

Abstract
Oncolytic virus (OV) therapy takes advantage of common cancer characteristics, such as defective type I interferon (IFN) signaling, to preferentially infect and kill cancer cells with viruses. Our recent study (Murphy et al., 2012, J. Virol., 86: 3073-87) found human pancreatic ductal adenocarcinoma (PDA) cells were highly heterogeneous in their permissiveness to vesicular stomatitis virus (VSV) and suggested at least some resistant cell lines retained functional type I IFN responses. Here we examine cellular responses to infection by the oncolytic VSV recombinant VSV-ΔM51-GFP by analyzing a panel of 11 human PDA cell lines for expression of 33 genes associated with type I IFN pathways. Although all cell lines sensed infection by VSV-ΔM51-GFP and most activated IFN-α and β expression, only resistant cell lines displayed constitutive high-level expression of the IFN-stimulated antiviral genes MxA and OAS. Inhibition of JAK/STAT signaling decreased levels of MxA and OAS and increased VSV infection, replication and oncolysis, further implicating IFN responses in resistance. Unlike VSV, vaccinia and herpes simplex virus infectivity and killing of PDA cells was independent of the type I IFN signaling profile, possibly because these two viruses are better equipped to evade type I IFN responses. Our study demonstrates heterogeneity in the type I IFN signaling status of PDA cells and suggests MxA and OAS as potential biomarkers for PDA resistance to VSV and other OVs sensitive to type I IFN responses.
INTRODUCTION
Oncolytic virus (OV) therapy utilizes viruses with naturally inherited or engineered properties enabling them to preferentially infect and kill cancer cells (Breitbach et al., 2010; Russell and Peng, 2007; Vähä-Koskela et al., 2007). This approach utilizes common cancer characteristics such as defective innate immune responses or abnormalities in regulation of mRNA translation or cellular signaling pathways to provide the needed cancer specificity.
Vesicular stomatitis virus (VSV) has been successfully used as an OV in preclinical models of a number of malignancies [reviewed in (Barber, 2004; Hastie and Grdzelishvili, 2012)]. As a result, a clinical trial using VSV against hepatocellular carcinoma is currently in progress (Clinicaltrials.gov, 2012, Trial ID: NCT01628640). A number of oncolytic VSV recombinants have been developed to address safety concerns relating to the use of wild-type (wt) VSV. In one of these, VSV-ΔM51-GFP, a deletion of the methionine at amino acid position 51 of the matrix (M) protein prevents shut down of cellular gene expression (Ahmed et al., 2003), providing enhanced safety, including an absence of neurotoxicity in vivo, while still demonstrating good oncolytic potential (Ahmed et al., 2008; Ebert et al., 2005; Goel et al., 2007; Kelly et al., 2010; Stojdl et al., 2003; Wollmann et al., 2010; Wu et al., 2008).
We recently tested wild-type (wt) VSV and two non-neurotropic VSV recombinants (including VSV-ΔM51-GFP), as well as recombinant Sendai virus, recombinant respiratory syncytial virus and two recombinant adenoviruses against a panel of human pancreatic ductal adenocarcinoma (PDA) cell lines (Murphy et al., 2012). PDAs are highly aggressive and metastatic (Stathis and Moore, 2010) and represent about 95% of pancreatic cancers. PDA is one of the most lethal abdominal malignancies (Farrow et al., 2008; Lindsay et al., 2005), and current treatments are largely ineffective (Stathis and Moore, 2010). Our study demonstrated VSV is a promising oncolytic agent against PDA, as the majority of PDA cell lines tested were highly susceptible to infection and killing by VSV recombinants (Murphy et al., 2012). However, five PDA cell lines as well as the non-malignant HPDE cell line were resistant to most VSV recombinants, (wt VSV, VSV-ΔM51-GFP, and VSV-p1-GFP), at least at low multiplicities of infection (MOI), the expected scenario in vivo.
Unlike permissive PDA cell lines, most resistant PDA cell lines were able to both secrete and respond to type I interferon (IFN), suggesting intact type I IFN responses contributed to their resistance phenotype (Murphy et al., 2012). While other mechanisms have been noted (Hastie and Grdzelishvili, 2012), type I IFN sensitivity is believed to be a major factor contributing to VSV’s oncoselectivity, as it is unable to efficiently infect healthy cells. In contrast, the majority of cancer cells are thought to be defective in type I IFN production and responses (Barber, 2004; Hastie and Grdzelishvili, 2012; Lichty et al., 2004), as IFN responses are generally anti-proliferative, anti-angiogenic and pro-apoptotic (Wang et al., 2011), conditions unfavorable for tumor formation. However, some cancer cells are known to produce and/or respond to type I IFN (Naik and Russell, 2009; Stojdl et al., 2000), including some mesotheliomas (Saloura et al., 2010), melanomas (Linge et al., 1995; Wong et al., 1997), lymphomas (Sun et al., 1998), bladder cancers (Matin et al., 2001), renal cancers (Pfeffer et al., 1996), and possibly other cancers (Stojdl et al., 2003).
Here we further analyze a panel of 11 clinically relevant human PDA cell lines for the presence of type I IFN response, determine the functionality of that response in resistance to VSV-ΔM51-GFP and attempt to identify an RNA and/or protein which presence or absence was well correlated with resistance to this virus. The cell lines most resistant to VSV-ΔM51-GFP infection were shown to constitutively express at least some interferon stimulated genes (ISGs), including the antiviral genes MxA and OAS. Inhibition of the JAK/STAT signaling pathways reduced ISG expression and improved VSV-ΔM51-GFP infectivity, replication and oncolysis, implicating IFN responses in resistance.
MATERIALS AND METHODS
Cell lines
The human PDA cell lines used in this study were: AsPC-1 (ATCC CRL-1682), Capan-1 (ATCC HTB-79), Capan-2 (ATCC HTB-80), CFPAC-1 (ATCC CRL-1918), HPAC (ATCC CRL-2119), HPAF-II (ATCC CRL-1997), Hs766T (ATCC HTB-134), MIA PaCa-2 (ATCC CRL-1420), Panc-1 (ATCC CRL-1469), Suit2 (Iwamura et al., 1987) and T3M4 (Okabe et al., 1983). In addition, a non-malignant human pancreatic duct epithelial (HPDE) cell line (Furukawa et al., 1996) was used and maintained in Keratinocyte-SFM (K-SFM, Gibco). This cell line was generated by introduction of the E6 and E7 genes of human papillomavirus 16 into normal adult pancreas epithelium, retains a genotype similar to pancreatic duct epithelium and is non-tumorigenic in nude mice (Furukawa et al., 1996). The mouse breast cancer cell line 4T1 (ATCC CRL-2539), baby hamster kidney BHK-21 fibroblasts (ATCC CCL-10) and African green monkey kidney Vero cells (ATCC CCL-81) were used to grow viruses and/or as controls. Capan-1, CFPAC-1, HPAC, MIA PaCa-2, Panc-1, Suit2, 4T1 and Vero cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Cellgro). AsPC-1, Capan-2 and T3M4 cells were maintained in RPMI 1640 (HyClone). HPAF-II and BHK-21 cells were maintained in modified Eagle’s medium (MEM, Cellgro). All cell growth media were supplemented with 9% fetal bovine serum (FBS, Gibco), 3.4 mM L-glutamine, 900 U/ml penicillin and 900 μg/ml streptomycin (HyClone). MEM was further supplemented with 0.3% glucose (w/v). K-SFM was never supplemented with serum. Cells were kept in a 5% CO2 atmosphere at 37°C. For all experiments, PDA cell lines were passaged no more than 10 times.
Viruses
The following viruses were used in this study: VSV-ΔM51-GFP, vaccinia virus expressing T7 (VVT7) and herpes simplex virus type 1 (HSV-1) (MacIntyre strain; ATCC, VR-539). VSV-ΔM51-GFP has a deletion of methionine at amino acid position 51 of the M protein and the green fluorescent protein (GFP) ORF inserted at position 5 of the viral genome (Wollmann G, 2010). VVT7 was created by integration of the bacteriophage T7 RNA polymerase into the vaccinia virus (strain Western Reserve) thymidine kinase gene (Fuerst et al., 1986). VSV-ΔM51-GFP was grown on BHK-21 while VVT7 and HSV-1 were grown on Vero. Viral titers were determined by standard plaque assay on 4T1 and/or Vero cells and expressed as cell infectious units (CIU) per ml.
RNA analysis
Cells were either mock-treated, infected with VSV-ΔM51-GFP at MOI of 10 CIU per cell (based on 4T1) or treated with 5,000 U/ml IFN-α (Calbiochem, 407294). Total RNA was extracted from cells at 4 or 12 hours (h) post-infection (p.i.) using the Quick-RNA Mini Prep kit in accordance with manufacturer instructions (Zymo Research). 0.5 μg of total RNA per reverse transcription (RT) reaction using SMARTScribe reverse transcriptase (Clontech) was used for the cDNA synthesis as per manufacturer’s protocol. PCR was carried out on this cDNA using the following conditions: denaturation at 94°C for 45 seconds (s), annealing at 57°C for 45 s, extension at 72°C for 45 s for either 25, 30 or 35 cycles and a finishing step at 72°C for 8 min. All primers for PCR are shown in the Supplementary Table 1 and were designed to not amplify genomic DNA. PCR products were electrophoresed on a 2% agarose gel with ethidium bromide and photographed using a GelDoc-It imager (UVP imaging, Upland, CA).
Western blot
Cellular lysates were prepared from cells either mock-treated, infected with VSV-ΔM51-GFP at MOI 10 CIU/cell (based on 4T-1) or treated with 5,000 U/ml IFN-α. At 1 h p.i., virus was aspirated and cells were extensively washed and incubated in growth media containing 5% FBS. Cells were harvested at 12 h p.i. and lysed in lysis buffer (pH 7.5) containing 1% Triton-X-100, 20mM Hepes, 0.15 M NaCl, 2 mM EDTA and supplemented with c-inhibitor (2X, Roche) and Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich). Total protein concentration was determined by Bradford assay. Twenty μg of total protein was separated by electrophoresis on 10% SDS-PAGE gels and electroblotted to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked using 5% non-fat powdered milk in TBS-T [0.5 M NaCl, 20 mM Tris (pH 7.5), 0.1%Tween20]. Membranes were incubated with 1:5000 rabbit polyclonal anti-VSV antibodies (raised against VSV virions), 1:1000 rabbit anti-MX1 (Sigma-Aldrich, SAB1100070), 1:200 rabbit anit-OAS2 (Santa Cruz, sc-99097), 1:3000 mouse anti-GFP (Rockland, 600-301-215), and the following antibodies from Cell Signaling (1:1000): anti-STAT1 (9172), Stat1-P (9171), Stat2 (4594), Stat2-P (4441), IRF3 (4302), IRF3-P (4947), eIF2α (5324), and eIF2α-P (3398) in TBS-T with 5% BSA. Detection was with 1:2000 goat anti-rabbit or 1:5000 goat anti-mouse horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, 111-035-003 and 115-035-003, respectively) using the Enhanced Chemiluminescence Plus (ECL+) protein detection system (GE Healthcare). Membranes were reprobed with mouse anti-actin antibody (clone C4) (Moyer et al., 1986) to verify sample loading.
JAK Inhibition
For new infectious particle production, 6-well plates were seeded such that they were approximately 80% confluent at the time of inhibitor treatment. Cells were pretreated with 0.5 or 2.5μM JAK inhibitor I (Jak Inh. I, Calbiochem) or vehicle (DMSO) only in cell culture media with 5% FBS (K-SFM was used without FBS) for 48 h prior to infection (media was removed and replaced with fresh drug/vehicle containing media after the first 24 h). Cells were then mock infected or infected with VSV-ΔM51-GFP in DMEM without FBS at an MOI of 10 CIU/cell (based on 4T1). Following a 1 h absorption period, the virus containing media was aspirated, cells were washed three times with PBS and growth media with 5% FBS containing either 0.5 or 2.5μM JAK Inh. 1 or vehicle was added to the wells. At 16 h p.i., media was collected and used for a standard plaque assay on BHK-21 cells. Cells treated and infected in the same manner were also used to prepare cellular lysates for western blotting as described above.
For plaque and cell viability assays, cells were seeded in 96-well plates such that they were approximately 80% confluent at the time of inhibitor treatment, and pretreated for 48 h with JAK Inh. I as described above. For the plaque assay, cells were then infected with 8-fold serial dilutions of VSV-ΔM51-GFP in DMEM without FBS. Following a 1 h absorption period, the virus containing media was aspirated and replaced with cell culture media containing 5% FBS and either JAK Inh. I or vehicle. At 17 h p.i., fluorescent foci were visualized and counted using fluorescent microscopy. For the cell viability assay, following pretreatment, cells were mock infected or infected with VSV-ΔM51-GFP in DMEM without FBS at a MOI of 1 CIU/cell (based on 4T1). Following a 1 h absorption period, the virus containing media was aspirated and replaced with growth media with 5% FBS containing either 0.5 or 2.5 μM JAK Inh. 1 or vehicle. GFP fluorescence was measured every 24 h using a CytoFluor multi-well plate reader (PerSeptive Biosystems) with excitation filter of 450/50nm, emission filter of 530/25nm and gain=50. At 5 days (d) p.i., cell viability was analyzed using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assay (Biotium) in accordance with manufacturer instructions.
Permissiveness of cells to different viruses
Cells were seeded in 24-well plates such that they were approximately 90% confluent at the time of infection. Cells were infected with 8-fold serial dilutions of VSV-ΔM51-GFP, VVT7, or HSV-1 in DMEM without FBS for 1 h. Virus was aspirated and cells were overlaid with the appropriate growth media containing 5% FBS and 0.5% BactoAgar. For VSV-ΔM51-GFP, fluorescent foci were visualized at 3 d p.i. by fluorescent microscopy. For all viruses, at 5 d p.i. cells were fixed by addition of 350 μl 10% neutral buffered formalin (Sigma-Aldrich) to each well. Following a 1 h incubation at room temperature, the agar was gently removed and cells stained with 1% crystal violet in 20% methanol to allow visualization of plaques.
Cell viability following infection with different viruses
Cells were seeded in 96-well plates such that they were approximately 80% confluent at the time of infection. Cells were mock infected or infected with VSV-ΔM51-GFP, VVT7, or HSV in SFM-MegaVir, at an MOI of 1 or 0.01 CIU/cell (based on Vero). Following a 1 h absorption period, the virus containing media was aspirated and replaced with growth media containing 5% FBS. At 5 d p.i., cell viability was analyzed using an MTT cell viability assay (Biotium) in accordance with manufacturer instructions.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism, version 5.03 for Windows (GraphPad Software, San Diego, California). Comparisons within a cell line following treatment were made using a one-way ANOVA with Bonferroni post-test.
RESULTS
Identification of antiviral genes constitutively expressed in PDA cells resistant to VSV
Our recent evaluation of VSV against human PDA cell lines revealed substantial diversity in their susceptibility to VSV-mediated oncolysis, which correlated with permissiveness to VSV infection (Murphy et al., 2012). Most resistant PDA cell lines were both sensitive to IFN-α treatment (which strongly inhibited VSV infection) and capable of secreting IFN-β following VSV infection (Murphy et al., 2012), suggesting resistant PDA cell lines may retain active type I IFN signaling. In this study, we assessed type I IFN related cellular responses to VSV-ΔM51-GFP infection on a molecular level in a panel of 11 clinically relevant human PDA cell lines (Table 1), and examined the role of the JAK/STAT signaling pathways in the observed resistance phenotypes.
Table 1.
Human pancreatic cell lines used in this study and a summary of VSV-ΔM51-GFP susceptibility and expression of select mRNAs.
| Human cell line | Origina | VSV-ΔM51 resistancec | IRF7 | IFN-α | IFN-β | IFN-λ | MxA | OAS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lowd | Highe | VSVf | Low | High | VSV | Low | High | VSV | Low | High | VSV | Low | High | VSV | Low | High | VSV | |||
| Capan-1 | liver metastasis | − | x | x | x | x | x | x | x | |||||||||||
| MIA PaCa-2 | primary PDA | − | x | x | x | x | x | x | x | x | ||||||||||
| Panc-1 | primary PDA | − | x | x | x | x | x | x | x | x | ||||||||||
| Capan-2 | primary PDA | − | x | x | x | x | x | x | x | x | ||||||||||
| AsPC-1 | ascites | − | x | x | x | x | x | x | x | x | x | |||||||||
| Suit2 | liver metastasis | − | x | x | x | x | x | x | x | x | x | x | ||||||||
| T3M4 | LN metastasisb | − | x | x | x | x | x | x | x | x | x | |||||||||
| HPDE | Non-malignant | + | x | x | x | x | x | x | x | x | ||||||||||
| CFPAC-1 | primary PDA | ++ | x | x | x | x | x | x | x | x | x | |||||||||
| HPAC | primary PDA | ++ | x | x | x | x | x | x | x | x | ||||||||||
| Hs766T | LN metastasis | +++ | x | x | x | x | x | x | x | x | ||||||||||
| HPAF-II | primary PDA | +++ | x | x | x | x | x | x | x | x | ||||||||||
All cell lines (except for non-malignant HPDE) have PDA origin
LN, lymph node
−, susceptible; +, intermediate; ++, resistant; +++ highly resistant
mRNA expression undetectable or barely detectable
mRNA robustly expressed
mRNA expression increases upon VSV-Δ M51-GFP infection
Expression of 33 human genes (Supplementary Table 1) responsible for sensing viral infection [e.g. retinoic-acid-inducible gene I (RIG-I)], activating production of type I IFNs [e.g. IFN-regulatory factor (IRF) 3], sensing IFNs [e.g. interferon alpha/beta receptor 1 (IFNAR1)] and inducing an antiviral state in cells [e.g. myxovirus (influenza) resistance 1 (MxA)] was assessed. Since we wanted to focus on a specific set of genes, this was done using semi-quantitative RT-PCR. Genes were selected based on their importance in type I IFN responses. Some of these genes, particularly ISGs, are expressed in response to type I IFNs, and are expected to be upregulated in cells with intact type I IFN signaling following VSV infection. Also, differential expression of RIG-I (Wilden et al., 2009), myeloid differentiation primary response protein MyD88 (MyD88) (Wongthida et al., 2011), IRF3 (Marozin et al., 2008; Wilden et al., 2009), IRF7 (Wilden et al., 2009) IFNAR1/2 (Saloura et al., 2010; Zhang et al., 2010), tyrosine-protein kinase JAK1 (JAK1) (Dunn et al., 2005), signal transducer and activator of transcription (STAT) 1 (Lee et al., 1997; Sun et al., 1998), IRF9 (Matin et al., 2001; Saloura et al., 2010), MxA (Monsurro et al., 2010; Paglino and van den Pol, 2011), 2′-5′-oligoadenylate synthetase (OAS) (Monsurro et al., 2010; Saloura et al., 2010), and interferon-induced, double-stranded RNA-activated protein kinase (PKR) (Saloura et al., 2010) has been previously reported to correlate to or be responsible for the virus or IFN resistance phenotypes of various cancer cell types.
PDA cells were mock infected or infected with VSV-ΔM51-GFP for 4 h, and total RNA was isolated and analyzed for expression of candidate genes. As shown in Figure 1, most analyzed genes (including all additional genes listed in Supplementary Table 1 but not shown in Figure 1) were expressed at similar levels in all cell lines regardless of the VSV resistance phenotype. However, several genes were clearly differentially expressed. Among genes associated with sensing viral infection, IRF7 was down regulated in Capan-1 and MIA PaCa-2, both highly susceptible to VSV-mediated cell death (Murphy et al., 2012). In agreement with a key role for IRF7 in the expression of type I IFNs following viral infection (Honda et al., 2005), these same two cell lines also lacked IFN-α and β gene expression even after VSV-ΔM51-GFP infection (Fig. 1). Interestingly, reduced expression of IRF-7 has recently been linked to increased metastasis in breast cancer, with disruption of IFN signaling identified as the primary cause (Bidwell et al., 2012).
Figure 1. mRNA expression of IFN related genes.
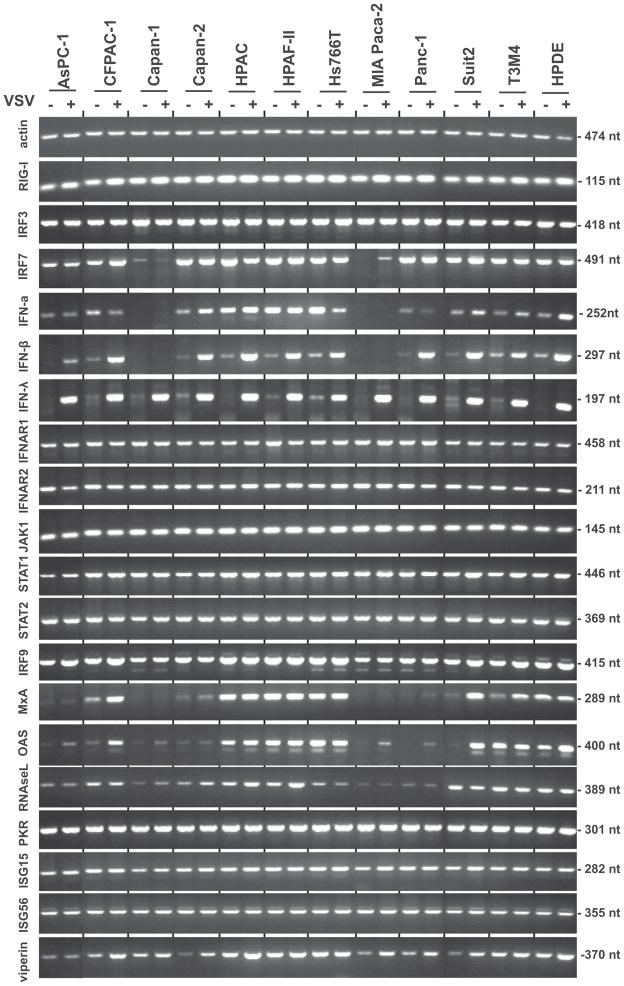
Cells were mock infected (−) or infected (+) with VSV-ΔM51-GFP at MOI 10 CIU/cell and harvested at 4 h p.i. Extracted mRNA was reverse transcribed and then analyzed by semi-quantitative PCR for the indicated genes. PCR product sizes are indicated on the right.
Although in our previous study only 4 cell lines (HPAF-II, HPAC, Hs766T and non-malignant HPDE) produced detectable levels of secreted IFN-β at 18 h p.i. (Murphy et al., 2012), here VSV-ΔM51-GFP at 4 h p.i. induced production of IFN-β mRNA in all cell lines except Capan-1 and MIA PaCa-2 (Fig. 1). Most cell lines (again, with the exception of Capan-1 and MIA PaCa-2) also showed constitutive expression of IFN-α mRNA that was only marginally increased upon VSV-ΔM51-GFP infection at 4 h p.i. The highest constitutive levels of IFN-α mRNA were seen in three of the resistant cell lines, HPAF-II, Hs766T and HPAC. While differences in IRF7, IFN-α and IFN-β mRNA expression were observed, these alone could not discriminate between the phenotypes of PDA cells based on their permissiveness to VSV.
As shown in Figure 1, differences were not seen in the expression of the IFN-α/IFN-β receptor (IFNAR1 and IFNAR2), or the major components of IFN type I signal transduction (JAK1, STAT1, STAT2 and IRF9) at the transcriptional level. However, there was great variability in the expression of two ISGs, MxA and OAS. Importantly, there was a correlation between virus resistance phenotype and the levels of MxA and OAS mRNA. MxA and OAS were at the highest levels in the resistant cell lines HPAF-II, Hs766T, HPAC and non-malignant HPDE; at lower levels (especially in the absence of infection) in less resistant Suit2, T3M4 and CFPAC-1; and minimally produced in the remaining cell lines, all of which are highly susceptible to VSV-ΔM51-GFP infection. Interestingly, while MxA and OAS had variable mRNA expression in different PDA cell lines a 4 h p.i., a similar pattern was not observed with other antiviral ISGs such as PKR, RNAse L, ISG56 and viperin.
To confirm the association of MxA and OAS expression with the resistance of PDA cells to VSV-ΔM51-GFP, we conducted a similar experiment with a representative group of 7 cell lines mock-treated, treated with IFN-α, or infected with VSV-ΔM51-GFP for 12 h to allow for accumulation of activated proteins. In this experiment (Fig. 2), we conducted not only mRNA analysis (as in Figure 1), but also western blot analysis to determine protein levels of the analyzed genes. In addition to the non-malignant HPDE, which has intermediate resistance, we analyzed two highly resistant (HPAF-II, Hs766T), one intermediate (HPAC) and three susceptible cell lines (MIA PaCa-2, AsPC-1, Suit2). Susceptibility to viral infection was confirmed based on virus-directed GFP expression, which we have previously shown correlates well with viral protein synthesis (Murphy et al., 2012). Importantly, among susceptible cell lines, we selected two cell lines (AsPC-1 and Suit2) previously shown to be responsive to IFN-α treatment and one cell line (MIA PaCa-2) which was completely resistant to IFN-α treatment (Murphy et al., 2012). As shown in Figure 2, a correlation was observed between MxA and OAS accumulation (mRNA and protein) and the resistance phenotype of PDA cell lines. Again, all resistant cell lines showed constitutive expression of MxA and OAS even in the absence of virus infection or IFN treatment. The expression of MxA and OAS is controlled via STAT1 and STAT2 activation. Although variations in mRNA levels were not observed for STAT1 and STAT2 (Fig. 1 and 2), we tested whether phosphorylation of the protein products differed in VSV permissive and resistant cell lines. As shown in Figure 2, although most cell lines had detectable levels of phosphorylated STAT1 protein following 12-h IFN-α treatment, the highest levels of STAT1 activation following VSV-ΔM51-GFP infection were observed in HPAF-II, Hs766T and HPAC, all of which are resistant. Also, lower but detectable levels of phosphorylated STAT1 in untreated cells were detected in these three cell lines and in HPDE, which may at least partially explain the constitutive expression of MxA and OAS in these cell lines. Among the susceptible cell lines, STAT1 was activated in AsPC-1 and to a much lesser degree in Suit2 upon infection, but not in MIA PaCa-2, although it was strongly activated in MIA PaCa-2 upon IFN treatment. A similar pattern of protein phosphorylation was observed for STAT2 (Fig. 2).
Figure 2. mRNA and protein expression of IFN related genes.
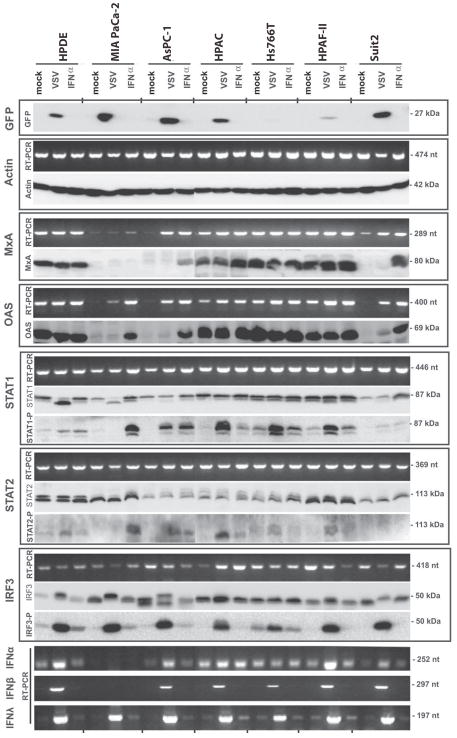
Cells were mock infected, infected with VSV-ΔM51-GFP (indicated as VSV) at MOI 10 CIU/cell or treated with 5,000 U/ml IFN-α. Cells were harvested at 12 h p.i. and mRNA was reverse transcribed and analyzed by semi-quantitative PCR or cell lysates were prepared and analyzed by western blot for the indicated protein. PCR (nt) and protein (kDa) product sizes are indicated on the right.
To test whether PDA cell lines susceptible to VSV-ΔM51-GFP are able to detect VSV infection, we analyzed phosphorylation of IRF3. Interestingly, all tested PDA cell lines showed increased IRF3 phosphorylation in response to infection (Fig. 2), suggesting the upstream components of the RIG-I pathway are functional. In agreement with this observation, type III IFN-λ mRNA was expressed in all cell lines in response to VSV-ΔM51-GFP infection (Fig. 1 and 2). However, at 12 h p.i., MIA Paca-2 still failed to activated IFN-α and IFN-β mRNA expression.
Key differences in mRNA and protein expression are summarized in Table 1 and Table 2 respectively. Together, our gene expression data suggest all tested PDA cell lines are capable of sensing VSV infection, most produce type I IFNs, and most are capable of sensing IFN. However, resistant cell lines were characterized by constitutive expression of at least two ISGs, MxA and OAS, which possibly contribute to their resistance phenotype.
Table 2.
Summary of VSV-ΔM51-GFP susceptibility and expression of select proteins.
| Human cell line | VSV-Δ M51 resistancea | IRF3-p | STAT1-p | MxA | OAS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lowb | Highc | VSVd | IFN-α e | Low | High | VSV | IFN-α | Low | High | VSV | IFN-α | Low | High | VSV | IFN-α | ||
| MIA PaCa-2 | − | x | x | x | x | x | x | x | |||||||||
| AsPC-1 | − | x | x | x | x | x | x | x | x | x | |||||||
| Suit2 | − | x | x | x | x | x | x | x | x | x | x | ||||||
| HPDE | + | x | x | x | x | x | x | x | |||||||||
| HPAC | ++ | x | x | x | x | x | x | x | x | ||||||||
| Hs766T | +++ | x | x | x | x | x | x | x | |||||||||
| HPAF-II | +++ | x | x | x | x | x | x | x | |||||||||
−, susceptible; +, intermediate; ++, resistant; +++ highly resistant
protein expression undetectable or barely detectable
protein robustly expressed
protein expression increases upon VSV-ΔM51-GFP infection
protein expression increases upon IFN-α treatment
Improved oncolysis of resistant PDA cells by combination treatment with JAK inhibitor I
Gene expression data showed a correlation between resistance to VSV-ΔM51-GFP infection and elevated expression of at least some ISGs. Therefore, a possible causative role for these and other ISGs in the resistance phenotype was tested by inhibition of the JAK/STAT signaling pathways responsible for activation of ISG expression, using a general inhibitor of JAKs, JAK Inh. I. First, the effectiveness of the JAK Inh I treatment was confirmed by western blot for p-STAT1, MxA and OAS. Twenty-four h treatment with 0.5 or 2.5μM JAK Inh I completely eliminated STAT1 phosphorylation and markedly reduced MxA and OAS protein levels (data not shown). The same treatments for 48 h, followed by mock infection or infection with VSV-ΔM51-GFP at MOI 10 (based on 4T-1) for 16 h, further reduced MxA and OAS protein to below detectable levels in uninfected cells and sharply reduced them in infected cells (Figure 3). In general, MxA and OAS expression in mock treated cells was consistent with that seen in Figure 2, despite different treatment conditions, although some variation was noted (e.g., MxA levels in HPAC cells).
Figure 3. Effect of JAK/STAT signaling inhibition on p-STAT1, MxA and OAS expression.
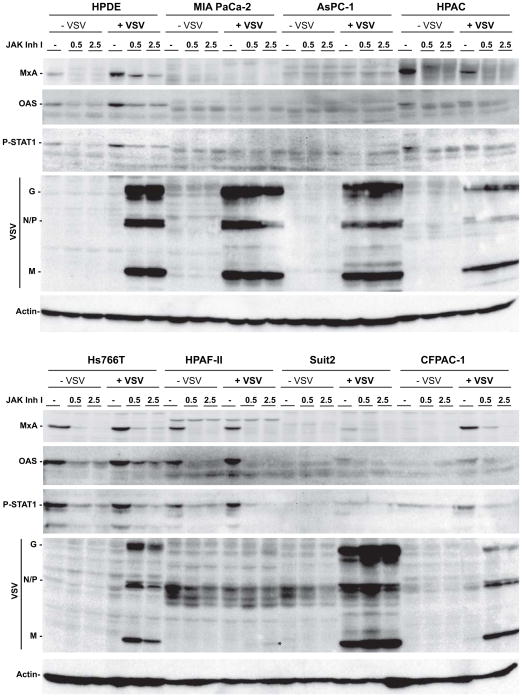
Cells were mock (DMSO) treated or treated with 0.5 or 2.5μM JAK Inh I for 48 h prior to infection with VSV-ΔM51-GFP at MOI 10 CIU/cell. Cells were harvested at 16h p.i. and cell lysates were prepared and analyzed by western blot for the indicated proteins. *, position of M protein in JAK Inh I treated HPAF-II cells
When new infectious virus particle production was determined following JAK Inh. I treatment, by collecting supernatants and titering them on BHK-21, increases were seen for the resistant cell lines (CFPAC, HPAC, HPAF-II, HPDE and Hs766t) (Table 3). Consistent with the improvement in virus production, a robust increase in viral protein accumulation was seen for the resistant cell lines CFPAC, HPAC, HPDE and Hs766T and a slight increase was seen for the resistant cell line HPAF-II. Treatment also caused a modest increase in viral protein accumulation in Suit2, a cell line we classify as susceptible. While titers were well correlated with viral protein as determined by western blot, this was not absolute, possibly due to low viral titers in some cell lines (especially HPAF-II) and cell line variations in virus replication kinetics and infectious/non-infectious particle ratios.
Table 3.
New infectious virus particle production at 16h p.i. after 48hr pre-treatment with JAK Inh. I
| PDA Cells | Treatmenta | Yieldb | Ratioc |
|---|---|---|---|
| AsPC-1 | VSV | 8.4×107 | 1 |
| VSV + 0.5 μM JAK Inh. I | 4.2×108 | 5 | |
| VSV + 2.5 μM JAK Inh. I | 2.6×108 | 3 | |
| CFPAC | VSV | 1.3×106 | 1 |
| VSV+ 0.5 μM JAK Inh. I | 2.6×107 | 20 | |
| VSV + 2.5 μM JAK Inh. I | 2.1×107 | 16 | |
| HPAC | VSV | 2.6×106 | 1 |
| VSV + 0.5 μM JAK Inh. I | 3.1×107 | 12 | |
| VSV + 2.5 μM JAK Inh. I | 7.9×106 | 3 | |
| HPAF-II | VSV | 2.0×104 | 1 |
| VSV + 0.5 μM JAK Inh. I | 1.3×106 | 64 | |
| VSV + 2.5 μM JAK Inh. I | 5.2×106 | 256 | |
| Hs766T | VSV | 4.1×104 | 1 |
| VSV + 0.5 μM JAK Inh. I | 2.1×107 | 512 | |
| VSV + 2.5 μM JAK Inh. I | 1.0×107 | 256 | |
| MIA PaCa-2 | VSV | 4.2×108 | 1 |
| VSV + 0.5 μM JAK Inh. I | 5.0×108 | 1.2 | |
| VSV + 2.5 μM JAK Inh. I | 2.1×108 | 0.5 | |
| Suit2 | VSV | 2.1×108 | 1 |
| VSV + 0.5 μM JAK Inh. I | 1.3×108 | 0.6 | |
| VSV + 2.5 μM JAK Inh. I | 8.4×107 | 0.4 | |
| HPDE | VSV | 6.1×104 | 1 |
| VSV + 0.5 μM JAK Inh. I | 1.3×108 | 2048 | |
| VSV + 2.5 μM JAK Inh. I | 1.3×108 | 2048 |
“VSV” indicates VSV-Δ M51-GFP
Virus was collected from the indicated cell line and titer was determined on BHK-21 cells
Ratio of virus yield on JAK Inh. I treated cells to mock treated cells
Our previous study showed a correlation between susceptibility of PDA cells to VSV-mediated cell death and the relative infectivity of VSV on different PDA cell lines (Murphy et al., 2012). To determine if inhibition of JAK/STAT signaling improved VSV-ΔM51-GFP infectivity in resistant cells, VSV-ΔM51-GFP was titered on mock treated cells or cells treated with JAK Inh. I for 48 h. Consistent with previous results (Murphy et al., 2012), titers were lower on resistant cell lines (CFPAC, HPAC, HPAF-II, HPDE, Hs766T) than susceptible cell lines (AsPC-1, MIA PaCa-2, Suit2). This difference remained even with JAK Inh. I treatment. However, some improvement in titer was observed on HPAC and HPAF-II with treatment (Figure 4), although it was statistically significant only for HPAF-II. What was observed for all resistant cell lines, with the possible exception of HPDE, was a clear increase in plaque size upon JAK Inh. I treatment (data not shown). An increase in plaque size was also observed on the susceptible AsPC-1 cell line at the highest inhibitor concentration. This data suggests that while activation of the JAK/STAT pathways contributes to resistance to VSV-ΔM51-GFP in most resistant cell lines, other factors may contribute to the differences in VSV titers in various PDA cell lines.
Figure 4. Effect of JAK/STAT signaling inhibition on PDA cell susceptibility to VSV-ΔM51-GFP.
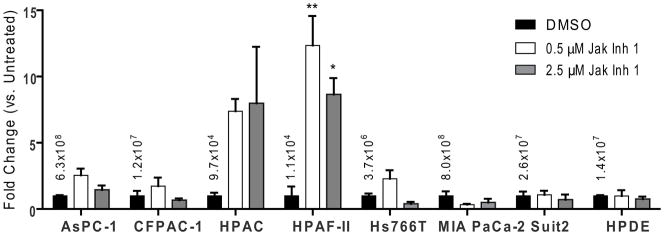
Cells were mock (DMSO) treated or treated with 0.5 or 2.5μM JAK Inh I for 48 h prior to infection with serial dilutions of VSV-ΔM51-GFP. At 17hpi, VSV-ΔM51-GFP fluorescent foci were counted to determine viral titers. Titers are expressed as a ratio to the mock treated titer, with the mock treated titer indicated. Titers were done in duplicate and data represent the mean ± standard error of mean. Treatments were compared using a 1-way ANOVA followed by the Bonferroni posttest for multiple comparisons. *=p<0.05; **=p<0.01
To determine if the improvement in new infectious particle production and plaque size translated into improved cell killing, cells were pretreated with JAK Inh I for 48 h and infected with VSV-ΔM51-GFP at MOI 1 (based on 4T-1). Virus-directed GFP expression was measured every 24 h and cell viability was determined at the end of five days by MTT assay. GFP expression increased upon JAK Inh I treatment for all five resistant cell lines plus the susceptible cell lines AsPC-1 and Suit2 (Figure 5). No increase was seen for the susceptible cell line MIA PaCa-2. When cell viability was determined (Figure 6A), a statistically significant increase in VSV-mediated cell death was seen with inhibitor treatment for HPDE, the expected outcome for a “normal’ (non-malignant) cell line. Importantly, a similar result was observed in the VSV-resistant CFPAC-1, HPAC and Hs766T cell lines at least at the highest concentration of inhibitor (2.5μM), suggesting an involvement of JAK/STAT signaling in the resistance phenotype of these cell lines. Treatment with JAK Inh. I alone generally did not cause a loss in cell viability, as measured by MTT, although a statistically significant decrease was seen for Hs766T at the lowest concentration only, HPDE at the highest concentration only and Suit2 at both concentrations. Consistent with the possible cytotoxic effects of JAK Inh. I on Suit2 and HPDE, virus-directed expression of GFP dropped at the higher concentration (2.5 μM) compared to the lower concentration (0.5 μM), although it still exceeded expression in untreated cells (Figure 5). Interestingly, in some cases, most notably HPAF-II, JAK Inh I caused a significant increase in MTT signal in uninfected cells. This may be due to an increase in the number of viable cells or due to an increase in mitochondrial activity (the actual parameter measured by the MTT assay), which can be caused by stress. To more specifically look at this question, the experiment was repeated with HPAF-II with a parallel plate used for cell counting. While less dramatic than in the previous experiment, treatment with 2.5μM JAK Inh I alone did cause a statistically significant increase in MTT signal (Figure 6B). However, this treatment did not change the number of viable cells number (Figure 6B). This experiment also confirmed that treatment of HPAF-II with 2.5 μM JAK Inh. I caused an increase in cell killing by VSV-ΔM51-GFP.
Figure 5. Effect of JAK/STAT signaling inhibition on virus-directed GFP expression.
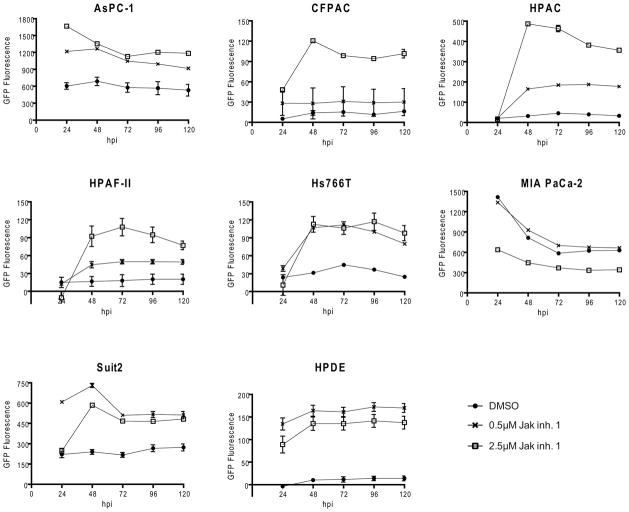
Cells were mock (DMSO) treated or treated with 0.5 or 2.5μM JAK Inh. I for 48 h prior to infection with VSV-ΔM51-GFP at MOI 1 CIU/cell. GFP fluorescence following infection was measured at 24 h intervals.
Figure 6. Effect of JAK/STAT signaling inhibition on PDA cell viability following infection.
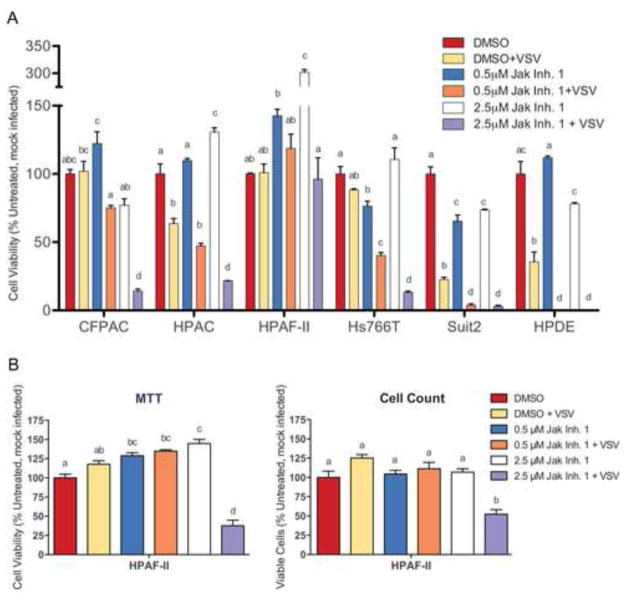
(A) Cells were mock (DMSO) treated or treated with 0.5 or 2.5μM JAK Inh. I for 48 h prior to infection with VSV-ΔM51-GFP at MOI 1 CIU/cell. Cell viability was analyzed by MTT assay at 5 d p.i. and is expressed as a percent of the DMSO only (mock) control. (B) The same assay was also performed in parallel with viable cell counts. The assays were done in triplicate and data represent the mean ± standard error of mean. Treatments were compared using a 1-way ANOVA followed by the Bonferroni posttest for multiple comparisons. Within each cell line, the presence of the same letter above a bar indicates treatments are not statistically different (cut-off p<0.05). For example, a bar marked “ab” does not differ from one marked “a” or “b” but is significantly different from one marked “c”.
Susceptibility of PDA cells to other viruses
The ability of VSV-ΔM51-GFP to initiate infection on the PDA and HPDE cell lines was assessed by performing a plaque assay using serial dilutions of virus Permissiveness was expressed as a ratio of the viral titer on PDA cell line to the titer on Vero cells with higher numbers indicating greater permissiveness and cell lines listed in order of permissiveness (Figure 7). In confirmation of our previous results, the five resistant cell lines (CFPAC, HPAC, HPAF-II, HPDE and Hs766T) showed the least susceptibility to VSV-ΔM51-GFP, in terms of both plaque size and number, and formed a distinct cluster from the susceptible cell lines. All of these highly resistant cells constitutively express at least some ISGs, including MxA and OAS, not seen in the more susceptible cell lines. In contrast, the two PDA cell lines where VSV-ΔM51-GFP formed the largest plaques (MIA PaCa-2 and Capan-1) are the only cell lines that fail to express both IFN-α and β following VSV-ΔM51-GFP infection (Figure 1 and 2). Previous studies showed that VSV is highly sensitive to type I IFN responses, an effect even more pronounced in VSV-ΔM51-GFP as a result of the methionine 51 deletion in the M protein (Black et al., 1993; Coulon et al., 1990; Stojdl et al., 2003). If indeed the susceptibility profile of these cells is at least in part determined by their type I IFN status, it would be expected that viruses capable of evading the host type I IFN response would display a different profile. To test this hypothesis, we used two large DNA viruses unrelated to VSV, recombinant vaccinia virus VVT7 (a poxvirus) and HSV-1 (a herpesvirus), both of which have been shown to evade type I IFN antiviral responses (Paladino and Mossman, 2009; Perdiguero and Esteban, 2009). While the specific VV and HSV-1 viruses used in this experiment are not used as OVs, other recombinants based on VV and HSV have been developed for that purpose. When VVT7 and HSV-1 were titered on PDA cell lines, there was not a correlation between the permissiveness of cells to these two viruses (Figure 7) and their type I IFN status (Figure 1 and 2), as indicated by the different ordering of these cells lines by permissiveness as compared to VSV-ΔM51-GFP. This is consistent with the greater abilities of VVT7 and HSV-1 to evade this pathway. The degree of curvature of the graphs in Figure 7 indicates the variability in cell line permissiveness to the viruses. The variability in cell line permissiveness was similar for VSV-ΔM51-GFP and HSV-1 although they differed in which cell lines were susceptible or resistant. In contrast, the range much smaller for VVT7 with the exception of highly resistant (to VVT7) Capan-2
Figure 7. Permissiveness of PDA cell lines to VSV-ΔM51-GFP, VVT7 and HSV-1.
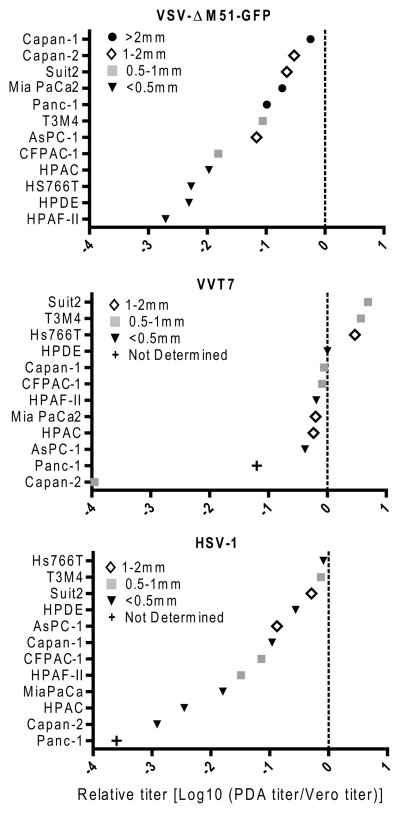
Cells were infected with serial dilutions of virus and, after a 1 h absorption, overlayed with media containing 0.5% BactoAgar. VSV-ΔM51-GFP foci were counted by fluorescent microscopy at 3 d p.i. VVT7 and HSV-1 plaques were counted after staining with crystal violet at 5 d p.i. Titers are expressed relative to those on Vero cells. A relative yield of 0 indicates that the PDA cell line and Vero are equally permissive to the virus, while negative numbers indicate reduced permissiveness on the PDA cell line. Plaque size was determined for all viruses at 5 d p.i. following crystal violet staining.
To determine if susceptibility of the PDA cells to these three viruses extended to cell killing, cell viability was determined by MTT assay at 5 d p.i. following infection with VSV-ΔM51-GFP, VVT7 or HSV-1 at MOI 1 or 0.01 as determined by titration on Vero cells that support robust replication of all three viruses (Fig. 8). For VSV-ΔM51-GFP, results closely mimicked those reported in our previous study (Murphy et al., 2012). However, all VSV-resistant cell lines (CFPAC-1, HPAC, Hs766T, HPAF-II and HPDE), were more effectively killed by HSV-1 and VVT7 than VSV-ΔM51-GFP at MOI 1, and this was also true for most of those cell lines at MOI 0.01. Importantly, HSV-1 and VVT7 did not demonstrate superior oncolytic abilities in all cell lines (eg. AsPC-1 and Capan-2 at MOI 0.01). Although we cannot rule out that other (IFN-unrelated) factors influenced cell susceptibility to these three viruses, the results are consistent with type I IFN responses being at least a factor in determining resistance to VSV-ΔM51-GFP infection.
Figure 8. PDA cell viability following infection with VSV-ΔM51-GFP, VVT7 and HSV-1.
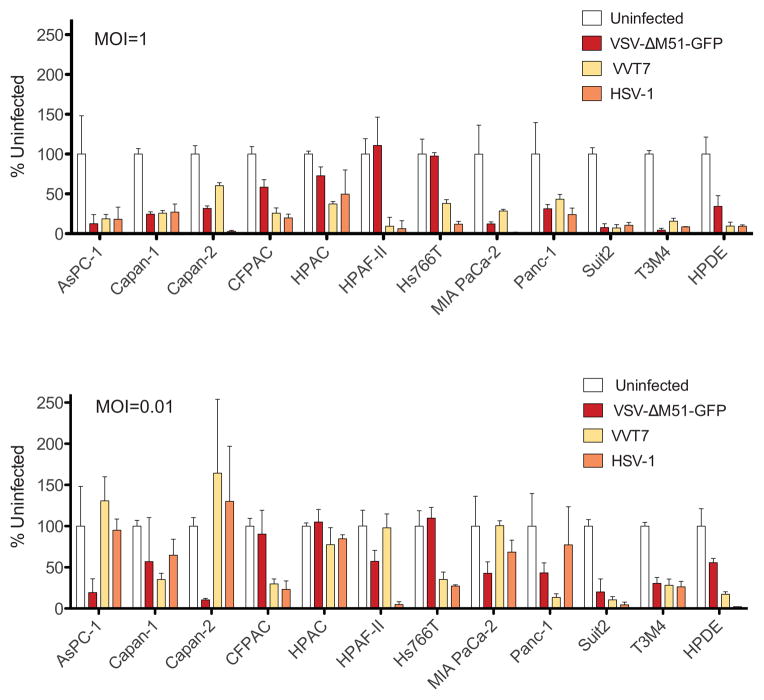
Cells were mock infected or infected with VSV-ΔM51-GFP, VVT7 or HSV-1 at MOI of 1 (A) or 0.01 (B) CIU/cell. Cell viability was analyzed by an MTT assay at 5 d p.i. and expressed as a percent of mock-infected controls. MTT assays were done in triplicate and the data represent the mean ± standard error of mean.
DISCUSSION
In this study, we demonstrated on a molecular level that the resistant cell lines tested have functional type I IFN responses similar to those observed in the non-malignant HPDE cell line. Importantly, we found that, unlike susceptible PDA cells, resistant cell lines constitutively expressed high levels of MxA and OAS, two important IFN-stimulated antiviral proteins.
We previously showed that while the majority of tested PDA cell lines were highly susceptible to VSV-ΔM51-GFP, five of these cell lines (BxPC-3, CFPAC-1, HPAC, HPAF-II and Hs766T) and the non-malignant pancreatic duct epithelial cell line HPDE were at least somewhat resistant to VSV-ΔM51-GFP mediated oncolysis (Murphy et al., 2012). The same pattern of resistance was also observed for wt VSV and VSV-p1-GFP, with a minor deviation for wt VSV in that HPDE and CFPAC were more susceptible than AsPC-1 and T3M4 (Murphy et al., 2012). These phenotypes were confirmed here for VSV-ΔM51-GFP (Fig. 7 and 8). However, it should be noted that BxPC-3, previously shown to be highly resistant to VSV (Murphy et al., 2012), was omitted from this study as it displayed an unstable phenotype, being generally resistant but occasionally highly susceptible to VSV-ΔM51-GFP. Even though passage numbers were limited for all tested cell lines, we observed that increased passage number tended to correlate with increased susceptibility of BxPC-3 to VSV-ΔM51-GFP suggesting alterations in cell biology may be responsible for this variability. BxPC-3 cells in culture have been shown to undergo an epithelial to mesenchymal transition (Roy et al., 2011). Interestingly, our previous study showed BxPC-3 differed from the other highly resistant cell lines in that secretion of IFN-β was not detectable following VSV infection (Murphy et al., 2012). Further studies are needed to better understand the BxPC-3 phenotype.
In order to mount an effective IFN mediated antiviral response, cells must first detect the virus. For RNA viruses replicating in the cytoplasm, such as VSV, detection occurs primarily through binding of single or double stranded viral RNA to RIG-I or melanoma differentiation associated gene 5 (MDA5) (Nakhaei et al., 2009; Shmulevitz et al., 2010). This initiates a signaling cascade resulting in phosphorylation of IRF 3 and 7 and formation of homo- and heterodimeric transcription factors necessary for expression of the type I IFNs α and β. Both of these secreted IFNs bind to the IFNAR1/2 receptor of the infected as well as surrounding non-infected cells, resulting in phosphorylation of the receptor by the Janus kinases JAK1 and TYK2. This results in recruitment and phosphorylation of STAT1 and STAT2 which together with IRF9 form a transcription factor, ISGF3, which recognizes IFN-stimulated response elements (ISRE) leading to transcription of ISGs, many of which have direct antiviral functions or contribute to the formation of an antiviral state. Several of these ISGs, including ISG15, MxA, OAS and PKR, have been shown to effectively inhibit replication of rhabdoviruses such as VSV (Sadler and Williams, 2008).
The high sensitivity of VSV to type I IFN responses is a major factor determining VSV’s oncoselectivity, as it is believed most cancer cells are defective in type I IFN responses (Naik and Russell, 2009; Stojdl et al., 2000). However, some cancers retain the ability to produce and/or respond to type I IFN (Naik and Russell, 2009; Stojdl et al., 2000). For example, PC3 prostate cancer cells (Ahmed et al., 2004; Carey et al., 2008), SW982 human sarcoma cells (Paglino and van den Pol, 2011), RT-4 and RT112 bladder cancer cells (Zhang et al., 2010), and multiple mesothelioma cells lines (Saloura et al., 2010) have been shown to be resistant to VSV infection at least in part due to IFN responsiveness and/or constitutive ISG expression. Furthermore, constitutive ISG expression was shown to be predictive of permissiveness of several PDA cell lines to adenovirus infection (Monsurro et al., 2010). Given this and our previous data demonstrating many resistant PDA cell lines both produce and respond to type I IFN (Murphy et al., 2012), as does the “normal” non-malignant HPDE cell line, we examined the responses of these cell lines to VSV-ΔM51-GFP at the molecular level. All tested cell lines appeared to be able to sense VSV-ΔM51-GFP as seen by production of IFN-λ mRNA and an increase in IRF3 phosphorylation following infection, even in cell lines where IFN-α and/or β transcription is not induced (Fig. 1 and 2).
STAT1 phosphorylation was detected in response to VSV-ΔM51-GFP infection in the resistant cell lines and in the susceptible cell lines AsPC-1 and Suit2, although the response in Suit2 was not robust. In the susceptible Mia PaCa-2 cells, STAT1 was phosphorylated in response to exogenously added IFN-α but not to VSV-ΔM51-GFP, suggesting the inability of this cell line to produce type I IFN (possibly due to poor IRF7 expression).
Importantly, MxA was constitutively expressed at both the mRNA and protein level in all resistant cell lines but in none of the susceptible cell lines. MxA has broad antiviral activity against a wide range of RNA and even some DNA viruses, regardless of subcellular site of replication (Haller and Kochs, 2011). It is believed that MxA GTPases recognize viral RNP complexes and form oligomeric rings around them, thereby blocking their function. MxA has also been implicated in the hyperphosphorylation of VSV P protein, which may interfere with its function (Schuster et al., 1996). Constitutive expression of MxA (270-fold over baseline) was observed in SW982 sarcoma cell line resistant to VSV (Paglino and van den Pol, 2011). Similarly, increased expression of MxA in PDA cells was recently associated with resistance to adenovirus-based OV (Monsurro et al., 2010). Furthermore, OAS was constitutively expressed in all the resistant cell lines as well as a few susceptible cell lines, at least at the mRNA level, although the highest levels were detected in resistant cell lines. OAS converts adenosine triphosphate into a series of 20–50 oligoadenylates (2-5A), which activate the latent ribonuclease (RNaseL). The activated OAS-RNaseL system promotes apoptosis, attenuates proliferation, degrades viral and cellular RNA, and inhibits protein synthesis (Justesen et al., 2000; Mandal et al., 2011). Previous studies showed increased expression of OAS in PDA cells resistant to adenoviruses (Monsurro et al., 2010), and human mesothelioma cells resistant to VSV (Saloura et al., 2010). As both MxA and OAS have been shown to have antiviral activity against VSV, they almost certainly contribute to the resistance phenotype of the PDA cells studied here. However, as there are hundreds of ISGs, a number of which also have known antiviral functions (Sadler and Williams, 2008), resistance is likely to involve more than just these two proteins.
It is unclear why these proteins are expressed constitutively at high levels. While low levels of phosphorylated STAT1 were detected in some uninfected resistant cell lines, it is uncertain whether these levels were sufficient for the observed expression of MxA and other ISGs in uninfected cells. However, the strong reduction in MxA and OAS protein levels upon inhibition of JAK/STAT signaling (Fig. 3) would argue the effect is at least partially mediated through this mechanism. It is also not clear why not all resistant cell lines constitutively express all of the ISGs tested, since the ISGs are all under the control of ISREs. One possibility is that alternative regulatory mechanisms influence expression of some of these genes. For example, RelA has been shown to regulate a subset of ISGs (Basagoudanavar et al., 2011). IFN-λ is also known to regulate ISG expression, although it regulates those genes through the same mechanisms as type I IFN and the resulting ISG expression profile is thought to be nearly identical (Donnelly and Kotenko, 2010). Expression of these genes may also be influenced by chromosome modification as treatment with a histone deacetylase inhibitor was shown to decrease ISG expression and increase VSV infectivity in the SW982 human sarcoma cell line (Paglino and van den Pol, 2011).
In our study, five of the 11 human PDA cell lines tested showed constitutive expression of the ISGs MxA and OAS. While this collection of cell lines is likely not representative of the clinical situation, a recent study showed that a significant subset of the bulk PDA tissues and xenografted primary PDA cells tested had an mRNA expression profile typical of an inflamed state including upregulation of ISGs such as MxA (Monsurro et al., 2010), demonstrating the existence of this phenotype in the patient population. In addition, we have identified a number of cell lines able to respond to type I IFN (Murphy et al., 2012) as well as produce type I IFN in response to VSV-ΔM51-GFP. While all of these cell lines were susceptible to VSV-ΔM51-GFP in vitro, results presented here suggest that the type I IFN responsiveness of these cells may lead to suboptimal VSV-ΔM51-GFP oncolysis. For example, virus-directed GFP expression increased in both AsPC-1 and Suit2 upon JAK Inh. I treatment (Figure 5) and AsPC-1 plaque size increased when the higher concentration of inhibitor was used (data not shown).
Given these phenotypes, our results suggest that high constitutive expression of ISGs may be useful biomarkers in identifying PDAs, and possibly other cancers, resistant to OV therapy with VSV or other viruses highly sensitive to IFN. In our study, the ISGs MxA and OAS were particularly well correlated with this phenotype. While PDAs with this profiles are unlikely to be successfully treated with IFN sensitive OVs such as VSV, use of alternative OVs (e.g. vaccinia virus or HSV-1), better equipped to evade IFN responses could be a better option. Alternatively, treatment with more than one OV (combined virotherapy) could also lead to enhanced oncolysis as was previously shown for VSV in combination with vaccinia virus (Le Boeuf et al., 2010). Furthermore, any future OV therapy will likely involve a combination of OV(s) and chemotherapy (Ottolino-Perry et al., 2010).
While we have demonstrated a role for type I IFN responses in the resistance of PDA cells to VSV-ΔM51-GFP infection, we cannot rule out the possible influence of other factors on susceptibility and/or oncolysis. For example, VSV has been shown to cause cell death via apoptosis (Cary et al., 2011; Gaddy and and Lyles, 2005, 2007; Sharif-Askari et al., 2007), and inhibition of apoptosis, a signature of many cancer cells (Hamacher et al., 2008), has the potential of limiting/delaying cell death following VSV infection. Studies are in progress examining the role apoptosis and other factors may have in contributing to the resistance of some PDAs to oncolytic VSV.
Supplementary Material
RESEARCH HIGHLIGHTS.
We examined cellular responses of human pancreatic adenocarcinomas (PDA) to VSV
11 PDA cell lines were examined for expression of type I IFN associated genes
All PDA cell lines, resistant and permissive, were able to sense VSV infection
Only resistant PDA cell lines displayed constitutive expression of MxA and OAS
Inhibition of JAK/STAT signaling both down regulated MxA and OAS and helped VSV
Acknowledgments
The authors are grateful to the following laboratories for kindly providing reagents for this project: Jack Rose (Yale University) for VSV-ΔM51-GFP; David McConky (MD Anderson Cancer Center) for CFPAC-1 and Hs766T cells; Randall Kimple (University of North Carolina at Chapel Hill) for Capan-2 and T3M4 cells; Timothy Wang (Columbia University) for AsPC-1 cells; Andrei Ivanov (University of Rochester Medical School) for HPAF-II cells; Michael Hollingsworth (University of Nebraska Medical Center) for Suit2 cells; Emmanuel Zervos (Tampa General Hospital) for HPAC cells. We thank Sue Moyer (University of Florida College of Medicine) for providing a range of reagents for this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MMT, Puckett S, Kock ND, Lyles DS. Immune response in the absence of neurovirulence in mice infected with m protein mutant vesicular stomatitis virus. J Virol. 2008;82:9273–9277. doi: 10.1128/JVI.00915-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunology. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- Basagoudanavar SH, Thapa RJ, Nogusa S, Wang J, Beg AA, Balachandran S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J Virol. 2011;85:2599–2610. doi: 10.1128/JVI.02213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, de Weerd NA, Gould J, Argani P, Moller A, Smyth MJ, Anderson RL, Hertzog PJ, Parker BS. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012 Jul 22; doi: 10.1038/nm.2830. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Black BL, Rhodes RB, McKenzie M, Lyles DS. The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. JVirol. 1993;67:4814–4821. doi: 10.1128/jvi.67.8.4814-4821.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach CJ, Reid T, Burke J, Bell JC, Kirn DH. Navigating the clinical development landscape for oncolytic viruses and other cancer therapeutics: no shortcuts on the road to approval. Cytokine Growth Factor Rev. 2010;21:85–89. doi: 10.1016/j.cytogfr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Carey BL, Ahmed M, Puckett S, Lyles DS. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol. 2008;82:12104–12115. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary ZD, Willingham MC, Lyles DS. Oncolytic Vesicular Stomatitis Virus Induces Apoptosis in U87 Glioblastoma Cells by a Type II Death Receptor Mechanism and Induces Cell Death and Tumor Clearance In Vivo. J Virol. 2011;85:5708–5717. doi: 10.1128/JVI.02393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon P, Deutsch V, Lafay F, Martinet-Edelist C, Wyers F, Herman RC, Flamand A. Genetic evidence for multiple functions of the matrix protein of vesicular stomatitis virus. J Gen Virol. 1990;71:991–996. doi: 10.1099/0022-1317-71-4-991. [DOI] [PubMed] [Google Scholar]
- Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res. 2010;30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005;65:3447–3453. doi: 10.1158/0008-5472.CAN-04-4316. [DOI] [PubMed] [Google Scholar]
- Ebert O, Harbaran S, Shinozaki K, Woo SL. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 2005;12:350–358. doi: 10.1038/sj.cgt.7700794. [DOI] [PubMed] [Google Scholar]
- Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, Fontoura BM. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell. 2005;17:93–102. doi: 10.1016/j.molcel.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Farrow B, Albo D, Berger DH. The role of the tumor microenvironment in the progression of pancreatic cancer. J Surg Res. 2008;149:319–328. doi: 10.1016/j.jss.2007.12.757. [DOI] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996;148:1763–1770. [PMC free article] [PubMed] [Google Scholar]
- Gaddy DF, Lyles DS. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J Virol. 2005;79:4170–4179. doi: 10.1128/JVI.79.7.4170-4179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddy DF, Lyles DS. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol. 2007;81:2792–2804. doi: 10.1128/JVI.01760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Carlson SK, Classic KL, Greiner S, Naik S, Power AT, Bell JC, Russell SJ. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110:2342–2350. doi: 10.1182/blood-2007-01-065573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O, Kochs G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res. 2011;31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64. doi: 10.1186/1476-4598-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie E, Grdzelishvili V. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol. 2012 Oct 9; doi: 10.1099/vir.0.046672-0. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Iwamura T, Katsuki TKI. Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen 19-9. Jpn J Cancer Res. 1987;78:54–62. [PubMed] [Google Scholar]
- Justesen J, Hartmann R, Kjeldgaard NO. Gene structure and function of the 2′-5′-oligoadenylate synthetase family. Cell Mol Life Sci. 2000;57:1593–1612. doi: 10.1007/PL00000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly EJ, Nace R, Barber GN, Russell SJ. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J Virol. 2010;84:1550–1562. doi: 10.1128/JVI.01788-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Boeuf F, Diallo JS, McCart JA, Thorne S, Falls T, Stanford M, Kanji F, Auer R, Brown CW, Lichty BD, Parato K, Atkins H, Kirn D, Bell JC. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18:888–895. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Bluyssen HA, Levy DE. Regulation of interferon-alpha responsiveness by the duration of Janus kinase activity. J Biol Chem. 1997;272:21872–21877. doi: 10.1074/jbc.272.35.21872. [DOI] [PubMed] [Google Scholar]
- Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. TRENDS in Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Lindsay TH, Jonas BM, Sevcik MA, Kubota K, Halvorson KG, Ghilardi JR, Kuskowski MA, Stelow EB, Mukherjee P, Gendler SJ, Wong GY, Mantyh PW. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain. 2005;119:233–246. doi: 10.1016/j.pain.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Linge C, Gewert D, Rossmann C, Bishop JA, Crowe JS. Interferon system defects in human malignant melanoma. Cancer Research. 1995;55:4099–4104. [PubMed] [Google Scholar]
- Mandal S, Abebe F, Chaudhary J. 2 ′-5 ′ oligoadenylate synthetase 1 polymorphism is associated with prostate cancer. Cancer. 2011;117:5509–5518. doi: 10.1002/cncr.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marozin S, Altomonte J, Stadler F, Thasler WE, Schmid RM, Ebert O. Inhibition of the IFN-beta response in hepatocellular carcinoma by alternative spliced isoform of IFN regulatory factor-3. Mol Ther. 2008;16:1789–1797. doi: 10.1038/mt.2008.201. [DOI] [PubMed] [Google Scholar]
- Matin SF, Rackley RR, Sadhukhan PC, Kim MS, Novick AC, Bandyopadhyay SK. Impaired alpha-interferon signaling in transitional cell carcinoma: lack of p48 expression in 5637 cells. Cancer Research. 2001;61:2261–2266. [PubMed] [Google Scholar]
- Monsurro V, Beghelli S, Wang R, Barbi S, Coin S, Di Pasquale G, Bersani S, Castellucci M, Sorio C, Eleuteri S, Worschech A, Chiorini JA, Pederzoli P, Alter H, Marincola FM, Scarpa A. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: potential relevance for adenoviral gene therapy. J Transl Med. 2010;8:10. doi: 10.1186/1479-5876-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer SA, Baker SC, Lessard JL. Tubulin: a factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc Natl Acad Sci USA. 1986;83:5405–5409. doi: 10.1073/pnas.83.15.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvili VZ. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol. 2012;86:3073–3087. doi: 10.1128/JVI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9:1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Okabe T, Yamaguchi NNO. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer. 1983;51:662–668. doi: 10.1002/1097-0142(19830215)51:4<662::aid-cncr2820510419>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther. 2010;18:251–263. doi: 10.1038/mt.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglino JC, van den Pol AN. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J Virol. 2011;85:9346–9358. doi: 10.1128/JVI.00723-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P, Mossman KL. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interferon Cytokine Res. 2009;29:599–607. doi: 10.1089/jir.2009.0074. [DOI] [PubMed] [Google Scholar]
- Perdiguero B, Esteban M. The interferon system and vaccinia virus evasion mechanisms. J Interferon Cytokine Res. 2009;29:581–598. doi: 10.1089/jir.2009.0073. [DOI] [PubMed] [Google Scholar]
- Pfeffer LM, Wang C, Constantinescu SN, Croze E, Blatt LM, Albino AP, Nanus DM. Human renal cancers resistant to IFN’s antiproliferative action exhibit sensitivity to IFN’s gene-inducing and antiviral actions. J Urol. 1996;156:1867–1871. [PubMed] [Google Scholar]
- Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SI, Gendler SJ, Mukherjee P. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30:1449–1459. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28:326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, Sterman DH, Harty RN, Okumura A, Barber GN, Vile RG, Federspiel MJ, Russell SJ, Litzky L, Albelda SM. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther. 2010;21:51–64. doi: 10.1089/hum.2009.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A, Johnston IC, Das T, Banerjee AK, Pavlovic J, ter MV, Schneider-Schaulies S. Expression of the human MxA protein is associated with hyperphosphorylation of VSV P protein in human neural Cells. Virology. 1996;220:241–245. doi: 10.1006/viro.1996.0308. [DOI] [PubMed] [Google Scholar]
- Sharif-Askari E, Nakhaei P, Oliere S, Tumilasci V, Hernandez E, Wilkinson P, Lin R, Bell J, Hiscott J. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology. 2007;365:20–33. doi: 10.1016/j.virol.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Research. 2010;70:4912–4921. doi: 10.1158/0008-5472.CAN-09-4676. [DOI] [PubMed] [Google Scholar]
- Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nature Reviews in Clinical Oncology. 2010;7:163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- Stojdl DFLB, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- Sun WH, Pabon C, Alsayed Y, Huang PP, Jandeska S, Uddin S, Platanias LC, Rosen ST. Interferon-alpha resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood. 1998;91:570–576. [PubMed] [Google Scholar]
- Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Letters. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BX, Rahbar R, Fish EN. Interferon: current status and future prospects in cancer therapy. J Interferon Cytokine Res. 2011;31:545–552. doi: 10.1089/jir.2010.0158. [DOI] [PubMed] [Google Scholar]
- Wilden H, Fournier P, Zawatzky R, Schirrmacher V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int J Oncol. 2009;34:971–982. doi: 10.3892/ijo_00000223. [DOI] [PubMed] [Google Scholar]
- Wollmann GRV, Simon I, Rose JK, van den Pol AN. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol. 2010;84:1563–1573. doi: 10.1128/JVI.02040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, Edmondson S, Devenish RJ, Ralph SJ. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem. 1997;272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, Vile R. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol Ther. 2011;19:150–158. doi: 10.1038/mt.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LHT, Meseck M, Altomonte J, Ebert O, Shinozaki K, García-Sastre A, Fallon J, Mandeli J, Woo SL. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Human Gene Therapy. 2008;19:635–647. doi: 10.1089/hum.2007.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KX, Matsui Y, Hadaschik BA, Lee C, Jia W, Bell JC, Fazli L, So AI, Rennie PS. Down-regulation of type I interferon receptor sensitizes bladder cancer cells to vesicular stomatitis virus-induced cell death. Int J Cancer. 2010;127:830–838. doi: 10.1002/ijc.25088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.