further release of the available vesicular pool.
Tonic activation of presynaptic NMDA receptors to enhance excitatory synaptic input has been suggested in other regions of the brain (Berretta and Jones 1996; Sjostrom et al. 2003). This autoreceptor-mediated facilitation may serve to enhance the overall gain of the glutamate response at particular synapse locations. As such, presynaptic NMDA receptor modulation could selectively enhance specific signals, for example those arriving from the NTS, as opposed to those arriving from other central areas. In the DMV, satiety signals from diencephalic brain areas converge, with inputs arising from NTS neurons relaying specific mechanical or chemical visceral signals pertaining to physiological state, especially from digestive system organs. Modulation of feed-forward glutamate tone via presynaptic NMDA receptors would be expected to increase the gain at active synapses, contributing to both temporal and spatial integration of synaptic signals in the DMV. In effect, appropriately timed central and peripheral satiety signals could thus augment each other by affecting summated glutamate release along specific physiologically relevant synaptic pathways driving central parasympathetic output.
In addition to enhancing glutamate release, presynaptic NMDA receptors have also been found to enhance GABA release in some regions of the brain (Duguid and Smart 2004; Glitsch and Marty 1999; Mathew and Hablitz 2011), and glutamate enhances GABA release in the DMV by acting at ionotropic and metabotropic receptors on presynaptic GABA terminals (Derbenev et al. 2006). GABA neurons are prominent in the medial NTS, which project inhibitory inputs to gastrointestinal-projecting regions of the DMV (Davis et al. 2004; Derbenev et al. 2004; Ferreira et al. 2001; Glatzer et al. 2007; Travagli et al. 1991). Inhibitory input to DMV cells has been found to be modulated through a variety of preterminal receptors, including those for monoamines, catecholamines, peptides, and lipids, in addition to glutamate receptors on GABA terminals (Browning and Travagli 2007; Derbenev et al. 2006; Glatzer et al. 2007; Travagli and Gillis 1995; Zsombok et al. 2011). Metabotropic glutamate receptors have been found to serve a complex integrative role to maintain both tonic tone of inhibitory neurotransmission and negatively modulate activity-dependent excitatory neurotransmission in synaptic connections between NTS and DMV neurons (Browning and Travagli 2007). Failure to observe a consistent change in mIPSC frequency in response to NMDA or AP-5 suggests that the glutamatergic heteroreceptors participating in the response of DMV neurons to transient receptor potential vanilloid-1 activation (Derbenev et al. 2006) are likely not predominantly NMDA receptors. It further suggests that glutamatergic terminals with presynaptic NMDA receptors are not in close proximity to heteroreceptors, since the increased glutamate released by presynaptic NMDA receptor activation might be expected to “spill over” and activate glutamate heteroreceptors on GABA terminals and, consequently, increase mIPSC frequency. This was not the case. It remains possible that presynaptic NMDA receptors exist on a subset of GABA terminals, but the absence of any predominant effect on GABA release suggests that presynaptic NMDA receptors are located preferentially on glutamatergic terminals, most likely including those arising from NTS afferents. Thus activation of presynaptic NMDA autoreceptors may represent a spatially distinct means of selectively augmenting glutamate release in the DMV.
Despite immunohistochemical evidence of their location in axons and the modulation of synaptic release by NMDA receptor activation in neocortex (Corlew et al. 2007), functional axonal NMDA receptors could not be detected in layer 5 pyramidal cells (Christie and Jahr 2009), suggesting that NMDA receptor-mediated facilitation of neurotransmitter release may involve indirect mechanisms in some cells. In cerebellar nuclei, preterminal depolarizations can result from voltage-gated calcium channel activation elicited through electrotonic spread of NMDA-mediated dendritic depolarization (Christie and Jahr 2008), suggesting that postsynaptic NMDA receptor activation can elicit effects on presynaptic terminals. These data challenge electrophysiological evidence and also light and electron microscopic imaging studies localizing NMDA receptors to axon terminals in several forebrain regions (Aoki et al. 1992, 1994; Bidoret et al. 2009; Charton et al. 1999; Woodhall et al. 2001; Yang et al. 2006; Zhang et al. 2009). The present results are consistent with the hypothesis that NMDA receptors located at presynaptic terminals regulate synaptic glutamate release in the DMV, since NMDA effects on mEPSCs persisted when the postsynaptic channels were blocked from inside the cell. However, the possibility that NMDA receptors located on dendrites contribute to presynatic modulation via a retrograde signal (e.g., electrotonic depolarization) remains to be explored. In summary, the results obtained in this study suggest that NMDA receptors are present on presynaptic terminals, and their activation positively modulates glutamate release onto DMV neurons, with little effect on GABA release. The presence of presynaptic NMDA receptors on GABAergic terminals cannot be entirely excluded, but, if present, their physiological function relative to synaptic regulation of DMV neurons is unclear. Presynaptic NMDA receptors on glutamate terminals facilitate the release of glutamate tonically, providing a means of downregulating or upregulating glutamate release, depending on ongoing synaptic activity. Their activation may serve to augment specific excitatory synaptic contacts in an autoregulatory fashion, acting to enhance specific inputs in a spatially and temporally distinct manner.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK056132 and R01 DK080901.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.C.B. and B.N.S. conception and design of research; E.C.B. performed experiments; E.C.B. analyzed data; E.C.B. and B.N.S. interpreted results of experiments; E.C.B. and B.N.S. prepared figures; E.C.B. and B.N.S. drafted manuscript; E.C.B. and B.N.S. edited and revised manuscript; E.C.B. and B.N.S. approved final version of manuscript.
REFERENCES
- Aoki C, Go CG, Wu K, Siekevitz P. Light and electron microscopic localization of alpha subunits of GTP-binding proteins, G(o) and Gi, in the cerebral cortex and hippocampus of rat brain. Brain Res 596: 189–201, 1992 [DOI] [PubMed] [Google Scholar]
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci 14: 5202–5222, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75: 339–344, 1996 [DOI] [PubMed] [Google Scholar]
- Bidoret C, Ayon A, Barbour B, Casado M. Presynaptic NR2A-containing NMDA receptors implement a high-pass filter synaptic plasticity rule. Proc Natl Acad Sci U S A 106: 14126–14131, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier DJ, Feldman DE. Synapse-specific expression of functional presynaptic NMDA receptors in rat somatosensory cortex. J Neurosci 28: 2199–2211, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Functional organization of presynaptic metabotropic glutamate receptors in vagal brainstem circuits. J Neurosci 27: 8979–8988, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Modulation of inhibitory neurotransmission in brainstem vagal circuits by NPY and PYY is controlled by cAMP levels. Neurogastroenterol Motil 21: e1309–e1126, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Neuropeptide Y and peptide YY inhibit excitatory synaptic transmission in the rat dorsal motor nucleus of the vagus. J Physiol 549: 775–785, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Plasticity of vagal brainstem circuits in the control of gastrointestinal function. Auton Neurosci 161: 6–13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton JP, Herkert M, Becker CM, Schroder H. Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res 816: 609–617, 1999 [DOI] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415: 793–798, 2002 [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron 60: 298–307, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Selective expression of ligand-gated ion channels in L5 pyramidal cell axons. J Neurosci 29: 11441–11450, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, BDP Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci 27: 9835–9845, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 14: 609–625, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SF, Derbenev AV, Williams KW, Glatzer NR, Smith BN. Excitatory and inhibitory local circuit input to the rat dorsal motor nucleus of the vagus originating from the nucleus tractus solitarius. Brain Res 1017: 208–217, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol 491: 163–176, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbenev AV, Monroe MJ, Glatzer NR, Smith BN. Vanilloid-mediated heterosynaptic facilitation of inhibitory synaptic input to neurons of the rat dorsal motor nucleus of the vagus. J Neurosci 26: 9666–9672, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbenev AV, Stuart TC, Smith BN. Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J Physiol 559: 923–938, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci U S A 94: 14843–14847, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18: 995–1008, 1997 [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci 7: 525–533, 2004 [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci 5: 135–145, 2004 [DOI] [PubMed] [Google Scholar]
- Ferreira M, Jr, Browning KN, Sahibzada N, Verbalis JG, Gillis RA, Travagli RA. Glucose effects on gastric motility and tone evoked from the rat dorsal vagal complex. J Physiol 536: 141–152, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Glatzer NR, Williams KW, Derbenev AV, Liu D, Smith BN. Morphological and electrophysiological features of motor neurons and putative interneurons in the dorsal vagal complex of rats and mice. Brain Res 1291: 40–52, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzer NR, Derbenev AV, Banfield BW, Smith BN. Endomorphin-1 modulates intrinsic inhibition in the dorsal vagal complex. J Neurophysiol 98: 1591–1599, 2007 [DOI] [PubMed] [Google Scholar]
- Glatzer NR, Smith BN. Modulation of synaptic transmission in the rat nucleus of the solitary tract by endomorphin-1. J Neurophysiol 93: 2530–2540, 2005 [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci 19: 511–519, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia M, Mesulam MM. Brain stem projections of sensory and motor components of the vagus complex in the cat. I. The cervical vagus and nodose ganglion. J Comp Neurol 193: 435–465, 1980a [DOI] [PubMed] [Google Scholar]
- Kalia M, Mesulam MM. Brain stem projections of sensory and motor components of the vagus complex in the cat. II. Laryngeal, tracheobronchial, pulmonary, cardiac, and gastrointestinal branches. J Comp Neurol 193: 467–508, 1980b [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. New York: McGraw-Hill, Health Professions Division, 2000, pp. xli, 1414 [Google Scholar]
- Ma QP, Hargreaves RJ. Localization of N-methyl-d-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience 101: 699–707, 2000 [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443–485, 1999 [DOI] [PubMed] [Google Scholar]
- Mathew SS, Hablitz JJ. Presynaptic NMDA receptors mediate IPSC potentiation at GABAergic synapses in developing rat neocortex. PLos One 6: e17311, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309: 261–263, 1984 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol 7: 39–47, 2007 [DOI] [PubMed] [Google Scholar]
- Paoletti PVA, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience 158: 126–136, 2009 [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654, 2003 [DOI] [PubMed] [Google Scholar]
- Travagli RA. The nucleus tractus solitarius: an integrative centre with “task-matching” capabilities. J Physiol 582: 471, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA. Effects of 5-HT alone and its interaction with TRH on neurons in rat dorsal motor nucleus of the vagus. Am J Physiol Gastrointest Liver Physiol 268: G292–G299, 1995 [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol Gastrointest Liver Physiol 260: G531–G536, 1991 [DOI] [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol 68: 279–305, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology 148: 1868–1881, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall G, Evans DI, Cunningham MO, Jones RS. NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J Neurophysiol 86: 1644–1651, 2001 [DOI] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci 26: 406–410, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Meng K, Li YH, Han TZ. NR2A-containing NMDA receptors are required for L-LTP induction and depotentiation in CA1 region of hippocampal slices. Eur J Neurosci 29: 2137–2144, 2009 [DOI] [PubMed] [Google Scholar]
- Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN. Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin-treated hyperglycemic mice. J Neurosci 31: 14024–14031, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]