

Abstract
Eight serotypes of human astroviruses (the classic human astroviruses) are causative agents of diarrhea. Recently, five additional astroviruses belonging to two distinct clades have been described in human stool, including astroviruses MLB1, MLB2, VA1, VA2 and VA3. We report the discovery in human stool of two novel astroviruses, astroviruses MLB3 and VA4. The complete genomes of these two viruses and the previously described astroviruses VA2 and VA3 were sequenced, affording 7 complete genomes from the MLB and VA clades for comparative analysis to the classic human astroviruses. Comparison of the genetic distance, number of synonymous mutations per synonymous site (dS), number of non-synonymous mutations per non-synonymous site (dN) and the dN/dS ratio in the protease, polymerase and capsid of the classic human, MLB and VA clades suggests that the protease and polymerase of the classic human astroviruses are under distinct selective pressure.
Keywords: Astrovirus discovery, diarrhea, nonstructural genes, constrained evolution
Introduction
Astroviruses are well-established causative agents of gastroenteritis in many mammalian and avian hosts. The first human astrovirus was identified in 1975 (Madeley and Cosgrove, 1975). Since then, 8 serotypes of human astrovirus (referred to hereafter as “classic human astroviruses”) have been identified and characterized (Kjeldsberg, 1994; Sakamoto et al., 2000) which together account for about 10% of sporadic diarrhea cases (Kirkwood et al., 2005; Soares et al., 2008). Very recently, two other phylogenetic clades of astroviruses have been identified in human stool. The first clade contains MLB1 and MLB2, which were initially identified in pediatric stool specimens from Australia (Finkbeiner et al., 2008) and India (Finkbeiner, 2009a), respectively. The second clade contains VA1, VA2 and VA3 (also known as HMO-A, HMO-B and HMO-C). VA1 was first identified in an unexplained outbreak of gastroenteritis (Finkbeiner et al., 2009b). VA2 and VA3 were identified in a cohort of children with diarrhea in India (Finkbeiner, 2009a); HMO-A, B, and C were simultaneously described in stools from Nigeria, Pakistan and Nepal (Kapoor et al., 2009). More recently, a virus very similar to astrovirus VA1/HMO-C was detected in brain tissue from an immunocompromised child with encephalitis (Quan et al., 2010). The discoveries of these viruses provide not only novel candidate agents of human disease, but also enable comparative genomic analyses that may shed insight into the evolutionary history and functional constraints of astroviruses.
Astroviruses are positive sense RNA viruses that typically range in size from 6.1–7.9 kb (Mendez and Arias, 2007). The genome contains two nonstructural genes, ORF1a and 1b, and a capsid gene, ORF2, as well as short 5′ and 3′ UTRs. The ORF1a of astroviruses contains a serine protease with a highly conserved catalytic amino acid triad (His, Asp, Ser). The ORF1b, which is translated via a ribosomal frameshifting mechanism, encodes a RNA dependent RNA polymerase (RdRp) with a conserved catalytic domain (Gly, Asp, Asp) (Steitz, 1999) that is responsible for replication and transcription of the viral RNA. Finally the astrovirus ORF2 encodes the viral capsid protein which is translated from a subgenomic RNA. The subgenomic RNA is transcribed from a highly conserved promoter sequence AUUUGGAGNGGNGGACCNAAN5-8AUGNC (the ORF2 start codon is italicized) (Mendez and Arias, 2007). In addition, in many astroviruses, a conserved stem loop structure of unknown function, referred to as stem-loop II (s2m) is present in the 3′UTR (Monceyron et al., 1997).
Here we describe the discovery and complete genome sequencing of two additional novel astroviruses during efforts to define the prevalence of the recently discovered astroviruses by consensus RT-PCR. Furthermore, the complete genomes of the previously described VA2 and VA3 were also sequenced, providing a total of four VA-clade genomes and three MLB-clade genomes for comparative analysis between and within clades. Using these genomic sequences and analyses of genetic distance and synonymous and non-synonymous mutations, we determined that the nonstructural genes of the classic human astrovirus clade are under evolutionary pressure that restricts their mutation rate compared to the corresponding regions in viruses in the newly described VA and MLB clades.
Material and methods
Stool samples from a cohort of Indian children
A previously described cohort (Holtz et al., 2011a) of 400 specimens collected from children experiencing acute diarrhea and 400 stool specimens collected from the same children while asymptomatic was analyzed using an astrovirus consensus RT-PCR assay (Finkbeiner et al., 2009). The 400 diarrhea samples were negative for the following enteric pathogens: rotavirus by ELISA and PCR; norovirus using PCR; bacterial pathogens (Vibrio cholerae, enteropathogenic Escherichia coli, Salmonella, Shigella, Aeromonas and Plesiomonas) by culture, biochemical reactions and serogrouping where appropriate; and parasitic pathogens using routine saline and iodine preparations and modified acid fast stain (Ajjampur et al., 2008). The 400 asymptomatic stool samples were screened for rotaviruses by ELISA.
Stool samples from a cohort of Nepalese children
Stool specimens were collected from children age 3 months to 5 years with acute diarrhea who were treated at a hospital in Nepal. All the stool specimens were tested using microscopy for ova and parasite; standard microbiology methods for common bacterial enteric pathogens (Salmonella, Shigella, Vibrio, Aeromonas, Plesiomonas) and commercially available EIA assays for Giardia, Cryptosporidium (ProSpecT, Oxoid, UK), rotavirus, astrovirus and adenovirus (Ridascreen, R-Biopharm, Darmstadt, Germany); diarrheagenic E. coli (EIEC, EPEC, ETEC, EAgg, STEC) using DNA hybridization; norovirus using RT-PCR. 196 stool specimens without any identifiable pathogen were screened for astrovirus using the same consensus RT-PCR as in the Indian cohort.
Screening of astroviruses from stool samples
For consensus RT-PCR screening of the Indian cohort, total nucleic acids were extracted from 200 μl of a ~20% fecal suspension using the Boom method (Boom et al., 1990) and were then eluted with 40 μl water. For the Nepalese cohort, nucleic acids were extracted from 300 μl of a 10% stool suspension using silica particles (NucliSens, bioMérieux) based on Boom chemistry and were then eluted in 75 μl of elution buffer. For all samples, primers SF0073 (5′-GATTGGACTCGATTTGATGG-3′) and SF0076 (5′-CTGGCTTAACCCACATTCC-3′) that target a conserved region of the RNA-dependent RNA polymerase were used as described (Finkbeiner et al., 2009). Positive PCR amplicons from Indian and Nepal samples were cloned into pCR4-TOPO vector (Invitrogen) and pSC-A-amp/kan vector (StrataClone), respectively, and were then sequenced using standard Sanger chemistry.
Full genome sequencing
For each virus, nucleic acids from selected samples positive in the RT-PCR screening were randomly amplified as previously described (Wang et al., 2003). The amplified products were subsequently sequenced by Roche/454 pyrosequencing. Sequence reads with detectable similarity to known astroviruses were identified using a computational pipeline as described (Finkbeiner et al., 2008). Contigs were generated using the Newbler assembler. For each virus (VA2, VA3, VA4 and MLB3), the complete genome was elaborated from the initial contigs by a combination of RT-PCR, 5′ rapid amplification of cDNA ends (RACE) and 3′RACE. High quality consensus genomes were defined by sequencing multiple overlapping RT-PCR amplicons.
5′ and 3′ rapid amplification of cDNA ends (RACE)
Stool filtrates were pretreated with 0.2 μg/ml proteinase K (Invitrogen) at 37 °C for 30 minutes before being extracted using RNAbee (Tel-Test, Inc.). Thermoscript RTase (Invitrogen) was used to reverse transcribe the viral RNA at 65 °C for 45 minutes and then inactivated by heating to 85 °C for 5 minutes. Viral cDNA was purified by a Zymo column (Zymo Research) to remove primers and free nucleotides. cDNA terminal poly(C) tailing was performed in 10 mM Tris-HCl (pH 8.0), 25 mM KCl, and 1.5 mM MgCl2 by incubating with terminal transferase enzyme (Thermo) and 2mM dCTP at 37 °C for 10 minutes. Terminal transferase enzyme activity was inactivated by heating for 10 min at 65 °C. PCR was performed by an abridged anchor primer (5′-GGCCACGCGTCGAC TAGTACGGGGGGGGGG-3′) and a viral gene specific primer (GSP2). Successful RACE PCR products were visible on a 1% agarose gel. 3′ RACE was performed by reverse transcription of the viral cDNA with a dT-adaptor primer (5′-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3′). Viral cDNA was also purified by Zymo column (Zymo Research) to remove primers and free nucleotides. Subsequently, the 3′ cDNA ends were amplified by PCR with a gene specific primer and an adaptor primer (5′-GGCCACGCGTCGACTAGTAC-3′). 5′ and 3′ RACE products were cloned and sequenced using standard Sanger chemistry.
Genome annotation
For each genome, ORF1a was predicted using NCBI ORF Finder. Because ORF1b is generated by ribosomal frameshifting, the conserved heptameric slippery sequence (AAAAAAC) (Jiang et al., 1993), which is perfectly conserved in all astroviruses, including MLB3, VA2, VA3 and VA4, was found by manually searching the full genome sequence. The nearest stop codon upstream of the slippery sequence in the -1 frame was identified, and the codon triplet immediately 3′ to the stop codon was selected as the start codon of ORF1b. For ORF2, the start codon was defined by manually identifying the broadly conserved promoter region of subgenomic RNA sequences (Mendez and Arias, 2007) (AUUUGGAGNGGNGGACCNAAN5-8AUGNC, start site for ORF2 is italicized) near the stop codon of ORF1b.
Sequence alignments and phylogenetic analysis
Nucleotide sequences from the 5′ and 3′ UTRs of the MLB-clade and VA-clade viruses were aligned using ClustalX (version 1.83). Complete protein sequences of ORF1a, ORF1b and ORF2 of astroviruses were downloaded from Genbank and aligned by MUSCLE. Best-fit models of astrovirus protein evolution were selected by Prottest (version 2.4)(Abascal et al., 2005) and maximum-likelihood trees were generated by Phyml (version 3.0)(Guindon et al., 2010). The number of bootstrap trials was set at 1000 replicates. Genetic p-distance, defined as the fraction of divergent amino acids, was calculated based on pairwise deletion (i.e. gaps were deleted in each pair of sequences compared) using MEGA5 (Tamura et al., 2011). The genetic distance matrix was made from those viruses for which both full length capsid and polymerase sequences from the same isolate were available. Statistical analysis of paired capsid to protease and capsid to polymerase genetic distance ratios among VA, MLB, avian and classical human astrovirus clades were performed by ANOVA test (SAS 8.0). Post hoc analysis was done using the SNK (Student-Newman-Keuls) test in SAS. The number of synonymous substitutions per synonymous site (dS) and nonsynonymous substitutions per nonsynonymous site (dN) from averaging over all sequence pairs were calculated using the Nei-Gojobori model (Nei and Gojobori, 1986) in MEGA5.
Sequence accession numbers
Complete genome sequences have been deposited in Genbank for VA3 (JX857868), VA4 (JX857869), and MLB3 (JX857870).
The sequences used to generate the phylogenetic trees are as follows:
Protease
Turkey Astrovirus 1: CAB95005; Turkey Astrovirus 2: NP_987086; Chicken Astrovirus: NC_003790; Ovine Astrovirus: CAB95002; Mink Astrovirus: NP_795334; Huma Astrovirus 8: AAF85962; Human astrovirus 6: ACV92105; Human Astrovirus 5: AAY46272; Human Astrovirus 4: AAY84777; Human astrovirus 3: AEN74892; Human Astrovirus 2: L13745; Human Astrovirus 1: NC_001943; Sea lion astrovirus 9: AEM37629; Sea lion astrovirus 5: AEM37617; Sea lion astrovirus 4: AEM37614; Bovine astrovirus B76: AED89607; Bovine astrovirus B18 AED89598; Porcine Astrovirus 5: AER30001; Porcine Astrovirus 4: AER30007; Porcine Astrovirus 2: AER29998; Mouse Astrovirus M-52: YP_004782205; Duck Astrovirus 1: ADB79805; Wild boar Astrovirus: AEZ67024; Astrovirus MLB1: YP_002290966; Astrovirus MLB2: YP_004934008; Astrovirus VA1: YP_003090287; Astrovirus VA2: ACX83590.
RNA dependent RNA polymerase
Turkey Astrovirus 1: CAB95006; Turkey Astrovirus 2: NP_987087; Duck astrovirus 1: ADB79809; Chicken astrovirus: AEE88304; Avian Nephritis: Q9JGF2; Ovine Astrovirus: CAB95003; Mink Astrovirus: NC_004579; Human Astrovirus 1: NP_059444; Human Astrovirus 2: L13745; Human Astrovirus 3: AEN74891; Human Astrovirus 4: AAY84778; Human Astrovirus 5: AAY46273; Human astrovirus 6: ADJ17722; Human Astrovirus 8: AAF85963; Sea lion Astrovirus 4: AEM37615; Sea lion Astrovirus 5: AEM37618; Sea lion Astrovirus 10: AEM37636; Bat Astrovirus AFCD337: ACF75864; Bat Astrovirus LD38: ACN88707; Bat Astrovirus LD71: ACN88711; Rat Astrovirus: ADJ38393; Wild boar Astrovirus: AEZ67025; Astrovirus MLB1: YP_002290967; Astrovirus MLB2: YP_004934009; Astrovirus VA1: NC_013060; Astrovirus VA2: ACX69838;
Capsid
Turkey Astrovirus 1: CAB95007; Turkey Astrovirus 2: NP_987088; Turkey Astrovirus 3: AAV37187; Chicken Astrovirus 1: NP_620618; Chicken astrovirus 2: BAB21617; Duck astrovirus: ACN82429; Fowl Astrovirus: AFF57968; Pigeon Astrovirus: CBY02488; Avian Nephritis Virus 2: AEB15604; Ovine Astrovirus: CAB95004; Mink Astrovirus: NP_795336; Human Astrovirus 8: AAF85964; Human Astrovirus 7: AAK31913; Human Astrovirus 6: CAA86616; Human Astrovirus 5: AAY46274; Human Astrovirus 4: AAY84779; Human Astrovirus 3: AAD17224; Human Astrovirus 2: L13745; Human Astrovirus 1: NP_059444; Porcine astrovirus: BAA90309; Dolphin Astrovirus: ACR54280; Sea lion Astrovirus 1: ACR54272; Sea lion Astrovirus 2: ACR54274; Feline astrovirus: AAC13556; Bat astrovirus LC03: ACN88720; Bat astrovirus LD71: ACN88712; Bat astrovirus LD38: ACN88708; Bat Astrovirus AFCD337: ACF75865; Porcine Astrovirus 2: AER30006; Swine Astrovirus: ADV16836; Canine Astrovirus: AEX00102; Bovine Astrovirus B76: AED89609; Rabbit Astrovirus: AEV92822; Wild boar Astrovirus: AEZ67026; Astrovirus MLB1: YP_002290968; Astrovirus MLB2: YP_004934010.1; Astrovirus VA1: YP_003090288; Astrovirus VA2: ACX83591.
Results
Identification and complete genome sequencing of MLB3 from children in India
Astrovirus consensus RT-PCR screening has identified multiple novel astrovirus species (Finkbeiner, 2009; Finkbeiner et al., 2009a). Here, consensus RT-PCR screening of 400 stool specimens from children in India with diarrhea and 400 stool samples from asymptomatic children yielded five positive samples with amplicons that, when sequenced, shared only ~81% nucleotide identity with astrovirus MLB2. Because of the extent of divergence in this locus located within the RdRp, this virus has tentatively been named MLB3. Four of the MLB3 positive stool samples were from patients with diarrhea while only one was from an asymptomatic child (Table 1). However, there was no statistically significant association of MLB3 with diarrheal cases (McNemar’s Test; OR 4.00, 95% CI 0.39 – 196.90, p=0.371).
TABLE 1.
Epidemiological data
| Sample ID | Virus | Country | Symptoms | Other pathogen testeda |
|---|---|---|---|---|
| 5461 | MLB3 | India | Diarrhea | Rotavirus, Norovirus, Bacterial pathogens and Parasites |
| 5463 | MLB3 | India | Diarrhea | Rotavirus, Norovirus, Bacterial pathogens and Parasites |
| 5462 | MLB3 | India | Diarrhea | Rotavirus, Norovirus, Bacterial pathogens and Parasites |
| 22077 | MLB3 | India | Diarrhea | Rotavirus, Norovirus, Bacterial pathogens and Parasites |
| 28054 | VA3 | India | Diarrhea | Rotavirus, Norovirus, Bacterial pathogens and Parasites |
| 26564 | MLB3 | India | Asymptomatic | Rotavirus |
| S5363 | VA4 | Nepal | Diarrhea | Rotavirus, Norovirus, Adenovirus, Astrovirus, Bacterial pathogens, Giardia, Cryptosporidium |
| S5362 | VA4 | Nepal | Diarrhea | Rotavirus, Norovirus, Adenovirus, Astrovirus, Bacterial pathogens, Giardia, Cryptosporidium |
Assays were performed individually in Nepal or India. Bacterial pathogens tested in India included Vibrio cholerae, enteropahtogenic Escheria coli, Salmonella, Shigella, Aeromonas and Plesiomonas; bacterial pathogens tested in Nepal included Salmonella, Shigella, Vibrio, Aeromonas, Plesiomonas and diarrheagenic E. coli (EIEC, EPEC, ETEC, EAgg, STEC).
One specimen positive for MLB3 was subjected to high-throughput Roche/454 pyrosequencing and the resulting reads were assembled to yield a contig of 6112 nucleotides (nt). To obtain the 5′ and 3′ ends of the virus, RACE was performed to generate a complete genome of 6124 nt (Table 2). Sequencing of a second MLB3 sample yielded two contigs that totaled approximately 4.5 kb; these contigs shared 99% nucleotide identity to the completely sequenced MLB3 strain (data not shown).
TABLE 2.
Genome comparison of the MLB clade and VA clade astroviruses
| Virus | Length (nt) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Full genome | 5′ UTR | ORF1a | ORF1b | ORF2 | 3′UTR (without polyA) | |
| MLB1 | 6171 | 14 | 2364 | 1536 | 2271 | 58 |
| MLB2 | 6119 | 14 | 2364 | 1536 | 2238 | 39 |
| MLB3 | 6124 | 14 | 2364 | 1536 | 2244 | 38 |
| VA1 | 6586 | 38 | 2661 | 1575 | 2277 | 98 |
| VA2 | 6531 | 42 | 2664 | 1587 | 2196 | 117 |
| VA3 | 6581 | 36 | 2670 | 1662 | 2268 | 95 |
| VA4 | 6518 | 40 | 2661 | 1581 | 2190 | 115 |
VA4 from children in Nepal
The same RT-PCR screening strategy applied to a cohort of 196 children with diarrhea in Nepal yielded two positive samples that shared ~75% nucleotide identity to VA2. This virus has tentatively been named VA4. Specimens positive for VA4 were subjected to high-throughput Roche/454 pyrosequencing and the resulting reads were assembled yielding a contig of 6327 nt. Finally, RACE was performed to obtain a complete genome of 6518 nt (Table 2).
Complete genome sequencing of VA2 and VA3
We had previously described the discovery of VA2 and VA3 (Finkbeiner, 2009a). However, we were only able to obtain partial sequences of VA2 and VA3 due to the limited specimen availability. One stool sample from a child in India in the current study was positive for VA3. High throughput sequencing generated a contig of 6135 nt for VA3, which was extended by 5′ and 3′ RACE to a complete genome of 6581 nt. We also extended the previously published partial VA2 sequence of 5977 nt (Genbank GQ502193) using 5′ and 3′ RACE to generate a complete genome of 6531 nt. Gene prediction generally yielded very similar results for each of the viruses. For VA3, the presence of an ATG near the very 5′ end of the genome yielded a slightly larger ORF1a than in the other VA-clade astroviruses. Based on the gene predictions in the other VA-clade astroviruses, we used the second ATG of VA3 as the putative start codon as this gave a predicted ORF that was more consistent with the other VA-clade astroviruses.
Phylogenetic analysis of novel astroviruses ORFs
To characterize the phylogenetic relationships of these newly identified astroviruses, we performed maximum likelihood phylogenetic analysis of all three ORFs. The phylogenies showed that MLB3 is most closely related to MLB2 with strong bootstrap support for a monophyletic MLB-clade. The MLB-clade was consistently most closely related to the classic human astroviruses and some sea lion astroviruses in ORF1a and ORF1b (Fig. 1). VA4 was most closely related to VA2, and this pair diverged from the VA1/VA3 branch consistently for all three ORFs. The four viruses in the VA-clade were most closely related to ovine astrovirus, mink astrovirus and bat astrovirus.
FIG. 1.
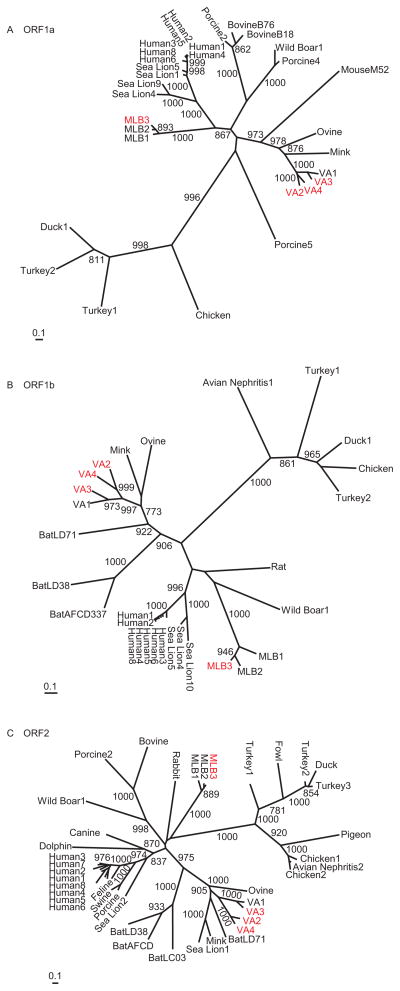
Phylogenetic analysis of the astrovirus open reading frames. Phylogenetic trees were generated by the maximum likelihood method with 1000 replicates based on amino acids alignments. Significant bootstrap values (>700) are shown. (A) ORF1a serine proteinase. (B) ORF1b RNA dependent RNA polymerase (RdRp). (C) ORF2 capsid protein. Scale bars represent the number of differences in each ORF.
The evolutionary pressures exerted on the protein coding sequence are highly indicative of the role and functions of viral genes. With the additional complete genome sequences generated in this study (MLB3, VA2, VA3 and VA4), it was possible to compare the extent of divergence within each clade. Visual inspection of the phylogenetic trees (Fig. 1) demonstrated that the branch lengths within the classic human astrovirus clade were shorter in ORF1a and ORF1b compared to ORF2, whereas both the MLB and VA clades had more similar branch lengths in ORF1a and ORF1b compared to ORF2. Hence, we rationalized that by comparing the genetic distance between the three genes, we would be able to determine if these viruses have been evolving under the same selective pressure. In order to quantify this difference, we calculated the pairwise sequence divergence for viruses within a clade, which was defined by using genetic p-distance for the MLB, VA, classic human astrovirus and avian astrovirus (composed of chicken, turkey 1, turkey 2, and duck) clades (Fig. 3). From each pairwise comparison, we then calculated the ratio of the genetic distance of the capsid to the genetic distance of the protease. We also calculated the ratio of the genetic distance of the capsid to the genetic distance of the polymerase. The average value of each ratio was then determined for each clade (Table 3). The magnitude of the average ratio varied considerably, and both the average ratio of capsid to protease genetic distance and the average ratio of capsid to polymerase genetic distance in the classic human astrovirus clade were statistically different (ANOVA, p<0.01) from the average ratios in the MLB, VA and avian astrovirus clades.
FIG. 3.
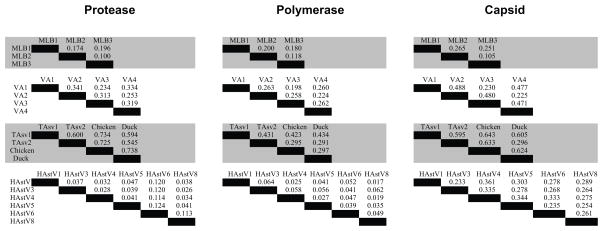
Pairwise genetic p-distance comparisons for MLB, VA, avian and classic human astroviruses. TAstV: turkey astrovirus, HAstV: human astrovirus. TABLE 1: Epidemiological data
TABLE 3.
Average genetic distance ratio of the capsid to the polymerase within the clades
| Clade | Genetic distance ratio (Mean±Standard deviation)
|
|
|---|---|---|
| capsid/protease | capsid/polymerase | |
| Classic human | 6.35±3.39 | 8.15±4.27 |
| MLB | 1.23±0.32 | 1.26±0.18 |
| VA | 1.29±0.28 | 1.59±0.39 |
| Avian | 0.86±0.17 | 1.59±0.44 |
To further evaluate factors that might contribute to the high genetic distance ratios observed in the classic human astrovirus clade, the overall mean number of synonymous mutations per synonymous site (dS), the number of non-synonymous mutations per nonsynomous site (dN) and the ratio of dN/dS in each protein in the classic human, MLB and VA clades were calculated (Table 4). Due to saturation of dS in the avian clade, we did not analyze the avian clade. In the capsid, the dN/dS ratio was similar in all three clades. However, in the polymerase, the classic human astrovirus clade had much lower values of dN/dS, dN and dS compared to the other clades. Similarly, the dN/dS, dN and dS values of the protease of the classic human clade were lower than in the other clades. These observations suggest that there has been limited evolution in the classic human astrovirus nonstructural genes.
TABLE 4.
Average values of dS, dN, and dN/dS within each clade
| Clade | Protease | Polymerase | Capsid | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| ds | dN | dN/dS | dS | dN | dN/dS | dS | dN | dN/dS | |
| Classic human | 0.745 | 0.035 | 0.047 | 0.797 | 0.023 | 0.029 | 1.708 | 0.233 | 0.136 |
| MLB | 1.784 | 0.120 | 0.067 | 1.118 | 0.113 | 0.101 | 1.375 | 0.157 | 0.114 |
| VA | 2.679 | 0.230 | 0.086 | 2.053 | 0.185 | 0.090 | 2.547 | 0.366 | 0.144 |
Conserved sequences in the 5′ and 3′ UTRs
The MLB3 5′ UTR was determined to be 14 nt in length (Table 2). Sequence alignments demonstrated that the 11 nt at the very 5′ end of MLB3 were perfectly conserved with the 5′ termini of MLB2 and MLB1 (Fig. 2A). The 5′ UTRs of the MLB-clade viruses were much shorter than that of the classic human astroviruses, which range from 80 to 85 nt in length; nevertheless, there were 8 out of 11 nt conserved at the very 5′ end between the three MLB-clade viruses and the 8 classic human astroviruses. By contrast, no sequence conservation in the 5′ UTR was detected among the four viruses in the VA-clade.
FIG. 2.
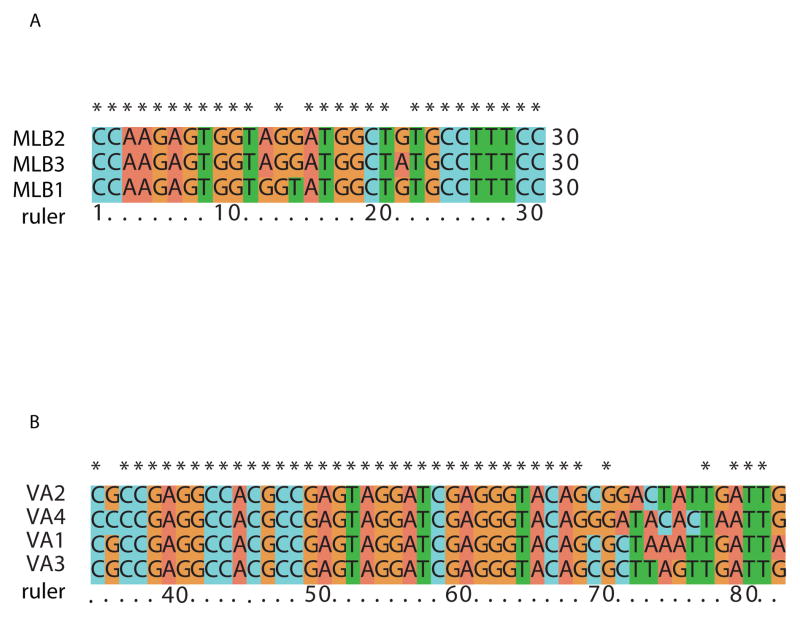
Conserved motifs in the 5′ UTR of MLB clade astroviruses and the 3′ UTR of VA clade astroviruses. (A) Multiple sequence alignment of the 30 nt at the very 5′ end of each MLB clade astrovirus genome. (B) 150 nt from the 3′ terminus of each of the VA clade astroviruses were aligned. The portion of the alignment containing a perfectly conserved 33 nt stretch is shown.
Viruses in the MLB clade had 3′ UTRs ranging from 38 to 58 nt (excluding the polyA tail) in length (Table 2). There was no clear conservation in sequence or secondary structure among different MLB viruses. However, the VA-clade astroviruses had much longer 3′ UTRs varying from 95 to 117 nt (excluding the polyA tail) in length. Within the 3′ UTR of the VA astroviruses there was a strictly conserved 33 nt sequence (Fig. 2B) which forms the conserved s2m RNA secondary structure that has been described in many astroviruses and other positive sense RNA viruses (Jonassen et al., 1998; Monceyron et al., 1997). Interestingly, none of the MLB-clade viruses possess the s2m motif.
Discussion
In this study, we identified two novel astroviruses, MLB3 and VA4, in pediatric stool specimens collected in India and Nepal. The full genome sequences of MLB3, VA4, and the previously reported viruses VA2 and VA3, were defined by using a combination of high-throughput 454 pyrosequencing and traditional Sanger sequencing. The presence of these viruses in stool samples from children with diarrhea raises the possibility that they may play roles in causing human diarrhea. In the Indian cohort, MLB3 was present in four diarrhea stools and one asymptomatic stool. Based on the sample size, there was, however, no statistically significant association of MLB3 with diarrhea. VA3 was identified in one diarrhea sample from India, and VA4 was found in two diarrhea samples from Nepal. Because stool is not a sterile site, it is not possible to directly conclude from the detection of VA4 and MLB3 in human stool samples that these viruses cause bona fide human infection. It is possible that these viruses are present due to dietary ingestion, for example. However, we note that previous studies have detected members of both the VA-clade and MLB-clade of astroviruses in sterile sites in the human body. MLB2 was detected in serum of a febrile child (Holtz et al., 2011b) while VA1/HMO-C was detected in brain tissue of a child with encephalitis (Quan et al., 2010). Serological evidence of human infection by VA1/HMO-C has also been described (Burbelo et al., 2011).
With the identification of MLB3 and VA4, there are now three members of the MLB-clade and four members of the VA-clade of astroviruses. Comparison of the sequences within and between the clades provided multiple interesting observations. First, there was conservation of the most terminal 11 nt in the 5′ UTR of the MLB-clade astroviruses but not in the 5′ UTR of the VA-clade astroviruses. This motif is also partially conserved with the 5′ UTR of the classic human astroviruses, suggesting that there is likely to be a functional role for this conserved element. Conserved elements in the 5′ and 3′ UTR of positive sense RNA viruses are frequently cis-acting elements important for viral replication, transcription or translation.
The VA-clade viruses, but not the MLB-clade viruses, shared a perfectly conserved stem loop motif in the 3′ UTR, the s2m motif. As with the conserved 5′ UTR sequence motif in the MLB-clade viruses, the possible role of the s2m structure in the life cycle of the VA-clade viruses needs to be further characterized.
Phylogenetic analysis demonstrated strong bootstrap support in all three ORFs for both a MLB-clade and a VA-clade of viruses. In ORF1a, the MLB-clade formed a monophyletic group with the classic human astroviruses and various sea lion astroviruses.
The VA-clade of the viruses clustered consistently with mink and ovine astroviruses. In ORF1b and ORF2, Bat astrovirus LD71 was also included within the clade (sequence of ORF1a of Bat astrovirus LD71 was unavailable). The distinct evolutionary relationship of the VA-clade and the MLB and classic human astrovirus clades suggests that multiple introductions of astrovirus into the human population occurred.
In the classic human astrovirus, MLB, VA and avian astrovirus clades, the average ratio of the genetic distance of the capsid to polymerase was greater than one, consistent with the notion that the RNA dependent RNA polymerase is more highly conserved than the capsid protein, presumably due to functional constraints on evolution of the polymerase. However, the magnitude of the ratio observed for the classic human astrovirus clade was much larger than that observed for the MLB, VA or avian astrovirus clades. This observation, coupled with the classic human astrovirus polymerase possessing the lowest dS, dN and dN/dS values among all the clades, suggests that there has been more limited rates of mutation in the polymerase gene rather than elevated levels of mutation of the capsid. Analysis of the protease yielded similar evidence of constrained evolution. It is unclear at this time the underlying root of the limited mutation rate in the protease and polymerase of the classic human astrovirus clade compared to the MLB and VA astrovirus clades. One possibility is that there may be cryptic overlapping ORFs or RNA secondary structures present in the protease and polymerase region of the classic human astroviruses that are absent in the same region of the other clades. Another possibility entails the selective sweep of a single or a few closely-related nonstructural genes in the classic human astroviruses which replaced the previously-existing, nonstructural genes, via recombination. We note that recombination near the classic human astrovirus polymerase and capsid junction has been reported (Walter et al., 2001). Mechanistically, the difference in the ratios may reflect distinct types of immune pressure on the various clades of virus, perhaps due to unique tissue and/or host tropisms.
Astroviruses are commonly accepted as causative agents of gastroenteritis of both mammalian and avian species. In this study, we identified two novel astroviruses, MLB3 and VA4, from pediatric stools, expanding the diversity of astroviruses that are known to be present in human stool specimens. The discovery of these two viruses and their complete genome sequences enabled us to better define characteristic properties of members of the MLB and VA astroviruses subclades and to compare them to the classic human astroviruses. These comparisons yielded the unexpected observation that the nonstructural region of the classic human astroviruses has undergone much more limited mutation than that of other clades. As exemplified here, the ever-increasing number of virus genomes being discovered and sequenced will greatly facilitate comparative genomic analysis that can generate novel insights into the function and evolution of viral proteins.
Highlights.
Discovered and sequenced complete genomes of two novel astroviruses in human stool.
Identification of these viruses enabled comparative analysis between astroviruses.
Classic human astrovirus nonstructural genes are evolutionarily constrained.
Acknowledgments
We thank Henry Huang and Efrem Lim for helpful discussions and critical reading of the manuscript. This work was supported in part by NIH grant U54 AI057160 to the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research and NIH grant R21 AI090199. D.W. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. LRH is supported by UL1 TR000448 sub award KL2 TR000450 from the NIH-National Center for Advancing Translational Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajjampur SS, Rajendran P, Ramani S, Banerjee I, Monica B, Sankaran P, Rosario V, Arumugam R, Sarkar R, Ward H, Kang G. Closing the diarrhoea diagnostic gap in Indian children by the application of molecular techniques. Journal of medical microbiology. 2008;57:1364–1368. doi: 10.1099/jmm.0.2008/003319-0. [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. Journal of clinical microbiology. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Ching KH, Esper F, Iadarola MJ, Delwart E, Lipkin WI, Kapoor A. Serological studies confirm the novel astrovirus HMOAstV-C as a highly prevalent human infectious agent. PloS one. 2011;6:e22576. doi: 10.1371/journal.pone.0022576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner SR. Detection of Newly Described Astrovirus MLB1 in Stool Samples from Children. Emerging infectious diseases. 2009;15:441–444. doi: 10.3201/1503.081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner SR, Allred AF, Tarr PI, Klein EJ, Kirkwood CD, Wang D. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS pathogens. 2008;4:e1000011. doi: 10.1371/journal.ppat.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner SR, Holtz LR, Jiang Y, Rajendran P, Franz CJ, Zhao G, Kang G, Wang D. Human stool contains a previously unrecognized diversity of novel astroviruses. Virology journal. 2009a;6:161. doi: 10.1186/1743-422X-6-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner SR, Li Y, Ruone S, Conrardy C, Gregoricus N, Toney D, Virgin HW, Anderson LJ, Vinje J, Wang D, Tong S. Identification of a novel astrovirus (astrovirus VA1) associated with an outbreak of acute gastroenteritis. Journal of virology. 2009b;83:10836–10839. doi: 10.1128/JVI.00998-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Holtz LR, Bauer IK, Rajendran P, Kang G, Wang D. Astrovirus MLB1 is not associated with diarrhea in a cohort of Indian children. PloS one. 2011a;6:e28647. doi: 10.1371/journal.pone.0028647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz LR, Wylie KM, Sodergren E, Jiang Y, Franz CJ, Weinstock GM, Storch GA, Wang D. Astrovirus MLB2 viremia in febrile child. Emerg Infect Dis. 2011b;17:2050–2052. doi: 10.3201/eid1711.110496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Monroe SS, Koonin EV, Stine SE, Glass RI. RNA sequence of astrovirus: distinctive genomic organization and a putative retrovirus-like ribosomal frameshifting signal that directs the viral replicase synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10539–10543. doi: 10.1073/pnas.90.22.10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen CM, Jonassen TO, Grinde B. A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. The Journal of general virology. 1998;79 ( Pt 4):715–718. doi: 10.1099/0022-1317-79-4-715. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Li L, Victoria J, Oderinde B, Mason C, Pandey P, Zaidi SZ, Delwart E. Multiple novel astrovirus species in human stool. The Journal of general virology. 2009;90:2965–2972. doi: 10.1099/vir.0.014449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood CD, Clark R, Bogdanovic-Sakran N, Bishop RF. A 5-year study of the prevalence and genetic diversity of human caliciviruses associated with sporadic cases of acute gastroenteritis in young children admitted to hospital in Melbourne, Australia (1998–2002) J Med Virol. 2005;77:96–101. doi: 10.1002/jmv.20419. [DOI] [PubMed] [Google Scholar]
- Kjeldsberg E. Serotyping of human astrovirus strains by immunogold staining electron microscopy. Journal of virological methods. 1994;50:137–144. doi: 10.1016/0166-0934(94)90170-8. [DOI] [PubMed] [Google Scholar]
- Madeley CR, Cosgrove BP. Letter: 28 nm particles in faeces in infantile gastroenteritis. Lancet. 1975;2:451–452. doi: 10.1016/s0140-6736(75)90858-2. [DOI] [PubMed] [Google Scholar]
- Mendez E, Arias CF. Astrovirus. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 1. Lippincott Williams & Wilkins; Phiadelphia: 2007. pp. 981–1000. [Google Scholar]
- Monceyron C, Grinde B, Jonassen TO. Molecular characterisation of the 3′-end of the astrovirus genome. Archives of virology. 1997;142:699–706. doi: 10.1007/s007050050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular biology and evolution. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Quan PL, Wagner TA, Briese T, Torgerson TR, Hornig M, Tashmukhamedova A, Firth C, Palacios G, Baisre-De-Leon A, Paddock CD, Hutchison SK, Egholm M, Zaki SR, Goldman JE, Ochs HD, Lipkin WI. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerging infectious diseases. 2010;16:918–925. doi: 10.3201/eid1606.091536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, Negishi H, Wang QH, Akihara S, Kim B, Nishimura S, Kaneshi K, Nakaya S, Ueda Y, Sugita K, Motohiro T, Nishimura T, Ushijima H. Molecular epidemiology of astroviruses in Japan from 1995 to 1998 by reverse transcription-polymerase chain reaction with serotype-specific primers (1 to 8) Journal of medical virology. 2000;61:326–331. [PubMed] [Google Scholar]
- Soares CC, Maciel de Albuquerque MC, Maranhao AG, Rocha LN, Ramirez ML, Benati FJ, Timenetsky do MC, Santos N. Astrovirus detection in sporadic cases of diarrhea among hospitalized and non-hospitalized children in Rio De Janeiro, Brazil, from 1998 to 2004. Journal of medical virology. 2008;80:113–117. doi: 10.1002/jmv.21053. [DOI] [PubMed] [Google Scholar]
- Steitz TA. DNA polymerases: structural diversity and common mechanisms. The Journal of biological chemistry. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JE, Briggs J, Guerrero ML, Matson DO, Pickering LK, Ruiz-Palacios G, Berke T, Mitchell DK. Molecular characterization of a novel recombinant strain of human astrovirus associated with gastroenteritis in children. Archives of virology. 2001;146:2357–2367. doi: 10.1007/s007050170008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Urisman A, Liu YT, Springer M, Ksiazek TG, Erdman DD, Mardis ER, Hickenbotham M, Magrini V, Eldred J, Latreille JP, Wilson RK, Ganem D, DeRisi JL. Viral discovery and sequence recovery using DNA microarrays. fPLoS biology. 2003;1:E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]