

Abstract
The structure of OmpF porin in complex with three common antibiotics (zwitterionic ampicillin, anionic ertapenem, and di-anionic carbenicillin) was determined using X-ray crystallography. The three antibiotics are found to bind within the extracellular and periplasmic pore vestibules, away from the narrow OmpF constriction zone. Using the X-ray structures as a starting point, non-equilibrium MD simulations with an applied membrane voltage show that ionic current through the OmpF channel is blocked with bound ampicillin, but not with bound carbenicillin. The susceptibility of E. coli expressing OmpF mutants to ampicillin and carbenicillin was also experimentally characterized using microbiological assays. These results show that general diffusion by OmpF porins allows for transfer of molecules with varied charged states and give new insights into the design of more efficient antibiotics. A better understanding of this mechanism will shed light on nature's way of devising channels able to enhance the transport of molecules through membranes.
Introduction
β-lactam antibiotics, the most widely used antibacterial agents, function by inactivating penicillin-binding proteins involved in cell wall synthesis within the periplasmic space of Gram-negative bacteria. The treatment of bacterial infections, however, is increasingly complicated by the ability of bacteria to develop resistance to antimicrobial agents, now prevalent in a number of key Gram-negative bacterial pathogens, including Escherichia coli, Salmonella, Klebsiella, Enterobacter, and Pseudomonas (Pagès et al., 2008). Bacterial resistance to β-lactam antibiotics can be caused by three factors: modification by inactivating enzymes, expulsion by multidrug efflux pumps, and reduction of outer membrane (OM) permeability, the latter being the least well-understood. The OM, unique to Gram-negative bacteria, acts as a selectivity barrier by providing an extra layer against harmful compounds in the environment. General diffusion porins, expressed at high levels (>105 copies per cell) in the OM (Delcour, 2009), allow non-specific transport of charged and zwitterionic nutrient molecules. E. coli produce three major general diffusion porins: OmpF, OmpC, and PhoE. The loss of OmpF and OmpC porins has been linked to antibiotic resistance in several reports, especially for E. coli and Salmonella typhimurium (Nikaido, 2003). Additional studies have also shown that mutations of key OmpF residues can substantially alter susceptibility to antibiotics (Bredin et al., 2002; Bredin et al., 2003; Simonet et al., 2000). Since a decrease in membrane permeability results in reduced accumulation of cellular antibiotic levels, it often functions synergistically with the other resistance mechanisms by allowing time for drug inactivation or efflux pump expression (Delcour, 2009). Therefore, a better understanding of how modification of membrane permeability triggers bacterial resistance is necessary for the development of new antibiotic therapy strategies.
Crystal structures for the E. coli OmpF and PhoE proteins (Cowan et al., 1995; Cowan et al., 1992), and more recently for the E. coli OmpC (Baslé et al., 2006), Klebsiella pneumoniae OmpK36 (Dutzler et al., 1999), and Salmonella typhi OmpF (Balasubramaniam et al., 2012) proteins reveal large homotrimers of β-barrels consisting of a conserved antiparallel 16-strand structure. The large number and configuration of β-strands allows each monomer to form a separate central pore, which functions independently of the others in terms of solute permeation. β-turn motifs connect the β-strands on the periplasmic side of the channel, while a number of long loops protrude out of the extracellular domain. One of these extracellular loops (L2) contacts the adjacent porin monomer to stabilize the trimer structure, while a second long loop (L3) folds back into the middle of the barrel forming the “constriction zone,” which defines the size exclusion limit for permeation. The E. coli OmpF constriction zone is marked by a cluster of acidic residues found on L3, which faces a cluster of basic residues on the adjacent β-barrel wall. This gives rise to a strong transverse electric field that is thought to affect ion permeation and solute transfer (Im and Roux, 2002a; Im and Roux, 2002b). A recent X-ray structure of OmpF porin from KCl or RbCl-soaked crystals has also provided experimental evidence for a specific ion pathway across the OmpF channel (Dhakshnamoorthy et al., 2010).
The high-resolution crystal structure of OmpF porin has enabled detailed molecular dynamics (MD) simulation studies of antibiotic permeation (Ceccarelli et. al., 2004; Danelon et al., 2006; Hajjar et al., 2010a; Hajjar et al., 2010b; Hajjar et al., 2010c; Kumar et al., 2010). Specifically, MD simulations showed that the zwitterionic ampicillin molecule remained stably bound when docked at the OmpF constriction zone. On the other hand, di-anionic carbenicillin moved toward the extracellular side of the OmpF constriction zone, reorienting parallel to the channel axis where it closely associated with the positively charged pore wall and blocked the channel lumen only partially (Danelon et al., 2006). Computational techniques have also been used to assess total antibiotic diffusion profiles across OmpF by employing a metadynamics biasing algorithm to overcome the long timescale of antibiotic translocation (Ceccarelli et al., 2004; Danelon et al., 2006; Hajjar et al., 2010a; Hajjar et al., 2010b; Hajjar et al., 2010c; Kumar et al., 2010). Such calculations suggested that ampicillin visits an energy minimum within the constriction region of the OmpF pore, and led to the proposal that interaction with residues in the OmpF constriction zone would help facilitate antibiotic transfer by partially compensating for the entropic and desolvation costs of drug confinement in this narrow region (Danelon et al., 2006). However, due to the relatively short length of the MD trajectories, these simulation studies remain somewhat limited.
Experimental data for antibiotic permeation through OM porins remains scarce. Measurement of antibiotic flux in whole cells reported rates on the order of ~10–50×10−5 cm/s for the permeation of zwitterionic drugs across OmpF, but were greatly reduced for anionic compounds (Nikaido et al., 1983). Evidence for a direct role of general diffusion porins in mediating the diffusion of β-lactams was further elucidated by liposome swelling assays, involving reconstitution of purified OmpF or OmpC into proteoliposomes. In these studies, zwitterionic compounds penetrated rapidly, flux of mono-anionic compounds was related to the hydrophobicity of the molecule, and di-anionic compounds penetrated at the slowest rates (Yoshimura and Nikaido, 1985). However, some 50–70% of swelling may be due to the influx of counter ions for mono-anionic and di-anionic compounds, respectively (Yoshimura and Nikaido, 1985). For this reason, direct evidence for the interaction of antibiotics with OmpF porin has been sought from electrophysiology measurements of OmpF inserted in a lipid bilayer. In these single channel recording experiments, brief interruptions of ionic current were observed in the presence of ampicillin and amoxicillin (Danelon et al., 2006; Nestorovich et al., 2002), but no such current interruptions were observed in the presence of three anionic compounds: carbenicillin, azlocillin, and piperacillin (Danelon et al., 2006), reflecting either a lack of binding or reduced binding affinity within the pore. Therefore, the single channel recording experiments, together with the results from MD simulations of antibiotic permeation, suggest that zwitterionic antibiotics—but not their anionic analogues—make specific and favorable interactions near the OmpF constriction zone. Given that liposome swelling assays report fast diffusion rates for zwitterionic antibiotics, this counter-intuitively implies that binding at the constriction zone is necessary to facilitate efficient antibiotic transfer across OmpF porin. However, antibiotic binding within the pore can only be detected in single channel recordings if the flow of ionic current through the channel is completely blocked or substantially reduced. It seems conceivable that some compounds may bind within the wide aqueous extracellular or periplasmic OmpF pores without actually altering the flow of ionic current. Ultimately, whether interactions in the pore facilitate or hinder efficient antibiotic transfer across OmpF porin remains unclear.
To address these issues, the OmpF channel was crystallized in complex with three common β-lactam antibiotics, ampicillin, carbenicillin, and ertapenem (Fig. 1), allowing determination of their binding sites within the lumen of the pore for the first time. The structures reveal a bound ampicillin oriented perpendicular to the pore axis on the extracellular side of the constriction zone, while the bound carbenicillin and ertapenem are oriented parallel to the pore axis near the periplasmic mouth or extracellular loops of the pore, respectively. In addition, all-atom MD simulations based on the X-ray structures show that ionic current through the OmpF channel is blocked with bound ampicillin, but not with bound carbenicillin. Lastly, the susceptibility of E. coli expressing mutants of OmpF porin to ampicillin and carbenicillin showed that disruption of key drug-protein interactions resulted in a general increase in antibiotic susceptibility. These results provide new evidence for the role of drug-protein interactions in bacterial permeation of antibiotics.
Figure 1.
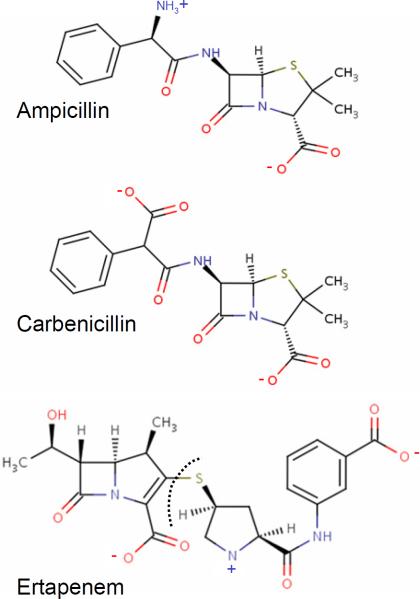
Three β-lactam antibiotics are considered in the present investigation. The zwitterionic ampicillin molecule (top) is neutral but carries a large dipole moment owing to the positively-charged amine group (NH3+) and the negatively charged carboxylate group (COO−). The di-anionic carbenicillin (middle) differs only by a substitution of ampicillin's NH3+ by a second negatively-charged carboxylate moiety located at the segment linked to the aromatic group (referred as a-COO− while the carboxylate linked to the penem group will be referred as p-COO−). The anionic ertapenem (bottom) is longer than ampicillin and carbenecillin and harbors one positively and two negatively-charged groups. The segment of ertapenem that is observed in the X-ray structure lies on the right of the dashed line.
Results
Ampicillin binds within the extracellular pore
The structure of OmpF in complex with ampicillin was solved at 2.0 Å resolution after subsequent ampicillin soaking of Mg2+-free OmpF crystals (Fig. 2A and Table 1). An Fobs – Fcalc difference map, with ligand omitted from the atomic model, showed a well-defined electron density in the OmpF pore, which was absent from crystals grown in the same condition without ampicillin. The ampicillin binding site was located ~6 Å above the OmpF constriction zone, within the larger extracellular pore vestibule. According to the electron density, ampicillin is oriented perpendicular to the channel axis, in a configuration that allows a number of stabilizing interactions with OmpF residues on both sides of the pore. Specifically, ampicillin's negatively charged COO− group is positioned in between two arginine residues (R167, R168) and is also within hydrogen-bonding distance of the S125 side chain OH and backbone nitrogen groups. Furthermore, a nearby ordered water molecule, found at this same position in the apo OmpF structure (PDB ID 2ZFG), extends the hydrogen-bonding network by forming a bridge between R168 and the ampicillin COO− group. At the other end of the ampicillin molecule, the positively charged NH3+ moiety is positioned between the G119 backbone oxygen atom, the side chains of D121 and Y32, and an additional water molecule. Finally, the phenyl ring of ampicilin is positioned in a hydrophobic pocket near several aromatic residues (Y22, Y32, F118), which could provide favorable pi stacking interactions. Comparison with the apo structure of OmpF indicates that the bound ampicillin does not induce a significant conformational change of the surrounding protein atoms; the total RMSD (including side chain atoms) for residues that interact with ampicillin is only 0.13 Å compared with the apo structure (PDB ID 2ZFG). However, the B-factor values for ampicillin (average of 59 Å2, compared with an average B-factor of 38 Å2 for all protein atoms and 33 Å2 for interacting residue atoms), suggest that ampicillin experiences thermal fluctuations within its binding site (Fig. S3). This is consistent with the weak equilibrium binding constant (1.9 M−1) of ampicillin to the OmpF pore (Nestorovich et al., 2002).
Figure 2.
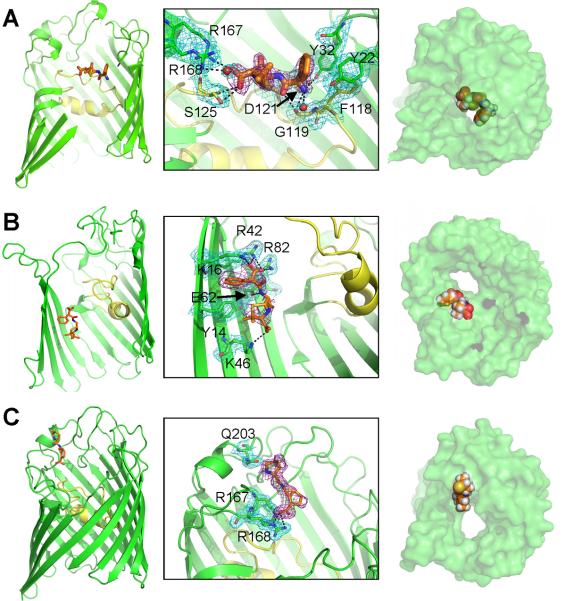
Crystal structure of OmpF porin in complex with three β-lactam antibiotic molecules: ampicillin (A), carbenicillin (B), or ertapenem (C). Left: Ribbon diagram depiction of a single OmpF monomer with the antibiotic (sticks) binding orientation shown (L3 colored yellow). Middle: Zoomed-in view of OmpF residues (green or yellow sticks) within hydrogen-bonding distance (3.3 Å) of the antibiotic molecule (orange sticks). Right: Space-filling view of the OmpF monomer with the bound antibiotic from the extracellular side (ampicillin and ertapenem) or from the periplasmic side (carbenicillin). Ampicillin is also coordinated by two water molecules shown as red spheres. The Fobs – Fcalc omit map at 2.0 σ cutoff (purple mesh) shows the electron density for the antibiotic and the 2Fobs – Fcalc density map at 1.0 σ cutoff (cyan mesh) shows the electron density for the modeled OmpF residues. L3 is colored yellow. Also see Figures S1, S2, and S3.
Table 1.
Intensity and refinement statistics: OmpF in complex with antibiotic.
| OmpF Structure | OmpF/ampicillin | OmpF/carbenicillin | OmpF/ertapenem |
|---|---|---|---|
| Crystal | |||
| Space group | P3 | P3 | P3 |
| Cell Constants | |||
| a, b (Å) | 116.6 | 116.7 | 116.8 |
| c (Å) | 53.3 | 53.9 | 52.8 |
| Data Collection | |||
| Resolution (Å) | 50.0–2.0 | 47.5–2.3 | 36.5–1.9 |
| Measured reflections | 245,574 | 224,254 | 471,080 |
| Unique reflections | 56,274 | 36,275 | 65,981 |
| Redundancy | 2.2 (2.1) | 2.1 (2.0) | 2.0 (1.9) |
| I/σ (I) | 16.8 (2.0) | 17.1 (2.0) | 9.5 (2.1) |
| Completeness (%) | 98.7 (97.9) | 98.3 (98.5) | 96.5 (96.9) |
| Rmerge | 0.071 (0.424) | 0.074 (0.413) | 0.085 (0.339) |
| Refinement | |||
| R | 0.202 | 0.219 | 0.188 |
| Rfree | 0.239 | 0.272 | 0.227 |
| RMSD from ideal | |||
| Bond lengths (Å) | 0.025 | 0.023 | 0.024 |
| Bond angles (°) | 2.12 | 2.06 | 2.16 |
Values in parentheses are for the outermost shell
Carbenicillin binds within the periplasmic pore
The crystal structure of OmpF in complex with carbenicillin (2.3 Å), a di-anionic molecule with a size and structure similar to ampicillin was also determined (Fig. 2B and Table 1). The OmpF-carbenicillin complex was also obtained by subsequent soaking OmpF crystals in antibiotic solution. An Fobs - Fcalc map, with carbenicillin absent in the atomic model, shows extra electron density within the OmpF pore as well, but far from the ampicillin binding site (Fig. 4A). Carbenicillin is oriented parallel to the channel axis and closely associates with the β-barrel wall in the intracellular OmpF pore vestibule, about 10 Å below the constriction zone. In its binding orientation, the phenyl group of carbenicillin faces upward (toward the extracellular side of the pore), and is positioned close to Y14. This allows the nearby COO− group, located at the linkage segment terminating at the aromatic group (referred as a-COO−), to make several contacts with basic residues (K16, R42, R82) just below the constriction zone. A hydrogen bond between a carbenicillin nitrogen moiety and E62 further stabilizes this binding orientation. On the other hand, only a single hydrogen bond, between the COO− attached to the penem group (referred as p-COO−) and K46, is observed in the periplasmic pore, allowing for considerable fluctuations of this half of carbenicillin. This is reflected in the B-factor values (Fig. S3), which are ~25% higher for atoms near the penem group (average B-factor of 86 Å2), compared with atoms on the phenyl side of the carbenicillin molecule (average B-factor of 63 Å2). Carbenicillin binding does not perturb the OmpF residues, as shown by an RMSD of 0.26 Å for residues that can interact with carbenicillin, when compared with the corresponding residues from the apo structure (PDB ID 2ZFG).
Figure 4.
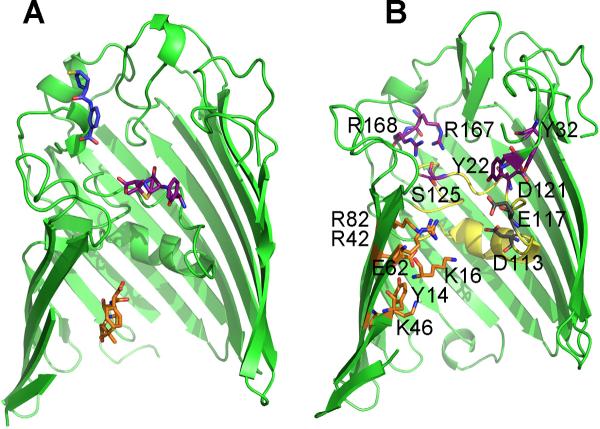
Antibiotic binding sites. (A) The crystallographic positions of ertapenem (blue sticks), ampicillin (purple sticks), and carbenicillin (orange sticks) are superimposed on a single OmpF monomer (ribbons with front β-strands removed for clarity). (B) OmpF residues assessed in this study are shown and colored based on those found in either the ampicillin (purple sticks) or carbenicillin (orange sticks) binding sites, or additional constriction zone residues (grey sticks). L3 is colored yellow.
Ertapenem binds in the extracellular loop region
The structure of OmpF in complex with ertapenem, a β-lactam molecule belonging to the carbapenem family and harboring one positive and two negative charges, was determined at 1.9 Å resolution after soaking OmpF crystals in ertapenem solution (Fig. 2C and Table 1). The Fobs – Fcalc omit map, with ertapenem unmodeled, showed clear density at the extracellular mouth of the OmpF pore, located ~17 Å above the constriction zone. The bound molecule is closely associated with the extracellular loops on one side of the OmpF pore. Although the experimental electronic density from the omit map was well defined for the ligand at this position, it could not be used to model the entire ertapenem molecule. Ertapenem consists of two rigid planar structures (including phenyl pyrroline rings on one side and the penem group on the other) brought together by a flexible bond at a sulfur atom (indicated by a dashed line in Fig. 1). Therefore, several rotameric states could exist for this molecule. It has been previously shown that ertapenem samples different isomerization states around this sulfur atom in the active sites of β-lactamase enzymes (Kalp and Carey, 2008). For example, a recent crystal structure of ertapenem in complex with a β-lactamase protein shows that two alternative ertapenem conformations could be modeled in the X-ray density due to isomerization at the sulfur atom (PDB IDs 3M6B and 3M6H) (Tremblay et al., 2010). Furthermore, in one β-lactamase binding orientation, only half of ertapenem (the segment lying on the right of the dashed line displayed in Fig. 1) was modeled, which was the same segment of ertapenem that was modeled in complex with OmpF. It was, however, still possible to predict the most probable orientation for ertapenem based on the partial experimental electron density. In this conformation, the ertapenem carboxylate group can hydrogen bond with R168, an interaction that was also identified for the ampicillin carboxylate moiety.
Molecular dynamics simulations of bound antibiotics
To assess the validity of the antibiotic binding sites for ampicilin and carbenicilin, MD simulations were carried out starting from the X-ray structures. The binding site of ertapenem was not simulated because the antibiotic is incomplete in the X-ray structure. To reduce the computational cost, a single OmpF monomer was considered. The monomer was embedded in a POPC lipid membrane and fully solvated, including 0.15 M KCl (Fig. S4). Despite fluctuations during the MD simulations, ampicillin did not leave its binding site as shown by a superposition of snapshots taken along the trajectory (Fig. 3A). In fact, the hydrogen bonds predicted from the X-ray structure (ampicillin COO− and OmpF residues S125, R167, and R168 and ampicillin NH3+ and OmpF residues Y32 and D121) were maintained along the trajectory. Furthermore, the molecule's dynamics were greatly reduced in the OmpF binding site when compared with free ampicillin simulated in a waterbox (Fig. 3A). Carbenicillin also remained bound in its X-ray orientation during the MD trajectory (Fig. 3B) and OmpF residues found to closely associate with carbenicillin in the crystal structure (carbenicillin a-COO− and OmpF residues K16, R42, and R82; carbenicillin nitrogen atom and OmpF residue E62; and carbenicillin p-COO− and OmpF residue K46) displayed hydrogen-bonding interactions along the MD trajectory. RMSD analysis also showed that while the phenyl side of carbenicillin underwent little fluctuation, the penem side displayed an increase in thermal motion, consistent with the large B-factor values also observed for these atoms in the crystal structure (Figs. 3B and S3).
Figure 3.
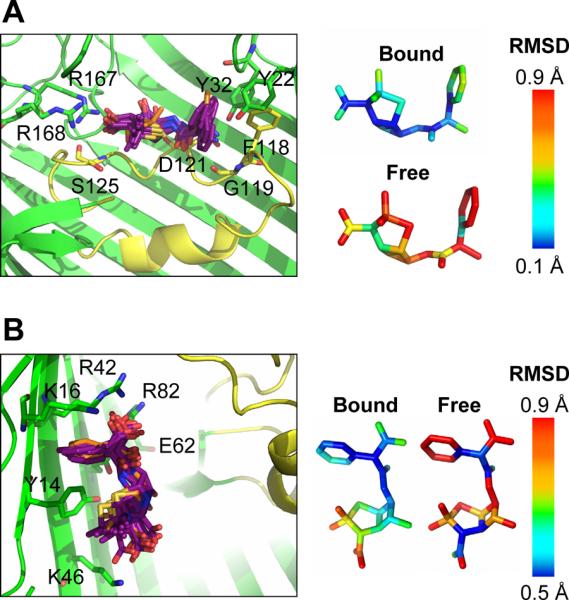
Antibiotic dynamics in the OmpF binding site. Left: A superposition of 1 ns snapshots shows ampicillin (A) or carbenicillin (B) conformations (purple sticks with the X-ray conformation shown in orange sticks) along the 10 ns MD trajectory. OmpF protein is depicted as ribbons with residues that form hydrogen bonds (within 3.3 Å) with the antibiotic shown as sticks. L3 is colored yellow. Right: The average RMSD along the MD trajectory for the antibiotic in its OmpF binding site was calculated and compared with the RMSD for free antibiotic simulated in a water box (10 ns). A snapshot of the MD system is shown in Figure S4.
Antibiotic effect on ion permeation and selectivity of OmpF porin
MD simulations were used to examine the effect of a bound antibiotic on ion permeation across the OmpF pore. Here the KCl concentration was increased to 1 M and a membrane potential of +/−150 mV was applied across the membrane along the Z-axis to reproduce the conditions used in single channel recordings. In a 50 ns trajectory, ion permeation was observed, corresponding to ionic conductance values of 0.43 nS and 0.58 nS for +/−150 mV simulations, respectively (Table 2). These ion conductance values are reduced by ~50% from experimental measurements and previous results from Brownian dynamics (BD) simulations (Im and Roux, 2002a). Presumably, the reduced conductance observed in the present MD reflects an increase in the ion permeation energy barrier (on the order of ~kBT) due to the loss of favorable interactions with the high dielectric regions from the two OmpF aqueous pores that were not included in the present simulations. Nonetheless, a slight cation selectivity was observed for the OmpF monomer (Fig. S5A), which has also been reported for the trimer channel using both experimental and computational approaches (Charrel et al., 1996; Gill et al., 1998; Goldstein et al., 1983; Nikaido, 1998).
Table 2.
Conductance (G) and ion selectivity (IK/ICl) of OmpF porin in 1 M KCl. Also see Figure S5.
| Vapp = +150 mV | Vapp = −150 mV | |||
|---|---|---|---|---|
|
|
||||
| G+ (nS) | IK/ICl | G− (nS) | IK/ICl | |
|
| ||||
| Apo | 0.43 | 1.00 | 0.58 | 1.46 |
| Ampicillin | 0.06 | 2.00 | 0 | 0 |
| Carbenicillin | 0.41 | - | 0.30 | 13.03 |
Alteration of ion permeation and selectivity for the OmpF pore was also investigated in the presence of antibiotic by all-atom MD. When ampicillin was restrained in its X-ray conformation, ion current through OmpF was dramatically reduced. Total ion conductance was either decreased over 7-fold (to 0.06 nS) or completely abolished for the +/−150 mV simulations in the presence of ampicillin (Fig. S5B and Table 2). This finding is consistent with previous electrophysiology recordings for OmpF channels inserted in a bilayer lipid membrane, which showed time-resolved current blockages across single channels in the presence of ampicillin (Danelon et al., 2006; Hajjar et al., 2010a; Hajjar et al., 2010c; Nestorovich et al., 2002). Interestingly, the binding of ampicillin in the extracellular vestibule even decreased the number of ions entering from the periplasmic side of the pore by 2-fold (Fig. S5B), providing evidence that long-range electrostatic properties of porins are not solely controlled by the narrow constriction zone. On the other hand, previous electrophysiology data has shown that, in the presence of carbenicillin and other anionic antibiotic molecules, ion permeation across the OmpF channel is not disrupted, suggesting that anionic molecules do not cross the OmpF channel (Danelon et al., 2006). MD simulations of ion transfer across the OmpF pore with carbenicillin bound in its X-ray site indicate that ions flow freely in the presence of the antibiotic molecule. During the 50 ns trajectory, the total ion conductance across the OmpF pore is only slightly decreased in the presence of carbenicillin (0.41 and 0.30 nS for the +/−150 mV simulations, respectively) compared with conductance values calculated in the absence of antibiotic (Table 2). However, carbenicillin binding induced a dramatic increase in cation selectivity, with zero Cl− crossing the pore in the +150 mV simulation and only a single Cl− permeation event for the −150 mV simulation (Fig. S5C). Most likely because of long-range electrostatic effects, the binding of carbenicillin in the periplasmic OmpF vestibule even reduces the number of anions that entered the extracellular vestibule on the other side of the pore.
Antibiotic diffusion pathways across OmpF
Analysis of the crystal structures suggests that interactions with specific residues may affect the binding and transfer rates of antibiotics through OmpF porin (Fig. 4). Therefore, non-equilibrium steered molecular dynamics (SMD) simulations were carried out to further examine the ampicillin and carbenicillin transfer paths. Here translocation of the molecule was driven through the pore with an applied external force, thereby accelerating this process from ~ 100 μs to a few ns. The initial state was defined by the coordinates from the antibiotic-bound OmpF crystal structures to ensure that the experimentally-located binding sites would be visited along the pathway. The antibiotic molecule was then dragged upward or downward from the starting position, and is subsequently discussed as a continuous permeation event from the extracellular side.
Results of the SMD trajectories show the interaction profiles within the OmpF pore for zwitterionic ampicillin and di-anionic carbenicillin. This was defined by mapping protein residues along the SMD trajectory within hydrogen-bonding distance (3.3 Å) of the translocating molecules. Along its transition pathway (Figs. 5A, 5C and S6A), ampicillin enters the extracellular mouth of the OmpF channel near residues E29, G33, T241, K243, and N246. It then continues to its extracellular binding site identified in the crystal structure, involving residues Y32, D121, S125, R167, and R168, where ampicillin is oriented perpendicular to the channel axis with the NH3+ moiety near the acidic L3 and the COO− group near the β-barrel wall lined with basic residues. The interaction with R167 and R168 was destabilized first, allowing the ampicillin COO− group to sample the area near the cluster of arginine residues (R42, R82, R132) in the narrow OmpF constriction. Establishing these new contacts, while maintaining the interaction between the ampicillin NH3+ group and D121, facilitates a reorientation of the ampicillin conformation from perpendicular to parallel relative to the pore axis (Figs. 5A and S6A). After crossing the constriction, ampicillin does not come within hydrogen-bonding distance of other residues until it exits the periplasmic side of the pore near residues D149, E182, and Y182.
Figure 5.
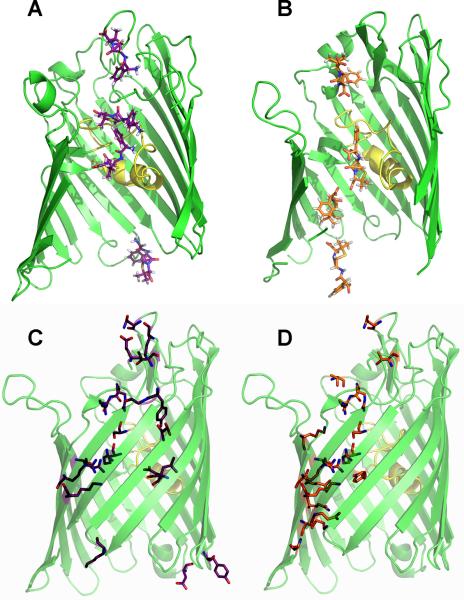
SMD simulation ofantibiotic transition pathways across the OmpF pore. Conformations of ampicillin (A) or carbenicillin (B) as well as OmpF residues that hydrogen-bond (within 3.3 Å) with either ampicillin (C) or carbenicillin (D) during the total antibiotic diffusion paths across the OmpF pore are shown as sticks. Also see Figure S6.
Along the translocation pathway determined from the SMD simulations (Figs. 5B, 5D and S6B), carbenicillin enters the extracellular mouth near a similar region as ampicillin, close to residues T165, N246, T247, and S248, but is immediately diverted to the β-barrel wall away from the acidic loop L3. In the extracellular pore, carbenicillin forms favorable interactions with S125, R167, and R168, contacts that were also identified for ampicillin. However, carbenicillin stays oriented parallel to the channel axis with its phenyl group pointed toward the extracellular mouth while it associates with the β-barrel wall and interacts with residues K80 and R132. Carbenicillin transfers across the constriction zone by forming favorable interactions with the arginine cluster and then settles into its high-affinity periplasmic binding site defined by interactions of the a-COO− moiety with K16, R42, and R82 and the p-COO− moiety with K46 in the periplasmic pore. Carbenicillin then continues along the β-barrel wall and exits the periplasmic mouth of the channel near residues A1 and Q339. The large number of contacts between carbenicillin and OmpF periplasmic residues is in sharp contrast from the lack of interactions observed with ampicillin in the periplasmic vestibule.
Antibiotic uptake of E. coli bacteria expressing OmpF mutant porins
Susceptibility of E. coli expressing mutations of OmpF to ampicillin and carbenicillin was assayed using disk diffusion and agar dilution tests. Results of both tests showed a general increase in both ampicillin and carbenicillin susceptibility values for E. coli bacteria expressing the mutant OmpF porins designed to disrupt antibiotic binding in the pore (Fig. 6). In the case of ampicillin (Fig. 6A), the largest increase in antibiotic susceptibility was observed by mutation of OmpF residues (D121N, R167S, R168S) that hydrogen-bond with the charged NH3+ and COO− ampicillin moieties. On the other hand, the Y22S mutant, involved only in hydrophobic interactions with the ampicillin phenyl group, showed no effect on antibiotic susceptibility measured by either microbiological test. Bacterial susceptibility to ampicillin was not affected by expression of OmpF mutants that disrupted the carbenicillin binding site in the periplasmic vestibule as shown by the disk diffusion test with no significant difference in ampicillin uptake for expression of Y14S, K16S, K46S, or E62Q OmpF proteins. Finally, mutations made to residues found in the narrow OmpF constriction zone showed varying levels of ampicillin susceptibility, with the R42S mutation, disrupting the conserved arginine cluster, displaying the largest effect.
Figure 6.
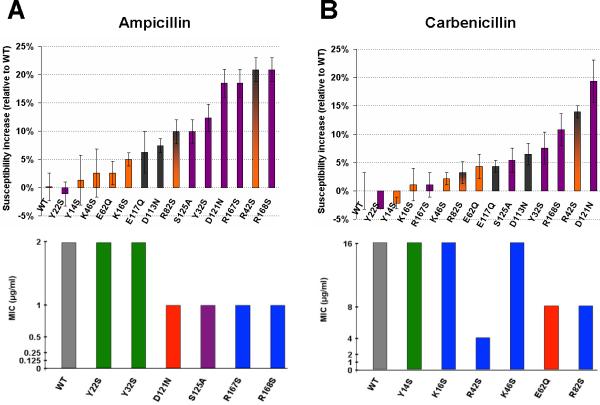
Microbial assaymeasurements of antibiotic susceptibility. The relative increase in measured bacterial growth inhibition zone diameters for expression of mutant OmpF porins (relative to WT) in the presence of either ampicillin (A) or carbenicillin (B) is shown by the disk diffusion assay (top). Bars are colored based on OmpF residues found in the extracellular ampicillin binding site (purple), periplasmic carbenicillin binding site (orange), constriction zone (grey), or both carbenicillin binding site and constriction zone (orange and grey). Error bars represent results from three different trials. Bottom:Minimal inhibitoryconcentrations (MICs) of ampicillin (A) or carbenicillin (B) are shown for bacteria expressing WT or mutant OmpF protein by the agar dilution assay. Bars are colored based on the WT residue characteristics: hydrophobic (green), hydrophilic (purple), acidic (red), and basic (blue). Results for bacteria expressing WT OmpF protein are colored grey. Data represents results from three different trials. Also see figure S7 and Table S1.
Bacterial susceptibility to carbenicillin also followed the general trend of increased antibiotic uptake with expression of OmpF mutants that disrupt its binding interactions (Fig. 6B). There was a significant increase in carbencillin uptake for for E. coli expressing the R42S mutant porin. Mutation of this residue, found in the middle of the three basic residues that hydrogen bond with the carbencillin a-COO− group (K16, R42, R82), would severely destabilize this binding site. On the other hand, single mutations of either OmpF residues K16 or R82 display less of an effect. Furthermore, mutation of K46, found near the carbenicillin p-COO− group, showed little difference in carbenicillin uptake when compared with bacteria expressing WT OmpF. Carbenicillin uptake was also affected by mutation of OmpF residues found in the extracellular vestibule, near the high-affinity ampicillin binding site. For example, there was a ~20% increase in carbenicillin susceptibility for bacteria expressing the D121N mutant OmpF porin as shown by the disk diffusion test. Finally, hydrophobic interactions did not affect bacterial uptake of carbencillin as the Y14S OmpF mutation did not alter carbenicillin susceptibility.
Discussion
Crystallization of the OmpF channel in complex with β-lactam antibiotics allowed visualization, for the first time, of their specific binding sites within the lumen of the large aqueous pore and showed wide variation for their preferred binding locations. In contrast with broadly held views, these results, summarized in Fig. 4A, show that the larger pore vestibular entryways of OmpF, rather than the narrow constriction zone, provide the lowest-energy binding sites for these common antibiotic molecules. Specifically, ampicillin in its zwitterionic form binds to the extracellular pore vestibule, oriented perpendicular to the pore axis where it simultaneously associates with both sides of the pore. The di-anionic carbenicillin binding site is located in the periplasmic pore vestibule, where carbenicillin is oriented parallel to the pore axis and closely associates with the β-barrel wall. Finally, harboring one positive and two negative charges, ertapenem is positioned within the extracellular vestibule, but located at the mouth of the extracellular vestibule near the extracellular loops. It adopts an orientation parallel to the pore axis, and on the same face of the OmpF pore as carbenicillin.
The ampicillin and carbenicillin binding sites identified by X-ray crystallography were further validated by MD simulations. Such simulations can offer an important test of the overall correctness of the antibiotic binding configurations, since these were determined principally from the limited information provided by omit maps and subsequently refined with B-factors higher than the surrounding protein atoms. It is thus encouraging to observe that, during the MD simulations, ampicillin and carbenicillin form stable hydrogen bonds with OmpF residues and remain in the specific configurations determined by X-ray crystallography. Interaction profiles of ampicillin and carbenicillin along the entire OmpF channel length were further identified by carrying out SMD simulations. While limited in scope, these SMD simulations are suggestive of the dynamical interactions during antibiotic diffusion through the pore. The results confirm that ampicillin adopts an orientation perpendicular to the pore axis while it is in the wide extracellular vestibule, most likely due to its large electric dipole. Similar interactions were observed in a previous metadynamics simulation study, which showed that the polar oxygens of ampicillin could form hydrogen bonds with extracellular residues R167 and R168 (Kumar et al., 2010). Furthermore, a reorientation of ampicillin from perpendicular to parallel with respect to the pore axis is observed for crossing the narrow constriction zone, as suggested previously (Ceccarelli et al., 2004; Danelon et al., 2006; Hajjar et al., 2010a; Hajjar et al., 2010b; Hajjar et al., 2010c; Kumar et al., 2010). Finally, ampicillin does not interact with specific residues in the periplasmic vestibule. The complete path identified from SMD for carbenicillin is very different. Carbenicillin maintains interactions with the β-barrel wall along its entire translocation path, including finding many favorable contacts in the periplasmic vestibule. A previous MD simulation study also observed similar interactions between carbenicillin and the β-barrel wall, allowing simultaneous interactions with the cluster of conserved arginine residues found in the constriction zone (R42, R82, R132) and two additional arginine residues (R167, R168) in the extracellular pore vestibule (Danelon et al., 2006). Therefore, while none of the previous simulation studies correctly predicted the binding site of ampicillin or carbenicillin in the OmpF pore, many observations were broadly consistent with the present results.
Previous antibiotic flux and microbial susceptibility assays for mutations of key charged OmpF constriction zone residues showed an increase in bacterial susceptibility to several β-lactams upon truncation of side chain residues (K16D, D113A, D121A, R132A, R132D) (Bredin et al., 2002; Bredin et al., 2003; Simonet et al., 2000; Vidal et al., 2005) and an increase in β-lactam resistance for substitution of a glycine residue for a protruding acidic side chain on L3 (G119D and G119E) (Bredin et al., 2003; Simonet et al., 2000). This led to the conclusion that charge alterations in the constriction zone substantially modify antibiotic susceptibility profiles. However, it is also possible that the impact of these mutations was caused by alteration in pore size. For example, a noticeable increase in cavity volume was observed for the R132A and R132D mutants by protein modeling (Bredin et al., 2002). Therefore, it remains to be shown whether charged residues within the narrow constriction zone play the largest role in determining total solute diffusion rates.
A recent study of clinical strains of multi-drug resistant E. coli identified sequence alterations of charged and polar residues within the pore of the related OmpC porin that altered antibiotic susceptibility profiles without affecting overall pore size (Lou et al., 2011). To achieve a more complete assessment of the role of charged OmpF residues in antibiotic uptake, microbial susceptibility assays were carried out for mutations of the residues directly interacting with either ampicillin or carbenicillin in the crystal structures. Results of microbial assays showed a general increase in both ampicillin and carbenicillin susceptibility for E. coli expressing OmpF mutations that disrupt electrostatic and hydrogen-bonding, but not hydrophobic, interactions in the X-ray binding sites. Although alteration of residues in the extracellular OmpF vestibule affects bacterial infection by both ampicillin and carbenicillin, these assays show that only carbenicillin susceptibility is affected by periplasmic pore mutations. Therefore, the rates of channel exit for zwitterionic and charged molecules may be completely altered by a single charge substitution, even in the large, aqueous OmpF periplasmic pore vestibule.
Ion permeation measurements through the OmpF channel in planar lipid bilayers has been an important technique to directly probe the interactions of antibiotics with the pore. These experiments revealed brief interruptions of ionic current in the presence of ampicillin (Danelon et al., 2006; Nestorovich et al., 2002), suggesting that the zwitterionic molecule can act as a pore blocker. Interestingly, no similar ion current blockage was observed in the presence of carbenicillin or other anionic molecules. This led to the suggestion that di-anionic molecules do not pass through the OmpF pore and that efficient translocation of antibiotics through the OmpF pore is associated with high affinity drug-protein interactions. However, the present X-ray structures suggest a different interpretation of these results. Visual inspection of space-filling models of the OmpF pore with the bound antibiotics (Fig. 2, right) indicates that ion current should be blocked by ampicillin, but not by carbenicillin or ertapenem. Furthermore, OmpF mutations that destabilize drug-protein interactions are shown here to increase uptake of both zwitterionic ampicillin and di-anionic carbenicillin by microbial assays. Results from non-equilibrium MD simulations carried out in the presence of a membrane potential show that, indeed, ion current through the channel is blocked when ampicillin is bound in the configuration from the crystal structure. On the other hand, ion current is not blocked when carbenicillin is bound as in the X-ray structure, showing that single channel recording was an unreliable approach for detecting the interaction of this molecule with OmpF. Interestingly, although carbenicillin has only a small effect on the magnitude of ion current, the bound di-anionic molecule yields a significant increase in cation selectivity. Therefore, it appears that the molecular charge of a diffusing antibiotic not only affects its own transfer pathway, but also that of the ions permeating through the channel.
The functional advantage for a bacterium to have weak-affinity antibiotic binding sites within the OmpF pore may be counter-intuitive. Previously proposed models suggest that an affinity site close to the mouth of the channel might favor the translocation of antibiotics across OmpF (Ceccarelli et al., 2012). One possibility is that, at a concentration approaching the equilibrium dissociation constant Kd of the binding site, the probability that the OmpF pore would be occupied and blocked by a bound molecule would increase such that the net flux would saturate to a finite maximum value independent of the surrounding antibiotic concentration. This imperfect defense barrier could provide an effective protection until slower mechanisms, such as regulation of porin expression or translation of catalytic enzymes or efflux pumps, are activated. One additional series of experiments were carried out to test this conjecture. It confirms that bacterial resistance against high concentrations of ampicillin from co-expression of the β-lactamase enzyme with OmpF is reduced two-fold upon mutation of key residues either found within the constriction zone or identified from the X-ray structure (Table S1).
It is then perhaps not surprising that mutation of single OmpF residues can show dramatic differences in the infection properties for ampicillin and carbenicillin, two antibiotics that are widely prescribed in the clinical setting. This has also been observed previously by a comparison of biophysical measurements of diffusing ampicillin across three different bacterial porins: E. coli OmpF, E. coli OmpC, and Enterobacter aerogenes Omp36 (Danelon et al., 2006; James et al., 2009; Nestorovich et al., 2002). Interestingly, these proteins show high sequence conservation within the narrow constriction zone, the previously assumed antibiotic binding site (Fig. 7A), but there was no ion conductance defect measured across OmpC and Omp36 in the presence of ampicillin (James et al., 2009). Analysis of the OmpC crystal structure shows a slightly more constricted pore (radius of 2.9 Å at the narrowest region, compared with 3.2 Å for OmpF) (Baslé et al., 2006), but examination of the K. pneumoniae OmpK36 crystal structure (Dutzler et al., 1999), which is 91% homologous to E. aerogenes Omp36, shows a slightly increased constriction zone (3.3 Å radius). Therefore, it seems likely that residues outside the constriction zone are responsible for providing the ampicillin binding site. Several residues found within the ampicillin binding site for E. coli OmpF are altered in the X-ray structures of OmpC and OmpK36, as well as in a recent crystal structure of the OmpF homologue from S. typhi (Balasubramaniam et al., 2012) (Fig. 7B). Specifically, binding interactions of the ampicillin carbonyl moiety would be destabilized in the OmpC, OmpK36, and S. typhi OmpF pores due to alteration of the proteins near the R167 and R168 residues. Furthermore, the OmpC, OmpK36, and S. typhi OmpF proteins each lack the Y32 residue, which is found in the hydrophobic pocket close to the ampicillin phenyl group and can also hydrogen-bond with the ampicillin NH3+ moiety. This not only provides an explanation for the surprising loss of pore-blocking by ampicillin for the OmpC and Omp36 proteins, but also gives further support that single point mutations within general diffusion porins can dramatically alter the uptake of antibiotics. It might therefore be technically possible to fine-tune the rate of antibiotic uptake by Gram-negative bacteria by controlling the porin residues that an antibiotic molecule contacts during its translocation.
Figure 7.
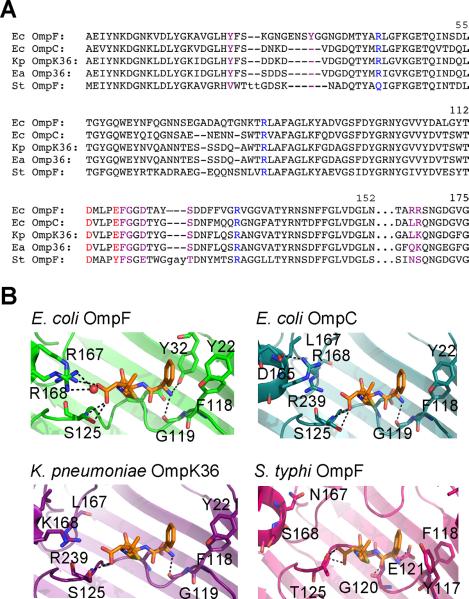
Comparison of E. coli OmpF, E. coli OmpC, K. pneumoniae OmpK36, and S. typhi OmpF. (A) Sequence alignment generated by the Dali server using E. coli OmpF (PDB ID 2OMF) as the query structure. Key constriction zone residues (acidic residues colored red and basic residues colored blue) and residues found within the ampicillin binding site (colored purple) are indicated. Lowercase letters represent residues without a match in the query structure. Numbering is based on the OmpF sequence. (B) Bound ampicillin, based on the OmpF-ampicillin crystal structure (top left), is superimposed with the structures of OmpC, OmpK36, and S. typhi OmpF and proposed hydrogen-bonding interactions (within 3.3 Å) are indicated.
Materials and Methods
Bacterial strains, media and expression of OmpF protein
OmpF was purified from E. coli strain BL21(DE3)Omp5, a porin-deficient mutant strain (Prilipov et al., 1998), which was kindly provided by Professor Ralf Koebnik. BL21(DE3)Omp5 cells were transformed with pRSF-1b plasmid (Novagen) encoding the OmpF protein sequence engineered with a His6-tag and a tobacco etch virus (TEV) cleavage site between the outer membrane targeting sequence and OmpF mature domain, leaving one glycine at the N-terminus after TEV cleavage (Fig. S1). This construct was engineered by the GenScript Corporation (GenScript USA Inc.) and is similar to that for PhoE (Gelder et al., 1996). Mutants were constructed using Quikchange site-directed mutagenesis (Agilent), Bacteria were grown at 37°C in Luria-Bertani (LB) broth at 37 °C, supplemented with kanamycin (30 g/ml).
Inner membrane solubilization and OmpF purification
Isolation of OM fractions was carried out as described previously (Taylor et al., 1998), which involves inner membrane solubilization by Triton X-100. OMs were extracted overnight (room temp) with 3% n-octyl-β-D-glucopyranoside (Anatrace) in 20 mM Tris-HCl, 1 mM EDTA, pH 8.0. Protein extractions were centrifuged for 30 min at 125,000×g (room temp). OmpF protein was purified using Ni affinity chromatography (Ni-NTA agarose, Qiagen) and eluted with 100 mM imidazole. To remove the N-terminal His6-tag, purified OmpF protein was incubated with TEV protease at a ratio of 20:1 (w/w) in 50 mM Tris-HCl, 0.5 mM EDTA, 1 mM DTT, pH 8.0 and dialyzed overnight (room temp) into 20 mM Tris-HCl, 0.1 M NaCl, 0.8% octyl-polyoxyethylene (OPOE), 0.5 mM PMSF, 1 mM sodium azide, pH 8.0. Final purification was conducted by gel-filtration chromatography (Superdex 200 10/300 GL, GE Healthcare) and eluted with 20 mM Tris-HCl, 0.1 M NaCl, 0.8% OPOE, pH 8.0. Protein was concentrated in Amicon Ultra spin filters (Millipore) for crystallization.
X-ray crystallography, data collection, and refinement
Crystallization conditions with high Mg2+ were inspired by a previous study of OmpF (Yamashita et al., 2008). However, since divalent cations are known to induce pKa shifts of key acidic OmpF residues and affect ion-selectivity of the channel (Queralt-Martin et al., 2011), a reduced concentration of MgCl2 (200 mM) was used, as previously reported (Reitz et al., 2009). Crystals of E. coli OmpF protein were obtained at 20 °C in a hanging drop (VDX plates, Hampton Research) with the reservoir solution, 100 mM sodium cacodylate, 200 mM MgCl2, 50% (w/v) PEG 200, pH 6.5 mixed in a 1:1 ratio with protein solution, 20 mM Tris-HCl, 0.1 M NaCl, 0.8% octyl-POE, pH 8.0; OmpF concentration 5–8 mg/ml. Crystals were transferred to mother liquor solution contaiing 1–2 M antibiotic and 25% glycerol for cryoprotection, but lacking MgCl2 for two overnight soaks to ensure that Mg2+ did not affect antibiotic binding (Fig. S2). X-ray diffraction data were collected at beamlines NE-CAT 24-ID at the Advanced Photon Source (Argonne National Lab). Processing of diffraction data was carried out with iMosflm (Battye et al., 2011). The structures were solved by the molecular replacement method using the program Phaser v2.1 (McCoy et al., 2007) implemented in the CCP4 suite (Winn et al., 2011). The structure of PDB 2ZFG was used as the starting model and refined using REFMAC (Murshudov et al., 1997). Model building was carried out using the program COOT (Emsley and Cowtan, 2004). Since β-lactams are flexible molecules that can adopt a number of different conformations in solution, conformers that fit within the experimental density were generated by AFITT, a fully automatic ligand fitting process distributed by OpenEye Scientific Software (Wlodek et al., 2006). The AFITT program was supplied with the OmpF coordinates and the Fobs – Fcalc electron map (without antibiotic), and the coordinates for the antibiotic molecule were supplied separately. The AFITT program searched for all unmodeled density that was the correct size and shape for the ligand and the lowest energy solution was used for REFMAC refinement.
Disk diffusion and agar dilution antibiotic susceptibility assays
E. coli BL21(DE3)Omp5 bacteria transformed with pRSF-1b plasmid encoding the WT or mutant OmpF protein sequence were incubated in LB broth supplemented with kanamycin (30 μg/ml), at 37 °C, 250 rpm. OmpF expression was induced with 1 mM IPTG for 3 hr once an OD600 of 0.6 was reached. For the disk diffusion tests, cultures were spread on Mueller-Hinton (MH) agar plates and either ampicillin (10 μg) or carbenicillin (100 μg) charged disks (Becton, Dickinson and Company) were placed on inoculated agar plates, supplemented with 30 μg/ml kanamycin. Slow diffusion of antibiotic into the agar surrounding the disk created an antibiotic gradient, which decreased in concentration outward from the disk. The diameter of the “inhibition zone” around each disk was measured after overnight incubation at 37 °C. For the agar dilution test, induced cultures were diluted 20-fold and spotted (5 μl) on MH-agar plates containing 30 μg/ml kanamycin as well as 2-fold serial dilutions of either ampicillin or carbenicillin (16, 8, 4, 2, 1, 0.5, 0.25, 0.125, and 0 μg/ml). In a separate agar dilution experiment, bacteria were co-transformed with pRSF-1b plasmid encoding OmpF and pGEM-3Z plasmid encoding β-lactamase (Ampr) (Promega), incubated in LB broth supplemented with kanamycin (30 μg/ml) and ampicillin (100 μg/ml), and spotted as above on plates containing 30 μg/ml kanamycin and serial dilutions of ampicillin (160, 80, 40, 20, 10, 5, 2.5, 1.25, and 0.1 mg/ml). Plates were incubated overnight at 37 °C. MICs were defined as the concentration of antibiotic, which prevented bacterial growth (less than 10 colonies per spot). WT and mutant porin expression was compared by Western blot quantification of E. coli lysates using an anti-His primary antibody (Qiagen) for OmpF and an antibody to DnaK (Abcam) as a loading control after induction (Fig. S7).
All-atom molecular dynamics simulations
The simulation model, constructed by CHARMM-GUI (Jo and Im, 2007; Jo et al., 2008), comprises one OmpF monomer, 103 palmitoyl oleoyl phosphatidyl choline (POPC) lipid molecules, and solvated with 9,190 water molecules in 150 mM KCl (39 K+ and 12 Cl−), for a total of ~47,000 atoms. Tetragonal periodic boundary conditions were applied with a distance of 70.29 Å in the XY-direction and 91.82 Å in the Z-direction. Electrostatic interactions were calculated using the Particle-Mesh Ewald (PME) method with a 75×75×100 grid. Protein residues E296, D312, and D127 were protonated (Im and Roux, 2002a; Im and Roux, 2002b; Varma et al., 2006). The simulations were performed at constant pressure (1 atm) and temperature (300 K) with a time step of 2 fs. Harmonic restraints (1 kcalmol−1Å−1) were applied to the Cα atoms at their crystallographic positions to prevent lateral movement of the protein. A single antibiotic molecule (ampicillin modeled in its zwitterionic form, or carbenicillin in its anionic form) was positioned in the pore using the coordinates extracted from crystallography. Restraints were initially applied to antibiotic atoms and then released to relax the system. For SMD, a dummy atom attached to the antibiotic center-of-mass (5 kcal/molÅ2 spring constant) was pulled at a constant velocity (0.00001 Å/timestep) along the +/− Z-axis until the molecule escaped from either the extracellular or periplasmisic mouths of the pore. The KCl concentration was increased to 1 M (92 K+ and 83 Cl−, respectively) for the ion permeation simulations to produce a larger conductance. An electric field corresponding to a transmembrane potential of ±150 mV was applied along the Z-direction (Roux, 2008), pressure control was removed to avoid distortion of the system by the electric field, and Langevin friction was removed for the ions. The all-atom CHARMM force field was used for protein (MacKerell et al., 1998) and lipids (Feller et al., 1997), and TIP3P (Jorgensen et al., 1983) for water. All the MD simulations were carried out using the NAMD scalable molecular dynamics program (Phillips et al., 2005). CHARMM residue topology and parameter files for the antibiotic molecules were constructed using Antechamber (Wang et al., 2006) with a restrained electrostatic potential (RESP) charge fitting procedure.
Supplementary Material
OmpF porin binds antibiotics in the extracellular and periplasmic pore vestibules.
MD simulations show that ionic current through OmpF is blocked by bound ampicillin.
Carbenicillin binding alters ion selectivity of OmpF but not total ionic current.
Disruption of binding increases the susceptibility of E. coli for antibiotics.
Acknowledgements
The help and advice from Balasundaresan Dhakshnamoorthy, Erin Adams, Eduardo Perozo, and Anthony Kossiakoff, and the support of Lydia Blachowicz are gratefully acknowledged. This work was supported by the National Institutes of Health through grant R01 GM062342 and is also based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by grants from the National Center for Research Resources (5P41RR015301-10) and the National Institute of General Medical Sciences (8 P41 GM103403-10) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers Atomic coordinates and structure factors for the reported crystal structures of OmpF in complex with amipicillin, carbenicillin, and ertapenem, have been deposited in the Protein Data Bank under accession codes 4GCP, 4GCQ, and 4GCS, respectively.
Supplemental information Supplemental Information includes seven figures and one table that can be found with this article
References
- Balasubramaniam D, Arockiasamy A, Kumar PD, Sharma A, Krishnaswamy S. Asymmetric pore occupancy in crystal structure of OmpF porin from Salmonella typhi. J. Struct. Biol. 2012;178:233–244. doi: 10.1016/j.jsb.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Baslé A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. Crystal structure of osmoporin OmpC from E. coli at 2.0 Å. J. Mol. Biol. 2006;362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Battye TGG, Johnson O, Powell HR, Leslie AGW. iMOSFLM : a new graphical interface for diffraction- image processing with MOSFLM. Acta Cryst. 2011;D67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredin J, Saint N, Malléa M, Dé E, Molle G, Pagès J-M, Simonet V. Alteration of pore properties of Escherichia coli OmpF induced by mutation of key residues in anti-loop 3 region. Biochem. J. 2002;363:521–528. doi: 10.1042/0264-6021:3630521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredin J, Simonet V, Iyer R, Delcour AH, Pagès J-M. Colicins, spermine and cephalosporins: a competitive interaction with the OmpF eyelet. Biochem. J. 2003;376:245–252. doi: 10.1042/BJ20030814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Danelon C, Laio A, Parrinello M. Microscopic mechanism of antibiotics translocation through a porin. Biophys. J. 2004;87:58–64. doi: 10.1529/biophysj.103.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Vargiu AV, Ruggerone P. A kinetic Monte Carlo approach to investigate antibiotic translocation through bacterial porins. J. Phys.: Condens. Matter. 2012;24:104012–104019. doi: 10.1088/0953-8984/24/10/104012. [DOI] [PubMed] [Google Scholar]
- Charrel RN, Pagès J-M, De Micco P, Malléa M. Prevalence of outer membrane porin alteration in β-lactam-antibiotic-resistant Enterobacter aerogenes. Antmicrob. Agents Chemother. 1996;40:2854–2858. doi: 10.1128/aac.40.12.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan SW, Garavito RM, Jansonius JN, Jenkins JA, Karlsson R, König N, Pai EF, Pauptit RA, Rizkallah PJ, Rosenbusch JP, et al. The structure of OmpF porin in a tetragonal crystal form. Structure. 1995;3:1041–1050. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP. Crystal structures explain functional properties of two E. coli porins. Nature. 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- Danelon C, Nestorovich EM, Winterhalter M, Ceccarelli M, Bezrukov SM. Interaction of zwitterionic penicillins with the OmpF channel facilitates their translocation. Biophys. J. 2006;90:1617–1627. doi: 10.1529/biophysj.105.075192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour AH. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshnamoorthy B, Raychaudhury S, Blachowicz L, Roux B. Cation-selective pathway of OmpF porin revealed by anomalous X-ray diffraction. J. Mol. Biol. 2010;396:293–300. doi: 10.1016/j.jmb.2009.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Rummel G, Albertí S, Hernandez-Allés S, Phale PS, Rosenbusch JP, Benedí VJ, Schirmer T. Crystal structure and functional characterization of OmpK36 , the osmoporin of Klebsiella pneumoniae. Structure. 1999;7:425–434. doi: 10.1016/s0969-2126(99)80055-0. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Feller SE, Yin D, Pastor RW, MacKerell AD., Jr Molecular dynamics simulation of unsaturated lipid bilayers at low hydration: parameterization and comparison with diffraction studies. Biophys. J. 1997;73:2269–2279. doi: 10.1016/S0006-3495(97)78259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MJ, Simjee S, Al-Hattawi K, Robertson BD, Easmon CSF, Ison CA. Gonococcal resistance to β-lactams and tetracycline involves mutation in loop 3 of the porin encoded at the penB Locus. Antimicrob. Agents Chemother. 1998;42:2799–2803. doi: 10.1128/aac.42.11.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein FW, Gutmann L, Williamson R, Collatz E, Acar JF. In vivo and in vitro emergence of simultaneous resistance to both β-lactam and aminoglycoside antibiotics in a strain of Serratia marcescens. Ann. Microbiol. (Paris) 1983;134A:329–337. [PubMed] [Google Scholar]
- Hajjar E, Bessonov A, Molitor A, Kumar A, Mahendran KR, Winterhalter M, Pagès J-M, Ruggerone P, Ceccarelli M. Toward screening for antibiotics with enhanced permeation properties through bacterial porins. Biochemistry. 2010a;49:6928–6935. doi: 10.1021/bi100845x. [DOI] [PubMed] [Google Scholar]
- Hajjar E, Kumar A, Ruggerone P, Ceccarelli M. Investigating reaction pathways in rare events simulations of antibiotics diffusion through protein channels. J. Mol. Model. 2010b;16:1701–1708. doi: 10.1007/s00894-010-0698-4. [DOI] [PubMed] [Google Scholar]
- Hajjar E, Mahendran KR, Kumar A, Bessonov A, Petrescu M, Weingart H, Ruggerone P, Winterhalter M, Ceccarelli M. Bridging timescales and length scales : from macroscopic flux to the molecular mechanism of antibiotic diffusion through porins. Biophys. J. 2010c;98:569–575. doi: 10.1016/j.bpj.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W, Roux B. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 2002a;322:851–869. doi: 10.1016/s0022-2836(02)00778-7. [DOI] [PubMed] [Google Scholar]
- Im W, Roux B. Ions and counterions in a biological channel: a molecular dynamics simulation of OmpF porin from Escherichia coli in an explicit membrane with 1M KCl aqueous salt solution. J. Mol. Biol. 2002b;319:1177–1197. doi: 10.1016/S0022-2836(02)00380-7. [DOI] [PubMed] [Google Scholar]
- James CE, Mahendran KR, Molitor A, Bolla J.-M.l, Bessonov AN, Winterhalter M, Pagès J-M. How β-lactam antibiotics enter bacteria : a dialogue with the porins. PLoS One. 2009;4:e5453. doi: 10.1371/journal.pone.0005453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI : a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Kalp M, Carey PR. Carbapenems and SHV-1 β-lactamase form different acyl-enzyme populations in crystals and solution. Biochemistry. 2008;47:11830–11837. doi: 10.1021/bi800833u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hajjar E, Ruggerone P, Ceccarelli M. Molecular simulations reveal the mechanism and the determinants for ampicillin translocation through OmpF. J. Phys. Chem. B. 2010;114:9608–9616. doi: 10.1021/jp9110579. [DOI] [PubMed] [Google Scholar]
- Lou H, Chen M, Black SS, Bushell SR, Ceccarelli M, Mach T, Beis K, Low AS, Bamford VA, Booth IR, et al. Altered antibiotic transport in ompC mutants isolated from a series of clinical strains of multi-drug resistant E . coli. PLoS One. 2011;6(10):e25825. doi: 10.1371/journal.pone.0025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nestorovich EM, Danelon C, Winterhalter M, Bezrukov SM. Designed to penetrate: time-resolved interaction of single antibiotic molecules with bacterial pores. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9789–9794. doi: 10.1073/pnas.152206799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H, Rosenberg EY, Foulds J. Porin channels in Escherichia coli: studies with β-lactams in intact cells. J. Bacteriol. 1983;153:232–40. doi: 10.1128/jb.153.1.232-240.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Antibiotic resistance caused by Gram-negative multidrug efflux pumps. Clin.Infect. Dis. 1998;27(Suppl 1):S32–41. doi: 10.1086/514920. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès J-M, James CE, Winterhalter M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008;6:893–903. doi: 10.1038/nrmicro1994. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilipov A, Phale PS, Van Gelder P, Rosenbusch JP, Koebnik R. Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbio. Lett. 1998;163:65–72. doi: 10.1111/j.1574-6968.1998.tb13027.x. [DOI] [PubMed] [Google Scholar]
- Queralt-Martín M, García-Giménez E, Mafé S, Alcaraz A. Divalent cations reduce the pH sensitivity of OmpF channel inducing the pKa shift of key acidic residues. Phys. Chem. Chem. Phys. 2011;13:563–569. doi: 10.1039/c0cp01325k. [DOI] [PubMed] [Google Scholar]
- Reitz S, Cebi M, Reiß P, Studnik G, Linne U, Koert U, Essen L-O. On the function and structure of synthetically modified porins. Angew. Chem. 2009;48:4853–4857. doi: 10.1002/anie.200900457. [DOI] [PubMed] [Google Scholar]
- Roux B. The membrane potential and its representation by a constant electric field in computer simulations. Biophys. J. 2008;95:4205–4216. doi: 10.1529/biophysj.108.136499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet V, Malléa M, Pagès J-M. Substitutions in the eyelet region disrupt cefepime diffusion through the Escherichia coli OmpF channel. Antimicrob. Agents Chemother. 2000;44:311–315. doi: 10.1128/aac.44.2.311-315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R, Burgner JW, Clifton J, Cramer WA. Purification and characterization of monomeric Escherichia coli vitamin B12 receptor with high affinity for colicin E3. J. Biol. Chem. 1998;273:31113–31118. doi: 10.1074/jbc.273.47.31113. [DOI] [PubMed] [Google Scholar]
- Tremblay LW, Fan F, Blanchard JS. Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems ertapenem and doripenem. Biochemistry. 2010;49:3766–3773. doi: 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder P, Steiert M, El Khattabi M, Rosenbusch JP, Tommassen J. Structural and functional characterization of a His-tagged PhoE pore protein of Escherichia coli. Biochem.Biophys. Res. Commun. 1996;229:869–875. doi: 10.1006/bbrc.1996.1894. [DOI] [PubMed] [Google Scholar]
- Varma S, Chiu S-W, Jakobsson E. The influence of amino acid protonation states on molecular dynamics simulations of the bacterial porin OmpF. Biophys. J. 2006;90:112–23. doi: 10.1529/biophysj.105.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Bredin J, Pagès J-M, Barbe J. β-lactam screening by specific residues of the OmpF eyelet. J. Med. Chem. 2005;48:1395–1400. doi: 10.1021/jm049652e. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Cryst. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodek S, Skillman AG, Nicholls A. Automated ligand placement and refinement with a combined force field and shape potential. Acta Cryst. 2006;D62:741–749. doi: 10.1107/S0907444906016076. [DOI] [PubMed] [Google Scholar]
- Yamashita E, Zhalnina MV, Zakharov SD, Sharma O, Cramer WA. Crystal structures of the OmpF porin: function in a colicin translocon. EMBO J. 2008;27:2171–2180. doi: 10.1038/emboj.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura F, Nikaido H. Diffusion of β-lactam antibiotics through the porin channels of Escherichia coli K-12. Antimicrob. Agents Chemother. 1985;27:84–92. doi: 10.1128/aac.27.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.