

Abstract
Phosphomevalonate kinase (PMK) phosphorylates mevalonate-5-phosphate (M5P) in the mevalonate pathway, which is the sole source of isoprenoids and steroids in humans. We have identified new PMK inhibitors with virtual screening, using Autodock. Promising hits were verified and their affinity measured using NMR-based 1H-15N Heteronuclear Single Quantum Coherence (HSQC) chemical shift perturbation and fluorescence titrations. Chemical shift changes were monitored, plotted, and fitted to obtain dissociation constants (Kd). Tight binding compounds with Kd’s ranging from 6–60 µM were identified. These compounds tended to have significant polarity and negative charge, similar to the natural substrates (M5P and ATP). HSQC crosspeak changes suggest that binding induces a global conformational change, such as domain closure. Compounds identified in this study serve as chemical genetic probes of human PMK, to explore pharmacology of the mevalonate pathway, as well as starting points for further drug development.
Keywords: Human phosphomevalonate kinase, inhibitors, virtual screening, molecular docking
Introduction
Phosphomevalonate kinase (PMK), a mevalonate pathway enzyme, catalyzes a key phosphorylation step in isoprenoid/sterol biosynthesis, and provides a variety of products that are necessary for normal cellular growth and signaling, such as cholesterol, bile acids, heme A, dolichol, and ubiquinone.[1] Due to the central role of isoprenoids, imbalances in isoprenoid synthesis and utilization can lead to cellular dysfunction and disease. [2–4] Inhibitors of two enzymes in this pathway are of particular clinical relevance. The statins, a large class of hydroxymethyl glutaryl-CoA reductase (HMG CoA) inhibitors, are widely used to treat elevated cholesterol levels.[5] And, bisphosphonate inhibitors of farnesylpyrophosphate synthase are used to treat metabolic bone disease.[6] However, the mechanism and pharmacology of other potentially druggable enzyme targets in this pathway are less well-characterized, so additional studies and pharmacological probes are needed.[7]
PMK in comprised of two domains. The larger ATP-binding domain (CORE, Fig. 1) is composed of a five stranded parallel β-sheet interweaved with three α-helices. The mevalonate- 5-phosphate (M5P) domain (Substrate, Fig. 1) is composed of loop regions and two-α-helices. PMK catalyzes the transfer of a phosphoryl group from adenosine-5'-triphosphate (ATP) to M5P, resulting in mevalonate-5-pyrophosphate and adenosine diphosphate (ADP). [8–12] While details of the protein motion that occurs during catalysis remain unclear, the LID (Fig. 1) region is likely involved in an opening and closing motion to permit the binding and release of substrates.[13–15] Herdendorf and Miziorko investigated the functional role of conserved basic residues in human PMK and suggested that R110 was important for human PMK catalysis.[16] R111 and R84 are situated close to the “Walker B” motif and seem to be involved in binding M5P.[16] Residues K100 and K101 are in close proximity to the ATP binding site, and likely stabilize the transition state for conversion of ATP to ADP.[17, 18]
Fig. 1.
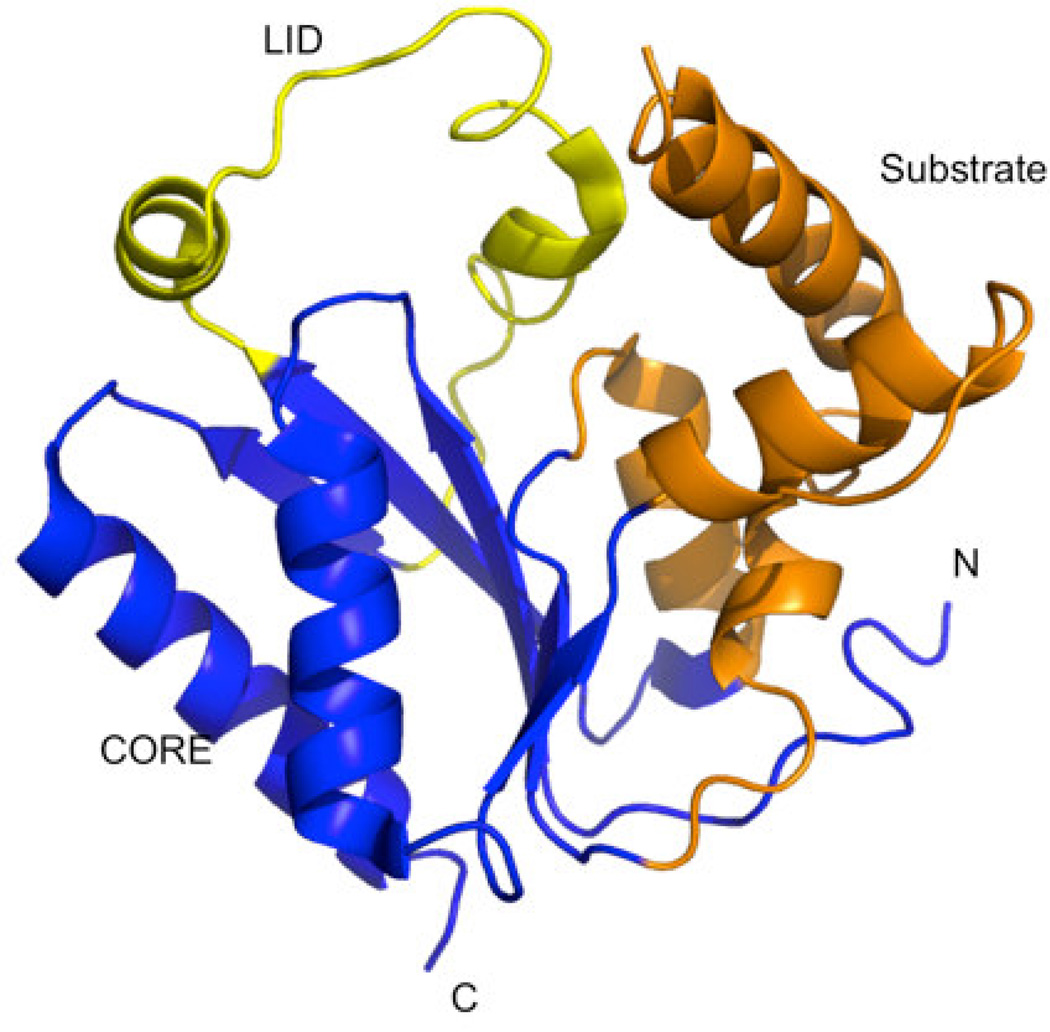
Crystal structure of human PMK (PDB ID 3ch4)[22] showing the different functionally relevant regions of the protein. The LID region caps the active site where phosphate transfer occurs, and contains catalytic residues needed for stabilizing the negative charge buildup in the phosphate transfer transition state. The CORE and Substrate domains are proposed to participate in significant domain rearrangements upon binding the substrates M5P and MgATP.[15]
In this study, we aim to identify novel inhibitors with high affinity for human PMK, based on our understanding of important protein-ligand interactions that involve the abovementioned basic residues. These inhibitors could provide clues as to the relative importance of binding interactions, and could themselves be useful as chemical probes of function (ex. in cellular assays or in vivo). Molecular docking has become an important early-stage method for finding novel inhibitors, when a protein structure is available.[19, 20] The process allows a large number of chemicals to be tested quickly in silicoand promising chemicals can then be prioritized for experimental verification of binding in vitro.[21] Given the high false-positive rate of docking predictions, it is crucial to verify predicted binding affinity using experimental assays. Two PMK structures have been published, allowing for docking-based identification of inhibitors.[2, 22]
NMR methods allow for a comprehensive monitoring of the effects of ligand binding, by providing chemical shift change measurements that reflect structural changes throughout the entire protein. Of particular interest, the 1H-15N HSQC experiment can provide an effective method for monitoring changes to a protein structure upon substrate or inhibitor addition, based on changes to 1H and 15N chemical shifts of all the protein backbone amides.[23] Since PMK is known to undergo substrate-induced structural changes[24], solution NMR techniques are well-suited to study interactions involving the human PMK/ligand complexes, which may also involve ligand-induced global structural rearrangements, such as the domain closure that substrate binding induces. Using molecular docking, solution NMR techniques, and fluorescence titrations, we identified novel inhibitors of human PMK. By monitoring 1H-15N HSQC chemical shift perturbations, site specific molecular interactions were identified and characterized.
Materials and Methods
Virtual Screening Protocol
Chemicals to be docked were sourced from two datasets: an in-house collection of 10,000 drug-like compounds, previously purchased or synthesized for kinase and oxidoreductase studies, and Chemiebase, a Thai Natural Products collection containing 1,000 structures.[25, 26] Before docking, ligands were adjusted to the proper protonation state at pH 7.4 by Pipeline Pilot, ver. 7.4 [27], and three-dimensional structures were calculated using Corina.[28] Molecular docking experiments were performed with AutoDock 4.2.[29–31]
The structure of human PMK (PDB ID: 3CH4) was obtained from the Protein Data Bank.[22] Gasteiger charges and hydrogens were added using AutoDock Tools (ADT). The docking grid, centered on the M5P binding pocket, was also prepared using ADT with a grid box created using 60 × 60 × 60 points and a resolution of 0.375 Å.
Protein Expression and Purification
Human PMK is a 192-residue protein with a molecular weight of 22.0 kDa. To aid in purification, a hexa-histidine tag was added at the N-terminus resulting in a total molecular weight of 24.2 kDa. Human PMK cDNA was purchased from OriGene, subcloned into the pET15b expression plasmid, expressed, and purified as described previously.[8] Briefly, the expression plasmid was transformed into Escherichia coli BL21 (DE3) Rosetta cells(Novagen) which encoded human PMK. Then, cells were grown in 1 L of Luria-Bertani(LB) media containing ampicillin (amp) and chloramphenicol (chl) until the optical cell densities at 600 nm (OD600) reached 0.7. The cells were harvested by centrifugation for 15 minutes at 6000xg. These cells were suspended in minimal media with amp-chl antibiotics supplemented with 15N-NH4Cl (Sigma Aldrich) as the lone source of nitrogen.[32] Cells were allowed to acclimate for an hour, and then they were induced with 1 mM IPTG. Cells were harvested 4h after induction by centrifugation. Cell pellets were re-suspended in a buffer containing 50 mM potassium phosphate, 5 mM imidazole, 5% glycerol, 1mM phenylmethanesulfonylfluoride (PMSF), and 300 mM NaCl at pH 7.8. Cells were lysed by passage through in a microfluidizer at ~17 kpsi. The lysate was clarified by centrifugation at 15,000 rpm for 30 minutes, and the supernatant loaded onto 1 mL of a Ni-Sepharose Fast-Flow resin (GE Healthcare). The column was washed with the 50 mM phosphate buffer until A280 < 0.005, and the protein was eluted using the same buffer supplemented with 300 mM imidazole. Protein concentration was determined spectrophotometrically using an extinction coefficient (ε280) of 32,290 M−1 cm−1 calculated with ExPASy ProtParam and the amino acid sequence.[33]
NMR Sample Preparation and Spectroscopy
Protein was concentrated to 400–600 µM by using a 10 kDa cutoff centricon (AMD Millipore) filter and exchanged into a buffer containing 20 mM potassium phosphate, 5 mM dithiothreitol (DTT), 100 mM potassium chloride, 10% glycerol, 0.02% sodium azide, and 10% D2O, at pH 6.5. All screened compounds were dissolved in d6-dimethyl sulfoxide (DMSO) to a concentration of 5 mM. Titrations were performed using 100 µM increments. Spectra were compared to control experiments of PMK and d6-DMSO (Supp. Fig. 2). Crosspeaks without a d6-DMSO effect 1H-15N HSQC chemical shifts were monitored and used to calculate Kd values. NMR experiments were performed at 25 °C using a Varian 600 MHz NMR system at 599.515 MHz using a triple resonance probe, with actively shielded Z-gradients. NMR data were processed and visualized by using NMRPipe[34] and analyzed with NMRview.[35] Dissociation constants (Kd) were calculated by from chemical shift changes resulting from conversion of free PMK, to the various bound states, as described previously.[15] The peaks that were monitored were in fast exchange in both 1H and 15N dimensions. Chemical-shift perturbations from NMR titrations were quantified using Eq. 1,
| (1) |
Then, the Kd value was determined by plotting and fitting (GraphPad Prism ver. 4.00[36]) the chemical shift changes (Δshiftobs) as a function of the concentration of protein and ligand, using the quadratic equation in Eq. 2,
| (2) |
where L0 and P0 are the total ligand concentration at a particular point and protein concentration, respectively, while Δshiftmax is the maximum chemical shift change observed for the particular peak of interest.
Fluorescence Titration
Fluorescence titrations were performed at 25 °C, using a Jasco FP-6500 spectrofluorotometer. The fluorescence emission of human PMK was measured in a total volume of 0.4 mL of buffer containing 20 mM potassium phosphate, 5 mM DTT, 100 mM potassium chloride, 10% glycerol at pH 6.5 and 5 µM human PMK. Excitation was performed at 295 nm, and emission was recorded at 300–400 nm. To determine the Kdthe difference in intensity between bound and free states at each data point were monitored and fitted as a function of ligand concentration to the one-site specific binding equation using GraphPad Prism ver. 4.00.[36]
Results
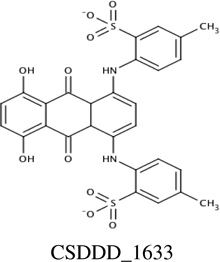
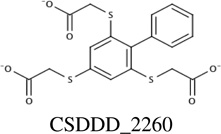
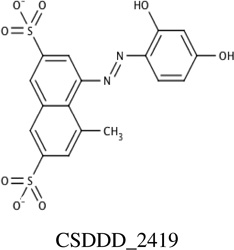
Chemicals from the two sources (synthetic and natural products) were prioritized for experimental screening based upon docking scores and cluster size, from Autodock 4.2 virtual screening. In total, 26 compounds were identified and then screened using NMR 1H-15N HSQC titration experiments; and, chemical shift changes were monitored upon addition of ligand aliquots. Four of these 26 chemicals (15%) showed measurable affinity for PMK in the NMR assay, so fluorescence titrations were also performed for these four compounds to confirm their binding to PMK. Binding affinities for these experiments are summarized in Table 1 and binding fits are provided in Supp. fig. S1 and S2.
Table 1.
Predicted and experimental binding affinity for compounds identified using Autodock 4.2 to dock compounds into human PMK. NMR and fluorescence techniques were then used to experimentally verify binding, and determine dissociation constants (Kd values).
| Chemical | Predicted Lowest Binding Energy (kcal/mol) |
Kd (µM) NMR [Residue used for data fitting] |
Kd (µM) Fluorescence |
|---|---|---|---|
 |
−11.6 | 55.4 ± 12.9 [G177] |
31 ± 8 |
 |
−10.34 | 6.3 ± 5.7 [T128] |
14.6 ± 4.6 |
 |
−11.8 | NDa [Q136] |
12 ± 2 |
 |
−9.2 | NDa [A172] |
61.0 ± 19.5 |
The NMR fit was not reliable because in addition to a specific binding event that seems to occur at low concentrations, there is also a non-specific effect that does not plateau.
In an attempt to illustrate the location of inhibitor binding, chemical changes were mapped onto the human PMK crystal structure (PDB ID: 3ch4). In Figure 2the 2D 1H-15N HSQC spectra and chemical shift mapping are shown for 3 the chemicals sourced from the in house collection of synthetic compounds. Figure 3 depicts this information for the hit obtained from the Chemibase collection of Thai natural products.
Fig. 2.
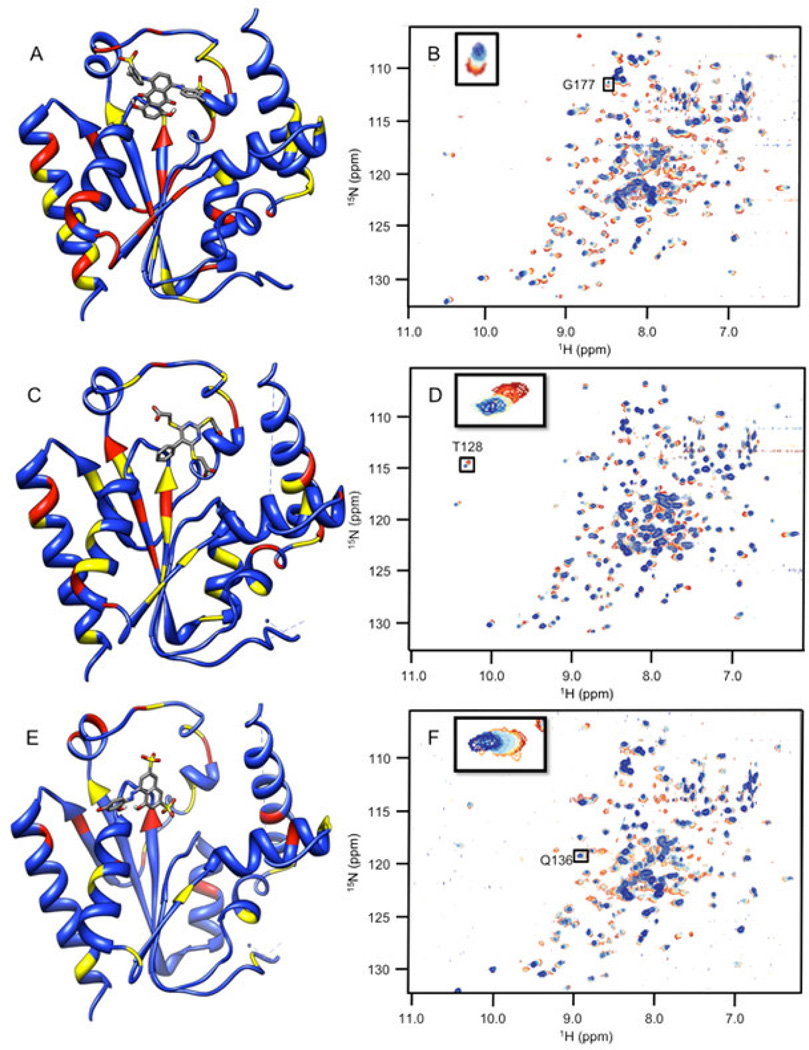
Chemical shift perturbations due to binding of the inhibitors identified from the synthetic compound collection. 1H-15N HSQC spectra chemical shifts are mapped onto the human PMK crystal structure [22], with the predicted binding pose from Autodock for each ligand; chemical shift changes are color-coded such that red is a large (> 0.09 ppm) chemical shift change, yellow is a medium (0.05–0.09 ppm) chemical shift change, and blue indicates small or no chemical shift change. Inset panels show chemical shift changes that were used to fit binding data (Eq. 1 and Eq. 2). Panels A and B are for compound CSDDD_1633, C and D are for compound CSDDD_2260, and E and F are for compound CSDDD_2419.
Fig. 3.
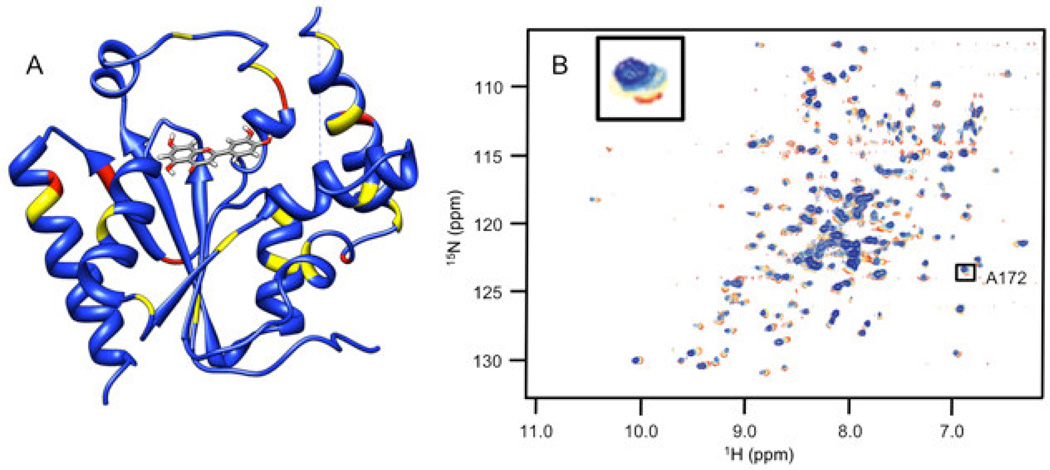
Chemical shift perturbations due to binding of an inhibitor (luteolin) from the Thai Natural Products compound collection, Chemiebase.[25, 26]. 1H-15N HSQC spectra chemical shifts (Panel B) are mapped onto the human PMK crystal structure (Panel A) [22], with the predicted binding pose from Autodock shown; chemical shift changes are color-coded such that red is a large (> 0.09 ppm) chemical shift change, yellow is a medium (0.05–0.09 ppm) chemical shift change, and blue indicates small or no chemical shift change. The inset panel shows chemical shift changes that were used to fit binding data (Eq. 1 and Eq. 2).
Discussion
The studies presented herein identified four novel inhibitors of PMK, which could lead to pharmacologically useful chemical probes or perhaps even drug leads that target the mevalonate pathway via inhibition of PMK. Molecular docking provided a prioritized listing of chemicals with potential affinity for PMK, with chemicals selected from two chemical collections, an inhouse collection of synthetic compounds, and Chemiebase[25, 26], a Thai natural product collection. Hits were identified from each chemical source and experimentally validated using NMR 1H-15N HSQC and fluorescence experiments. Of compounds predicted to bind to PMK using Autodock, 15% were verified by two methods (NMR and fluorescence) as being actual ligands for PMK. It is noteworthy that NMR titration methods are inherently limited in that they do not provide accurate measures of Kd values, when the Kd value is much less than the protein concentration that must be used in the experiment (which is itself limited by experimental sensitivity of the HSQC experiment). In these situations, data fitting to determine Kd must be done using a quadratic equation (Eq. 2), and errors increase as Kd≪[protein]. But, NMR chemical shift perturbation data will confirm binding and provide an upper limit for a Kd even in these situations, and more accurate Kd values can then be obtained with fluorescence titrations.
Docking and subsequent titrations (NMR and fluorescence) identified three synthetic compounds from the in-house compound collection (CSDDD library) with Kd values in the 12–30 µM range (fluorescence), Table 1. A common feature of these compounds is the presence of negative charges, which could mimic the negative charge on the phosphate groups of the natural substrates, ATP and M5P. Indeed, Autodock 4.2 placed these ligands (Fig. 2A, 2C, 2E) near the LID and CORE regions of the protein, close to the highly basic region of the known binding site for the natural substrates. Likely, these inhibitors are taking advantage of the strong electrostatic interactions with positively charged amino acids in the vicinity, including K17, R18, K19, K22, R138, and R141. Each of the three in-house collection inhibitors bind tightly to human PMK (Table 1) and these affinities favorably compare to those of the natural substrates ATP (29 ± 6 µM) and ADP (19.9 ± 9.0 µM).[15] When plotting the chemical shift perturbations for two chemicals (Supp. Fig. S3), complex binding was observed that could not be accounted for by the quadratic binding model used herein. In the case of CSDDD_2419, (Supp. fig. S3A) the binding fit showed a nonspecific effect in addition to an initial binding event that appears to be saturable. These additional effects can be detected using NMR titrations, because NMR is adept at detecting weakly interacting ligands; thus, there is a need for additional fluorescence titrations to verify the presence of saturable ligand-protein binding, and to obtain more accurate Kd values when Kd≪[protein].
NMR titrations can also provide structural information, based on location of the perturbed amino acid in the 3D structure of the protein, which corresponds to the crosspeak that is perturbed (if chemical shift assignments are available for the protein). Monitoring of chemical shift perturbations of crosspeaks in 2D 1H-15N HSQC spectra of PMK indicates there has been substantial movement of residues in the CORE and LID regions shown in Fig 2.[15] CSDDD_1633 and CSDDD_2260 both perturb the chemical shift of D163, an important hinge residue between the LID and CORE regions.[15] Also, ligand-PMK binding produced chemical shift perturbations outside of the binding site, suggesting a conformational changes, such as those that could result from motion around the hinge residue between domains, as we have reported previously.[15] Similar chemical shift perturbations are seen in this study to those that we have reported previously for ATP binding, both at the hinge and outside the ATP binding site, further suggesting that the in-house synthetic compounds are binding similarly to ATP.
Consistent with their binding in a highly basic pocket (intended to accommodate phosphates), all three of the compounds identified have at least two formal negative charges, either on sulfonate groups (CSDDD_1633 and CSDDD_2419) or carboxylic acids (CSDDD_2260). Recently, we observed that molecules with sulfonates could function as biomimics for naturally occurring negatively charged ligands with phosphates, sulfates, or posttranslationally modified residues (either phosphorylated or sulfated).[37] Compounds CSDDD_1633 and CSDDD_2419 also contain phenolic groups that would be expected to have low pKa’s, based on potential for resonance delocalization of negative charge (though an azo group or into a quinine carbonyl). This would put an additional negative charge on these molecules. Thus, all inhibitors are highly charged, making them suitable mimics of the PMK substrates which are also rich with negative charges.
Docking and subsequent titration studies also identified a natural product inhibitor, luteolin, a flavanoid, from the Chemibase collection. Luteolin has modest affinity for PMK, with a Kd of 61 µM. While it appears that luteolin exhibits no charge, this is probably not accurate. As for two of the compounds reported above, luteolin contains phenolic groups that are likely to be readily ionized, due to resonance delocalization into the quinine carbonyls. Indeed, the phenolic quinone substructure in in CSDDD_1633 is similar to the corresponding substructure in luteolin. Chemical shift perturbation mapping for this compound (Fig. 3) is strikingly different from the in-house compounds. It is therefore possible that luteolin is binding differently than the in-house synthetic chemicals. But, luteolin does cause a large chemical shift perturbation to the active site D163, as expected.[15] Interestingly, luteolin has been reported to show a wide range of biological effects including cardiovascular protection [38], but it is not clear what protein targets are responsible for this effect. It is possible that binding to PMK may at least contribute to the favorable cardiovascular effects of luteolin.
Conclusion
In summary, we have identified novel inhibitors of PMK, which possess significant negative charge that is suitable for binding to the highly basic PMK active site. Using virtual screening to identify potential inhibitors, and NMR and fluorescence experiments to verify binding, we found that docking yielded a hit rate of 15% and identifies reasonably potent PMK inhibitors with Kd values in the 6–60 µM range. These molecules could be useful to investigators as chemical genetic probes or as starting points in further drug and inhibitor design studies.
Supplementary Material
Highlights.
Natural and synthetic inhibitors of human phosphomevalonate kinase identified.
Virtual screening yielded a hit rate of 15%, with inhibitor Kd’s of 10–60 µM.
NMR studies indicate significant protein conformational changes upon binding.
Acknowledgements
This work was supported by a grant from the Thailand Research Fund (RTA5380010), and P.B. is grateful to The Royal Golden Jubilee PhD Program (PHD/0262/2549) for a scholarship. The National Center of Excellence in Petroleums, Petrochemical Technology and Advanced Materials and KURDI, and Kasetsart University are gratefully acknowledged for providing research facilities. D.S.S. acknowledges support from the American Heart Association (05303072) and an NIH Instrumentation Grant (S10 RR019012). H.M. acknowledges support from NIH (DK53766). Finally, Dr.Chak Sangma is gratefully acknowledged for providing access to Chemiebase.
Abbreviations
- PMK
phosphomevalonate kinase
- M5P
mevalonate-5-phosphate
- HSQC
heteronuclear single quantum coherence
- 1HMG CoA
hydroxymethyl glutaryl-CoA reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chambliss KL, Slaughter CA, Schreiner R, Hoffmann GF, Gibson KM. Molecular cloning of human phosphomevalonate kinase and identification of a consensus peroxisomal targeting sequence. J. Biol. Chem. 1996;271:17330–17334. doi: 10.1074/jbc.271.29.17330. [DOI] [PubMed] [Google Scholar]
- 2.Andreassi JLI, Vetting MW, Bilder PW, Roderick SL, Leyh TS. Structure of the ternary complex of phosphomevalonate kinase: The enzyme and its family. Biochemistry. 2009;48:6461–6468. doi: 10.1021/bi900537u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bliznakov EG. Diabetes and the role of isoprenoid biosynthesis. FEBS Lett. 2002;525:169–170. doi: 10.1016/s0014-5793(02)03062-4. [DOI] [PubMed] [Google Scholar]
- 4.Elson CE. Suppression of mevalonate pathway activities by dietary isoprenoids: Protective roles in cancer and cardiovascular disease. J. Nutr. 1995;125:1666S–1672S. doi: 10.1093/jn/125.suppl_6.1666S. [DOI] [PubMed] [Google Scholar]
- 5.Istvan ES. Structural mechanism for statin inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Am. Heart J. 2002;144:S27–S32. doi: 10.1067/mhj.2002.130300. [DOI] [PubMed] [Google Scholar]
- 6.Rodan GA, Balena R. Bisphosphonates in the treatment of metabolic bone diseases. Ann. Med. 1993;25:373–378. doi: 10.3109/07853899309147299. [DOI] [PubMed] [Google Scholar]
- 7.Buhaescu I, Izzedine H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Bioche. 2007;40:575–584. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Herdendorf TJ, Miziorko HM. Phosphomevalonate kinase: Functional investigation of the recombinant human enzyme. Biochemistry. 2006;45:3235–3242. doi: 10.1021/bi052231u. [DOI] [PubMed] [Google Scholar]
- 9.Bazaes S, Beytia E, Jabalquinto AM, Solis de Ovando F, Gomez I, Eyzaguirre J. Pig liver phosphomevalonate kinase. 1. purification and properties. Biochemistry. 1980;19:2300–2304. doi: 10.1021/bi00552a003. [DOI] [PubMed] [Google Scholar]
- 10.Bloch K, Chaykin S, Phillips AH, De Waard A. Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J. Biol. Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 11.Ferrand S, Tao J, Shen X, Mcguire D, Schmid A, Glickman JF, Schopfer U. Screening for mevalonate biosynthetic pathway inhibitors using sensitized bacterial strains. J. Biomol. Screening. 2011;16:637–646. doi: 10.1177/1087057111403927. [DOI] [PubMed] [Google Scholar]
- 12.Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011;505:131–143. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller CW, Schlauderer GJ, Reinstein J, Schulz GE. Adenylate kinase motions during catalysis: An energetic counterweight balancing substrate binding. Structure. 1996;4:147–156. doi: 10.1016/s0969-2126(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 14.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 15.Olson AL, Yao H, Herdendorf TJ, Miziorko HM, Hannongbua S, Saparpakorn P, Cai S, Sem DS. Substrate induced structural and dynamics changes in human phosphomevalonate kinase and implications for mechanism. Proteins: Struct. , Funct. , Bioinf. 2009;75:127–138. doi: 10.1002/prot.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herdendorf TJ, Miziorko HM. Functional evaluation of conserved basic residues in human phosphomevalonate kinase. Biochemistry. 2007;46:11780–11788. doi: 10.1021/bi701408t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 18.Koonin EV. A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol. 1993;229:1165–1174. doi: 10.1006/jmbi.1993.1115. [DOI] [PubMed] [Google Scholar]
- 19.Irwin JJ, Shoichet BK. ZINC - A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavasotto CN, Orry AJW. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007;7:1006–1014. doi: 10.2174/156802607780906753. [DOI] [PubMed] [Google Scholar]
- 21.Shoichet BK. Virtual screening of chemical libraries. Nature Reviews. Molecular Cell Biology. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang Q, Yan X, Gu S, Liu J, Liang D. Crystal structure of human phosphomavelonate kinase at 1.8 Å resolution. Proteins: Structure, Function, and Bioinformatics. 2008;73:254–258. doi: 10.1002/prot.22151. [DOI] [PubMed] [Google Scholar]
- 23.Sem DS, Pellecchia M. NMR in the acceleration of drug discovery. Curr. Opin. Drug Discov. Devel. 2001;4:479–492. [PubMed] [Google Scholar]
- 24.Olson AL, Cai S, Herdendorf TJ, Miziorko HM, Sem DS. NMR dynamics investigation of ligand-induced changes of main and side-chain arginine N-H's in human phosphomevalonate kinase. J. Am. Chem. Soc. 2010;132:2102–2103. doi: 10.1021/ja906244j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sangma C, Chuakheaw D, Jongkon N, Gadavanij S. Computer techniques for drug development from thai traditional medicine. Curr. Pharm. Des. 2010;16:1753–1784. doi: 10.2174/138161210791164054. [DOI] [PubMed] [Google Scholar]
- 26.Thai medicinal plants. Http://Chemiebase.ku.ac.th. [Google Scholar]
- 27.Accelrys Software. Pipeline pilot, ver. 7.4. [Google Scholar]
- 28.Molecular Networks. CORINA, ver. 2.4. [Google Scholar]
- 29.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 30.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J. Computational Chemistry. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 32.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 33.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook. 2005:571–607. [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 36.GraphPad Software. ver. 4.0. [Google Scholar]
- 37.Olson AL, Neumann TS, Boonsri P, Ziarek JJ, Peterson FC, Cai S, Volkman B, Hannongbua S, Sem DS. Aryl-sulfonates as biomimetics for negatively charged ligands: Application to phosphomevalonate kinase and the CXCL12 chemokine. Med. Chem. Comm. (in preparation) [Google Scholar]
- 38.Lopez-Lazaro M. Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 2009;9:31–59. doi: 10.2174/138955709787001712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.