

Abstract
Previously we demonstrated that sphingosine 1-phosphate receptor 1 (S1PR1) played a prominent, but not exclusive, role in enhancing the excitability of small-diameter sensory neurons, suggesting that other S1PRs can modulate neuronal excitability. To examine the potential role of S1PR2 in regulating neuronal excitability we used the established selective antagonist of S1PR2, JTE-013. Here we report that exposure to JTE-013 alone produced a significant increase in excitability in a time- and concentration-dependent manner in 70–80% of recorded neurons. Internal perfusion of sensory neurons with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) via the recording pipette inhibited the sensitization produced by JTE-013 as well as prostaglandin E2. Pretreatment with pertussis toxin or the selective S1PR1 antagonist W146 blocked the sensitization produced by JTE-013. These results indicate that JTE-013 might act as an agonist at other G protein-coupled receptors. In neurons that were sensitized by JTE-013, single-cell RT-PCR studies demonstrated that these neurons did not express the mRNA for S1PR2. In behavioral studies, injection of JTE-013 into the rat's hindpaw produced a significant increase in the mechanical sensitivity in the ipsilateral, but not contralateral, paw. Injection of JTE-013 did not affect the withdrawal latency to thermal stimulation. Thus JTE-013 augments neuronal excitability independently of S1PR2 by unknown mechanisms that may involve activation of other G protein-coupled receptors such as S1PR1. Clearly, further studies are warranted to establish the causal nature of this increased sensitivity, and future studies of neuronal function using JTE-013 should be interpreted with caution.
Keywords: sensitization, mechanical sensitivity
the lysophospholipid sphingosine 1-phosphate (S1P) is an important signaling molecule between cells but also plays an important role as an intracellular second messenger (Hannun and Obeid 2008; Hla 2004; Hla and Maciag 1990; Spiegel and Milstien 2003; Takabe et al. 2008; Van Brocklyn et al. 1998). S1P interacts with a family of five G protein-coupled receptors (S1PR1–5, previously known as Edg receptors) and has a significant impact on the development and activation of different cell types (Anliker and Chun 2004; Meyer zu Heringdorf and Jakobs 2007; Rosen et al. 2009; Sanchez and Hla 2004). Upon activation, a variety of immunocompetent cells (e.g., mast cells and platelets) release S1P where it can function as either an autocrine or paracrine signaling molecule (Goetzl and Rosen 2004; Olivera and Rivera 2005; Rivera et al. 2008; Weigart et al. 2009). In particular, interaction between S1P and the S1P receptor S1PR1 plays a critical role in regulating many aspects of the inflammatory response.
Upon activation of immunocompetent cells, the release of multiple mediators can heighten the sensitivity of sensory neurons to a variety of stimuli (reviewed by DeLeo and Yezierski 2001; Miller et al. 2009; Milligan and Watkins 2009; Scholz and Woolf 2007; Thacker et al. 2007; White et al. 2005). The impact of these different mediators on neuronal sensitivity is poorly understood. Our previous work showed that S1P enhanced the generation of current-evoked action potential (AP) firing in small-diameter capsaicin-sensitive sensory neurons (Zhang et al. 2006), in which S1PR1 played a prominent although not exclusive role in directly augmenting excitability of sensory neurons (Chi and Nicol 2010). These results suggest that other S1PRs also are capable of regulating excitability. In this study, we attempted to investigate the role of S1PR2 through the use of the selective S1PR2 antagonist JTE-013 (Arikawa et al. 2003; Osada et al. 2002). However, we found that JTE-013 increased the excitability of small-diameter sensory neurons independently of its interaction with S1PR2 and that in vivo JTE-013 was capable of enhancing the behavioral sensitivity to mechanical stimulation of the hindpaw.
MATERIALS AND METHODS
Isolation and maintenance of adult rat sensory neurons.
Sensory neurons were isolated from young adult male Sprague-Dawley rats (100–150 g) by procedures developed by Lindsay (1998), with slight modifications (Chi and Nicol 2007). Briefly, the young rats were killed by placing them in a chamber filled with CO2. The dorsal root ganglia (DRGs) were collected in a culture dish filled with sterilized Puck's solution. The ganglia were transferred to a conical tube with F-12 medium containing papain (20 U/ml) and incubated for 15 min at 37°C, followed by incubation with 1 mg/ml collagenase IA and 2.5 mg/ml dispase for 10 min at 37°C. The suspension was centrifuged for 30 s (1,000 g), after which the enzyme-containing supernatant was removed. The pellet was resuspended in F-12 medium supplemented with nerve growth factor (30 ng/ml) (Harlan Bioproducts, Indianapolis, IN) and mechanically dissociated with fire-polished pipettes. Isolated cells were plated onto plastic coverslips previously coated with poly-d-lysine (100 μg/ml) and laminin (5 μg/ml). Isolated cells were maintained in culture at 37°C and 3% CO2 for 18–24 h before electrophysiological recordings were obtained. All procedures were approved by the Animal Use and Care Committee of Indiana University School of Medicine.
Electrophysiology.
Recordings were made with the whole cell patch-clamp technique (Chi and Nicol 2007; Hamill et al. 1981). Briefly, a coverslip with sensory neurons was placed in a recording chamber where neurons were bathed in normal Ringer solution of the following composition (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH at 7.4 with NaOH. Recording pipettes were pulled from disposable borosilicate glass tubing and typically had resistances of 2–5 MΩ when filled with the following solution (in mM): 140 KCl, 5 MgCl2, 4 ATP, 0.3 GTP, 2.5 CaCl2, 5 EGTA (calculated free Ca2+ concentration of ∼100 nM), and 10 HEPES, at pH 7.3 with KOH. Whole cell voltages were recorded with an Axopatch 200 or Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The whole cell recording configuration was established in normal Ringer solution. The data were acquired and analyzed with pCLAMP 6.04 or pCLAMP 9.0 (Molecular Devices). All drugs were applied by external superfusion of the recording chamber with a VC-8 bath perfusion system (Warner Instruments, Hamden, CT). To assess excitability in the current-clamp experiments, neurons were held at their resting potentials (range between −45 and −65 mV) and ramps of depolarizing currents (1 s in duration) were used to evoke 2–4 APs under control conditions. The same ramp was then used throughout the recording period for each individual neuron. The current-clamp traces were filtered at 5 kHz and sampled at 1 kHz. At the end of each recording, the neuron was exposed to 400 nM capsaicin for ∼30 s. This neurotoxin was used to distinguish capsaicin-sensitive sensory neurons, as these neurons are believed to transmit nociceptive information (Holzer 1991). However, the correlation between the idea that a neuron is a nociceptor and capsaicin sensitivity is not absolute. Some nociceptive neurons are insensitive to capsaicin, and some capsaicin-sensitive neurons are not nociceptors (see Petruska et al. 2000). Therefore this agent was used to define a population of small-diameter sensory neurons that could serve a nociceptive function. The results reported in the following text were obtained from only capsaicin-sensitive neurons except as noted. All experiments were performed at room temperature (∼23°C).
Single-cell reverse transcription-polymerase chain reaction (RT-PCR).
The presence of gene transcripts for S1PR2 and HPRT was detected with techniques described by Chi and Nicol (2010) with modification. Briefly, after the effects of JTE-013 on AP firing were established, a small (≤25 μm)-diameter sensory neuron was aspirated into a sterilized micropipette (baked at 250°C for 2 h) containing diethylpyrocarbonate (DEPC)-treated water. The contents of the micropipette were forced into a 0.2-ml microtube with 5 μl of DEPC water, and the RNA was reverse transcribed with the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) for cDNA synthesis in 20-μl reactions according to the manufacturer's instructions. The cDNA was stored at −20°C before PCR detection. Amplification of S1PR2 (accession no. NM_017192.1) used the forward primer (5′ to 3′, 180..198 bp) TGAGAAGGTTCAGGAACAC and the reverse primer (5′ to 3′, 632..614 bp) AGAATCAGCGATATCAGCC (product size 453 bp). Amplification of hypoxanthine-guanine phosphoribosyltransferase (HPRT, accession no. NM_012583.2) used the forward primer (5′ to 3′, 495..514 bp) GCAGACTTTGCTTTCCTTGG and the reverse primer (5′ to 3′, 772..753 bp) TACTGGCCACATCAACAGGA (product size 278 bp). Amplification was performed with Platinum PCR Supermix (Invitrogen, Carlsbad, CA). These PCR reactions were run on a PTC-100 programmable thermal controller (MJ Research, Watertown, MA); for S1PR2 47 cycles were run at 94°C for 15 s, 47°C for 40 s, and 72°C for 1 min, and for HPRT 45 cycles were run at 94°C for 15 s, 58°C for 30 s, and 72°C for 40 s. The specificity of these amplifications, i.e., the detection of a single band at the appropriate size, was verified by electrophoresis on a 1% agarose gel.
Behavioral measurements and analysis.
All procedures were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati. Sprague-Dawley rats (male, 200 g; Harlan, Indianapolis IN) were tested for mechanical sensitivity with von Frey filaments exerting bending forces between 0.4 and 15 g, using the up-and-down method (Chaplan et al. 1994; Dixon 1965). Animals not responding to the stiffest von Frey filament were assigned a cutoff value of 15 g. Animals received baseline testing on two separate days prior to injection of 20 μl of 2 μM JTE-013 in HEPES-buffered extracellular solution (in mM: 150 NaCl, 5 KCl, 1 MgCl2, 2.5 CaCl2, 10 HEPES, and 10 glucose, pH 7.4 adjusted with NaOH). JTE-013 was made up as a 40 mM stock solution in DMSO, purged with N2. Dilution to the final concentration and sonication were done just before injection. Control animals received vehicle injections (0.005% DMSO in the extracellular solution). The injection, done with a tuberculin syringe with a 31-gauge needle, was subcutaneous in the heel region used for mechanical and thermal testing. Slight redness during the first hour after injection was noted in JTE-013-injected but not control animals. Thermal testing was conducted by the Hargreaves method (Hargreaves et al. 1988) in which animals were placed on a glass surface maintained at 30°C, a radiant heat source was applied to the heel region from beneath, and the latency to paw withdrawal was measured. The heat source was adjusted to give baseline withdrawal times in control animals of ∼10 s. A 20-s cutoff time was set; however, no rats reached this value. Values presented are the average of four trials. Baseline thermal testing was conducted on two different days prior to the paw injection, which were not the same days as baseline mechanical testing. The thermal testing after JTE-013 or vehicle injection was conducted 1 h after injection, between the first and second rounds of mechanical testing.
Data analysis.
Data are presented as means ± SE. The excitability parameters described in Table 1 were determined, in part, by differentiating the voltage trace (dV/dt) in the current-clamp recordings. The voltage and time at which the first AP was fired were taken as the point that exceeded the baseline value of dV/dt by >20-fold. The baseline value of dV/dt was determined by averaging the points between the onset of the ramp and the next 100 ms (66–166 ms). The rheobase was measured as the amount of ramp current at the firing threshold. The resistance at threshold (RTh) was calculated as the difference between the firing threshold and the resting membrane potential divided by the rheobase current. Statistical differences between the control recordings and those obtained under various treatment conditions were determined by using either an ANOVA or a repeated-measures (RM) ANOVA whenever appropriate. When a significant difference was obtained, post hoc analyses were performed with a Holm-Sidak all pairwise test. If the data set failed the normality test, a Kruskal-Wallis analysis of variance on ranks was performed with a Dunn's all pairwise test. For the behavioral measurements, statistical significance was determined by a two-way RM ANOVA with a Bonferroni posttest. Values of P < 0.05 were judged to be statistically significant.
Table 1.
Effects of JTE-013 on parameters of AP firing
| JTE-013 |
||||
|---|---|---|---|---|
| Control | 5 min | 10 min | 15 min | |
| RMP, mV | −53.0 ± 0.8 | −51.4 ± 2.1 | −50.1 ± 2.2 | −49.0 ± 2.6 |
| FT, mV | −17.7 ± 3.0 | −18.8 ± 1.9 | −18.5 ± 1.9 | −18.0 ± 2.9 |
| Rheo, pA | 271 ± 108 | 222 ± 101 | 185 ± 75 | 195 ± 87 |
| NRheo | 1.0 | 0.73 ± 0.08* | 0.70 ± 0.08* | 0.62 ± 0.06* |
| RTh, MΩ | 399 ± 91 | 532 ± 114 | 526 ± 112 | 505 ± 118 |
| NRTh | 1.0 | 1.34 ± 0.10* | 1.37 ± 0.10* | 1.45 ± 0.10* |
| n | 12 | 12 | 12 | 10 |
Values are means ± SE for n neurons. AP, action potential; RMP, resting membrane potential; FT, firing threshold; Rheo, rheobase; NRheo, normalized rheobase; RTh, resistance at threshold; NRTh, normalized RTh.
P < 0.05 ANOVA, Holm-Sidak all pairwise posttest.
Wound healing assay.
B16 mouse melanoma cells, a generous gift of Dr. Jeffrey B. Travers (Dept. of Dermatology, Indiana University School of Medicine), were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen) in 75-cm2 cell culture flasks. Cells were plated into the wells of a six-well culture dish that had been coated with 10 μg/ml fibronectin. Cells were maintained at 37°C and 5% CO2, allowing them to adhere onto the substrate. When the cells reached 70–80% confluence, the cell monolayer was scraped in a straight line with a 200-μl pipette tip to create a “wound.” To remove the debris and smooth the edge of the wound, cells were washed twice with 1 ml of phosphate-buffered saline followed by 1 ml of migration medium, which contained serum-free F-12 medium (Invitrogen) and 0.1% fatty acid-free bovine serum albumin. There were four different treatment groups: the untreated control group with migration medium alone, cells exposed to 100 nM S1P, cells exposed to 1 μM JTE-013, and cells exposed to 100 nM S1P and 1 μM JTE-013. A mark on the plate was placed near the wound site and used as a reference point. Images were acquired with a ×10 objective at both 0 and 48 h after creation of the wound.
Chemicals.
JTE-013, W140, and W146 were purchased from Cayman Chemical (Ann Arbor, MI). Pertussis toxin (PTX) was purchased from EMD Bioscience (San Diego, CA). S1P was obtained from Avanti Polar Lipids (Alabaster, AL) and dissolved according to instructions provided by the supplier (www.avantilipids.com/SyntheticSphingosine-1-phosphate.asp). Tissue culture supplies were purchased from Fisher (Pittsburgh, PA). All other chemicals were obtained from Sigma Chemical (St. Louis, MO). For the electrophysiology experiments, capsaicin and JTE-013 were dissolved in 1-methyl-2-pyrrolidinone (MPL, HPLC grade). These stock solutions were then diluted with Ringer solution to yield the appropriate concentration. The vehicle MPL was typically used at 1,000- to 5,000-fold dilutions; we demonstrated previously that the vehicle MPL has no effect on AP firing or the activation of either TTX-resistant INa or IK (Zhang et al. 2002). For paw injection experiments JTE-013 was dissolved in DMSO and made up just before use in bath solution as described above.
RESULTS
JTE-013 increases excitability of small-diameter sensory neurons.
We previously demonstrated that sensory neurons of the DRG expressed the mRNA for S1P receptors 1–4 (S1PR1–4) and that S1PR1 played a prominent but not exclusive role in enhancing the excitability of small-diameter capsaicin-sensitive sensory neurons (Chi and Nicol 2010; Zhang et al. 2006). To elucidate the possible role of S1PR2 in regulating the S1P-induced increase in excitability of sensory neurons, these cells were pretreated with 100 nM JTE-013, which has been described as a selective antagonist of S1PR2 (Arikawa et al. 2003; Osada et al. 2002). Surprisingly, the initial exposure to JTE-013 alone produced a significant time-dependent increase in the number of APs evoked by the ramp of current in these neurons. As illustrated in Fig. 1A, a representative recording from a sensory neuron shows that under control conditions the ramp evoked 3 APs, whereas after a 10-min exposure to 100 nM JTE-013 the number of APs was increased to 10 (Fig. 1A, right) although there was no change in the resting membrane potential. The increase in AP firing produced by JTE-013 is summarized in Fig. 1B, where 11 of 16 neurons exhibited increased AP firing; there was a significant twofold increase in the number of APs after only a 5-min exposure compared with control values (P < 0.001, Kruskal-Wallis ANOVA, Dunn's all pairwise test). However, 5 of the 16 neurons failed to show any increase in excitability after exposure to 100 nM JTE-013 over the 15-min recording period (see below). The number of APs at each time point was then normalized to the respective control values, and this analysis demonstrated that JTE-013 produced a rapid and significant increase in excitability in ∼70% of the sensory neurons compared with their control values (summarized in Fig. 1C; P < 0.001, Kruskal-Wallis ANOVA, Dunn's all pairwise test). In addition, there was no significant difference in the number of APs obtained after 5-, 10-, and 15-min exposures to JTE-013 (P = 0.33, ANOVA). The effects of JTE-013 on the parameters of excitability are summarized in Table 1. Treatment with JTE-013 had no effect on the resting membrane potential (P = 0.72, ANOVA), firing threshold (P = 0.99, ANOVA), rheobase (P = 0.55 ANOVA), or RTh (P = 0.80 ANOVA, see Table 1). To reduce the variance in the rheobase and RTh, changes in these values after exposure to JTE-013 were then normalized to their respective control values. After normalization, the JTE-013-induced increase in AP firing was associated with a significant decrease in the values of the normalized rheobase and an increase in the normalized RTh compared with the controls (P < 0.001 and P = 0.003, respectively, ANOVA, Holm-Sidak all pairwise test); there was no difference between the values obtained at 5, 10, and 15 min.
Fig. 1.
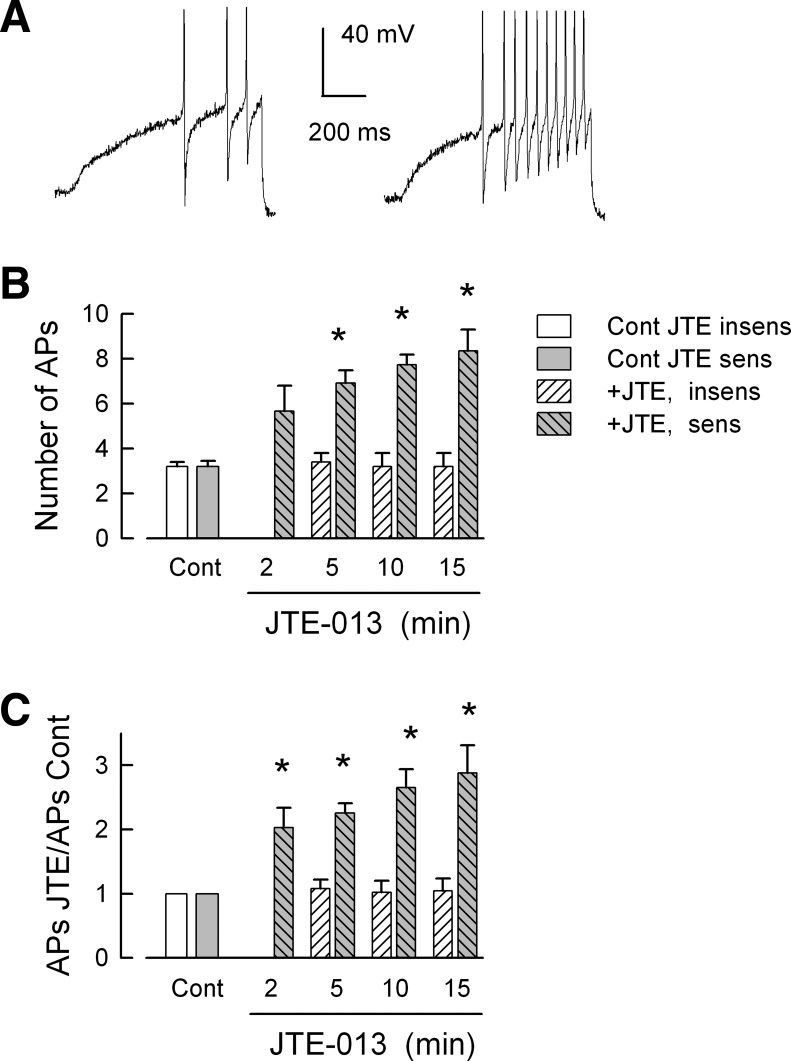
JTE-013 enhances the excitability of capsaicin-sensitive small-diameter sensory neurons. A: a representative recording in which the ramp of depolarizing current evoked 3 action potentials (APs) under control conditions, whereas after a 10-min exposure to 100 nM JTE-013 the number of APs increased to 10 (right). B summarizes the sensitizing actions of JTE-013 over a 15-min recording period. There was no significant difference between the number of APs at the 2, 5, 10, and 15 min time points. The number of neurons at each time point are as follows: control 11, 2 min 6, 5 min 11, 10 min 11, and 15 min 9. C: the number of evoked APs after exposure to JTE-013 normalized to their respective control values; these are the same neurons as shown in B. Note that there were no recordings obtained at 2 min for JTE-013-insensitive neurons. *Significant difference compared with the control condition (P < 0.001, ANOVA on ranks).
In addition to those sensory neurons that exhibited increased AP firing, 5 of the 16 sensory neurons were not sensitized by 100 nM JTE-013 (see Fig. 1B). For these insensitive neurons 3.2 ± 0.2 APs were generated under control conditions, and this remained unchanged after a 15-min exposure to JTE-013, where 3.2 ± 0.6 APs were evoked by the ramp (n = 5, P = 0.98 ANOVA). Also, there was no significant change in the resting membrane potential in these neurons after exposure to JTE-013 (control −56.3 ± 2.3 mV vs. after 15 min −53.4 ± 5.4 mV, n = 5, P = 0.71 RM ANOVA, data not shown for 5 and 10 min). Neurons insensitive to JTE-013 were not included in the analysis shown in Table 1. In recordings from six capsaicin-insensitive neurons (ranging in diameter from 20 to 50 μm), four were sensitized by JTE-013 where under control conditions 2.8 ± 0.5 APs were evoked; after 2-, 5-, and 10-min exposures to JTE-013, the numbers of APs were increased to 7.0 ± 1.0, 8.0 ± 1.3, and 7.3 ± 1.5 APs, respectively (data not shown). The values obtained at 5 and 10 min, but not 2 min, were significantly different from the control values (P = 0.03, ANOVA Holm-Sidak all pairwise test). Two of the six capsaicin-insensitive neurons were unaffected by 10 nM JTE-013; for the control conditions both fired 2 APs, and after 10 min one fired 2 and the other 1 AP. Another series of experiments examined whether the sensitizing actions of JTE-013 could be reversed. In recordings from four neurons, a 10-min exposure significantly increased the number of APs from a control value of 3.8 ± 0.5 APs to 9.5 ± 1.3 APs. After this 10-min exposure time, the neurons were then superfused with normal Ringer solution for an additional 15 min in which the number of evoked APs remained unchanged at 10.8 ± 1.3 APs (data not shown), indicating that the actions of JTE-013 were not readily reversed in this time period. Therefore, these results indicate that the S1PR2 antagonist JTE-013 by itself is capable of augmenting the excitability of some, but not all, small-diameter capsaicin-sensitive and -insensitive sensory neurons.
To further characterize the effects of JTE-013 on the capacity of sensory neurons to fire APs, the concentration-response relation was determined. Figure 2A summarizes the effects of different concentrations of JTE-013 (from 1 to 1,000 nM) at different time points on the average number of APs evoked by the current ramp. These results indicate that there are both time- and concentration-dependent effects of JTE-013 on excitability. Figure 2B summarizes the effects of JTE-013 at the different times and concentrations after normalization to their respective values obtained under control conditions. The normalization was performed on those neurons represented in Fig. 2A. Treatment with either 1 or 3 nM JTE-013 did not alter the number of evoked APs over the 15-min recording period (1 nM: n = 4, control 4.3 ± 0.3 APs vs. 15 min 5.5 ± 0.9 APs, P = 0.38 ANOVA; 3 nM: n = 5, control 4.0 ± 0 APs vs. 15 min 5.5 ± 0.6 APs, P = 0.15 ANOVA on ranks). Similar results were obtained for exposure to vehicle (MPL, 5,000-fold dilution) where after 10-min exposure 4.2 ± 0.6 APs were evoked compared with the control value of 3.4 ± 0.2 (n = 5, P = 0.20 RM ANOVA). In addition, neurons insensitive to JTE-013 exhibited no significant change in AP firing over the 15-min recording period, demonstrating that AP firing is stable over time (Chi and Nicol 2010). In contrast, exposure to 10 nM JTE-013 produced a significant increase in excitability in 10 of 12 neurons (2 were JTE-013 insensitive). The number of evoked APs was significantly different for exposures of 5, 10, and 15 min compared with their respective control values (P < 0.001 ANOVA on ranks); there was no difference between the values obtained at 5, 10, and 15 min. For 100 nM JTE-013, 11 of 16 neurons (as shown in Fig. 1) exhibited a significant increase in excitability after 5-, 10-, and 15-min exposures (P < 0.001 ANOVA on ranks). Finally, 8 of 11 sensory neurons exposed to 1,000 nM JTE-013 demonstrated significant increases in excitability after 5-, 10-, and 15-min exposures (P < 0.001 ANOVA on ranks). Three neurons were insensitive to 1,000 nM JTE-013, where after 15 min 3.7 ± 0.7 APs were evoked compared with 3.7 ± 0.9 APs for the untreated control (P = 0.95 ANOVA). These results are similar to those obtained for neurons insensitive to 10 or 100 nM JTE-013 and reflect the consistency of these recordings over time. In Fig. 2C, the number of evoked APs normalized to their respective controls for the 10 and 15 min time points are plotted as a function of JTE-013 concentration. At 10 nM JTE-013 there was a significant threefold increase in the number of APs for both the 10 and 15 min time points compared with either 1 or 3 nM (P < 0.001 ANOVA on ranks). There was no significant difference for the increased AP firing produced by 10, 100, or 1,000 nM JTE-013 at the 10 min time point (ANOVA). These results demonstrate that JTE-013 enhanced the excitability of sensory neurons in both a time- and a concentration-dependent manner. However, the activation range was quite narrow and may reflect the nonlinear aspects of AP firing as a response measure.
Fig. 2.
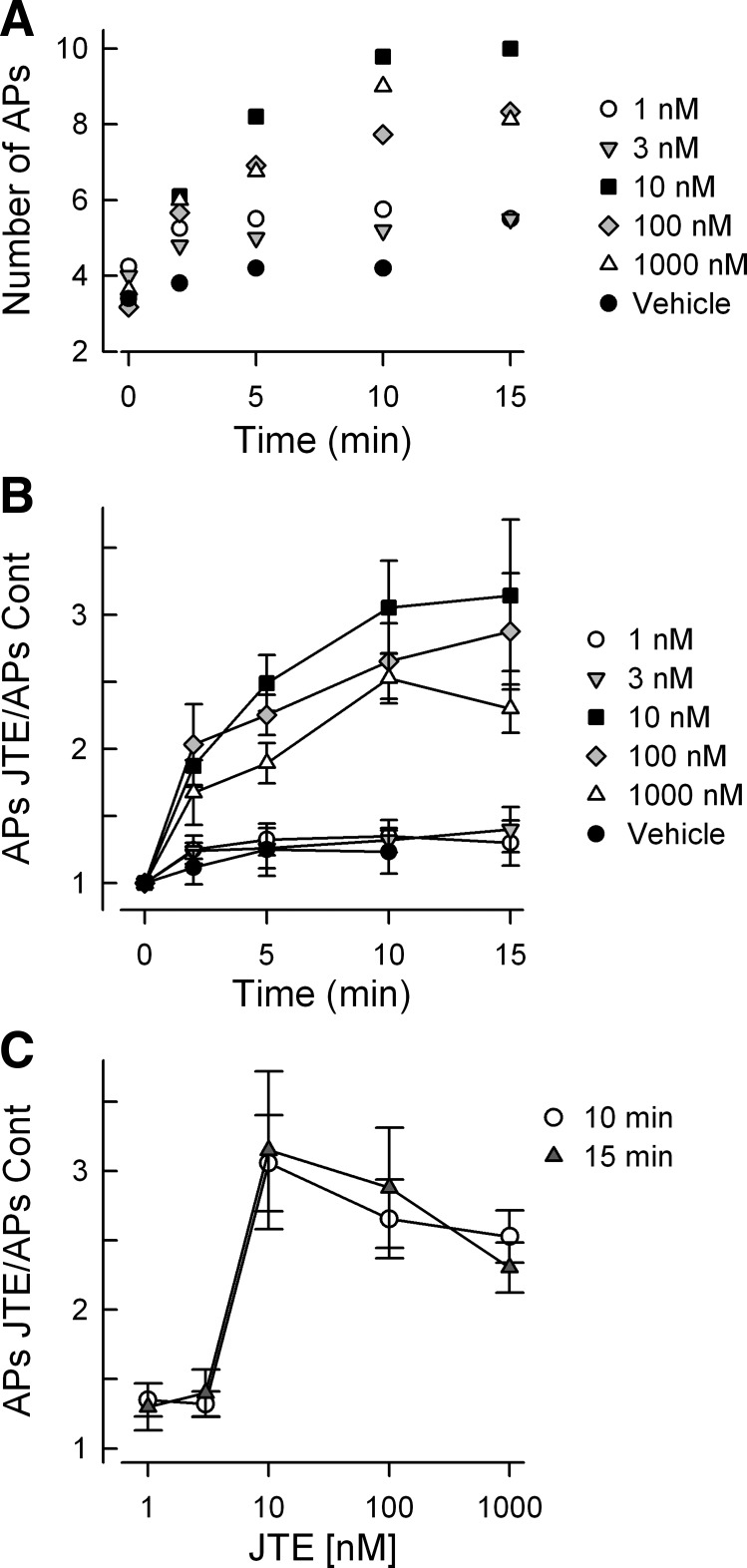
JTE-013 augments excitability in a time- and concentration-dependent manner. A: number of evoked APs for the different times and concentrations of JTE-013. The data shown represent only the mean values; the SE is not shown for clarity of presentation. The number of neurons comprising the results for each concentration are as follows: vehicle 5, 1 nM 4, 3 nM 5, 10 nM 10, 100 nM 11, 1,000 nM 8. B: number of evoked APs obtained for the different times and concentrations of JTE-013 normalized to their respective control values. The data were obtained from the neurons shown in A and represent means ± SE. There was no difference in either the number of evoked APs or the normalized number of evoked APs over the recording periods for the vehicle (P = 0.20 and P = 0.24 for the number and the normalized number, respectively, ANOVA), for 1 nM JTE-013 (P = 0.38 and P = 0.27 for the number and the normalized number, respectively, ANOVA), and for 3 nM JTE-013 (P = 0.16 for both for the number and the normalized number, ANOVA on ranks). There was a significant difference in both the number and normalized number of APs for treatment times at 5, 10, and 15 min for 10, 100, and 1,000 nM JTE-013 compared with their control values (P < 0.001 ANOVA on ranks, Dunn's all pairwise test). C summarizes the increase in the normalized number of APs as a function of JTE-013 concentration for treatment times of 10 and 15 min.
The unexpected result that JTE-013 augmented AP firing in sensory neurons raises the question as to whether treatment with JTE-013 alters the neuronal response to S1P. In a separate set of experiments shown in Fig. 3, treatment with 100 nM JTE-013 enhanced AP firing in a manner similar to that described above. After the recordings were obtained at 15 min, the neurons were then exposed to 1 μM S1P via bath superfusion and AP production was measured over the next 10 min. These results indicate that S1P did not further increase AP firing over that obtained after 15 min of JTE-013 (n = 7, P = 0.99, ANOVA on ranks). As the effects of JTE-013 were not readily reversed, it is difficult to know whether JTE-013 masked any potential effects of S1P due to a ceiling effect on AP firing. Also, a lower concentration of JTE-013, i.e., 10 nM, produced maximal sensitization that was unaffected by S1P (see Fig. 5A). JTE-013 at 3 nM had no effect; thus titrating an effective concentration was difficult. This remains a question for future studies elucidating the specific target as well as the mechanism of action for JTE-013.
Fig. 3.
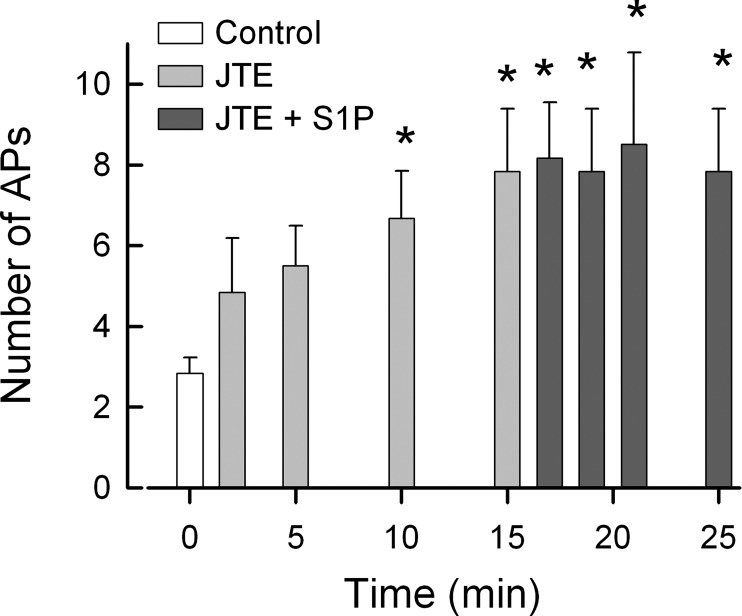
Sphingosine 1-phosphate (S1P) does not cause a further increase in AP firing after treatment with JTE-013. In a separate series of experiments, 7 sensory neurons were exposed to 100 nM JTE-013 over a 15-min recording period. After the recoding at 15 min, these neurons were exposed to 1 μM S1P and recordings were obtained over the next 10 min. The data represent means ± SE. *Significant difference from the control values [P < 0.001, repeated-measures (RM) ANOVA].
Fig. 5.
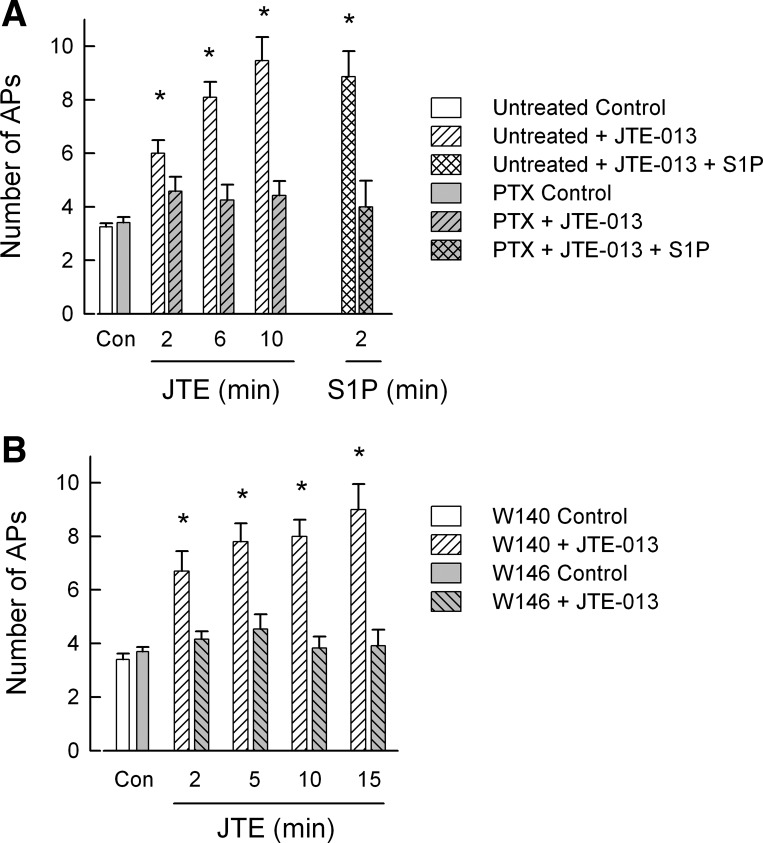
Pertussis toxin (PTX) and the S1P receptor 1 (S1PR1) antagonist W146 block the JTE-013-induced increase in excitability. A: 1 group of sensory neurons were treated with 200 ng/ml PTX for 24 h, while the untreated neurons were composed of 2 groups: 1 group was obtained from neurons isolated from the same tissue harvests as those for the PTX neurons, and another group was derived from the concentration-response experiments for 10 nM JTE-013. These 2 groups were not significantly different for all time points (P > 0.05, ANOVA) and were combined. The numbers of neurons in the untreated group were as follows: control 12, JTE-013 12, 12, and 11 at 2, 6, and 10 min, respectively, and S1P 7. The numbers of neurons in the PTX-treated group were as follows: control 12, JTE-013 12, and S1P 6. *Significant difference between the control and after exposure to JTE-013 within the untreated group (P < 0.001, ANOVA Holm-Sidak all pairwise). To compare the untreated and PTX-treated neurons, an ANOVA was used. There was a significant difference (P < 0.001, Holm-Sidak all pairwise test) between the untreated neurons at 6 and 10 min from all PTX-treated time points. Untreated neurons at 2 min were significantly different from the PTX-treated controls but not PTX-treated neurons at 2, 6, and 10 min. The untreated and PTX-treated controls were not different. B: pretreatment with the selective S1PR1 antagonist W146, but not its inactive analog W140, blocks the effects of JTE-013. A within-group statistical analysis indicated that in the presence of 1 μM W140 JTE-013 significantly increased AP firing for the recordings obtained at 2, 5, 10, and 15 min compared with the control (P < 0.001, ANOVA Holm-Sidak all pairwise test), whereas there was no difference between the number of APs recorded at 2, 5, 10, and 15 min. The numbers of neurons in the W140-treated group were as follows: control 10, JTE-013 2 min 10, 5 min 10, 10 min 7, and 15 min 5. In contrast, a within-group analysis showed that JTE-013 had no effect on the number of evoked APs in the presence of 1 μM W146 (P = 0.66 ANOVA). The numbers of neurons in the W146-treated group were as follows: control 13, JTE-013 2 min 13, 5 min 13, 10 min 12, and 15 min 12. A comparison between the W140 and W146 groups indicated that in the presence of W140 JTE-013 significantly increased the number of evoked APs at 5, 10, and 15 min compared with all time points for W146 treatment (P < 0.001, ANOVA Holm-Sidak all pairwise test). For the W140 2 min recordings, the difference was significant for the W146 control, 2, 10, and 15 min but not 5 min time points. There was no difference between the W140 and W146 control groups.
Guanosine 5′-O-(2-thiodiphosphate) blocks the increased excitability produced by JTE-013.
We have shown that exposure to JTE-013 increased the number of evoked APs. JTE-013 may act as a agonist in addition to its established role as an S1PR2 antagonist. To explore this possibility, sensory neurons were internally perfused via the recording pipette with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) to block activation of G protein-coupled receptors (Eckstein et al. 1979). To determine the effectiveness of GDP-β-S to block activation of G protein-coupled receptors, sensory neurons were initially exposed to the inflammatory prostaglandin PGE2, which is known to increase neuronal excitability through activation of the Gs-cAMP pathway (Cui and Nicol 1995; Hingtgen et al. 1995; Taiwo et al. 1989). As summarized in Fig. 4A, under normal recording conditions, a 2-min exposure to 1 μM PGE2 produced a significant increase in the number of evoked APs that was maintained through the 10-min recording period compared with the control value (P < 0.001, ANOVA, Holm-Sidak all pairwise test). In contrast, in neurons that were internally perfused with 3 mM GDP-β-S, exposure to PGE2 failed to increase the number of evoked APs. Similarly, internal perfusion with GDP-β-S for 10 min prevented the increase in excitability produced by exposure to 100 nM JTE-013 (see Fig. 4B). Thus, in the presence of GDP-β-S, the increases in AP firing produced by PGE2 and JTE-013 were blocked, suggesting that JTE-013 is capable of increasing excitability via a mechanism that is dependent on G protein activation.
Fig. 4.
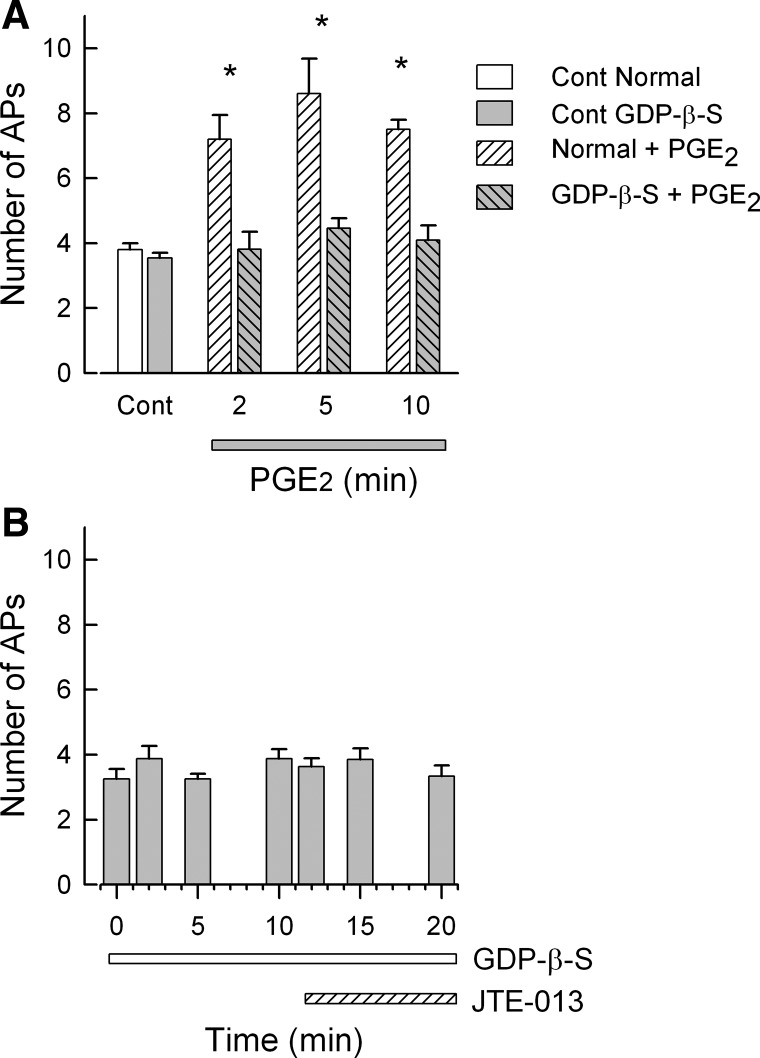
Internal perfusion with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) blocks the increased excitability produced by JTE-013. A demonstrates that internal perfusion with 3 mM GDP-β-S prevents the increase in excitability produced by 1 μM PGE2, which is known to act via the Gs-cAMP-PKA pathway. For the normal control condition, n = 5; for internal perfusion with GDP-β-S, n = 11. B shows that internal perfusion with GDP-β-S blocks the increase in AP firing produced by 100 nM JTE-013. The numbers of neurons at each time point are as follows: control through 12 min 8 neurons, 15 min 7, and 20 min 6. *Significant difference compared with the control condition (P < 0.05, RM ANOVA).
Pretreatment with either pertussis toxin or selective S1PR1 antagonist W146 blocks the effects of JTE-013.
To test the idea that JTE-013 might be acting via Gi/Go types of G proteins, isolated sensory neurons were treated with 200 ng/ml PTX for 24 h, after which recordings were obtained. As illustrated in Fig. 5A, PTX blocked the capacity of JTE-013 to augment AP firing. In recordings from 12 PTX-treated neurons, 10 nM JTE-013 failed to enhance the excitability over a 10-min recording period (P = 0.44, ANOVA on ranks). Additionally, eight of these neurons were then exposed to 1 μM S1P. In six of the eight neurons S1P did not alter AP firing; however, in two of those eight neurons, AP firing went from 5 and 6 APs to 12 and 11 APs, respectively, after a 2-min exposure to S1P. It is possible that these recordings were obtained from only JTE-013-insensitive neurons. However, as indicated by our results described above, JTE-013-sensitive neurons comprise ∼70–80% of recorded neurons. Thus it seems unlikely that all 12 neurons from which these recordings were obtained were insensitive to JTE-013 (P = 0.002, Fisher's exact test). A parallel set of untreated isolated sensory neurons obtained from these same tissue harvests were combined with those measurements obtained from other neurons used in the concentration-response experiments (the effects of JTE-013 on AP production were not significantly different for these 2 groups of neurons). In this combined set, 10 nM JTE-013 produced a significant increase in AP firing after only 2 min compared with the untreated controls (P < 0.001, RM ANOVA Holm-Sidak all pairwise test). Furthermore, the JTE-013-induced increase in AP firing in the untreated neurons was significantly different for the 6 and 10 min time points compared with all time points for PTX-treated neurons (see Fig. 5A legend for details). In the parallel untreated neurons obtained from the same tissue harvests used for PTX treatment, a 2-min exposure to 1 μM S1P after the 10-min exposure to 10 nM JTE-013 produced no further increase in AP firing (n = 7); these results are similar to those described above for Fig. 3. Thus these results indicate that JTE-013 may be acting via a Gi/Go-type G protein. In addition, sensitization produced by S1P was prevented by treatment with PTX, indicating that S1P receptor activation (e.g., S1PR1) of a Gi/Go-type G protein may play a prominent role in the S1P-mediated increase in excitability. However, in some neurons, S1P may act via PTX-insensitive G proteins, for example, Gq (see Rosen et al. 2009). This result is consistent with our previous findings in which siRNA knockdown of S1PR1 expression prevented S1P-induced sensitization in most, but not all, sensory neurons (Chi and Nicol 2010).
In light of our previous findings that S1PR1 plays a prominent role in enhancing excitability of sensory neurons (Chi and Nicol 2010) and that S1PR1 activation of downstream signaling pathways is mediated by Gi-type G proteins (see reviews by Obinata and Hla 2012; Rosen et al. 2009; Strub et al. 2010), we examined the possibility that JTE-013 acted via S1PR1 by using the selective S1PR1 antagonist W146 (Sanna et al. 2006). Sensory neurons were pretreated with either 1 μM W146 or its inactive analog W140 for 30 min prior to recording. For both W140 and W146 treatment groups, the effects of 10 and 100 nM JTE-013 were examined, as W146 is a competitive antagonist of S1PR1 (Sanna et al. 2006). An ANOVA indicated that there were no significant differences between the responses elicited by 10 and 100 nM JTE-013. For example, W146 treatment was equally effective for 10 or 100 nM JTE-013 (P = 0.98). Therefore, the AP recordings from 10 and 100 nM JTE-013 were combined. These results (see Fig. 5B) demonstrate that pretreatment with W146 blocked the sensitizing action of JTE-013 where the number of APs was not changed over the 15-min recording period [control 3.7 ± 0.2 APs (n = 13) vs. 15-min JTE-013 3.9 ± 0.6 APs (n = 12), P = 0.66 ANOVA]. In contrast, pretreatment with 1 μM W140 had no effect on JTE-013-induced increase in excitability; after a 15-min exposure to JTE-013 the number of APs was increased to 9.0 ± 0.9 (n = 5) from a control value of 3.4 ± 0.2 APs (n = 10, P < 0.001 ANOVA Holm-Sidak all pairwise). These results were similar to those observed in untreated neurons (e.g., Fig. 5A). Taken together, these results suggest that JTE-013 may be acting as an agonist for S1PR1 via a G protein signaling pathway that is sensitive to PTX.
Neurons sensitized by JTE-013 do not express mRNA for S1PR2.
The findings with GDP-β-S and PTX suggest that JTE-013 may act as an agonist for G protein-coupled receptors. One possibility is that JTE-013 may activate rather than block S1PR2 in sensory neurons. To assess whether the sensitizing actions of JTE-013 could result from S1PR2 activation, neurons (3 total) that exhibited increased AP firing after JTE-013 were aspirated and then processed by RT-PCR to detect the presence of mRNA for S1PR2. Representative results from two individual neurons are illustrated in Fig. 6A, in which under control conditions the current ramp evoked 3 and 4 APs from cell 1 and cell 2, respectively. After a 10-min exposure to 100 nM JTE-013, the number of APs was increased to 8 in both cells. The genetic material obtained from these individual neurons underwent reverse transcription, after which the cDNA was split into two samples, one for the detection of S1PR2 and the other for the reference gene, HPRT. Surprisingly, neither cell 1 nor cell 2 expressed the mRNA for S1PR2 but did express the mRNA for HPRT (shown in Fig. 6B). As additional positive controls, samples were isolated from rat intact DRG and liver, in which both S1PR2 and HPRT were detected at their appropriate sizes. Thus these results indicate that JTE-013 was capable of augmenting the excitability of sensory neurons that did not express the mRNA for S1PR2 and demonstrate that JTE-013 sensitizes sensory neurons independently of this particular receptor.
Fig. 6.
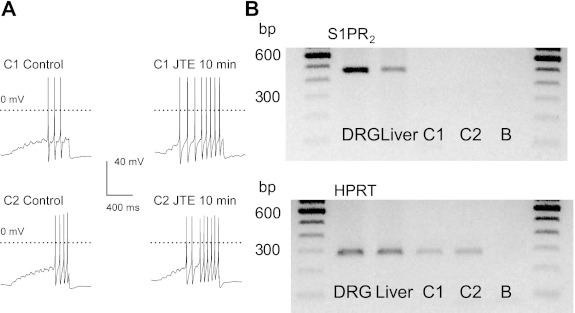
Single-cell RT-PCR demonstrates that neurons sensitized by JTE-013 do not express the mRNA for S1PR2. A: current-clamp recordings obtained from 2 representative neurons, cell 1 (C1) and cell 2 (C2), in which a 10-min exposure to 100 nM JTE-013 increased the number of APs evoked by the current ramp. B: the RT-PCR results obtained for these 2 individual neurons. Neither C1 nor C2 expressed the mRNA for S1PR2 (top), but they did express HPRT (bottom). As positive controls S1PR2 and HPRT were detected in mRNA isolated from intact dorsal root ganglia (DRG) and liver. Lanes labeled B (blank) represent reactions performed in the absence of any template.
JTE-013 enhances mechanical but not thermal sensitivity in vivo.
Our observations in isolated sensory neurons would suggest that JTE-013 could enhance nocifensive in vivo behaviors when injected into the hindpaw of a rat. To test this idea, rats underwent both mechanical stimulation with von Frey filaments and thermal stimulation with a Hargreaves apparatus (described in materials and methods). Baseline sensitivities were established with two measurements on separate days prior to the injection of 20 μl of 2 μM JTE-013. As summarized in Fig. 7A, injection of JTE-013 produced a significant increase in the mechanical sensitivity measured in the ipsilateral paw at the first time point tested (30 min); this heightened sensitivity was maintained for >2 h (n = 5, P < 0.001 at all postinjection time points, 2-way RM ANOVA with Bonferroni posttest). In contrast, no change in mechanical sensitivity was detected in the contralateral paw after injection of JTE-013 (n = 5). Injection of the vehicle (0.005% DMSO in the extracellular solution) had no effect on either ipsilateral or contralateral responses (n = 5 for each group). In contrast to the mechanical response, injection of JTE-013 caused no significant changes in thermal sensitivity 1 h after paw injection for ipsilateral or contralateral paws (Fig. 7B). Consistent with the increased excitability observed in isolated neurons, JTE-013 augmented the sensitivity of the rat's hindpaw to mechanical stimulation.
Fig. 7.
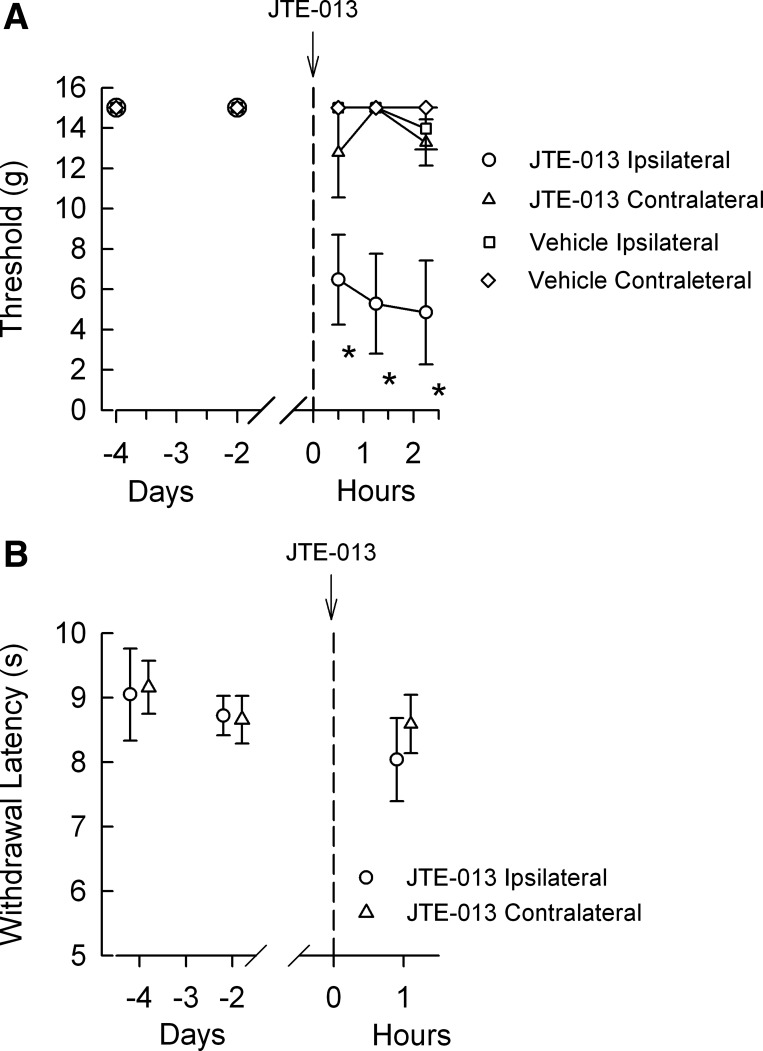
JTE-013 increases the sensitivity to mechanical but not thermal stimulation of the hindpaw. A: results obtained from 5 rats undergoing mechanical stimulation of the hindpaw with von Frey filaments. The time of JTE-013 injection is represented as the dashed line at time 0. B: results obtained for the thermal testing before and after injection of JTE-013. *Significant difference compared with the untreated control conditions at 4 and 2 days prior to injection (P < 0.05, 2-way RM ANOVA).
JTE-013 suppresses S1P-mediated cellular migration.
Previous studies demonstrated that S1P could stimulate or inhibit migration or invasion of cells depending on the particular cellular model system. In B16 melanoma cells, activation of S1PR1 and S1PR3 promotes cell migration whereas activation of S1PR2 inhibits cellular migration (Arikawa et al. 2003; Okamoto et al. 2000). The suppression of B16 migration could be reversed by pretreatment with the S1PR2 antagonist JTE-013 (Arikawa et al. 2003). Therefore, to confirm that our JTE-013 was effective, we performed an in vitro wound healing study as described by Arikawa et al. (2003) to test whether JTE-013 prevented the S1P-mediated inhibition of B16 migration. As shown in two representative experiments (see Fig. 8, A and B), under untreated control conditions B16 cells migrated back into the wound region 48 h after creating the wound. In cells treated with 100 nM S1P this migration was suppressed, suggesting S1P activation of S1PR2. However, cells treated with 1 μM JTE-013 and 100 nM S1P exhibited increased migration compared with S1P alone, indicating that JTE-013 was capable of preventing the S1P-mediated inhibition of cell migration. Treatment with JTE-013 alone had little effect on cell migration in the wound healing assay and was similar to the untreated control cells. Similar qualitative results were obtained in five additional wound healing assays. These results are quite similar to those reported previously by Arikawa et al. (2003) and demonstrate that JTE-013 can suppress the actions of S1PR2 in the B16 melanoma cell line.
Fig. 8.

S1P-induced inhibition of B16 cell migration is blocked by JTE-013 in the wound healing assay. A and B: 2 representative results (from a total of 7 assays). Panels labeled Control show the untreated controls at 0 and 48 h after the wound. Panels labeled S1P show the inhibition of migration produced by 100 nM S1P 48 h after the wound. Panels labeled S1P + JTE-013 demonstrate that 100 nM JTE-013 blocks the inhibition of cell migration produced by S1P after 48 h. Panels labeled JTE-013 show that cell migration after 48 h is not affected by exposure to 100 nM JTE-013 alone.
DISCUSSION
To our knowledge, the use of the selective antagonist JTE-013 to establish a role for S1PR2 in modulating the activity of peripheral neurons has not been investigated. In this report, we demonstrate that this putatively selective antagonist for S1PR2 can increase the number of APs evoked by a ramp of depolarizing current. The increase in AP firing produced by JTE-013 appeared to be receptor mediated, since antagonism of G protein-coupled receptor activation by GDP-β-S prevented the JTE-013-induced increase in excitability as well as the increase in excitability produced by PGE2. However, JTE-013 is not acting via S1PR2, as neurons sensitized by JTE-013 did not express the mRNA for S1PR2. Thus these results suggest that in sensory neurons JTE-013 has the capacity to modulate excitability by mechanisms that are mediated by a G protein pathway and the activation is independent of S1PR2. Consistent with these ideas, additional experiments demonstrated that the sensitization produced by JTE-013 was blocked by a 24-h treatment with PTX, an established antagonist of Gi/Go-type G protein activation. Surprisingly, the increase in excitability was also blocked by pretreatment with W146, a selective antagonist of S1PR1, whereas the inactive analog W140 did not alter the increased excitability. Previous work showed that W146 had a Ki of 70–80 nM for S1PR1 and exhibited no activity for S1PR2, -R3, or -R5 when expressed in a heterologous expression system (Sanna et al. 2006). Therefore, the parsimonious interpretation of these findings suggests that JTE-013 may be acting as an agonist at a Gi/Go protein-coupled receptor(s), perhaps S1PR1. It is well established that S1PR1 acts uniquely via a Gi/Go-type G protein (Obinata and Hla 2012; Rosen et al. 2009) and would thereby be sensitive to the actions of PTX. However, this notion may be confounded by the fact that possible off-target actions for W146 remain to be established.
Early studies indicated that JTE-013 could serve as a selective antagonist of S1PR2. JTE-013 blocked the binding of S1P to S1PR2 expressed in CHO cells with an IC50 of ∼20 nM, whereas JTE-013 had little to no effect on S1P binding to either S1PR1 or S1PR3 even at concentrations up to 10 μM (Osada et al. 2002). Functionally, JTE-013 blocked the increase in intracellular Ca2+ concentration produced by S1P both in aortic smooth muscle cells (Osada et al. 2002) and in CHO cells expressing S1PR2 but not S1PR1 or S1PR3 (Arikawa et al. 2003). These observations are in contrast to our finding that the JTE-013-induced increase in excitability was blocked by an S1PR1 antagonist. This distinction may suggest significant differences in the regulation/modulation of S1PR1 activation between CHO cells and sensory neurons such that a neuronal system permits JTE-013 to behave as a functional agonist. Another possibility is that in the neuronal membrane there exist other receptors or binding partners, which are not found in CHO cells, that form an association with S1PR1 that regulates its activation. For example, studies have demonstrated important interactions between the receptor tyrosine kinase and G protein-coupled receptor pathways that are distinct from the process of transactivation (Alderton et al. 2001; Pyne et al. 2007; Waters et al. 2005). This work demonstrated that S1PR1 appears to form an integrative signaling complex with the PDGF-β receptor that facilitates activation of p42/p44 MAPK. Perhaps association with some undefined signaling partner in sensory neurons permits functional activation of S1PR1 by JTE-013. This would be an important area of future investigation to elucidate potential binding partners in the activation of S1PR1 in neurons as well as contributing factors in the mechanisms of action for JTE-013.
In the CNS, S1P produces vasoconstriction; treatment with JTE-013 blocked this effect in both the spiral modiolar artery (Kono et al. 2007) and the cerebral basilar artery (Salomone et al. 2008). Taken together, results obtained in nonneuronal tissues indicate that JTE-013 can block the functional outcomes of S1PR2 activation by S1P. However, recent findings indicate that JTE-013 may not be as selective as previous studies indicated. Salomone et al. (2008) observed that vasoconstriction of the basilar artery produced by elevated KCl, U46619 (a thromboxane A1 agonist), or endothelin-1 was suppressed by JTE-013. Surprisingly, these authors also demonstrated that JTE-013 was capable of blocking S1P-mediated vasoconstriction in S1PR2−/− knockout mice. These results are consistent with our observations that JTE-013 enhances excitability in neurons that did not express mRNA for S1PR2. In another study, a 6-day treatment with JTE-013 significantly reduced the incidence of diabetes produced by streptozotocin (Imasawa et al. 2010); however, it was previously reported that administration of FTY720 could prevent and also reverse the incidence of diabetes in a nonobese diabetic mouse model (Maki et al. 2005). The actions of JTE-013 in reducing the incidence of diabetes need further investigation, since FTY720 is an activating ligand for four of the five S1PRs but not for S1PR2 (Brinkmann 2009; Brinkmann et al. 2002; Brinkmann and Lynch 2002). Furthermore, in MDA-MB-453 breast cancer cells JTE-013 was demonstrated to be an antagonist at S1PR4 (Long et al. 2010). Issues regarding the selectivity of JTE-013 have been reviewed recently by Pyne and Pyne (2011) and Salomone and Waeber (2011). Taken together, early studies indicated that JTE-013 specifically blocked S1PR2; however, more recent work is beginning to suggest that this antagonist may have actions at other S1PRs and also targets that presently remain undefined.
Similar to our findings obtained from isolated sensory neurons, injection of JTE-013 into the hindpaw of a rat produced enhanced sensitivity to mechanical stimulation; however, there was no effect on the thermal sensitivity. The AP recordings were obtained from both capsaicin-insensitive and -sensitive neurons; these results are consistent with the JTE-013-induced increased mechanical sensitivity. Studies have demonstrated that sensory neurons other than heat nociceptors express the capsaicin receptor TRPV1, and TRPV1 can be activated or sensitized by many stimuli in addition to heat (Nilius et al. 2007; Ro et al. 2009; Venkatachalam and Montell 2007; Yamamoto et al. 2008). Additionally, in vivo studies have shown that mechanical hypersensitivity and allodynia in several different pain models can be modulated by agonists and antagonists of TRPV1 (Huang et al. 2006; Kanai et al. 2007; Patwardhan et al. 2009; Ro et al. 2009; Watabiki et al. 2011; Yu et al. 2008). Thus it is possible that JTE-013 is capable of augmenting the sensitivity of both TRPV1-expressing and TRPV1-nonexpressing mechanoreceptive sensory neurons. Exploring the exact mechanism of action for JTE-013 is an interesting question for future studies.
In conclusion, our results indicate that JTE-013, independently of its actions at S1PR2, is capable of increasing the excitability of sensory neurons. In in vivo behavioral studies, JTE-013 can enhance sensitivity to mechanical stimulation. Our findings demonstrate that JTE-013 could be acting as an agonist at other G protein-coupled receptors. To our knowledge, the metabolism of JTE-013 is poorly understood; it is possible that a metabolic product could be responsible for the enhancement of neuronal excitability. The exact mechanisms of JTE-013's actions remain undefined, and thus the potential role of S1PR2 in regulating neuronal function will be difficult to establish until better pharmacological approaches are developed.
GRANTS
This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR-015481-01 from the National Center for Research Resources [National Institutes of Health (NIH)]. This work was supported by National Institute of Neurological Disorders and Stroke (NIH) Grants R01 NS-046084 and R01 NS-060853.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.L., X.X.C., W.X., J.A.S., J.-M.Z., and G.D.N. conception and design of research; C.L., X.X.C., and W.X. performed experiments; C.L., X.X.C., W.X., J.A.S., and G.D.N. analyzed data; C.L., W.X., J.A.S., J.-M.Z., and G.D.N. interpreted results of experiments; C.L., J.A.S., J.-M.Z., and G.D.N. drafted manuscript; C.L., X.X.C., W.X., J.A.S., J.-M.Z., and G.D.N. edited and revised manuscript; C.L., X.X.C., W.X., J.A.S., J.-M.Z., and G.D.N. approved final version of manuscript; J.A.S. and G.D.N. prepared figures.
REFERENCES
- Alderton F, Rakhit S, Kong KC, Palmer T, Sambi B, Pyne S, Pyne NJ. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J Biol Chem 276: 28578–28585, 2001 [DOI] [PubMed] [Google Scholar]
- Anliker B, Chun J. Cell surface receptors in lysophospholipid signaling. Semin Cell Dev Biol 15: 457–465, 2004 [DOI] [PubMed] [Google Scholar]
- Arikawa K, Takuwa N, Yamaguchi H, Sugimoto N, Kitayama J, Nagawa H, Takehara K, Takuwa Y. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J Biol Chem 278: 32841–32851, 2003 [DOI] [PubMed] [Google Scholar]
- Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol 158: 1173–1182, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277: 21453–21457, 2002 [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Lynch KR. FTY720: targeting G-protein-coupled receptors for sphingosine 1-phosphate in transplantation and autoimmunity. Curr Opin Immunol 14: 569–575, 2002 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63, 1994 [DOI] [PubMed] [Google Scholar]
- Chi XX, Nicol GD. Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J Neurophysiol 98: 2683–2692, 2007 [DOI] [PubMed] [Google Scholar]
- Chi XX, Nicol GD. The sphingosine 1-phosphate receptor, S1PR1, plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol 104: 2741–2748, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience 66: 459–466, 1995 [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 90: 1–6, 2001 [DOI] [PubMed] [Google Scholar]
- Dixon W. The up-and-down method for small samples. J Am Stat Assoc 60: 967–978, 1965 [Google Scholar]
- Eckstein F, Cassel D, Levkovitz H, Lowe M, Selinger Z. Guanosine 5′-O-(2-thiodiphosphate). An inhibitor of adenylate cyclase stimulation by guanine nucleotides and fluoride ions. J Biol Chem 254: 9829–9834, 1979 [PubMed] [Google Scholar]
- Goetzl EJ, Rosen H. Regulation of immunity by lysosphingolipids and their G protein-coupled receptors. J Clin Invest 114: 1531–1537, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch-clamp techniques for high-resolution current recordings from cells and cell free membrane patches. Pflügers Arch 391: 85–100, 1981 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150, 2008 [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88, 1988 [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci 15: 5411–5419, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T. Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol 15: 513–520, 2004 [DOI] [PubMed] [Google Scholar]
- Hla T, Maciag T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J Biol Chem 265: 9308–9313, 1990 [PubMed] [Google Scholar]
- Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev 43: 143–201, 1991 [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol 4: 197–206, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imasawa T, Koike K, Ishii I, Chun J, Yatomi Y. Blockade of sphingosine 1-phosphate receptor 2 signaling attenuates streptozotocin-induced apoptosis of pancreatic beta-cells. Biochem Biophys Res Commun 392: 207–211, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Hara T, Imai A, Sakakibara A. Differential involvement of TRPV1 receptors at the central and peripheral nerves in CFA-induced mechanical and thermal hyperalgesia. J Pharm Pharmacol 59: 733–738, 2007 [DOI] [PubMed] [Google Scholar]
- Kono M, Belyantseva IA, Skoura A, Frolenkov GI, Starost MF, Dreier JL, Lidington D, Bolz SS, Friedman TB, Hla T, Proia RL. Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem 282: 10690–10696, 2007 [DOI] [PubMed] [Google Scholar]
- Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci 8: 2394–2405, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JS, Fujiwara Y, Edwards J, Tannahill CL, Tigyi G, Pyne S, Pyne NJ. Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J Biol Chem 285: 35957–35966, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki T, Gottschalk R, Ogawa N, Monaco AP. Prevention and cure of autoimmune diabetes in nonobese diabetic mice by continuous administration of FTY720. Transplantation 79: 1051–1055, 2005 [DOI] [PubMed] [Google Scholar]
- Meyer zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta 1768: 923–940, 2007 [DOI] [PubMed] [Google Scholar]
- Miller RJ, Jung H, Bhangoo SK, White FA. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol 194: 417–49, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10: 23–36, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 87: 165–217, 2007 [DOI] [PubMed] [Google Scholar]
- Obinata H, Hla T. Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol 34: 73–91, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Takuwa N, Yokomizo T, Sugimoto N, Sakurada S, Shigematsu H, Takuwa Y. Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol Cell Biol 20: 9247–9261, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera A, Rivera J. Sphingolipids and the balancing of immune cell function: lessons from the mast cell. J Immunol 174: 1153–1158, 2005 [DOI] [PubMed] [Google Scholar]
- Osada M, Yatomi Y, Ohmori T, Ikeda H, Ozaki Y. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochem Biophys Res Commun 299: 483–487, 2002 [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc Natl Acad Sci USA 106: 18820–18824, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruska JC, Napaporn J, Johnson RD, Gu JG, Cooper BY. Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol 84: 2365–2379, 2000 [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Selectivity and specificity of sphingosine 1-phosphate receptor ligands: “off-targets” or complex pharmacology? Front Pharmacol 2: 26, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Waters CM, Long JS, Moughal NA, Tigyi G, Pyne S. Receptor tyrosine kinase-G-protein coupled receptor complex signaling in mammalian cells. Adv Enzyme Regul 47: 271–280, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol 8: 753–763, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro JY, Lee JS, Zhang Y. Activation of TRPV1 and TRPA1 leads to muscle nociception and mechanical hyperalgesia. Pain 144: 270–277, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem 78: 743–768, 2009 [DOI] [PubMed] [Google Scholar]
- Salomone S, Potts EM, Tyndall S, Ip PC, Chun J, Brinkmann V, Waeber C. Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br J Pharmacol 153: 140–147, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomone S, Waeber C. Selectivity and specificity of sphingosine-1-phosphate receptor ligands: caveats and critical thinking in characterizing receptor-mediated effects. Front Pharmacol 2: 9, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem 92: 913–922, 2004 [DOI] [PubMed] [Google Scholar]
- Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2: 434–441, 2006 [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10: 1361–1368, 2007 [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4: 397–407, 2003 [DOI] [PubMed] [Google Scholar]
- Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol 688: 141–155, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience 32: 577–580, 1989 [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev 60: 181–195, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg 105: 838–847, 2007 [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Lee MJ, Menzeleev R, Olivera A, Edsall L, Cuvillier O, Thomas DM, Coopman PJ, Thangada S, Liu CH, Hla T, Spiegel S. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol 142: 229–240, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 76: 387–417, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabiki T, Kiso T, Kuramochi T, Yonezawa K, Tsuji N, Kohara A, Kakimoto S, Aoki T, Matsuoka N. Amelioration of neuropathic pain by novel transient receptor potential vanilloid 1 antagonist AS1928370 in rats without hyperthermic effect. J Pharmacol Exp Ther 336: 743–750, 2011 [DOI] [PubMed] [Google Scholar]
- Waters CM, Connell MC, Pyne S, Pyne NJ. c-Src is involved in regulating signal transmission from PDGFbeta receptor-GPCR(s) complexes in mammalian cells. Cell Signal 17: 263–277, 2005 [DOI] [PubMed] [Google Scholar]
- Weigert A, Weis N, Brüne B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology 214: 748–760, 2009 [DOI] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 4: 834–844, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto W, Sugiura A, Nakazato-Imasato E, Kita Y. Characterization of primary sensory neurons mediating static and dynamic allodynia in rat chronic constriction injury model. J Pharm Pharmacol 60: 717–722, 2008 [DOI] [PubMed] [Google Scholar]
- Yu L, Yang F, Luo H, Liu FY, Han JS, Xing GG, Wan Y. The role of TRPV1 in different subtypes of dorsal root ganglion neurons in rat chronic inflammatory nociception induced by complete Freund's adjuvant. Mol Pain 4: 61, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol 96: 1042–1052, 2006 [DOI] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J Physiol 544: 385–402, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]