

Abstract
The chemokine stromal cell-derived factor-1α (SDF-1α) has multiple effects on neuronal activity, survival, and death under conditions that generate a proinflammatory microenvironment within the brain, via signaling through C-X-C-type chemokine receptor 4 (CXCR4), although the underlying cellular/molecular mechanisms are unclear. Using rat hippocampal neurons, we investigated distinct modifications in the voltage-gated K+ (Kv) channel Kv2.1 in response to short- and long-term SDF-1α/CXCR4-mediated signaling as an underlying mechanism for CXCR4-dependent regulation of neuronal survival and death. Acute exposure of neurons to SDF-1α led to dynamic dephosphorylation and altered localization of Kv2.1 channel, resulting in enhanced voltage-dependent activation of Kv2.1-based delayed-rectifier Kv currents (IDR). These changes were dependent on CXCR4- and/or NMDA receptor-mediated activation of calcineurin and provide neuroprotection. However, prolonged SDF-1α treatment leads to CXCR4-mediated activation of p38 mitogen-activated protein kinase, resulting in phosphorylation of Kv2.1 at S800 and enhanced surface trafficking of the channel protein, resulting in increased IDR/Kv2.1 current density. This, in combination with sustained dephosphorylation-induced enhancement of the voltage-dependent activation of IDR/Kv2.1, predisposed neurons to excessive K+ efflux, a vital step for the neuronal apoptotic program. Such apoptotic death was dependent on CXCR4 and Kv2.1 function and was absent in cells expressing the Kv2.1–S800A mutant channel. Furthermore, similar modifications in Kv2.1 and CXCR4/Kv2.1-dependent apoptosis were observed following treatment of neurons with the human immunodeficiency virus-1 (HIV-1) glycoprotein gp120. Therefore, distinct modifications in Kv2.1 in response to short- and long-term CXCR4-mediated signaling could provide a basis for neuroprotection or apoptosis in neuropathologies, such as neuroinflammation, stroke, brain tumors, and HIV-associated neurodegeneration.
Introduction
The chemokine stromal cell-derived factor-1α (SDF-1α) and its receptor C-X-C-type chemokine receptor 4 (CXCR4) participate in many developmental and pathological processes, including neuronal migration and axon pathfinding in the developing brain (Lazarini et al., 2003; Pujol et al., 2005; Mithal et al., 2012). CXCR4 is expressed in neurons, astrocytes, and microglia (Banisadr et al., 2002), in which it can be activated by SDF-1α released during neuroinflammation, ischemic stroke, and brain tumor growth, as well as by the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein gp120, resulting in elevated [Ca2+]i and induction of diverse signaling cascades (Lo et al., 1992; Meucci et al., 1998; Guyon and Nahon, 2007). Enhanced CXCR4 signaling has been implicated in neuronal death associated with ischemic stroke (Hill et al., 2004), glioblastoma (Rempel et al., 2000), and HIV-associated neurodegeneration (HAND) (Kaul and Lipton, 1999). SDF-1α-mediated cytotoxicity in neurons operates via an NMDA receptor-mediated elevation in [Ca2+]i (Guyon and Nahon, 2007; Li and Ransohoff, 2008) and p38 mitogen-activated protein kinase (MAPK)-dependent induction of apoptosis (Hesselgesser et al., 1998; Kaul and Lipton, 1999). In contrast, SDF-1α signaling has also been shown to promote neuronal survival by increasing the expression of Rb transcription repressor (Khan et al., 2008), downregulating expression of the NR2B subunit of the NMDA receptor, and decreasing Ca2+ influx (Nicolai et al., 2010). However, the distinct mechanism(s) underlying SDF-1α-mediated regulation of neuronal excitability, survival, and death are poorly understood.
Proper functioning and survival of neurons is maintained by homeostatic control of intrinsic excitability (Stuart et al., 1997). Voltage-gated K+ (Kv) channels exert such control by limiting the onset, frequency, and backpropagation of action potential firing (Johnston et al., 2010). Kv2.1, the principal Kv channel underlying delayed-rectifier currents (IDR) in most mammalian brain neurons (Murakoshi and Trimmer, 1999; Malin and Nerbonne, 2002), regulates excitability during periods of high-frequency firing (Du et al., 2000). The extensive and constitutive phosphorylation of Kv2.1 (Park et al., 2006) regulates the characteristic clustered localization and functional properties of the channel (Misonou et al., 2005a). Increased excitatory activity and neuromodulatory stimuli lead to Ca2+/calcineurin-dependent dephosphorylation of Kv2.1, resulting in lateral dispersion of channel clusters and an ∼20 mV hyperpolarizing shift in the voltage dependence of channel activation (Misonou et al., 2004; Mohapatra and Trimmer, 2006). This enhances Kv2.1 channel opening with minimal depolarization, limiting firing frequency and increasing the refractory period duration, thereby providing neuroprotection during the early phases of hyperexcitable assaults (Misonou et al., 2005b; Mohapatra et al., 2009). In contrast, p38 MAPK-mediated phosphorylation of Kv2.1 increases current density and K+ efflux, resulting in the induction of neuronal apoptosis (Pal et al., 2003; Redman et al., 2007). However, the contribution of Kv2.1 to induction of neuronal survival/death by specific pathophysiological mediators (such as SDF-1α) is not known.
Here, we report bimodal regulation of neuronal excitability and cell death via distinct SDF-1α-mediated signaling events that lead to differential modifications in Kv2.1 channel localization and function. Calcineurin-mediated Kv2.1 dephosphorylation upon acute SDF-1α/CXCR4 signaling is neuroprotective, whereas sustained SDF-1α/CXCR4 signaling leads to p38 MAPK-mediated increased surface trafficking of Kv2.1, causing pathological K+ efflux and neuronal apoptosis.
Materials and Methods
All experiments involving the use of rats and the procedures followed therein were approved by the University of Iowa Institutional Animal Care and Use Committee and were in strict accordance with the Guide for the Care and Use of Laboratory Animals described by the National Institutes of Health. Every effort was made to minimize the number of rats used and their suffering.
Chemicals and reagents.
Recombinant mouse SDF-1α, WZ811 (N1,N4-Di-2-pyridinyl-1,4-benzenedimethanamine), AMD3100 (1,1′-[1,4-phenylenebis-(methylene)]-bis-(1,4,8,11-tetraazacyclotetradecane) octahydrochloride), (+)-MK-801 [(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate], FK506 [(3S,4R,5S,8R, 9E,12S,14S,15R,16S,18R,19R,26aS)-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[(1E)-2-[(1R,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propen-1-yl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)tetrone], and SB203580 (4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine) were purchased from R & D Systems/Tocris Bioscience; purified HIV-1 IIIB gp120 was from ImmunoDiagostics; recombinant stromatoxin-1 (ScTx-1), recombinant heteropodatoxin-2 (HpTx-2), trypsin, glutamate, and 6,7-dinitroquinoxaline-2,3-dione (DNQX) were from Sigma-Aldrich; and fura-2-AM, HBSS, B27, DMEM, GlutaMAX, penicillin/streptomycin, and Neurobasal media were from Invitrogen. All other chemicals used in this study were purchased from Sigma, Bio-Rad, Roche Applied Science, VWR, and Thermo Fisher Scientific. All NeuroMab antibodies were purchased from the University of California, Davis/National Institutes of Health NeuroMab Facility through Antibodies Inc.
Primary culture of rat hippocampal neurons.
Hippocampal neurons from rat embryos were isolated and cultured as described previously (Mohapatra et al., 2008). Briefly, hippocampi from Sprague Dawley rat embryos of either sex at stage E18 (Harlan) were removed and incubated in HBSS with trypsin (1 mg/ml) for 12 min at 37°C. The hippocampi were then washed three times with HBSS, followed by trituration to dissociate cells. After centrifugation at 1100 rpm for 3 min, the dissociated cells were resuspended in Neurobasal medium containing B27, 0.6 mm glutamine, and 5% horse serum. Cells were then counted and plated onto poly-l-lysine-coated glass coverslips at a density of 100,000 cells per 35 mm dish (for immunocytochemical and electrophysiological experiments) and into poly-l-lysine-coated six-well culture dishes at a density of 200,000 cells per well (for biochemical experiments). The cells were incubated in a 5% CO2 incubator at 37°C for 3–4 h, after which the media were replaced with serum-free Neurobasal/B27/glutamine media and maintained at 37°C in a 5% CO2 incubator. One-third of the medium was exchanged weekly. All experiments were performed on neurons that were cultured for 16–18 d in vitro (DIV). Using immunostaining with mitogen-activated protein 2 (MAP2) and nuclear marker of cultured hippocampal neurons, it is estimated that ∼40–50% of the cells in our mixed neuronal culture at 16–18 DIV are neurons, with the remainder being astrocytes and microglia.
Site-directed mutagenesis and transfection of human embryonic kidney 293 cells.
Recombinant rat Kv2.1 and rat Kv1.4 cDNAs in pRBG4 plasmids were generously provided by Dr. James S. Trimmer (University of California, Davis, Davis, CA). Mutagenesis was performed on the pRBG4–rKv2.1 plasmid to generate the rKv2.1–S800A mutant using the quick-change site-directed mutagenesis kit (Agilent Technologies). The human CXCR4 cDNA (Missouri cDNA Resource Center) was subcloned into the enhanced yellow fluorescent protein (pEYFP)–N1 plasmid (Clontech), using standard protocols. Human embryonic kidney 293 (HEK293) cells (American Type Culture Collection) were cultured in DMEM with 1× GlutaMAX, 1× penicillin/streptomycin, and 10% fetal bovine serum (HyClone) and maintained at 37°C in a 5% CO2 incubator. Cells were transiently transfected with wild-type (WT) or S800A mutant Kv2.1 plasmids (0.5 μg), either with the reporter plasmid (0.5 μg of pEYFP–N1) or with the hCXCR4–pEYFP–N1 plasmid (0.5 μg) using Lipofectamine 2000 (Invitrogen), as per the instruction of the manufacturer. Transfected cells were used for experiments within 36–48 h.
Immunohistochemical staining of rat brain sections.
Adult rats were anesthetized with pentobarbital and immediately perfused intracardially with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4. Sagittal brain sections (40 μm thickness) were generated with a cryostat (CM3050S; Leica Microsystems) and stained as described previously (Misonou et al., 2004, 2005b). Briefly, free-floating sections were blocked and permeabilized with 10% goat serum and 0.3% Triton X-100 in 0.1 m PB and incubated overnight with mouse monoclonal anti-Kv2.1 (K89/34, 5 μg/ml; NeuroMab) and rabbit polyclonal anti-CXCR4 (5 μg/ml; Imgenex). The sections were then incubated in species-specific Alexa Fluor 488- and 555-conjugated secondary antibodies (1:1000; Invitrogen) for 3 h, followed by mounting onto SuperFrost Plus microscope slides (Thermo Fisher Scientific) with ProLong Gold antifade mounting medium (Invitrogen). Images were captured using a BX61WI microscope equipped with the Fluoview 300 laser-scanning confocal imaging system (Olympus) with a 10× and 60× UPlanSApo objective [numerical aperture (NA) 0.25, 10×; NA 0.17, 60×; Olympus]. Z-stack images at 60× were compiled from 11 individual images taken through 20 μm in the z-axis at 2 μm increments and merged using the same Fluoview image acquisition and analysis software.
Immunocytochemical staining of cultured neurons and mammalian cell lines.
Immunocytochemical staining of cells were performed as described previously (Mohapatra et al., 2008). Cultured rat hippocampal neurons and HEK293 cells on glass coverslips were fixed for 30 min at 4°C with 3% PFA in 0.1 m PB, pH 7.3 (additionally with 4% sucrose for neurons). Cells were then permeabilized and blocked in a 4% solution of nonfat dry milk in Tris-buffered saline, containing 0.1% Triton X-100 (Blotto). The primary antibodies [rabbit polyclonal anti-MAP2B (1:1000; Sigma), rabbit polyclonal anti-CXCR4 (1:250; Imgenex), mouse monoclonal anti-Kv2.1 (1:1000; clone K89/34; NeuroMab), and rabbit polyclonal Alexa Fluor-488-conjugated anti-cleaved-caspase-3 (c-caspase-3) (1:100; Cell Signaling Technology)] were incubated for 1 h in 4% Blotto, followed by species-specific Alexa Fluor 488-, 555-, and 633-conjugated secondary antibodies (1:2000) for 1 h in 4% Blotto. Coverslips were then mounted onto glass slides with ProLong Gold antifade mounting medium. Immunofluorescence images were captured by MRc-5 digital camera connected to a Carl Zeiss AxioImager epifluorescence microscope, using the AxioVision software (Carl Zeiss). Images were taken with a 63X Plan-Apochromat objective (NA 1.4; Carl Zeiss). All the images were transferred to Photoshop software (Adobe Systems) as TIFF files. Slides containing HEK293 cells or hippocampal neurons stained with Kv2.1 were coded such that counting was conducted in a blinded manner. Cells expressing Kv2.1 were arbitrarily divided into three localization patterns/groups: “clustered,” in which the vast majority of Kv2.1 immunoreactivity is confined to distinct surface clusters; “intermediate,” in which some Kv2.1 clustering remains, but there is also a substantial pool of diffuse Kv2.1 staining; and “dispersed,” in which all or the vast majority of Kv2.1 immunoreactivity is diffused across the membrane. Results were obtained by counting ≥500 cells for each treatment condition, from four or more independent batches of cultures.
Ratiometric Ca2+ imaging.
Functional Ca2+ imaging on cultured rat hippocampal neurons was performed as described previously (Loo et al., 2012). Neurons on glass coverslips were incubated at room temperature (22°C) for 30 min with 5 μm of the AM form of the Ca2+-sensitive dye fura-2 (Invitrogen). The coverslip was then placed in the recording chamber mounted on the stage of an inverted IX-71 microscope (Olympus) and perfused for 10 min with the standard extracellular HEPES–HBSS (HH buffer) composed of the following (in mm): 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 NaHPO4, 3 NaHCO3, 10 glucose, and 10 HEPES, pH 7.4, with NaOH (310 mOsm/kg with sucrose). Fluorescence was alternately excited at 340 and 380 nm (both 12 nm bandpass) using the Polychrome IV monochromator (T.I.L.L. Photonics), via a 20× objective (NA 0.75; Olympus). Emitted fluorescence was collected at 510 (80) nm using an IMAGO CCD camera (T.I.L.L. Photonics). Pairs of 340/380 nm images were sampled at 1 Hz. Bath application of glutamate (10 μm, 5 min) or SDF-1α (100 nm, 5 min) or WZ811 plus SDF-1α (100 nm each, 5 min) or MK-801 (10 μm) plus DNQX (10 μm) plus SDF-1α (100 nm; 5 min) was performed in HH buffer, followed by washing with HH buffer for 5 min. The fluorescence ratio (r = F340/F380) values over time were processed and analyzed using TILLvisION 4.0.1.2 (T.I.L.L. Photonics) and Origin 7.0 (OriginLab) software. All the Ca2+ imaging experiments with or without drug treatments were performed in multiple batches of rat hippocampal neuron cultures.
Biochemical analysis of Kv2.1 and p38 MAPK.
SDS-PAGE (7.5% for Kv2.1 and 9% for p38 MAPK) and immunoblotting of Triton X-100-soluble protein extracts from cultured rat hippocampal neurons and transfected HEK293 cells (without or with drug treatments) were performed as described previously (Misonou et al., 2005b; Mohapatra and Trimmer, 2006; Mohapatra et al., 2008). For alkaline phosphatase (AP) treatment of neuronal lysates, aliquots of lysates were incubated with calf intestinal AP (100 U/ml) in lysis buffer containing 0.1% SDS for 2 h at 37°C. As a control, lysate aliquots without AP were incubated for 2 h at 37°C. Proteins transferred to nitrocellulose membrane were blocked with 4% Blotto (without Triton X-100) and incubated with primary antibodies [mouse monoclonal anti-Kv2.1 (1:1000; clone K89/34; NeuroMab) or rabbit polyclonal anti-p38 MAPK or rabbit monoclonal anti-phospho-p38 MAPK (1:800; both from Cell Signaling Technology)]. After washing three times with Tris-buffered saline, blots were incubated with HRP-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibodies (1:10,000; Antibodies Inc.), followed by enhanced chemiluminescence reagent (PerkinElmer Life and Analytical Sciences), and immunoreactive bands were visualized by exposure to x-ray films (BioMax; Eastman Kodak). x-Ray signal intensities of immunoreactive bands were quantified using the NIH Image J software, as described previously (Misonou et al., 2005b). All quantifications were performed on a minimum of three independent experimental samples. For quantification of phosphorylation-dependent mobility of Kv2.1 in SDS-PAGE/immunoblots, based on molecular weight, the signal intensity of the Kv2.1 band in each individual sample was normalized to the maximal intensity and plotted as a function of protein molecular weight. Data are plotted as mean signal intensity distribution across a molecular weight range of ∼160 to ∼90 kDa. Quantification was performed on four to eight independent experimental samples.
For surface biotinylation assays, cultured neurons and transfected HEK293 cells were treated with the mentioned drugs in the presence of boc-aspartyl(OMe)-fluoromethylketone (BAF; 10 μm), a cell-permeable cysteine protease inhibitor, to prevent apoptosis. Cells were then treated with EZ-Link Sulfo-NHS-SS-biotin from a cell-surface protein isolation kit (Thermo-Pierce) according to the instructions of the manufacturer. After cell lysis and immediately before pull-down of biotinylated surface proteins, an aliquot of each sample was taken for total protein assessment using a BCA assay kit (Thermo-Pierce). This value was then used to normalize the level of biotinylated Kv2.1 subsequently detected in immunoblot and quantified by NIH Image J software, as described previously. Total lysates demonstrated strong immunoreactivity for intracellular proteins (tubulin, mortalin) and were not present in biotinylated fractions, indicating that biotinylated proteins were exclusively from the cell surface (data not shown).
Immunocytochemistry-based cell death assay.
For the quantification of cell death, an immunofluorescence-based Live–Dead cell viability assay kit (Invitrogen) was used, as described previously (Misonou et al., 2005b), following the instructions of the manufacturer with slight modifications. Briefly, cells were incubated with 1 μm ethidium homodimer (EthD-1) in 0.1 m PBS, pH 7.4, for 30 min after various drug treatments and immediately before fixation with 3% PFA. Cells were then permeabilized and blocked in a 4% Blotto containing 0.1% Triton X-100, followed by incubation with 4% Blotto containing rabbit polyclonal anti-MAP2B (1:1000; Sigma) for 1 h and subsequently with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (1:2000) for 1 h in 4% Blotto. After mounting onto slides, immunofluorescence images were captured by an MRc-5 digital camera connected to a Carl Zeiss AxioImager epifluorescence microscope (Carl Zeiss). Images were taken with a 10× Plan-Apochromat objective (NA 0.25; Carl Zeiss). All the images were transferred to PhotoShop software (Adobe Systems) as TIFF files. Cell counting was conducted in a blinded manner, taking into account only MAP2-positive neurons or GFP-positive HEK293 cells. Results were obtained by counting ≥500 cells for each treatment condition, from four or more independent batches of neuron/HEK293 cell cultures.
Electrophysiology and data analyses.
Currents were recorded from cultured rat hippocampal neurons and transfected HEK293 cells at room temperature with the whole-cell configuration of the patch-clamp technique, as described previously (Mohapatra et al., 2009). Patch pipettes were pulled from borosilicate glass tubes (TW150F-4; World Precision Instruments) and heat-polished at the tip using a microforge (MF200; World Precision Instruments) to give a resistance of 3–6 MΩ when filled with the pipette solution consisting of the following (in mm): 140 KCl, 5 NaCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 10 EGTA, and 0.4 Na-ATP, pH 7.3. Cells were bathed in extracellular buffer consisting of the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4. For recordings in neurons, 0.5 μm tetrodotoxin was added to the extracellular buffer to block fast-activating Nav currents. Cells were treated with SDF-1α in culture media for 30 min or 3 h at 37°C and in the bath solution before the beginning of recording. For 3 h treatment groups, 10 μm BAF was included in the culture media to prevent apoptosis. Currents were recorded with an Axopatch 200B patch-clamp amplifier connected to a Digidata 1440A data acquisition system (Molecular Devices), with a sampling rate of 10 kHz and filtering at 2 kHz using a digital Bessel filter. pClamp 10 software (Molecular Devices) was used for the acquisition of currents, and Clampfit 10 (Molecular Devices) and Origin 7.0 (OriginLab) software were used for the analysis of currents and preparing traces/figures. For voltage-dependent activation experiments on neuronal IDR or Kv2.1 currents in HEK293, cells were held at −80 mV and step depolarized to +80 mV for 250 ms with +10 mV increments. For neurons, a prepulse at −10 mV for 30 ms was given before each test pulse to inactivate the majority of IA. For voltage-dependent steady-state inactivation experiments, neurons were held at −100 mV and step depolarized to +40 mV for 10 s with +10 mV increments (conditioning pulses), followed by depolarization to +20 mV for 100 ms (test pulse). For inactivation experiments, 5 mm 4-AP was included in the extracellular buffer to inhibit the A-type K+ currents. All currents were capacitance and series resistance compensated, and leak subtractions were performed offline. The Nernst K+ equilibrium potential (EK) was calculated as −85 mV. For neuronal recordings, peak outward currents from the last 150 ms depolarized test pulses were taken as IDR, and peak outward Kv currents from the entire duration of test pulses were taken as Kv2.1 currents in HEK293 cells, for calculating current density and voltage-dependent activation/conductance. Current density was calculated by dividing the peak currents (picoamperes) at each individual depolarizing test pulses with the cell capacitance (picofarads). For steady-state inactivation, peak currents from the test pulse after each conditioning voltage pulses were taken for analysis. Voltage-dependent activation and steady-state inactivation curves were generated as described previously (Mohapatra et al., 2009), and the half-maximal voltage-dependent activation potentials (G½) and steady-state inactivation potentials (V½) are mentioned in the respective figures. Data are presented as means ± SEM or fitted value ± SE of the Boltzmann's equation fit for the conductance–voltage (G–V) and voltage dependence of inactivation curves.
Statistical analyses.
Data were analyzed using one-way ANOVA with post hoc Bonferroni's correction to test the statistical significance of the differences between various treatment groups. For densitometry analysis of phospho-p38 MAPK levels and electrophysiological analysis of voltage-dependent gating, data were analyzed using Student's t test. p < 0.05 in each set of data comparisons were considered statistically significant. All statistical analyses were conducted using SPSS version 19 software (IBM).
Results
Acute SDF-1α treatment leads to dephosphorylation and altered localization of Kv2.1
Expression of CXCR4 in hippocampal neurons and the downstream signaling cascades have been described previously (Banisadr et al., 2002; Guyon and Nahon, 2007). First, we verified the expression of CXCR4 and Kv2.1 in mammalian brain neurons. Like Kv2.1 (Vacher et al., 2008), CXCR4 is expressed in most rat brain neurons. Coimmunostaining of CXCR4 and Kv2.1 in rat brain sections revealed extensive coexpression of both proteins throughout the brain, particularly in the hippocampal formation (Fig. 1A,B). Kv2.1 staining is restricted to neuronal somata and proximal dendrites, whereas CXCR4 is more widely distributed throughout neurons, non-neuronal cells, and neuropil as a whole (Fig. 1A,B). These staining patterns or subcellular localization patterns of CXCR4 and Kv2.1 are maintained in cultured rat hippocampal neurons (Fig. 1C). We next verified the functional expression of CXCR4 in cultured rat hippocampal neurons by ratiometric Ca2+ imaging. Neurons were treated with 10 μm glutamate as a positive control, which rapidly increased [Ca2+]i (Fig. 2). Application of SDF-1α (100 nm) caused a comparable increase in [Ca2+]i with an initial time delay, which could be blocked by coapplication of the CXCR4 antagonist WZ811 [100 nm (Zhu et al., 2010)] and by coapplication of the selective noncompetitive NMDA receptor antagonist MK-801 and AMPA receptor antagonist DNQX (10 μm each; Fig. 2). These results are in agreement with previous reports that SDF-1α-mediated Ca2+ influx in neurons is dependent on NMDA receptor activation (Kaul and Lipton, 1999; Zheng et al., 1999; Kaul et al., 2001; Guyon and Nahon, 2007; Li and Ransohoff, 2008), and such trans-glutamate signaling explains the initial delay in [Ca2+]i elevation in response to SDF-1α compared with glutamate-induced [Ca2+]i elevation.
Figure 1.

Kv2.1 and CXCR4 are coexpressed in rat hippocampal neurons. A, Photomicrographs of rat brain sections (40 μm, sagittal) immunostained with anti- Kv2.1 (red) and anti-CXCR4 (green) antibodies. Kv2.1 is expressed in the soma and proximal dendrites in CA1, CA3, and dentate gyrus of the hippocampal formation. CXCR4 (green) is expressed in the same pyramidal cells in CA1 and CA3 and to a lesser extent in the stratum granulosum. CXCR4 is also expressed in the outer molecular layer of the dentate gyrus. Scale bars, 250 μm. B, Confocal images taken in the stratum pyramidale of CA1 (left) and CA3 (right) at marked rectangular area in A, displaying coexpression of Kv2.1 (red) and CXCR4 (green) within the same neurons. Kv2.1 is expressed on somata and proximal dendrites, whereas the majority of CXCR4 puncta appear to be confined to somata. Scale bars, 50 μm. C, Photomicrographs of cultured rat hippocampal neurons immunostained with anti-MAP2 (blue), anti-Kv2.1 (red), and anti-CXCR4 (green) antibodies. Kv2.1 maintains the similar somatodendritic distribution in the form of clusters, as seen in vivo, and CXCR4 expression is still predominantly restricted to the soma. Scale bars, 25 μm.
Figure 2.

Acute SDF-1α exposure leads to CXCR4 and NMDA/AMPA receptor-dependent [Ca2+]i elevation in hippocampal neurons. Representative traces of agonist-induced [Ca2+]i elevation in cultured rat hippocampal neurons, as quantified in ratiometric Ca2+ imaging with the fura-2 AM Ca2+-sensitive dye. Traces from six neurons (somata and dendrites up to ∼100 μm length) are presented in the form of F340/F380 fluorescence ratios, showing SDF-1α-induced (100 nm) elevation in [Ca2+]i that is blocked by coapplication of the CXCR4 inhibitor WZ811 (WZ; 100 nm) and by the NMDA/AMPA receptor blockers MK-801 and DNQX (MK/DN; 10 μm each). Glutamate-induced (10 μm) elevation in [Ca2+]i is shown as a positive control.
Given that SDF-1α application elevates [Ca2+]i in hippocampal neurons, we next tested whether it could result in dephosphorylation of Kv2.1 protein by the Ca2+/calmodulin-dependent protein phosphatase calcineurin, leading to changes in the subcellular localization and function of the channel. Kv2.1 in cultured hippocampal neurons is detected as a ∼125 kDa protein in SDS-PAGE (hyperphosphorylated), which during AP digestion shifts to ∼97 kDa, the deduced molecular weight from the amino acid sequence, indicating a fully dephosphorylated protein (Fig. 3A–C). Acute SDF-1α treatment (100 nm) shifted the electrophoretic mobility of Kv2.1 to an intermediate level, between untreated control and AP-treated conditions (Fig. 3A–C). This shift was blocked by WZ811 (100 nm), MK-801 (10 μm), and the calcineurin inhibitor FK506 (10 μm; Fig. 3A–C), suggesting that SDF-1α/CXCR4-induced elevations in [Ca2+]i result in calcineurin-dependent dephosphorylation of the Kv2.1 channel protein.
Figure 3.

Acute SDF-1α exposure alters Kv2.1 channel phosphorylation and localization in hippocampal neurons. A, Immunoblot analysis of lysates from cultured hippocampal neurons, showing constitutive hyperphosphorylation of Kv2.1 protein (control, ∼125 kDa; AP digestion, ∼97 kDa). Treatment of neurons with SDF-1α (100 nm, 30 min) before lysis led to Kv2.1 dephosphorylation to an intermediate phosphorylated state, which was inhibited by pretreatment with the calcineurin inhibitor FK506 (10 μm). B, Immunoblot analysis of lysates from cultured hippocampal neurons, showing that SDF-1α-induced increase in Kv2.1 mobility was inhibited by pretreatment with the CXCR4 inhibitor WZ811 (100 nm) and the NMDA receptor blocker MK-801 (10 μm). Numbers on the left in A and B denote the approximate molecular weight. C, Quantification of extent of Kv2.1 phosphorylation from immunoblot experiments as shown in A and B, as detailed in Materials and Methods. Traces are presented as mean normalized Kv2.1-immunoreactive band intensities across the shown molecular weight. The higher molecular weight corresponds to phosphorylated and the lower molecular weight corresponds to dephosphorylated Kv2.1 channel protein. Data were calculated from the following independent number of experiments: control (8), +AP (5), +SDF-1α (8), FK506+SDF-1α (4), WZ811+SDF-1α (5), and MK-801+SDF-1α (4). Numbers on the left in A and B denote the approximate molecular weight. D, Representative photomicrographs of cultured hippocampal neurons immunostained with anti-MAP2 (blue) and anti-Kv2.1 (red) antibodies, showing distinct clustered localization of Kv2.1 in the soma and proximal dendrites. SDF-1α treatment (100 nm, 30 min) of neurons led to breakdown and lateral dispersal of Kv2.1 clusters, which was attenuated by pretreatment with WZ811 (100 nm) and FK506 (10 μm). Glutamate (10 μm, 30 min) was used as a positive control for the dispersal of Kv2.1 clusters (Misonou et al., 2004). Scale bars, 25 μm. E, Quantification of the alterations in localization pattern of Kv2.1 during treatment of various drugs, from experiments shown in D, and additionally with the pretreatment of MK-801 (10 μm). Data are presented as mean ± SEM (n = >500 neurons from 4 independent cultures). ***p < 0.001 versus respective patterns in control; ###p < 0.001 versus respective patterns in SDF-1α treatment (one-way ANOVA with post hoc Bonferroni's correction).
Kv2.1 dephosphorylation is associated with changes in the subcellular distribution of the channel protein. Constitutive phosphorylation of Kv2.1 is associated with clustered localization of the channel protein in the soma and proximal dendrites, whereas dephosphorylation coincides with dispersal of channel clusters across the plasma membrane (Misonou et al., 2004, 2005b; Mohapatra and Trimmer, 2006; Mohapatra et al., 2007). Consistent with the immunoblot results, the majority of neurons immunostained with Kv2.1 displayed a clustered distribution under control conditions, whereas most neurons showed dispersed staining after glutamate treatment (10 μm; Fig. 3D,E). Acute SDF-1α treatment (100 nm) also induced disruption and lateral diffusion of Kv2.1 clusters, thereby significantly shifting to a predominantly intermediate and dispersed localization pattern (Fig. 3D,E). Coapplication of WZ811, MK-801, or FK506 inhibited SDF-1α-induced disruption and dispersal of Kv2.1 clusters (Fig. 3E), indicating that SDF-1α-induced Kv2.1 dephosphorylation and altered channel localization are mediated exclusively through CXCR4- and NMDA receptor-dependent intracellular Ca2+/calcineurin signaling.
Our previous studies showed a direct link between clustered/phosphorylated Kv2.1 and channels requiring stronger depolarizing potentials for activation and between dispersed/dephosphorylated Kv2.1 and channels requiring minimal depolarizing potentials for activation (Mohapatra and Trimmer, 2006; Mohapatra et al., 2007, 2009). Furthermore, elegant work by O'Connell et al. (2010) demonstrated that dispersal of Kv2.1 clusters reflects a transition from relatively nonconducting (clustered) to conducting (dispersed) channel configurations. Altogether, these previous observations and our results concerning the rapid SDF-1α-induced dephosphorylation of Kv2.1 leading to the dispersal of channel clusters are presumably indicative of a shift from a less-conducting to a more- and fast-conducting channel configuration, which we investigated further.
Acute SDF-1α treatment leads to enhancement of IDR in hippocampal neurons
Calcineurin-mediated dephosphorylation of Kv2.1 is also associated with hyperpolarizing shifts in the voltage-dependent activation of Kv2.1 currents (Mohapatra and Trimmer, 2006; Park et al., 2006; Mohapatra et al., 2007, 2009), which constitute the majority (>70%) of IDR in hippocampal neurons (Murakoshi and Trimmer, 1999; Mohapatra et al., 2009). Consistent with our previous reports (Mohapatra and Trimmer, 2006; Park et al., 2006; Mohapatra et al., 2009), as well as observations from our immunoblot and immunocytochemical analysis (Fig. 3), IDR from cultured rat hippocampal neurons exhibited a G–V relationship with a G½ potential of +16.20 ± 1.07 mV (Fig. 4A,C) and a V½ potential of −24.2 ± 0.5 mV. Acute SDF-1α treatment (100 nm, 30 min) led to an increase in the current density of IDR at less depolarizing potentials (Fig. 4B), a significant ∼12 mV hyperpolarizing shift in the voltage dependence of IDR activation (Fig. 4C), and a significant ∼19 mV hyperpolarizing shift in the voltage dependence of steady-state inactivation (Fig. 4D). Because IDR currents in hippocampal neurons are contributed by several Kv channels (Du et al., 2000; Malin and Nerbonne, 2002; Lai and Jan, 2006; Vacher et al., 2008), we next determined whether the SDF-1α-induced increase in the current density and voltage dependence of IDR is specific for Kv2.1 or not. Neurons were incubated with 100 nm SDF-1α for 30 min, with 100 nm ScTx-1, a Kv2.1 channel-blocking toxin (Shiau et al., 2003), added for the final 10 min of this period. ScTx-1 at 100 nm was included in the extracellular buffer during these voltage-clamp recordings. A significant decrease in IDR current density was observed during ScTx-1 application, and no increase in the current density was observed during SDF-1α treatment in the presence of ScTx-1 (Fig. 4E). Furthermore, no hyperpolarizing shift in the voltage-dependent activation of IDR from cultured rat hippocampal neurons was observed during SDF-1α treatment in the presence of ScTx-1. The G½ potential of ScTx-1-resistant IDR was +4.21 ± 0.29 mV, which during SDF-1α treatment led to an ∼5 mV depolarizing shift that was statistically insignificant (Fig. 4F). These data suggest that, in addition to disruption of clustered localization and dephosphorylation of Kv2.1 channel, SDF-1α treatment enhances activation of IDR in hippocampal neurons that is specifically contributed by hyperpolarizing voltage-dependent activation of Kv2.1. Such modifications in Kv2.1 and IDR have been suggested to induce suppression of neuronal firing and provide neuroprotection (Misonou et al., 2005b; Mohapatra et al., 2009).
Figure 4.
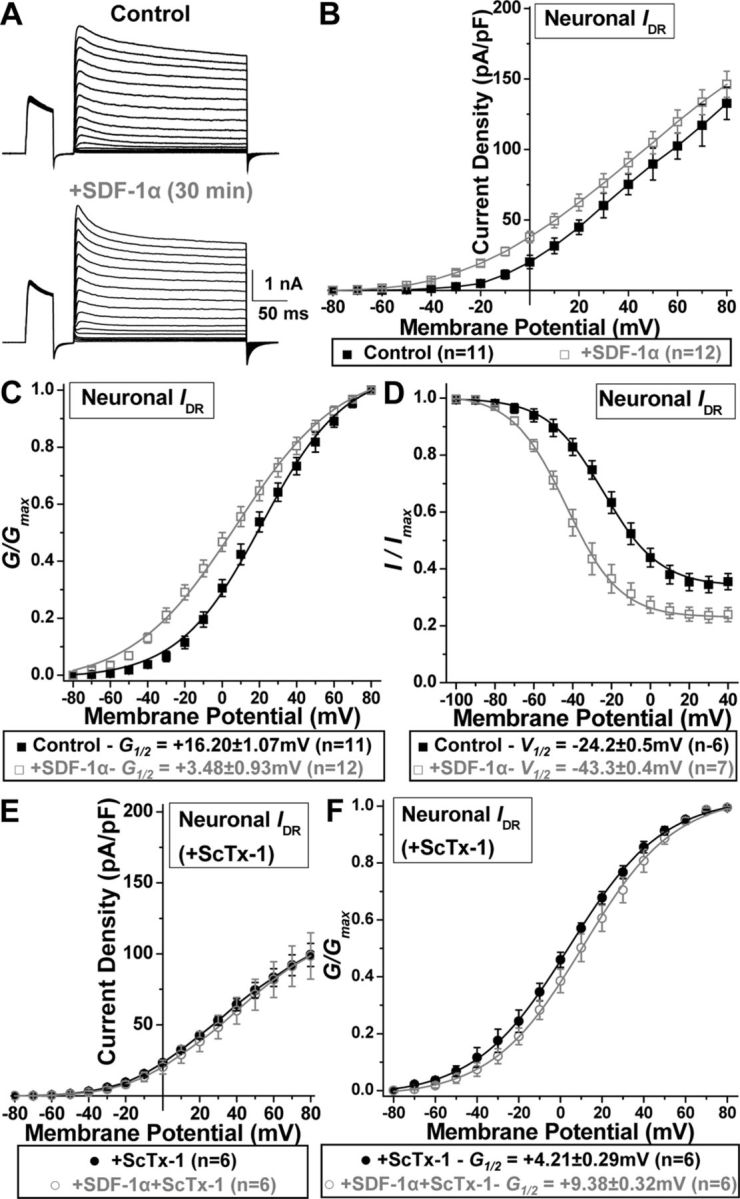
Acute SDF-1α exposure leads to enhanced voltage dependence of IDR that is specifically contributed by Kv2.1 in hippocampal neurons. A, Representative traces of IDR recorded from control and SDF-1α-treated (100 nm, 30 min) cultured rat hippocampal neurons. B, Current density plots of IDR from control and SDF-1α-treated neurons, showing increased current density at less depolarizing voltages. C, G–V relationship plot, showing increased IDR conductance during SDF-1α treatment of neurons. D, Plot showing voltage dependence of steady-state inactivation of IDR, showing hyperpolarizing shift in the voltage dependence of IDR steady-state inactivation during SDF-1α treatment of neurons. Hyperpolarizing shifts in the voltage dependence of IDR activation and inactivation are specific to Kv2.1. E, Current density plots of IDR from control and SDF-1α-treated neurons during application of the Kv2.1 channel-blocking toxin ScTx-1 (100 nm), showing decreased current density compared with control IDR shown in B. No additional change in IDR density was observed during application of ScTx-1 to SDF-1α-treated neurons. F, G–V relationship plot showing no hyperpolarizing shift in the IDR conductance during SDF-1α treatment of neurons during the application of ScTx-1. Data are presented as mean ± SEM for B–F and fitted value ± SE of Boltzmann equation fits. The G½ and V½ potentials of IDR for control and SDF-1α treatment groups are mentioned in the boxes below the respective plots. For details on voltage-pulse protocols for IDR recordings and calculations of current density, G–V, and steady-state inactivation plots, see Materials and Methods.
Direct SDF-1α/CXCR4 signaling underlies altered localization and function of Kv2.1
The effects of SDF-1α on Kv2.1 channel phosphorylation, localization, and function in neurons could occur either directly through neuronal CXCR4 and/or indirectly through trans-signaling via CXCR4 expressed in astrocytes and microglia (Kaul et al., 2001; Banisadr et al., 2002; Guyon and Nahon, 2007). Our results show that acute SDF-1α-induced disruption and dispersal of Kv2.1 clustering and dephosphorylation in neurons was dependent on NMDA receptor activity (Fig. 3B,C,E). However, this cannot rule out the possible involvement of glial/astrocytic CXCR4 signaling-induced increased release of glutamate and subsequent AMPA/NMDA receptor activation (Guyon and Nahon, 2007) involved in SDF-1α-induced modifications in Kv2.1. As such, we next established that the modulatory effects of SDF-1α on Kv2.1 could also be mediated by direct CXCR4 signaling, for which we used HEK293 cells transfected with Kv2.1 and CXCR4–YFP. Acute SDF-1α treatment (100 nm, 30 min) caused widespread internalization of CXCR4 and disruption/dispersal of Kv2.1 clusters in HEK293 cells, which was sensitive to WZ811 and FK506 (Fig. 5A,B). These observations are comparable with those in hippocampal neurons with SDF-1α treatment (Fig. 3D,E). It should be noted here that heterologous overexpression of CXCR4 in mammalian cells has been shown to induce Ca2+ release from intracellular stores and activation of calcineurin (Rigo et al., 2012). Furthermore, in cells cotransfected with CXCR4–YFP, SDF-1α application (100 nm, 30 min) led to an increase in the density of Kv2.1 currents at less depolarizing potentials (Fig. 5C,D) and a significant ∼13 mV hyperpolarizing shift in the voltage dependence of channel activation (Fig. 5E), changes that are consistent with observations in neuronal IDR (Fig. 4). However, application of SDF-1α (100 nm, 30 min) did not alter the current density and voltage-dependent activation of Kv2.1 channel in HEK293 cells cotransfected with YFP alone (Fig. 5F,G). These data indicate that SDF-1α/CXCR4 signaling could also directly modulate the localization and voltage-dependent activation of neuronal Kv2.1 channels.
Figure 5.
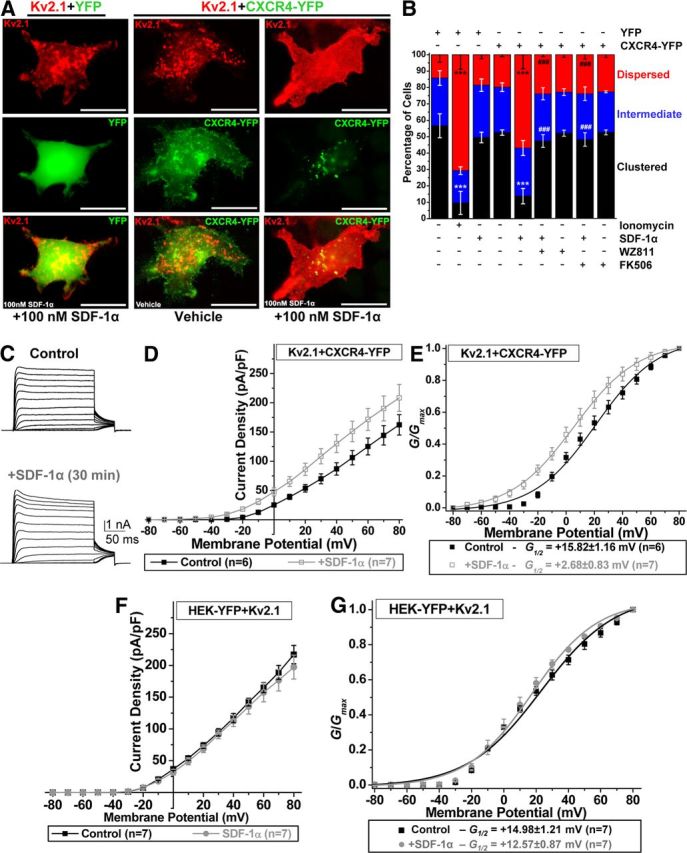
CXCR4 is required for SDF-1α-mediated disruption of Kv2.1 clusters and changes in the voltage dependence of channel activation. A, Representative photomicrographs of HEK293 cells expressing Kv2.1 (red), along with YFP (green; left column) or CXCR4–YFP (green; middle and right columns). Treatment of HEK293 cells expressing YFP with SDF-1α (100 nm, 30 min) does not alter Kv2.1 localization. However, SDF-1α application (100 nm, 30 min) to cells expressing Kv2.1 and CXCR4–YFP causes CXCR4 internalization and lateral diffusion of Kv2.1 clusters. Scale bars, 25 μm. B, Quantification of altered localization patterns of Kv2.1 in HEK293 cells expressing either YFP or CXCR4–YFP (mentioned above the graph) during different drug treatment conditions (mentioned below the graph). SDF-1α-induced (100 nm, 30 min) dispersal of Kv2.1 clusters was attenuated by pretreatment with WZ811 (100 nm) and FK506 (10 μm), indicating that CXCR4-mediated activation of calcineurin is responsible for SDF-1α-induced dispersal of Kv2.1 clusters. Ionomycin-induced (10 μm, 30 min) dispersal of Kv2.1 clusters was used as a positive control, as reported previously (Mohapatra and Trimmer, 2006). Data are presented as mean ± SEM (n = >500 cells from 4 independent experiments). ***p < 0.001 versus respective localization patterns in control; ###p < 0.001 versus respective patterns in SDF-1α treatment group (one-way ANOVA with post hoc Bonferroni's correction). C–E, SDF-1α exposure leads to enhancement in voltage-dependent activation of Kv2.1 channel in HEK293 cells expressing Kv2.1 and CXCR4–YFP. C, Representative traces of Kv2.1 currents from control and SDF-1α (100 nm, 30 min)-treated cells. D, Current density plots of Kv2.1 currents from control and SDF-1α-treated cells, showing increased current density. E, G–V relationship plots showing increased Kv2.1 conductance after SDF-1α treatment. F, G, SDF-1α does not influence Kv2.1 currents in HEK293 cells expressing YFP alone. F, Current density plot of Kv2.1 expressed in HEK293 cells under control and SDF-1α treatment (100 nm, 30 min), showing no change during SDF-1α treatment. G, G–V relationship plots of currents shown in F, showing no change in the voltage-dependent activation of Kv2.1 during SDF-1α treatment (100 nm, 30 min). For D–G, data are presented as mean ± SEM. The G½ potentials of Kv2.1 for control and SDF-1α treatment groups are mentioned in the box below the plot in E and G. For details on voltage-pulse protocols for Kv2.1 current recordings and calculations of current density and G–V plots, see Materials and Methods.
Calcineurin-dependent modulation of Kv2.1 provides neuroprotection to acute SDF-1α treatment
Calcineurin-dependent dephosphorylation and hyperpolarizing voltage-dependent activation of Kv2.1-based IDR has been suggested to be neuroprotective during hyperexcitable assaults (Misonou et al., 2005b; Mohapatra et al., 2009). We next tested whether similar phenomena operate during increased SDF-1α/CXCR4 activation in neurons. Treatment with SDF-1α (100 nm, 30 min) did not lead to any increase in the number of neurons (MAP2-immunostained) with EthD-1-positive nuclei, an indicator of dead or dying cells (Fig. 6A,B). However, coapplication of 100 nm SDF-1α with calcineurin inhibitor (10 μm FK506) or Kv2.1 blocking toxin ScTx-1 (100 nm), but not the Kv4.2 blocking toxin HpTx-2 [100 nm (Zarayskiy et al., 2005)], led to a significant increase in the number of EthD1-positive neurons (Fig. 6B). Previous studies with prolonged SDF-1α treatment of neurons have shown to induce apoptotic cell death via activation of p38 MAPK (Kaul et al., 2001; Vlahakis et al., 2002; Mattson et al., 2005; Guyon and Nahon, 2007; Chen et al., 2011). Interestingly, coapplication of 100 nm SDF-1α with the p38 MAPK inhibitor SB203580 (1 μm) did not lead to any change in EthD-1-positive neurons (Fig. 6B). To this end, we found that acute SDF-1α treatment led to the activation of p38 MAPK in neurons, as determined by increased phospho-p38 MAPK levels (Fig. 6C,D). Furthermore, we immunostained neurons with a c-caspase-3-specific antibody, which is an indicator of neuronal apoptosis (Srinivasan et al., 1998). Our data show no change in the number of c-caspase-3-stained neurons in response to SDF-1α plus FK506 and SDF-1α plus ScTx-1 treatment compared with ScTx-1 treatment alone. These results suggest that calcineurin-dependent modulation of Kv2.1 provides neuroprotection in response to acute SDF-1α treatment, which could otherwise induce significant non-apoptotic neuronal death.
Figure 6.

Acute SDF-1α treatment provides neuroprotection via calcineurin-dependent modulation of Kv2.1. A, Representative images of cultured hippocampal neurons immunostained with anti-MAP2 (green) antibody and EthD-1 (red), showing increased EthD-1-positive nuclei, indicative of dead neurons, during coapplication of the Kv2.1 channel-blocking toxin ScTx-1 (100 nm) or the calcineurin inhibitor FK506 (10 μm) along with SDF-1α (100 nm, 30 min). However, under control and SDF-1α-treated (100 nm, 30 min) conditions, no significant increase in EthD-1-positive neuronal nuclei was observed. B, Quantification of neuronal death in response to various drug treatments, as determined by percentage of EthD-1-positive neurons. Cotreatment of ScTx-1 (100 nm) or the calcineurin inhibitor FK506 (10 μm), but not the Kv4.2 blocking toxin HpTx-2 (100 nm) or the p38 MAPK inhibitor SB203580 (1 μm), along with SDF-1α (100 nm; 30 min) led to a significant increase in neuronal death. Data are presented as mean ± SEM (n = >500 neurons/cells from 4 independent experiments). ***p < 0.001 versus untreated conditions; ###p < 0.001 versus only SDF-1α treatment (one-way ANOVA with post hoc Bonferroni's correction). C, D, SDF-1α treatment (100 nm, 30 min) increased phosphorylation of p38 MAPK in cultured hippocampal neurons, as determined by immunoblot analysis (C) and quantification by densitometry from three independent experiments (D). **p < 0.01 versus control (Student's t test). E, Increased neuronal death during cotreatment of neurons with acute SDF-1α and ScTx-1 or FK506 is not apoptotic in nature. Quantification of c-caspase-3-positive neurons, as an indication of apoptotic cell death, after various drug treatments. None of the treatments led to any increase in the number of neurons with c-caspase-3 staining. Data are presented as mean ± SEM (n = >500 neurons from 4 independent experiments). Scale bars (in A), 50 μm.
Sustained SDF-1α exposure leads to apoptotic neuronal death via complex intracellular signaling involving CXCR4 and Kv2.1
Because acute SDF-1α/CXCR4 signaling leads to neuroprotective modifications in Kv2.1 channels, we next tested whether such modifications prevail during sustained activation of CXCR4. Prolonged application of SDF-1α (100 nm, 3 h) led to sustained dephosphorylation of Kv2.1, which was attenuated by the coapplication of FK506 (Fig. 7A). However, we observed a decrease in the number of neurons during such prolonged SDF-1α application, which was clearly evident during visualization under microscope, an observation that is consistent with a decrease in the overall immunoblot signal intensity for Kv2.1 during prolonged SDF-1α application (Fig. 7A). In addition, we observed a significant increase in the number of neurons (MAP2-immunostained) with EthD-1-positive nuclei, indicating increased neuronal death (Fig. 7B,C). This prolonged SDF-1α-induced neuronal death was attenuated by coapplication of the CXCR4 antagonist WZ811, the NMDA receptor blocker MK-801 (10 μm), the nonspecific IDR blocker tetraethylammonium (TEA; 70 mm), and the Kv2.1 channel-blocking toxin ScTx-1 (100 nm; Fig. 7C). However, in addition to Kv2.1, ScTx-1 also exhibits inhibitory activity for Kv4.2 channels (Shiau et al., 2003; Chen et al., 2010). Thus, we further used the Kv4.2-specific blocking toxin HpTx-2 (100 nm), which during coapplication with SDF-1α did not significantly attenuate the prolonged SDF-1α-induced neuronal death, suggesting that sustained enhancement of Kv2.1 conductance is primarily responsible for the induction of apoptosis in response to sustained SDF-1α treatment (Fig. 7C).
Figure 7.

Sustained SDF-1α treatment results in the induction of neuronal apoptosis that is dependent on CXCR4–calcineurin/p38 MAPK–Kv2.1 signaling. A, Immunoblot analysis showing dephosphorylation of Kv2.1 protein during sustained SDF-1α treatment (100 nm, 3 h) of cultured rat hippocampal neurons, which was attenuated by pretreatment with FK506 (10 μm). B, Representative images of cultured hippocampal neurons immunostained with anti-MAP2 (green) antibody and EthD-1 (red), showing increased EthD-1-positive nuclei, indicative of dead neurons, during sustained SDF-1α treatment (100 nm, 3 h). Scale bars, 50 μm. C, Quantification of neuronal death in response to various drug treatments, as determined by percentage of EthD-1-positive neurons. Sustained SDF-1α-induced neuronal death was significantly attenuated by pretreatment with WZ811 (100 nm), the nonspecific K+ channel blocker TEA (70 mm), and the Kv2.1 channel-blocking toxin ScTx-1 (100 nm) but not by the Kv4.2 channel-blocking toxin HpTx-2 (100 nm). D, Sustained SDF-1α-induced neuronal death was significantly attenuated by pretreatment with FK506 (10 μm) and the p38 MAPK inhibitor SB203580 (1 μm). E, Sustained SDF-1α-induced neuronal death is apoptotic in nature and is dependent on p38 MAPK–Kv2.1 signaling. Quantification of c-caspase-3-positive neurons after various drug treatments. Sustained SDF-1α-induced (100 nm, 3 h) increase in the number of c-caspase-3 nuclei-stained neurons was significantly attenuated by pretreatment with SB203580 (1 μm) and ScTx-1 (100 nm). All data in C–E are presented as mean ± SEM (n = >500 neurons from 4 independent experiments). *p < 0.05, **p < 0.01, and ***p < 0.001 versus untreated conditions; #p < 0.05 and ###p < 0.001 compared with only SDF-1α treatment (one-way ANOVA with post hoc Bonferroni's correction).
We next determined the role of sustained calcineurin-mediated dephosphorylation of Kv2.1 (Fig. 7A) in prolonged SDF-1α-induced neuronal death. Coapplication of FK506 with SDF-1α significantly but not completely attenuated prolonged SDF-1α-induced neuronal death (Fig. 7D); however, such cell death was completely attenuated by the p38 MAPK inhibitor SB203580 (1 μm; Fig. 7D). Furthermore, we determined whether or not this prolonged SDF-1α-induced neuronal death was apoptotic in nature. Indeed, sustained SDF-1α application (100 nm, 3 h) significantly increased the number of neurons with c-caspase-3 stained nuclei, which was significantly attenuated by coapplication of SB203580 and ScTx-1 (Fig. 7E). Furthermore, we investigated the direct role of Kv2.1 function in sustained SDF-1α-induced cell death by using HEK293 cells expressing recombinant rat Kv2.1 and CXCR4. Prolonged SDF-1α application (100 nm, 3 h) led to increased cell death but only when both Kv2.1 and CXCR4 were expressed (Fig. 8). Similar to our observations in neurons, the prolonged SDF-1α-induced cell death in HEK293 cells coexpressing Kv2.1 and CXCR4 was completely attenuated by coapplication of CXCR4 inhibitors WZ811 (100 nm) and AMD3100 (1 μm), TEA (70 mm), and SB203580 (1 μm; Fig. 8). These results indicate that distinct signaling cascades originating from CXCR4 activation modify Kv2.1 channel function and govern increased neuronal apoptosis during sustained exposure to SDF-1α.
Figure 8.

Sustained SDF-1α/CXCR4/p38 MAPK/Kv2.1 signaling leads to enhanced cell death in HEK293 cells expressing Kv2.1 and CXCR4 heterologously. Treatment of cells with SDF-1α (100 nm, 3 h) led to enhanced cell death in HEK293 cells only when both Kv2.1 and CXCR4 are expressed and was completely attenuated by pretreatment with the CXCR4 inhibitors WZ811 (100 nm) and AMD3100 (1 μm), the nonspecific Kv2.1 channel blocker TEA (70 mm), and the p38 MAPK inhibitor SB203580 (1 μm). Data are presented as mean ± SEM (n = >500 cells from 4 independent experiments). ***p < 0.001 versus untreated conditions; ###p < 0.001 compared with only SDF-1α treatment (one-way ANOVA with post hoc Bonferroni's correction).
HIV-1 gp120 induces CXCR4-mediated changes in Kv2.1 and neuronal apoptosis
The HIV-1 glycoprotein gp120 has been shown to induce neuronal degeneration/death via chemokine receptor signaling, wherein CXCR4 (or CCR5) serves as a coreceptor (along with CD4) for gp120 (Hesselgesser et al., 1998; Kaul and Lipton, 1999; Zheng et al., 1999; Kaul et al., 2001, 2007; Mattson et al., 2005). Thus, we next tested whether modifications in Kv2.1 are key to gp120-induced neuronal death. Similar to our observations with SDF-1α, prolonged application of gp120 (1 and 10 nm for 3 h) also significantly increased neuronal apoptosis, as measured by quantifying the number of neurons with EthD-1- and c-caspase-3-stained nuclei, which were significantly attenuated by coapplication of WZ811, MK-801, FK506, SB203580, and ScTx-1 (Fig. 9A,B). These observations suggest that distinct CXCR4-induced modifications in Kv2.1 by calcineurin and p38 MAPK are presumably critical to gp120-associated HAND.
Figure 9.

HIV-1 IIIB envelope glycoprotein gp120 induces neuronal apoptosis that is dependent on CXCR4/calcineurin/p38 MAPK/Kv2.1 signaling. A, Quantification of neuronal death in response to prolonged treatment with HIV-1 IIIB gp120 (1 and 10 nm, 3 h) and various drug cotreatments, as determined by percentage of EthD-1-positive neurons. Sustained gp120-induced neuronal death was significantly attenuated by pretreatment with the CXCR4 inhibitor WZ811 (100 nm), the NMDA receptor blocker MK-801 (10 μm), the calcineurin inhibitor FK506 (10 μm), and the Kv2.1 channel-blocking toxin ScTx-1 (100 nm). B, Sustained gp120-induced increased number of nuclear c-caspase-3 immunostained neurons was significantly attenuated by pretreatment with SB203580 (1 μm) and ScTx-1 (100 nm). Data in both the panels are presented as mean ± SEM (n = >500 neurons from 4 independent cultures). ***p < 0.001 versus untreated conditions; #p < 0.05 and ###p < 0.001 versus gp120 treatment group (one-way ANOVA with post hoc Bonferroni's correction).
CXCR4-mediated neuronal apoptosis is dependent on p38 MAPK phosphorylation of Kv2.1 at S800
Kv2.1 protein has been shown to be phosphorylated at 24 amino acid residues spanning the cytoplasmic N and C terminus (Park et al., 2006, 2007; Song et al., 2012), of which the residue S800 has been shown to be phosphorylated by p38 MAPK (Redman et al., 2007), which contributes to its surface insertion and induction of apoptosis (Redman et al., 2007). We also observed that inhibition of p38 MAPK and Kv2.1 attenuated sustained SDF-1α-induced neuronal apoptosis (Figs. 7, 8). Therefore, we next investigated whether sustained SDF-1α-induced neuronal apoptosis is mediated by increased plasma membrane insertion of Kv2.1. Prolonged SDF-1α application (100 nm, 3 h) significantly increased phosphorylated-p38 MAPK levels (Fig. 10A,B) in cultured rat hippocampal neurons, wherein a significant increase in surface Kv2.1 protein levels was also observed (Fig. 10C). This increase in surface Kv2.1 protein levels was attenuated by the coapplication of SB203580 (Fig. 10C). We also performed similar quantification of surface Kv2.1 protein levels in HEK293 cells coexpressing CXCR4, which during prolonged SDF-1α treatment led to an increase in surface Kv2.1 levels that could also be attenuated by the coapplication of SB203580 (Fig. 10D). However, sustained application of SDF-1α did not increase surface Kv2.1 protein levels in HEK293 cells coexpressing the Kv2.1–S800A mutant and CXCR4 (Fig. 10D). Accordingly, sustained SDF-1α exposure also failed to induce cell death in HEK293 cells coexpressing CXCR4 with Kv2.1–S800A mutant, as opposed to SDF-1α-induced increase in cell death with HEK293 cells expressing Kv2.1–WT (Fig. 10E). We also performed similar experiments in HEK293 cells coexpressing CXCR4 and another Kv channel, Kv1.4, in which prolonged SDF-1α treatment did not lead to any cell death (Fig. 10E), indicating the specific role of Kv2.1 in the induction of cell death by sustained SDF-1α application.
Figure 10.
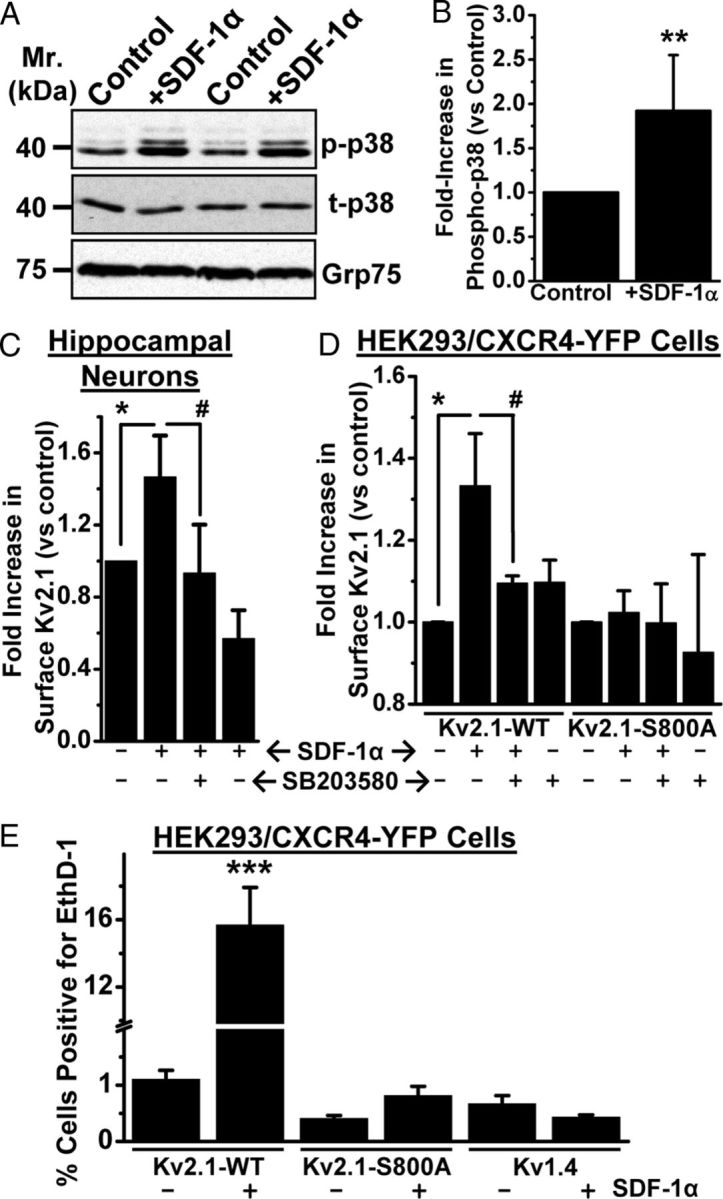
Sustained SDF-1α treatment leads to increased surface expression of Kv2.1 that is dependent on p38 MAPK phosphorylation at residue S800, which governs the induction of cell death. A, B, Prolonged treatment with SDF-1α (100 nm, 3 h) led to increased phosphorylation of p38 MAPK in cultured rat hippocampal neurons, as determined by immunoblot analysis (A) and quantification by densitometry from three independent experiments (B). **p < 0.01 versus control (Student's t test). C, D, Increase in the level of surface biotinylated Kv2.1 protein, as determined by immunoblot analysis, from cultured rat hippocampal neurons (C) and HEK293 cells expressing Kv2.1–WT and CXCR4–YFP (D) during SDF-1α treatment (100 nm, 3 h), which can be attenuated by pretreatment with SB203580 (1 μm). No increase in the level of surface biotinylated Kv2.1 protein was observed during SDF-1α treatment of HEK293 cells expressing Kv2.1–S800A mutant and CXCR4–YFP (D). For details of biotinylation experiments and densitometry analysis, see Materials and Methods. Data are presented as mean ± SEM from three independent experiments. *p < 0.05 versus untreated conditions; #p < 0.05 versus SDF-1α treatment group (one-way ANOVA with post hoc Bonferroni's correction). E, Increased cell death during sustained SDF-1α treatment (100 nm, 3 h) was absent in HEK293 cells expressing CXCR4–YFP and Kv2.1–S800A mutant, as well as in HEK293 cells expressing another Kv channel, Kv1.4. Data are presented as mean ± SEM of percentage of cells positive for EthD-1 (n = >500 cells from 4 independent experiments). *p < 0.05 and ***p < 0.001 versus untreated conditions; #p < 0.05 versus SDF-1α treatment group (one-way ANOVA with post hoc Bonferroni's correction).
To further verify whether the sustained SDF-1α-induced increase in surface Kv2.1 protein leads to enhanced IDR/Kv2.1 channel function, we performed whole-cell voltage-clamp experiments on cultured rat hippocampal neurons and HEK293 cells coexpressing Kv2.1 and CXCR4. Cells were treated with SDF-1α (100 nm, 3 h) along with the cysteine protease inhibitor BAF (10 μm), to prevent apoptosis. Sustained exposure of hippocampal neurons to SDF-1α significantly increased IDR density (Fig. 11A), which was accompanied by an ∼8 mV hyperpolarizing shift in the voltage-dependent activation (Fig. 11B). Similarly, a significant increase in the current density (Fig. 11C) and a ∼10 mV hyperpolarizing shift in the voltage-dependent activation of Kv2.1 currents (Fig. 11D) were observed during sustained SDF-1α treatment of HEK293 cells coexpressing CXCR4. However, no increase in Kv2.1 current density was observed during sustained SDF-1α treatment of HEK293 cells coexpressing CXCR4 and the Kv2.1–S800A mutant (Fig. 11E), which is in agreement with our observations that there was no increase in the protein levels of this mutant channel at the cell surface under these conditions (Fig. 10D). In contrast, the Kv2.1–S800A mutant channel still exhibited a sustained SDF-1α-induced ∼11 mV hyperpolarizing shift in voltage-dependent activation (Fig. 11F), suggesting that an increased number of Kv2.1 channels on the cell surface as well as hyperpolarizing voltage dependence of activation are both required to predispose neurons to apoptotic cell death.
Figure 11.

Sustained SDF-1α exposure leads to increased current density and enhanced conductance of IDR/Kv2.1 currents. Cells were treated with either the cysteine protease inhibitor BAF (10 μm, vehicle) or BAF (10 μm) plus SDF-1α (100 nm) for 3 h immediately before recordings. Current density (A, C, E) and G–V plots (B, D, F) of IDR from vehicle and SDF-1α-treated cultured rat hippocampal neurons (A, B) and Kv2.1 currents from control, vehicle, and SDF-1α-treated HEK293 cells expressing CXCR4–YFP and Kv2.1–WT (C, D) or Kv2.1–S800A (E, F). Sustained SDF-1α treatment led to increased current density of IDR and Kv2.1–WT but not Kv2.1–S800A currents. However, sustained SDF-1α treatment led to enhanced voltage-dependent activation of IDR, Kv2.1–WT, and Kv2.1–S800A currents. Data are presented as mean ± SEM. The G½ potentials for each treatment group are mentioned in the boxes below the plots. For details on calculations of current density and G–V plots, see Materials and Methods.
Altogether, our results indicate that distinct modifications in Kv2.1 channel originating from activation of the chemokine receptor CXCR4 can exert both neuroprotective and apoptotic effects in a sequential and time-dependent manner (Fig. 12).
Figure 12.

Scheme depicting distinct intracellular signaling events that regulate neuronal survival-death dynamics via CXCR4 modulation of Kv2.1 channel. Short-term CXCR4 signaling leads to calcineurin-dependent acute enhancement of Kv2.1 currents that leads to homeostatic suppression of hyperexcitability and neuronal survival. However, prolonged CXCR4 signaling leads to p38 MAPK- and calcineurin-mediated sustained K+ efflux through Kv2.1 channel, leading to activation of caspase-3 and neuronal apoptosis. + sign in green circles indicates activation. SOC, Store-operated calcium channel; ER, endoplasmic reticulum; NMDAR, NMDA receptor; Cav, voltage-gated calcium channel; CaM, calmodulin; pS800, phospho-serine at residue 800.
Discussion
The mechanism(s) by which enhanced CXCR4 signaling influences neuronal survival and/or apoptosis are poorly understood. Our results constitute a novel mechanism underlying CXCR4-dependent regulation of neuronal survival/death dynamics. Interestingly, this regulation occurs through two distinct modifications in the same protein, a major somatodendritic Kv channel, Kv2.1. Our study demonstrates that SDF-1α acts on neuronal CXCR4 to induce an NMDA receptor-dependent elevation in [Ca2+]i, leading to calcineurin-dependent dephosphorylation of Kv2.1, dispersal of channel clusters, and enhancement of voltage-dependent activation of Kv2.1/IDR currents. In the short term, these changes decrease neuronal excitability and provide neuroprotection. However, prolonged CXCR4 activation leads to p38 MAPK-induced phosphorylation and surface delivery of Kv2.1 protein, coupled with sustained calcineurin-mediated channel dephosphorylation, resulting in enhanced IDR/Kv2.1 current density and hyperpolarized voltage dependence of channel activation. These changes enable sustained K+ efflux and caspase-dependent apoptosis, which can be attenuated by inhibition of CXCR4, calcineurin, p38 MAPK, or Kv2.1 channel function. In addition, gp120 produces similar short- and long-term changes in Kv2.1 and neuronal apoptosis that can be attenuated by the same pharmacological inhibitors.
SDF-1α- and gp120-induced [Ca2+]i elevation in mammalian brain neurons have been reported to be mediated indirectly through increased glutamate release from presynaptic terminals and microglia and subsequent activation/modulation of postsynaptic NMDA receptors and N-type Cav channels (Zheng et al., 1999; Catani et al., 2000; Kaul et al., 2001; Oh et al., 2002; Guyon and Nahon, 2007). Indeed, our results showing attenuation of SDF-1α-induced [Ca2+]i elevation and calcineurin-dependent dephosphorylation of Kv2.1 by the NMDA receptor antagonist MK-801 are consistent with these reports. It has also been reported that SDF-1α activation of CXCR4 could lead to Gαi–βγ-mediated Ca2+ release from intracellular stores (Lo et al., 1992; Zheng et al., 1999; Guyon and Nahon, 2007), which could subsequently activate calcineurin (Rigo et al., 2012). Our observations on calcineurin-dependent dephosphorylation and dispersal of Kv2.1 clusters and hyperpolarizing shifts in the voltage dependence of channel activation during SDF-1α treatment of HEK293 cells overexpressing Kv2.1 and CXCR4 also suggest the existence of direct CXCR4/Gαi–βγ-mediated Ca2+/calcineurin signaling. NMDA-mediated increased [Ca2+]i has been suggested to induce apoptosis in response to SDF-1α/gp120 (Zheng et al., 1999; Catani et al., 2000; Kaul et al., 2001; Guyon and Nahon, 2007). However, recent reports have suggested that SDF-1α/CXCR4 signaling promotes survival of postmitotic neurons, via enhancing the expression/activity of the Rb transcriptional repressor, thereby suppressing expression of the NMDAR NR2B subunit (Khan et al., 2008; Nicolai et al., 2010). In contrast, a large body of evidence points toward the activation of p38 MAPK, downstream of CXCR4 activation in neurons, as a key factor for SDF-1α/gp120-induced neuronal apoptosis (Meucci et al., 1998; Kaul and Lipton, 1999; Kaul et al., 2001, 2007; Guyon and Nahon, 2007). Indeed, our results suggest the key role of NMDA receptor activation, as well as p38 MAPK in SDF-1α/gp120-induced neuronal apoptosis. Furthermore, our study indentified the dynamicity of the SDF-1α/CXCR4-induced regulation of neuronal survival and apoptosis that is mediated through distinct modifications in the same neuronal Kv channel protein, Kv2.1. Similar bidirectional SDF-1α/CXCR4 signaling has been shown to induce both survival and apoptotic pathways in CD4+ T-cells (Vlahakis et al., 2002).
Kv channels are implicated in HAND, with conflicting reports on the modulation of IA and IDR currents by gp120 as underlying mechanisms (Keblesh et al., 2009; Chen et al., 2011). Kv2.1 contributes the majority of IDR in mammalian brain neurons and is a key regulator of intrinsic neuronal excitability (Murakoshi and Trimmer, 1999; Du et al., 2000; Malin and Nerbonne, 2002; Mohapatra et al., 2009). Our previous studies have shown that Kv2.1 protein undergoes dynamic calcineurin-dependent dephosphorylation in response to increased excitatory activity, ischemic stroke, and neuromodulations, resulting in dispersal of Kv2.1 clusters and hyperpolarizing shifts in voltage-dependent channel gating properties. These changes cause homeostatic suppression of neuronal firing, thereby providing neuroprotection (Surmeier and Foehring, 2004; Misonou et al., 2005b; Mohapatra et al., 2009). Indeed, our present study shows that acute SDF-1α/CXCR4-mediated activation of calcineurin leads to Kv2.1 dephosphorylation, dispersal of channel clusters, increased current density at less depolarizing potentials, and enhanced voltage-dependent activation of IDR/Kv2.1 currents. These changes provide protection against acute non-apoptotic neuronal death, as evidenced by increased neuronal death during coapplication of SDF-1α with the calcineurin inhibitor FK506 and Kv2.1 channel-blocking toxin ScTx-1. This is consistent with our previous observations on regulation of intrinsic excitability and homeostatic plasticity that provide neuroprotection (Mohapatra et al., 2009). Our results also provide evidence for both trans-glutamate/NMDA receptor activation, as well as direct CXCR4–[Ca2+]i–calcineurin signaling, which mediate prolonged SDF-1α-induced modulation of Kv2.1 and induction of neuronal apoptosis. Our observations in cultured neurons suggest the predominant role of a trans-glutamate/NMDA receptor signaling cascade, presumably through enhanced glutamate release from presynaptic terminals and microglia/astrocytes, in combination with increased p38 MAPK activity, in Kv2.1 channel modulation and induction of neuronal apoptosis. This is in agreement with previous reports suggesting that SDF-1α activation of CXCR4 in microglia and astrocytes leads to glutamate release and enhanced excitatory activity, which results in excitotoxic neuronal death (Kaul et al., 2001; Guyon and Nahon, 2007). Furthermore, our experiments using heterologous expression of CXCR4/Kv2.1 suggest that a direct interaction between SDF-1α/CXCR4 signaling and modulation of Kv2.1 could also induce neuronal apoptosis.
Despite the initial protective effects of enhanced IDR/Kv2.1 currents during hyperexcitable conditions (Mohapatra et al., 2009), excessive K+ efflux has been shown to be a prerequisite for neuronal apoptosis (Yu et al., 1997; Pal et al., 2003; Zhao et al., 2006; Redman et al., 2007; Yao et al., 2009). Although many Kv channels are expressed in mammalian brain neurons (Vacher et al., 2008), our results show that CXCR4 signaling-induced neuronal death was completely attenuated by the nonspecific IDR blocker TEA and the Kv2.1/Kv4.2 blocker ScTx-1. Furthermore, blockade of Kv4.2 by HpTx-2 did not attenuate this increase in neuronal death, suggesting a specific role for Kv2.1. Prolonged application of SDF-1α also increased surface delivery of Kv2.1 protein via p38 MAPK-mediated phosphorylation at S800 (Redman et al., 2007), leading to enhanced IDR/Kv2.1 current density. Numerous studies have shown that p38 MAPK is activated downstream of SDF-1α/gp120-activation of neuronal CXCR4/Gαi (Kaul et al., 2001; Vlahakis et al., 2002; Mattson et al., 2005; Guyon and Nahon, 2007; Chen et al., 2011), a finding replicated here. The mechanism by which p38 MAPK phosphorylation at S800 promotes Kv2.1 insertion into the plasma membrane remains unclear; however, the involvement of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins SNAP-25 and syntaxin could be predicted (Leung et al., 2003). In particular, syntaxin associates with the C terminus of Kv2.1 (Leung et al., 2003), raising the possibility that S800 phosphorylation facilitates SNARE-mediated insertion of Kv2.1 into the plasma membrane, a hypothesis that warrants additional investigation. Interestingly, pS800 is not dephosphorylated by calcineurin and does not participate in the graded regulation of Kv2.1 channel gating (Park et al., 2006). Indeed, our previous studies, as well as our present study on the S800A mutant Kv2.1, show similar hyperpolarizing shifts in the voltage dependence of channel activation during calcineurin-mediated dephosphorylation. Together, these two modifications in Kv2.1 increase the number of channels in the neuronal membrane that can be activated with minimal membrane depolarization, thus leading to sustained (possibly pathological) K+ efflux and induction of apoptosis. Although the prolonged SDF-1α-induced increase in IDR/Kv2.1 current density seems modest, it should be noted that there is also a significant hyperpolarizing shift in the voltage-dependent activation of these currents, which altogether could lead to sustained K+ efflux over a longer time period. Future studies will focus on identifying the precise mechanisms underlying the sustained K+ efflux-induced mitochondrial dysfunction and subsequent activation of caspase-3 in neuronal apoptosis.
We propose that, under conditions of initial SDF-1α/CXCR4-induced elevation in [Ca2+]i, dephosphorylation of Kv2.1 can occur within minutes, as an “acute-phase response” that acts to limit excitability of neurons in the surrounding network. However, if the initial insult/perturbation is not transient and signaling through CXCR4 persists for longer durations, increased Kv2.1 current density, in combination with hyperpolarized shifts in the voltage dependence of channel activation, predispose neurons to a sustained, pathological reduction in [K+]i. This eventually leads to caspase-3 cleavage and execution of the neuronal apoptotic program. This mechanism is consistent with both the neurotoxic effect of HIV-1 gp120 (Kaul and Lipton, 1999; Zheng et al., 1999; Kaul et al., 2001, 2005, 2007; Mattson et al., 2005; Guyon and Nahon, 2007) and the propensity of glioblastoma cells to secrete or fail to uptake glutamate (Sontheimer, 2008), an effect that would enhance the proapoptotic effects of chemokine signaling such as those induced by SDF-1α. It remains to be seen if (should the neuron survive) signaling induced by CXCR4 can induce changes in gene expression [for example, via CREB (Joo et al., 2004)] that could alter susceptibility to excitotoxicity. Developing a more detailed understanding of the events at the level of gene expression and posttranslational modification could represent an effective strategy to boost neuronal survival in a diverse array of neuropathologies. In this regard, our study delineates a precise mechanistic basis underlying the regulation of neuronal survival and death, under conditions of neuroinflammation and HAND.
Footnotes
This work was funded by startup funds from the University of Iowa Office of the Vice President of Research, Epilepsy Foundation and American Epilepsy Society Research Grant Pc-190423, and National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS069898 (D.P.M.). A.D.M. was supported by Predoctoral Fellowship NS045549 through an institutional T32 grant from National Institutes of Health/National Institute of Neurological Disorders and Stroke. We thank Dr. Yuriy M. Usachev for his critical review and constructive criticisms throughout the course of this investigation and Dr. Donna L. Hammond for her continuous support and encouragement.
References
- Banisadr G, Fontanges P, Haour F, Kitabgi P, Rostène W, Mélik Parsadaniantz S. Neuroanatomical distribution of CXCR4 in adult rat brain and its localization in cholinergic and dopaminergic neurons. Eur J Neurosci. 2002;16:1661–1671. doi: 10.1046/j.1460-9568.2002.02237.x. [DOI] [PubMed] [Google Scholar]
- Catani MV, Corasaniti MT, Navarra M, Nisticò G, Finazzi-Agrò A, Melino G. gp120 induces cell death in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors. J Neurochem. 2000;74:2373–2379. doi: 10.1046/j.1471-4159.2000.0742373.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu J, Xu C, Keblesh J, Zang W, Xiong H. HIV-1gp120 induces neuronal apoptosis through enhancement of 4-aminopyridine-sensitive outward K+ currents. PLoS One. 2011;6:e25994. doi: 10.1371/journal.pone.0025994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Kellett WF, Petkov GV. Voltage-gated K+ channels sensitive to stromatoxin-1 regulate myogenic and neurogenic contractions of rat urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol. 2010;299:R177–R184. doi: 10.1152/ajpregu.00036.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell-derived factor-1alpha on neuronal activity. J Mol Endocrinol. 2007;38:365–376. doi: 10.1677/JME-06-0013. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8:595–598. doi: 10.1016/s0960-9822(98)70230-1. [DOI] [PubMed] [Google Scholar]
- Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, Maeda M, Fagan SC, Carroll JE, Conway SJ. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63:84–96. doi: 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- Johnston J, Forsythe ID, Kopp-Scheinpflug C. Going native: voltage-gated potassium channels controlling neuronal excitability. J Physiol. 2010;588:3187–3200. doi: 10.1113/jphysiol.2010.191973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo EK, Broxmeyer HE, Kwon HJ, Kang HB, Kim JS, Lim JS, Choe YK, Choe IS, Myung PK, Lee Y. Enhancement of cell survival by stromal cell-derived factor-1/CXCL12 involves activation of CREB and induction of Mcl-1 and c-Fos in factor-dependent human cell line MO7e. Stem Cells Dev. 2004;13:563–570. doi: 10.1089/scd.2004.13.563. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14:296–305. doi: 10.1038/sj.cdd.4402006. [DOI] [PubMed] [Google Scholar]
- Keblesh J, Hu D, Xiong H. Voltage-gated potassium channels in human immunodeficiency virus type-1 (HIV-1)-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2009;4:60–70. doi: 10.1007/s11481-008-9106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ. 2008;15:1663–1672. doi: 10.1038/cdd.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–148. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- Leung YM, Kang Y, Gao X, Xia F, Xie H, Sheu L, Tsuk S, Lotan I, Tsushima RG, Gaisano HY. Syntaxin 1A binds to the cytoplasmic C terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem. 2003;278:17532–17538. doi: 10.1074/jbc.M213088200. [DOI] [PubMed] [Google Scholar]
- Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol. 2008;84:116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo TM, Fallert CJ, Piser TM, Thayer SA. HIV-1 envelope protein evokes intracellular calcium oscillations in rat hippocampal neurons. Brain Res. 1992;594:189–196. doi: 10.1016/0006-8993(92)91125-x. [DOI] [PubMed] [Google Scholar]
- Loo L, Shepherd AJ, Mickle AD, Lorca RA, Shutov LP, Usachev YM, Mohapatra DP. The C-type natriuretic peptide induces thermal hyperalgesia through a noncanonical Gβγ-dependent modulation of TRPV1 channel. J Neurosci. 2012;32:11942–11955. doi: 10.1523/JNEUROSCI.1330-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2 α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22:10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Trimmer JS. Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology. 2005a;26:743–752. doi: 10.1016/j.neuro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005b;25:11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithal DS, Banisadr G, Miller RJ. CXCL12 signaling in the development of the nervous system. J Neuroimmune Pharmacol. 2012 doi: 10.1007/s11481-011-9336-x. Advance online publication. Retrieved October 30, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Trimmer JS. The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J Neurosci. 2006;26:685–695. doi: 10.1523/JNEUROSCI.4620-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Park KS, Trimmer JS. Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation. Biochem Soc Trans. 2007;35:1064–1068. doi: 10.1042/BST0351064. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Siino DF, Trimmer JS. Interdomain cytoplasmic interactions govern the intracellular trafficking, gating, and modulation of the Kv2.1 channel. J Neurosci. 2008;28:4982–4994. doi: 10.1523/JNEUROSCI.0186-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J, Meucci O. CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis. 2010;1:e33. doi: 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Loftus R, Tamkun MM. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci U S A. 2010;107:12351–12356. doi: 10.1073/pnas.1003028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SB, Endoh T, Simen AA, Ren D, Miller RJ. Regulation of calcium currents by chemokines and their receptors. J Neuroimmunol. 2002;123:66–75. doi: 10.1016/s0165-5728(01)00485-4. [DOI] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Trimmer JS. Proteomic analyses of K(v)2.1 channel phosphorylation sites determining cell background specific differences in function. Channels (Austin) 2007;1:59–61. doi: 10.4161/chan.4388. [DOI] [PubMed] [Google Scholar]
- Pujol F, Kitabgi P, Boudin H. The chemokine SDF-1 differentially regulates axonal elongation and branching in hippocampal neurons. J Cell Sci. 2005;118:1071–1080. doi: 10.1242/jcs.01694. [DOI] [PubMed] [Google Scholar]
- Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci U S A. 2007;104:3568–3573. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel SA, Dudas S, Ge S, Gutiérrez JA. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res. 2000;6:102–111. [PubMed] [Google Scholar]
- Rigo A, Gottardi M, Damiani E, Bonifacio M, Ferrarini I, Mauri P, Vinante F. CXCL12 and [N33A]CXCL12 in 5637 and HeLa cells: regulating HER1 phosphorylation via calmodulin/calcineurin. PLoS One. 2012;7:e34432. doi: 10.1371/journal.pone.0034432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau YS, Huang PT, Liou HH, Liaw YC, Shiau YY, Lou KL. Structural basis of binding and inhibition of novel tarantula toxins in mammalian voltage-dependent potassium channels. Chem Res Toxicol. 2003;16:1217–1225. doi: 10.1021/tx0341097. [DOI] [PubMed] [Google Scholar]
- Song MY, Hong C, Bae SH, So I, Park KS. Dynamic modulation of the Kv2.1 channel by Src-dependent tyrosine phosphorylation. J Proteome Res. 2012;11:1018–1026. doi: 10.1021/pr200770v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105:287–295. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Häusser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Foehring R. A mechanism for homeostatic plasticity. Nat Neurosci. 2004;7:691–692. doi: 10.1038/nn0704-691. [DOI] [PubMed] [Google Scholar]
- Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahakis SR, Villasis-Keever A, Gomez T, Vanegas M, Vlahakis N, Paya CV. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol. 2002;169:5546–5554. doi: 10.4049/jimmunol.169.10.5546. [DOI] [PubMed] [Google Scholar]
- Yao H, Zhou K, Yan D, Li M, Wang Y. The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J Neurochem. 2009;108:909–919. doi: 10.1111/j.1471-4159.2008.05834.x. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- Zarayskiy VV, Balasubramanian G, Bondarenko VE, Morales MJ. Heteropoda toxin 2 is a gating modifier toxin specific for voltage-gated K+ channels of the Kv4 family. Toxicon. 2005;45:431–442. doi: 10.1016/j.toxicon.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Zhao YM, Sun LN, Zhou HY, Wang XL. Voltage-dependent potassium channels are involved in glutamate-induced apoptosis of rat hippocampal neurons. Neurosci Lett. 2006;398:22–27. doi: 10.1016/j.neulet.2005.12.073. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Zhu A, Zhan W, Liang Z, Yoon Y, Yang H, Grossniklaus HE, Xu J, Rojas M, Lockwood M, Snyder JP, Liotta DC, Shim H. Dipyrimidine amines: a novel class of chemokine receptor type 4 antagonists with high specificity. J Med Chem. 2010;53:8556–8568. doi: 10.1021/jm100786g. [DOI] [PMC free article] [PubMed] [Google Scholar]