

Abstract
Previous work demonstrated that hyperthermia (43°C for 2 h) results in delayed, apoptotic-like death in striatal neuronal cultures. We investigated early changes in mitochondrial function induced by this heat stress. Partial depolarization of the mitochondrial membrane potential (ΔΨm) began about 1 h after the onset of hyperthermia and increased as the stress continued. When the heat stress ended, there was a partial recovery of ΔΨm, followed hours later by a progressive, irreversible depolarization of ΔΨm. During the heat stress, O2 consumption initially increased but after 20–30 min began a progressive, irreversible decline to about one-half the initial rate by the end of the stress. The percentage of oligomycin-insensitive respiration increased during the heat stress, suggesting an increased mitochondrial leak conductance. Analysis using inhibitors and substrates for specific respiratory chain complexes indicated hyperthermia-induced dysfunction at or upstream of complex I. ATP levels remained near normal for ∼4 h after the heat stress. Mitochondrial movement along neurites was markedly slowed during and just after the heat stress. The early, persisting mitochondrial dysfunction described here likely contributes to the later (>10 h) caspase activation and neuronal death produced by this heat stress. Consistent with this idea, proton carrier-induced ΔΨm depolarizations comparable in duration to those produced by the heat stress also reduced neuronal viability. Post-stress ΔΨm depolarization and/or delayed neuronal death were modestly reduced/postponed by nicotinamide adenine dinucleotide, a calpain inhibitor, and increased expression of Bcl-xL.
Keywords: heat stress, hyperthermia, mitochondria, respiration, ATP, apoptosis, neuron, mitochondrial membrane potential
hyperthermia can cause brain damage and exacerbates the damage produced by stroke, traumatic brain injury, and certain drugs (Brown and Kiyatkin 2004; Castillo et al. 1998; Jones et al. 1994; Simon 1993; reviewed by White et al. 2007). After a 2-h, 43°C heat stress, cultured neurons exhibit a delayed (>10 h) death with apoptotic characteristics including shrunken and fragmented nuclei, caspase-3 activation, and cytochrome c release (e.g., White et al. 2003, 2007). These findings may also have relevance in vivo, since some hyperthermia-induced neuronal death occurs after a delay of more than 10 h (e.g., Uney et al. 1993; Vogel et al. 1997) and cytochrome c release has been reported in the brain of heat-stressed murine embryos (Mirkes and Little 2000).
The present study investigated earlier consequences of heat stress, focusing on changes in mitochondrial function, since studies in nonneuronal cells and isolated mitochondria demonstrate hyperthermia-induced disruption of mitochondrial energy production. For example, in isolated mitochondria hyperthermia increases the permeability of the mitochondrial inner membrane and impairs oxidative phosphorylation (Qian et al. 2004; Willis et al. 2000). Hyperthermia above 42°C induces mitochondrial uncoupling in rat cardiomyocytes (Qian et al. 2004) and in mitochondria isolated from heart cells (Zukiene et al. 2010). Other signs of damage/dysfunction in mitochondria isolated from rat hearts following in vivo hyperthermia (rectal temperature reaching 42°C) include decreased ATP synthesis, a decreased respiratory control ratio, a greater tendency to open the mitochondrial permeability transition pore (mPTP) when exposed to moderate Ca2+ loads (Qian et al. 2004), mitochondrial swelling, and a reduced number of cristae (Song et al. 2000). Mouse embryo fibroblasts stressed at 43°C show B cell lymphoma/leukemia 2 (Bcl-2)-associated protein X (BAX) oligomerization and release of apoptotic factors from mitochondria that is promoted by Bcl-2 homology 3 (BH3)-only family proteins and reduced by Bcl-2 (Pagliari et al. 2005).
Consistent with these studies in nonneuronal tissues, we present evidence that hyperthermia produces a severe disruption of mitochondrial function in cultured rat central neurons, including a transiently reversible depolarization of membrane potential across the mitochondrial inner membrane (ΔΨm) and an irreversible reduction in O2 consumption. These changes in mitochondrial function occurred before, and thus may contribute to, the caspase-3 activation and neuronal death produced by this heat stress. To probe underlying mechanisms, we also tested whether inhibitors of various stress-activated pathways could reduce the heat-induced ΔΨm depolarization. Caspase inhibitors had no significant effect, but a calpain inhibitor and addition of nicotinamide adenine dinucleotide (NAD+) offered some protection.
MATERIALS AND METHODS
Neuronal cultures.
Striatal (and in some cases septal) tissue was dissected from embryonic day 15 (E15) rats, dissociated, and plated on poly-l-lysine-coated 72-well Terasaki plates (Nalge Nunc, Rochester, NY) or glass coverslips at a density of ∼1,000–1,800 cells/mm2. Cultures were grown in a basic nutrient medium (N5; Kawamoto and Barrett, 1986) or in Neurobasal medium (Invitrogen, Carlsbad, CA), supplemented with l-alanyl-l-glutamine (0.5 mM, GlutaMAX-1; Invitrogen) and a 55-kDa serum fraction that supports prolonged neuronal survival with minimal proliferation of nonneuronal cells (Nonner et al. 2001; Yan and Barrett 1998). Cultures were maintained for 5–14 days in 5.5% CO2-94.5% air at 36–37°C before use. Fresh medium was added every 5–7 days.
For some respiration measurements, we used neurosphere cultures prepared from striatum or cerebral cortex, dissected and pooled from one E15 rat litter. Dissociated cells were suspended in 10 ml of medium and plated into untreated 100-mm Optilux tissue culture dishes (Becton Dickinson, Franklin Lakes, NJ) for 5–14 days, during which time neurospheres formed (similar to those described by White et al. 1999).
Heat stress protocol.
Cultures were stressed for 2 h at 43°C on aluminum plates within a tissue culture incubator to ensure temperature uniformity. This is the minimum temperature and minimum duration that reliably produced delayed neuronal death (White et al. 2003). After the stress, cultures were transferred back to the standard 36–37°C incubator.
Cultures imaged during the heat stress were plated in glass-bottom dishes (MatTek, Ashland, MA) and heated in a small incubator on the microscope stage (5% CO2 in air). The oil-immersion microscope objective was also heated to 43°C using hot air from a hairdryer powered via a variable AC transformer (ISE, Cleveland, OH). The temperature close to the cells was monitored using a very small thermistor. The room was heated to ∼30°C to reduce temperature gradients and so allow better temperature control.
Measurements of mitochondrial membrane potential.
Changes in mitochondrial membrane potential (ΔΨm) produced by the heat stress were measured using tetramethylrhodamine methyl ester (TMRM), a membrane-permeable, positively charged dye that accumulates within the mitochondrial matrix in a Nernstian manner dependent on ΔΨm. Some of these experiments used high-[K+] bath solutions to completely depolarize the plasma membrane, thereby preventing changes in cytosolic TMRM concentrations caused by changes in the plasma membrane potential (in mM: 145 K-gluconate, 10 NaCl, 1 MgCl2, 5.5 glucose, and 1 pyruvate). In some experiments, 145 mM KCl was used in place of K-gluconate and 80 mM sucrose was added to prevent the cell swelling otherwise seen in this KCl solution.
For assays of ΔΨm performed using a plate reader (Wallac 1420 Victor; PerkinElmer, Boston, MA), cells were exposed to 1 μM TMRM for 20 min and then stressed in the continued presence of TMRM (see Figs. 1, A and B, and 7A). At different post-stress intervals, cultures were washed twice rapidly with the same medium used for incubation (normal or high [K+]) but lacking TMRM, after which cells were solubilized/lysed with 50% DMSO in distilled water to release TMRM from cells/mitochondria. Fluorescence was then assayed (535 nm excitation, >590 nm emission) in this lysis solution, with ΔΨm depolarization detected as a decrease in TMRM fluorescence. Because TMRM fluorescence was assayed only after TMRM was released from the cells, and the concentration of TMRM in the lysis solution was <1 nM (far below the TMRM concentration at which quenching occurs), this assay avoided errors due to self-quenching effects of high [TMRM] within mitochondria. Some culture wells were treated with a proton carrier, carbonyl cyanide m-chlorophenylhydrazone (CCCP; 10 μM), to thoroughly depolarize ΔΨm, permitting measurement of background fluorescence.
Fig. 1.
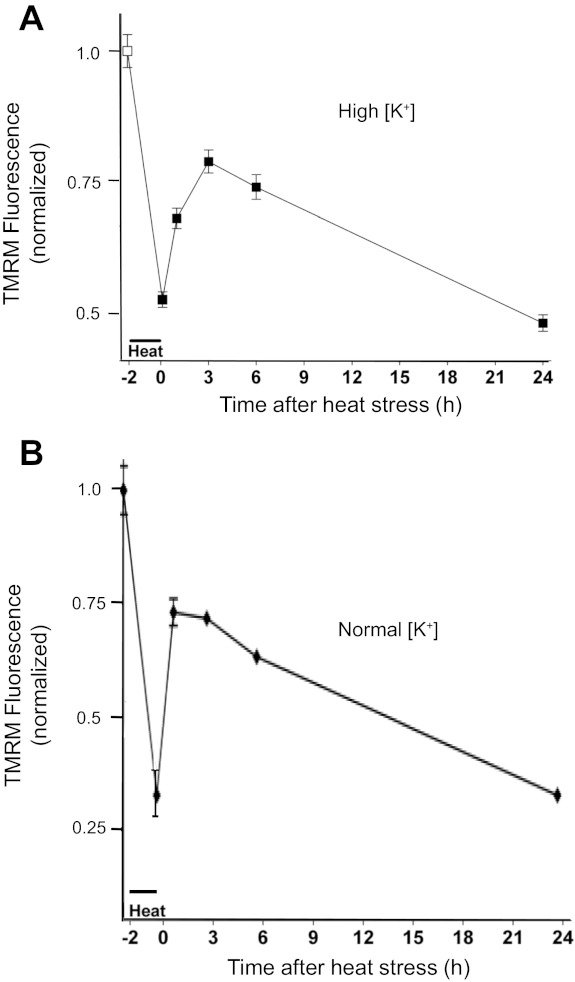
Early and delayed phases of mitochondrial membrane potential (ΔΨm) depolarization associated with a 2-h, 43°C heat stress, recorded with a plate reader in high-[K+] (A) and normal [K+] medium (B). A: tetramethyl rhodamine methyl ester (TMRM) fluorescence before (−2 h) and after the heat stress, recorded in a high-K+ (145 mM), nominally zero-Ca2+ medium. ΔΨm partially depolarized during the heat stress, recovered partially following the stress, and then exhibited a later, slow, irreversible depolarization. Values are means ± SE for 30 culture wells. This biphasic pattern of heat stress-induced ΔΨm depolarization was observed in >10 additional experiments. B: a similar pattern of ΔΨm changes was recorded in normal culture medium (2 mM Ca2+, 155 mM Na+). In both A and B, TMRM fluorescence was normalized to values recorded before stress application (−2 h).
Fig. 7.
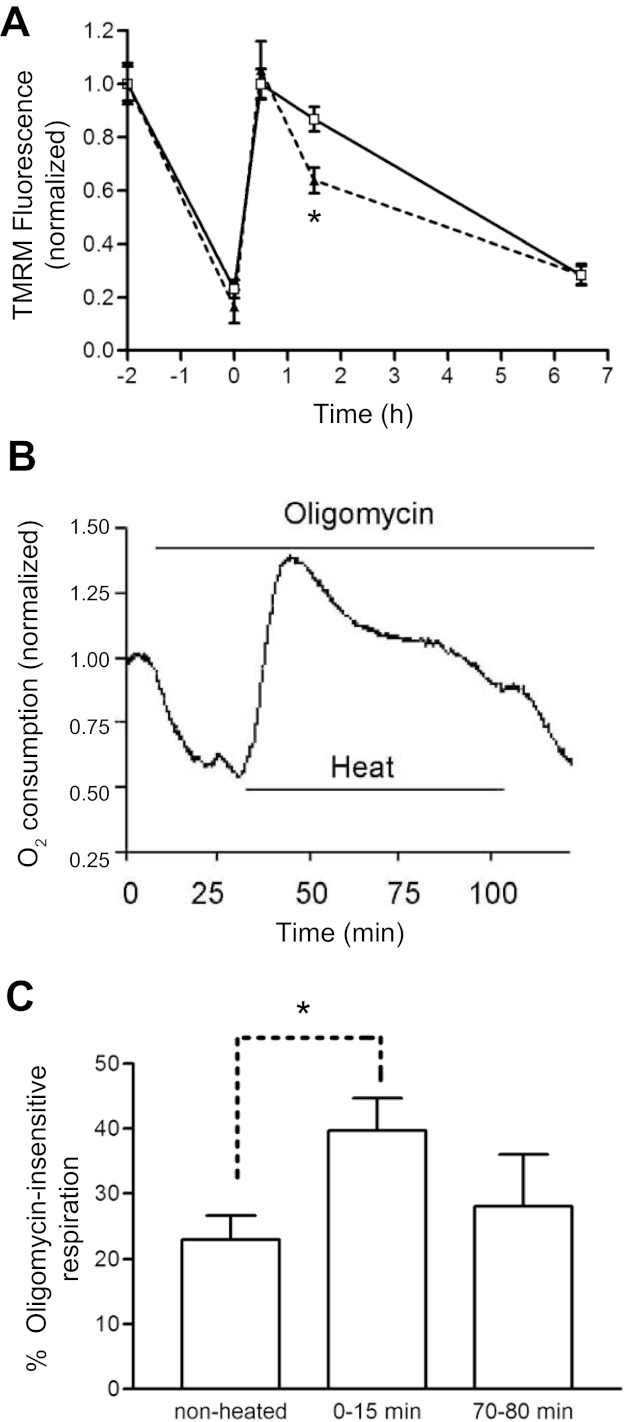
Hyperthermia-induced changes in ΔΨm and O2 consumption persist in oligomycin (5 μg/ml). A: oligomycin (dashed line) did not block the ΔΨm depolarization during the heat stress or the transient ΔΨm repolarization on return to 37°C. Measurements were made on sister striatal cultures in high-[K+] medium using TMRM, as in Fig. 1A. TMRM signals were normalized to those recorded before the heat stress; n = 8 culture wells for each time point. Similar results were obtained when this experiment was repeated in normal [K+] medium. B: effect of oligomycin on O2 consumption before, during, and immediately after a heat stress, normalized to pre-stress/pre-drug value. Before the stress, oligomycin reduced O2 consumption to ∼60% of control. Oligomycin did not block the hyperthermia-induced increase in O2 consumption. O2 consumption remained elevated above the pre-stress oligomycin value throughout the 75-min heat stress. C: percentage of oligomycin-insensitive respiration in neurospheres before the heat stress and at two times following the 2-h heat stress, calculated by comparing O2 consumption in cell aliquots in the absence and presence of oligomycin. The larger percentage of oligomycin-insensitive respiration before the heat stress in B may have occurred because the effect of oligomycin had not yet reached a steady state when the heat stress was applied or because of some damage to the cultures before the heat stress. *P < 0.05. O2 consumption in B was measured as in Fig. 3B; O2 consumption in C was measured as in Fig. 3A.
In the platereader TMRM assay described above, [TMRM] in the original solution should not exceed 1 μM, to avoid effects on mitochondrial respiration. It was also important that the washes with TMRM-free medium (prior to extraction of TMRM by cell lysis) be done consistently and quickly to minimize changes in intracellular [TMRM]. The half-time for TMRM exit from cells was ∼10 min, probably limited by the rate at which TMRM crosses the cell plasmalemma (based on surface-to-volume considerations, TMRM likely redistributes more rapidly between mitochondria and cytoplasm than between cytoplasm and the extracellular medium).
The platereader assay described above gives information about average changes in ΔΨm in cell cultures. To gain information about ΔΨm changes in individual neurons, we also assayed ΔΨm by microscopic imaging of single neurons. These assays used a lower concentration of TMRM (10–30 nM) to avoid self-quenching so that the TMRM fluorescence would be monotonically related to ΔΨm. TMRM was added 20 min before the imaging was begun and remained present throughout the experiment, allowing transiently depolarized mitochondria to reaccumulate the dye as they recovered their membrane potential. Images of TMRM fluorescence were collected using a Nikon TE2000 inverted microscope (Nikon, Melville, NY) with an Olympus ×60, 1.45-NA objective (Olympus, Center Valley, PA). Images were taken at a rate of one per minute with a 500- to 800-ms exposure time. A Cascade 512B charge-coupled device (CCD) camera (Roper Scientific, Trenton, NJ) and a shuttered monochromator (Photon Technology International, Birmingham, NJ) with a slit attenuator enabled the use of low-excitation light intensities to minimize photodamage to dyes and cells.
Respiration measurements.
Respiration was measured both in cultures of free-floating (unattached) neurospheres and in standard cultures with cells attached to the substrate. After heat stress was applied to neurospheres, cultures were gently centrifuged (5 min at 250 g) in 10-ml tapered conical tubes. The pellet was resuspended in 400 μl of culture medium or (if to be permeabilized) in respiration medium (in mM: 220 d-mannitol, 60 sucrose, 3 KCl, 2 KH2PO4, 2 MgCl2, and 2 HEPES) in a prewarmed (37°C) recording chamber with constant stirring. The oxygen pressure (Po2) in the chamber was monitored continuously using a Clarke-type O2 electrode, with respiration measured as the rate of O2 consumption. Additional O2 was introduced if chamber O2 fell below 10–50 nmol/ml. Pyruvate (2 mM) and malate (2 mM) were added to ensure that respiration was not limited by substrates. To evaluate the function of individual electron transport chain (ETC) complexes, 2 mM ADP was added (to ensure state 3 respiration) and cells were permeabilized with 16 μM digitonin (e.g., Barrientos 2002; separate experiments demonstrated that this [digitonin] rendered cells permeable to the normally impermeable propidium iodide [5 μM] within 2 min). The rate of O2 consumption was then measured after each of the following sequential additions: 2 μM rotenone, to inhibit complex I; 5 mM succinate and 5–10 mM glycerol-3-phosphate (G3P), to measure complex II- and complex III-driven respiration; 1 μM antimycin A, to inhibit complex III; 500 μM TMPD (N,N,N′,N′-tetramethyl-1,4-phenylenediamine) and 1 mM ascorbate, to measure complex IV-driven respiration; and finally, 12 mM KCN or NaCN, to block complex IV, yielding a background rate (nonmitochondrial) of O2 consumption. CCCP (2 μM) or the ATP synthase inhibitor oligomycin (5 μg/ml = 6 μM) was used in experiments examining proton leak. In some experiments, neurospheres were heat-stressed in the chamber itself, with temperature measured by a miniature thermistor.
To determine whether the heat stress induced similar changes in respiration in cells that remained attached to the polylysine substrate, O2 consumption was measured using a modification of the flow-through technique of Jekabsons and Nicholls (2004). The heat stress was applied to cultures in a thin, O2-impermeable chamber (10 × 40 × 0.1 mm) perfused with medium whose temperature was controlled by a Peltier device. Chamber temperature was monitored using two miniature thermistors. Fine quartz tubing (inner diameter 0.1 mm) connected the chamber inlet and outlet with the O2 sensors. These O2 sensors were located in a constant-temperature 37°C water bath “decoupled” from the temperature of the chamber by a 15-cm loop of the quartz tubing within the water bath. The perfusing culture medium was first heated to 44°C for 5 min to prevent bubble formation due to escape of dissolved gas from the heated culture chamber. This preheating helped ensure that the amount of dissolved O2 and hence the concentration of O2 passing over the cells in the chamber remained constant during the heat stress. The rate of O2 consumption was measured as the flow rate (60 μl/min) multiplied by the difference in Po2 values (converted to nanomolar oxygen) measured at the inlet and outlet tubes with flow-through Clarke-type O2 sensors (Microelectrodes, Bedford, NH). The output of the O2 sensors was sampled at a rate of 0.5 Hz using a 16-bit analog-to-digital converter. With the use of this system, the rate of O2 consumption at the normal 37°C remained constant for many hours.
Measurements of ATP, glutathione, and intracellular [Ca2+].
ATP levels were measured using the CellTiter-Glo luminescent assay (Promega, Madison, WI), according to the manufacturer's protocol. Glutathione levels were measured in intact cells using monochlorobimane (50 μM), a cell-permeant, nonfluorescent compound that, via a reaction catalyzed by glutathione S-transferases, forms a fluorescent glutathione-monochlorobimane adduct that can be measured fluorometrically (excitation 355 nm/emission 460 nm; e.g., Chatterjee et al. 1999). Initial fluorescence was subtracted from fluorescence read 30 min later using a plate reader.
Cytosolic free calcium ([Ca2+]i) was measured by using fura 2 loaded as the acetoxymethyl ester (30 min of incubation with 5 μM fura 2-AM). Pairs of images measured at different excitation wavelengths [340 and 380 nm, provided using a TILL Photonics monochromator (Victor, NY); emissions monitored at 515 ± 20 nm] were used to calculate the 340/380 emission ratio, which increases with [Ca2+]i. [Ca2+]i changes were calculated using the equation [Ca2+] = Kd(R − Rmin)/(Rmax − R) (Grynkiewicz et al. 1985), where Rmin is the 340/380 ratio when the cells are in zero-Ca2+ medium (no added Ca2+ with 2 mM BAPTA) and Rmax is the maximal ratio measured in normal medium after addition of 100 μM ionomycin. The ionomycin solution was prepared freshly from a DMSO stock because it becomes inactive after just a few hours in water.
Assays for cell survival and mitochondrial reducing ability.
Cell survival was measured with a live/dead imaging assay, described in White et al. (2003), using propidium iodide (PI; 15 μM) to label the nuclei of dead cells and Hoechst (bisbenzimide) 33342 (16 μM) to label the nuclei of all cells.
Cellular/mitochondrial reducing activity, an assay of cell viability, was measured using Alamar blue (Biosource International, Camarillo, CA). The fluorescence of this water-soluble, membrane-permeable indicator increases when resazurin is reduced to resorufin by mitochondrial respiratory chain activity (as well as other cellular reductase systems). Alamar blue was added at a 1:20 dilution of the commercial stock solution, and its fluorescence was measured using a plate reader (excitation 535 nm/emission 590 nm) before and after 2 h of incubation at 37°C.
Measurement of mitochondrial movement.
Movement of Mitotracker green-labeled mitochondria (20 min of incubation in 5 μM) along neurites was measured using a spinning disk confocal (Yokogawa CSU10; Solamere Technology Group, Salt Lake City, UT) equipped with a Cascade 512B CCD camera (Roper Scientific; excitation 488 nm, emission 535 nm). In most of these experiments, the heat stress was applied in an incubator and the culture dish was then transferred to the microscope stage for imaging at 37°C. Images were collected at 0.5 Hz with the use of a ×60, 1.45-NA Olympus objective; movement was detected by subtracting successive images. The average velocity of each mitochondrion was calculated over a 20-s interval.
Transfection.
Neuronal cultures (10–12 days in vitro) were transfected as described by Panickar et al. (2005), using plasmids containing cDNA for green fluorescent protein (pIRES-2EGFP; Clontech, Palo Alto, CA) and Bcl-xL (gift from Dr. Larry Boise; Boise et al. 1995). Stresses were applied 48 h after transfection.
Reagents.
The pan-caspase inhibitor quinolyl-valyl-O-methylaspartyl-(2,6-difluorophenoxy)methyl ketone (qVD-OPH) was obtained from Enzyme Systems/ICN (Livermore, CA). The calpain inhibitor PD150606 was obtained from Calbiochem/EMD Biosciences (La Jolla, CA). Fluorescent indicators, monochlorobimane, and the antioxidant N-tert-butyl-α-phenylnitrone (PBN) were obtained from Molecular Probes/Invitrogen (Eugene, OR). Other chemicals were obtained from Sigma (St. Louis, MO).
Statistics.
Averages are means ± SE. To compare multiple groups with a common control, data were analyzed using a one-way analysis of variance followed by Dunnett's test. The Student-Newman-Keuls test was used to compare all groups. Prism and InStat software (GraphPad Software, San Diego, CA) were used.
RESULTS
Heat stress induces two phases of mitochondrial depolarization.
Figure 1A shows that in striatal cultures, the heat stress (2 h, 43°C) partially depolarized ΔΨm, measured using TMRM fluorescence. After the return to 37°C, ΔΨm partially repolarized but subsequently began a second, slow phase of depolarization that was irreversible. Figure 2A shows TMRM fluorescence changes tracked by imaging at higher frequency in a representative neuron, demonstrating that the initial ΔΨm depolarization developed mainly during the second hour of the heat stress and occurred in both soma and proximal processes. Because the measurements in Figs. 1A and 2A were made in high-[K+] medium (145 mM K+ with low Ca2+, see materials and methods), one can be certain that the stress-induced loss of TMRM fluorescence was due to depolarization of ΔΨm rather than to a reduction in cytosolic [TMRM] resulting from depolarization of the plasma membrane potential.
Fig. 2.
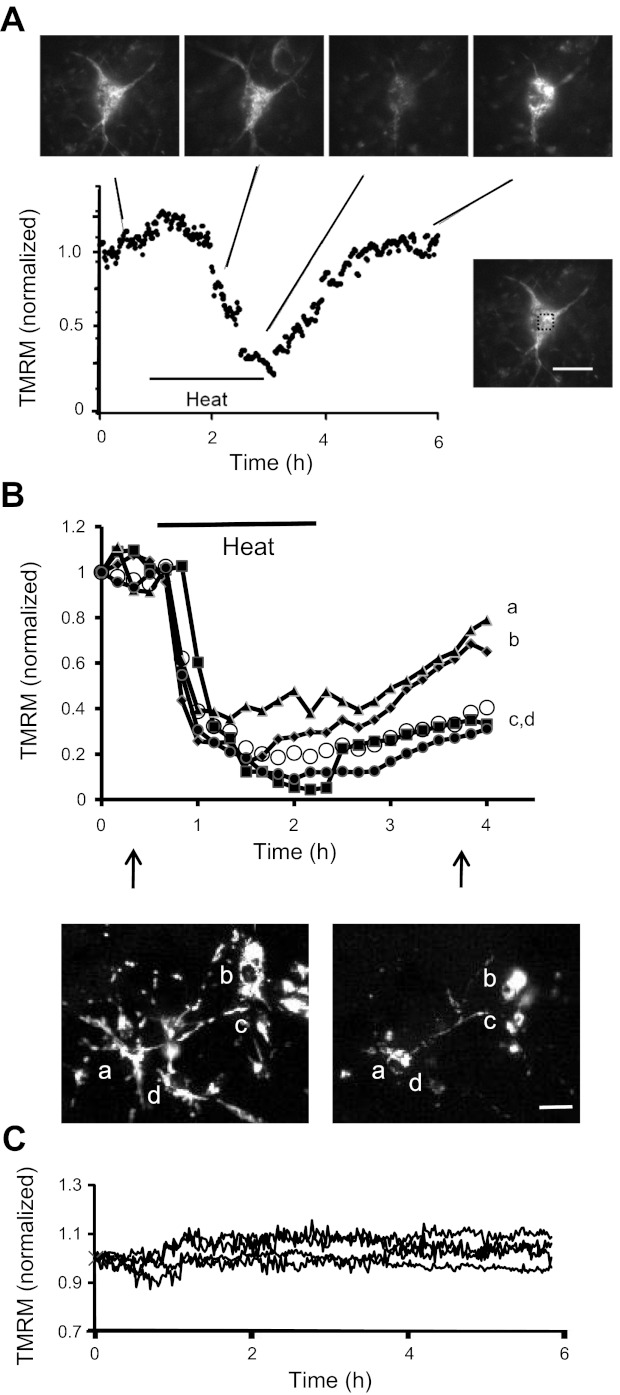
Time course of changes in TMRM fluorescence in individual neurons heat-stressed in high-[K+] (A), heat-stressed in normal [K+] (B), or nonstressed (C). A: TMRM fluorescence changes averaged over the soma of a representative neuron, demonstrating that ΔΨm depolarization developed with a delay during the heat stress and showed partial recovery after the stress. Micrographs show fluorescence images taken at the indicated times. Image at bottom right shows the location of the region whose fluorescence was plotted. B: changes in TMRM fluorescence in 4 neurons labeled a–d; vertical arrows indicate the timing of the 2 illustrated micrographs. Open circles plot the average fluorescence from 14 neurons in this culture (6 days in vitro); a similar pattern of fluorescence changes was observed in >10 other cultures. All cells exhibited a decrease in TMRM fluorescence during the heat stress and most exhibited a partial recovery of TMRM fluorescence after the end of the stress. The degree of depolarization and recovery showed considerable cell-to-cell variation. Magnification was lower than that in A to allow ΔΨm changes to be recorded simultaneously in several neurons in the same microscope field. C: maintenance of TMRM fluorescence in 4 neurons in a nonstressed sister culture maintained at 37°C throughout. Measurements in A and B were normalized to pre-stress controls. TMRM was present throughout (30 nM in A, 10 nM in B and C). Calibration bars: 20 μm in A, 100 μm in B.
The pattern of stress-induced changes in TMRM fluorescence was similar in experiments performed in normal culture medium, as shown by the plate reader assays in Fig. 1B. A similar pattern was also evident in the normal [K+] time-lapse TMRM imaging experiments in Fig. 2B. All cells showed stress-induced partial ΔΨm depolarization, and most showed some degree of (transient) recovery post-stress. However, there was considerable cell-to-cell variation in the magnitudes of the ΔΨm depolarization and recovery (compare single cell and averaged results in Fig. 2B). There appeared to be a tendency for more differentiated neurons like that in Fig. 2A to show more (transient) post-stress ΔΨm recovery than some of the less differentiated neurons in Fig. 2B, consistent with an apparent tendency for younger cultures (5–6 days in vitro) to exhibit more heat-induced damage than older cultures (13–14 days in vitro). Future studies are needed to investigate these possible age/differentiation-related differences more systematically. Figure 2C shows that TMRM fluorescence remained stable for many hours in nonstressed cultures.
Heat stress transiently increases, then irreversibly decreases, mitochondrial respiration.
Figure 3A plots changes in the rate of O2 consumption of striatal neurosphere cultures during the heat stress. During the first hour the respiration rate was increased, reaching peak levels ∼30% above pre-stress (control) levels by ∼30 min. This initial hyperthermia-induced increase in O2 consumption has also been noted in other cells (e.g., cardiac myocytes, Sammut et al. 2001; Sammut and Harrison 2003). Following this initial increase, O2 consumption fell, dropping to ∼50% of control by the end of the 2-h stress. Figure 3B shows that a similar pattern of heat stress-induced changes in O2 consumption occurred in cells that remained attached to substrate, measured using the flow-through technique described in materials and methods. The decrease in respiration at the end of the heat stress persisted for many hours following return to 37°C (Fig. 3C).
Fig. 3.
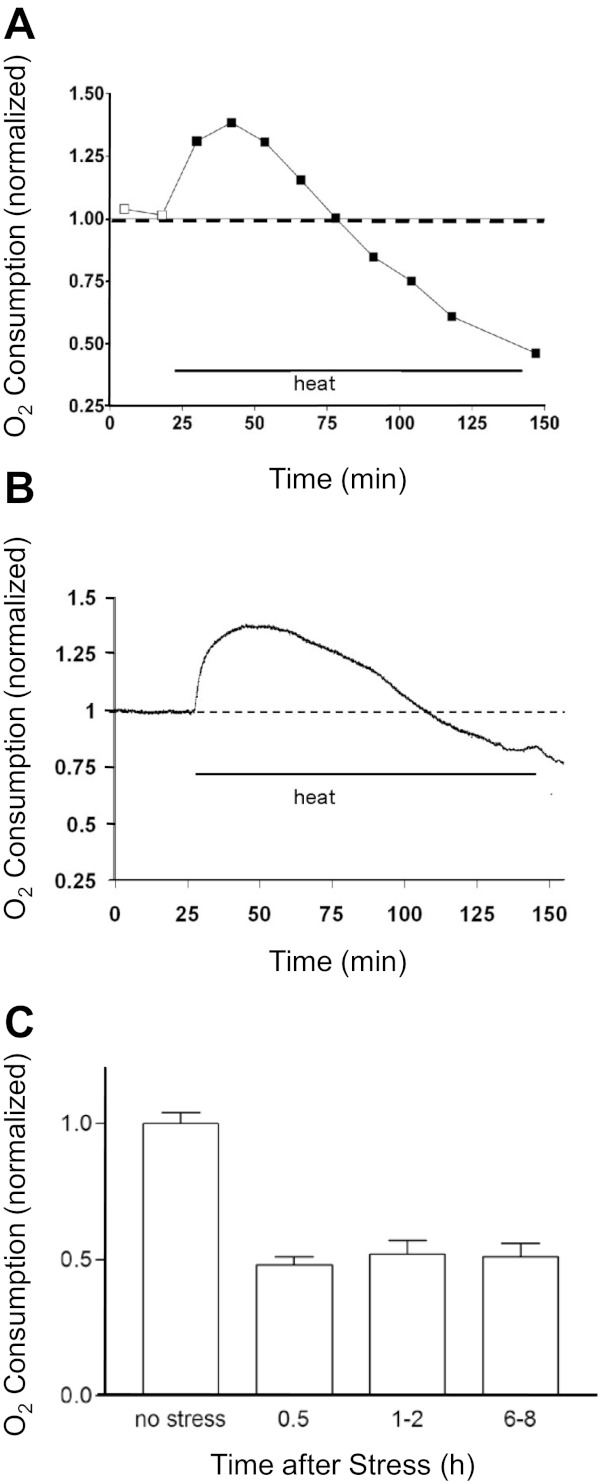
Heat stress first increases and then decreases O2 consumption. A–C: measurements of O2 consumption during the heat stress applied to stirred suspensions of neurospheres (A and C) or substrate-attached cultures (B). Data in A came from sister preparations analyzed after the indicated durations of heat exposure. Data in B reflect continuous measurements on the same culture, made using the flow-through O2 measurement system described in materials and methods. Data in C were obtained by exposing the cultures to the heat stress in a 43°C incubator and then returning them to 37°C for the indicated times before assaying O2 consumption with a Clarke electrode. At all the indicated times, post-stress O2 consumption was significantly less than that measured in nonstressed cultures (P < 0.001). All data were normalized to the O2 consumption measured before exposure to heat.
Hyperthermia reduces pyruvate/malate-dependent respiration.
This persisting decrease in O2 consumption suggests damage to the mitochondrial respiratory chain. The function of specific respiratory chain complexes was investigated by measuring O2 consumption in neurosphere cultures following sequential addition of substrates and inhibitors specific to particular complexes (see materials and methods). Figure 4 plots measurements made in nonstressed (left) and stressed (right) sister cultures (15–30 min post-stress). The only significant difference occurred in the pyruvate/malate solution, where stressed neurospheres consumed O2 at about one-half the rate measured in nonstressed neurospheres (52.1 ± 4.9% of control). The nearly normal respiration measured in the presence of succinate/G3P and in TMPD/ascorbate indicates that the function of complexes II, III, and IV was not significantly altered at this early post-stress time. Thus the main hyperthermia-induced dysfunction of the respiratory chain probably occurred at (or upstream of) complex I. The similar magnitudes (∼50%) of the heat stress-induced reduction in O2 consumption in pyruvate/malate in Fig. 4 and the reduction in overall O2 consumption in Fig. 3 suggest that the digitonin-induced permeabilization used in Fig. 4 did not by itself significantly alter mitochondrial function.
Fig. 4.
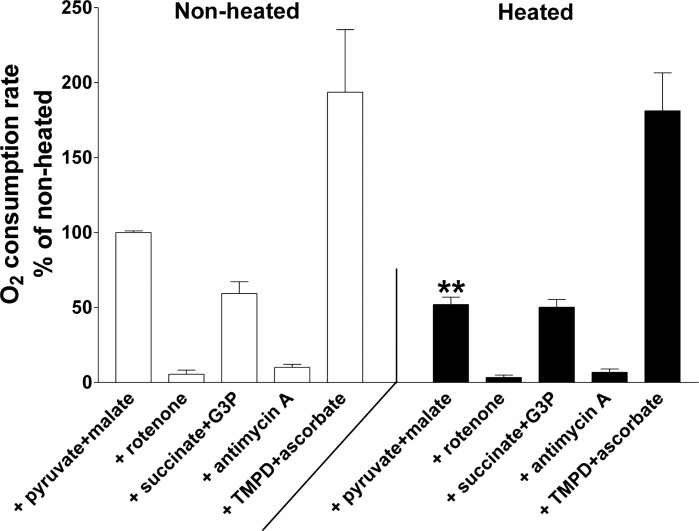
Measurements of O2 consumption in digitonin-permeabilized neurospheres. Measurements were made on sister nonheated (left) and heated (right) neurospheres beginning 15–30 min after heat stress termination, using the sequence and drug concentrations described in materials and methods. Pyruvate and malate were added to measure respiration driven by complex I (with 2 mM ADP to ensure state 3 respiration). Rotenone (1 μM) was added to inhibit complex I. Succinate and glycerol 3-phosphate (G3P) were added to measure respiration mediated via complexes II and III. Antimycin A inhibits complex III. Addition of substrate TMPD plus ascorbate measures respiration mediated by complex IV. All O2 consumption measurements were normalized to the O2 consumption in the presence of pyruvate + malate in the nonheated condition. The only significant difference was in pyruvate + malate-driven respiration, indicating damage to complex I (and/or reactions upstream of complex I). **P < 0.01, significant difference from nonheated cultures (2-tailed t-test).
Hyperthermia reduces mitochondrial movement and produces a delayed decrease in ATP.
Figure 5A shows that movement of Mitotracker green-labeled mitochondria along neurites was markedly slowed during the first hour following the heat stress (P < 0.001). Partial recovery of movement occurred ∼3 h after the end of the heat stress (not shown). ΔΨm depolarization produced by a proton carrier also causes cessation of mitochondrial movement (e.g., Bantseev and Sivak 2005).
Fig. 5.
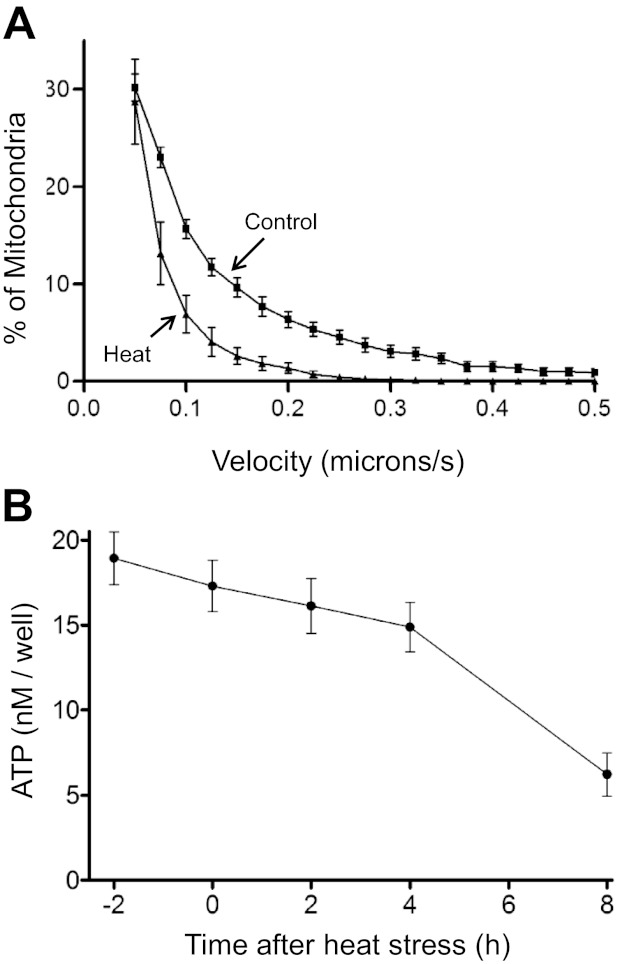
Hyperthermia slows mitochondrial movement (A) and produces a delayed decrease in cellular ATP (B). A: cumulative plots of the percentage of Mitotracker green-labeled mitochondria in neurites with velocities equal to or greater than the x-axis value in heat-stressed (triangles) and nonheated (squares) sister cultures. Measurements were made at 37°C, with >400 mitochondria assayed in each condition. Measurements in stressed cultures were made during the first hour post-stress. Differences between heated and nonheated cultures were significant at all movement velocities >0.1 μm/s. Similar results were documented in >10 additional experiments; post-stress recovery of movement was slow and incomplete. B: time course of hyperthermia-induced changes in ATP following the heat stress. Time 0 on the x-axis indicates the end of the heat stress, when the temperature was returned to 37°C. ATP was measured using the luminescence assay described in materials and methods. Each point represents the average of 6 substrate-attached sister cultures. Three additional experiments yielded similar results.
Figure 5B shows that total cellular ATP levels remained at or above 75% of nonstressed control levels for at least 4 h following the stress. Consistent with this temporary maintenance of ATP levels, some cells exhibited continued ruffling movements of lamellipodia membranes (not shown) even when mitochondrial movement was greatly decreased. These results suggest that the rapid reduction in mitochondrial movement following the heat stress was not caused by generalized depletion of ATP, although ATP depletion in localized microdomains cannot be ruled out. Perhaps these young neurons have sufficient glycolytic potential to maintain ATP levels even when mitochondria are partially depolarized.
Proton carrier-induced ΔΨm depolarization reduces cell viability.
To test whether the ΔΨm depolarization measured here (Figs. 1 and 2) is relevant to the delayed cell death measured after the hyperthermia stress (White et al. 2003), we tested whether ΔΨm depolarization produced by a different technique would also reduce cell viability. Figure 6 shows results of an experiment in which sister cultures were exposed to the proton carrier CCCP (20 μM) for 5–60 min and then assayed for Alamar blue reducing activity 2–4 days later. When CCCP was added to normal culture medium (open bars in Fig. 6), exposures longer than 20 min reduced viability assayed 4 days later. The 60-min CCCP exposure would be expected to produce a ΔΨm depolarization similar in duration to that produced by the heat stress, since the heat stress-induced ΔΨm depolarization did not become marked until the second hour of the stress (Fig. 2A). Because exposure to CCCP can also cause cytoplasmic acidification due to H+ influx driven by the plasma membrane potential (Tretter et al. 1998), we also tested the effects of CCCP exposure in medium containing high [K+] to depolarize the plasma membrane. Hatched bars in Fig. 6 show that a 60-min exposure to CCCP in high-[K+] medium reduced cell viability assayed 2 days later. These results indicate that a CCCP-imposed ΔΨm depolarization of about the same duration as that produced during the heat stress does indeed reduce cell viability assayed several days later, consistent with the hypothesis that ΔΨm depolarization (and/or associated downstream effects) contribute to hyperthermia-induced neuronal death. Similar results were obtained with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, 5 μM; not shown).
Fig. 6.
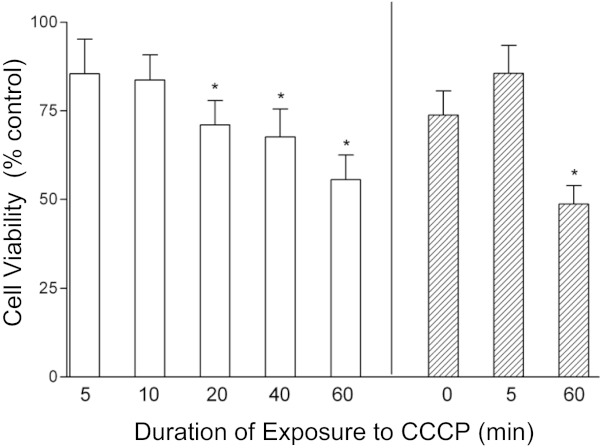
Exposure to a proton carrier (CCCP; 20 μM) reduces cell viability assayed 2–4 days later. Viability was measured as Alamar blue reducing activity (see materials and methods) normalized to values measured in sister cultures not exposed to CCCP or high [K+]. For results indicated by open bars, sister cultures were exposed for 5–60 min to CCCP in normal medium and assayed 4 days later. For results represented by hatched bars, sister cultures were all exposed to a high-[K+] medium for 60 min (in mM: 145 KCl, 20 NaHCO3, 5 glucose, 80 sucrose, and 1 BAPTA), during which some cultures were also exposed to CCCP for the indicated times. Cultures were washed thoroughly with normal medium at the end of the CCCP and/or high-[K+] exposure and assayed 2 days later. *P < 0.05, significant difference from sister cultures not exposed to CCCP or high [K+] (Dunnett's test for multiple comparisons). The illustrated findings were confirmed in 2 additional experiments.
Early hyperthermia-induced ΔΨm depolarization is not mediated by increased ATP consumption or caspase activation.
In an effort to determine the major mechanisms underlying heat stress-induced cell damage, we tested whether inhibitors of various mitochondria-associated functions might alter stress-induced changes in ΔΨm.
Oligomycin blocks ATP synthesis by inhibiting the F1,F0-ATP synthetase. The experiment represented in Fig. 7A tested oligomycin's effect on hyperthermia-induced ΔΨm changes. Except for the point at 1.5 h post-stress, the pattern of ΔΨm changes was similar in the presence or absence of oligomycin. We also tested the effect of oligomycin on O2 consumption, because one possible cause of the initial hyperthermia-induced increase in O2 consumption (Fig. 3) is acceleration of ATP consumption. If this were true, then most of the O2 consumption during the heat stress should be ATP-dependent, and thus sensitive to oligomycin. However, Fig. 7B shows that oligomycin did not block the large, early hyperthermia-induced increase in O2 consumption, and Fig. 7C shows that immediately following the heat stress, O2 consumption became more insensitive to oligomycin compared with nonstressed cultures. These findings, together with the finding that ATP levels were not immediately depleted following the heat stress (Fig. 5B), suggest that changes in ΔΨm and O2 consumption during the heat stress were not linked solely to changes in ATP consumption. Another possible cause of the transient increase in respiration during the heat stress is an increase in leak current across the inner mitochondrial membrane (i.e., uncoupling, see discussion). By 70–80 min post-stress, the percentage of oligomycin-insensitive respiration was no longer significantly elevated, suggesting that any hyperthermia-induced increase in leak current was at least partially reversible.
A possible cause of increased mitochondrial leak as well as delayed cell death is opening of the mPTP (e.g., Qian et al. 2004; Yasuda et al. 2006). Data summarized in Table 1 show that an inhibitor of mPTP opening, cyclosporin A (CsA, 1 μM), did not reduce ΔΨm depolarization during or following the heat stress. This finding does not completely rule out a role for mPTP opening, however, because for central nervous system mitochondria CsA may not be sufficient to block mPTP opening by severe stresses (Brustovetsky and Dubinsky 2000).
Table 1.
Inhibition of mPTP opening (cyclosporin A) or caspase activation (qVD-OPH) does not prevent ΔΨm depolarization following heat stress
| TMRM Fluorescence, % no-stress control |
||
|---|---|---|
| Treatment | Cyclosporin A | No drug |
| No stress | 83.8 ± 6.3 | 100 ± 7.5 |
| Time post-stress, h | ||
| 0.2 | 55.3 ± 2.6 | 56.8 ± 1.44 |
| 6.0 | 59.4 ± 3.2 | 70.6 ± 4.0 |
| 12.0 | 30.8 ± 2.9 | 32.6 ± 2.3 |
| 24.0 | 31.1 ± 2.3 | 39.2 ± 2.6 |
| TMRM Fluorescence, % no-stress control |
||
|---|---|---|
| Treatment | qVD-OPH | No drug |
| No stress | 88.0 ± 6.4 | 100 ± 3.9 |
| Time post-stress, h | ||
| 1.5 | 83.0 ± 2.3 | 84.2 ± 2.7 |
| 4.5 | 41.3 ± 2.3 | 34.8 ± 2.47 |
| 7.0 | 49.9 ± 4.8 | 41.6 ± 3.92 |
| 24.0 | 21.5 ± 4.6 | 29.7 ± 8.7 |
Platereader measurements of TMRM fluorescence in sister cultures in the presence or absence of the indicated drug. N = 12–18 culture wells. 1 μM cyclosporin A or 20 μM qVD-OPH was added 30 min before the stress; cyclosporin A was also supplemented 2 h before each post-stress measurement. Cyclosporin A findings were confirmed in 3 additional experiments; qVD-OPH findings were confirmed in 2 additional experiments.
Table 1 also shows that the pan-caspase inhibitor qVD-OPH (20 μM), which delays neuronal death following this heat stress (White et al. 2003), also did not significantly alter the post-stress time course of ΔΨm depolarization. Thus the early hyperthermia-induced changes in ΔΨm studied here appear to be independent of and upstream of caspase activation, consistent with the finding that caspase-3 activation was first detected 10 h following this heat stress (White et al. 2003).
Oxidative damage and Ca2+ may contribute to hyperthermia-induced neuronal death.
Heat stress can increase the levels of reactive oxygen species (ROS; e.g., Venkataraman et al. 2004), and oxidative damage depletes NAD(H) in stressed mitochondria (Di Lisa and Ziegler 2001; Du et al. 2003). An attempt to minimize this depletion by pretreatment with 1–10 mM NAD+ beginning 24 h before the heat stress significantly increased TMRM fluorescence assayed 6.5 h post-stress, and pretreatment with 5 mM NAD+ produced a modest but significant increase in cell survival (Table 2). NAD+ had no significant effect on TMRM fluorescence or cell survival in nonstressed cultures. Stress-protective effects of applied NAD+ or nicotinamide have also been reported in other neurons (e.g., transected dorsal root ganglion axons, Conforti et al. 2007; Wang et al. 2005; cultures stressed with glutamate and in vivo brain ischemia models, Liu et al. 2009).
Table 2.
Effects of NAD+ and γGC on post-stress survival and/or TMRM fluorescence
| Cell survival, % |
||||
|---|---|---|---|---|
| Treatment | No stress | No stress + drug | Stress alone | Stress + drug |
| γGC (500 μM) | 93.7 ± 0.4 (12) | 93 ± 0.6 (12) | 32 ± 0.7 (24) | 30.3 ± 0.8 (12) |
| NAD+ (5 mM) | 85.9 ± 0.8 (23) | 81.3 ± 1 (12) | 40.9 ± 0.8 (23) | 50.5 ± 1† (12) |
| TMRM Fluorescence, % no-stress control |
|||
|---|---|---|---|
| [NAD+] | No stress + NAD+ | Stress alone | Stress + NAD+ |
| 1 mM | 96 ± 3 (12) | 13 ± 1 (12) | 45 ± 2* (12) |
| 10 mM | 94 ± 3 (12) | 13 ± 1(12) | 40 ± 4* (12) |
Values are means ± SE, with number of culture wells shown in parentheses. Cell survival was assayed 50 h post-stress for γ-glutamyl cysteine (γGC) and 40 h poststress for NAD+, using the live/dead assay described in materials and methods. TMRM fluorescence was assayed 6.5 h after stress termination. NAD+ was applied 24 h before heat stress.
P < 0.001;
P < 0.05 compared with stress alone (sister cultures, 1-way ANOVA with Dunnett's multiple comparison test). Similar results were obtained in 2 additional experiments.
Oxidative damage can also deplete glutathione. The heat stress decreased glutathione levels in neuronal cultures to 50 ± 2% of pre-stress control levels 6 h post-stress and 48 ± 2% 18 h post-stress, suggesting a persisting depletion of endogenous ROS defenses (measured by monochlorobimane fluorescence, see materials and methods, n = 12 cultures each). γ-Glutamyl cysteine (γGC; 500 μM), a precursor for reduced glutathione, increased glutathione concentrations for 48 h in both stressed and nonstressed cultures (not shown) but did not increase survival measured 2 days post-stress (Table 2; live/dead assay in materials and methods). White et al. (2007) present evidence that other antioxidants [500 μM PBN, 10 μM N-acetyl-cysteine, 100 μM Mn(III)tetrakis(4-benzoic acid)porphyrin chloride] were likewise ineffective at improving post-stress survival. Thus there is evidence for oxidative damage following the heat stress, but only one of the tested antioxidative treatments (bath-applied NAD+) significantly increased post-stress survival, and it is likely that NAD+ acted via a mechanism independent of oxidative stress (see discussion).
Imaging of cells loaded with the Ca2+ indicator fura 2 revealed that just after the end of the heat stress, the average fura 2 340/380 ratio was 1.28 ± 0.06 times that measured in nonstressed sister cultures, corresponding to a 20–50 nM increase in [Ca2+]i (n = 50 cells, P < 0.001). However, in each of 4 experiments, reduction of bath [Ca2+] to 0–100 μM failed to prevent or reduce the ΔΨm depolarization during the heat stress (see Figs. 1A and 2A for records in low-[Ca2+] medium).
Elevated [Ca2+]i can activate calpain proteases, some present in cytosol, others in mitochondria (e.g., Arrington et al. 2006). PD150606 inhibits a mitochondrial calpain (e.g., Mizukoshi et al. 2010) as well as cytosolic calpains. In 4 of 6 experiments, PD150606 (100 μM) produced significant reductions in the ΔΨm depolarization recorded 4–6 h post-stress. Table 3 summarizes an experiment in which TMRM fluorescence measured 5 h post-stress was twofold higher in cultures treated with PD150606 than in stressed controls. PD150606 also transiently increased post-stress neuronal survival (Table 3). These results suggest that increased [Ca2+]i and calpain activation may contribute to, but are not required for, hyperthermia-induced neuronal damage.
Table 3.
Effect of calpain inhibitor PD150606 (100 μM) on poststress TMRM fluorescence and survival
| TMRM Fluorescence 5 h post-stress, % no-stress control | |
|---|---|
| Stress alone | Stress + PD150606 pretreat |
| 12.5 ± 1.8 (12) | 24.9 ± 2.6* (12) |
| Cell survival, % | ||
|---|---|---|
| Stress alone | Stress + PD150606 pretreat | Stress + PD150606 stress onset |
| 26 ± 3 | 50 ± 3* | 52.5 ± 4* |
Values are means ± SE, with number of culture wells shown in parentheses. Pretreat indicates that PD150606 was present 2 h before, during, and after the stress; stress onset indicates drug presence during and after the stress. TMRM fluorescence was measured 5 h post-stress (high-[K+] medium throughout). Cell survival was measured 24 h post-stress (live/dead assay, see materials and methods).
P < 0.001 compared with cultures stressed without drug. The protection afforded by PD150606 did not extend to 48 h post-stress, when survival measured in both PD150606-treated and nontreated cultures was similar (18-21%), a result repeated in 2 additional experiments. Long-term exposure to this drug may itself be toxic, because in nonheated cultures a 48-h exposure to PD150606 reduced survival from 90 to 39.5% as measured by propidium iodide exclusion.
Transgenic expression of Bcl-xL delays post-stress neuronal degeneration.
If mitochondrial damage contributes importantly to the delayed neuronal death following the heat stress, then enhanced expression of a mitochondria-protective, antiapoptotic protein might be predicted to reduce post-stress neuronal death. Figure 8 shows results of an experiment in which striatal cultures were cotransfected to (over)express Bcl-xL and green fluorescent protein (GFP) by using techniques detailed in Panickar et al. (2005). Fluorescence micrographs (Fig. 8A) taken before and 42 h after the heat stress show increased survival and more intact processes in stressed, GFP-labeled neurons transfected with Bcl-xL than in stressed neurons expressing GFP alone. Figure 8B shows that expression of Bcl-xL increased the number of surviving neurons more than twofold when assayed 1 day post-stress. However, neurons continued to die; Bcl-xL transfection postponed, but did not prevent, hyperthermia-induced neuronal death.
Fig. 8.
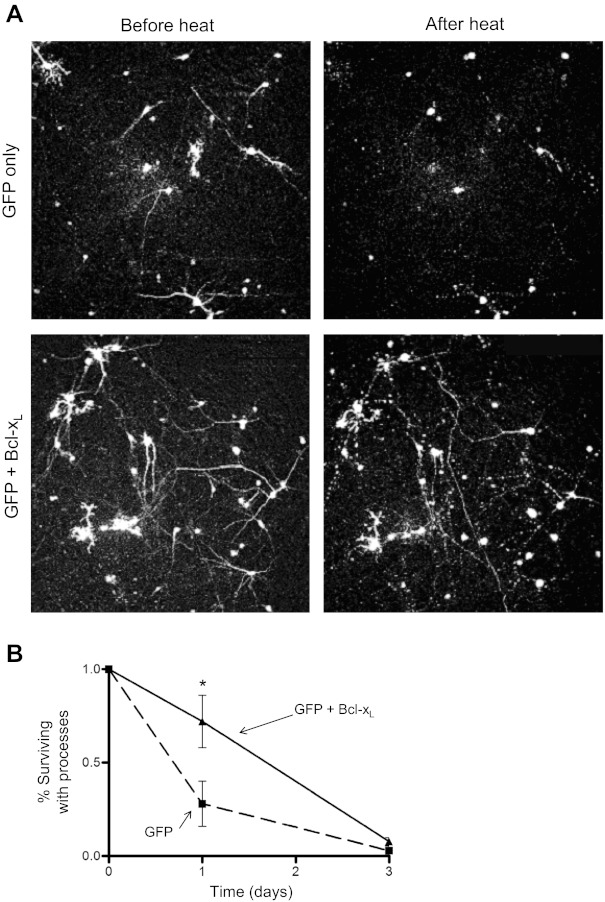
Transgenic expression of Bcl-xL delays neuronal death following heat stress. A: cultures were transfected to express green fluorescent protein (GFP) alone (top row) or GFP plus Bcl-xL (bottom row). For each group a representative microscope field is shown before (left) and 42 h after (right) the heat stress. B: comparison of post-stress neuronal survival in GFP-positive cells with or without Bcl-xL. Bcl-xL increased survival of GFP-positive neurons with processes on the first post-stress day (P < 0.05, n > 10 culture wells), but by the third post-stress day most GFP-labeled process-bearing cells in both groups had disappeared.
DISCUSSION
Results presented here show that striatal neurons exposed to a 2-h, 43°C heat stress exhibited mitochondrial dysfunction more than 10 h before the onset of caspase-3 activation and neuronal death described by White et al. (2003, 2007). This discussion considers possible mechanisms underlying these early mitochondrial changes.
Heat stress damages mitochondrial respiration and increases mitochondrial leak conductance.
During the first hour of the heat stress, O2 consumption was increased and ΔΨm depolarization was minimal. Heat-induced acceleration of metabolism probably contributes to this early increase in O2 consumption, but the data in Fig. 7B suggest that mitochondrial leak conductance also increases within 10–20 min of hyperthermia onset, which would also increase O2 consumption. A 1-h heat stress at 43°C produced no neuronal death (White et al. 2003), suggesting that any heat-induced damage during the first hour was reversible on return to normal temperature.
During the second hour of the heat stress, there was partial ΔΨm depolarization accompanied by a ∼50% decrease in O2 consumption. In nonstressed cells, ΔΨm depolarization is usually accompanied by an increase in mitochondrial respiration to maintain the electrochemical proton gradient across the mitochondrial membrane (e.g., Brand and Nicholls 2011). Our finding that the hyperthermia-induced partial depolarization of ΔΨm was associated with decreased O2 consumption suggests damage to mitochondrial respiratory complexes/enzymes, and indeed, measurements made immediately following stress termination disclosed dysfunction at and/or upstream of complex I (see Fig. 4). Hyperthermia has been hypothesized to inactivate complex I in a manner that can be reversed by NADH (Grivennikova et al. 2001). Our finding that pretreatment with NAD+ increased posthyperthermia ΔΨm and cell survival is consistent with this mechanism, assuming that some of the exogenously applied NAD+ was converted to NADH within the cells. Another possibility is that protection was afforded by NAD+ itself. NAD+ applied nasally (Ying et al. 2007) or intraperitoneally (Zheng et al. 2012) has been reported to reduce brain damage in mice in transient brain ischemia models. One possible mechanism for NAD+-mediated protection is that NAD+ acts as a substrate for sirtuin 3, which can increase complex I activity by deacetylating certain components of complex I (Ahn et al. 2008).
Inhibition of poly ADP-ribose polymerase (PARP) reduces organ damage after thermal injury (Avlan et al. 2005), and exogenous NAD(H) can protect neurons from stresses involving PARP activation (Alano et al. 2010). However, this mechanism seems unlikely, because a PARP inhibitor did not protect from this heat stress (White et al. 2007).
Other possible mechanisms underlying hyperthermia-induced dysfunction of mitochondrial respiration include depletion/oxidation of cardiolipin (a component of the mitochondrial inner membrane that enhances ETC function, Fry and Green 1981; Paradies et al. 2004) or damage to enzymes that would affect complex I function (e.g., yeast aconitase is sensitive to hyperthermia, Bender et al. 2011).
The ΔΨm depolarization that developed during the second hour of the heat stress may have been due in part to the decrease in respiration discussed above, but this is unlikely to be the sole mechanism, because on termination of the heat stress the ΔΨm depolarization was partially (albeit transiently) reversible, whereas the decrease in respiration was not. The increase in oligomycin-insensitive respiration measured during and immediately after the heat stress suggests that the reversible component of the ΔΨm depolarization was attributable to a hyperthermia-induced increase in leak current across the mitochondrial inner membrane (uncoupling). This hypothesized increased leak was probably not due solely to high-conductance openings of the mPTP, because stress-induced changes in ΔΨm were not reduced by CsA. “Recycling” of Ca2+ and Na+ across the mitochondrial membrane can cause an apparent increase in mitochondrial leak (reviewed by Castaldo et al. 2009), but in this heat stress neither Ca2+ nor Na+ was essential for the ΔΨm depolarization during the heat stress (Figs. 1A and 2A). The increased leak conductance is more likely related to the reversible, hyperthermia-induced increase in proton conductance reported in isolated mitochondria (e.g., Willis et al. 2000; Zukiene et al. 2010).
The hyperthermia-induced ΔΨm depolarization likely contributes to the eventual neuronal death, because a proton carrier-induced ΔΨm depolarization of comparable duration also reduced cell viability. CCCP-induced cell death in Jurkat and FL5.12 cells is also preceded by signs of mitochondrial damage and apoptosis (de Graaf et al. 2004). However, ΔΨm depolarization-induced death is likely to involve additional susceptibility factors, since some cells are not readily killed by CCCP (e.g., osteosarcoma cells, Lim et al. 2001).
Slow, post-stress ΔΨm depolarization precedes apoptotic death.
Immediately following termination of the heat stress, there was a partial recovery of ΔΨm. Because this post-stress repolarization occurred in the absence of an increase in respiration (suggesting persisting mitochondrial damage), this transient recovery of ΔΨm was likely due to a decrease in the mitochondrial leak conductance.
A slowly developing, irreversible depolarization of ΔΨm followed this brief repolarization phase. The causes of this slow depolarization are not yet resolved, but our findings rule out some mechanisms. For example, the findings that this ΔΨm depolarization was not significantly altered by CsA or by a pan-caspase inhibitor (qVD-OPH) that delays post-stress neuronal death suggest that the depolarization occurs independently of mPTP opening or caspase activation. Milleron and Bratton (2006) reported that a different pan-caspase inhibitor [Z-Val-Ala-Asp(OMe)-fluoromethylketone (zVAD-fmk)] reduced the ΔΨm depolarization in Jurkat T cells stressed by a 2-h exposure to 45°C. Because the early post-stress depolarization was not accompanied by activation of any identified caspase, they suggested that heat stress activates an as-yet unidentified zVAD-fmk-inhibited protease. A candidate for this upstream protease is a calpain-like enzyme, since μ-calpain is inhibited by zVAD-fmk (Bizat et al. 2005). Consistent with this possibility, we found that a calpain inhibitor (PD150606) reduced the ΔΨm depolarization measured 5 h after the heat stress and temporarily prolonged post-stress neuronal survival. The actual heat-sensitive target(s) of this inhibitor remain(s) to be resolved, but a mitochondrial calpain could contribute both to mitochondrial damage and indirectly to the later caspase activation. One possibility is that calpain-mediated cleavage of Bax could contribute to mitochondrial damage, as found for damage to oligodendrocytes induced by excitotoxicity (Sánchez-Gómez et al. 2011).
The slow ΔΨm depolarization began before the onset of marked ATP depletion, suggesting that this late ΔΨm depolarization was not initiated by ATP depletion. However, the eventual ATP depletion, combined with reduced glutathione and reduced respiration, would make mitochondria more susceptible to further damage, perhaps tilting the balance between pro- and antiapoptotic mechanisms. Pagliari et al. (2005) found that heat-stressed mitochondria showed Bax oligomerization that was enhanced by proapoptotic proteins and reduced by the antiapoptotic protein Bcl-2. We found that transgenic expression of the antiapoptotic protein Bcl-xL increased survival measured 1–3 days following the heat stress but only delayed (rather than prevented) the eventual neuronal death, similar to the temporary protection afforded by caspase inhibition with qVD-OPH (White et al. 2003). Perhaps cellular damage from the heat stress triggers apoptotic pathways, but stress-induced damage to essential cellular components eventually results in death even when apoptotic mechanisms are inhibited.
In summary, measurements of mitochondrial function in cultured central neurons demonstrate ΔΨm depolarization and reduction in respiration both during and after a 2-h, 43°C heat stress. An early, rapid phase of ΔΨm depolarization, likely due to increased leak conductance, is partially reversible on return to normal temperature, but a later slow phase of post-stress ΔΨm depolarization is irreversible. Mitochondrial damage is likely to be an upstream event that helps bring about the delayed neuronal death that follows this heat stress.
GRANTS
This work was supported by National Institutes of Health Grants R01 NS 12207, R01 EY 010804, R01 NS 12404, and R01 AG036871, the South Florida Chapter of the American Heart Association, the Muscular Dystrophy Association, and the University of Miami. M. G. White was supported by NIH Training Grant 2T32 NS 07044 and was a Lois Pope Leaders In Furthering Education Fellow.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.G.W., O.S., D.N., E.F.B., C.T.M., and J.N.B. conception and design of research; M.G.W., O.S., D.N., and J.N.B. performed experiments; M.G.W., O.S., D.N., and J.N.B. analyzed data; M.G.W., O.S., D.N., E.F.B., C.T.M., and J.N.B. interpreted results of experiments; M.G.W., O.S., D.N., E.F.B., and J.N.B. prepared figures; M.G.W., E.F.B., and J.N.B. drafted manuscript; M.G.W., E.F.B., and J.N.B. edited and revised manuscript; M.G.W., E.F.B., C.T.M., and J.N.B. approved final version of manuscript.
REFERENCES
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105: 14447–14452, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30: 2967–2978, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol 291: C1159–C1171, 2006 [DOI] [PubMed] [Google Scholar]
- Avlan D, Unlü A, Ayaz L, Camdeviren H, Nayci A, Aksöyek S. Poly(ADP-ribose) synthetase inhibition reduces oxidative and nitrosative organ damage after thermal injury. Pediatr Surg Int 21: 449–455, 2005 [DOI] [PubMed] [Google Scholar]
- Bantseev V, Sivak JG. Confocal laser scanning microscopy of dynamic TMRE movement in the mitochondria of epithelial and superficial cortical fiber cells of bovine lenses. Mol Vis 11: 518–523 2005 [PubMed] [Google Scholar]
- Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods 26: 307–316, 2002 [DOI] [PubMed] [Google Scholar]
- Bender T, Lewrenz I, Franken S, Baitzel C, Voos W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell 22: 541–554, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffman SN, Hantraye P, Blum D, Brouillet E. Neuroprotective effect of zVAD against the neurotoxin 3-nitroproprionic acid involves inhibition of calpain. Neuropharmacology 49: 695–702, 2005 [DOI] [PubMed] [Google Scholar]
- Boise LH, Mime AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xl. Immunity 3: 87–98, 1995 [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PL, Kiyatkin EA. Brain hyperthermia induced by MDMA (ecstasy): modulation by environmental conditions. Eur J Neurosci 20: 51–58, 2004 [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci 20: 8229–8237, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaldo P, Cataldi M, Magi S, Lariccia V, Arcangeli S, Amoroso S. Role of the mitochondrial sodium/calcium exchanger in neuronal physiology and in the pathogenesis of neurological diseases. Prog Neurobiol 87: 58–79, 2009 [DOI] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Marrugat J, Noya M. Timing for fever-related brain damage in acute ischemic stroke. Stroke 29: 2455–2460, 1998 [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Noack H, Possel H, Keilhoff G, Wolf G. Glutathione levels in primary glial cultures: monochlorobimane provides evidence of cell type-specific distribution. Glia 27: 152–161, 1999 [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD+ and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ 14: 116–127, 2007 [DOI] [PubMed] [Google Scholar]
- De Graaf AO, van den Heuvel LP, Dijkman HB, de Abreu RA, Birkenkamp KU, de Witte T, van der Reijden BA, Smeitink JA, Jansen JH. Bcl-2 prevents loss of mitochondria in CCCP-induced apoptosis. Exp Cell Res 299: 533–540, 2004 [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD. FEBS Lett 492: 4–8, 2001 [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SB, Graham SH, Carcillo JA, Szabó C, Clark RSB. Intra-mitochondrial poly (ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem 278: 18426–18433, 2003 [DOI] [PubMed] [Google Scholar]
- Fry M, Green DE. Cardiolipin requirement for electron transport in complex I and III of the mitochondrial respiratory chain. J Biol Chem 256: 1874–1880, 1981 [PubMed] [Google Scholar]
- Grivennikova VG, Kapustin AN, Vinogradov AD. Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. Evidence for the slow active/inactive transition. J Biol Chem 276: 9038–9044, 2001 [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 1985 [PubMed] [Google Scholar]
- Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem 279: 32989–33000, 2004 [DOI] [PubMed] [Google Scholar]
- Jones PA, Andrews PJ, Midgley S, Anderson SI, Piper IR, Tocher JL, Housley AM, Corrie JA, Slattery J, Dearden NM, Miller JD. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesthesiol 6: 4–14, 1994 [PubMed] [Google Scholar]
- Kawamoto JC, Barrett JN. Cryopreservation of primary neurons for tissue culture. Brain Res 384: 84–93, 1986 [DOI] [PubMed] [Google Scholar]
- Lim ML, Minamikawa T, Nagley P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Lett 503: 69–74, 2001 [DOI] [PubMed] [Google Scholar]
- Liu L, Xing D, Chen WR. Micro-calpain regulates caspase-dependent and apoptosis inducing factor-mediated caspase-independent apoptotic pathways in cisplatin-induced apoptosis. Int J Cancer 125: 2757–2766, 2009 [DOI] [PubMed] [Google Scholar]
- Milleron RS, Bratton SB. Heat shock induces apoptosis independently of any known initiator caspase-activating complex. J Biol Chem 281: 16991–17000, 2006 [DOI] [PubMed] [Google Scholar]
- Mirkes PE, Little SA. Cytochrome c release from mitochondria of early postimplantation murine embryos exposed to 4-hydroperoxycyclophosphamide, heat shock, and staurosporine. Toxicol Appl Pharmacol 162: 197–206, 2000 [DOI] [PubMed] [Google Scholar]
- Mizukoshi S, Nakazawa M, Sato K, Osaki T, Metoki T, Ishiguro S. Activation of mitochondrial calpain and release of apoptosis-inducing factor from mitochondria in RCS rat retinal degeneration. Exp Eye Res 91: 353–36, 2010 [DOI] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Kaplan P, Barrett JN. Bone morphogenetic proteins (BMP6 and BMP7) enhance the protective effect of neurotrophins on cultured septal cholinergic neurons during hypoglycemia. J Neurochem 76: 1–10, 2001 [DOI] [PubMed] [Google Scholar]
- Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, Green DR. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA 102: 17975–17980, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panickar KS, Nonner D, Barrett JN. Overexpression of Bcl-xl protects septal neurons from prolonged hypoglycemia and from acute ischemia-like stress. Neuroscience 135: 73–80, 2005 [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94: 53–59, 2004 [DOI] [PubMed] [Google Scholar]
- Qian L, Song X, Ren H, Gong J, Cheng S. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones 9: 281–293, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammut IA, Harrison JC. Cardiac mitochondrial complex activity is enhanced by heat shock proteins. Clin Exp Pharmacol Physiol 30: 110–115, 2003 [DOI] [PubMed] [Google Scholar]
- Sammut IA, Jayakumar J, Latif N, Rothery S, Severs NJ, Smolenski RT, Bates TE, Yacoub MH. Heat stress contributes to the enhancement of cardiac mitochondrial complex activity. Am J Pathol 158: 1821–1831, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Alberdi E, Pérez-Navarro E, Alberch J, Matute C. Bax and calpain mediate excitotoxic oligodendrocyte death induced by activation of both AMPA and kainate receptors. J Neurosci 31: 2996–3006, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HB. Hyperthermia. N Engl J Med 329: 483–487, 1993 [DOI] [PubMed] [Google Scholar]
- Song XL, Qian LJ, Li FZ. Injury of heat stress on rat cardiomyocytes. Chin J Appl Physiol 16: 227–230, 2000 [Google Scholar]
- Tretter L, Chinopoulos C, Adam-Vizi V. Plasma membrane depolarization and disturbed Na+ homeostasis induced by the protonophore carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazon in isolated nerve terminals. Mol Pharmacol 53: 734–741, 1998 [DOI] [PubMed] [Google Scholar]
- Uney JB, Kew JN, Staley K, Tyers P, Sofroniew MV. Transfection-mediated expression of human hsp70i protects rat dorsal root ganglion neurones and glia from severe heat stress. FEBS Lett 334: 313–316, 1993 [DOI] [PubMed] [Google Scholar]
- Venkataraman S, Wagner BA, Jiang X, Wang HP, Schafer FQ, Ritchie JM, Patrick BC, Oberley LW, Buettner GR. Overexpression of manganese superoxide dismutase promotes the survival of prostate cancer cells exposed to hyperthermia. Free Radic Res 38: 1119–1132, 2004 [DOI] [PubMed] [Google Scholar]
- Vogel P, Dux E, Wiessner C. Evidence of apoptosis in primary neuronal cultures after heat shock. Brain Res 764: 205–213, 1997 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 170: 349–355, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MG, Emery M, Nonner D, Barrett JN. Caspase activation contributes to delayed death of heat-stressed striatal neurons. J Neurochem 87: 958–968, 2003 [DOI] [PubMed] [Google Scholar]
- White MG, Hammond RR, Sanders VJ, Bonaroti EA, Mehta AP, Wang G, Wiley CA, Achim CL. Neuron-enriched second trimester human cultures: growth factor response and in vivo graft survival. Cell Transplant 8: 59–73, 1999 [DOI] [PubMed] [Google Scholar]
- White MG, Luca LE, Nonner D, Saleh O, Hu B, Barrett EF, Barrett JN. Cellular mechanisms of neuronal damage from hyperthermia. Prog Brain Res 162: 347–371, 2007 [DOI] [PubMed] [Google Scholar]
- Willis WT, Jackman MR, Bizeau ME, Pagliassotti MJ, Hazel JR. Hyperthermia impairs liver mitochondrial function in vitro. Am J Physiol Regul Integr Comp Physiol 278: R1240–R1246, 2000 [DOI] [PubMed] [Google Scholar]
- Yan J, Barrett JN. Purification from bovine serum of a survival-promoting factor for cultured central neurons, and its identification as selenoprotein-P. J Neurosci 18: 8682–8691, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda O, Fukuo K, Sun X, Nishitani M, Yotsui T, Higuchi M, Suzuki T, Rakugi Smithies O, Maeda N, Ogihara T. Apop-1, a novel protein inducing cyclophilin D-dependent but Bax/Bak-related channel-independent apoptosis. J Biol Chem 281: 23899–23907, 2006 [DOI] [PubMed] [Google Scholar]
- Ying W, Wei G, Wang D, Wang Q, Tang X, Shi J, Zhang P, Lu H. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci 2: 2728–2734, 2007 [DOI] [PubMed] [Google Scholar]
- Zheng C, Han J, Xia W, Shi S, Liu J, Ying W. NAD+ administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett 512: 67–71, 2012 [DOI] [PubMed] [Google Scholar]
- Zukiene R, Nauciene Z, Ciapaite J, Mildaziene V. Acute temperature resistance threshold in heart mitochondria: febrile temperature activates function but exceeding it collapses the membrane barrier. Int J Hyperthermia 26: 56–66, 2010 [DOI] [PubMed] [Google Scholar]