

Abstract
APOE is the major known genetic risk factor for late-onset Alzheimer's disease (AD). While relationships between APOE-encoded apoE and β-amyloid are increasingly well described, mounting evidence supports wide-ranging effects of APOE on the brain. Specifically, APOE appears to impact brain network activity and closely related neuroenergetic functions that may be involved in vulnerability to neurodegenerative pathophysiology. These effects highlight the salience of further investigation into the diverse influences of APOE. Therefore, this article reviews the interplay between APOE and neuroenergetics and proposes areas for further investigation. This research may lead to the identification of novel therapeutic targets for the treatment and/or prevention of AD.
Keywords: APOE, apolipoprotein E, mitochondria, neuroenergetics, brain imaging, fMRI, FDG PET, cytochrome oxidase, biomarkers, Alzheimer's disease, energy metabolism, neurodegeneration
1. Introduction
Despite decades of intense research, the causes of Alzheimer's disease (AD) remain poorly understood and truly effective therapies remain out of reach. AD is expected to become markedly more prevalent over the next half-century (Ferri et al., 2005), which further intensifies the need to develop therapies as soon as possible. Since the initial reports linking APOE to Alzheimer's disease (AD) in the early 1990s (Corder et al. 1993; Strittmatter et al., 1993), considerable research has focused on elucidating the mechanisms by which the gene contributes to risk for the disease. Current evidence supports APOE-encoded apolipoprotein E (apoE) isoforms differentially modulating β-amyloid aggregation and clearance (Bu, 2009; Holtzman et al., 2012; Kim et al., 2009). Genetically, APOE ε4 is associated with dramatically increased risk, APOE ε3 is associated with neutral risk, and APOE ε2 is associated with decreased risk (Bertram and Tanzi, 2008; Gomez-Isla et al., 1996). APOE-related risk is gene-dose dependent: in the US, when compared to persons homozygous for risk-neutral APOE ε3, APOE ε4 homozygotes have up to 15 times and APOE ε4 heterozygotes up to 4 times the risk for developing AD (Ashford and Mortimer, 2002; Raber et al., 2004). Therefore, ameliorating APOE ε4's powerful effects may be an viable strategy to decrease AD incidence – delaying the average age of onset by 5 years could reduce the number of cases by over 50% and save nearly $300 billion in Medicare spending in coming years (Sperling et al., 2011).
While findings regarding apoE and its interactions with β-amyloid are essential to the current understanding of AD pathophysiology, it is important to further develop knowledge of the potential for APOE to impact brain function in a manner that may precede or be independent of β-amyloid pathology. Notably, apoE has known effects on cholesterol transport, inflammation, neurodevelopment, and synaptic plasticity, and study in these contexts clearly represents vital avenues of research. Mitochondrial energy metabolism and cellular bioenergetics in the brain (i.e., neuroenergetics) have also begun to be linked to the genetic risk conferred by APOE. Therefore, the intent of this article is to review the brain imaging background and potential cellular and molecular mechanisms for this emerging avenue of approach, with the hope that this rapidly evolving knowledge may stimulate innovative research approaches and the identification of tractable therapeutic targets for treatment and/or prevention of disease.
2. Metabolic brain imaging in Alzheimer's disease
Long-standing efforts have focused on the relevance of neuroenergetics in AD both as a mediator of β-amyloid induced changes and as an independent driver (Reddy and Beal, 2008; Smith et al., 2002; Swerdlow et al., 2010; Yao et al., 2011). The energetic needs of the human brain are remarkable; despite comprising only 2% of gross body mass, the brain accounts for 20% of the body's glucose and oxygen consumption (Jolivet et al., 2009). The provision of energy to the synapse is vital for the signaling function of neurons. Adenosine triphosphate (ATP) can be generated to meet this need primarily via the metabolism of glucose by glycolysis followed by the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, which is the most efficient method (36+ net ATP/glucose), or by glycolysis without subsequent oxidative phosphorylation which is faster yet relatively inefficient (2 net ATP/glucose). Despite the limited ATP yield of glycolysis without subsequent oxidative phosphorylation, the temporal dynamics of synaptic signaling make it an important source of energy because of its relative speed. Functional brain imaging has provided a wealth of information on the alterations in neuroenergetics and brain network activity that exist in AD. Brain energy metabolism is most often studied in human subjects by fluorodeoxyglucose (18F) positron emission tomography (FDG PET), which results in the calculation of the cerebral metabolic rate for glucose (CMRgl) for each region of interest. Early FDG PET studies of AD patients found progressive reductions in measurements of CMRgl in the parietal, temporal, and frontal association cortices (Friedland et al., 1985). FDG PET studies of subjects with mild cognitive impairment (MCI) demonstrate similar reductions (Boyle et al., 2006), and subjects who ultimately convert to AD show specific reductions in the prefrontal cortex and progressive decrements in posterior cingulate cortex (PCC; Drzezga et al., 2003, 2005). The PCC has been consistently noted as a region of particular significance in the metabolic alterations in AD, as it shows very early and comparatively large reductions in CMRgl (Minoshima et al., 1994) and sits at the convergence point of multiple metabolic covariance networks (Salmon et al., 2009). PCC CMRgl reductions in AD patients are thought to represent true changes in glucose metabolism and are not simply the result of local disease-related atrophy (Chételat et al., 2008; Ibáñez et al., 1998). In this context, CMRgl reductions have been interpreted as an indicator of altered synaptic function and energy metabolism, possibly as a consequence of deafferentiation (Chételat et al., 2009; Villain et al., 2008), although another local process (e.g., a primary energy metabolism defect) has not been ruled out. Via its functional neuroanatomy, the PCC is a key integration node between the medial temporal lobe and medial prefrontal subsystems in the default mode network (DMN), a brain system that is active when subjects are engaged in internal cognition and unengaged with the external world (Buckner et al., 2008; Raichle et al., 2001). Certain regions involved in the DMN are key sites of β-amyloid deposition and AD-related atrophy (Buckner et al., 2005), possibly due to conducive metabolic conditions and the linkages between synaptic activity and β-amyloid metabolism (Bero et al., 2011; Cirrito et al., 2005, 2008). Therefore, the PCC may have a particular and unique vulnerability to perturbations of energy metabolism in AD and AD risk.
3. Metabolic brain imaging and APOE in older populations
The use of brain imaging to investigate APOE's effects is rooted in the idea of utilizing APOE-related changes in CMRgl as an endophenotype – a quantitative, genetically-based biomarker associated with disease risk (Reiman, 2007). Thus, we have proposed CMRgl as an endpoint in the evaluation of AD treatments and/or preventive therapies, with the underlying assertion being that region-specific CMRgl alterations correlate with disease-risk (Reiman et al., 2001), perhaps as a measure of cognitive reserve (Cohen et al., 2009), such that elevated basal energy metabolism may enhance ability to resist pathological insult (Stranahan and Mattson, 2012), and/or represent an early manifestation of a related parallel pathogenic process. While not all APOE ε4 carriers will develop AD (likely reflecting additional covariates underlying disease processes) APOE genotype strongly correlates with overall AD risk as well as age of symptomatic onset. We and others have used APOE ε4 gene dose to detect and track the brain and cognitive changes associated with the three levels of genetic risk for AD. Shortly after the initial reports linking APOE to AD, we used FDG PET to compare CMRgl in cognitively normal late-middle age (50-65 year old) APOE ε4 homozygotes and non-carrier controls. APOE ε4 homozygotes displayed significant reductions in CMRgl in the same parietal, temporal, and prefrontal regions demonstrating CMRgl reductions in probable AD patients (Reiman et al., 1996). Notably, PCC displayed the largest and most significant deficit in CMRgl. In follow-up studies, cognitively-normal middle-aged APOE ε4 heterozygotes showed similar regional CMRgl reductions and also exhibited longitudinal (2 year) declines in CMRgl (Reiman et al., 2001). Further FDG PET study of cognitively-normal middle-aged subjects identified a gene-dose effect in APOE (i.e., APOE ε4 homozygotes exhibited the lowest values and non-carriers exhibited the highest values, with ε4 heterozygotes falling between these extremes) on CMRgl in the same AD-related brain regions identified in previous FDG PET studies (Reiman et al., 2005). Additionally, using a genome-wide association study (GWAS) we have identified in GAB2 a common neutral, less common protective, and rare neutral haplotype associated with AD risk in APOE ε4 carriers (Reiman et al., 2007). In cognitively-normal late-middle age APOE ε4 carriers, the putatively protective GAB2 haplotype was associated with elevated CMRgl (in comparison to both APOE ε4 carriers without the protective haplotype and APOE ε4 non-carriers with the protective haplotype), again in regions that overlap those previously found in AD patients and APOE ε4 carriers (Liang et al., 2011). While the cellular physiology underlying this association is not well-established, study centers on the role of the GAB2-encoded Gab2 protein as an activator of the phosphatidyl inositol kinase (PI3K) pathway.
4. Brain imaging and APOE in young adults
In addition to studies of middle- and late-middle age individuals, we have utilized FDG PET to study even earlier effects of APOE ε4 on brain functional measures. In a study of cognitively normal 20-39 year olds, APOE ε4 carriers exhibited significantly decreased CMRgl in the PCC and other cortical regions associated with metabolic defects in older APOE ε4 carriers and AD patients, in this case several decades before the potential onset of dementia, and also several decades before any apparent pathology (Reiman et al., 2004). Our further studies investigating functional mitochondrial activity via cytochrome oxidase histochemistry, which measures the functional enzymatic activity of Complex IV of the electron transport chain (ETC), in young-adult APOE ε4 carriers discovered a mitochondrial activity deficit in the PCC, specifically localized to the superficial layers (I & II) of the cortical lamina (Valla et al., 2010). This result mirrored our earlier study of AD patients, who showed superficial laminar metabolic deficits across the neocortex, and most significantly in the PCC (Valla et al., 2001, 2007). These superficial layers are rich in the dendritic tufts of deeper neurons (e.g., layer III pyramidals), and these reductions may relate to synaptic declines or localized metabolic dysfunction. These findings indicate that functional mitochondrial changes may be an early indicator of AD-related risk and physiological change that is preferentially manifest in APOE ε4 carriers.
While studies in older populations display mixed results likely due to methodological differences (Trachtenberg et al., 2012a), fMRI studies of 20-35 year-old APOE ε4 carriers have found that the DMN exhibits decreased deactivation during memory retrieval tasks in APOE ε4 carriers, but displays increased coactivation at rest (Dennis et al., 2010; Filbey et al., 2006; Filippini et al., 2009). Similarly, H215O PET studies have indicated alterations in at-rest and task-activated cerebral blood flow in similarly-young APOE ε4 carriers (Scarmeas et al., 2003, 2005). Interestingly, diffusion tensor imaging has revealed that APOE modulates white matter integrity in the young-adult brain, with APOE ε4 carriers displaying reduced fractional anisotropy, which may be considered a marker of pathology/vulnerability (Heise et al., 2011). Additionally, a recent fMRI study identified that APOE ε2 carriers and APOE ε4 carriers both display increased task-related activation of the DMN in comparison to APOE ε3 carriers (Trachtenberg et al., 2012b), in addition to differences in resting functional architecture (Trachtenberg et al., 2012c). Unfortunately, the effects of APOE ε2 are not frequently studied; the inherent difficulties in populating cohorts for human subject studies may be exacerbated by the rareness (<10%) of APOE ε2 in the population and its association with decreased risk for AD. Thus, how APOE ε2 effects carryover to FDG PET studies of metabolic alterations remain unknown.
5. Limitations and future directions of metabolic brain imaging
Importantly, both FDG PET and CO studies have an inherent limitation in not being able to identify with certainty whether neurons or glial cells (particularly astrocytes) are the cellular source of the metabolic signal. While the brain contains different cell types with different bioenergetic profiles, the compartmentalization of these bioenergetic processes has often been ignored, in part due to the limited resolution of brain imaging. For example, the astrocyte-neuron lactate shuttle hypothesis proposes that astrocytes are largely glycolytic and, under high energy demand, may provide vital energetic support (lactate) to neurons which rely more heavily on oxidative phosphorylation (Pellerin et al., 2007) and can produce ATP from reducing equivalents derived from such provided TCA cycle substrates. The CO histochemistry signal is thought to be related primarily to neuronal oxidative metabolism (Wong-Riley, 1989); the FDG PET signal is more heavily debated, but it is hypothesized that astrocytes play the key role (Barros et al., 2005). More research is needed to address the relative roles of astrocytes and neurons in neuroenergetic processes, and the respective impact on disease risk. Additionally, metabolic signal alterations may be related to other factors, including the density of cells or synapses. Notably, mice expressing human apoE4 display decreased dendritic arborization and spine density (Dumanis et al., 2009; Ji et al., 2003). While further research in human tissue is needed to clarify these issues, an MRI study of children shows an interesting association between APOE genotype and cortical thickness, with APOE ε4 carriers having thinner entorhinal cortices (Shaw et al., 2007). Additional MRI work in young-adults demonstrates APOE ε4 carriers having smaller hippocampal volumes than APOE ε2 carriers (Alexopoulos et al., 2011), but this effect was not found in a study comparing APOE ε4 carriers to non-carriers (Richter-Schmidinger et al., 2011).
Adding another potential confound to FDG PET studies, recent reports using a novel brain imaging approach in young-adults showed that aerobic glycolysis, in this case defined as glucose utilization in excess of that used for oxidative phosphorylation despite sufficient oxygen, is differentially present in the medial and lateral parietal and prefrontal cortices, which are also preferentially vulnerable to β-amyloid pathology (Vaishnavi et al., 2010; Vlassenko et al., 2010). Glucose that is not fully metabolized via oxidative phosphorylation may be converted to lactate, an important and oft-debated brain fuel (Nehlig and Coles, 2007), or shunted to the pentose phosphate pathway. Previous studies have viewed FDG PET largely as a marker of cell-autonomous energy metabolism and ignored the complication of glycolysis followed (or not followed) by oxidative phosphorylation; detailed examination of the metabolic fate of glucose may provide even stronger insight. FDG PET allows for the measurement of only the first step of the metabolism of glucose (phosphorylation to glucose-6-phosphate by hexokinase) and cannot by itself be used to identify the subsequent metabolic fate of glucose. Further research is needed to determine the relative importance of each use of glucose in the brain and how it may be relevant to disease risk, especially given the alterations in glucose uptake apparent in both young and aged APOE ε4 carriers and the overlap of these regions with increased aerobic glycolysis and β-amyloid pathology.
6. Cellular functions of apoE in the brain
While brain imaging can provide a great deal of insight to the effects of APOE, an understanding of cellular function and mechanisms conferring risk is essential for any effective therapeutic development. The primary function of apoE in the brain is to traffic cholesterol and other lipids. While it is expressed in several peripheral tissues, apoE is most highly expressed in the liver and brain (Elshourbagy et al., 1985). In the brain, apoE is the primary apolipoprotein that associates with HDL-like lipoproteins (Pitas et al., 1987a), the only lipoprotein assembly, meaning that apoE plays a vital role in maintaining neural functions dependent on cholesterol, in addition to other roles. Indeed, a significant body of work supports apoE involvement in synaptogenesis (Mauch et al., 2001), neurite outgrowth (Bellosta et al., 1995; Holtzman et al., 1995; Nathan et al., 1994, 1995, 2002; Qiu et al., 2004) and dendritic arborization (Dumanis et al., 2009; Ji et al., 2003), modulation of synaptic plasticity (Herz and Chen, 2006; Klein et al., 2010; Korwek et al., 2009), neurogenesis (Li et al., 2009; Yang et al., 2011), and neuroinflammation (Bales et al., 2000), often with isoform-specific efficacy.
Under normal conditions astrocytes are the primary adult brain source of apoE, with apoE comprising up to 3% of the protein secreted from these cells (Pitas et al., 1987b), while microglia and neurons produce much smaller amounts of apoE (Boyles et al., 1985; Xu et al., 1999). Once synthesized, apoE is secreted by astrocytes and loaded further with cholesterol and other lipids via the ATP-binding cassette transporter (ABCA1) to form lipoprotein particles before being endocytosed by neurons, primarily via low-density lipoprotein (LDL) receptor and LDL-related protein 1 (LRP1), both members of the low-density lipoprotein receptor (LDLR) family (Bu, 2009). While produced only in low amounts at baseline, neuronal apoE expression can be significantly increased, particularly as a response to injury or stress (Aoki et al., 2003; Boschert et al., 1999; Xu et al., 2006, 2008). Considering the known differences in neuronal processing of apoE (described below), this response may be of particular relevance to neural pathophysiology. In the peripheral circulation, lipid trafficking and metabolism are differentially impacted by the structurally-dependent lipid- and ligand-binding abilities of apoE isoforms, evident in the pathophysiology of multiple diseases (Mahley, 1988; Mahley and Rall, 2000). Unfortunately, isoform-specific knowledge of these processes in the brain remains insufficient to draw conclusions on potential disease relationships.
7. ApoE structure and its relevance to disease
Aspects of apoE structure are thought to be a driving force in its role in AD risk. ApoE is a 34kDa, 299 amino acid glycoprotein with two major functional domains: the N-terminal domain exists as a four helix bundle and contains the apoE receptor binding region at residues 136-150; the C-terminal domain is highly α-helical and contains the major lipid binding region at residues 244-272 (Aggerbeck et al., 1988; Dong et al., 1994; Wetterau et al., 1988; Wilson et al., 1991). When unbound by lipid, the N-terminal and C-terminal domains are linked by a flexible hinge region consisting approximately of residues 165-215 (Wetterau et al., 1988; for comprehensive reviews of apoE structure see Hatters et al., 2006a; Zhong and Weisgraber, 2009).
In humans, APOE is located on chromosome 19 and encodes three common alleles: APOE ε2 (protective; US frequency ~10%), APOE ε3 (neutral; ~70%), and APOE ε4 (risk-associated; ~20%) (Mahley et al., 2006). While the three isoforms differ at only two amino acid positions—apoE2 has cysteine at residues 112 & 158, apoE3 has cysteine at residue 112 & arginine at residue 158, and apoE4 has arginine at residues 112 &158 (Weisgraber et al., 1981)—these amino acid changes have a profound impact on the structure of the protein and are therefore thought to play a fundamental role in the association of apoE with AD risk. In particular, apoE4 is much more likely to exhibit a compacting phenomenon known as domain interaction due to the presence of arginine at residue 112 (Xu et al., 2004). The presence of this arginine residue results in the side chain of the arginine at residue 61 (located in the N-terminal domain) interacting with the glutamate at residue 255 (located in the C-terminal domain) via the formation of a salt bridge (Dong and Weisgraber, 1996). Notably, human apoE is the only form of the protein with arginine at residue 61. The arginine side chain is less available in both apoE2 and apoE3, making them much less likely to undergo domain interaction. It is thought that domain interaction is responsible for many of apoE4's neurotoxic effects (discussed in part below) and may partially underlie its association with AD. Additionally, apoE4 has a propensity to exist as a molten globule (Morrow et al., 2002) and form soluble aggregates (Hatters et al., 2006b) under physiological conditions, both of which are potentially neurotoxic. Physiologically, the differential influence of apoE4 on AD risk may manifest as a gain of toxic function or a loss (limitation) of normal function when compared to apoE3. In the case of neural repair, apoE4 likely represents a loss of normal function, as apoE4 is thought to be less effective at these roles (Mahley et al., 2006).
In addition to influences on such functions as synaptic plasticity and neuroinflammation, growing evidence from cell culture, animal model, and pathology studies suggests effects of apoE, most with isoform-specificity, that are linked at the cellular level to dysfunction in neuroenergetics-relevant processes. Therefore, these effects may provide potential mechanisms for the alterations in energy metabolism and synaptic function seen in human brain imaging studies of APOE ε4 carriers and may be validated as such given further study in vivo in model systems or in appropriate post-mortem tissues. When coupled with the neurodevelopmental effects of APOE these processes may serve as “second hits” to alter brain function to increase risk for AD.
8. Mechanisms of apoE cleavage and processing
As previously emphasized, the cellular source of apoE is highly regulated, and neuronal production of apoE appears to be driven by astrocytic signaling mechanisms (Harris et al., 2004a), particularly as a result of neural injury. It has been demonstrated in post-mortem human samples that apoE4 undergoes neuron-specific proteolysis (Huang et al., 2001); this dramatic increase in intracellular cleavage of apoE4 compared to apoE3 and apoE2 is thought to be due to apoE4's much greater tendency to exhibit domain interaction. Studies of transgenic mice expressing the human isoform have determined that apoE4 produced in neurons is cleaved by a chymotrypsin-like serine protease termed apoE cleaving enzyme (AECE; Harris et al., 2003). Further, although neurons take up apoE secreted by astrocytes, as demonstrated in transgenic mice, the proteolysis occurs in the neuronal secretory pathway and not in an internalization pathway, indicating cell-source specificity to this potentially toxic event (Brecht et al., 2004). C-terminal segments of apoE4 have been shown to be vital for structural integrity and the protein's ability to alter its conformation (Chroni et al., 2008; Tanaka et al., 2006). Notably, apoE4 cleaved by AECE and missing residues 272-299 (Δ272–299) is capable of translocating into the cytosol to escape the secretory pathway. Reports from Neuro-2a and mouse primary hippocampal neuron cultures show that apoE4 trafficking is impaired throughout the endoplasmic reticulum and Golgi apparatus (Brodbeck et al., 2011). Interestingly, this effect can be eliminated with small-molecule apoE structure correctors that make apoE4's structure more similar to apoE3 and thus decreases domain interaction and subsequent cleavage (Brodbeck et al., 2011). Studies in Neuro-2a mouse neuroblastoma cultures demonstrated that the LDL-receptor binding region (residues 136-150) is required for escape from the secretory pathway, as it is rich in the positively-charged amino acids arginine, lysine, and histidine (Chang et al., 2005).
9. Effects of apoE on cytoskeletal components and intracellular trafficking
ApoE cleavage fragments have been shown to have a number of effects on the cytoskeleton and related intracellular trafficking functions. In Neuro-2a cells, expressed apoE4(Δ272–299) has been found to interact with cytoskeletal proteins to form neurofibrillary tangle-like structures containing phosphorylated tau (Huang et al., 2001). Mice expressing high levels of apoE4(Δ272–299) in neurons display AD-like neurofibrillary tangles and die at 2-4 months. With lower levels of expression the mice display learning and memory deficits at 6-7 months (Harris et al., 2003). Interestingly, neuronal apoE4(Δ272–299) expression has also been linked to GABAergic interneuron dysfunction and consequential learning and memory deficits mediated by tau (Andrews-Zwilling et al., 2010). Deleterious effects of apoE4 fragments on GABAergic interneurons have also been linked to impaired neurogenesis via tau (Li et al., 2009).
Full-length apoE4 expressed in Neuro-2a cultures acts along with zinc to phosphorylate tau via the ERK pathway (Harris et al., 2004b). Additionally, apoE3 seems to be effective at binding the microtubule-binding repeat region of tau, which is responsible for the formation of the paired helical filaments that compose neurofibrillary tangles; apoE4 does not bind this region and thus may allow more aberrant fibrillization (Strittmater et al., 2000). Proper cytoskeletal structure is required for the appropriate distribution and trafficking of mitochondria, with improper distribution having a number of deleterious effects (Detmer and Chan, 2007), including impaired energy metabolism. The necessity of effective mitochondrial distribution is intensified in neurons due to their morphology and highly-localized energy use (MacAskill and Kittler, 2010). Therefore, alterations and/or disruptions due to apoE4 fragment interactions with cytoskeletal components may impair the ability to maintain proper energy supply to the synapse, which requires a dynamic population of mitochondria (Li et al., 2004). Overexpression of tau in Neuro-2a cultures has been shown to inhibit mitochondrial transport (Ebneth et al., 1998). Similarly, transgenic mice expressing human apoE4 display impaired axonal transport, with mitochondria accumulating in bulb-like dilations (Tesseur et al., 2000). Interestingly, prior studies of human post-mortem tissue and AD mouse models have found axonal swellings containing organelles that appear to precede the formation of AD pathology (Stokin et al., 2005). Additionally, a recent study in differentiated PC12 cells either expressing or incubated with apoE found that apoE4 impairs mitochondrial motility in relation to apoE3 (Chen et al., 2012). This effect was ameliorated by apoE structure correctors. Additionally, microtubule depolymerization is thought to at least partially underlie the deleterious effects of apoE4 on neurite outgrowth (Nathan et al., 1995).
10. ApoE and mitochondrial function
In addition to its deleterious effects on intracellular transport, apoE4 has been shown to have direct effects on mitochondria. In an early study, apoE4 was shown to bind the alpha and beta subunits of the F1 portion of ATP synthase in liver (Mahley et al., 1989). While direct effects on enzyme function were not assessed in that study, neuronal apoE4 fragments have subsequently been shown to perturb mitochondrial function. In Neuro-2a cultures expressing apoE4(Δ272–299), apoE4 fragments that escape the secretory pathway can cause mitochondrial dysfunction via an unknown mechanism that requires the lipid-binding region (residues 244-272; Chang et al., 2005). It has since been shown in Neuro-2a cultures that apoE4(Δ272–299) binds the subunits ubiquinol cytochrome c reductase core protein 2 (UQCRC2) and cytochrome CI of Complex III (ubiquinol:cytochrome c oxidoreductase) and cytochrome c oxidase subunit 4 isoform 1 (COX IV 1) of Complex IV (cytochrome c:oxygen oxidoreductase) of the ETC and served to significantly reduce respiratory function of both complex III and complex IV (Nakamura et al., 2009). This is the precise pattern of ETC dysfunction we observed in peripheral tissue (platelet) mitochondria isolated from AD patients (Valla et al., 2006). Analysis of ETC protein expression in apoE4-expressing Neuro-2a and mouse primary neuron cultures demonstrated reduction in expression of subunits for all ETC complexes, and notably, in a bigenomic manner (Chen et al., 2011)--ETC protein expression is a highly-regulated process with subunits encoded from genes on both the nuclear and mitochondrial genomes (Hock and Kralli, 2009). Complex IV functional activity was also significantly decreased (Chen et al., 2011). As with the previously mentioned studies on intracellular trafficking, small-molecule structure correctors that make apoE4's structure more similar to that of apoE3 were able to alleviate these deficits. Given the fact that cytoplasmic toxicity of neuronal apoE is apparent after its cleavage by AECE, further knowledge of the enzyme would be useful to enable the design of pharmaceuticals to modulate AECE activity as a complementary mechanism to altering apoE4's structure (Huang, 2010).
Identification of protein signatures in APOE mice found that mitochondrially-enriched fractions prepared from hippocampal tissue of apoE4 and apoE3 expressing mice differed in levels of several proteins involved in such capacities as mitochondrial function, oxidative stress response, and organelle transport (James et al., 2011). Our prior multi-region gene expression study utilizing laser capture microdissection to select only neurons in AD patients found reductions in ETC gene expression, with PCC displaying the most prominent effects (Liang et al., 2008). Further, a recent study utilizing postmortem tissue from the middle temporal gyrus to compare gene expression profiles in middle-aged APOE ε4 carriers to non-carrier controls found significant differential expression in 70 transcripts, 30 of which are involved in oxidative mitochondrial function (Conejero-Goldberg et al., 2011). Unfortunately, this study lacked the ability to resolve cell-type specific alterations in gene expression, which may be important given the respective roles of astrocytes and neurons in neuroenergetic processes (Allaman et al., 2011). Future study along these lines, especially in young-adults and including analysis of astrocytes (see next section), may prove valuable in elucidating cell-type specific roles in ε4-related functional changes.
ApoE isoforms expressed in b12 cells display differing anti-oxidant ability in a manner correlated with disease-risk (apoE2>apoE3>apoE4; Miyata and Smith, 1996). It is possible therefore, that detrimental or beneficial effects of different apoE isoforms are at least in part due to their relative ability to control the levels of reactive oxygen species, of which mitochondria are a primary endogenous source (Lin and Beal, 2006), including AD, in which oxidative stress is thought to be a very early hallmark of pathophysiology (Hirai et al., 2001; Nunomura et al., 2001).
11. ApoE impacts on astrocytes
Beyond the effects shown in neurons, apoE4 also appears to alter function in astrocytes. ApoE4 induces endoplasmic reticulum stress in astrocytes (Zhong et al., 2009) that does not occur in neurons (Brodbeck et al., 2011). However, again pointing to the cell-type specific importance of apoE expression, mouse primary astrocyte cultures expressing apoE4 do not show significant changes in ETC gene expression (Chen et al., 2011). Additionally, transgenic mice that express apoE4 under the control of a GFAP promoter (astrocyte-specific) display severe deficits in working memory without any evident β-amyloid pathology (Hartman et al., 2001). Recent reports have demonstrated that deleterious effects of β-amyloid internalization by astrocytes in turn impacts neuronal viability through effects on astrocytic energy metabolism (Allaman et al., 2010). It is possible that apoE4 acts in a similar manner to this astrocytic β-amyloid, serving as a double-hit to impair neuronal function both directly, through escape from the secretory pathway and subsequent toxic effects on the mitochondria, and indirectly, through deleterious effects on astrocytes, serving to attenuate their ability to provide essential metabolic support (Zhong et al., 2009). Notably, based on current understanding of apoE production, loss of expression due to astrocyte endoplasmic reticulum stress may lead to an increase in potentially pathogenic (and non-functional if truncated) neuronal apoE (Zhong et al., 2009), in addition to the consequences of loss of functional astrocytic apoE. Interestingly, apoE4 has been shown recently to contribute to breakdown of the blood brain barrier via effects on cyclophilin A, which could further impact energetics due to disruption of nutrient transport (Bell at al., 2012).
12. Potential Roles for TOMM40
APOE is located in a region of linkage disequilibrium on chromosome 19 that also encompasses TOMM40 and APOC1. TOMM40, which encodes Tom40, the pore-forming subunit of the translocase of the outer mitochondrial membrane, has now been proposed as a potential genetic risk factor for AD (Roses et al., 2010). However, the exact nature of the relationship, at least partly, but perhaps not entirely attributable, to linkage disequilibrium, is not yet understood as the initial findings have not been consistently replicated (Chu et al., 2011; Cruchaga et al., 2011). Functionally, the translocase is responsible for the import of proteins into the mitochondria—99% of mitochondrial proteins must be imported (Bolender et al., 2008). Deep sequencing and phylogenetic analysis of this chromosome 19 LD region identified a variable-length poly-T polymorphism at the rs10524523 (‘523) locus in intron 6 of TOMM40 that may be able to further refine the age of onset distribution for AD. The mechanism of how TOMM40 intronic polymorphisms may potentially influence age of onset is unclear, but given the extent of neuroenergetic defects both in AD and in at-risk ε4 carriers, the possibility of involvement is of great interest.
Suggested hypotheses with a direct impact on mitochondrial function include effects of the polymorphism on splicing of Tom40 transcripts resulting in isoforms (Roses et al., 2010) with differential functionality or even propensity to bind apoE. In the latter case the pathological action would be synergistic between Tom40 and apoE, and correspondingly TOMM40 and APOE. Additionally, genome wide pathway analysis has implicated genes involved in intracellular protein transport, particularly TOMM40, in AD (Hong et al., 2010). Other potential hypotheses include effects on apoE transcription; previous AD GWAS have shown that TOMM40 single nucleotide polymorphisms influence apoE levels in the cerebrospinal fluid (Bekris et al., 2010), yet the ‘523 poly-T repeat does not appear to alter CSF levels of β-amyloid in aged controls, indicating that the mechanism influencing age of onset may not exert itself via a β-amyloid-linked process (Pomara et al., 2011). A recent study utilizing fibroblast cultures found no apparent effects of the ‘523 poly-T repeats on expression levels of Tom40 protein and mRNA, Tom40 mRNA splicing, or mitochondrial function and morphology (Hedskog et al., 2012). Structural MRI and neuropsychological data shows that cognitively-normal late-middle aged APOE ε3 homozygotes who are also homozygous for longer ‘523 poly-T repeats have significant declines in learning and memory function and gray matter volume in the ventral PCC and medial ventral precuneus when compared to other APOE ε3 homozygotes (Johnson et al., 2011). The detection of presymptomatic changes in the PCC serves again to highlight the apparent differential vulnerability of the region to processes linked to vulnerability to neurological disease. Additional investigation to clarify the possible effects of TOMM40 genotype may help to explain disease onset variation in at-risk populations and may also provide an impetus to further examine neuroenergetic dysfunction.
13. Conclusions
The strong association between APOE and AD has been known for nearly two decades, during which time significant advances have been made in understanding how APOE may contribute to disease risk. While considerably more research is needed to establish the mechanistic effects of APOE on disease processes, mounting evidence linking APOE to alterations in neuroenergetics has illuminated exciting new areas for research. Due to the apparently early nature of these functional changes (i.e., often apparent in young-adults), modulation of related cellular and molecular processes may provide viable targets for therapies aiming to prevent and/or treat AD, and potentially a number of other neurological disorders with possible links to APOE (Verghese et al., 2011). While no apoE-directed therapies (e.g., small-molecule structure correctors) have yet been utilized in humans, bioenergetically-relevant therapies have undergone clinical trials. Most prominent have been efforts using intranasal insulin (Craft et al., 2012; Reger et al., 2006, 2008a, 2008b), ketogenic medium chain triglycerides (Henderson et al., 2009), and PPAR-γ agonists (Geldmacher et al., 2011; Gold et al., 2010; Risner et al., 2006, Sato et al., 2011, Watson et al., 2005). It remains to be clarified whether clinically-affected APOE ε4 carriers and non-carriers may respond differently to amyloid-targeting therapies. A recent Phase II trial of the amyloid antibody therapy bapineuzumab found less benefit in carriers than in non-carriers with AD dementia (Salloway et al., 2009), but the subsequent Phase III studies failed to demonstrate a clear benefit in AD dementia patients whether they were carriers or non-carriers (not yet published). It has also been suggested that these treatments may be associated with a greater risk of vasogenic edema and cerebral microhemorrhage in clinically-affected carriers than non-carriers (Salloway et al., 2009), perhaps related to blood-brain barrier breakdown in APOE ε4 carriers (Bell et al., 2012). If a differential response to these and other treatments are confirmed in APOE ε4 carriers and non-carriers, it could be attributable to the apoE4 protein or, perhaps more likely, underlying disease severity (since each copy of the ε4 allele in a person's APOE genotype is associated with a younger average age at onset, it is possible that the treated AD dementia patients who carry the APOE ε4 allele have greater disease severity than the treated non-carriers of the same age).
An alternative, or perhaps synergistic, interpretation of the APOE ε4-associated deficits in AD, given the early functional (Reiman et al., 2004; Valla et al., 2010) and morphological changes (Shaw et al., 2007; Alexopoulos et al., 2011) reported in APOE ε4 carriers, is that these alterations do not reflect progressive disease-related changes in the brain but rather APOE-related neurodevelopmental alterations. As such, APOE ε4 may convey a developmental limitation that provides a foothold for the regional vulnerability that contributes to an earlier age of clinical onset decades later. Such a model would fit well with the concept of cognitive reserve in that APOE ε4 carriers may not demonstrate equivalent synaptic number or dendritic complexity, as in apoE expressing mice (Dumanis et al., 2009; Ji et al., 2003), and thus far reported to manifest as reduced entorhinal cortical thickness in young human subjects (Shaw et al., 2007).
Additionally, further understanding of the role of TOMM40 polymorphisms may provide insight. Hopefully, the extension of current knowledge can lead to continued improvement in elucidating the causes of disease and correspondingly the development of critically needed therapies.
Fig. 1.
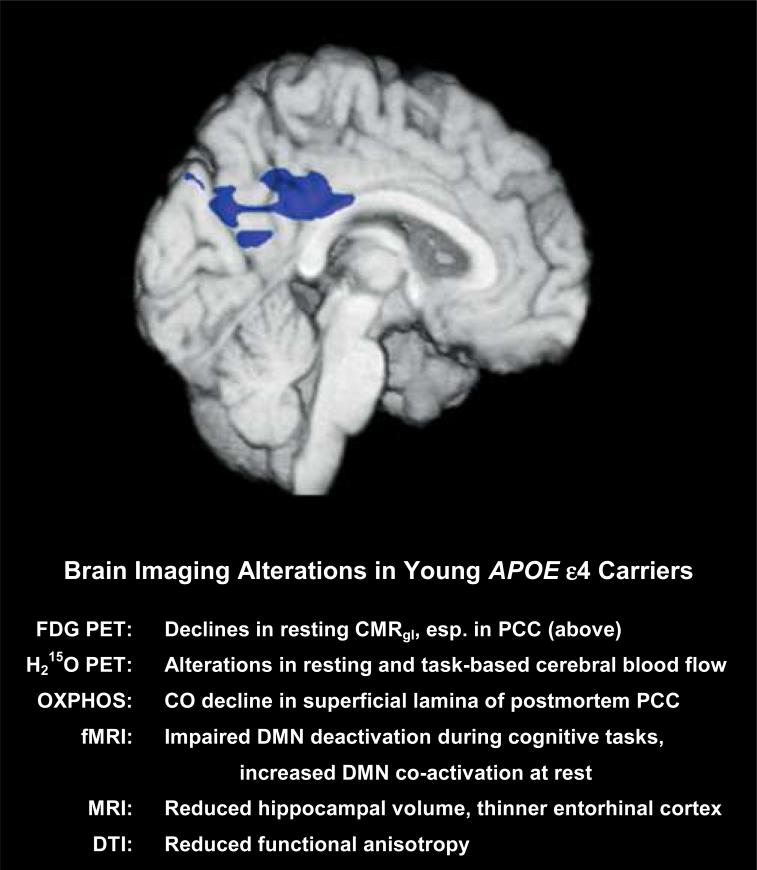
Medial view localizing abnormally low CMRgl (blue) in the PCC in young adult carriers of the APOE ε4 allele, and a summary of brain imaging results thus far reported in similarly-aged young adults and children (citations in text). Widespread changes in neuroenergetics and brain structure point to a neurodevelopmental role for APOE and early interference by the APOE ε4 allele, but this is yet to be confirmed. CMRgl=cerebral metabolic rate for glucose; PCC=posterior cingulate cortex; OXPHOS=oxidative phosphorylation; CO=cytochrome oxidase, Complex IV of the ETC; DMN=default mode network; DTI=diffusion tensor imaging
Fig. 2.
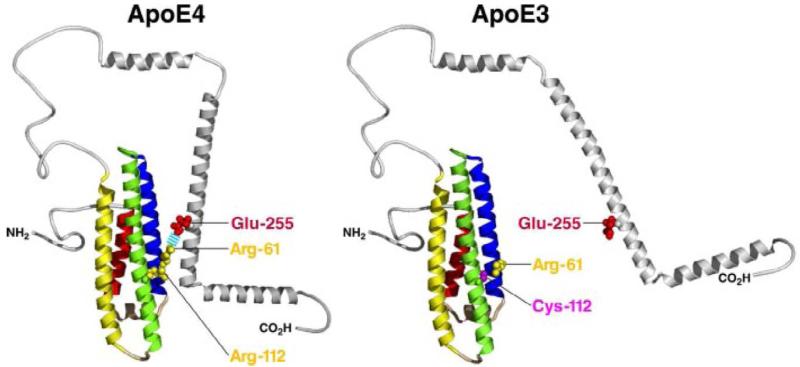
Structural difference between apoE3 and apoE4. Domain interaction prevalent in apoE4 is predicated on the substitution of Arg for Cys at position 112 (Zhong & Weisgraber, 2009; reprinted with permission. © 2009 The American Society for Biochemistry and Molecular Biology. All rights reserved.)
Fig. 3.
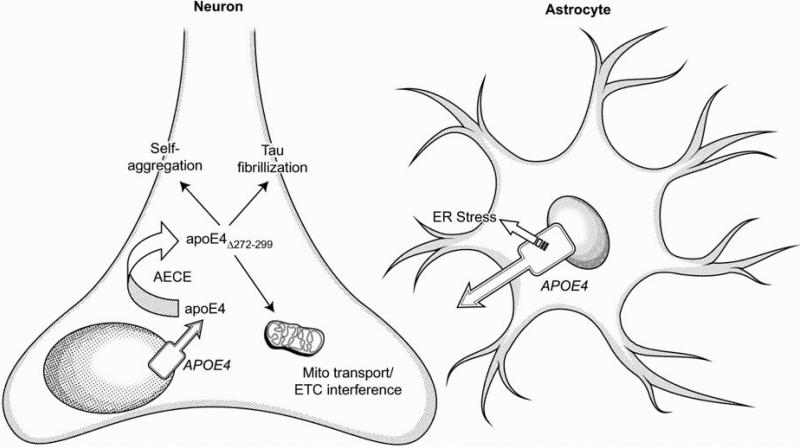
Summary of the effects of cell-type specific expression and aberrant processing of apoE4 in the brain. Astrocyte-expressed apoE is internalized by neurons, but apoE4 expression in astrocytes has been associated with ER stress that may impair astrocytes’ ability to contribute to brain energy flux. A smaller proportion of brain apoE expression is attributed to neurons: apoE4 expressed in neurons (but not that internalized from astrocytes) is highly susceptible to cleavage by AECE (apoE cleaving enzyme) in the secretory pathway becoming apoE(Δ272–299). ApoE4 cleaved by AECE is capable of escaping the secretory pathway, self-aggregating in the cytosol, increasing the fibrillization of tau, and interfering with mitochondrial transport and function.
Acknowledgments
This work was supported by the Arizona Alzheimer's Consortium, the State of Arizona, and the Arizona Alzheimer's Disease Core Center (P30 AG19610).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures Statement
Each author declares no actual or potential conflicts of interest.
References
- Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CC, Lindgren FT. Human apolipoprotein E3 in aqueous solution. II. properties of the amino- and carboxyl-terminal domains. J. Biol.Chem. 1988;263:6249–6258. [PubMed] [Google Scholar]
- Alexopoulos P, Richter-Schmidinger T, Horn M, Maus S, Reichel M, Sidiropoulos C, Rhein C, Lewczuk P, Doerfler A, Kornhuber J. Hippocampal volume differences between healthy young apolipoprotein E ε2 and ε4 carriers. J. Alzheimer's Dis. 2011;26:207–210. doi: 10.3233/JAD-2011-110356. [DOI] [PubMed] [Google Scholar]
- Allaman I, Gavillet M, Bélanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci. 2010;30:3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Bélanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Uchihara T, Sanjo N, Nakamura A, Ikeda K, Tsuchiya K, Wakayama Y. Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke. 2003;34:875–880. doi: 10.1161/01.STR.0000064320.73388.C6. [DOI] [PubMed] [Google Scholar]
- Ashford JW, Mortimer JA. Non-familial Alzheimer's disease is mainly due to genetic factors. J. Alzheimer's Dis. 2002;4:169–177. doi: 10.3233/jad-2002-4307. [DOI] [PubMed] [Google Scholar]
- Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflammation and Alzheimer's disease: critical roles for cytokine/Abeta-induced glial activation, NF-kappaB, and apolipoprotein E. Neurobiol. Aging. 2000;21:427–432. doi: 10.1016/s0197-4580(00)00143-3. [DOI] [PubMed] [Google Scholar]
- Barros LF, Porras OH, Bittner CX. Why glucose transport in the brain matters for PET. Trends Neurosci. 2005;28:117–119. doi: 10.1016/j.tins.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Bekris LM, Galloway NM, Montine TJ, Schellenberg GD, Yu C-E. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. Am J Med Genet B Neuropsychiatr Genet. 2010;153:409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta S, Nathan BP, Orth M, Dong LM, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J. Biol. Chem. 1995;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Bolender N, Sickmann A, Wagner R, Meisinger C, Pfanner N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep. 2008;9:42–49. doi: 10.1038/sj.embor.7401126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschert U, Merlo-Pich E, Higgins G, Roses AD, Catsicas S. Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol. Dis. 1999;6:508–514. doi: 10.1006/nbdi.1999.0251. [DOI] [PubMed] [Google Scholar]
- Boyle PA, Wilson RS, Aggarwal NT, Tang Y, Bennett DA. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology. 2006;67:441–445. doi: 10.1212/01.wnl.0000228244.10416.20. [DOI] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, Fish JD, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong D-E, Pleiss M, McComas C, Hess F, Witter D, Peterson S, Childers M, Goulet M, Liverton N, Hargreaves R, Freedman S, Weisgraber KH, Mahley RW, Huang Y. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011;286:17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann. NY Acad. Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Chang S, Ran Ma T, Miranda R Dennis, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-K, Ji Z-S, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer's disease. J. Biol.Chem. 2011;286:5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-K, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, Pleiss MA, Ji ZS, Balestra ME, Walker DW, Xu Q, Jeong DE, Budamagunta MS, Voss JC, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Small-molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 2012;287:5253–5266. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chroni A, Pyrpassopoulos S, Thanassoulas A, Nounesis G, Zannis VI, Stratikos E. Biophysical analysis of progressive C-terminal truncations of human apolipoprotein E4: insights into secondary structure and unfolding properties. Biochemistry. 2008;47:9071–9080. doi: 10.1021/bi800469r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, Landeau B, Mézenge F, Poline JB, de la Sayette V, Viader F, Eustache F, Baron J-C. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer's disease. Brain. 2008;131:60–71. doi: 10.1093/brain/awm288. [DOI] [PubMed] [Google Scholar]
- Chételat G, Villain N, Desgranges B, Eustache F, Baron J-C. Posterior cingulate hypometabolism in early Alzheimer's disease: what is the contribution of local atrophy versus disconnection? Brain. 2009;132:e133. doi: 10.1093/brain/awp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SH, Roeder K, Ferrell RE, Devlin B, Demichele-Sweet MAA, Kamboh MI, Lopez OL, Sweet RA. TOMM40 poly-T repeat lengths, age of onset and psychosis risk in Alzheimer disease. Neurobiol. Aging. 2011;32:2328.e1–9. doi: 10.1016/j.neurobiolaging.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang J-E, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, Nebes RD, Saxton JA, Snitz BE, Aizenstein HA, Wolk DA, DeKosky ST, Mathis CA, Klunk WE. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J. Neurosci. 2009;29:14770–14778. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C, Hyde TM, Chen S, Dreses-Werringloer U, Herman MM, Kleinman JE, Davies P, Goldberg TE. Molecular signatures in post-mortem brain tissue of younger individuals at high risk for Alzheimer's disease as based on APOE genotype. Mol. Psychiatry. 2011;16:836–847. doi: 10.1038/mp.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Nowotny P, Kauwe JSK, Ridge PG, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM. Alzheimer's Disease Neuroimaging Initiative. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch. Neurol. 2011;68:1013–1019. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis NA, Browndyke JN, Stokes J, Need A, Burke JR, Welsh-Bohmer KA, Cabeza R. Temporal lobe functional activity and connectivity in young adult APOE4 carriers. Alzheimer's Dement. 2010;6:303–311. doi: 10.1016/j.jalz.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- Drzezga A, Grimmer T, Riemenschneider M, Lautenschlager N, Siebner H, Alexopoulus P, Minoshima S, Schwaiger M, Kurz A. Prediction of individual clinical outcome in MCI by means of genetic assessment and (18)F-FDG PET. J. Nucl. Med. 2005;46:1625–1632. [PubMed] [Google Scholar]
- Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, Schwaiger M, Kurz A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer's disease: a PET follow-up study. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:1104–1113. doi: 10.1007/s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M, Global prevalence of dementia: a Delphi consensus study Alzheimer's Disease International. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbey FM, Slack KJ, Sunderland TP, Cohen RM. Functional magnetic resonance imaging and magnetoencephalography differences associated with APOE e4 in young healthy adults. Neuroreport. 2006;17:1585–1590. doi: 10.1097/01.wnr.0000234745.27571.d1. [DOI] [PubMed] [Google Scholar]
- Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc. Natl. Acad. Sci. USA. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland RP, Brun A, Budinger TF. Pathological and positron emission tomographic correlations in Alzheimer's disease. Lancet. 1985;1:228. doi: 10.1016/s0140-6736(85)92074-4. [DOI] [PubMed] [Google Scholar]
- Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011;68:45–50. doi: 10.1001/archneurol.2010.229. [DOI] [PubMed] [Google Scholar]
- Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, Craft S, Landreth G, Linnamägi U, Sawchak S. Rosiglitazone monotherapy in mild-to-moderate Alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 2010;30:131–146. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E e4 in Alzheimer's disease. Ann. Neurol. 1996;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley RW, Huang Y. Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer's disease. J. Biol. Chem. 2004a;279:3862–3868. doi: 10.1074/jbc.M309475200. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y. Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J. Biol. Chem. 2004b;279:44795–447801. doi: 10.1074/jbc.M408127200. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L, Holtzman DM. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp. Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 2006a;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J. Mol. Bio. 2006b;361:932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- Heise V, Filippini N, Ebmeier KP, Mackay CE. The APOE ε4 allele modulates brain white matter integrity in healthy adults. Mol. Psychiatry. 2011;16:908–916. doi: 10.1038/mp.2010.90. [DOI] [PubMed] [Google Scholar]
- Hedskog L, Brohede J, Wiehager B, Pinho CM, Revathikumar P, Lilius L, Glaser E, Graff C, Karlström H, Ankarcrona M. Biochemical studies of poly-T variants in the Alzheimer's disease associated TOMM40 gene. J. Alzheimer's Disease. doi: 10.3233/JAD-2012-120580. Published online May 16, 2012. PMID: 22596268. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. USA. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M-G, Alexeyenko A, Lambert J-C, Amouyel P, Prince JA. Genomewide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. J. Hum. Genet. 2010;55:707–709. doi: 10.1038/jhg.2010.92. [DOI] [PubMed] [Google Scholar]
- Huang Y. Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer's disease. Trends Mol. Med. 2010;16:287–294. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. USA. 2001;98:8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez V, Pietrini P, Alexander GE, Furey ML, Teichberg D, Rajapakse JC, Rapoport SI, Schapiro MB, Horwitz B. Regional glucose metabolic abnormalities are not the result of atrophy in Alzheimer's disease. Neurology. 1998;50:1585–1593. doi: 10.1212/wnl.50.6.1585. [DOI] [PubMed] [Google Scholar]
- James R, Searcy JL, Le Bihan T, Martin SF, Gliddon CM, Povey J, Deighton RF, Kerr LE, McCulloch J, Horsburgh K. Proteomic analysis of mitochondria in APOE transgenic mice and in response to an ischemic challenge. J. Cereb. Blood. Flow. Metab. 2012;32:164–176. doi: 10.1038/jcbfm.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience. 2003;122:305–315. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, Bendlin BB, Hogan KJ, Roses AD, Saunders AM, Lutz MW, Asthana S, Green RC, Sager MA. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOEε3/ε3 genotype. Alzheimer's Dement. 2011;7:456–465. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivet R, Magistretti PJ, Weber B. Deciphering neuron-glia compartmentalization in cortical energy metabolism. Front. Neuroenergetics. 2009;1:4. doi: 10.3389/neuro.14.004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RC, Mace BE, Moore SD, Sullivan PM. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience. 2010;171:1265–1272. doi: 10.1016/j.neuroscience.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korwek KM, Trotter JH, Ladu MJ, Sullivan PM, Weeber EJ. ApoE isoform-dependent changes in hippocampal synaptic function. Mol. Neurodegener. 2009;4:21. doi: 10.1186/1750-1326-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, Halabisky B, Deng C, Mahley RW, Huang Y. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–645. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K-I, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Chen K, Lee W, Sidhar K, Corneveaux JJ, Allen AN, Myers A, Villa S, Meechoovet B, Pruzin J, Bandy D, Fleisher AS, Langbaum JB, Huentelman MJ, Jensen K, Dunckley T, Caselli RJ, Kaib S, Reiman EM. Association between GAB2 haplotype and higher glucose metabolism in Alzheimer's disease-affected brain regions in cognitively normal APOEε4 carriers. NeuroImage. 2011;54:1896–1902. doi: 10.1016/j.neuroimage.2010.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Hui DY, Innerarity TL, Beisiegel U. Chylomicron remnant metabolism. role of hepatic lipoprotein receptors in mediating uptake. Arteriosclerosis. 1989;9:114–118. [PubMed] [Google Scholar]
- Mahley RW, Rall SC. Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. of Genomics Hum. Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller E, Otto A, Pfrieger F. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Foster NL, Kuhl DE. Posterior cingulate cortex in Alzheimer's disease. Lancet. 1994;344:895. doi: 10.1016/s0140-6736(94)92871-1. [DOI] [PubMed] [Google Scholar]
- Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. a potential basis for its association with disease. J. Biol. Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009;4:35. doi: 10.1186/1750-1326-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Chang KC, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE. The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J. Biol.Chem. 1995;270:19791–19799. doi: 10.1074/jbc.270.34.19791. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Jiang Y, Wong GK, Shen F, Brewer GJ, Struble RG. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 2002;928:96–105. doi: 10.1016/s0006-8993(01)03367-4. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Coles JA. Cellular pathways of energy metabolism in the brain: is glucose used by neurons or astrocytes?. Glia. 2007;55:1238–1250. doi: 10.1002/glia.20376. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj E, Jones P, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore A-K, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem. 1987a;262:14352–14360. [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta. 1987b;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- Pomara N, Bruno D, Nierenberg JJ, Sidtis JJ, Martiniuk FT, Mehta PD, Zetterberg H, Blennow K. TOMM40 poly-T variants and cerebrospinal fluid amyloid beta levels in the elderly. Neurochem. Res. 2011;36:1124–1128. doi: 10.1007/s11064-011-0459-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Hyman BT, Rebeck GW. Apolipoprotein E receptors mediate neurite outgrowth through activation of p44/42 mitogen-activated protein kinase in primary neurons. J. Biol. Chem. 2004;279:34948–34956. doi: 10.1074/jbc.M401055200. [DOI] [PubMed] [Google Scholar]
- Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod A, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc. Natl. Acad. Sci. USA. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Frey WH, Baker LD, Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR, Schellenberg GD, Cherrier MM, Craft S. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging. 2006;27:451–458. doi: 10.1016/j.neurobiolaging.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH, Craft S. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer's Dis. 2008a;13:323–331. doi: 10.3233/jad-2008-13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JC, DeGroodt W, Mehta P, Craft S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008b;70:440–448. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun L, Chen K, Bandy D, Minoshima S, Thibodeau S, Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the ε 4 allele for apolipoprotein E. N. Engl. J. Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E ε 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2001;98:3334–3339. doi: 10.1073/pnas.061509598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc. Natl. Acad. Sci. USA. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E ε4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. USA. 2005;102:8299–82302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM. Linking brain imaging and genomics in the study of Alzheimer's disease and aging. Ann. NY Acad. Sci. 2007;1097:94–113. doi: 10.1196/annals.1379.011. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL, Joshipura KD, Pearson JV, Hu-Lince D, Huentelman MJ, Craig DW, Coon KD, Liang WS, Herbert RH, Beach T, Rohrer KC, Zhao AS, Leung D, Bryden L, Marlowe L, Kaleem M, Mastroeni D, Grover A, Heward CB, Ravid R, Rogers J, Hutton ML, Melquist S, Petersen RC, Alexander GE, Caselli RJ, Kukull W, Papassotiropoulos A, Stephan DA. GAB2 alleles modify Alzheimer's risk in APOE ε4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Schmidinger T, Alexopoulos P, Horn M, Maus S, Reichel M, Rhein C, Lewczuk P, Sidiropoulos C, Kneib T, Perneczky R, Doerfler A, Kornhuber J. Influence of brain-derived neurotrophic-factor and apolipoprotein E genetic variants on hippocampal volume and memory performance in healthy young adults. J. Neural Transm. 2011;118:249–257. doi: 10.1007/s00702-010-0539-8. [DOI] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD, Rosiglitazone in Alzheimer's Disease Study Group Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J. 2010;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon E, Kerrouche N, Perani D, Lekeu F, Holthoff V, Beuthienbaumann B, Sorbi S, Lemaire C, Collette F, Herholz K. On the multivariate nature of brain metabolic impairment in Alzheimer's disease. Neurobiol. Aging. 2009;30:186–197. doi: 10.1016/j.neurobiolaging.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, Habeck CG, Stern Y, Anderson K. APOE genotype and cerebral blood flow in healthy young individuals. JAMA. 2003;290:1581–1582. doi: 10.1001/jama.290.12.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Habeck CG, Hilton J, Anderson KE, Flynn J, Park A, Stern Y. APOE related alterations in cerebral activation even at college age. J. Neurol. Neurosurg. Psychiatry. 2005;76:1440–1444. doi: 10.1136/jnnp.2004.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, Clasen L, Evans A, Rapoport JL, Giedd JN. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6:494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- Smith MA, Drew KL, Nunomura A, Takeda A, Hirai K, Zhu X, Atwood CS, Raina AK, Rottkamp CA, Sayre LM, Friedland RP, Perry G. Amyloid-beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs?. Neurochem. Int. 2002;40:527–531. doi: 10.1016/s0197-0186(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Metabolic reserve as a determinant of cognitive aging. J. Alzheimer's Dis. 2012;30:S5–S13. doi: 10.3233/JAD-2011-110899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance MA, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance MA, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA. 2000;91:11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J. Alzheimer's Dis. 2010;20:S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Vedhachalam C, Sakamoto T, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H. Effect of carboxyl-terminal truncation on structure and lipid interaction of human apolipoprotein E4. Biochemistry. 2006;45:4240–4247. doi: 10.1021/bi060023b. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A, Van Leuven F. Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am. J. Pathol. 2000;157:1495–1510. doi: 10.1016/S0002-9440(10)64788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg AJ, Filippini N, Mackay CE. The effects of APOE-ε4 on the BOLD response. Neurobiol Aging. 2012a;33:323–334. doi: 10.1016/j.neurobiolaging.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Trachtenberg AJ, Filippini N, Cheeseman J, Duff EP, Neville MJ, Ebmeier KP, Karpe F, Mackay CE. The effects of APOE on brain activity do not simply reflect the risk of Alzheimer's disease. Neurobiol. Aging. 2012b;33:618.e1–618. doi: 10.1016/j.neurobiolaging.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Trachtenberg AJ, Filippini N, Ebmeier KP, Smith SM, Karpe F, Mackay CE. The effects of APOE on the functional architecture of the resting brain. NeuroImage. 2012c;59:565–572. doi: 10.1016/j.neuroimage.2011.07.059. [DOI] [PubMed] [Google Scholar]
- Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA. 2010;107:17757–17762. doi: 10.1073/pnas.1010459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer's patients: superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001;21:4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Schneider LE, Niedzielko TL, Coon KD, Caselli RJ, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Schneider L, Small A, Gonzalez-Lima F. Quantitative cytochrome oxidase histochemistry: applications in human Alzheimer's disease and animal models. J. Histotechnol. 2007;30:235–247. [Google Scholar]
- Valla J, Yaari R, Wolf AB, Kusne Y, Beach TG, Roher AE, Corneveaux JJ, Huentelman MJ, Caselli RJ, Reiman EM. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset Alzheimer's susceptibility gene. J. Alzheimer's Dis. 2010;22:307–313. doi: 10.3233/JAD-2010-100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villain N, Desgranges B, Viader F, de la Sayette V, Mézenge F, Landeau B, Baron J-C, Eustache F, Chételat G. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J. Neurosci. 2008;28:6174–6181. doi: 10.1523/JNEUROSCI.1392-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ ) deposition. Proc. Natl. Acad. Sci. USA. 2010;107:17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am. J. Geriatr. Psychiatry. 2005;13:950–958. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- Weisgraber KH, Rall SC, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- Wetterau JR, Aggerbeck LP, Rall SC, Weisgraber KH. Human apolipoprotein E3 in aqueous solution. I. evidence for two structural domains. J. Biol. Chem. 1988;263:6240–6248. [PubMed] [Google Scholar]
- Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- Wong-Riley M. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol. 1999;154:601–611. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 Domain Interaction Occurs in Living Neuronal Cells as Determined by Fluorescence Resonance Energy Transfer . J. Biol. Chem. 2004;279:25511–25516. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Walker D, Bernardo A, Brodbeck J, Balestra ME, Huang Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J. Neurosci. 2008;28:1452–1459. doi: 10.1523/JNEUROSCI.3253-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-P, Gilley JA, Zhang G, Kernie SG. ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development. 2011;4362:4351–4362. doi: 10.1242/dev.065540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer's disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med. 2011;32:247–257. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. J. Biol. Chem. 2009;284:6027–6031. doi: 10.1074/jbc.R800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. J. Biol. Chem. 2009;284:27273–27280. doi: 10.1074/jbc.M109.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]