

Abstract
Y-family polymerases help cells tolerate DNA damage by performing translesion synthesis opposite damaged DNA bases, yet they also have a high intrinsic error rate. We constructed chimeras of two closely related Y-family polymerases that display distinctly different activity profiles and found that the polypeptide linker that tethers the catalytic polymerase domain to the C-terminal DNA-binding domain is a major determinant of overall polymerase activity, nucleotide incorporation fidelity, and abasic-site bypass ability. Exchanging just three out of the 15 linker residues is sufficient to inter-convert the polymerase activities tested. Crystal structures of four chimeras show that the conformation of the protein correlates with the identity of the inter-domain linker sequence. Thus, residues that are more than 15 Å away from the active site are able to influence many aspects of polymerase activity by altering the relative orientations of the catalytic and DNA-binding domains.
INTRODUCTION
Polymerases belonging to the Y-family are the main enzymes that help cells to tolerate DNA damage, by allowing DNA synthesis to continue across from lesions that stall replicative polymerases (Ohmori et al., 2001). The importance of these polymerases became apparent when the variant form of xeroderma pigmentosum (XPV) was found to be caused by a lack of polymerase (pol) eta, which was shown to have the ability to bypass cis-syn cyclopyrimidine dimers (CPD) accurately (McDonald et al., 1999; Washington et al., 1999).
While providing a benefit through their translesion synthesis ability, the Y-family polymerases have a 10- to 1000-fold higher mutational rate than replicative polymerases (Kunkel, 2009). In some specific instances, mispair formation is even favored over correct nucleotide insertion (Tissier et al., 2000). The members of the Yfamily are grouped by sequence similarity into six classes (Ohmori et al., 2001). Many organisms have more than one type of Y-family polymerase, with humans having four: pol eta, pol iota, Rev1 and pol kappa. The polymerases within a class display similar lesion-bypass and mutational specificities, although there can be significant variation even within one class.
The Y-family polymerases contain two domains that are required for full polymerase activity: an N-terminal catalytic polymerase domain (comprised of fingers, palm and thumb subdomains), and a C-terminal “little finger” or polymerase-associated domain (LF/PAD) that assists with binding DNA. The conserved sequence motifs that define the Y-family of polymerases are all contained within the catalytic domain. Both domains are conserved in structure across the entire family, even though the LF/PAD domains share little sequence identity.
The spectrum and frequency of mutations that occur during translesion DNA synthesis is highly dependent on which polymerase replicates past a lesion. This is a key issue when multiple translesion polymerases are present in a cell. The sources of mutational specificity are still not well defined, at least in part because the Y-family polymerases have diverged highly in sequence, typically having just 30% sequence identity between different classes and 40% or less identity within a class (Ohmori et al., 2001). The importance of understanding the differences in Y-family polymerase specificity is evident in XPV cells where, in the absence of pol eta, CPD lesions are bypassed by pol kappa, pol iota and pol zeta (Dumstorf et al., 2006; Gueranger et al., 2008; Wang et al., 2007; Ziv et al., 2009). Translesion synthesis by these alternative polymerases is inaccurate, increasing the mutation rate in XPV cells and greatly increasing the likelihood of skin cancer in XPV patients.
The first Y-family polymerase crystal structures (Ling et al., 2001; Silvian et al., 2001; Trincao et al., 2001; Zhou et al., 2001) suggested that the ability to bypass damaged template bases arises from a very open & solvent accessible active site, a feature that also allows highly error-prone DNA synthesis to occur. Subsequent Y-family polymerase structures have supported this original proposal and have also been able to 5 identify a few protein-DNA interactions that facilitate bypass of specific DNA lesions, but given the wide range of specificities displayed by the Y-family polymerases, there are clearly more specificity determinants that remain to be discovered.
Dpo4, from Sulfolobus solfataricus, is the most characterized Y-family polymerase (Ling et al., 2001), with well over 50 structures deposited in the Protein Data Bank (PDB). Dbh, from S. acidocaldarius, is a close relative of Dpo4, sharing 54% amino acid identity overall (Boudsocq et al., 2004; Kulaeva et al., 1996). Both are homologs of E. coli DinB and human pol kappa, all of which belong to the only class of Y-family polymerases that has been found in all domains of life. Sequence identity is much higher in the polymerase domain (60% identity), which contains all of the sequence motifs that define the Y-family, than in the remainder of the protein (41% identity).
Despite the close relationship between Dbh and Dpo4, the two polymerases display distinct activities in vitro (Boudsocq et al., 2004). Dpo4 is the more active enzyme, but is also more prone to making base-substitution errors; Dbh inserts nucleotides with higher fidelity, but creates a larger fraction of deletion mutations (Boudsocq et al., 2004). The polymerases also differ in translesion specificity: Dpo4 bypasses cis-syn thymidine dimers and abasic sites relatively efficiently while Dbh does not (Boudsocq et al., 2004).
The source of these differences was investigated by creating chimeric versions of Dpo4 and Dbh and demonstrating that differences in activity could be attributed to differences in the C-terminal part of the protein, outside of the catalytic polymerase domain (Boudsocq et al., 2004). Differences between electrostatic potential and curvature of the LF/PAD domains of Dbh and Dpo4 were proposed to account for the differences in activity. An implicit assumption in this proposal was that the LF/PAD of Dbh would adopt a Dpo4-like conformation upon binding DNA, suggesting that sequence variation in the LF/PAD would be responsible for the differences in activity. However, when we determined structures of three complexes of Dbh bound to DNA (Wilson and Pata, 2008), we found that Dbh did not adopt the typical conformation observed for Dpo4 (Figure 1A,B). Instead, it retained a conformation close to the one observed for Dbh apoenzyme (Silvian et al., 2001).
Figure 1. Parental Y-family Polymerases Used to Construct Chimeric Enzymes.
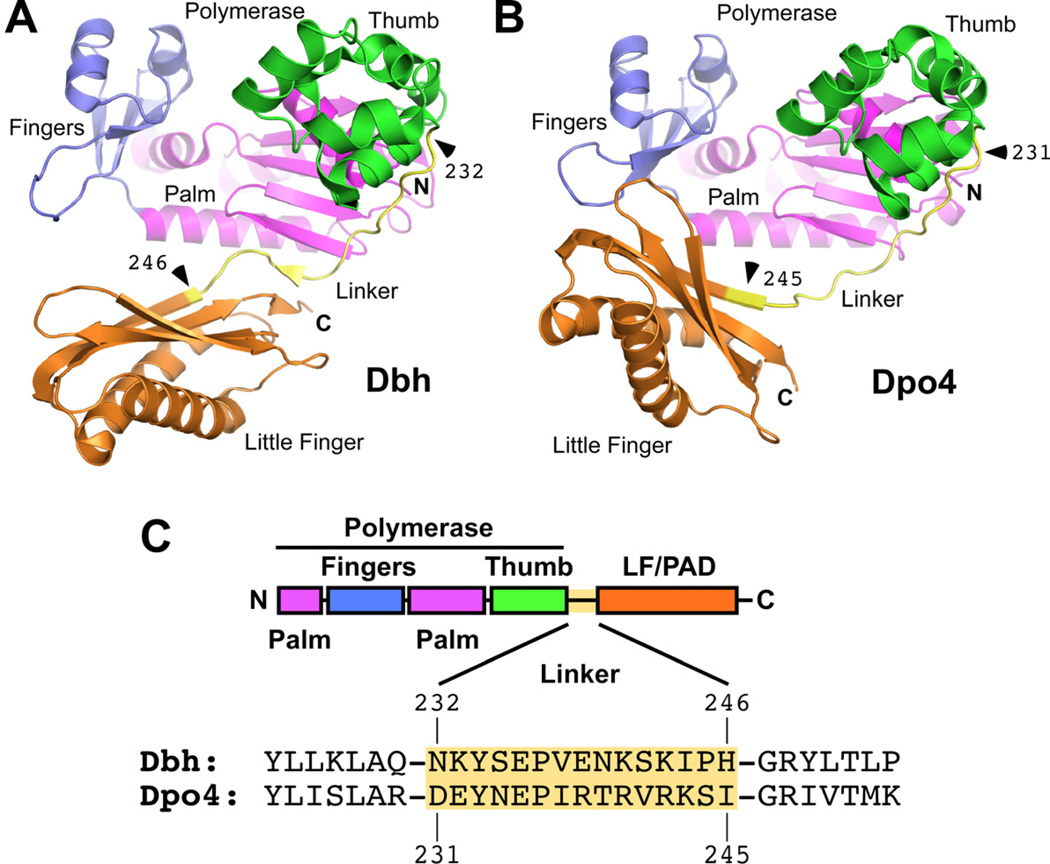
(A) Structure of Dbh showing overall protein conformation and junctions (marked with arrowheads) before and after the linkers that were exchanged in chimeric polymerases. Note the short β-strand in the linker that contacts both the palm and the LF/PAD domains. Made using coordinates from PDB code 3BQ1 (Wilson and Pata, 2008), a ternary complex that contained incoming dNTP and primer-template DNA with an extrahelical nucleotide in the template strand three nucleotides to the 3’ side of the templating base (substrates not shown). Colored by domain: blue (fingers), magenta (palm), green (thumb), yellow (linker), and orange (LF/PAD). (B) Structure of Dpo4. Note the contact between the LF/PAD and fingers domains. Domains are colored as in A and junctions are marked with arrowheads. Made using coordinates from PDB code 3QZ7 (Wu et al., 2011). (C) Diagram showing the linear organization of the polymerase domains and the protein sequences of Dbh and Dpo4 in the region of the linker. Domains are colored as in A.
Thus, the positioning of the LF/PAD domain, rather than any specific sequence differences within the domain, could be responsible for the differing activities of Dbh and Dpo4. The inter-domain linker sequence that connects the polymerase and LF/PAD domains stood out as a candidate for determining the domain orientation (Figure 1). In particular, the Dbh linker forms a short β-strand that interacts with both the polymerase palm and the LF/PAD, which could interfere with changes in the relative orientation of the two domains (Figure 1A). The Dpo4 linker, in contrast, does not form this short β-strand and has many fewer contacts in this region with the catalytic and LF/PAD domains (Figure 1B). The linker was included as part of the LF/PAD in the chimeras made previously (Boudsocq et al., 2004). We hypothesized that the linker sequences, rather than the LF/PAD itself, are important for determining the orientation of the LF/PAD domain and that differences in domain orientation determine differences in activity.
Here we show that the inter-domain linker is indeed the key region that controls specificity. Chimeric polymerases that contain the Dpo4 linker behave like Dpo4 while those that contain the Dbh linker behave like Dbh, regardless of the source of the sequences in the polymerase and LF/PAD domains. Furthermore, the overall conformation of the chimeric polymerases depends on the identity of the linker sequence. Thus, by controlling the enzyme conformation, the linker is a major determinant of specificity, even though it is located more than 15 Å away from the active site.
RESULTS
Experimental design
To test the hypothesis that the inter-domain linker sequences are important for activity, we made chimeric polymerases with all six possible combinations of polymerase, linker and LF/PAD domains. Constructs are named by the parental source of each domain, in order from N- to C-terminus. If the linker were the key determinant of activity, switching after the linker (Dbh-Dbh-Dpo4 and Dpo4-Dpo4-Dbh) would give the opposite result as switching before the linker (Dbh-Dpo4-Dpo4 and Dpo4-Dbh-Dbh). Furthermore, swapping the linker alone (Dbh-Dpo4-Dbh and Dpo4-Dbh-Dpo4) should switch the activity of the otherwise parental protein.
We defined the linker as shown in Figure 1C, starting immediately after the final helix in the thumb and running through the first two residues in the first β-strand in the LF/PAD: residues 232–246 in Dbh and 231–245 in Dpo4. We decided not to use the boundary of the first β-strand in the LF/PAD as the boundary for the sequence exchange because the properties of Dbh residues Pro245 and His246 are very different from the structurally equivalent residues in Dpo4, Ser244 and Ile 245.
Polymerase activity is determined by the linker sequence
We compared all six chimeras and the two parental enzymes and found that in assays to measure primer extension on undamaged and abasic-site containing tempate DNA and single nucleotide incorporation fidelity, the identity of the linker domain is the major determinant of polymerase activity (Figure 2). In each experiment, the enzymes were compared under identical conditions. Quantitation of these assays is provided in Figure S2.
Figure 2. Single and Multiple Nucleotide Incorporation by Chimeric Polymerases on Undamaged and Abasic Site DNA.
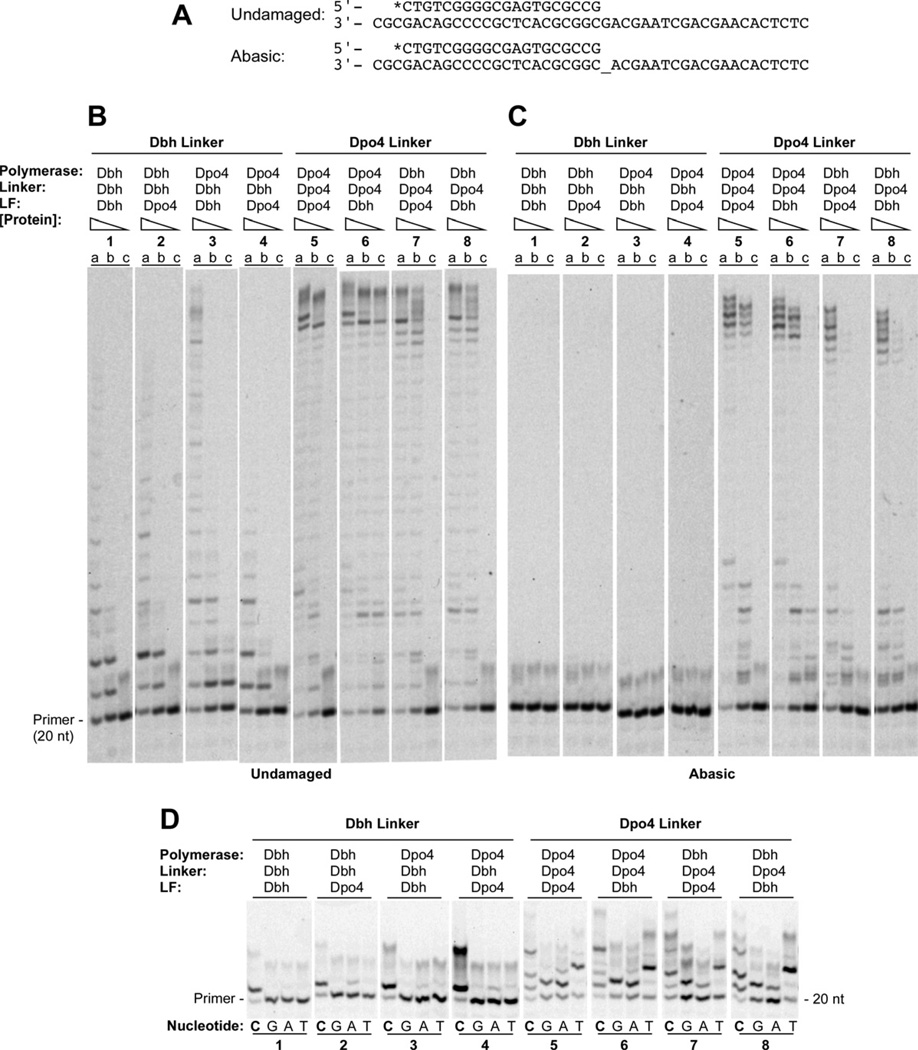
(A) Primer-template sequences showing undamaged template, top, and template containing an abasic site (denoted by _), bottom. Primers were labeled at the 5’ end during synthesis with 6-FAM (denoted by *). (B) Polymerase assays on undamaged primer-template DNA containing (a) 160 nM, (b) 40 nM, or (c) 10 nM protein with 40 nM DNA and 1 mM each dATP, dCTP, dGTP and dTTP. Reactions were incubated at 60°C for 5 min. (C) Polymerase assays on primer-template DNA containing abasic site in the template strand immediately adjacent to the terminal basepair. Protein and substrate concentrations were the same as in B. Reactions were incubated at 60°C for 10 min. (D) The undamaged primer-template DNA shown in A was used as the substrate in reactions that separately contained 1mM dCTP (lanes C), dGTP (lanes G), dATP (lanes A), or dTTP (lanes T) with 1µM enzyme, and 40nM primer-template DNA. Reactions were incubated at 37°C for 5 min. See Figure S2 for quantitation.
In primer-extension assays that contained equal concentrations of all four dNTPs, we found that each of the chimeric polymerases behaves predominantly like the parental polymerase that contributed the linker domain (Figure 2A,B and Figure S2A). All of the chimeras containing the Dbh linker sequence behaved most like the Dbh parent (Figure 2B, reactions 1, 3, 6 and 8, and Figure S2A), which extends the primer by just a few nucleotides even at the highest concentration of enzyme. Similarly, all of the chimeras containing the Dpo4 linker sequence behaved like the Dpo4 parent (Figure 2B, reactions 2, 4, 5 and 7, and Figure S2A), which was able to fully extend the primer to the end of the template at all but the lowest enzyme concentration.
An even stronger correlation of activity with the linker sequence identity was found in assays measuring the bypass of an abasic site located immediately following the primer terminus (Figure 2A,C and Figure S2B). Neither Dbh nor any chimera containing the Dbh linker sequence was able to bypass the abasic site, even at the highest concentration of enzyme used (Figure 2C, reactions 1, 3, 6 and 8, and Figure S2B). In contrast, Dpo4 and all of the chimeras containing the Dpo4 linker sequence were able to bypass the lesion and extend to the end of the template at the highest enzyme concentration (Figure 2C, reactions 2, 4, 5 and 7, and Figure S2B).
In both of these multiple-nucleotide incorporation assays, the predominant factor in the enzyme activity is the linker sequence, but in some cases there is a smaller contribution from other domains. For example, at the highest polymerase concentration, the Dpo4-Dbh-Dbh chimera is more active than Dbh and the other Dbh linker-containing chimeras, 68% vs. less than 36% primer extension (Figure 2B lane 3a vs. lanes 1a, 2a, and 4a and Figure S2A). Also, at the lowest polymerase concentration, the Dpo4-Dpo4-Dbh chimera is more active than Dpo4 and the other Dpo4 linker-containing chimeras, 65% vs. less than 12% primer extension (Figure 2B, lane 6c vs. lanes 4c, 7c and 8c, and Figure S2).
In assays where each of the four dNTPs was included individually in a reaction to measure polymerase fidelity, the linker domain was also the major determinant of nucleotide misincorporation activity (Figure 2D and Figure S2C). Dbh and all of the Dbh linker-containing chimeras displayed the highest accuracy, predominantly incorporating only dCTP, the correct incoming nucleotide, opposite a guanosine templating base, with at least 77% primer extension with dCTP compared to at most 30% primer extenstion with any of the incorrect nucleotides (Figure 2D, reactions 1, 3, 6, and 8 and Figure S2C). In contrast, Dpo4 and all of the Dpo4 linker-containing chimeras were able to incorporate each of the three incorrect nucleotides to a significant extent, with 89–93% primer extension with dCTP compared to 46–88% primer extension with the incorrect nucleotides (Figure 2D, reactions 2, 4, 5 and 7). In fact, in many of the reactions, enzymes containing the Dpo4 linker were able to incorporate multiple copies of the single nucleotide provided, and to a much greater extent than any of the enzymes containing the Dbh linker.
Again, protein sequences outside the linker contribute some to the activity, but much less than the linker sequences. For example, the Dpo4-Dbh-Dpo4 chimera is somewhat less accurate, incorporating two dCTPs efficiently (95% primer extension, about equally distributed between the two products), in contrast to Dbh, Dbh-Dbh-Dpo4 and Dpo4-Dbh-Dbh, which predominantly incorporate just one (Figure 2D, lane 8C vs. lanes 1C, 3C, and 6C and Figure S2C), indicating that the polymerase domain of Dpo4 does contribute somewhat to the increased error rate when it is paired with the Dbh linker and Dpo4 LF. This effect, however, is quite small compared to the contributions from the linker.
The linker sequence determines the protein conformation
We have determined five crystal structures of four out of the six chimeric proteins, at resolutions ranging from 1.9 to 2.35 Å (Table 1 and Figure 3), and we have determined an additional structure of Dbh (Supplemental Data). All of the chimeras crystallized under similar conditions, conditions that also allow growth of the Dbh and Dpo4 parental enzyme crystals. The structures include all three of the chimeras containing the Dpo4 linker sequence (Dbh-Dpo4-Dpo4, Figure 3A,B; Dbh-Dpo4-Dbh, Figure 3C; Dpo4-Dpo4-Dbh, Figure 3D) and one of the chimeras containing the Dbh linker sequence (Dbh-Dbh-Dpo4; Figure 3E). Three of the structures are ternary complexes, containing protein, primer-template DNA and incoming dNTP (Figure 3A–C) while two are binary complexes, without incoming dNTP (Figure 3D–E). Three of the structures have two complexes in the asymmetric unit, with minimal differences between complexes (RMSDs of 0.11 Å, 0.53 Å and 0.85 Å, repectively, for Dbh-Dbh-Dpo4 #1, Dbh-Dpo4-Dbh and Dpo4-Dpo4-Dbh). Unless otherwise noted, the descriptions that follow apply to both complexes in an asymmetric unit, as most structural features are identical. Despite many attempts, we were unable to crystallize the chimeric polymerases with the same primer-template DNA. The sequences used for each crystal are shown in Figure 3.
Table 1.
Data Collection and Refinement Statistics
| Dbh-Dpo4- Dpo4 #1 |
Dbh-Dpo4- Dpo4 #2 |
Dbh-Dpo4- Dbh |
Dpo4-Dpo4- Dbhb |
Dbh-Dbh- Dpo4 |
|
|---|---|---|---|---|---|
| PDB Code |
4F4W | 4F4X | 4F4Y | 4F4Z | 4F50 |
| Data Collectiona | |||||
| Space Group | P21 | P21212 | P21 | P1 | P41212 |
| Complexes per a.s.u. | 2 | 1 | 2 | 2 | 1 |
| Unit Cell Dimensions (Å) Lengths: a, b, c (Å); Angles |
52.82, 99.66, 101.63; β=90.16° |
101.12, 102.31, 52.88 |
53.31, 103.76, 112.99; β=101.76° |
50.97, 51.20, 98.84; α=75.6°, (β=82.97° γ=70.14° |
124.48, 124.48, 70.47 |
| Beamline | X2 | X25 | X25 | X25 | X25 |
| Wavelength (Å) | 1.075 | 1.1 | 1.1 | 0.9795 | 0.9795 |
| Resolution Range (Å) (outer shell) |
30-1.90 (1.93-1.90) |
30-2.04 (2.08-2.04) |
30-2.35 (2.39-2.35) |
30-2.3 (2.34-2.30) |
30-2.22 (2.26-2.22) |
| Reflections Measured | 278,972 | 170,978 | 132,008 | 40,538 | 262,511 |
| Average redundancy | 3.5 (2.5) | 4.9 (2.2) | 2.8(1.7) | 1.8(1.4) | 9.7 (3.8) |
| Completeness (%) | 96.5(81.1) | 97.3 (80.9) | 95.3 (80.7) | 81.1 (50.5) | 97.4 (79.9) |
| Rmeroe(%) | 3.9 (16.6) | 7.0 (33.3) | 8.4 (24.5) | 3.0(19.0) | 5.1 (35.3) |
| I/sig(I) |
28.5 (3.8) | 24.4 (5.1) | 16.5(3.9) | 21.3(2.0) | 31.9(2.2) |
| Refinement | |||||
| Number of reflections | 80,155 | 35,101 | 47,570 | 32,443 | 27,377 |
| Resolution Range (Å) | 30-1.9 | 30-2.04 | 30-2.3 | 30-2.3 | 30-2.2 |
| Rwork (%) / Rfree (%) | 18.7/21.9 | 21.8/25.0 | 22.2/25.9 | 18.5/26.0 | 21.2/26.9 |
| Molecular composition: | |||||
| # Amino acid residues | 684 | 342 | 686 | 684 | 337 |
| # Nucleotides | 56 | 32 | 68 | 47 | 24 |
| # Incoming dCTP | 2 | 1 | 2 | 0 | 0 |
| #Ca2+lons | 4 | 4 | 4 | 3 | 1 |
| # Water molecules | 708 | 186 | 252 | 91 | 74 |
| Average B-factors (A2) | |||||
| Macromolecules | 32.7 | 43.0 | 49.3 | 69.3 | 81.3 |
| Solvent | 40.5 | 42.8 | 41.2 | 53.6 | 58.8 |
| RMS deviations | |||||
| Bonds (Å) | 0.011 | 0.008 | 0.006 | 0.007 | 0.008 |
| Angles (°) | 1.493 | 1.323 | 1.110 | 1.142 | 1.227 |
| Ramachandran Plot | |||||
| Favored (%) | 98.4 | 98.0 | 94.1 | 95.3 | 95.2 |
| Allowed (%) | 1.6 | 2.0 | 5.1 | 4.6 | 4.8 |
| Outliers (%) | 0.0 | 0.0 | 0.8 | 0.1 | 0.0 |
Values for outermost resolution shells are given in parentheses.
Diffraction from these crystals was highly anisotropic. The high resulution limit used was based on a signal-to-noise ratio of 2.0 in the outer resolution shell, even though the completeness of the data was low (50% in the outer shell; 81 % overall). At a high resolution limit of 2.75 A, the signal-to-noise ratio was 7.0 in the outer shell, with a completeness of 79.0% (91.6% overall) and a merging R-factor of 8.6% (2.8% overall).
Figure 3. Structural Overview of Chimeric Polymerases.
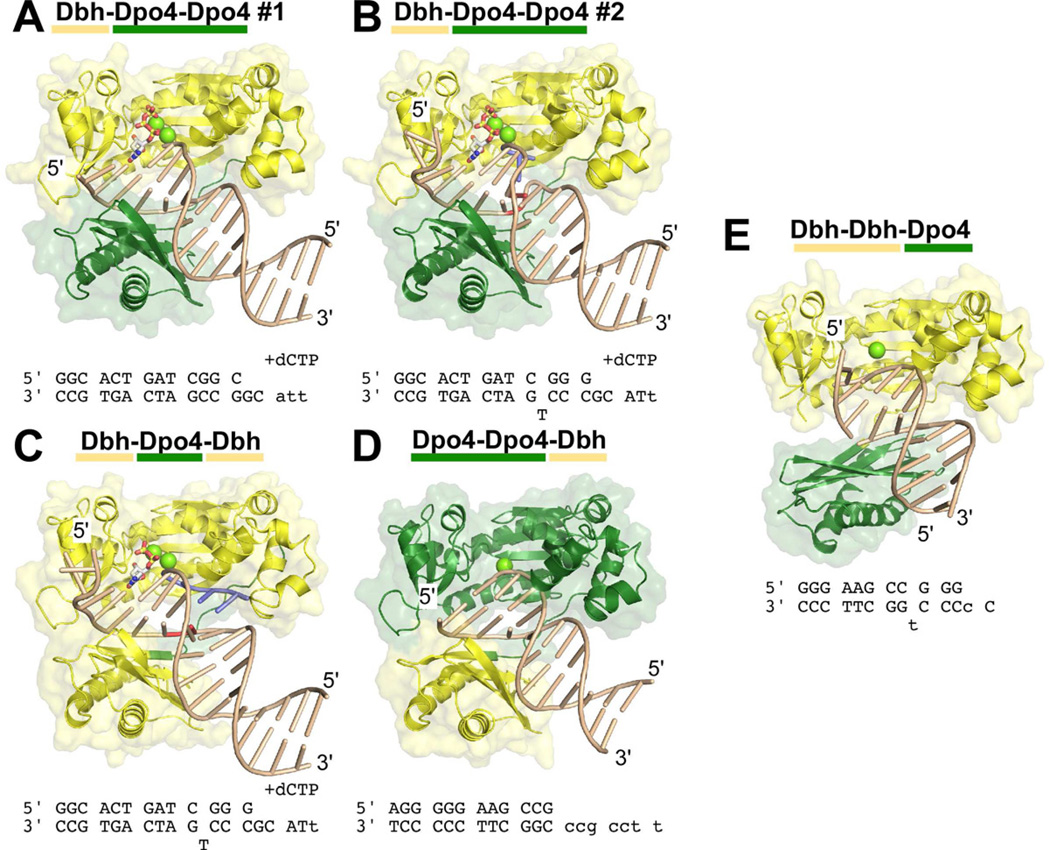
The structures of five complexes between chimeric polymerases and DNA are shown: (A) Dbh-Dpo4-Dpo4, complex #1, (B) Dbh-Dpo4-Dpo4, complex #2, (C) Dbh-Dpo4-Dbh, (D) Dpo4-Dpo4-Dbh, and (E) Dbh-Dbh-Dpo4. The protein is shown with a molecular surface representation and is colored by the parental sequence: Dpo4 (green), Dbh (yellow). DNA primer-template sequences and nucleotide (if any) that were included during co-crystallization are shown below each structure; sequences shown in lower case were not visible in the electron density maps. DNA is shown in a ladder representation. Nucleotides shown in red (in B and C) were designed to be unpaired; nucleotides shown in blue (in B and C) were added to the primer-terminus during co-crystallization (see Supplemental Material, Figure S3 D and E). Calcium ions at the active site are shown as chartruse spheres; incoming nucleotides are shown in stick representation, colored by atom: carbon (white), oxygen (red), nitrogen (blue), phosphate (yellow). See Figures S3 and S4 for additional details.
All of the structures with the Dpo4 linker were solved by molecular replacement using full-length Dpo4 as a search model, but no significant solutions were identified using full-length Dbh. The structure of Dbh-Dbh-Dpo4 was solved by molecular replacement using the polymerase domain of Dbh as a search model; the LF/PAD was placed manually as refinement proceeded and electron density for this domain became clear.
The most significant aspect of the structures is that they demonstrate that the identity of the linker sequence is the major determinant of the overall polymerase conformation: chimeras containing the Dpo4 linker are in the same conformation as Dpo4 and the chimera containing the Dbh linker is in a Dbh-like conformation (Figure 4). The structures of the Dpo4-linker chimeras superimpose on Dpo4 with an average root-mean-square deviation (RMSD) of 1.15 Å (range: 0.98 to 1.27 Å over 340 CA atoms; Figure 4A). In contrast, these chimeras superimpose on Dbh with an average RMSD of 4.95 Å (range: 4.65–5.22 Å over 340 CA atoms). With the polymerase domains aligned, the LF/PAD of these chimeras would need to rotate approximately 50° around an axis roughly parallel to the DNA in order to line up with the LF/PAD of Dbh (Figure 4A). Conversely, over all CA atoms, the Dbh-Dbh-Dpo4 chimera superimposes on Dbh with an RMSD of 2.97 Å compared to 5.21 Å for Dpo4 (PDB code 2AGQ(Vaisman et al., 2005)). Rotating the LF/PAD of the chimera by 30° a round an axis nearly perpendicular to the DNA axis would bring the domains into alignment (Figure 4B).
Figure 4. Chimeric polymerase conformation depends on the identity of the linker sequence.
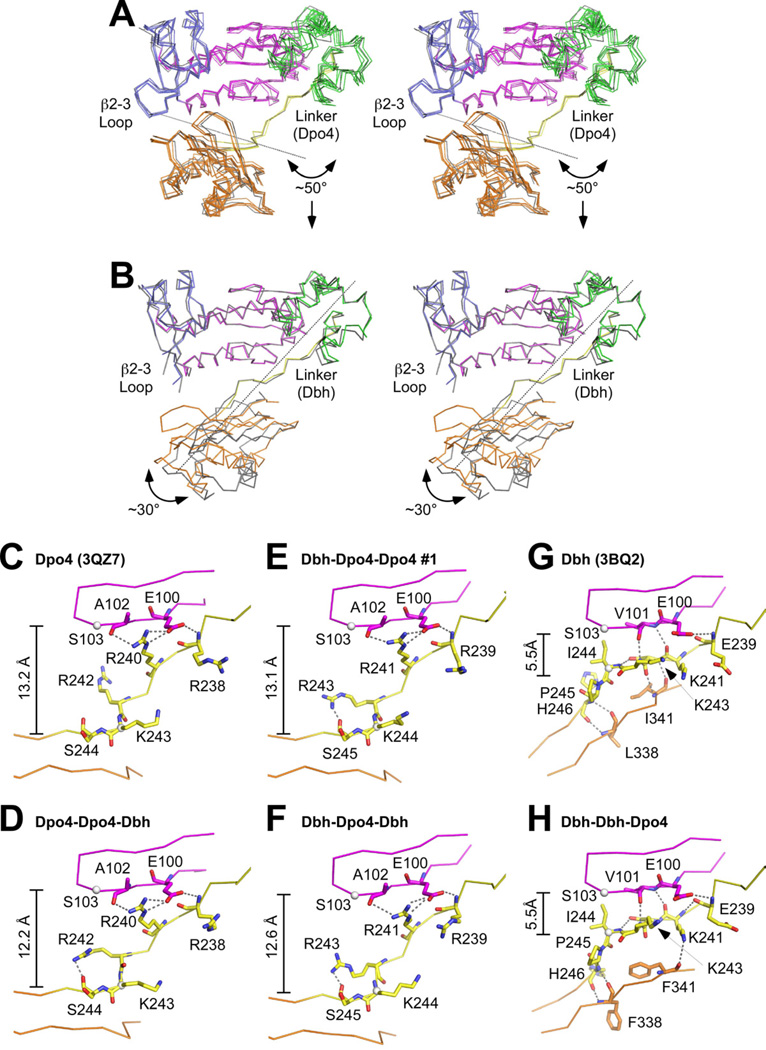
(A) Stereo diagram of the Dpo4-linker-containing chimeric polymerases (Figure 3A–D) superimposed on Dpo4 (PDB code 2AGQ (Vaisman et al., 2005))). Individual proteins superimpose with RMSDs ranging from 0.80 to 1.82 Å over 340 CA atoms. Chimeric proteins are colored as in Figure 1A; Dpo4 is colored gray. Arrow and dotted line show the magnitude and axis of rotation that would be needed to align the LF/PAD of these proteins onto the LF/PAD of Dbh. (B) Stereo diagram of the Dbh-linker-containing chimera, Dbh-Dbh-Dpo4 (Figure 3E), superimposed on Dbh (PDB code 3BQ2 (Wilson and Pata, 2008)). RMSD 2.97 Å over 337 CA atoms. Chimeric proteins are colored as in Figure 1A; Dbh is colored gray. Arrow and dotted line show the magnitude and axis of rotation that would be needed to align the LF/PAD of Dbh-Dbh-Dpo4 with the LF/PAD of Dbh. (C–H) Close-up views of the linker (yellow) and nearby sequences in the palm (magenta) and LF/PAD (orange) domains of (C) Dpo4 (PDB code 3QZ7), (D) Dpo4-Dpo4-Dbh, (E) Dbh-Dpo4-Dpo4 #1, (F) Dbh-Dpo4-Dbh, (G) Dbh (PDB code 3BQ2), and (H) Dbh-Dbh-Dpo4. Residues discussed in the text are show in stick representation. Dotted lines indicate hydrogen bonds.
Because the chimeras did not all crystallize with the identical DNA substrate, it is possible that the conformation is influenced by the DNA instead of (or in addition to) the linker, but we do not think this is the case. For Dpo4, we have previously shown that DNA containing a bulged base in the template strand does not cause Dpo4 to adopt the Dbh conformation (Wu et al., 2011). For Dbh, the conformation is nearly identical in all of the structures that have been determined (RMSDs between 0.69 Å and 2.22 Å; Figure S2D): three independent views of the apo-enzyme (Silvian et al., 2001), three structures in complex with DNA containing a bulged template base with and without incoming nucleotide (Wilson and Pata, 2008), and one ternary complex without a bulged base (Supplemental Data).
In all of the chimeras, the linker sequence retains the conformation of the parental enzyme (Figure 4 and Supplemental Figure S1D). The largest structural differences between the Dbh and Dpo4 linkers are located in the last six amino acids of the linker (Figure 4C–H; residues Lys241-His246 in Dbh and Arg240-Ile245 in Dpo4; equivalent to residues Arg241-Ile246 in the Dbh-Dpo4-Dpo4 and Dbh-Dpo4-Dbh chimeras). The Dpo4 linker adopts a more extended conformation (Figure 4C), while the Dbh linker maintains closer contacts with both the palm and LF/PAD (Figure 4D). The distance from the CA atom of Ser103 (in the palm of both Dbh and Dpo4) to the CA atom of Lys244 in the Dpo4 linker ranges from 12.2 to 13.2 Å (Figure 4C–F), while the distance to the CA atom of Ile 244 in the Dbh linker (the structurally equivalent residue) is just 5.5 Å (Figure 4G,H).
In all of the chimeras that contain the Dpo4 linker, the LF/PAD spans the major groove of the primer-template DNA duplex (Figure 3A–D), with the outer strands of the β-sheet forming a series of hydrogen bonds with the DNA backbone on both strands. Additionally, the β2–3 loop on the side of the fingers contacts the LF/PAD and becomes well ordered, even in the three chimeric protein structures that contain the Dbh polymerase domain (Figures 3A–C and 4A). The β2–3 loop has never been well-ordered in structures of the Dbh parental enzyme, but in these chimeras it adopts the same conformation as in Dpo4 (Figure 4A and Figure S3A). Arg36 in the loop forms hydrogen bonds with two residues in the LF/PAD: the side chain oxygen (OD1) of Asn255 and the backbone carbonyl of residue Leu252 in Dbh (Met251 in Dpo4). In this conformation, two conserved arginines in the LF/PAD (Arg332 and Arg333 in Dbh, Arg331 and Arg332 in Dpo4) are able to contact the phosphate of the templating base and Thr251 can hydrogen bond to the phosphate immediately 3’ to the templating base (Figure S3A).
In contrast to the Dpo4 linker, the Dbh linker appears to restrict the movement of the LF/PAD, preventing it from fully docking in the DNA duplex and making contact with the β2–3 loop (Figures 3E and 4B). In the Dbh-Dbh-Dpo4 chimera, the outer strand of the LF/PAD β-strand points into the major groove of the DNA and the β2–3 loop on the side of the fingers is disordered (Figures 3E and 4B). In structures of the Dbh parent (Silvian et al., 2001; Wilson and Pata, 2008), the short β-strand in the linker forms backbone hydrogen bonds with β-strands in both the polymerase domain and the LF/PAD, holding the two domains together (not shown). In the Dbh-Dbh-Dpo4 chimera, however, the interactions with the polymerase palm are maintained, but the interactions with the LF/PAD are disrupted (Figure 4H vs 4G). These are replaced in the chimera by two weaker hydrogen bonds (~3.5 Å long) that form between the side chain nitrogen (NZ) of Lys241 (in the Dbh linker) and the backbone carbonyl of Phe341 (in the Dpo4 LF/PAD) and between the side chain nitrogen of Arg280 (in the Dpo4 LF/PAD; not shown) and the backbone carbonyl of Ile244 (in the Dbh linker).
To further define the linker residues that are important for the differences between Dbh and Dpo4, we exchanged the six amino acids that adopt different conformations and found that overall activity, ability to bypass an abasic site, and base-substitution fidelity were exchanged as well (Figure 5; quantitation shown in Figure S2). Dbh containing the Dpo4 linker residues (Dbh-RVRKSI) behaves like Dpo4 (Figure 5, reaction set 1 vs. Figure 2, reaction set 2) while Dpo4 containing the Dbh linker residues (Dpo4-KSKIPH) behaves like Dbh (Figure 5, reaction set 3 vs. Figure 2, reaction set 1). Exchanging three of these six amino acids, residues 242–244 (Arg-Lys-Ser) of Dpo4 and residues 243–245 (Lys-Ile-Pro) of Dbh, gives a similar result (Figure 5, reaction set 2, Dbh-RKS and reaction set 4, Dpo4-KIP).
Figure 5. Nucleotide Incorporation on Undamaged and Abasic Site DNA by Chimeric Polymerases with 3 or 6 Linker Residues Exchanged.
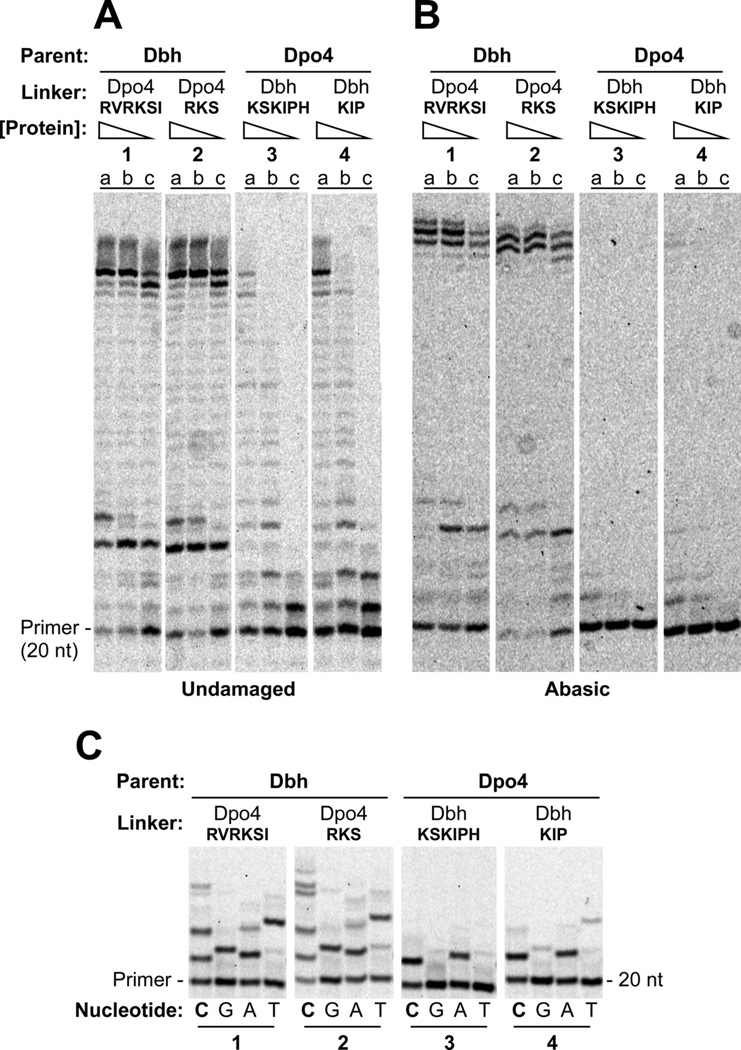
(A) Polymerase assays on undamaged primer-template DNA containing (a) 160 nM, (b) 40 nM, or (c) 10 nM protein with 40 nM DNA and 1 mM each dATP, dCTP, dGTP and dTTP. Reactions were incubated at 60°C for 5 min. DNA sequences shown in Figure 2A. (B) Polymerase assays on primer-template DNA containing abasic site in the template strand immediately adjacent to the terminal basepair. Protein and substrate concentrations were the same as in A. Reactions were incubated at 60°C for 10 min. (C) The undamaged primer-template DNA used in A was used as the substrate in reactions that separately contained 1mM dCTP (lanes C), dGTP (lanes G), dATP (lanes A), or dTTP (lanes T) with 1µM enzyme, and 40nM primer-template DNA. Reactions were incubated at 37°C for 5 min. See Figure S2 for quantitation.
No single point mutation in the linker is sufficient to completely change the enzyme activity in all the assays (Figure 6; quantitation shown in Figure S2). Of the residues tested, the S244P mutation in Dpo4 has the most significant effect on its own: dramatically decreasing bypass of an abasic site (Figure 6B, Dpo4 parent, reaction set 5 vs. reaction set 1) and increasing the single nucleotide incorporation fidelity somewhat (Figure 6C, Dpo4 parent, reaction set 5 vs. reaction set 1). The complementary mutation in Dbh, P245S, has little effect on lesion bypass (Figure 6B, Dbh parent, reaction set 5 vs. reaction set 1), but it does decrease the fidelity of Dbh to the extent that it is comparable to Dpo4 (Figure 6C, Dbh parent, reaction set 5 vs. Dpo4 parent, reaction set 1). The other point mutations tested (Dbh K241R, K243R and I244K; Dpo4 R240K, R242K and K243) had minimal effects on the activities of the parental polymerases (Figure 6 and Figure S2).
Figure 6. Single and Multiple Nucleotide Incorporation by Dbh and Dpo4 with Individual Point Mutations in the Linker.
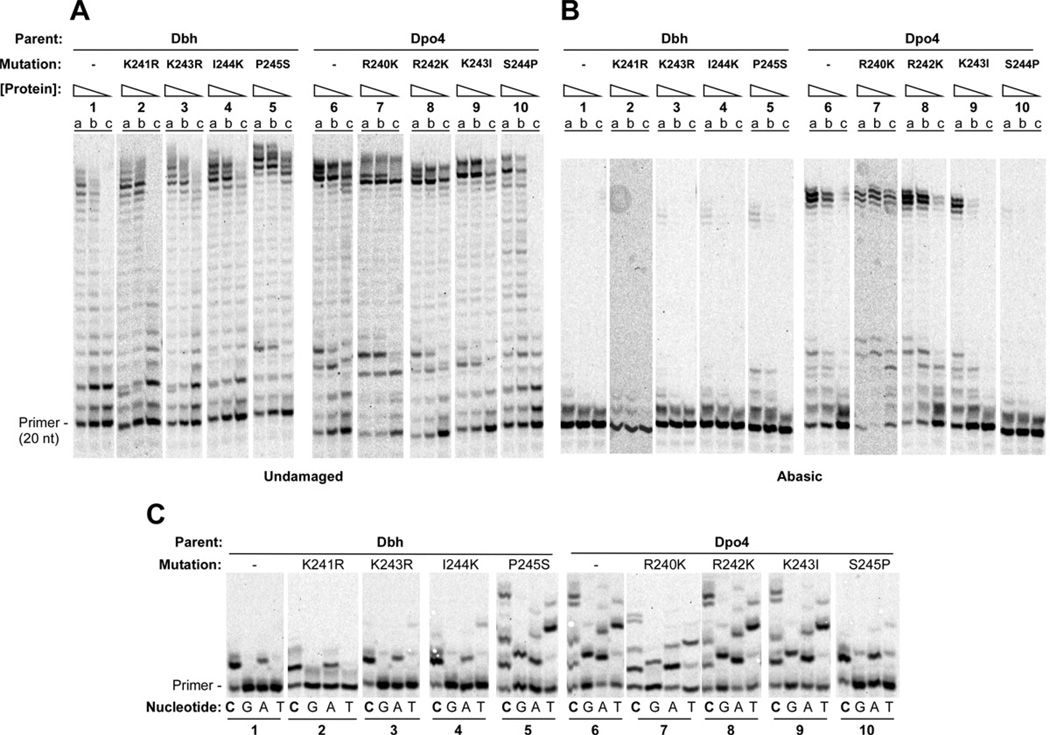
Primer-template sequences same as shown in Figure 2A. (A) Polymerase assays on undamaged primer-template DNA containing (a) 160 nM, (b) 40 nM, or (c) 10 nM protein with 40 nM DNA and 1 mM each dATP, dCTP, dGTP and dTTP. Reactions were incubated at 60°C for 5 min. (B) Polymerase assays on primer-template DNA containing abasic site in the template strand immediately adjacent to the terminal basepair. Protein and substrate concentrations were the same as in A. Reactions were incubated at 60°C for 10 min. (C) The undamaged primer-template DNA was used as the substrate in reactions that separately contained 1mM dCTP (lanes C), dGTP (lanes G), dATP (lanes A), or dTTP (lanes T) with 1µM enzyme, and 40nM primer-template DNA. Reactions were incubated at 37°C for 5 min. See Figure S2 for quantitation.
DISCUSSION
Long distance effects of the linker on catalytic activity
The data presented here demonstrate that the linker sequence connecting the catalytic and C-terminal DNA binding domains of the archaeal Y-family polymerases is a major determinant of catalytic activity on undamaged DNA, ability to bypass an abasic site and nucleotide incorporation fidelity. Furthermore, the linker is also a primary determinant of the overall polymerase conformation.
The most striking example of the importance of the linker is the Dbh-Dpo4-Dbh chimera: the polymerase and LF/PAD domains are both derived from Dbh, yet the conformation and catalytic activities are like those of Dpo4, the source of the linker. This chimera clearly establishes that the Dpo4 linker is sufficient to allow the rest of the protein, which is derived from Dbh, to adopt a Dpo4-like conformation and to behave like Dpo4. Crystals of this chimera even show Dpo4-like features of error-prone DNA synthesis (Supplemental Data).
The key differences between the Dbh and Dpo4 linkers are located in three amino acids near the junction with the LF/PAD (Lys243-Ile244-Pro245 in Dbh; Arg242-Lys243-Ser244 in Dpo4). The significance of these differences appears to be two-fold: the Arg-Lys-Ser residues in Dpo4 all make direct polar contacts to the DNA backbone, while the proline in Dbh seems to restrict the linker from moving into a position where it could directly contact the DNA. Of the single point mutations, Dpo4 S244P had the greatest effect, essentially eliminating the ability of Dpo4 to bypass an abasic site (Figure 6B, reaction set 10). The reciprocal mutation, Dbh P245S, was not able to substantially increase abasic site bypass (Figure 6B, reaction set 5), but it did increase nucleotide misincorporation of Dbh to a level comparable to that of Dpo4 (Figure 6C, reaction set 5 vs. 6). The next most important mutations were Dbh I244K and Dpo4 K243I, both of which had relatively modest effects compared to Dpo4 S244P.
Conformational differences in the linker are communicated to the active site via the LF/PAD domain. The extended conformation of the Dpo4 linker sequence allows the LF/PAD to move into a position where Arg36 in the β2–3 loop on the side of the fingers can contact the LF/PAD and form a shallow groove for the template DNA as it enters the active site. Two conserved arginines in the LF/PAD (Arg331/Arg332 in Dpo4; Arg332/Arg333 in Dbh) form hydrogen bonds with the phosphate on the 5’ side of the MANUSCRIPT 18 templating base, and a conserved threonine (Thr250 in Dpo4; Thr251 in Dbh) hydrogen bonds with the phosphate on the 3’ side of the templating base.
The position of the LF/PAD defines one edge of the nascent basepair binding pocket while the catalytic residues define the other. With the Dpo4 linker, the pocket is just wide enough to fit a standard Watson-Crick basepair and the substrates are thus positioned well for catalysis. With the Dbh linker, however, the LF/PAD is not able to reach the β2–3 loop. The templating base is therefore not constrained along the phosphate backbone and it is positioned further away from the active site compared to structures with the Dpo4 linker. The base paired with the templating base is consequently pulled further away from the active site. This provides an explanation for why, in the ternary complex of Dbh (Wilson and Pata, 2008), the incoming nucleotide and primer terminus are not positioned optimally for catalysis and consequently why Dbh (and the Dbh-linker-containing chimeras) perform the nucleotidyl transferase reaction much more slowly than Dpo4 (and the Dpo4-linker-containing chimeras).
Different polymerase conformations, arising from alterations in the linker, result in the observed differences in the shape and size of the nascent basepair binding pocket. These structural differences thus provide an explanation for how translesion synthesis and nucleotide incorporation fidelity could be so dramatically impacted by sequence changes so far away from the active site.
Conformational dynamics and the evolution of enzyme specificity
Extensive structural, kinetic and mutational studies of the A-, B-, and X-family DNA polymerases have identified close steric restraints around the nascent basepair binding pocket as a key source of high replication fidelity (Beard and Wilson, 2003). In these polymerase families, the fingers domain undergoes a large conformational change that is stabilized in the closed conformation by binding of the correct, but not incorrect, incoming nucleotide. The importance of the dynamics of the conformational change to fidelity have recently been demonstrated for T7 DNA polymerase (Jin and Johnson, 2011): mutating two glycine residues to alanines in a hinge sequence at the base of the fingers reduced the mobility of the domain and simultaneously decreased polymerase fidelity. The key to achieving high fidelity synthesis is that binding of only the correct nucleotide slows the rate of fingers opening, committing the correct ternary complex to catalysis (Johnson, 2008).
The Y-family polymerases do not display the same large-scale conformational change in the fingers domain as in the other DNA polymerase families, and instead have a relatively rigid, preformed active site that is in position to catalyze nucleotide addition when substrates are bound correctly. Rather than movement of the fingers, movement of the LF/PAD appears to alter fidelity and specificity because of the ease or difficulty of positioning the DNA and dNTP substrates at the active site. Thus, conformational changes affect the fidelity of Y-family polymerases in a distinctly different way than in other DNA polymerase families: flexibility of the LF/PAD allows both correct and incorrect nucleotides to be positioned at the active site for catalysis while restricting this flexibility results in more accurate nucleotide incorporation.
Two questions raised by the structures presented here are: how are the substrates positioned and what is the protein conformation when Dbh and the Dbh-linker-containing chimeras catalyze nucleotide addition? Further experiments will be required to address these questions definitively, but we favor the idea that the primer-template DNA, dNTP and LF/PAD all move together to transiently adopt a conformation similar to that observed in structures of Dpo4 and the Dpo4-linker-containing chimeras, for the following reasons. First, mutation of the “steric gate” residue (Phe12) of Dbh to alanine reduces the steric selection against ribonucleotide incorporation (DeLucia et al., 2006) and the also reduces the specificity that Dbh shows for bypassing N2-furfuryl-dG (Jarosz et al., 2006). In the ternary complex structure of Dbh, the incoming nucleotide has no contact with the steric gate residue, so it is difficult to imagine how the mutation could affect specificity unless there exists another state where they are in contact. Second, LF/PAD residues Arg331, Arg332, and Thr250, which are all involved in direct contacts to the templating DNA in Dpo4 crystal structures, are strictly conserved in Dbh, even though they do not make the same contacts as in Dpo4. Similarly, Arg36 in the β2–3 loop and Asn254 in the LF/PAD, which form the major contact between the fingers and LF/PAD domains in Dpo4, are identical in Dbh, even though this contact does not exist in the structures of Dbh.
The significance of the linker conformation in Dbh may be that it reduces the mobility of the LF/PAD. In contrast, Dpo4 appears relatively flexible, since the LF/PAD of Dpo4 has been found in several different positions: in the apo-enzyme structure (Wong et al., 2008), in one abasic-site containing DNA complex (Ling et al., 2004) and in the cocrystal structure of Dpo4 with PCNA (Xing et al., 2009), even though most DNA-bound structures of Dpo4 are in the conformation shown in Figure 1B, irrespective of having a bound nucleotide, a bulged base, or a variety of different lesions.
Flexibility of the inter-domain linker may allow efficient binding of DNA containing bulky or distorting lesions, and it may allow a wide range of lesions to be substrates, but those advantages may come at the cost of an increased error rate, as seen in the comparison of Dpo4 and Dbh. Dpo4 is able to bypass an especially wide range of lesions, including ones such as thymidine dimers (Boudsocq et al., 2004) that are not generally preferred substrates for DinB homologs. An active site than can be readily adapted to different substrates would be especially useful in organisms that have just one Y-family polymerase.
Many of the eukaryotic Y-family polymerases have sequence insertions that form additional contacts between the core polymerase and LF/PAD domains that could also influence the LF/PAD conformation. Based on the work presented here, it appears that reducing inter-domain mobility would increase specificity by reducing the variability in the geometry and size of the substrate binding pocket, which may also increase fidelity. Polymerases kappa, eta and Rev1p all contain sequences insertions that contact both the polymerase and LF/PAD domains and thereby reduce domain mobility (Alt et al., 2007; Lone et al., 2007; Nair et al., 2005; Silverstein et al., 2010; Swan et al., 2009; Trincao et al., 2001; Uljon et al., 2004).
Interestingly, Pol iota, which preferentially incorporates dGTP opposite a templating T, is missing the β2–3 loop on the side of the fingers domain (Nair et al., 2004). Because the loop is missing, the LF/PAD is located ~2 Å closer to the catalytic residues, narrowing the nascent basepair binding pocket, and which was proposed to explain for pol iota’s preference for forming Hoogsteen rather than standard Watson-Crick basepairs (Kirouac and Ling, 2009; Nair et al., 2004). Exchanging the fingers domains of Dpo4 and pol iota caused Dpo4 to have misincorporation specificity similar to pol iota (Kirouac & Ling, 2009), consistent with the proposal. This same study did not find a role for the linker in the specificity of these enzymes, however the linkers of both Dpo4 and pol iota extend away from the palm domain (unlike Dbh), so we would not predict that the linker would be a source of the difference between these two Y-family polymerases (Kirouac & Ling, 2009). Given the large evolutionary distance between the two enzymes, it may not be surprising that the sources of specificity are more complex.
The role of conformational changes in enzyme specificity has been controversial (Johnson, 2008). Examples can be difficult to identify because of the need to detect multiple conformational states and to demonstrate that changing the conformation causes a change in specificity. Recent work on the p53 tumor suppressor demonstrates that a conformational switch in a loop that directly contacts DNA is important for binding specific vs. non-specific DNA sequences by slowing the dissociation rate of p53 from the specific target site (Petty et al., 2011). In another case, the substrate specificity of human breast cancer resistance protein, a drug efflux pump, was changed by a single proline to alanine mutation in a transmembrane helix; structural modeling suggested that a change in helix flexibility cause the change in specificity (Ni et al., 2011). Thus conformational changes that are initiated both close to and far away from the substrate binding site can significantly impact specificity.
The Y-family of polymerases is distinctive in having an extraordinarily wide range of lesion specificity and nucleotide incorporation fidelity. Within this polymerase family, Dpo4 and Dbh have relatively high sequence conservation, but still have diverged enough so that they have acquired different specificities. Finding that at most three amino acid changes are required to change enzyme specificity provides a very clear example of how conformational dynamics can contribute to the evolution of enzyme specificity, even when the residues involved are quite distant from the active site. Finally, one intriguing implication of these findings is that the conformation of Y-family polymerases could be modulated allosterically, through interactions with other molecules or through post-translational modifications, thus providing a potential mechanism for regulating Y-family DNA polymerase activity and specificity in cells.
EXPERIMENTAL METHODS
Protein expression and purification
Dbh and Dpo4 were expressed and purified as described previously (Wilson and Pata, 2008; Wu et al., 2011). Chimeras were generated by overlap PCR from the parental Dbh and Dpo4 plasmids, simultaneously adding a C-terminal hexahistidine tag, and then cloning into the expression vector pKKT7. The intrinsic activities of Dpo4 (Fiala & Suo, 2004) and Dbh (unpublished data) are not altered by the presence of a tag at the C-terminus. All plasmids were transformed into E. coli BLR(DE3)pLysS cells (Novagen) and grown in 2xYT medium at 37°C. The cells were then induced with 0.5 mM IPTG and the temperature was reduced to 20°C for overnight expression.
For the chimeras, cell pellets were resuspended in lysis buffer (20 mM HEPES at pH 7.5, 500 mM NaCl). Cells were lysed by sonication and heated at 75°C for 20 min. Subsequent steps were performed at 4°C. Cleared ly sates were loaded onto HiTrap chelating HP columns (2 × 5 ml; GE Healthcare) charged with nickel sulfate and chimeras were eluted using a linear gradient of 50 mM to 1 M imidazole in 20 mM HEPES at pH 7.5, and 500 mM NaCl. Pooled fractions were dialyzed into storage buffer (20 mM HEPES at pH 7.5, 100 mM NaCl, 0.5 mM EDTA, and 5 mM DTT) and kept at 4°C. Concentrations were determined by UV absorbanc e at 280 nm using a calculated extinction coefficient of 22,350 M−1 cm−1.
Polymerase assays
Primers and templates were synthesized by Integrated DNA Technologies, with a 5’-6-carboxyfluorescein (6-FAM) label on the primers, and were annealed in 10 mM HEPES (pH 7.5) and 50 mM NaCl by heating for 2 min at 95°C, incubating for 5 min at 55°C, and then slowly cooling to 25°C. Reactions were performed in a solution containing 20 mM HEPES (pH 8.0), 85 mM NaCl, 5 mM MgCl2, 1 mM DTT, 40 nM annealed primer-template DNA. Incubation temperatures (22°C, 37°C or 60 °C), incubation times (0 to 20 min), and polymerase concentrations (10 nM to 4 µM) are given in figure legends. Each nucleotide added to the reactions was present at 1 mM. Reactions were quenched by the addition of an equal volume of 80% formamide containing 50 mM EDTA, with bromophenol blue and xylene cyanol dyes. Samples were incubated at 95°C for 5 min just prior to electrophoresis on a 17.5% polyacrylamide (19:1), 7.5 M urea, 1xTBE gel that was preheated and run at 50°C. Gels were imaged using a Typhoon 9400 scanner and quantified using ImageQuant software (GE Healthcare).
Crystallization and structure determination
Primer and template DNA oligonucleotides (sequences shown in Figure 3) were synthesized by Integrated DNA Technologies and annealed in a solution containing 10 mM HEPES (pH 7.5) and 50 mM NaCl by heating for 2 min at 95°C, incubating for 5 min at 55°C, and then slowly cooling to 25°C. Complexes were prepared at room temperatu re by combining 200 µM protein and 240 µM DNA in 25 mM HEPES (pH 7.0), 5 mM Ca(OAc)2, 85 mM NaCl, and 1 mM DTT (final concentrations). For ternary complexes, 1 mM dCTP was added. Crystals were grown at room temperature by hanging-drop vapor diffusion after mixing 2 µl of the complex with 2 µl of well solution. The well solutions for all chimeras contained 9–21% PEG-3350, 100 mM MES-Tris or HEPES (pH 5.9–7.1), 100 mM Ca(OAc)2, and 2.5% glycerol, with the exception that Dbh-Dpo4-Dbh also included 250 mM sucrose. Crystals were stabilized and cryoprotected by the addition of a solution containing 20% PEG-3350, MES-Tris (pH 6.5), 100 mM Ca(OAc)2, 25% v/v glycerol or 20% w/v sucrose, and 1 mM dCTP (for ternary complexes). Crystals were flash cooled in liquid nitrogen.
X-ray diffraction data were collected at Brookhaven National Laboratory (BNL), National Synchrotron Light Source (NSLS) beamlines X25 and X29, and were processed and scaled using HKL2000 (Otwinowski and Minor, 1997). The structures were solved by molecular replacement using Phaser (McCoy et al., 2005) as implemented in PHENIX (Adams et al., 2010). Search models used are described in the text. Structures were refined using PHENIX (Adams et al., 2010), alternating with cycles of manual rebuilding using Coot (Emsley et al., 2010) and PyMOL (Schrodinger, 2010). Structure figures were made using PyMOL (Schrodinger, 2010).
Supplementary Material
HIGHLIGHTS.
Y-family polymerases differ in fidelity and translesion synthesis specificity
Polymerase fidelity and specificity are controlled by the inter-domain linker
Three amino acids in the inter-domain linker are sufficient to determine conformation
Differences in polymerase conformation determine differences
ACKNOWLEDGEMENTS
We thank Satwik Kametkar, Indrajit Lahiri and Purba Mukherjee for critical reading of the manuscript, and Joachim Jaeger for insightful discussions. We thank the staff at BNL NSLS beamlines X25 and X29 for assistance during data collection. We acknowledge use of the Wadsworth Center’s Macromolecular Crystallography and Applied Genomic Technologies Cores. This work was funded by NIH grant #R01-GM080573 to J.D.P. Use of the BNL NSLS was supported by the U.S. Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank (www.pdb.org) with accession codes: 4F4W, Dbh-Dpo4-Dpo4 #1; 4F4X, Dbh-Dpo4-Dpo4 #2; 4F4Y, Dbh-Dpo4-Dbh; 4F4Z, Dpo4-Dpo4-Dbh; and 4F50, Dbh-Dbh-Dpo4.
AUTHOR CONTRIBUTIONS
J.D.P. designed the research; R.C.W. and M.J. performed the research; R.C.W. and J.D.P. analyzed the data; R.W. and J.D.P. wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D. Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure. 2003;11:489–496. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W, Woodgate R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem. 2004;279:32932–32940. doi: 10.1074/jbc.M405249200. [DOI] [PubMed] [Google Scholar]
- DeLucia AM, Chaudhuri S, Potapova O, Grindley ND, Joyce CM. The properties of steric gate mutants reveal different constraints within the active sites of Y-family and A-family DNA polymerases. The Journal of biological chemistry. 2006;281:27286–27291. doi: 10.1074/jbc.M604393200. [DOI] [PubMed] [Google Scholar]
- Dumstorf CA, Clark AB, Lin Q, Kissling GE, Yuan T, Kucherlapati R, McGregor WG, Kunkel TA. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc Natl Acad Sci U S A. 2006;103:18083–18088. doi: 10.1073/pnas.0605247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala KA, Suo Z. Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- Gueranger Q, Stary A, Aoufouchi S, Faili A, Sarasin A, Reynaud CA, Weill JC. Role of DNA polymerases eta, iota and zeta in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair (Amst) 2008;7:1551–1562. doi: 10.1016/j.dnarep.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- Jin Z, Johnson KA. Role of a GAG hinge in the nucleotide-induced conformational change governing nucleotide specificity by T7 DNA polymerase. The Journal of biological chemistry. 286:1312–1322. doi: 10.1074/jbc.M110.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA. Role of induced fit in enzyme specificity: a molecular forward/reverse switch. The Journal of biological chemistry. 2008;283:26297–26301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac KN, Ling H. Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase iota. The EMBO journal. 2009;28:1644–1654. doi: 10.1038/emboj.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Koonin EV, McDonald JP, Randall SK, Rabinovich N, Connaughton JF, Levine AS, Woodgate R. Identification of a DinB/UmuC homolog in the archeon Sulfolobus solfataricus. Mutat Res. 1996;357:245–253. doi: 10.1016/0027-5107(96)00164-9. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion: structural basis for base substitutions and frame-shifts. Molecular Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- Lone S, Townson SA, Uljon SN, Johnson R, Brahma A, Nair D, Prakash S, Prakash L, Aggarwal A. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta crystallographica Section D. Biological crystallography. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- McDonald JP, Rapic-Otrin V, Epstein JA, Broughton BC, Wang X, Lehmann AR, Wolgemuth DJ, Woodgate R. Novel human and mouse homologs of Saccharomyces cerevisiae DNA polymerase eta. Genomics. 1999;60:20–30. doi: 10.1006/geno.1999.5906. [DOI] [PubMed] [Google Scholar]
- Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- Ni Z, Bikadi Z, Shuster DL, Zhao C, Rosenberg MF, Mao Q. Identification of proline residues in or near the transmembrane helices of the human breast cancer resistance protein (BCRP/ABCG2) that are important for transport activity and substrate specificity. Biochemistry. 2011;50:8057–8066. doi: 10.1021/bi200573t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Methods in Enzymology, Volume 276, Macromolecular Crystallography, Part A. New York: Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Petty TJ, Emamzadah S, Costantino L, Petkova I, Stavridi ES, Saven JG, Vauthey E, Halazonetis TD. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. The EMBO journal. 2011;30:2167–2176. doi: 10.1038/emboj.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. [Google Scholar]
- Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, Aggarwal AK. Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature. 2010;465:1039–1043. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat Struct Biol. 2001;8:984–989. doi: 10.1038/nsb1101-984. [DOI] [PubMed] [Google Scholar]
- Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure of the human Rev1-DNA-dNTP ternary complex. J Mol Biol. 2009;390:699–709. doi: 10.1016/j.jmb.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier A, McDonald JP, Frank EG, Woodgate R. poliota, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- Uljon SN, Johnson R, Edwards TA, Prakash S, Prakash L, Aggarwal A. Crystal structure of the catalytic core of human DNA polymerase kappa. Structure. 2004;12:1395–1404. doi: 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. Embo J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash S, Prakash L. Fidelity and processivity of Saccharomyces cerevisiae DNA polymerase eta. J Biol Chem. 1999;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Pata JD. Structural insights into the generation of single-base deletions by the Y family DNA polymerase dbh. Mol Cell. 2008;29:767–779. doi: 10.1016/j.molcel.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Wong JH, Fiala KA, Suo Z. Snapshots of a Y-family DNA polymerase in replication: substrate-induced conformational transitions and implications for fidelity of Dpo4. J Mol Biol. 2008;379:317–330. doi: 10.1016/j.jmb.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wilson RC, Pata JD. The Y-family DNA polymerase Dpo4 uses a template slippage mechanism to create single-base deletions. J Bacteriol. 2011;193:2630–2636. doi: 10.1128/JB.00012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing G, Kirouac K, Shin YJ, Bell SD, Ling H. Structural insight into recruitment of translesion DNA polymerase Dpo4 to sliding clamp PCNA. 2009 doi: 10.1111/j.1365-2958.2008.06553.x. [DOI] [PubMed] [Google Scholar]
- Zhou BL, Pata JD, Steitz TA. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc Natl Acad Sci U S A. 2009;106:11552–11557. doi: 10.1073/pnas.0812548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.