

Abstract
The nitric oxide (NO)-cyclic guanosine monophosphate (cGMP) signaling system is a well-characterized modulator of cardiovascular function, in general, and blood pressure, in particular. The availability of mice mutant for key enzymes in the NO-cGMP signaling system facilitated the identification of interactions with other blood pressure modifying pathways (e.g. the renin-angiotensin-aldosterone system) and of gender-specific effects of impaired NO-cGMP signaling. In addition, recent genome-wide association studies identified blood pressure-modifying genetic variants in genes that modulate NO and cGMP levels. Together, these findings have advanced our understanding of how NO-cGMP signaling modulates blood pressure. In this review, we will summarize the results obtained with mice with disrupted NO-cGMP signaling and highlight the relevance of this pathway as a potential therapeutic target for the treatment of hypertension.
Keywords: Nitric oxide-cyclic guanosine monophosphate, NO-cGMP, Blood pressure, Hypertension, Cardiovascular function, Soluble guanylate cyclase, Nitric oxide, NO, Mutant mice, Genetic variants, Renin-angiotensin-aldosterone signaling, RAAS, Gender, S-nitrosylation, Therapeutics
Introduction: Prevalence and etiology of hypertension
High blood pressure or hypertension (HTN) is the most prevalent modifiable risk factor for cardiovascular disease, including coronary heart disease, congestive heart failure, stroke, end-stage renal disease, and peripheral vascular disease, in both women and men[1]. Over one billion people worldwide and 1 in 3 US adults have high blood pressure, with an additional 8% of US adults estimated to have undiagnosed hypertension[1]. Just as the risks associated with HTN are clear, so too are the benefits of blood pressure reduction: treatment of HTN is associated with significant reductions in cardiovascular events[2]. However, while extensive research, development of blood pressure medications, and widespread patient education have reduced mortality and morbidity rates associated with hypertension, it is estimated that in at least half of the US adults with HTN optimal blood pressure targets are not achieved[3].
Blood pressure is a complex heritable trait regulated by the integrated signaling from renal, neural, vascular, and hormonal inputs with multiple environmental and genetic influences[4, 5] . In the vast majority of cases, it is difficult to identify specific mechanisms that disrupt these control systems, and the etiology of hypertension remains unknown. Molecular and genetic analyses of specific, highly-heritable and rare forms of HTN point to the involvement of a small number of genes, many of which are associated with the regulation of renal salt handling[4, 6].
The concept that abnormalities in renal salt handling[7] are a major contributor to the etiology of hypertension has informed the development of treatments for HTN focused on altering dietary sodium. However, multiple clinical studies have indicated that altering salt intake does not necessarily affect the risk for the development of hypertension or mortality associated with cardiovascular disease[8]. More recently, the hypothesis that primary abnormalities of vascular smooth muscle cell (VSMC) proteins that regulate vascular tone can cause hypertension has emerged[9]. For example, ample evidence suggests that impaired vascular NO-cGMP signaling, a key pathway in regulating vascular relaxation, is associated with high blood pressure[10–12].
In this review, we will provide a brief introduction of the NO-cGMP signaling system and an overview of the blood pressure phenotypes of mouse models in which key enzymes of the NO-cGMP signaling system are mutated. We furthermore will summarize how reciprocal interaction between NO/sGC signaling and the renin-angiotensin-aldosterone system modulates blood pressure, and how recent information from population genetic studies have illustrated the clinical relevance of NO/sGC signaling in the development of hypertension. We will highlight how gender affects the impact on blood pressure of modulating the activity of the NO/sGC signaling system, and how sGC-independent effects of NO (such as S-nitrosylation) can mediate some of the cardiovascular effects of NO. Finally, we will briefly discuss available compounds that activate sGC and their development as promising therapeutic targets in the treatment of hypertension.
The nitric oxide-soluble guanylate cyclase signaling system
NO is synthesized from L-arginine by a family of three enzymes referred to as NO synthases (NOSs)[13]. Three forms of the enzyme exist: endothelial NOS (eNOS or NOS3), neuronal NOS (nNOS or NOS1), and inducible NOS (iNOS or NOS2). eNOS and nNOS are constitutively expressed, producing low levels of NO, while iNOS expression is induced by inflammatory conditions leading to the production of large amounts of NO. Upon Ca2+ binding, calmodulin (CaM) binds to a regulatory site on the constitutively-expressed nNOS and eNOS, triggering NO production. CaM binds irreversibly to iNOS, resulting in a continuous Ca2+-independent NO production. eNOS can be activated independently of Ca2+ upon phosphorylation by Akt kinase in response to stimuli such as shear stress, estrogens and insulin[14]. Recently, another important in vivo source of bioactive NO was identified: the circulating anion nitrite (NO2−), previously considered a stable and inert oxidation product of NO metabolism, serves as an endocrine transporter and storage pool of NO[15].
NO has numerous targets, reacting with a variety of intracellular and extracellular molecules, typically via thiol groups or transition metal centers[16, 17]. NO’s multiple targets contribute to the wide variety and often-conflicting effects of this molecule. The best-characterized target of NO in the cardiovascular system is soluble guanylate cyclase (sGC). NO stimulates sGC, an obligate heterodimer composed of an α and a β subunit, to synthesize the intracellular second messenger cGMP[18]. Although 2 isoforms of each subunit have been identified (α1, α2, β1, β2), only sGCα1β1 and sGCα2β1 appear to function in vivo[19]. The N-terminal domain of the sGCβ1 subunit contains a proximal histidine ligand (His105) required for binding the heme moiety[20]. Binding of NO to the heme results in a conformational change and activation of the catalytic domain. The catalytic center of sGC is formed by the conserved C-terminal cyclase homology domains of the α and β subunit. In cardiac and aortic tissue, the sGCα1 subunit is expressed at higher levels than the sGCα2 subunit[21], suggesting that the sGCα1β1 heterodimer is the principal cardiovascular sGC isoform. However, low levels of cGMP, generated by sGCα2β1, are sufficient to mediate many of NO’s cardiovascular effects[12, 22–27]. cGMP interacts with a variety of effector proteins, including cGMP-dependent protein kinases (PKGs), cGMP-regulated phosphodiesterases (PDEs), and ion channels. It is important to note that cGMP can also be generated by membrane guanylate cyclases (pGCs) that are activated by natriuretic peptides[28]. However, cGMP produced by sGC and by pGCs may have differential effects, possibly due to differential spatiotemporal distributions of cGMP produced by the two guanylate cyclase families [29– 31].
NO-sGC mediated regulation of blood pressure: mouse models
The role of NO-cGMP signaling in VSMC relaxation and the regulation of blood pressure is well characterized, both in humans[32, 33] and in animal models[34], and ample evidence suggests that impaired NO signaling is involved in the pathogenesis of hypertension. NO is a key regulator of smooth muscle tone, in large part by controlling sGC-mediated production of cGMP[35–37]. Increased levels of cGMP subsequently activate PKGI, which in turn phosphorylates and modulates the activity of numerous downstream targets. PKGI activation mediates a number of physiological signaling mechanisms in VSMC, including decreases in intracellular Ca2+ levels, membrane hyperpolarization, and inhibition of myosin light chain phosphorylation, which all contribute to vasorelaxation (Figure 1).
Figure 1. Nitric oxide (NO) - cGMP signaling in endothelium-dependent relaxation of vascular smooth muscle cells (VSMC).
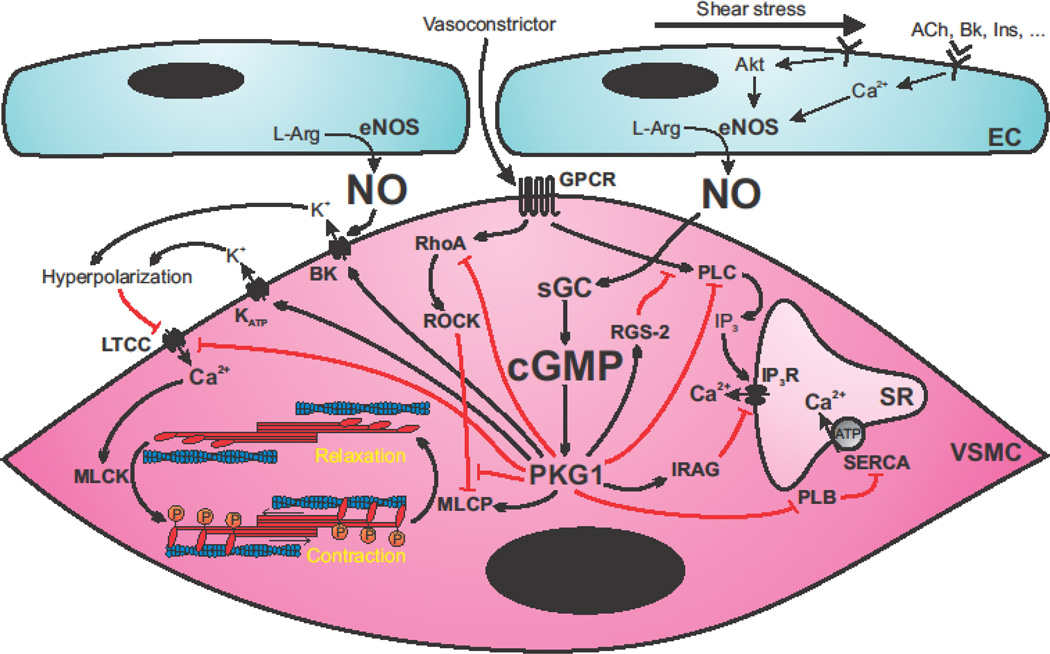
Endothelial cells (EC) respond to agonist stimulation by increasing intracellular Ca2+ concentration and activity of endothelial nitric oxide (NO) synthase (eNOS). Shear stress also leads to eNOS activation via Akt-dependent phosphorylation. NO diffuses into adjacent VSMC and activates soluble guanylate cyclase (sGC), leading to increased cGMP production and protein kinase G I (PKGI) activation. PKGI activation promotes vasorelaxation via multiple mechanisms. PKGI interferes with vasoconstrictor signaling via G protein-coupled receptors (GPCR) by activating regulator of G protein signaling-2 (RGS-2), inhibiting phospholipase C (PLC), and stimulating inositol triphosphate (IP3) receptor-associated PKG substrate (IRAG), which ultimately leads to inhibition of Ca2+ release from the sarcoplasmic reticulum (SR) via the IP3 receptor (IP3R). Phosphorylation by PKGI also inhibits phospholamban (PLB), leading to increased Ca2+ reuptake into the SR by sarco/endoplasmic reticulum ATPase (SERCA). On the other hand, PKGI interferes with RhoA and rho-associated protein kinase (ROCK)-dependent inhibition of myosin light chain phosphatase (MLCP). Together with a direct activation of MLCP, this leads to increased dephosphorylation of the regulatory light chain of myosin, which promotes vasorelaxation by blocking actin-myosin interaction. PKGI also reduces extracellular Ca2+ influx directly by inhibiting the L-type Ca2+ channel (LTCC). Activation of the large conductance Ca2+-activated K+ channel (BK), which can also be directly stimulated by NO, and the ATP-dependent K+ channel (KATP) cause VSMC hyperpolarization leading to inhibition of the LTCC. ACh: acetylcholine, Bk: bradykinin, Ins: insulin, L-Arg: L-Arginine, MLCK: myosin light chain kinase.
During the last decade, many genes involved in NO-cGMP signaling (including genes encoding NOS, sGC, and PKG subunits and/or isoforms) have been targeted by gene deletion (conventional or conditional), overexpression, or mutations in residues or domains crucial for normal enzyme function (Table 1). Hypertension is a recurring phenotype in mouse models of impaired NO-cGMP signaling. Below we will address the cardiovascular phenotype of these transgenic mouse models in greater detail.
Table 1.
Overview of the different mutant mouse strains in NO-cGMP signaling that have been generated, including their effect on blood pressure regulation.
| Mutant | Genotype | Blood Pressure Phenotype | Ref |
|---|---|---|---|
| nNOS−/− | nNOSβ splice variant present | Normotensive | [124] |
| nNOS−/− | Null mutation | Normotensive | [125] |
| eNOS−/− | Null mutation | Hypertensive | [38,39,126,127]’’’ |
| iNOS−/− | Null mutation | Normotensive | [128–130]’’ |
| n/iNOS−/− | nNOSβ splice variant present | Normotensive | [131,132]’’ |
| n/eNOS−/− | nNOSβ splice variant present | Hypertensive | ’[131–133] |
| e/iNOS−/− | nNOSβ splice variant present | Hypertensive | [131,132]’ |
| e/i/nNOS−/− | nNOSβ splice variant present | Hypertensive | [132] |
| nNOS transgenic | Conditional myocardial overexpression | Normotensive | [134,135] |
| Brain overexpression | Normotensive | [136] | |
| eNOS transgenic | Myocardial overexpression | Normotensive | [137,138] |
| Endothelial overexpression | Hypotensive | [139,140]’ | |
| iNOS transgenic | Conditional myocardial overexpression | Normotensive | [141] |
| Myocardial overexpression | Normotensive | [142] | |
| Pancreatic β cell overexpression | Normotensive | [143] | |
| sGCα1−/− | Null mutation | Mild Hypertensive | [25] |
| sGCα1−/− | Truncated inactive protein | Normotensive on C57Bl/6 Hypertensive on 129S6 |
[22] |
| sGCα2−/− | Null mutation | Normotensive | [25] |
| sGCβ1−/− | Null mutation | Hypertensive | [12] |
| sGCβ1H105F | NO-insensitive mutant protein | Hypertensive | [54] |
| PKG1−/− | Null mutation | Hypertensive at 6W Normotensive at >= 8W |
[57] |
| PKG1 SMα rescue | PKG1α only in SMC of PKG1−/− | Normotensive | [55] |
| PKG1 SMβ rescue | PKG1β only in SMC of PKG1−/− | Normotensive | [55] |
| PKG-LZM | Mutant protein | Hypertensive at >=12W | [11] |
| PKG1αC42S | H2o2-insensitive mutant protein | Hypertensive | [61] |
| PKG2−/− | Null mutation | Normotensive | [144] |
| PDE5 transgenic | Myocardial overexpression | Normotensive | [145] |
NOS mutant mice
The crucial role of endogenously produced NO in blood pressure regulation was confirmed by the presence of hypertension in mice deficient in eNOS (eNOS−/− mice). Depending on the genetic background and the method used to measure blood pressure, eNOS−/− mice have blood pressures ranging from 7 to 63 mmHg higher than those in control mice (Table 1)[26, 38–43]. In contrast, blood pressure was not affected in mice deficient in nNOS[44] or iNOS[45] (Table 1). Moreover, blood pressure in mice lacking both eNOS and nNOS, or in mice deficient in all NOS isoforms, was not greater than that in eNOS-deficient mice (Table 1)[46, 47]. Together, these data suggest that eNOS is the predominate source of vaso-active NO.
Acetylcholine (ACh)–induced vasorelaxation was blocked in aortic and carotid artery rings and in the pulmonary vasculature of eNOS−/− mice, indicating that eNOS is required for ACh-induced vasodilation[38, 48, 49] and suggesting that the hypertension associated with the lack of endothelial NO can primarily be attributed to increased vascular resistance. However, in small resistance arterioles, that are thought to primarily control systemic blood pressure, other endothelium-derived vasodilators (such as prostacyclin and endothelium-derived hyperpolarizing factor (EDHF)) appear to compensate for the loss of eNOS. For example, in mesenteric arteries of eNOS−/− mice, ACh-induced relaxation was preserved. Together, these findings suggest that, although NO is a central mediator of vasorelaxation and blood pressure control, other endothelial vasodilators (such as EDHF or prostacyclins)[50, 51] can mediate vasorelaxation in small resistance vessels in the absence of eNOS-derived NO.
sGC mutant mice
Increasing evidence, both from genetic association studies and from work in mutant mice, illustrates the central role for sGC in the regulation of blood pressure. In sGCα1−/− mice, basal and NO-stimulated cGMP production was significantly reduced in the cardiovascular system[22, 23, 25]. As a result, the ability of endothelial-derived NO to induce vasorelaxation was impaired[23, 25, 52], highlighting the importance of sGCα1β1-derived cGMP in the relaxation of vascular smooth muscle by NO. Nevertheless, NO donor compounds could still modulate hemodynamic parameters in sGCα1−/− mice, albeit to a lesser extent[22, 53], most likely reflecting the ability of sGCα2β1 to modulate the hemodynamic effects of NO in the absence of sGCα1β1. Interestingly, varying blood pressure phenotypes have been reported in mice deficient in the sGCα1 subunit of sGC (sGCα1−/− mice), ranging from a normotensive phenotype[53] to an increase in blood pressure of 7 mmHg[25] to 22 mmHg[22] (Table 1). Differences in genetic background and gender likely contribute to this discrepancy: male but not female sGCα1−/− mice on a 129S6 (S6) background (sGCα1−/−S6) develop hypertension[22], and no hypertension was observed in either male or female sGCα1−/− mice on the C57BL/6 (B6) background (sGCα1−/−B6)[53]. These findings suggest the existence of genetic modifiers of the hypertensive effects associated with sGCα1-deficiency (see below).
The importance of sGC in regulating blood pressure and vascular tone was further supported by the observation that blood pressure was higher in mice deficient in sGCβ1 (sGCβ1 −/− mice) than in WT mice[12] (Table 1). Similarly, smooth muscle cell-specific sGCβ1 −/− mice developed hypertension[34] (Table 1). In addition, in sGCβ1−/− mice, administration of NO donor compounds did not reduce blood pressure and the vasodilatory effect of NO in isolated aortic rings was abrogated[12], suggesting that NO-inducible sGC activity is required for NO to mediate its vasorelaxing effects in these vessels. The observed differences in phenotype between sGCα1−/− and sGCβ1−/− mice suggest that sGCα1 accounts for only part of the sGC-dependent vasorelaxing effects of NO in mice. In sGCα1−/− mice, NO stimulation likely leads to a small increase in cGMP production, attributable to sGCα2β1. This low level of sGC activity appears to be sufficient to mediate NO-induced vasorelaxation in sGCα1−/− mice. On the other hand, deletion of the sGCα2 subunit was associated with only a slight reduction in sGC enzyme activity in aortic tissue, and did not appear to influence vasorelaxation in response to NO-donor compounds[25] (Table 1). The relatively minor impact of sGCα2-deficiency (in the context of intact sGCα1β1 activity) on the ability of NO to mediate vasorelaxation is most likely due to the fact that sGCα2β1 is expressed at low levels in the vasculature[21]. Additional studies in available sGC mutant mice and recently developed mice expressing only the heme-free form of sGC (apo-sGC) (Table 1)[54] are required to further characterize the role of sGC in maintaining cardiovascular homeostasis.
PKG mutant mice
PKG1 is generally accepted to be the principal mediator of the hemodynamic effects of NO-cGMP signaling. VSMCs express two alternative splice variants of PKG1, α and β, both of which mediate VSMC relaxation[55, 56]. Dilation of both large conductance and small resistance arteries in response to NO/cGMP signaling are severely impaired in mice deficient for both PKG1 isoforms (PKG1−/−), indicating that PKG1 mediates, at least in part, the vasorelaxant effects of NO and other cGMP-elevating agents[57, 55, 58, 59].
PKG1−/− mice are hypertensive early in life, but blood pressure normalizes by 8 weeks of age[57, 60] (Table 1). However, PKG1 deficiency is associated with severe gastro-intestinal dysfunction and mortality greater than 50% by 6 weeks of age. Therefore, it is conceivable that non-cardiovascular effects of interfering with NO-cGMP signaling confound the blood pressure recordings obtained at 8 weeks. To avoid the confounding effects of the increased morbidity and mortality associated with complete loss of PKG1 activity, a mouse strain was generated which expresses a mutant PKG1α leucine zipper (LZ) (LZM mice[11]) (Table 1). The LZ mediates the interaction of PKG with its downstream targets RhoA and myosin light chain phosphatase (MLCP) (Figure 1). LZM mice express a PKG1α protein with preserved catalytic activity but which is incapable of making LZ-dependent interactions with these PKG1α target proteins. Homozygous LZM mice displayed a marked age-dependent hypertension, which developed at 10–12 weeks of age[11]. ACh-dependent relaxations were attenuated but not abolished in isolated large conductance arteries and resistance arteries from LZM mice. Together, these findings suggest that LZ-dependent PKG1α effects are not the only mechanisms by which PKG1 mediates the vasorelaxing effect of NO[11]. Recently it was shown that PKG1 is also a direct mediator of NO-independent EDHF-dependent vasorelaxation via its direct activation by H2O2 (Table 1)[61]. Other studies have indicated that PKG1 inhibits vasoconstrictor-induced Ca2+ release from the sarcoplasmic reticulum (SR) in several ways: 1) by inhibiting G protein signaling via activation of the regulator of G protein signaling-2[60], 2) by inhibition of inositol 1,4,5-triphosphate (IP3) production by direct phosphorylation of phospholipase C[62], and 3) by inhibition of the IP3 receptor via the IP3 receptor-associated PKG substrate[63]. In addition, PKG1-mediated inhibition of phospholamban increases Ca2+ reuptake into the SR via sarco/endoplasmic reticulum ATPase[64]. Finally, PKG1 activation has also been shown to inhibit Ca2+ entry into VSMC directly by phosphorylation of the L-type Ca2+ channel[65] and indirectly by activation of the big conductance Ca2+-activated K+ channel[66] and the ATP-sensitive K+ channel[67], leading to membrane hyperpolarization and subsequent inhibition of the voltage-dependent L-type Ca2+ channel (Figure 1). In summary, PKG1 is an important mediator of both NO-dependent and NO-independent vasodilation via its association with and phosphorylation of downstream targets that eventually lead to a multitude of signaling cascades involved in inhibition of contraction and activation of vasorelaxation.
Taken together, these studies using genetically modified mice underscore the important role of NO-sGC-PKG1 signaling in blood pressure control. Disrupting this signaling cascade at any point leads to impaired vasodilation, which in most cases is associated with a hypertensive phenotype.
NO/sGC signaling and its reciprocal interaction with the renin-angiotensin-aldosterone system
Multiple signaling mechanisms regulate blood pressure. Both NO-cGMP signaling and activity of the renin-angiotensin-aldosterone system (RAAS) play a central role in homeostatic control of systemic blood pressure[7, 68]. Increasing evidence suggests that NO-cGMP signaling regulates activity of the RAAS and vice versa. Different reports have suggested that NO-cGMP signaling either decreases or increases RAAS activity[69]. For example, plasma renin concentration (PRC) was elevated in eNOS−/− mice[39], suggesting that NO-cGMP inhibits renin secretion. NO also downregulates the synthesis of angiotensin I-converting enzyme (ACE) in the endothelium[70], as well as the expression of the angiotensin II (Ang II) type 1 receptor (AT1R) in VSMC, attenuating both Ang II production as well as its downstream actions[71, 72]. Other studies indicated that NO-cGMP signaling can stimulate renin secretion[73] and aldosterone production[74]. Together, these data illustrate that, although the interaction between the NO-cGMP signaling system and the RAAS remains controversial, there is evidence that NO-cGMP signaling modulates RAAS activity.
The impact of sGCα1-deficiency on blood pressure is dramatically impacted by the genetic background of the mice studied. sGCα1−/−S6 but not sGCα1−/−B6 develop hypertension, offering an opportunity to consider the existence of genetic modifiers of blood pressure in sGCα1−/− mice. Linkage analysis in sGCα1−/− mice revealed a quantitative trait locus (QTL) for blood pressure on chromosome 1 (designated Hsgcq10]), syntenic with previously identified hypertension-related QTLs in the human and rat genome[10]. Within this region, renin was identified to be a strong biological candidate modifier gene of hypertension. The role of renin in the hypertension associated with impaired NO-cGMP signaling was confirmed by the observation that interfering with activity of the RAAS with a renin inhibitor, an ACE inhibitor, or an aldosterone receptor antagonist, normalized blood pressure in male sGCα1−/−S6 mice[10]. In addition, an ACE inhibitor attenuated the vascular dysfunction in conductance and resistance arteries isolated from sGCα1−/−S6 mice[10]. Taken together, these results suggest that at least part of the increased blood pressure seen in eNOS−/− (see above) and sGCα1−/−S6 mice compared to WT mice could be due to higher activity of the RAAS in the mutant mice. These observations illustrate the intricate interplay between the RAAS and NO-cGMP signaling and highlight the interaction between two well-known blood pressure regulating pathways. Further characterization of how established blood pressure-modifying signaling mechanisms (such as NO-cGMP and RAAS signaling) reciprocally interact to modulate blood pressure (e.g. by studying the mutant mice listed in Table 1), is anticipated to improve our understanding of the role of those signaling pathways in blood pressure regulation, which may result in the identification of novel strategies to treat hypertension.
Role of sGC in the genetic control of HTN
Animal, population, and epidemiological studies have demonstrated that genetic factors contribute to the pathogenesis of essential hypertension, a polygenic trait influenced by both autosomal and sex-linked genes. Dozens of distinct genetic variants that are associated with blood pressure have been identified in GWAS and in candidate gene association studies[5, 32, 75–78]. In humans, an estimated 60% of blood pressure variation is attributable to additive genetic factors[77].
Among the common variant loci identified to date, 4 are involved in the NO/NP-sGC/pGC-cGMP system highlighting a central role of cGMP in blood pressure regulation. No other pathway includes more than 1 locus with a recognized common variant. Variants were identified in or near the genes encoding eNOS (NOS3)[79–82], sGC (GUCY1A3 and GUCY1B3)[32], and cardiac NP genes (NPPA and NPPB)[78, 83] and their clearance receptor (NPR3)[32, 76, 83, 84]. The GUCY1A3-GUCY1B3 variant that was identified in 200,000 individuals of European ancestry (designated rs13139571) is located in an intron of GUCY1A3 and is associated with a significant increase of 0.3 mmHg in systolic and diastolic blood pressure[32]. Understanding the mechanisms by which these genetic variants (e.g. in genes encoding sGC subunits) contribute to the development of hypertension may not only improve our understanding of the etiology of hypertension but may also identify novel therapeutic targets for the treatment of hypertension.
Gender-specific cardiovascular effects of impaired NO-cGMP signaling
Epidemiologic studies have identified male gender as an important risk factor for developing hypertension[85, 86]. However, after the onset of menopause, women are exposed to a unique set of risks and are at higher risk for developing hypertension than are men[87]. Several studies suggest that NO-cGMP signaling has a greater role in regulating blood pressure in male than in female mice. Although blood pressure was elevated in both genders in an independently generated eNOS−/− strain[39], eNOS−/− mice generated by Huang et al.[38] were reported to display hypertension in males only[88]. Similarly, in mice lacking both cyclooxygenase-1 (COX-1) and eNOS, only male mice were hypertensive, and ACh could relax resistance arteries only in female but not in male mice. These results were interpreted to suggest that EDHF is the principal endothelial-derived relaxing factor in females, while in males NO and prostacyclin are the predominant vasorelaxing mediators[88].
Male but not female sGCα1−/−S6 mice develop hypertension[22]. Orchiectomy or treatment with an androgen receptor antagonist prevented hypertension in male sGCα1−/−S6 mice, and chronic testosterone treatment increased blood pressure in ovariectomized female sGCα1−/−S6 mice[22]. Downstream of cGMP, PKG1 deficiency also increased blood pressure in male but not female mice[57, 58]. Together, these data illustrate that NO-cGMP signaling has gender-specific and testosterone-dependent cardiovascular effects. The exact mechanism underlying this gender-specific role of NO-cGMP signaling on cardiovascular function remains to be elucidated but may importantly impact the clinical development of compounds that modulate activity of the NO-cGMP signaling system, possibly in a gender-specific manner.
sGC-independent effects of NO: role of protein S-nitrosylation in cardiovascular function
An alternative pathway by which NO can affect the vasculature is the direct modification of protein thiols, resulting in the formation of S-nitrosothiols. Although NO-induced VSMC relaxation was reported to be abolished in mice lacking sGCβ1, this observation does not preclude the possibility that NO has other, non-sGC dependent effects on blood pressure, since only isolated aortic rings were examined in this study[12]. S-nitrosylation might play a more prominent role in smaller resistance vessels, which have a larger contribution to determining systemic vascular resistance and blood pressure than do large conductance arteries. In support of this hypothesis, mice deficient for S-nitrosoglutathione reductase (GSNOR, an enzyme promoting protein denitrosylation) were found to be in a persistent state of systemic vasodilation[89] and became hypotensive when anesthetized[90]. Combined with the observation that GSNOR inhibition causes relaxation of murine aortic rings[91], these results suggest that increased levels of protein S-nitrosylation are associated with improved vasodilation and lower blood pressure. Multiple possible targets of S-nitrosylation have been identified in smooth muscle, including structural and contractile proteins[92, 93]. For example, the direct activation of VSMC Ca2+-dependent K+ channels by S-nitrosylation[94] was proposed as a potential mechanism of cGMP-independent vasorelaxation[95]. Interestingly, sGC itself has also been identified as a target for S-nitrosylation. S-nitrosylation of two cysteines, Cys243 in sGCα and Cys122 in sGCβ, leads to desensitization of sGC to activation by binding of NO to its heme cofactor[96]. This might represent a mechanism responsible for the development of tolerance to NO.
sGC as a therapeutic target in hypertension
Several mechanisms, including genetic variation (see above) and oxidative stress[97] can modulate NO-cGMP signaling. Cardiovascular diseases, including hypertension, are often associated with oxidative stress caused by increased endogenous production of reactive oxygen species[98]. These strong oxidants not only scavenge NO, reducing its bioavailability[99, 100], but also oxidize sGC leading to loss of the heme cofactor, rendering sGC unresponsive to NO stimulation[97, 101, 102]. The suggestion that the level of heme-oxidized or apo-sGC is elevated in disease conditions[97, 103] highlights the physiological relevance of oxidative modification of sGC. Therefore, compounds that can activate sGC via an NO-independent mechanism hold great therapeutic potential for the treatment of cardiovascular diseases[100, 104, 105], including hypertension[106].
Two classes of compounds have been developed that can directly stimulate and increase cGMP formation in a setting where the use of classical NO donors may be problematic because of the development of tolerance or when sGC is oxidized: heme-dependent NO-independent sGC stimulators and heme-independent NO-independent sGC activators[104]. Preclinical studies confirmed the ability of the first class of heme-dependent NO-independent stimulators of sGC (YC-1, BAY 41-2272, BAY 41-8543, BAY 63-2521 or riociguat) to lower systemic and pulmonary arterial pressure, relax isolated vessels in a variety of organ systems, and reverse the cardiovascular complications associated with hypertension[106–112].
The second class of NO-independent, heme-independent sGC activators, only activates the oxidized and/or heme-deficient sGC enzyme, which cannot be stimulated by the first class of heme-dependent stimulators[97, 113, 114]. The sGC activator HMR1766 (ataciguat) did not affect blood pressure in humans or animals[115], although this compound did have beneficial effects in chronic renal disease[116]. In contrast, BAY 58-2667 (cinaciguat) and BAY 60-2770 were shown to be potent vasodilators, lowering systemic blood pressure in preclinical trials[117–119] . Cinaciguat reduced oxidative stress and attenuated cardiac dysfunction in a rat model of myocardial infarction[120]. In addition, cinaciguat improved cardiac contractility, coronary blood flow, and endothelium-dependent vasodilation to acetylcholine when given prior to cardiac ischemia-reperfusion[121]. In a Phase I clinical trial, cinaciguat exhibited dose-dependent hemodynamic effects on diastolic blood pressure, mean arterial blood pressure and heart rate. In addition, cinaciguat reduced both preload and after load in the heart[122]. In a phase II study in patients suffering from acute decompensated heart failure, cinaciguat significantly reduced systemic vascular resistance[123]. Together, these results illustrate the clinical potential of sGC stimulators and activators as modulators of blood pressure. Together with other compounds that modulate NO-cGMP signaling (e.g. PDE5 inhibitors), sGC activators and stimulators may provide an opportunity to manipulate the cGMP system and treat hypertension.
Conclusion
The studies described in this review illustrate how knowledge of the role of NO-cGMP signaling in the regulation of blood pressure has advanced over the last decade and demonstrate that the presence of a functional NO-cGMP signaling system is imperative for the maintenance of blood pressure homeostasis. Studies in animal models and human data illustrate that disruption of the NO-cGMP signaling cascade (e.g. genetically, pharmacologically, or in response to increased oxidative stress) leads to the development of hypertension. The identification of genetic variants in key NO-cGMP modulating enzymes and the further development and investigation of animal models of impaired NO-cGMP signaling will undoubtedly facilitate our further understanding of the role of NO-cGMP signaling and its interacting pathways in the development of hypertension.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
- 1. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Executive Summary: Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125(1):188–197. doi: 10.1161/CIR.0b013e3182456d46. The 2012 AHA Statistical Update, a comprehensive overview of cardiovascular health and disease in the population, illustrates how big a burden on society uncontrolled hypertension is.
- 2.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42(6):1206–1152. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 3.Kung H-C, Hoyert DL, Xu J, Murphy SL. Deaths: Final Data for 2005. National Vital statistics Report. 2008;56(10):1–121. [PubMed] [Google Scholar]
- 4.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 5.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41(6):666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamm LL, Hering-Smith KS. Pivotal role of the kidney in hypertension. Am J Med Sci. 2010;340(1):30–32. doi: 10.1097/MAJ.0b013e3181e590f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruilope LM. Hypertension in 2010: Blood pressure and the kidney. Nat Rev Nephrol. 2011;7(2):73–74. doi: 10.1038/nrneph.2010.189. [DOI] [PubMed] [Google Scholar]
- 8.Taylor RS, Ashton KE, Moxham T, Hooper L, Ebrahim S. Reduced dietary salt for the prevention of cardiovascular disease: a meta-analysis of randomized controlled trials (cochrane review) . Am J Hypertens. 2011;24(8):843–853. doi: 10.1038/ajh.2011.115. [DOI] [PubMed] [Google Scholar]
- 9.Mendelsohn ME. In hypertension, the kidney is not always the heart of the matter. J Clin Invest. 2005;115(4):840–844. doi: 10.1172/JCI24806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buys ES, Raher MJ, Kirby A, Mohd S, Baron DM, Hayton SR, et al. Genetic modifiers of hypertension in soluble guanylate cyclase alpha1-deficient mice. The Journal of clinical investigation. 2012;122(6):2316–2325. doi: 10.1172/JCI60119. Studies in sGC mutant mice illustrate that sGC signaling has gender-specific cardiovascular effects, identified quantitative trait loci linked to mean arterial pressure (MAP) in the context of sGC deficiency, and identified the RAAS as a blood pressure-modifying mechanism in a setting of impaired NO/cGMP signaling.
- 11. Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, et al. High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci U S A. 2008;105(18):6702–6707. doi: 10.1073/pnas.0802128105. In this study it was shown that mice with a mutant PKGI develop hypertension due to dysfunctional NO-induced vascular relaxation. This observation confirms that hypertension can arise due to a primary vascular defect.
- 12.Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci U S A. 2007;104(18):7699–7704. doi: 10.1073/pnas.0609778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 14.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt- dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 15.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 16.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106(6):675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 17.Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annual review of pharmacology and toxicology. 2001;41:203–236. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- 18.Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- 19.Hobbs AJ. Soluble guanylate cyclase: the forgotten sibling. Trends Pharmacol Sci. 1997;18(12):484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]
- 20.Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, Bohme E, et al. Mutation of His-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1994;91(7):2592–2596. doi: 10.1073/pnas.91.7.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mergia E, Russwurm M, Zoidl G, Koesling D. Major occurrence of the new alpha(2)beta(1) isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 2003;15(2):189–195. doi: 10.1016/s0898-6568(02)00078-5. [DOI] [PubMed] [Google Scholar]
- 22. Buys ES, Sips P, Vermeersch P, Raher MJ, Rogge E, Ichinose F, et al. Gender-specific hypertension and responsiveness to nitric oxide in sGC{alpha}1 knockout mice. Cardiovasc Res. 2008;79(1):179–186. doi: 10.1093/cvr/cvn068. This report indicated that loss of sGCa1 has a gender-specific effect on blood pressure, while showing that even low levels of sGC activation are still sufficient to produce NO-dependent vasorelaxation.
- 23.Nimmegeers S, Sips P, Buys E, Brouckaert P, Van de Voorde J. Functional role of the soluble guanylyl cyclase alpha(1) subunit in vascular smooth muscle relaxation. Cardiovasc Res. 2007;76(1):149–159. doi: 10.1016/j.cardiores.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, et al. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation. 2007;116(8):936–943. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- 25.Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest. 2006;116(6):1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stauss HM, Godecke A, Mrowka R, Schrader J, Persson PB. Enhanced blood pressure variability in eNOS knockout mice. Hypertension. 1999;33(6):1359–1363. doi: 10.1161/01.hyp.33.6.1359. [DOI] [PubMed] [Google Scholar]
- 27.Sips PY, Brouckaert P, Ichinose F. The alpha1 isoform of soluble guanylate cyclase regulates cardiac contractility but is not required for ischemic preconditioning. Basic Res Cardiol. 2011;106(4):635–643. doi: 10.1007/s00395-011-0167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garbers DL, Chrisman TD, Wiegn P, Katafuchi T, Albanesi JP, Bielinski V, et al. Membrane guanylyl cyclase receptors: an update. Trends Endocrinol Metab. 2006;17(6):251–258. doi: 10.1016/j.tem.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su J, Scholz PM, Weiss HR. Differential effects of cGMP produced by soluble and particulate guanylyl cyclase on mouse ventricular myocytes. Exp Biol Med (Maywood) 2005;230(4):242–250. doi: 10.1177/153537020523000403. [DOI] [PubMed] [Google Scholar]
- 30.Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113(18):2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piggott LA, Hassell KA, Berkova Z, Morris AP, Silberbach M, Rich TC. Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J Gen Physiol. 2006;128(1):3–14. doi: 10.1085/jgp.200509403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–109. doi: 10.1038/nature10405. This genome-wide association study of systolic and diastolic blood pressure identified sixteen novel loci in 200,000 individuals of European descent, including a variant in the GUCY1A3/GUCY1B3 locus, encoding the sGC 1 and sGC 1 subunits. This study provides new insights into the genetics and biology of blood pressure, and suggest potential novel therapeutic pathways for cardiovascular disease prevention.
- 33.Panza JA, Quyyumi AA, Brush JE, Jr, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. The New England journal of medicine. 1990;323(1):22–27. doi: 10.1056/NEJM199007053230105. [DOI] [PubMed] [Google Scholar]
- 34.Friebe A, Koesling D. The function of NO-sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide. 2009;21(3–4):149–156. doi: 10.1016/j.niox.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006;176(Pt 1):213–254. doi: 10.1007/3-540-32967-6_7. [DOI] [PubMed] [Google Scholar]
- 36.Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52(3):352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- 37.Ignarro LJ. Endothelium-derived nitric oxide: actions and properties. Faseb J. 1989;3(1):31–36. doi: 10.1096/fasebj.3.1.2642868. [DOI] [PubMed] [Google Scholar]
- 38.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377(6546):239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 39.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93(23):13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beierwaltes WH, Potter DL, Shesely EG. Renal baroreceptor-stimulated renin in the eNOS knockout mouse. Am J Physiol Renal Physiol. 2002;282(1):F59–F64. doi: 10.1152/ajprenal.0144.2001. [DOI] [PubMed] [Google Scholar]
- 41.Kurihara N, Alfie ME, Sigmon DH, Rhaleb NE, Shesely EG, Carretero OA. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension. 1998;32(5):856–861. doi: 10.1161/01.hyp.32.5.856. [DOI] [PubMed] [Google Scholar]
- 42.Duplain H, Burcelin Rm, Sartori C, Cook Sp, Egli M, Lepori M, et al. Insulin Resistance, Hyperlipidemia, and Hypertension in Mice Lacking Endothelial Nitric Oxide Synthase. Circulation. 2001;104(3):342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 43.Yang X-P, Liu Y-H, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial Nitric Oxide Gene Knockout Mice. Hypertension. 1999;34(1):24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- 44.Nelson RJ, Demas GE, Huang PL, Fishman MC, Dawson VL, Dawson TM, et al. Behavioural abnormalities in male mice lacking neuronal nitric oxide synthase. Nature. 1995;378(6555):383–386. doi: 10.1038/378383a0. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Carretero OA, Xu J, Rhaleb N-E, Yang JJ, Pagano PJ, et al. Deletion of Inducible Nitric Oxide Synthase Provides Cardioprotection in Mice With 2-Kidney, 1-Clip Hypertension. Hypertension. 2009;53(1):49–56. doi: 10.1161/HYPERTENSIONAHA.108.121822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakata S, Tsutsui M, Shimokawa H, Suda O, Morishita T, Shibata K, et al. Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms. Circulation. 2008;117(17):2211–2223. doi: 10.1161/CIRCULATIONAHA.107.742692. [DOI] [PubMed] [Google Scholar]
- 47.Barouch LA, Cappola TP, Harrison RW, Crone JK, Rodriguez ER, Burnett AL, et al. Combined loss of neuronal and endothelial nitric oxide synthase causes premature mortality and age-related hypertrophic cardiac remodeling in mice. J Mol Cell Cardiol. 2003;35(6):637–644. doi: 10.1016/s0022-2828(03)00079-8. [DOI] [PubMed] [Google Scholar]
- 48.Lamping KG, Faraci FM. Role of sex differences and effects of endothelial NO synthase deficiency in responses of carotid arteries to serotonin. Arterioscler Thromb Vasc Biol. 2001;21(4):523–528. doi: 10.1161/01.atv.21.4.523. [DOI] [PubMed] [Google Scholar]
- 49.Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, et al. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res. 2002;90(8):904–910. doi: 10.1161/01.res.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- 50.Ding H, Kubes P, Triggle C. Potassium- and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 2000;129(6):1194–1200. doi: 10.1038/sj.bjp.0703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scotland RS, Chauhan S, Vallance PJ, Ahluwalia A. An endothelium-derived hyperpolarizing factor-like factor moderates myogenic constriction of mesenteric resistance arteries in the absence of endothelial nitric oxide synthase-derived nitric oxide. Hypertension. 2001;38(4):833–839. doi: 10.1161/hy1001.092651. [DOI] [PubMed] [Google Scholar]
- 52.Atochin DN, Yuzawa I, Li Q, Rauwerdink KM, Malhotra R, Chang J, et al. Soluble guanylate cyclase alpha1beta1 limits stroke size and attenuates neurological injury. Stroke. 2010;41(8):1815–1819. doi: 10.1161/STROKEAHA.109.577635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buys ES, Cauwels A, Raher MJ, Passeri JJ, Hobai I, Cawley SM, et al. sGC(alpha)1(beta)1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am J Physiol Heart Circ Physiol. 2009;297(2):H654–H663. doi: 10.1152/ajpheart.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thoonen R, Buys E, Cauwels A, Rogge E, Nimmegeers S, Hemel M, et al. NO-insensitive sGCbeta1 H105F knockin mice: if NO has no place to go. BMC Pharmacology. 2009;9(Suppl 1):S41. [Google Scholar]
- 55.Weber S, Bernhard D, Lukowski R, Weinmeister P, Worner R, Wegener JW, et al. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ Res. 2007;101(11):1096–1103. doi: 10.1161/CIRCRESAHA.107.154351. [DOI] [PubMed] [Google Scholar]
- 56.Feil R, Gappa N, Rutz M, Schlossmann J, Rose CR, Konnerth A, et al. Functional reconstitution of vascular smooth muscle cells with cGMP-dependent protein kinase I isoforms. Circ Res. 2002;90(10):1080–1086. doi: 10.1161/01.res.0000019586.95768.40. [DOI] [PubMed] [Google Scholar]
- 57.Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17(11):3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U, et al. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension. 2004;44(6):952–955. doi: 10.1161/01.HYP.0000147661.80059.ca. [DOI] [PubMed] [Google Scholar]
- 59.Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, et al. Mechanisms of NO/cGMPdependent vasorelaxation. Circ Res. 2000;87(9):825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 60.Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med. 2003;9(12):1506–1512. doi: 10.1038/nm958. [DOI] [PubMed] [Google Scholar]
- 61.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18(2):286–290. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia C, Bao Z, Yue C, Sanborn BM, Liu M. Phosphorylation and regulation of G-protein-activated phospholipase C-beta 3 by cGMP-dependent protein kinases. J Biol Chem. 2001;276(23):19770–19777. doi: 10.1074/jbc.M006266200. [DOI] [PubMed] [Google Scholar]
- 63.Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 2000;404(6774):197–201. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- 64.Cornwell TL, Pryzwansky KB, Wyatt TA, Lincoln TM. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Mol Pharmacol. 1991;40(6):923–931. [PubMed] [Google Scholar]
- 65.Liu H, Xiong Z, Sperelakis N. Cyclic nucleotides regulate the activity of L-type calcium channels in smooth muscle cells from rat portal vein. J Mol Cell Cardiol. 1997;29(5):1411–1421. doi: 10.1006/jmcc.1997.0379. [DOI] [PubMed] [Google Scholar]
- 66.Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91(16):7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy ME, Brayden JE. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATPsensitive potassium channels. J Physiol. 1995;486(Pt 1):47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 69.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86(1):1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 70.Takemoto M, Egashira K, Usui M, Numaguchi K, Tomita H, Tsutsui H, et al. Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in rats. J Clin Invest. 1997;99(2):278–287. doi: 10.1172/JCI119156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ichiki T, Usui M, Kato M, Funakoshi Y, Ito K, Egashira K, et al. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension. 1998;31(1 Pt 2):342–348. doi: 10.1161/01.hyp.31.1.342. [DOI] [PubMed] [Google Scholar]
- 72.Usui M, Ichiki T, Katoh M, Egashira K, Takeshita A. Regulation of angiotensin II receptor expression by nitric oxide in rat adrenal gland. Hypertension. 1998;32(3):527–533. doi: 10.1161/01.hyp.32.3.527. [DOI] [PubMed] [Google Scholar]
- 73.Beierwaltes WH. cGMP stimulates renin secretion in vivo by inhibiting phosphodiesterase-3. Am J Physiol Renal Physiol. 2006;290(6):F1376–F1381. doi: 10.1152/ajprenal.00209.2005. [DOI] [PubMed] [Google Scholar]
- 74.Gambaryan S, Butt E, Marcus K, Glazova M, Palmetshofer A, Guillon G, et al. cGMP-dependent protein kinase type II regulates basal level of aldosterone production by zona glomerulosa cells without increasing expression of the steroidogenic acute regulatory protein gene. J Biol Chem. 2003;278(32):29640–29648. doi: 10.1074/jbc.M302143200. [DOI] [PubMed] [Google Scholar]
- 75.Johnson AD, Newton-Cheh C, Chasman DI, Ehret GB, Johnson T, Rose L, et al. Association of Hypertension Drug Target Genes With Blood Pressure and Hypertension in 86 588 Individuals. Hypertension. 2011;57(5):903–910. doi: 10.1161/HYPERTENSIONAHA.110.158667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43(6):531–538. doi: 10.1038/ng.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 2009;41(3):348–353. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson T, Gaunt TR, Newhouse SJ, Padmanabhan S, Tomaszewski M, Kumari M, et al. Blood pressure loci identified with a gene-centric array. Am J Hum Genet. 2011;89(6):688–700. doi: 10.1016/j.ajhg.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kathiresan S, Larson MG, Vasan RS, Guo CY, Vita JA, Mitchell GF, et al. Common genetic variation at the endothelial nitric oxide synthase locus and relations to brachial artery vasodilator function in the community. Circulation. 2005;112(10):1419–1427. doi: 10.1161/CIRCULATIONAHA.105.544619. [DOI] [PubMed] [Google Scholar]
- 81.Mitchell GF, Guo CY, Kathiresan S, Vasan RS, Larson MG, Vita JA, et al. Vascular stiffness and genetic variation at the endothelial nitric oxide synthase locus: the Framingham Heart study. Hypertension. 2007;49(6):1285–1290. doi: 10.1161/HYPERTENSIONAHA.106.085266. [DOI] [PubMed] [Google Scholar]
- 82.Salvi E, Kutalik Z, Glorioso N, Benaglio P, Frau F, Kuznetsova T, et al. Genomewide association study using a high-density single nucleotide polymorphism array and case-control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension. 2012;59(2):248–255. doi: 10.1161/HYPERTENSIONAHA.111.181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fedorowski A, Franceschini N, Brody J, Liu C, Verwoert GC, Boerwinkle E, et al. Orthostatic hypotension and novel blood pressure-associated gene variants: Genetics of Postural Hemodynamics (GPH) Consortium. Eur Heart J. 2012 doi: 10.1093/eurheartj/ehs058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu X, Young JH, Fox E, Keating BJ, Franceschini N, Kang S, et al. Combined admixture mapping and association analysis identifies a novel blood pressure genetic locus on 5p13: contributions from the CARe consortium. Hum Mol Genet. 2011;20(11):2285–2295. doi: 10.1093/hmg/ddr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kannel WBWP, Garrison RJ. Some risk factors related to the annual incidence of cardiovascular disease and death using pooled repeated biennial measurements: Framingham Heart Study 30 year followup. Springfield, MA: National Technical Information Service; 1987. [Google Scholar]
- 86.Burt VL, Whelton P, Roccella EJ, Brown C, Cutler JA, Higgins M, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995;25(3):305–313. doi: 10.1161/01.hyp.25.3.305. [DOI] [PubMed] [Google Scholar]
- 87.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension. 2001;37(5):1199–1208. doi: 10.1161/01.hyp.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 88.Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111(6):796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- 89.Beigi F, Gonzalez DR, Minhas KM, Sun QA, Foster MW, Khan SA, et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci U S A. 2012;109(11):4314–4319. doi: 10.1073/pnas.1113319109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116(4):617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 91.Sanghani PC, Davis WI, Fears SL, Green SL, Zhai L, Tang Y, et al. Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J Biol Chem. 2009;284(36):24354–24362. doi: 10.1074/jbc.M109.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Greco TM, Hodara R, Parastatidis I, Heijnen HF, Dennehy MK, Liebler DC, et al. Identification of Snitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103(19):7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ulrich C, Quillici DR, Schegg K, Woolsey R, Nordmeier A, Buxton IL. Uterine smooth muscle Snitrosylproteome in pregnancy. Mol Pharmacol. 2012;81(2):143–153. doi: 10.1124/mol.111.075804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lang RJ, Harvey JR, McPhee GJ, Klemm MF. Nitric oxide and thiol reagent modulation of Ca2+- activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. J Physiol. 2000;525(Pt 2):363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calciumdependent potassium channels in vascular smooth muscle. Nature. 1994;368(6474):850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 96.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(30):12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, H SA, Meurer S, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116(9):2552–2561. doi: 10.1172/JCI28371. This study proved for the first time the pharmacological principle of sGC activators under conditions of increased oxidative stress.
- 98.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44(3):248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 99.Chirkov YY, Horowitz JD. Impaired tissue responsiveness to organic nitrates and nitric oxide: a new therapeutic frontier? Pharmacol Ther. 2007;116(2):287–305. doi: 10.1016/j.pharmthera.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 100.Franco V, Oparil S. Is there a new treatment for hypertensive disease in the horizon? Role of soluble guanylate cyclase. Hypertension. 2006;48(5):822–823. doi: 10.1161/01.HYP.0000241432.82676.4f. [DOI] [PubMed] [Google Scholar]
- 101.Foerster J, Harteneck C, Malkewitz J, Schultz G, Koesling D. A functional heme-binding site of soluble guanylyl cyclase requires intact N-termini of alpha 1 and beta 1 subunits. Eur J Biochem. 1996;240(2):380–386. doi: 10.1111/j.1432-1033.1996.0380h.x. [DOI] [PubMed] [Google Scholar]
- 102.Zhao Y, Brandish PE, DiValentin M, Schelvis JP, Babcock GT, Marletta MA. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39(35):10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- 103.Ahrens I, Habersberger J, Baumlin N, Qian H, Smith BK, Stasch JP, et al. Measuring oxidative burden and predicting pharmacological response in coronary artery disease patients with a novel direct activator of haem-free/oxidised sGC. Atherosclerosis. 2011;218(2):431–434. doi: 10.1016/j.atherosclerosis.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 104.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature reviews Drug discovery. 2006;5(9):755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Doggrell SA. Clinical potential of nitric oxide-independent soluble guanylate cyclase activators. Curr Opin Investig Drugs. 2005;6(9):874–878. [PubMed] [Google Scholar]
- 106.Stasch JP, Dembowsky K, Perzborn E, Stahl E, Schramm M. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vivo studies. Br J Pharmacol. 2002;135(2):344–355. doi: 10.1038/sj.bjp.0704483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Straub A, Benet-Buckholz J, Frode R, Kern A, Kohlsdorfer C, Schmitt P, et al. Metabolites of orally active NO-independent pyrazolopyridine stimulators of soluble guanylate cyclase. Bioorg Med Chem. 2002;10(6):1711–1717. doi: 10.1016/s0968-0896(02)00034-2. [DOI] [PubMed] [Google Scholar]
- 108.Badejo AM, Jr, Nossaman VE, Pankey EA, Bhartiya M, Kannadka CB, Murthy SN, et al. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase stimulator, BAY 41- 8543, are modulated by nitric oxide. Am J Physiol Heart Circ Physiol. 2010;299(4):H1153–H1159. doi: 10.1152/ajpheart.01101.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410(6825):212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 110.Masuyama H, Tsuruda T, Kato J, Imamura T, Asada Y, Stasch JP, et al. Soluble guanylate cyclase stimulation on cardiovascular remodeling in angiotensin II-induced hypertensive rats. Hypertension. 2006;48(5):972–978. doi: 10.1161/01.HYP.0000241087.12492.47. [DOI] [PubMed] [Google Scholar]
- 111.Zanfolin M, Faro R, Araujo EG, Guaraldo AM, Antunes E, De Nucci G. Protective effects of BAY 41-2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J Cardiovasc Pharmacol. 2006;47(3):391–395. [PubMed] [Google Scholar]
- 112.Stasch JP, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, Minuth T, et al. Pharmacological actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vitro studies. Br J Pharmacol. 2002;135(2):333–343. doi: 10.1038/sj.bjp.0704484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113(2):286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 114.Boerrigter G, Costello-Boerrigter LC, Cataliotti A, Lapp H, Stasch JP, Burnett JC., Jr Targeting heme-oxidized soluble guanylate cyclase in experimental heart failure. Hypertension. 2007;49(5):1128–1133. doi: 10.1161/HYPERTENSIONAHA.106.083832. [DOI] [PubMed] [Google Scholar]
- 115.Oberwittler H, Hirschfeld-Warneken A, Wesch R, Willerich H, Teichert L, Lehr KH, et al. Significant pharmacokinetic and pharmacodynamic interaction of warfarin with the NOindependent sGC activator HMR1766. J Clin Pharmacol. 2007;47(1):70–77. doi: 10.1177/0091270006294540. [DOI] [PubMed] [Google Scholar]
- 116.Benz K, Orth SR, Simonaviciene A, Linz W, Schindler U, Rutten H, et al. Blood pressureindependent effect of long-term treatment with the soluble heme-independent guanylyl cyclase activator HMR1766 on progression in a model of noninflammatory chronic renal damage. Kidney Blood Press Res. 2007;30(4):224–233. doi: 10.1159/000104091. [DOI] [PubMed] [Google Scholar]
- 117.Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M, et al. NO- and haemindependent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136(5):773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Knorr A, Hirth-Dietrich C, Alonso-Alija C, Harter M, Hahn M, Keim Y, et al. Nitric oxideindependent activation of soluble guanylate cyclase by BAY 60-2770 in experimental liver fibrosis. Arzneimittelforschung. 2008;58(2):71–80. doi: 10.1055/s-0031-1296471. [DOI] [PubMed] [Google Scholar]
- 119.Pankey EA, Bhartiya M, Badejo AM, Jr, Haider U, Stasch JP, Murthy SN, et al. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase activator, BAY 60-2770, are not dependent on endogenous nitric oxide or reduced heme. Am J Physiol Heart Circ Physiol. 2011;300(3):H792–H802. doi: 10.1152/ajpheart.00953.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Korkmaz S, Radovits T, Barnucz E, Hirschberg K, Neugebauer P, Loganathan S, et al. Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation. 2009;120(8):677–686. doi: 10.1161/CIRCULATIONAHA.109.870774. [DOI] [PubMed] [Google Scholar]
- 121.Radovits T, Korkmaz S, Miesel-Groschel C, Seidel B, Stasch JP, Merkely B, et al. Pre-conditioning with the soluble guanylate cyclase activator Cinaciguat reduces ischaemia-reperfusion injury after cardiopulmonary bypass. Eur J Cardiothorac Surg. 2011;39(2):248–255. doi: 10.1016/j.ejcts.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 122.Frey R, Muck W, Unger S, Artmeier-Brandt U, Weimann G, Wensing G. Pharmacokinetics, pharmacodynamics, tolerability, and safety of the soluble guanylate cyclase activator cinaciguat (BAY 58-2667) in healthy male volunteers. J Clin Pharmacol. 2008;48(12):1400–1410. doi: 10.1177/0091270008322906. [DOI] [PubMed] [Google Scholar]
- 123. Lapp H, Mitrovic V, Franz N, Heuer H, Buerke M, Wolfertz J, et al. Cinaciguat (BAY 58-2667) improves cardiopulmonary hemodynamics in patients with acute decompensated heart failure. Circulation. 2009;119(21):2781–2788. doi: 10.1161/CIRCULATIONAHA.108.800292. This clinical study showed that cinaciguat reduces systemic vascular resistance in patients with heart failure, demonstrating the proof of concept that specific activation of oxidized sGC has the expected hemodynamic effects in patients.
- 124.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75(7):1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 125.Gyurko R, Leupen S, Huang PL. Deletion of exon 6 of the neuronal nitric oxide synthase gene in mice results in hypogonadism and infertility. Endocrinology. 2002;143(7):2767–2774. doi: 10.1210/endo.143.7.8921. [DOI] [PubMed] [Google Scholar]
- 126.Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, et al. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82(2):186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 127.Gregg AR, Schauer A, Shi O, Liu Z, Lee CG, O'Brien WE. Limb reduction defects in endothelial nitric oxide synthase-deficient mice. Am J Physiol. 1998;275(6 Pt 2):H2319–H2324. doi: 10.1152/ajpheart.1998.275.6.H2319. [DOI] [PubMed] [Google Scholar]
- 128.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 129.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A. 1995;92(23):10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81(4):641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 131.Tranguch S, Huet-Hudson Y. Decreased viability of nitric oxide synthase double knockout mice. Mol Reprod Dev. 2003;65(2):175–179. doi: 10.1002/mrd.10274. [DOI] [PubMed] [Google Scholar]
- 132.Morishita T, Tsutsui M, Shimokawa H, Sabanai K, Tasaki H, Suda O, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 2005;102(30):10616–10621. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, et al. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87(6):1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 134.Burkard N, Rokita AG, Kaufmann SG, Hallhuber M, Wu R, Hu K, et al. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res. 2007;100(3):e32–e44. doi: 10.1161/01.RES.0000259042.04576.6a. [DOI] [PubMed] [Google Scholar]
- 135.Loyer X, Gomez AM, Milliez P, Fernandez-Velasco M, Vangheluwe P, Vinet L, et al. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation. 2008;117(25):3187–3198. doi: 10.1161/CIRCULATIONAHA.107.741702. [DOI] [PubMed] [Google Scholar]
- 136.Packer MA, Hemish J, Mignone JL, John S, Pugach I, Enikolopov G. Transgenic mice overexpressing nNOS in the adult nervous system. Cell Mol Biol (Noisy-le-grand) 2005;51(3):269–77. [PubMed] [Google Scholar]
- 137.Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104(25):3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- 138.Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, et al. Cardiomyocytespecific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res. 2004;94(9):1256–1262. doi: 10.1161/01.RES.0000126497.38281.23. [DOI] [PubMed] [Google Scholar]
- 139.Ohashi Y, Kawashima S, Hirata K, Yamashita T, Ishida T, Inoue N, et al. Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest. 1998;102(12):2061–2071. doi: 10.1172/JCI4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.van Haperen R, de Waard M, van Deel E, Mees B, Kutryk M, van Aken T, et al. Reduction of blood pressure, plasma cholesterol, and atherosclerosis by elevated endothelial nitric oxide. J Biol Chem. 2002;277(50):48803–48807. doi: 10.1074/jbc.M209477200. [DOI] [PubMed] [Google Scholar]
- 141.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, et al. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002;109(6):735–743. doi: 10.1172/JCI13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Heger J, Godecke A, Flogel U, Merx MW, Molojavyi A, Kuhn-Velten WN, et al. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res. 2002;90(1):93–99. doi: 10.1161/hh0102.102757. [DOI] [PubMed] [Google Scholar]
- 143.Takamura T, Kato I, Kimura N, Nakazawa T, Yonekura H, Takasawa S, et al. Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic beta cells develop insulin-dependent diabetes without insulitis. J Biol Chem. 1998;273(5):2493–2496. doi: 10.1074/jbc.273.5.2493. [DOI] [PubMed] [Google Scholar]
- 144.Pfeifer A, Aszodi A, Seidler U, Ruth P, Hofmann F, Fassler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 1996;274(5295):2082–2086. doi: 10.1126/science.274.5295.2082. [DOI] [PubMed] [Google Scholar]
- 145.Pokreisz P, Pellens M, Van den Bergh A, Gillijns H, Bito V, Lenaers I, et al. Cardiomyocytespecific overexpression of type 5 phosphodiesterase impairs postinfarction myocardial function and left ventricular remodeling. Circulation. 2007;116(16):45–46. [Google Scholar]