

Abstract
Angiotensin II (Ang II) is the principal effector of the renin-angiotensin-aldosterone system (RAAS). It initiates myriad processes in multiple organs integrated to increase circulating volume and elevate systemic blood pressure. In the kidney, Ang II stimulates renal tubular water and salt reabsorption causing antinatriuresis and antidiuresis. Activation of RAAS is known to enhance activity of the epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron. In addition to its well described stimulatory actions on aldosterone secretion, Ang II is also capable to directly increase ENaC activity. In this brief review, we discuss recent findings about non-classical Ang II actions on ENaC and speculate about its relevance for renal sodium handling.
Keywords: Renin-angiotensin-aldosterone system, RAAS, intrarenal RAS, aldosterone, aldosterone-sensitive distal nephron, collecting duct, connecting tubule, mineralocorticoid receptors, hypertension, salt-sensitive hypertension, sodium reabsorption, AT1 receptors, aldosterone paradox
Introduction
As the major contributor to extracellular osmolarity, total amount of Na+ in the body determines circulatory volume and sets chronic blood pressure. Sodium homeostasis is under mastering control of the kidneys which adjust urinary sodium excretion to match with dietary sodium intake [1]. Renin-angiotensin-aldosterone system (RAAS) and its principal driving force Angiotensin II (Ang II) protect against decreases in systemic blood pressure and circulatory collapse, in part, by stimulating sodium reabsorption in the kidney [2]. In fact, we are programmed genetically to effectively conserve salt and excrete virtually Na+-free urine when faced dietary sodium restriction [3,4]. In contrast, drastic elevation of sodium content in Western diets often overuses antinatriuretic power of incompletely suppressed RAAS leading to expansion of the circulating volume and hypertension [5,6]. Multiple studies suggest positive correlation between habitual sodium intake and arterial blood pressure in different ethnic populations (reviewed in [6])
Distal part of the renal nephron is the final site where sodium reabsorption occurs. “Distal nephron” is a loose definition which is commonly used to describe tubular segments located distally to the macula densa. For simplistic reasons, we use this term as a combined reference to the connecting tubule (CNT) and the cortical collecting duct (CCD). Sodium reabsorption in the distal convoluted tubule (DCT) will be also briefly discussed below. Apically localized epithelial Na+ channels (ENaC) in principal cells of the CNT and CCD determine sodium reabsorption in the distal nephron [•7–14]. Proper ENaC function is a critical component of sodium handling by the kidney [12–16]. At the daily timescale, ENaC processes the amount of sodium greater than that present in the circulation. The importance of ENaC to blood pressure control in humans is carved by the fact that monogenic forms of blood pressure disorders such as Liddle’s syndrome and Type I pseudohypoaldosteronism (PHA) arise from gain-of-function and loss-of-function ENaC mutations, respectively [14,17–22]. ENaC-mediated electrogenic sodium entry also provides driving force for luminal potassium exit via renal outer medullary potassium (ROMK) and maxi-K (BK) channels (reviewed in [•7]). This is why distortions in ENaC-mediated sodium reabsorption are often associated with altered renal potassium secretion [19].
Aldosterone-Dependent and -Independent Regulation of ENaC
ENaC-mediated sodium reabsorption in the distal nephron is inversely related to dietary sodium intake [•7,23,24]. This is because ENaC, being a critical target of the RAAS, is involved in final adjustments of sodium handling by the kidney. The long-standing classical model of sodium balance regulation in the distal nephron postulates that decreases in effective circulation volume or reduced dietary sodium intake lead to production of the potent antinatriuretic hormone Ang II from its inactive precursor Angiotensinogen (AGT) via subsequent cleavages by renin and angiotensin converting enzyme (ACE) [2]. Ang II, in turn, promotes secretion of the principal ENaC regulator, aldosterone from zona glomerulosa in the adrenal gland. Aldosterone, through its binding to mineralocorticoid receptors (MR) in principal cells, stimulates Na+ reabsorption at the distal nephron by increasing ENaC activity at both functional and transcriptional levels [10–14,16,25]. Indeed, hyperaldosteronism is linked to enhanced sodium reabsorption in the distal nephron whereas aldosterone insufficiency leads to reduced ENaC activity and urinary sodium wasting [26,27]. In the literature, the distal part of renal nephron expressing MR is also often referred as aldosterone-sensitive distal nephron (ASDN).
ENaC-mediated Na+-reabsorption creates a favorable driving force for K+ secretion from principal cells [7]. Historically, hypokalemia was thought to be a prerequisite of primary hyperaldosteronism, but it is now recognized that most patients with primary hyperaldosteronism might not manifest low serum K+ levels [28]. This is likely related to so-called “aldosterone paradox” where comparable elevations in circulating aldosterone levels during volume depletion and hyperkalemia result in distinct physiological outcomes. Specifically, volume contraction leads to effective Na+ conservation with no prominent K+ wasting. In contrast, hyperkalemia causes primarily kaliuresis with little sodium retention (reviewed in [29]). This argues that regulation of sodium handling and specifically ENaC activity in the distal nephron by systemic physiological stimuli is not solely regulated by aldosterone. Indeed, substantial experimental and clinical evidence suggest a role for aldosterone-independent mechanisms in regulation of ENaC in vivo. For instance, mice lacking mineralocorticoid receptors possess significant (up to 30%) residual ENaC-mediated Na+ reabsorption in the ASDN [30]. Recent report demonstrates that mice with surgically removed adrenal gland have no detectable circulating aldosterone but possess robust ENaC activity in the distal nephron [•31]. Finally, ENaC blocker amiloride produces a significantly higher drop in blood pressure compared to MR inhibition in hypertensive patients [32].
As discussed above, decreases in effective circulating volume and elevations of plasma [K+] are potent physiological stimuli for aldosterone secretion. However, hyperkalemia, in contrast to hypovolemia, does not lead to activation of the renin-angiotensin axis. This may indicate that profound sodium retention in the distal nephron during hypovolemia is the result of combined actions of the fully activated RAAS and more specifically Ang II, whereas ENaC activation during hyperkalemia is mediated solely by aldosterone.
Direct Actions of Ang II on ENaC
Ang II is well recognized for its antinatriuretic actions in the kidney. Ang II can act through two different types of receptors: AT1 and AT2 receptors. The majority of Ang II actions on the renal sodium transport is mediated by AT1 receptors. Mice with global knockout of the major subtype of AT1 receptors, (AT1a) develop lifetime hypotension [33] and increased urinary sodium excretion particularly during low sodium diet [34]. Using cross-transplantation strategy, Crowley and colleagues [35] convincingly demonstrated that genetic ablation of renal AT1 receptors in the kidney abolishes Ang II-induced hypertension and urinary sodium retention. The follow-up recent study identifies that the antinatriuretic effect of Ang II in the kidney is mainly salt-dependent and attributed to the AT1 receptor-mediated increased sodium reabsorption [36].
Ang II receptors are abundantly expressed at both apical and basolateral sides of epithelial cells along the whole length of the renal nephron from the proximal tubule to the collecting duct [37–39]. Unlike the case in the proximal tubule, where AT1a receptor deletion disturbs pressure-natriuresis and decreases blood pressure [40], much less is known about contribution of the distal nephron AT1 receptors to renal sodium reabsorption. An important advance in our understanding of the direct role of Ang II in the distal nephron was provided upon development of mice lacking AT1a receptor. These mice exhibit a marked reduction in αENaC abundance in the kidney despite normal or even slightly elevated aldosterone levels [41]. This suggests that AT1aR-mediated signaling is a critical component of regulation of ENaC levels in the distal nephron that cannot be substituted by aldosterone-MR axis. Surprisingly, collecting duct-specific deletion of AT1a receptor (driven by Hoxb7 promoter) does not change abundance of ENaC subunits (α, β, and γ) upon normal conditions whereas it compromises urinary concentrating ability by reducing AQP-2 levels [••42]. This, though, does not disqualify Ang II from being a direct regulator of ENaC abundance. One possible explanation is that putative decreases in ENaC expression in the CCD might be masked by a respective upregulation of ENaC levels in the CNT. Alternatively, global deletion of AT1a receptor also alters release of other hormones, such as insulin and vasopressin via AT1-dependent pathways [43,44]. Both hormones were reported to stimulate ENaC activity in the distal nephron [45,46]. It does not seem that mice with AT1a receptor deletion in the collecting duct are volume depleted under normal salt intake and, thus, there is little need to stimulate ENaC-mediated sodium reabsorption in the distal nephron. Stressing these mice with low sodium diet would be useful with this regard. Furthermore, conventional Western blotting used to assess ENaC abundance has no precision to detect changes in subcellular ENaC distribution. Using physiologically relevant tissue, freshly isolated split-opened distal nephrons, we did observe prominent ENaC translocation from cytosolic compartments to the apical plasma membrane in response to direct application of Ang II [••47]. At the functional level, prolonged Ang II treatment increases the number of active ENaC in split-opened distal nephrons [••47]. Future studies are necessary to test if Ang II-facilitated ENaC trafficking does occur during hypovolemic states. Alternatively, such mechanism might contribute to the excessive renal sodium retention in pathological states, including diabetes, associated with strong upregulation of intrarenal RAS in the distal nephron (see below).
While the mechanism and importance of Ang II regulation of ENaC expression and trafficking remains largely obscure, our understanding of the functional consequences of the direct Ang II actions on ENaC in the distal nephron is much more advanced. The first observation of the acute Ang II effect on the sodium handling in the distal nephron was provided by Peti-Peterdi and colleagues [48]. They found that application of Ang II to perfused rabbit CCDs increased luminal sodium transport and this effect was prevented by AT1 receptor antagonist losartan. Such acute action indicates direct regulation of ENaC activity by Ang II. Indeed, using patch clamp electrophysiology in split-opened murine distal nephron [••47], our group directly demonstrated that Ang II via AT1 receptors stimulates ENaC activity by affecting channel gating (open probability, Po). Similar observation was also made in rat cortical collecting ducts [••49]. Figure 1A summarizes indirect and direct Ang II actions on ENaC. In contrast to its common action in vasculature cells [50,51], Ang II does not seem to stimulate Gq/11-PLC-[Ca2+]i pathway in principal cells. In contrast, a critical role of NADPH oxidase, possibly NOX2 [52], and subsequent generation of reactive oxygen species, possibly superoxide and peroxide, as a downstream of Ang II signaling cascade was identified in rat and mouse distal nephrons [••47, ••49]. Ang II-induced generation of H2O2 may, in part, stimulate ENaC by diminishing inhibitory actions of the arachidonic acid (AA) [••49]. Indeed, several AA metabolites, including 11,12-EET; 8,9-EET and 14,15-EET, were demonstrated to inhibit ENaC in the cortical collecting duct and cultured mpkCCDc14 cells [53–55]. Alternatively, peroxide can also affect ENaC via AA-independent mechanisms that involve activation of c-Src – PI3-kinase – PI(3,4,5)P3 cascade in Xenopus A6 distal nephron cells [•56]. However, inhibition of either PLA2 or PI3-kinase does not abolish stimulatory actions of Ang II on ENaC [••47]. It is possible that the mechanism of Ang II activation of ENaC is complex and involves several synergistic components. This may explain why pharmacological ablation of a single pathway fails to preclude stimulation of ENaC by Ang II.
Figure 1. (A) Principal scheme of direct stimulatory effect of Ang II and aldosterone on ENaC in the distal nephron.
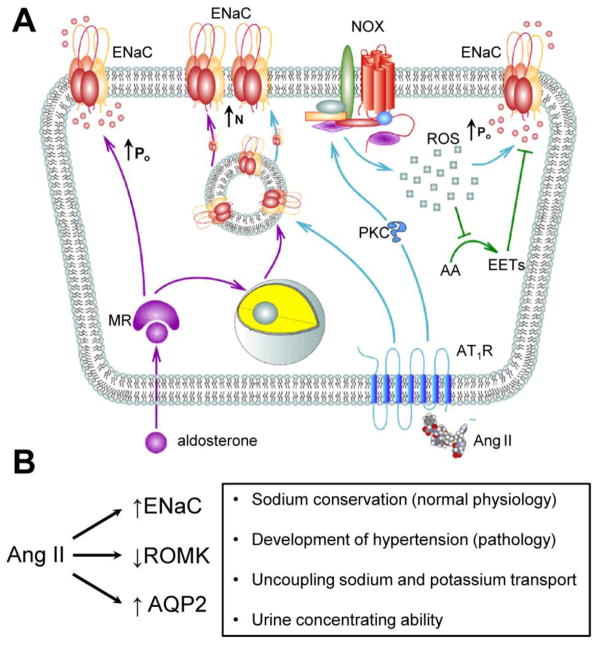
MR – mineralocorticoid receptors; AT1R – Angiotensin II type 1 receptors, PKC – protein kinase C; NOX – NADPH oxidase complex; ROS – reactive oxygen species; AA – arachidonic acid; EETs – Epoxyeicosatrienoic acids. ↑Po – increased ENaC open probability; ↑N – increased ENaC expression at the apical plasma membrane. This scheme is modified from Mamenko M, Zaika O, Ilatovskaya DV, et al. (2012) Angiotensin II Increases Activity of the Epithelial Na+ Channel (ENaC) in Distal Nephron Additively to Aldosterone. J Biol Chem 287: 660–671. © the American Society for Biochemistry and Molecular Biology. (B) Ang II actions on major transport proteins in the distal nephron. Physiological consequences of these actions are outlined on the right.
Currently, there is no direct experimental evidence that Ang II also facilitates the basolateral Na+ extrusion via Na+/K+ pump in the distal nephron. However, this is a highly probable scenario since Ang II is known to stimulate activity of the Na+/K+ pump in the proximal tubule [57].
Non-Redundancy of Ang II Actions in the Distal Nephron
Sufficient experimental evidence suggests that direct stimulatory actions of Ang II on ENaC occur in an aldosterone-independent manner. Thus, the effect of Ang II on ENaC persists upon saturation [••47] and inhibition [••49] of MR signaling. The time courses of ENaC activation by aldosterone and Ang II are also remarkably different. Ang II is able to stimulate ENaC within several minutes whereas aldosterone is ineffective at this timeframe [••47]. Indeed, aldosterone operates at the timescale of hours to affect ENaC [58] and the earliest detectable ENaC activation is approximately 30–60 min [59]. In addition, the primary and the most defined actions of aldosterone on ENaC are related to regulation of channel transcription and ENaC residency on the apical plasma membrane via stimulation of SGK-1 and release of Nedd4-2-mediated ENaC ubiquitination (for more details see excellent recent reviews [60–62]). Such separation in the mechanisms of actions (expression vs. gating) and time-dependencies (hours vs. minutes) between aldosterone and Ang II indicates that these two physiologically relevant antinatriuretic hormones are perfectly positioned to complementary stimulate ENaC in the distal nephron during volume depleted conditions.
Role of Ang II in Uncoupling Na+ and K+ Transport in the Distal Nephron
Recent experimental evidence favors the scenario in which coordinated actions of aldosterone and Ang II are able to separately tune interconnected sodium and potassium transport in the distal nephron to achieve appropriate physiological outcome. In this regard, a rare inherited salt-sensitive hypertension known as pseudohypoaldosteronism type II (PHA II; also known as familial hyperkalemic hypertension or Gordon syndrome) recently received a lot of attention. At the systemic levels this disease is characterized by the simultaneous retention of sodium and potassium leading to arterial hypertension, hyperkalemia, and metabolic acidosis [63,64]. Seminal discovery of the genetic basis of PHA type II revealed an important role of atypical with-no-Lysine kinases, WNKs (specifically WNK1 and WNK4) acting as a molecular switches to regulate sodium and potassium transporting systems depending on physiological demands [64]. Importantly, both Ang II and aldosterone utilize WNKs in their signal transduction pathways (reviewed in [29]). Since regulation of sodium and potassium handling in the distal nephron by WNKs was recently highlighted by several excellent reviews [65–68], we will not cover this aspect in the current manuscript.
To appreciate a role of Ang II in distinct regulation of renal sodium and potassium handling, both electrogenic sodium transport in the CNT/CCD and electroneutral sodium reabsorption in the distal convoluted tubule (DCT) need to be considered in tandem. DCT is the shortest tubular segment that reabsorbs 8–10% of total amount of filtered sodium. DCT is divided by the early (DCT1) and late (DCT2) segments. Activity of the luminal thiazide-sensitive sodium chloride cotransporter (NCC) is mainly responsible for the electroneutral sodium transport in this part of the nephron [69–71]. NCC expression gradually decays in DCT2 whereas ENaC expression becomes detectable in DCT2 [69]. MR and AT1 receptors are abundantly expressed at the both apical and basolateral sides although DCT1 is considered to be insensitive to aldosterone due to the lack of expression of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) which protects MR from activation by more abundant glucocorticosteroids. Similarly to the CNT/CCD, DCT expresses ROMK and BK that are responsible for potassium secretion at this site (reviewed in [•7]).
Elevated dietary potassium intake is known to be a physiological stimulus, whilst not as a potent as volume depletion, for aldosterone secretion from the adrenal gland [72]. Despite the fact that aldosterone stimulates both NCC- [73–75] and ENaC-mediated [60] sodium reabsorption via SGK-1 mediated pathways, increased renal secretion of potassium in response to hyperkalemia is not accompanied with renal sodium retention. Unexpectedly, NCC abundance and phosphorylation are greatly diminished during high potassium diet [75,76] suggesting that functional status of NCC is no longer regulated by aldosterone during this condition. The mechanism of this dissociation remains poorly understood. Furthermore, moderately increased aldosterone levels during high K+ intake in the absence of the activation of renin-Ang II axis do not cause robust ENaC activation resulting in a limited renal capacity to retain sodium. Increased delivery of Na+ to the ENaC-containing nephron segments creates favorable conditions for potassium exit via aldosterone-inducible potassium channel ROMK (discussed in [29]). In addition, elevations in tubular flow during high potassium intake stimulate flow-dependent K+ secretion by activating luminal BK channels in both principal and intercalated cells [77], though the mechanism of augmented basolateral K+ influx particularly in intercalated cells, which possess very low expression levels of Na+-K+-pump, remains elusive.
Low salt intake obligates the kidney to conserve virtually all filtered amount of sodium avoiding life-threatening hypovolemia. This requires maximal activation of the complete renin-angiotensin-aldosterone cascade which, in turn, increases the expression and activity of NCC and ENaC. Importantly, antinatriuretic actions of aldosterone and Ang II do not seem to duplicate each other. Indeed, recent studies [78,79] provide compelling evidence of the stimulatory Ang II actions on the sodium-chloride cotransporter in the DCT and this effect occurs additively to aldosterone-induced phosphorylation of NCC [80,81]. Similarly, activation of ENaC by Ang II does not depend on the status of aldosterone signaling [••47, ••49]. It is reasonable to speculate that Ang II is necessary for aldosterone-independent stimulation of the major sodium transporting proteins in the distal nephron, ENaC and NCC, to achieve maximally efficient renal sodium conservation specifically during volume depletion. Strikingly, antinatriuresis during low Na+ intake does not lead to appreciable potassium loses. It is proposed that augmented NCC activity results in decreased delivery of sodium to the ENaC-containing CNT/CCD which, in turn, reduces the driving force for ROMK-mediated potassium secretion. However, when hypotonic protourine exits from the water-impermeable DCT to the water permeable CNT/CCD the concentration of sodium will increase substantially due to AQP2-mediated water uptake. Avid sodium reabsorption in the CNT/CCD via maximally stimulated ENaC could create a sufficient electrochemical gradient for augmented potassium secretion via ROMK. Indeed, infusion of the ENaC blocker, amiloride produces robust natriuresis when rats placed on low sodium regimen [82]. This argues for involvement of additional mechanisms uncoupling sodium and potassium transport in the distal nephron. Importantly, recent evidence from Wang’s laboratory demonstrated that Ang II inhibits activity of ROMK1 during conditions of volume depletion [•83] and potassium restriction [84] and the signaling pathway likely involves activation of NADPH oxidase [84]. Recall, Ang II also stimulates ENaC activity in NADPH oxidase-dependent manner [••47, ••49]. Common signaling mechanism of Ang II regulation of sodium and potassium transport argues for an important role of Ang II in uncoupling sodium reabsorption by stimulating ENaC from potassium secretion by inhibiting ROMK in ASDN.
Intrarenal RAS in Controlling of Sodium Handling in the Distal Nephron
In experimental animal models of Ang II-induced hypertension [85,86], intrarenal Ang II levels become much higher than plasma Ang II levels. This suggests that the hormone can be synthesized in the kidney (reviewed in [87]). Indeed, all components of the functional local RAS are present in the distal nephron. Principal cells in the CNT and CCD abundantly express renin [88,89] which, in turn, cleaves Angiotensinogen (AGT) synthesized by the proximal tubule to Ang I [90]. ACE is present at the apical membrane of distal nephron cells allowing paracrine generation of Ang II from Ang I [89,91,92]. Luminal perfusion of isolated rabbit CCD with Ang I stimulates apical sodium transport in ACE-dependent manner [92]. Since principal cells express angiotensin receptors at both apical and basolateral sides, it is possible that circulating and tubular Ang II can separately regulate ENaC-mediated sodium reabsorption during different physiological conditions. In contrast to the systemic RAS where elevations in circulating Ang II inhibit renin secretion from the granular cells in the juxtaglomerular apparatus, augmented distal nephron Ang II further increases renin mRNA and protein levels [93]. It is proposed that this positive feedback leads to inappropriate elevation of the sodium reabsorption in the distal nephron playing a major role in the development and maintenance of high blood pressure [93,94]. While elevated dietary salt intake causes suppression of the systemic RAS, the intrarenal RAS is greatly upregulated in Dahl salt-sensitive (SS) rats leading to increased abundance of ROS-generating and proinflammatory/profibrosis proteins and an inability to raise antioxidant enzymes [95,96]. Consistently, both β and γENaC mRNA expression and protein abundance were significantly increased despite low plasma and kidney aldosterone content in Dahl SS rats on high-salt diet [97]. Substantial experimental evidence suggest that similar paradoxical upregulation of the intrarenal RAS also occurs during Diabetes mellitus likely contributing to hypertension and renal tissue injury (reviewed in [98]). The critical role of a succinate receptor, GPR91 in stimulating of (pro)renin production in CNT/CCD was recently proposed [99]. This will lead, in turn, to activation of the intrarenal RAS.
Conclusions
During past several years our understanding of the mechanisms and physiological relevance of the direct Ang II actions on water and electrolyte transport in the distal nephron is greatly advanced (Figure 1B). Cumulative evidence demonstrates that Ang II possesses multiple stimulatory actions on ENaC involving the effect on channel gating, trafficking and possibly expression. Combined stimulatory actions of aldosterone and Ang II on ENaC allow maximizing sodium reabsorption in the distal nephron under conditions of volume depletion to minimize sodium loss with urine. Ang II appears to be a critical factor that allows separate control of distal nephron sodium and potassium transport in response to physiological clues. In contrast, regulation of ENaC by Ang II might also have detrimental consequences on sodium balance under pathological conditions that cause overstimulation of the intrarenal RAS, such as salt-sensitive hypertension and Diabetes mellitus.
Acknowledgments
We thank Daria Ilatovskaya (Medical College of Wisconsin) for help in preparation of the figure. The research is supported by the American Heart Association SDG2230391 (to OP), the Carl W. Gottschalk research scholar grant from the American Society of Nephrology (to OP) and National Institutes of Health DK09502 (to OP) and HL108880 (to AS).
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
Contributor Information
Oleg Zaika, Email: Oleg.L.Zaika@uth.tmc.edu, Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, 6431 Fannin str., Houston, TX 77030 USA; Ph. (713) 500-6342; Fx. (713) 500-6342.
Mykola Mamenko, Email: Mykola.Mamenko@uth.tmc.edu, Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, 6431 Fannin str., Houston TX 77030, USA; Ph. (713) 500-6342; Fx. (713) 500-6342.
Alexander Staruschenko, Email: staruschneko@mcw.edu, Department of Physiology, Medical College of Wisconsin, 8701 Watertown Plank Rd., Milwaukee, WI 53226, USA; Ph. (414) 955-8475; Fx. (414) 955-6546.
Oleh Pochynyuk, Email: Oleh.M.Pochynyuk@uth.tmc.edu, Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, 6431 Fannin str., Houston TX 77030, USA; Ph. (713) 500-7466; Fx. (713) 500-7455.
References
Papers of articular interest, published recently, have been highlighted as:
• of importance
•• of major importance
- 1.Guyton AC. Blood pressure control-special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 2.Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res. 2012;318:1049–1056. doi: 10.1016/j.yexcr.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konner M, Eaton SB. Paleolithic nutrition: twenty-five years later. Nutr Clin Pract. 2010;25:594–602. doi: 10.1177/0884533610385702. [DOI] [PubMed] [Google Scholar]
- 4.Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med. 1985;312:283–289. doi: 10.1056/NEJM198501313120505. [DOI] [PubMed] [Google Scholar]
- 5.Khawaja Z, Wilcox CS. Role of the kidneys in resistant hypertension. Int J Hypertens. 2011;2011:143471. doi: 10.4061/2011/143471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 7•.Staruschenko A. Regulation of Transport in the Connecting Tubule and Cortical Collecting Duct. Compr Physiol. 2012;2:1541–1584. doi: 10.1002/cphy.c110052. This comprehensive review manuscript provides up-to-date overview of mechanisms controlling channels and transporters in the distal part of renal nephron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, Zhang JN, Zhao D, et al. Role of the epithelial sodium channel in salt-sensitive hypertension. Acta Pharmacol Sin. 2011;32:789–797. doi: 10.1038/aps.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem. 2010;285:30363–30369. doi: 10.1074/jbc.R110.155341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton DC, Malik B, Saxena NC, et al. Mechanisms of aldosterone’s action on epithelial Na+ transport. J Membr Biol. 2001;184:313–319. doi: 10.1007/s00232-001-0098-x. [DOI] [PubMed] [Google Scholar]
- 11.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 12.Hummler E. Implication of ENaC in salt-sensitive hypertension. J Steroid Biochem Mol Biol. 1999;69:385–390. doi: 10.1016/s0960-0760(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 13.Hummler E. Epithelial sodium channel, salt intake, and hypertension. Curr Hypertens Rep. 2003;5:11–18. doi: 10.1007/s11906-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 14.Schild L. The epithelial sodium channel: from molecule to disease. Rev Physiol Biochem Pharmacol. 2004;151:93–107. doi: 10.1007/s10254-004-0023-7. [DOI] [PubMed] [Google Scholar]
- 15.Schild L, Kellenberger S. Structure function relationships of ENaC and its role in sodium handling. Adv Exp Med Biol. 2001;502:305–314. doi: 10.1007/978-1-4757-3401-0_20. [DOI] [PubMed] [Google Scholar]
- 16.Stockand JD. New ideas about aldosterone signaling in epithelia. Am J Physiol Renal Physiol. 2002;282:F559–F576. doi: 10.1152/ajprenal.00320.2001. [DOI] [PubMed] [Google Scholar]
- 17.Hansson JH, Nelson-Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 18.Hansson JH, Schild L, Lu Y, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A. 1995;92:11495–11499. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 20.Schild L. The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie. 1996;17:395–400. [PubMed] [Google Scholar]
- 21.Shimkets RA, Warnock DG, Bositis CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 22.Schafer JA. Abnormal regulation of ENaC: syndromes of salt retention and salt wasting by the collecting duct. Am J Physiol Renal Physiol. 2002;283:F221–F235. doi: 10.1152/ajprenal.00068.2002. [DOI] [PubMed] [Google Scholar]
- 23.Palmer LG, Frindt G. Regulation of apical membrane Na and K channels in rat renal collecting tubules by aldosterone. Semin Nephrol. 1992;12:37–43. [PubMed] [Google Scholar]
- 24.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol. 2009;297:F1249–F1255. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verrey F. Transcriptional control of sodium transport in tight epithelial by adrenal steroids. J Membr Biol. 1995;144:93–110. doi: 10.1007/BF00232796. [DOI] [PubMed] [Google Scholar]
- 26.Warnock DG. Aldosterone-related genetic effects in hypertension. Curr Hypertens Rep. 2000;2:295–301. doi: 10.1007/s11906-000-0013-3. [DOI] [PubMed] [Google Scholar]
- 27.Makhanova N, Lee G, Takahashi N, et al. Kidney function in mice lacking aldosterone. Am J Physiol Renal Physiol. 2006;290:F61–F69. doi: 10.1152/ajprenal.00257.2005. [DOI] [PubMed] [Google Scholar]
- 28.Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1:621–636. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- 29.Arroyo JP, Ronzaud C, Lagnaz D, et al. Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology (Bethesda ) 2011;26:115–123. doi: 10.1152/physiol.00049.2010. [DOI] [PubMed] [Google Scholar]
- 30.Berger S, Bleich M, Schmid W, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Mironova E, Bugaj V, Roos KP, et al. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci U S A. 2012;109:10095–10100. doi: 10.1073/pnas.1201978109. This manuscript convincingly demonstrates that aldosterone is sufficient but not necessary for ENaC activity in the distal nephron. Ablation of adrenal gland results in compensatory regulation of ENaC by vasopressin. This shifts the role of ENaC from protecting Na+ balance to promoting water reabsorption. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16:3154–3159. doi: 10.1681/ASN.2005050460. [DOI] [PubMed] [Google Scholar]
- 33.Ito M, Oliverio MI, Mannon PJ, et al. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci U S A. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliverio MI, Best CF, Smithies O, Coffman TM. Regulation of sodium balance and blood pressure by the AT1A receptor for angiotensin II. Hypertension. 2000;35:550–554. doi: 10.1161/01.hyp.35.2.550. [DOI] [PubMed] [Google Scholar]
- 35.Crowley SD, Gurley SB, Herrera MJ, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crowley SD, Zhang J, Herrera M, et al. Role of AT1 receptor-mediated salt retention in angiotensin II-dependent hypertension. Am J Physiol Renal Physiol. 2011;301:F1124–F1130. doi: 10.1152/ajprenal.00305.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison-Bernard LM, Navar LG, Ho MM, et al. Immunohistochemical localization of ANG II AT1 receptor in adult rat kidney using a monoclonal antibody. Am J Physiol. 1997;273:F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 38.Ozono R, Wang ZQ, Moore AF, et al. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30:1238–1246. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- 39.Miyata N, Park F, Li XF, Cowley AW., Jr Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277:F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 40.Gurley SB, Riquier-Brison AD, Schnermann J, et al. AT1a angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brooks HL, Allred AJ, Beutler KT, et al. Targeted proteomic profiling of renal Na+ transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002;39:470–473. doi: 10.1161/hy02t2.102959. [DOI] [PubMed] [Google Scholar]
- 42••.Stegbauer J, Gurley SB, Sparks MA, et al. AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol. 2011;22:2237–2246. doi: 10.1681/ASN.2010101095. This study provides first evidence that collecting duct AT1a receptors are necessary for normal kidney function. While it does not affect ENaC abundance under normal sodium intake, disruption of AT1a in the principal cells impairs urinary concentrating ability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saavedra JM, Benicky J, Zhou J. Angiotensin II: multitasking in the brain. J Hypertens Suppl. 2006;24:S131–S137. doi: 10.1097/01.hjh.0000220418.09021.ee. [DOI] [PubMed] [Google Scholar]
- 44.Leung PS. The physiology of a local renin-angiotensin system in the pancreas. J Physiol. 2007;580:31–37. doi: 10.1113/jphysiol.2006.126193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol. 2009;297:F1411–F1418. doi: 10.1152/ajprenal.00371.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staruschenko A, Pochynyuk O, Vandewalle A, et al. Acute Regulation of the Epithelial Na+ Channel by Phosphatidylinositide 3-OH Kinase Signaling in Native Collecting Duct Principal Cells. J Am Soc Nephrol. 2007;18:1652–1661. doi: 10.1681/ASN.2007010020. [DOI] [PubMed] [Google Scholar]
- 47••.Mamenko M, Zaika O, Ilatovskaya DV, et al. Angiotensin II Increases Activity of the Epithelial Na+ Channel (ENaC) in Distal Nephron Additively to Aldosterone. J Biol Chem. 2012;287:660–671. doi: 10.1074/jbc.M111.298919. Using physiologically relevant tissue: split-opened distal nephrons of mice, this study demonstrates that Ang II directly controls ENaC activity. This regulation is complex and includes an acute activation of functional ENaC via generation of ROS as well as more chronic effect on ENaC trafficking to the apical plasma membrane. Furthermore, the effect of Ang II on ENaC is independent of aldosterone status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. J Am Soc Nephrol. 2002;13:1131–1135. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 49••.Sun P, Yue P, Wang WH. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol. 2012;302:F679–F687. doi: 10.1152/ajprenal.00368.2011. Synchronous with Ref 47, this study demonstrates that Ang II stimulates ENaC in rat CCDs through a Ca2+-independent PKC pathway, activation of NOX and superoxide generation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 51.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 52.Babilonia E, Lin D, Zhang Y, et al. Role of gp91phox -containing NADPH oxidase in mediating the effect of K restriction on ROMK channels and renal K excretion. J Am Soc Nephrol. 2007;18:2037–2045. doi: 10.1681/ASN.2006121333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun P, Lin DH, Yue P, et al. High potassium intake enhances the inhibitory effect of 11,12-EET on ENaC. J Am Soc Nephrol. 2010;21:1667–1677. doi: 10.1681/ASN.2009111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei Y, Lin DH, Kemp R, et al. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–727. doi: 10.1085/jgp.200409140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pavlov TS, Ilatovskaya DV, Levchenko V, et al. Effects of cytochrome P-450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC) Am J Physiol Renal Physiol. 2011;301:F672–F681. doi: 10.1152/ajprenal.00597.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56•.Ma HP. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase dependent pathway. J Biol Chem. 2011;286(37):32444–53. doi: 10.1074/jbc.M111.254102. This study provides the first evidence that ROS and particularly H2O2 directly activate ENaC. As discussed in this manuscript, ROS could serve as downstream signaling molecules for direct effect of Ang II. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bharatula M, Hussain T, Lokhandwala MF. Angiotensin II AT1 receptor/signaling mechanisms in the biphasic effect of the peptide on proximal tubular Na+, K+-ATPase. Clin Exp Hypertens. 1998;20:465–480. doi: 10.3109/10641969809053225. [DOI] [PubMed] [Google Scholar]
- 58.Pacha J, Frindt G, Antonian L, et al. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol. 1993;102:25–42. doi: 10.1085/jgp.102.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loffing J, Zecevic M, Feraille E, et al. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol. 2001;280:F675–F682. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- 60.Pearce D, Kleyman TR. Salt, sodium channels, and SGK1. J Clin Invest. 2007;117:592–595. doi: 10.1172/JCI31538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rotin D, Staub O. Role of the ubiquitin system in regulating ion transport. Pflugers Arch. 2011;461:1–21. doi: 10.1007/s00424-010-0893-2. [DOI] [PubMed] [Google Scholar]
- 62.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 63.Kahle KT, Rinehart J, Giebisch G, et al. A novel protein kinase signaling pathway essential for blood pressure regulation in humans. Trends Endocrinol Metab. 2008;19:91–95. doi: 10.1016/j.tem.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 64.Wilson FH, sse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 65.Arroyo JP, Gamba G. Advances in WNK signaling of salt and potassium metabolism: clinical implications. Am J Nephrol. 2012;35:379–386. doi: 10.1159/000337479. [DOI] [PubMed] [Google Scholar]
- 66.Hoorn EJ, Ellison DH. WNK kinases and the kidney. Exp Cell Res. 2012;318:1020–1026. doi: 10.1016/j.yexcr.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol. 2011;22:605–614. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol. 2008;70:329–355. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 69.Loffing J, Loffing-Cueni D, Valderrabano V, et al. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol. 2001;281:F1021–F1027. doi: 10.1152/ajprenal.0085.2001. [DOI] [PubMed] [Google Scholar]
- 70.Loffing J, Kaissling B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am J Physiol Renal Physiol. 2003;284:F628–F643. doi: 10.1152/ajprenal.00217.2002. [DOI] [PubMed] [Google Scholar]
- 71.Obermuller N, Bernstein P, Velazquez H, et al. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol. 1995;269:F900–F910. doi: 10.1152/ajprenal.1995.269.6.F900. [DOI] [PubMed] [Google Scholar]
- 72.LeHoux JG, Bird IM, Rainey WE, et al. Both low sodium and high potassium intake increase the level of adrenal angiotensin-II receptor type 1, but not that of adrenocorticotropin receptor. Endocrinology. 1994;134:776–782. doi: 10.1210/endo.134.2.7507836. [DOI] [PubMed] [Google Scholar]
- 73.Kim GH, Masilamani S, Turner R, et al. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A. 1998;95:14552–14557. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nielsen J, Kwon TH, Masilamani S, et al. Sodium transporter abundance profiling in kidney: effect of spironolactone. Am J Physiol Renal Physiol. 2002;283:F923–F933. doi: 10.1152/ajprenal.00015.2002. [DOI] [PubMed] [Google Scholar]
- 75.Vallon V, Schroth J, Lang F, et al. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol. 2009;297:F704–F712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol. 2010;299(4):F890–7. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pluznick JL, Sansom SC. BK channels in the kidney: role in K+ secretion and localization of molecular components. Am J Physiol Renal Physiol. 2006;291:F517–F529. doi: 10.1152/ajprenal.00118.2006. [DOI] [PubMed] [Google Scholar]
- 78.San-Cristobal P, Pacheco-Alvarez D, Richardson C, et al. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, et al. ANG II provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am J Physiol Renal Physiol. 2007;293:F662–F669. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 80.van der LN, Lim CH, Meima ME, et al. Aldosterone does not require angiotensin II to activate NCC through a WNK4-SPAK-dependent pathway. Pflugers Arch. 2012;463:853–863. doi: 10.1007/s00424-012-1104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van der LN, Lim CH, Fenton RA, et al. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 82.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol. 2009;297:F389–F396. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83•.Yue P, Sun P, Lin DH, et al. Angiotensin II diminishes the effect of SGK1 on the WNK4-mediated inhibition of ROMK1 channels. Kidney Int. 2011;79:423–431. doi: 10.1038/ki.2010.380. By demonstrating that Ang II inhibits ROMK channels in the cortical collecting duct of rats on a low sodium diet, this study suggests that Ang II has an important role in suppressing potassium secretion during volume depletion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei Y, Zavilowitz B, Satlin LM, Wang WH. Angiotensin II inhibits the ROMK-like small conductance K channel in renal cortical collecting duct during dietary potassium restriction. J Biol Chem. 2007;282:6455–6462. doi: 10.1074/jbc.M607477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gonzalez-Villalobos RA, Seth DM, Satou R, et al. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol. 2008;295:F772–F779. doi: 10.1152/ajprenal.00019.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez-Villalobos RA, Satou R, Ohashi N, et al. Intrarenal mouse renin-angiotensin system during ANG II-induced hypertension and ACE inhibition. Am J Physiol Renal Physiol. 2010;298:F150–F157. doi: 10.1152/ajprenal.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39:316–322. doi: 10.1161/hy0202.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rohrwasser A, Morgan T, Dillon HF, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34:1265–1274. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 89.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–186. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am J Nephrol. 2010;31:541–550. doi: 10.1159/000313363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Casarini DE, Boim MA, Stella RC, et al. Angiotensin I-converting enzyme activity in tubular fluid along the rat nephron. Am J Physiol. 1997;272:F405–F409. doi: 10.1152/ajprenal.1997.272.3.F405. [DOI] [PubMed] [Google Scholar]
- 92.Komlosi P, Fuson AL, Fintha A, et al. Angiotensin I conversion to angiotensin II stimulates cortical collecting duct sodium transport. Hypertension. 2003;42:195–199. doi: 10.1161/01.HYP.0000081221.36703.01. [DOI] [PubMed] [Google Scholar]
- 93.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, et al. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–229. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prieto-Carrasquero MC, Botros FT, Pagan J, et al. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hypertensive rats. Hypertension. 2008;51:1590–1596. doi: 10.1161/HYPERTENSIONAHA.108.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chandramohan G, Bai Y, Norris K, et al. Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-kappaB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol. 2008;28:158–167. doi: 10.1159/000110021. [DOI] [PubMed] [Google Scholar]
- 96.De MC, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1136–R1142. doi: 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, Tomita K. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens. 2009;27:1679–1689. doi: 10.1097/HJH.0b013e32832c7d23. [DOI] [PubMed] [Google Scholar]
- 98.Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int. 2010;78:1214–1217. doi: 10.1038/ki.2010.333. [DOI] [PubMed] [Google Scholar]
- 99.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, Peti-Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118:2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]