

Abstract
Mutations of human αB-crystallin cause congenital cataract and cardio-myopathy by protein aggregation and cell death. How mutations of αB-crystallin become pathogenic is poorly understood. To better understand the cellular events related to protein aggregation and cell death, we transfected cataract and cardio-myopathy causing mutants, R11H, P20S, R56W, D109H, R120G, D140N, G154S, R157H and A171T in HeLa cells and assessed protein aggregation and apoptosis by laser scanning confocal microspy (LSCM) and flow cytometry. Cells individually transfected with the mutants, D109H, R120G, D140N and R157H significantly showed more aggregates. Cells overexpressed with HspB1 (Hsp27) significantly sequestered aggregates in all mutants and suppressed apoptosis in mutants, P20S, D109H and A171T. Significant increases of apoptotic cells as stained with Annexin V were observed in mutants, D109H and A171T transfected cells. Cells positive for active caspase-3 was increased in the mutant, D109H. Thus the previously recognized anti-apoptotic functions of αB-crystallin were compromised in these mutants.
Keywords: αB-crystallin, HspB1, aggregates, apoptosis, caspase-3
Introduction
The αB-crystallin (HspB5) is a key member of the small heat shock protein (sHsp) family. It is constitutively expressed in many cells, known to be multi-functional and involved in many diseases [1–3]. Upon stress, αB-crystallin shows enhanced synthesis in cells [4]. In mammals, αB-crystallin is a major polypeptide of ocular lens where it is associated with its partner molecule, αA-crystallin (HspB4) to form a large globular heteropolymers with a molecular size ~700 kDa [5]. The highest concentration of αB-crystallin in non-ocular tissues was found in the heart and slow-twitch muscles [6–8]. Massive accumulation of αB-crystallin was reported in brains of patients with Alexander’s disease, functions as oncoprotein in breast cancer and in neurofibrillary tangles from patients of Alzheimer’s disease [8–10]. Abundant expression of αB-crystallin has been reported in various types of stress in cardiac [11] and skeletal muscles [12] as well as in the brain [13]. These studies suggest that in these organs αB-crystallin function as molecular chaperones suppressing the aggregation of specific client polypeptides.
Missense mutations identified in αB-crystallin which led to the development of congenital cataract were R11H [14], P20S [15], R56W [16], D109H [17] R120G [18], D140N [19] and A171T [20]. The mutants, R11H and P20S are autosomal dominant whereas the R56W is recessive [16] and all three of these mutations occur in the N-terminal region. The cataract phenotype implies that presence of mutant αB-crystallin is more harmful than its absence. αB-crystallin is expressed in striated muscle and truncated mutations cause muscular diseases [21]. Mutations of αB-crystallin contributing to cardio-myopathy were: D109H [17]; R120G [18] and R157H [22]. Recently, a mutant G154S has been reported in late-onset distal myopathy [23]. All these mutations (D109H, R120G, D140N, G154S and R157H) occur in the ‘α-crystallin domain’ and are autosomal dominant mutants. The well recognized mutant, R120G causes both dominant cataract and adult-onset desmin related myopathy [18]. The newly identified mutant, D109H has been described with the multi-system phenotype similarly to R120G [17]. The autosomal dominant mutant, A171T is located in the C-terminal domain.
HspB1 (Hsp27) is a small heat shock protein and a molecular chaperone which can participate in folding of partially unfolded proteins under both stressed and unstressed conditions [24, 25]. Moreover, HspB1 and αB-crystallin proteins often accumulate as inclusion bodies in many protein conformation diseases [26]. Previously, a study [25] has been demonstrated that HspB1 suppresses the formation of inclusion bodies formed by a mutant of αB-crystallin, R120G, in HeLa cells. However, there is no report on anti-aggregate function of HspB1 overexpression in other missense mutants of αB-crystallin involved in cardio-myopathy and congenital cataract.
In this study we used biochemical and cell biological methods to investigate the function of missense mutants of αB-crystallin.
Materials and Methods
Generation of YFP-tagged αB-crystallin mutants
QuickChange II site-directed mutagenesis kit (Agilent Technologies, CA) was used to generate mutated YFP-tagged αB-crystallin DNA constructs. Previously generated YFP-(N-terminus)-tagged αB-crystallin wild-type DNA [27] was used as template for PCR with the following conditions: 95°C for 15 seconds, 95°C for 30 seconds, 55°C for 1 minute and 68°C for 5 minutes for 16 cycles. The un-mutated parental DNA molecules from the PCR reaction was digested with Dpn I for 1 hour at 37°C and 1 μl of digested DNA was used for transformation. The transformants were selected on LB-agar plates containing 50 μg/ml kanamycin and validated by DNA sequencing.
Cell culture and transfection
HeLa cells, purchased from ATCC, Manassas, VA were grown in 35 mm glass bottom dishes (Mat Tek, MA) and transfected with Lipofectamine 2000 (Life Technologies, CA). For individual transfection experiments, 1 μg of YFP-tagged αB-crystallin-wt or its mutants and 1 μg of empty vector were transfected. In co-transfection experiments, 1 μg each of plasmid DNA; pCMV6-Myc-DDK-tagged-HspB1 (Hsp27) (Origene, MD) were co-transfected with YFP-tagged mutated αB-crystallin constructs. After 48 hours, transfected cells showing aggregates were counted at x40 magnification in a laser scanning confocal microscope. Fields were randomly chosen and 300 cells were counted per experiment and repeated in three independent experiments.
Laser scanning confocal microscopic studies
An LSM 510 confocal microscope (Carl Zeiss Inc., Thornwood, NY) with 63x oil-immersion objective (plan Apochromat, NA 1.4) (University of Arkansas for Medical Sciences core facility) was utilized. To visualize YFP fluorescence, cells expressing fluorescent proteins were excited at appropriate laser beam and filtered with both dichromatic band pass filters, captured at 12 bit 512 × 512, multi-track channel images with CCD camera. For YFP channel, the cells were excited with 514 nm filter by argon-ion laser and the emission intensity was collected using BP 530–600 nm filters.
Flow cytometry for apoptosis assay
Cells were individually transfected with 1 μg of YFP-tagged αB-crystallin-wt and its mutants. After 48 h transfection, cells were dislodged with accutase and first stained with Fixable Viability Dye eFlour780 (eBioscience, CA) for 30 min at 4°C. Cells were washed twice with PBS and subsequently stained with second stain, Annexin V-APC (BD Biosciences, CA) for 15 min at room temperature in the dark and gated the triple positive cells, i.e. YFP, Fixable Viability Dye eFlour780 and Annexin V-APC by BD FACSAria-IIu using Cell Quest software (BD Biosciences, CA) (University of Arkansas for Medical Sciences Core Facility).
Flow cytometry assay for detection of active caspase-3
For detection of active caspase-3, cells were transfected with 1 μg of DDK-tagged αB-wt and mutants, D109H and R120G. In one set of experiments, DDK-tagged αB-crystallin-wt, D109H, and R120G alone were transfected. In another group of experiment, transfected cells were treated with 50 nM of staurosporine (ST) (Cell Signaling, MA) for 14 hours. The ST treatment was started after 34 h transfection and appropriate amount of vehicle (DMSO) was added in control groups. After 48 h transfection, cells were fixed and permeabilized with BD Fix/Permeabilization solution (BD Biosciences, CA) for 20 minutes on ice and washed with Fix/Permeabilization washing buffer for two times and subsequently stained with PE-conjugated active caspase-3 (BD Biosciences, catalog # 557091) and DDK-DyLight 488 (Origene, catalog # TA 150021) for one hour in the dark at room temperature and washed with Fix/Permeabilization washing buffer for two times. Cells were resuspended with FACS buffer (1x PBS supplemented with 1% BSA and 0.1% sodium azide) and subjected to flow cytometry. Cells were gated out for double positive cells [i.e. DDK-DyLight 488 (FL 1) and PE-active caspase-3 (FL2)] by BD FACS Calibur (UAMS core facility) and analyzed by Cell Quest software (BD Biosciences, CA).
SDS-PAGE and Western blot analysis
After 48 hours transfection, cells were lysed with lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.02% sodium azide, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate and 0.1 mM EDTA supplemented with cock-tail protease inhibitors. For each sample, 5 μg of total protein was loaded into 12% SDS-PAGE and electro blotted to PVDF membrane. The blots were blocked with 5% non-fat dry milk prepared in TBST (Tris-buffered saline supplemented with 0.1% Tween 20) and subsequently incubated with primary monoclonal antibody for DDK-tag (Origene, TA5011), αB-crystallin (rabbit polyclonal, Abcam, ab13497, 1:2000) for one hour at room temperature. Blots were washed with TBST for three times and incubated with appropriate HRP-conjugated secondary antibodies (1 in 10000, Santa Cruz Biotechnology Inc., CA) for one hour at room temperature. Enhanced Chemiluminescence substrate was used and the signal was detected by exposing the blots on X-ray films. For loading control, blots were stripped with Restore Western Blot stripping buffer (Thermo Scientific Inc., IL) and re-probed with a rabbit polyclonal antibody against β-actin (Abcam, ab8227, 1: 10000) for 1 hour at room temperature.
Statistical analyses
Student’s t-test was used to calculate the significance between the wild-type and the mutant group. For each group, three independent experiments were conducted and the results were expressed as mean ± SD. The p < 0.05 was considered as statistically significant.
Results
Expression of YFP-tagged αB-crystallin mutants in HeLa cells
To determine whether mutants of αB-crystallin could generate aggregates in cells, cells were individually transfected with YFP-tagged αB-crystallin mutants. After 48 h transfection, cells were examined with a laser scanning confocal microscope (LSCM) and counted the number of cells with aggregates as described in Methods. The ‘aggregates’ were referred to as the YFP-fused mutant protein expression seen as scattered clump particles localized in the cytoplasm. Cells having more than four such particles are considered as positive for cells containing aggregates and scored. A homogenous expression of YFP-tagged αB-wt protein was seen in the cytoplasm. There was no or fewer number of cells with aggregates in the αB-wt transfected cells. The cellular morphology was significantly altered and distorted in mutants, D109H, R120G and D140N (Fig. 1). The percent of cells with aggregates were higher in mutants, D109H, R120G, D140N and R157H. Mutants, R11H, P20S, G154S and A171T showed a moderate number of cells with aggregates. Cells transfected with the mutant, R56W showed the least number of cells with aggregates. The percent of cells (Mean ± SD) with aggregates were: 2 ± 1; 20.6 ± 4.6; 20.6 ± 4; 4 ± 2; 44 ± 9.1; 77.6 ± 8.7; 46.6 ± 10.6; 24.6 ± 6.4; 48 ± 8; 15.3 ± 3 for αB-wt, R11H, P20S, R56W, D109H, R120G, D140N, G154S, R157H and A171T respectively (Fig. 2).
Figure 1.
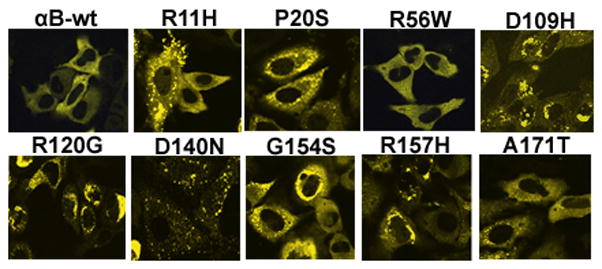
Expression of individually transfected YFP-tagged αB-wt and its mutant constructs in HeLa cells: Cells were transfected with 1 μg each of YFP-tagged αB-wt and its mutant constructs, R11H, P20S, R56W, D109H, R120G, D140N, G154S, R157H and A171T with 1 μg of empty vector. After 48 h transfection, cells were observed under a confocal microscope.
Figure 2.
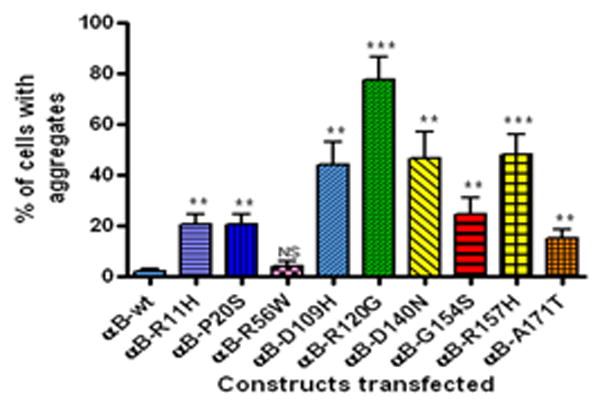
Percent of cells with aggregates in individually transfected YFP-tagged αB-wt and its mutants: The graph depicts the percent of cells with aggregates after 48 h transfection computed from Fig. 1. The mutants, D109H, R120G, D140N and R157H showed more cells with aggregates. The mutants, R11H, P20S, G154S and A171T showed modest number of cells with aggregates. The p value is * < 0.05; ** < 0.005 and *** < 0.0005 compared to wild-type. NS= not significant.
Co-expression of YFP-tagged αB-crystallin mutants with Myc-DDK-tagged HspB1 construct in HeLa cells
To investigate whether exogenous overexpression of HspB1 can inhibit the aggregates caused by αB-crystallin mutants, cells were co-transfected with Myc-DDK-tagged HspB1 with the mutants and scored the percent of cells with aggregates after 48 h transfection. The transfection efficiency of overexpressed Myc-DDK-tagged HspB1 was nearly equal as determined by flow cytometry (not shown). Interestingly, co-expression of HspB1 significantly suppressed the aggregates caused by all the mutants of αB-crystallin (Fig. 3). The percent of cells (Mean ± SD) with aggregates decreased significantly as given in the following data: 1 ± 0.01; 9.3 ± 3; 8 ± 2; 1.6 ± 1.1; 18 ± 2.6; 13.67 ± 2.082; 19 ± 3.6; 10 ± 2; 9 ± 2.6; 3.6 ± 1.5 for αB-wt + HspB1; R11H+HspB1; P20S+HspB1, R56W+HspB1, D109H+HspB1, R120G+HspB1, D140N+HspB1, G154S+HspB1, R157H+HspB1 and A171T+HspB1 respectively (Fig. 4A).
Figure 3.
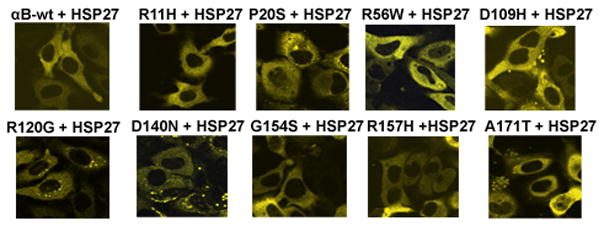
Co-expression of Myc-DDK-tagged-HspB1 inhibited aggregates formed by the mutants of αB: Mutants of αB-crystallin were co-transfected with Myc-DDK-tagged HspB1. After 48 h transfection, HspB1 co-expression significantly suppressed the cells with aggregates caused by the mutants. The cells were examined and images were captured with a laser scanning confocal microscope.
Figure 4.
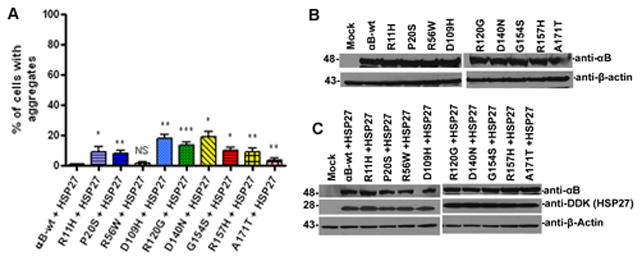
HspB1 co-expression sequestered aggregates. Figure 4A: Percent of cells with aggregates in YFP-tagged αB-wt and its mutants co-expressed with HspB1. The graph depicts the percent of cells with aggregates after 48 h transfection computed from Fig. 3. The p value is * p < 0.05; ** p < 0.005; *** p< 0.0005 compared to mutant alone. NS indicates the value is not significant. Figure 4B: Western blot analysis. Cells were individually transfected with YFP-tagged αB-wt and its mutant constructs. After 48 h transfection, cell lysates were subjected to immunoblot probed with anti-αB-crystallin polyclonal antibody. The actin blot was shown as loading control. Figure 4C: Western blot analyses for cells co-expressed with Myc-DDK-tagged HspB1 in YFP-tagged-αB-wt and its mutant constructs. The blot was probed with polyclonal αB-crystallin antibody (top panel). The same blot was stripped and re-probed with a monoclonal anti-DDK antibody to detect HspB1 (middle panel). The actin blot was shown as loading control (lower panel).
Level of expression of YFP-tagged αB-crystallin-wt and its mutants in HeLa cells
To validate the level of expression of individually transfected YFP-tagged αB-crystallin-wt and its mutant constructs, cells were lysed after 48 h transfection and analyzed by western blot. The level of expression was nearly equal in each of the individually transfected construct (Fig. 4B) suggest that aggregates caused by the mutants did not alter the level of YFP-tagged protein expression. In mutants co-expressed with Myc-DDK-tagged-HspB1, the blots were probed with αB-crystallin antibody and the same blot was reprobed with anti-DDK monoclonal antibody to detect DDK-tagged HspB1. The level of overexpressed YFP-αB-crystallin was nearly equal in all of the transfected groups. (Fig. 4C) and the level of overexpressed HspB1 were also nearly equal in the transfected groups (Fig. 4C). The YFP-tagged mutant of αB-crystallin gives rise to an expected molecular mass ~48 kDa was observed in blot probed with αB-crystallin antibody.
Increase in apoptotic cells in individually expressed αB-crystallin mutants
The αB-crystallin possesses anti-apoptotic properties [28]. To examine whether the mutants of αB-crystallin have their anti-apoptotic function compromised, cells were individually transfected with the YFP-tagged αB-crystallin mutants under the same culture conditions. After 48 h transfection, cells were assessed for apoptosis by staining with Annexin V-APC and Fixable Viability dye eFlour780. The change in Annexin V-positive/Fixable Viability dye eFlour780-positive cell rates indicates apoptosis [29]. Mutants D109H and A171T transfected cells showed significant increase of cells positive for apoptosis compared to wild-type control and higher or nearly equal to positive control mutants, R120G [30] and P20S [15] which has been demonstrated previously to induce apoptosis in rat neonatal cardiomyocytes (RCN) and human lens epithelial cells (HLE), respectively (Table 1).
Table 1.
Percent of apoptosis (Mean ± SD) detected in individually expressed αB-wt and its mutants and or co- expressed with HspB1.
| Constructs | % of apoptosis | |
|---|---|---|
| No HspB1 | with HspB1 | |
| αB-wt | 8.7 ± 0.5 | 8.3 ± 0.3NS |
| R11H | 9.5 ± 0.5NS | 8.6 ± 0.2NS |
| P20S | 12.5 ± 0.6** | 9.9 ± 0.7** |
| R56W | 8.6 ± 0.5NS | 8.2 ± 0.2NS |
| D109H | 18.1 ± 1.9** | 13.3 ± 0.4** |
| R120G | 20.4 ± 2.3** | 14.1 ± 1.4** |
| D140N | 10.6 ± 1.5NS | 9.9 ± 0.8NS |
| G154S | 9.8 ± 0.4NS | 9.1 ± 0.8NS |
| R157H | 10.1 ± 1.1NS | 9.9 ± 1.2NS |
| A171T | 11.8 ± 0.3** | 7.9 ± 0.3** |
indicates p < 0.005.
NS indicates the values are not significant. The individually expressed mutant groups were compared to αB-wt. The mutant co-expressed with HspB1 was compared to corresponding individually expressed mutant.
HspB1 overexpression protects cells from apoptosis only in some mutants of αB-crystallin
To investigate whether overexpression of exogenous HspB1 can suppress the number of apoptotic cells in mutants of αB-crystallin, cells were co-transfected with Myc-DDK-tagged HspB1 (1 μg) in YFP-tagged αB-crystallin (1 μg) mutants. After 48 h transfection, cells were stained with Annexin V-APC and eFluor780 and subjected to multi-color flow cytometry. Interestingly, our results indicate that overexpression of HspB1 suppressed the number of cells positive for apoptosis in mutants P20S, D109H and A171T (Table 1). In other mutants, there was no appreciable decrease in the number of cells positive for apoptosis.
Active caspase-3 was increased in αB-crystallin mutants
To examine whether the apoptotic pathway is modulated by caspase-3 in mutants of αB-crystallin, cells were transfected with DDK-tagged αB-wt and its mutants D109H and R120G. After 48 h transfection, cells were stained with active caspase-3 antibody and assessed by flow cytometry as described in Methods. The mutants D109H and R120G showed a two-fold increase of cells positive for active caspase-3 compared to αB-wt (Table 2). Furthermore, to determine whether the mutant, D109H can resist staurosporine-induced caspase-3 activation, transfected cells were treated with 50 nM of staurosporine (ST) for 14 hours. Increased number of cells positive for active caspase-3 was observed in D109H mutant suggests that anti-apoptotic properties were compromised in D109H mutant due to its partial impairment.
Table 2.
Percent of active caspase-3 positive cells in αB-wt and its mutant constructs transfected cells in the presence or absence of staurosporine (Mean ± SD).
| Constructs | % of active caspase-3 positive cells | |
|---|---|---|
| No staurosporine | with staurosporine | |
| αB-wt | 1.2 ± 0.2 | 1.48 ± 0.16 |
| D109H | 2.5 ± 0.2** | 3.69 ± 0.31** |
| R120G | 2.6 ± 0.2** | 3.02 ± 0.27** |
The ** indicates p < 0.005. The individually transfected mutant groups were compared to αB-wt without staurosporine treatment. The mutants expressing cells treated with staurosporine were compared to αB-wt expressing cells treated with staurosporine.
Discussion
In this study, we have demonstrated that all the αB-crystallin mutants were showing different level of protein aggregates and some of the mutants showing apoptosis in HeLa cells, a highly transfectable cell line. Previously [27, 31], we showed that there was no endogenous expression of αB-crystallin in these cells and therefore, we evaluated the function of αB-crystallin mutants by transient transfection. The question arises as to what makes the missense mutants of αB-crystallin to aggregate in mammalian cells. Lack of protein stability and conformational changes could be the two major factors influencing the formation of aggregates. Our recent studies (unpublished data) have shown that P20S, D109H and R120G have altered secondary structure as shown by increased α-helical and decreased β-sheet contents. The tertiary structure (tertiary fold) also appeared to be different as compared to αB-wt. Our results in the present study suggest that mutations located within the ‘α-crystallin’ domain (D109H, R120G and D140N) showed higher level of aggregates than mutations occur in the N-terminal domain. It is possible that higher level of aggregates formed by the mutants from the core ‘α-crystallin’ domain could be due to decreased chaperone function as reported previously [32]. And also when these mutants were expressed individually in the present study, they formed aggregates probably due to cellular stress in the absence of additional protective mechanism such as presence of an active small heat-shock protein like HspB1. Interestingly, HspB1 overexpression significantly suppressed the level of aggregates because HspB1 is a powerful chaperone and these results agree with a previous study with the R120G mutant [25]. A protective effect of HspB1 by sequestration of aggregates formed by the mutants is probably due to the formation of heterooligomers of the mutants with HspB1 [33, 34]. Anti-apoptotic effect was observed in some of the mutants; P20S, D109H and A171T with co-expression of HspB1 suggest that over-expression of HspB1 protects through its anti-apoptotic mechanism [35, 36]. In other mutants, HspB1 failed to protect cells from apoptosis possibly due to other mechanisms such as post-translational modifications like phosphorylation of HspB1 did not occur appropriately [37].
In this report, we provided evidence for apoptosis due to loss of anti-apoptotic properties in some mutants of αB-crystallin mutants. The mutants D109H and A171T expressed cells led to more apoptotic cell death. The cell death due to apoptosis could be one of the mechanisms for pathogenesis of cataract and cardio-myopathy. In a previous study by another research group [28] strong evidence has been presented that native αB-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. Also in the same study, the αB-crystallin mutant (R120G) showed only partial impairment in its cytoprotective function. Our results suggest that partially impaired anti-apoptotic effect was observed in the mutant, D109H. It is quite possible that altered protein conformational changes in αB-crystallin mutants result in inadequate binding and thus enabling activated caspase-3 formation. The future studies will be focused on interaction between caspase-3 and the mutants of αB-crystallin.
Highlights.
Mutants of αB-crystallin expression in cells cause aggregates and apoptosis.
HspB1 co-expression suppresses aggregates in all mutants.
HspB1 co-expression decreases apoptosis in P20S, D109H, R120G and A171T.
Acknowledgments
This study was supported by Grant EY11352 from National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A. 1992;89:10449–53. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narberhaus F. Alpha-crystallin-type heat shock proteins: socializing minichaperones in the context of a multichaperone network. Microbiol Mol Biol Rev. 2002;66:64–93. doi: 10.1128/MMBR.66.1.64-93.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Montfort R, Slingsby C, Vierling E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv Protein Chem. 2001;59:105–56. doi: 10.1016/s0065-3233(01)59004-x. [DOI] [PubMed] [Google Scholar]
- 4.Kegel KB, Iwaki A, Iwaki T, et al. AlphaB-crystallin protects glial cells from hypertonic stress. Am J Physiol. 1996;270:C903–9. doi: 10.1152/ajpcell.1996.270.3.C903. [DOI] [PubMed] [Google Scholar]
- 5.Augusteyn RC. alpha-crystallin: a review of its structure and function. Clin Exp Optom. 2004;87:356–66. doi: 10.1111/j.1444-0938.2004.tb03095.x. [DOI] [PubMed] [Google Scholar]
- 6.Longoni S, James P, Chiesi M. Cardiac alpha-crystallin. I. Isolation and identification. Mol Cell Biochem. 1990;99:113–20. [PubMed] [Google Scholar]
- 7.Bhat SP, Nagineni CN. alpha B subunit of lens-specific protein alpha-crystallin is present in other ocular and non-ocular tissues. Biochem Biophys Res Commun. 1989;158:319–25. doi: 10.1016/s0006-291x(89)80215-3. [DOI] [PubMed] [Google Scholar]
- 8.Iwaki T, Kume-Iwaki A, Liem RK, et al. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell. 1989;57:71–8. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- 9.Moyano JV, Evans JR, Chen F, et al. AlphaB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest. 2006;116:261–70. doi: 10.1172/JCI25888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjorkdahl C, Sjogren MJ, Zhou X, et al. Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J Neurosci Res. 2008;86:1343–52. doi: 10.1002/jnr.21589. [DOI] [PubMed] [Google Scholar]
- 11.van de Klundert FA, Smulders RH, Gijsen ML, et al. The mammalian small heat-shock protein Hsp20 forms dimers and is a poor chaperone. Eur J Biochem. 1998;258:1014–21. doi: 10.1046/j.1432-1327.1998.2581014.x. [DOI] [PubMed] [Google Scholar]
- 12.Kato K, Ito H, Kamei K, et al. Innervation-dependent phosphorylation and accumulation of alphaB-crystallin and Hsp27 as insoluble complexes in disused muscle. FASEB J. 2002;16:1432–4. doi: 10.1096/fj.02-0129fje. [DOI] [PubMed] [Google Scholar]
- 13.Iwaki T, Tateishi J. Immunohistochemical demonstration of alphaB-crystallin in hamartomas of tuberous sclerosis. Am J Pathol. 1991;139:1303–8. [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, Ma J, Yan M, et al. A novel mutation in CRYAB associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2009;15:1359–65. [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Li C, Lu Q, et al. Cataract mutation P20S of alphaB-crystallin impairs chaperone activity of alphaA-crystallin and induces apoptosis of human lens epithelial cells. Biochim Biophys Acta. 2008;1782:303–9. doi: 10.1016/j.bbadis.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Khan AO, Abu Safieh L, Alkuraya FS. Later retinal degeneration following childhood surgical aphakia in a family with recessive CRYAB mutation (p.R56W) Ophthalmic Genet. 2010;31:30–6. doi: 10.3109/13816810903452047. [DOI] [PubMed] [Google Scholar]
- 17.Sacconi S, Feasson L, Antoine JC, et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord. 2012;22:66–72. doi: 10.1016/j.nmd.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–5. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Zhang X, Luo L, et al. A novel alphaB-crystallin mutation associated with autosomal dominant congenital lamellar cataract. Invest Ophthalmol Vis Sci. 2006;47:1069–75. doi: 10.1167/iovs.05-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devi RR, Yao W, Vijayalakshmi P, et al. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–70. [PMC free article] [PubMed] [Google Scholar]
- 21.Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54:804–10. doi: 10.1002/ana.10767. [DOI] [PubMed] [Google Scholar]
- 22.Inagaki N, Hayashi T, Arimura T, et al. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–86. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 23.Reilich P, Schoser B, Schramm N, et al. The p.G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul Disord. 2012;20:255–9. doi: 10.1016/j.nmd.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Chavez Zobel AT, Loranger A, Marceau N, et al. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G alphaB-crystallin mutant. Hum Mol Genet. 2003;12:1609–20. doi: 10.1093/hmg/ddg173. [DOI] [PubMed] [Google Scholar]
- 25.Ito H, Kamei K, Iwamoto I, et al. Hsp27 suppresses the formation of inclusion bodies induced by expression of R120G alpha B-crystallin, a cause of desmin-related myopathy. Cell Mol Life Sci. 2003;60:1217–23. doi: 10.1007/s00018-003-3024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 27.Raju I, Kumarasamy A, Abraham EC. Multiple aggregates and aggresomes of C-terminal truncated human alphaA-crystallins in mammalian cells and protection by alphaB-crystallin. PLoS One. 2011;6:e19876. doi: 10.1371/journal.pone.0019876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamradt MC, Chen F, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–63. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 29.Cornelissen M, Philippe J, De Sitter S, et al. Annexin V expression in apoptotic peripheral blood lymphocytes: an electron microscopic evaluation. Apoptosis. 2002;7:41–7. doi: 10.1023/a:1013560828090. [DOI] [PubMed] [Google Scholar]
- 30.Maloyan A, Sanbe A, Osinska H, et al. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–61. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raju I, Abraham EC. Congenital cataract causing mutants of alphaA-crystallin/sHSP form aggregates and aggresomes degraded through ubiquitin-proteasome pathway. PLoS One. 2011;6:e28085. doi: 10.1371/journal.pone.0028085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muchowski PJ, Wu GJ, Liang JJ, et al. Site-directed mutations within the core “alpha-crystallin” domain of the small heat-shock protein, human alphaB-crystallin, decrease molecular chaperone functions. J Mol Biol. 1999;289:397–411. doi: 10.1006/jmbi.1999.2759. [DOI] [PubMed] [Google Scholar]
- 33.Kato K, Shinohara H, Goto S, et al. Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J Biol Chem. 1992;267:7718–25. [PubMed] [Google Scholar]
- 34.Kato K, Goto S, Hasegawa K, et al. Responses to heat shock of alpha B crystallin and HSP28 in U373 MG human glioma cells. Biochim Biophys Acta. 1993;1175:257–62. doi: 10.1016/0167-4889(93)90214-a. [DOI] [PubMed] [Google Scholar]
- 35.Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis. 2003;8:61–70. doi: 10.1023/a:1021601103096. [DOI] [PubMed] [Google Scholar]
- 36.Parcellier A, Gurbuxani S, Schmitt E, et al. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun. 2003;304:505–12. doi: 10.1016/s0006-291x(03)00623-5. [DOI] [PubMed] [Google Scholar]
- 37.Rane MJ, Pan Y, Singh S, DW, et al. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. 2003;278:27828–35. doi: 10.1074/jbc.M303417200. [DOI] [PubMed] [Google Scholar]