

Abstract
We investigated whether soluble EBV gH/gL (sgH/gL) functions in fusion and made a series of truncations of gH/gL domains based on the gH/gL crystal structure. We found sgH/gL failed to mediate cell-cell fusion both when co-expressed with the other entry glycoproteins and when added exogenously to fusion assays. Interestingly, sgH/gL inhibited cell-cell fusion in a dose dependent manner when co-expressed. sgH/gL from HSV was unable to inhibit EBV fusion, suggesting the inhibition was specific to EBV gH/gL. sgH/gL stably binds gp42, but not gB nor gH/gL. The domain mutants, DI/gL, DI-II/gL and DI-II-III/gL were unable to bind gp42. Instead, DI-II/gL, DI-II-III/gL and sgH/gL but not DI/gL decreased the expression of gp42, resulting in decreased overall fusion. Overall, our results suggest that domain IV may be required for proper folding and the transmembrane domain and cytoplasmic tail of EBV gH/gL are required for the most efficient fusion.
Introduction
Epstein-Barr virus (EBV), a member of the herpesvirus family, is transmitted orally and infects the majority of the adult population. Infant and young childhood infections are asymptomatic or subclinical whereas adulthood infections typically result in infectious mononucleosis (Rickinson, 2007). Significantly, EBV is associated with lymphoid malignancies as well as epithelial malignancies, for which immunodeficient patients are at particular risk (Bieging et al., 2010; Longnecker, 1998; Rickinson, 2007; Takada, 2001; Thompson and Kurzrock, 2004; Wei and Sham, 2005).
Members of the herpesvirus family are large enveloped DNA viruses, which use a conserved set of glycoproteins for the complex process of viral fusion with the host cell membrane either at the plasma membrane or following endocytosis (Connolly et al., 2011). The majority of herpesviruses use two or more receptors to dictate cell tropism and spread. EBV uses different glycoprotein and receptor combinations to infect oral epithelial cells than it does to infect B cells where it establishes latency until reactivation. In B cells, HLA class II receptor binding by gH/gL/gp42 complexes triggers conformational changes that result in activation of gB-dependent fusion between the viral envelope and a cellular membrane (Haan et al., 2000; Li et al., 1997; McShane et al., 2003; Spriggs et al., 1996). In epithelial cells, which typically lack HLA class II, binding to an epithelial receptor, such as integrins by gH/gL, presumably triggers conformational changes that result in activation of gB and fusion (Chen et al., 2012; Chesnokova and Hutt-Fletcher, 2011; Chesnokova et al., 2009). EBV virions exiting B cells primarily contain the bipartite complex gH/gL and infect epithelial cells efficiently, whereas virions exiting epithelial cells primarily contain the tripartite complex gH/gL/gp42 and infect B cells efficiently (Borza and Hutt-Fletcher, 2002). When added exogenously, sgp42 enhanced B cell fusion but inhibited epithelial fusion (Kirschner et al., 2006). The cytoplasmic domains for both gp42 and the HSV functional homolog, gD, are not required for cell-cell fusion, since the soluble forms of both gp42 and gD are functional in fusion (Atanasiu et al., 2010; Sorem et al., 2009).
The recently solved x-ray crystallographic structures of gH/gL from EBV, and HSV-2 as well as the partial gH structure of psuedorabies virus (PRV) suggest striking conservation of gH/gL within the herpesvirus family (Backovic et al., 2010; Chowdary et al., 2010; Matsuura et al., 2010). The EBV structure suggests four sequential semiautonomous domains: Domain I, II, III and IV (DI, DII, DIII and DIV) (Matsuura et al., 2010). EBV gL forms a stable heterodimer with the N-terminal residues of gH to form DI. gL is an integral part of the DI structure and is required for the export of gH to the cellular membrane (Pulford et al., 1995; Yaswen et al., 1993). DI is composed of mixed parallel/antiparallel β sheets (five strands from gL and two strands from gH) supported by a layer of three helices. The three helix layer of DI forms a wall of charged and polar residues which may interact with other viral proteins involved in the core fusion machinery. Between DI and DII there is a single α-helix linker which has been proposed to act as a “hinge”. DII is composed of an eight stranded anti-parallel β sheet (“picket fence”) followed by an anti-parallel five helix bundle. Projecting from the picket fence is a prominent KGD loop that is implicated in binding gp42, as well as a gH/gL epithelial receptor, presumably integrin αvβ6, αvβ8 or αvβ5 (Chen et al., 2012; Chesnokova et al., 2009). DIII consists of a total of nine helices, the first five of which spiral down DII and the last four form a distinct subdomain bundle. DIV consists of two connected anti-parallel β sheets forming a “β-sandwich”. Elucidation of the gH/gL structure provides a basis for deletion and mutagenesis studies aimed at understanding the function of gH in the core fusion machinery.
The N-terminal residues as well as the C-terminal residues of EBV gH/gL have been implicated in promoting membrane fusion. When EBV gL residues 54 and 94 were replaced with those of rhesus lymphocryptovirus (Rh-LCV) gL, the mutant gL showed wild-type expression but decreased fusion activity. Fusion could be restored by replacing EBV gB with Rh gB, suggesting gL interacts with gB in a species-specific manner to activate membrane fusion (Plate et al., 2009). Interestingly, a single substitution in the C-terminal Domain IV, G594A, resulted in complete abrogation of fusion with both B cells and epithelial cells, whereas the substitution E595A reduced fusion with epithelial cells but greatly enhanced fusion with B cells (Wu and Hutt-Fletcher, 2007).
Mutational studies of herpes simplex virus type 1 (HSV-1) gH have been more extensive and suggest that the transmembrane domain as well as the cytoplasmic tail of gH/gL plays an important role in membrane fusion. Harman et al. found that gH lacking the authentic transmembrane or cytoplasmic tail were unable to mediate cell-cell fusion (Harman et al., 2002). In addition, a conserved glycine residue at position 812 within the transmembrane domain was found to be crucial for fusion (Harman et al., 2002). Interestingly, fusion is unaffected by deletion of the final six residues of the gH cytoplasmic tail (residues 832 to 838), however further deletions decreased polykaryocyte formation by a syncytial HSV strain (Browne et al., 1996; Wilson et al., 1994). Maintenance of the valine at position 831 within the serine-valine-proline motif of the cytoplasmic tail was of particular importance for fusion (Wilson et al., 1994). The addition of a linker insertion at residue 824, which borders the junction between the transmembrane and the cytoplasmic tail, completely abrogated fusion (Jackson et al., 2010). gD-glycophosphatidylinositol (gDgpi), in which the transmembrane and cytoplasmic tail of gD is replaced with a gpi linkage, promoted fusion to near wild-type levels (Jones and Geraghty, 2004). However, gHgpi and gBgpi were unable to promote fusion, suggesting glycoprotein specific transmembrane and cytoplasmic tails are required for gH and gB. In contrast, more recent work has shown that purified HSV-2 sgH/gL protein, lacking the transmembrane and cytoplasmic tail, can induce a modest (2.1% of wild type) level of fusion of nectin-1-bearing C10 cells expressing gB and gD (Atanasiu et al., 2010). Interestingly, EBV sgH/gL purified from insect cells does not promote fusion when added exogenously (Kirschner et al., 2006).
To rigorously rule out that the source of the sgH/gL is not playing any role in the observed functional differences between the HSV and EBV studies, we expressed and tested sgH/gL in mammalian cells a in cell-cell fusion assay. We generated a soluble form of gH/gL and either co-expressed it with the other entry glycoproteins or added it exogenously to the core fusion machinery. Our results indicate that sgH/gL does not function efficiently in fusion and instead inhibits fusion. We generated domain deletion mutants of gH to try to map the region responsible for inhibition of fusion. We found only full length sgH/gL was able to bind gp42, suggesting DIV of gH is required for gp42 binding. Surprisingly, however, we found DI-II/gL, DI-II-III/gL and the full length sgH/gL all reduce gp42 expression levels. Overall, our results support previous observations that anchoring of gH in the membrane is essential for most efficient fusion.
Results
Construction of gH/gL FLAG-tagged mutants
Recent x-ray crystallography results suggest gH/gL is composed of four semi-autonomous domains: DI, DII, DIII and DIV. To study whether sgH/gL could mediate fusion, we constructed soluble C-terminal deletion variants of gH, incorporating the FLAG epitope at the C-terminus of each (Fig. 1). Mutant sgH (consisting of the full ectodomain: DI-II-III-IV) was truncated at amino acid 679, immediately upstream of the transmembrane domain and cytoplasmic tail which was replaced with the FLAG tag followed by a stop codon. Mutant DI-II-III, DI-II and DI were truncated at residues 529, 344 and 65 respectively, and C-terminally tagged with the FLAG epitope followed by a stop codon. We chose to add the FLAG epitope at the C-terminus of the truncations to allow us to readily monitor expression of the proteins and verify they were full length.
Fig. 1. Schematic representation of wild type gH, FLAG tagged sgH, DI-II-III, DI-II and DI gH mutants, and gL.
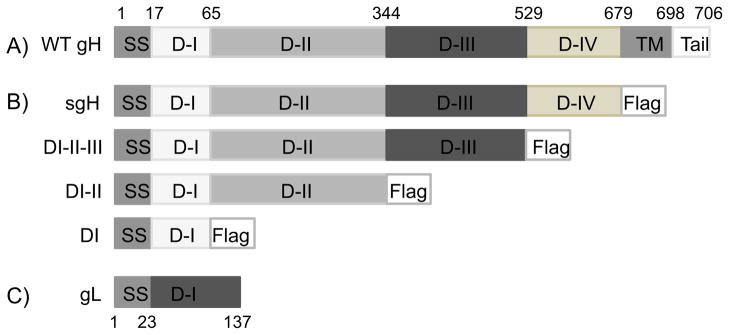
(A) Approximate location of gH functional domains are shown including the signal sequence (residues 1–17), DI (residues 18–65), DII (residues 66–344), DIII (residues 345–529), DIV (residues 530–679), transmembrane domain (residues 680–698) and the cytoplasmic tail (residues 699–706). (B) FLAG tagged mutants: sgH, DI-II-III, DI-II, DI are all c-terminally tagged with the FLAG epitope followed by a stop codon, as indicated. None of the mutants contain the transmembrane domain nor the cytoplasmic tail of gH. (C) Wild type gL includes the signal sequence (residues 1–23) followed by a single domain (residues 24–137).
Expression and secretion of soluble gH/gL domain mutants
To determine whether the FLAG- tagged gH mutants were expressed and produced as soluble forms, plasmids encoding each of the mutants together with gL were transfected into CHO-K1 cells and protein expression was analyzed 48 hrs post-transfection by Western Blot. We found that all of the mutants were expressed in cellular extracts at the approximate expected molecular weight (DI, 7 kDa; DI-II, 38 kDa; DI-II-III, 58 kDa; sgH 74 kDa), as determined by probing with an anti-FLAG antibody (Fig. 2A). However, only DI and sgH were detected as distinct bands in cell culture supernatants (Fig. 2B). DI, DI-II, DI-II-III, as well as sgH all tended to form higher molecular weight aggregates in cell lysates (intracellularly) and in culture supernatants (extracellularly), although DI to a lesser extent. The aggregates (indicated by an arrow, Fig 2B) could not be resolved by changing the denaturation temperature, the reducing agent, or cell expression system prior to loading on SDS-PAGE gels (data not shown).
Fig. 2. sgH/gL mutants are expressed.
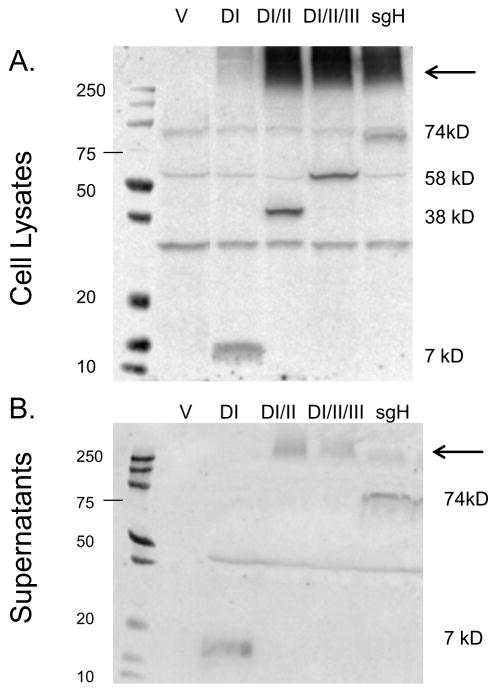
CHO-K1 cells were transiently transfected with (A) empty vector (pCAGGS), wild type gp42 with DI/gL, wild type gp42 with DI-II/gL, wild type gp42 with DI-II-III/gL and wild type gp42 with sgH/gL. Forty eight hours post transfection cells were lysed in triton X-100 lysis buffer containing protease inhibitors and analyzed by 12% SDS-PAGE and Western blotting with polyclonal anti-FLAG antibody (F7425-Sigma). (B) empty vector (pCAGGS), DI/gL, DI-II/gL, DI-II-III/gL and sgH/gL. Forty eight hours post transfection supernatant was collected and analyzed by 4–20% SDS-PAGE and Western blotting with polyclonal anti-FLAG antibody (F7425-Sigma). Size markers in kDa are noted to the right and left of the blots. Aggregates are indicated by an arrow.
sgH/gL is secreted, binds gp42, and is therefore properly folded
To study whether the expressed sgH/gL domain mutants were functional, we analyzed their ability to bind other EBV glycoproteins involved in viral entry and fusion by cELISA (Cell-based Enzyme-Linked Immunosorbent Assay). We co-transfected CHO-K1 cells with plasmids encoding sgH/gL and either vector, gB, gp42 or wild type gH/gL and detected sgH/L binding using an anti-FLAG antibody. We found sgH/gL was efficiently tethered to the cell surface by virtue of binding gp42, but not gB, nor wild type gH/gL, as expected (Fig. 3A). We next tested the ability of exogenously-expressed sgH/gL domain mutants to bind gp42 using a monolayer binding assay. CHO-K1 cells were co-transfected with plasmids encoding DI/gL, DI-II/gL, DI-II-III/gL or sgH/gL. Supernatants, harvested after two days, were overlaid for 1 hr at 4°C onto CHO-K1 monolayers transfected with wild type gp42. Following extensive washing, binding to gp42 was detected by cELISA using an anti-FLAG antibody. We found that none of the sgH/L protein supernatants bound CHO-K1 cells transfected with vector alone. Only sgH/gL containing all four gH/gL domains and none of the domain deletion mutants was able to bind gp42 (Fig. 3B). It is possible that the DI/gL mutant was unable to bind gp42 because gp42 binds at the groove formed by both DI/gL and DII. This would suggest that DI/gL and DII are functionally non-autonomous. Unexpectedly, however, the DI-II/gL and DI-II-III/gL mutants also failed to bind gp42 suggesting that misfolding or aggregation of the protein may in part be the cause of loss of gp42 binding for these mutants. Alternatively, our data suggests that all four domains may be required for proper folding or solubility of gH/gL in order to bind gp42.
Fig. 3. sgH/gL binds gp42 but not gB nor gH when co-expressed.
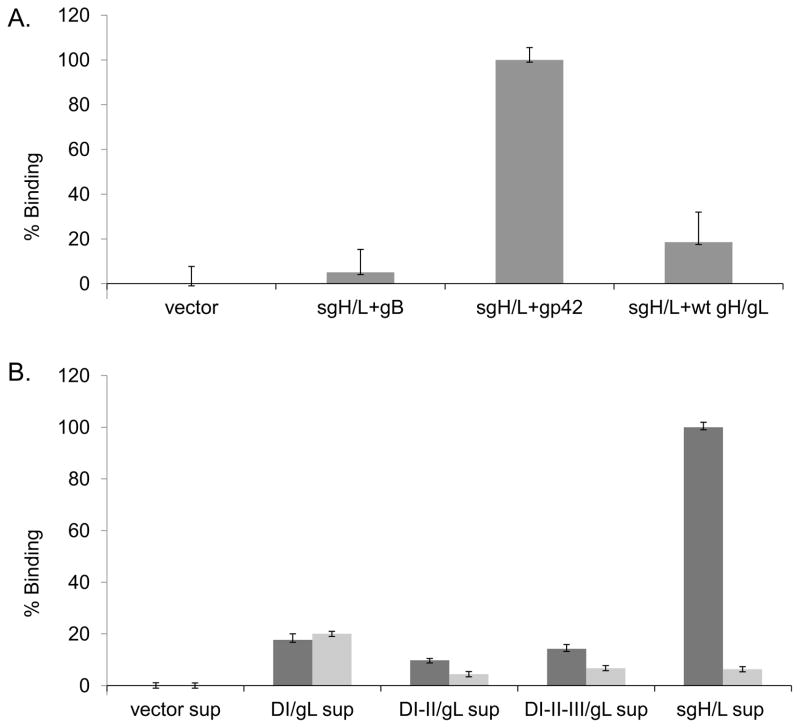
(A) CHO-K1 cells were co-transfected with sgH/gL and either vector alone, gB, gp42, or gH/gL. Binding of sgH/gL was measured by cELISA using anti-FLAG-M2 (F1804; Sigma), secondary biotinylated anti-mouse IgG antibody, tertiary streptavidin-HRP and TMB substrate. Color development was measured by absorbance at 370nm. Binding was normalized to gp42 binding levels, which were set to 100%. Data shown are representative results of three independent experiments. (B) CHO-K1 cells were co-transfected with DI-gL, DI-II/gL, DI-II-III/gL and sgH/gL. Forty eight hours post tranfection, protein supernatants were collected and overlaid on CHO-K1 cells transiently transfected with either vector alone (gray bars) or gp42 (dark gray bars) for 48 hours. Binding was normalized to gp42 binding levels, which were set to 100%. Data shown are representative results of three independent experiments. Error bars represent standard deviations for the normalized values.
To ensure that the lack of binding of DI/gL, DI-II/gL, DI-II-III/gL was not due to insufficient quantities of soluble protein, we quantitated the protein bands on the Western Blot (Figure 2B) and normalized them against sgH/gL using Licor software. We then repeated the binding assay using equal or three times excess amounts of the DI/g, DI-II/gL, DI-II-III/gL compared with sgH/gL. We found that DI/g, DI-II/gL, DI-II-III/gL were unable to bind gp42 even in these excess amounts (data not shown). We confirmed our results using independent supernatants collected on a different day and quantitated protein bands on Western Blot by licor and found the similar results.
sgH/gL does not efficiently mediate fusion
To assess the ability of sgH/gL to mediate fusion, we performed a cell-cell fusion assay in which CHO-K1 effector cells co-transfected with plasmids encoding the EBV glycoproteins and luciferase under control of the T7 promoter were overlaid with CHO-K1 target cells co-transfected with the gp42 receptor HLA class II and T7 polymerase. Eighteen to twenty-four hours after overlay, fusion activity was determined by quantification of luciferase activity. We found that without HLA class II, no fusion occurs, as expected (Fig. 4, lane 1, 2). When HLA class II is present in target cells and the effector cells express wild type gH/gL, gB, and gp42; the cells fuse efficiently. This wild type condition was set to 100% (Fig. 4, lane 3). However, when sgH/gL is co-transfected in place of wild type gH/gL, fusion is undetectable (Fig. 4, lane 4), suggesting that although sgH/gL is capable of binding gp42, it is not functional for fusion. In addition, in the absence of wild type gH/gL, exogenously added sgH/gL protein is not able to mediate fusion (data not shown).
Fig. 4. sgH/gL does not function in fusion when co-transfected or when added exogenously.
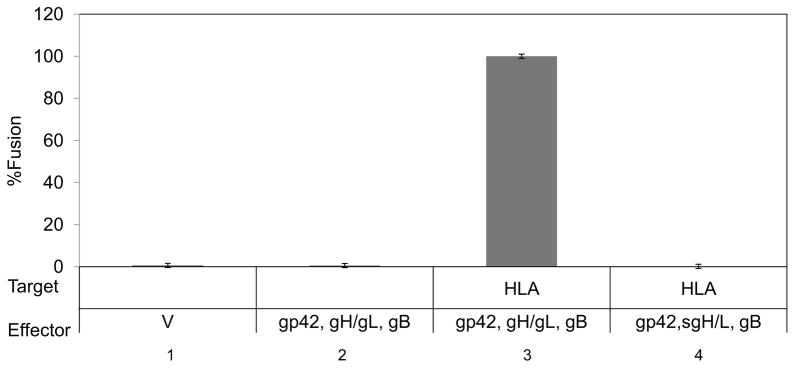
CHO-K1 target cells were transiently co-transfected with HLA-DR and T7 polymerase. CHO-K1 effector cells were transiently co-transfected with gp42, gB and either wild type gH/gL (lane 2, 3) or sgH/gL (lane 4). Cells were detached twenty four hours post-tranfection and target cells were overlaid onto effector cells. Fusion activity was accessed eighteen to twenty four hours post overlay by the addition of passive lysis buffer followed by the addition of luciferase substrate. Luciferase activity was measured with a Perkin-Elmer Victor plate reader. Data shown are representative results of three independent experiments. Error bars represent standard deviations for the normalized values.
SgH/gL inhibits fusion in a dose dependent manner
We next investigated whether soluble gH/gL could inhibit fusion induced by wild type gH in a dose dependent manner. For this cell-cell fusion experiment all target cells were co-transfected with HLA-DR and T7 polymerase (lanes 1–8). Effector cells were transfected with vector alone (lane 1), co-transfected with wild type gH (0.5 μg), excess gL (2.5 μg), gB (0.5 μg) and gp42 (2 μg) (lane 2–8), or co-transfected with increasing amounts of wild type gH or sgH (lanes 3–5 and lanes 6–8, respectively). All of the target cells were co-transfected with HLA-DR and T7 polymerase. We found that increasing the amount of wild type gH/gL decreased the amount of fusion initially but then fusion remained relatively constant (around 50% of maximal levels) (Fig 5, compare lane 2 with lanes 3–5). In contrast, when levels of sgH/gL were increased, fusion levels decreased in a dose dependent manner (Fig 5, compare lane 3–5 with lanes 6–8). gH/gL expression decreased with increased amounts of sgH/gL, however, whereas the gH/gL expression decreased to 70–80% of wild type, fusion decreased to 40–80% of wild type (Fig. 5, lanes 6–8) providing data that EBV sgH/gL inhibits fusion. We found the amount of gH expression does not increase with increasing amounts of wild type gH/gL. It is possible that the cELISA results are not on a linear scale or alternatively the gH/gL expression is dependent on overall glycoprotein stoichiometry.
Fig. 5. sgH/gL xpressed endogenously inhibits cell-cell fusion in a dose dependent manner.
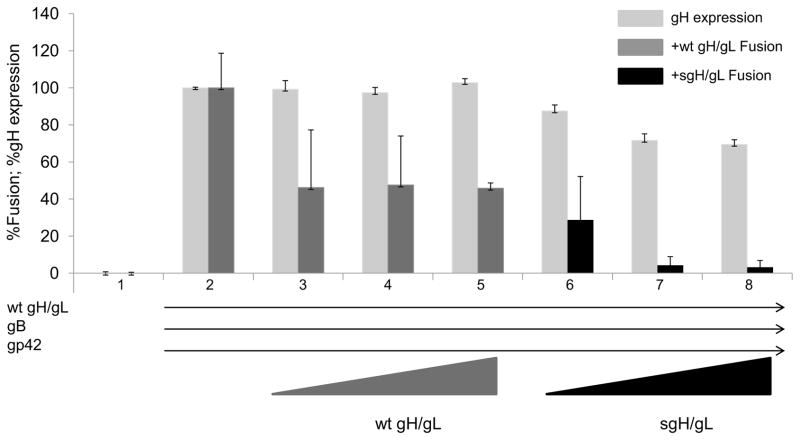
All CHO-K1 target cells were transiently co-transfected with HLA-DR and T7 polymerase. Lane 1: CHO-K1 effector cells were transfected with vector alone. Lane 2–8: All effector cells contain wild type gH (0.5 μg), excess gL (2.5 μg), gB (0.5 μg) and gp42 (2 μg). Lanes 3–5: Effector cells contain additional wild type gH (0.5, 1.0 and 1.5 μg respectively). Lanes 6–8: Effector cells contain additional sgH (0.5, 1.0 and 1.5 μg). Cells were detached twenty four hours post-tranfection and target cells were overlaid onto effector cells. Fusion activity was accessed for wt gH/gL (dark gray bars) and sgH/gL (black bars) eighteen to twenty four hours post overlay by the addition of passive lysis buffer followed by the addition of luciferase substrate. Luciferase activity was measured with a Perkin-Elmer Victor plate reader. gH expression (light gray bars) was determined by cELISA using anti-gH antibody E1D1. Data shown are representative results of three independent experiments. Error bars represent standard deviations for the normalized values.
DI/gL is insufficient for inhibition of fusion
Our previous data demonstrated that only full-length sgH/L bound gp42. We sought to determine if binding gp42 was the mechanism by which sgH/L inhibits fusion. We hypothesized that if this was true, only sgH/L would be capable of inhibiting fusion and the truncation mutants (DI/gL, DI-II/gL and DI-II-III/gL) would not be capable of inhibiting fusion. As a negative control, we also assessed the ability of a soluble form of HSV-1 gH/gL to inhibit EBV fusion. As predicted, DI/gL failed to inhibit fusion (Figure 6, lanes 4). However, to our surprise, DI-II/gL, DI-II-III/gL and EBV sgH/gL all inhibited fusion to a significant degree (Figure 6, lanes 5–7). HSV sgH/gL failed to inhibit fusion suggesting the inhibition with DI-II/gL DI-II-III/gL and EBV sgH/gL was species specific (Figure 6, lane 8). This suggests that sgH/gL binding to gp42 is not the sole mechanism by which sgH/L inhibits fusion.
Fig. 6. DI-II/gL, DI-II-III/gL and sgH/gL expressed endogenously inhibits cell-cell fusion.
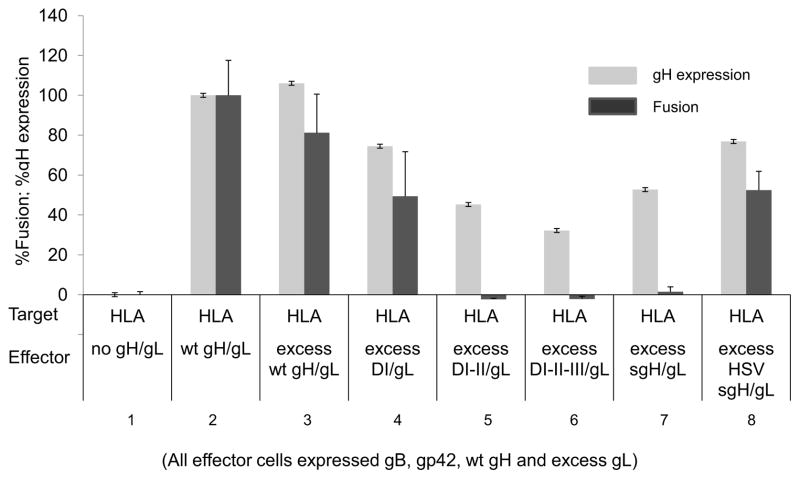
CHO-K1 target cells were transiently co-transfected with HLA-DR and T7 polymerase. CHO-K1 effector cells were transiently co-transfected with gp42, gB, gH, excess gL and either excess wild type gH, HSV gH, DI/gL, DI-II/gL, DI-II-III/gL or sgH/gL. Cells were detached twenty four hours post-transfection and effector cells were overlaid onto target cells. Fusion activity (dark gray bars) was accessed eighteen to twenty four hours post overlay by the addition of passive lysis buffer followed by the addition of luciferase substrate. Luciferase activity was measured with a Perkin-Elmer Victor plate reader. gH/gL expression (light gray bars) was determined by cELISA using anti-gH antibody E1D1. Data shown are representative results of three independent experiments. Error bars represent standard deviations for the normalized values.
gp42 glycoprotein expression is decreased in the presence of sgH/L
To determine if gp42 glycoprotein expression was affected by DI/gL, DI-II/gL, DI-II-III/gL or sgH/L co-expression, we co-transfected CHO-K1 cells with each of the gH domain mutants and wild type gp42 and analyzed gp42 expression by western blot using an anti-gp42 antibody. Wild-type gp42 is cleaved and secreted from cells when expressed alone, however when wild type gH/gL is expressed on the cell surface, the functional, soluble form of gp42 binds back to cells by virtue of binding gH/gL (Figure 7A, lane 7). When wild type gp42 was expressed alone or with DI/gL, the majority of the gp42 was cleaved and secreted (Figure 7B, lane 2 and 3). We did not see a significant difference in gp42 expression in cell lysates or in supernatants when we compared gp42 alone with gp42 co-transfected with DI/gL. However, when gp42 was co-expressed with DI-II/gL, DI-II-III/gL and sgH/gL, gp42 expression was decreased in the cell lysates as well as in the supernatants (Figure 7A and B, compare lane 2 with lanes 4–6). Our results suggest that insufficient soluble gp42 expression may account for the inhibition of fusion seen with sgH/gL, as well as, DI-II/gL, DI-II-III/gL co-expression.
Fig. 7. gp42 expression is decreased when co-expressed with sgH/gL.
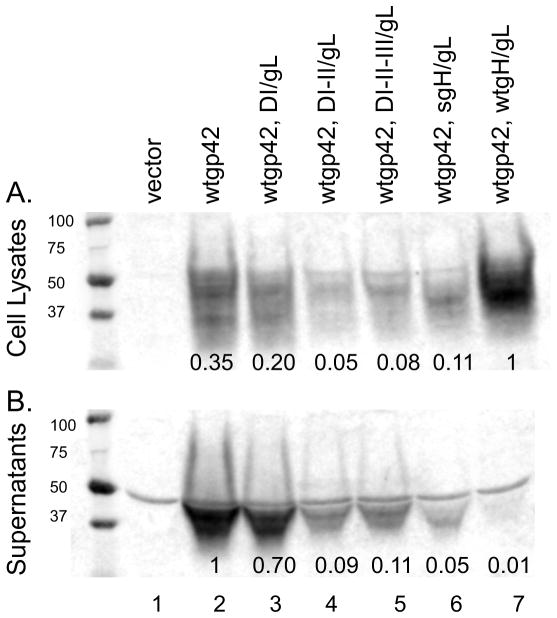
CHO-K1 cells were transiently transfected with empty vector, gp42 or gp42 with DI, DI-II/gL, DI-II-III/gL, sgH/gL or wild type gH/gL respectively. Forty eight hours post transfection, supernatants (B) were collected and cells (A) were lysed in triton X-100 lysis buffer containing protease inhibitors and analyzed by 12% SDS-PAGE and Western blotting with polyclonal anti-gp42 antibody serum (PB114). Size markers in kDa are noted to the left of the blots. Gp42 expression was normalized to wt gp42, wt gH/gL for cell lysates (panel A, lane 7) and to wt gp42 for supernatants (panel B, lane1).
Discussion
Recent data has shown that purified HSV-2 sgH/gL protein, lacking the transmembrane and cytoplasmic tail, can induce a modest (2.1% of wild type) level of fusion of nectin-1-bearing C10 cells expressing gB and gD (Atanasiu et al., 2010). Interestingly, studies with cytomegalovirus (CMV) have found that membrane bound forms of gH/gL can function in trans with gB when gH/gL was expressed on ARPE cells and gB was expressed on HeLa cells, however, the reverse was not true suggesting an additional cellular factor may be required (Vanarsdall et al., 2008). Importantly, CMV deletions that produced soluble gH/gL did not function in fusion. The authors proposed that both gB and gH/gL must be anchored in the membrane to promote fusion (Vanarsdall et al., 2008). Our finding that EBV sgH/gL does not enhance fusion when added exogenously is consistent with our previously published results (Kirschner et al., 2006). The differences in function of soluble gH/gL from the different herpesviruses may reflect significant differences between the fusion complexes of gB/gH/gL/gp42 of EBV and gB/gD/gH/gL of HSV that have not yet been identified or possibly the experimental techniques used to analyze fusion function. For EBV and CMV, the partnering of gH/gL with additional viral proteins (EBV gp42, CMV UL128-131, or CMV gO) can alter tropism. Such stable complexes have not been identified for HSV gH/gL. Perhaps this difference contributes to the inability of soluble forms of EBV and CMV gH/gL to promote fusion. Nevertheless, the fusion mediated by soluble HSV gH/gL was much reduced compared to wild-type levels (Atanasiu et al., 2010), and our data as well as the accumulated evidence described in the introduction suggest the transmembrane and cytoplasmic tail of gH/gL is critical for gB mediated activation of fusion for EBV.
Interestingly, soluble forms of both gp42 and gD function in fusion and both undergo conformational changes upon receptor binding (Carfi et al., 2001; Di Giovine et al., 2011; Kirschner et al., 2009; Krummenacher et al., 2005; Mullen et al., 2002). How this change alters gH/gL binding and how this change is ultimately translated to the fusogen, gB, is not completely understood. Studies in HSV and CMV have suggested there is a direct interaction of the ectodomains of gH/gL with gB using bi-molecular complementation (Atanasiu et al., 2007) or co-immunoprecipitation (Vanarsdall et al., 2008). Whether the cytoplasmic tail of gH/gL interacts with that of gB and whether this is important for activation of fusion is not understood. Alternatively, the gH/gL tail may be required for gH/gL conformation and/or stability, gH/gL association with viral or cellular proteins within the lipid bilayer or cytoplasm, or gH/gL multimerization as has been previously proposed (Jackson et al., 2010; Wilson et al., 1994).
We observed significant inhibition of cell-cell fusion when sgH/gL was co-expressed. The observation that sgH/gL was able to bind gp42, caused us to initially hypothesize that the mechanism of inhibition of fusion was competitive inhibition with wild type gH/gL for binding to gp42. Since the functional form of gp42 is soluble, sgH/gL bound to sgp42 would be expected to no longer be membrane bound and would not be able to mediate B cell fusion. In epithelial cells, sgH/gL can bind a cellular receptor and prevent gH/gL from binding to the receptor, since blocking of epithelial infection by sgH/gL has been shown (Chen et al., 2012; Chesnokova and Hutt-Fletcher, 2011; Chesnokova et al., 2009). Interestingly, HSV sgH/gL produced from insect cells binds to cells independent of avb3 integrin, inhibits virus infection and is able to activate NF-κb by binding Toll-like receptor 2 (TLR2) (Leoni et al., 2012). In addition, recent data on HCMV suggests gH/gL/gO may mislocalize or block entry mediators in cytoplasmic membranes of fibroblasts whereas gH/gL/UL128-131 blocks epithelial cell receptors (Ryckman et al., 2008; Vanarsdall et al., 2011). Specifically, gO promoted the export of gH/gL from the endoplasmic reticulum and the accumulation of gH/gL in the trans-Golgi network. We found DI-II/gL and DI-II-III/gL inhibited fusion but were not able to bind gp42 when added exogenously. However, we believe their propensity to form aggregates may be obscuring their potential for binding exogenously and that they may still bind gp42 intracellularly. DI/gL does not appear to form aggregates in supernatants, does not bind gp42 and also does not decrease gp42 expression. sgH/gL clearly bound gp42 when added exogenously and inhibited fusion when expressed endogenously. DI-II/gL, DI-II-III/gL and sgH/gL all decreased gp42 glycoprotein expression. Thus, we propose that the mechanism of inhibition of fusion by DI-II/gL, DI-II-III/gL and sgH/gL is via intracellular binding to gp42 in the endoplasmic reticulum and targeting it to degradation pathways, leading to a decrease in secreted and cell associated gp42. Similarly, during B cell infection, EBV gp42 binds HLA class II intracellularly. Replacement of the invariant chain with gp42 has been suggested to target the gp42-HLA class II complex and any associated proteins to a degradative pathway and prevent gp42 recognition by T cell receptors (Borza and Hutt-Fletcher, 2002).
When we tried to map the specific gH/gL domain responsible for binding gp42, we found all four domains are required for proper folding of sgH/gL and binding to gp42. Although the x-ray crystallographic structures of EBV gH/gL, PRV gH and HSV gH/L suggested four semiautonomous domains (Backovic et al., 2010; Chowdary et al., 2010; Matsuura et al., 2010) our truncation mutants suggest that there are likely interdomain interactions that are important for gH/gL folding, processing, and stability. Further mutational analysis is necessary to test whether the large groove between DI and DII adjacent to the gH/gL KGD motif, thought to be the receptor binding domain of gH/gL, is a site for gp42 binding. Maintenance of the overall protein conformation may be required for proper binding of viral as well as cellular proteins to gH/gL. It will be interesting to see if PRV and HSV gH/gL have similar multi-domain interaction requirements.
Methods
Cells and Antibodies
Chinese Hamster Ovary cells (CHO-K1) were grown in 75-cm2 cell culture flasks (Corning) in Ham’s F-12 medium (BioWhittaker) supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin-streptomycin (BioWhittaker). Trypsin-Versene (BioWhittaker) was used to detach adherent cells. Polyclonal anti-gp42 antibody serum (PB114) was used as previously described (McShane et al., 2003). Monoclonal antibody 3H3 (anti-gp42) was obtained as previously described (Kirschner et al., 2006). Monoclonal anti-FLAG M2 antibody (F1804) and polyclonal anti-FLAG antibody (F7425) were obtained from Sigma-Aldrich Chemical Company.
Plasmids
FLAG tagged EBV DI, DI-II, DI-II-III and sgH were generated by PCR amplification from wild type gH in pCAGGS (Haan et al., 2001) using the N-terminal primer 5′-CCGCTCGAGCGGGACCATGCAGTTGCTCTGTG-3′ and the following C-terminal FLAG tagged stop containing primers: DI primer 5′-GGAAGATCTCTACTTGTCGTCATCGTCTTTGTAGTCCAAATCTTCGGTGACATTTGCC-3′, DI-II primer 5′-GGAAGATCTCTACTTGTCGTCATCGTCTTTGTAGTCTTGCATGCCCTTGACAGTGG-3′, DI-II-III primer 5′-GGAAGATCTCTACTTGTCGTCATCGTCTTTGTAGTCGAGATGCCACGCATCCCTGTC-3′ and sgH primer 5′-CGAAGATCTCTACTTGTCGTCATCGTCTTTGTAGTCGTGTGCTCTTTCTTCATAC-3′. The PCR products were purified by Qiagen PCR purification kit, digested with Xho I and Bgl II, purified by Qiagen gel extraction kit and ligated into the gel purified Xho I- and Bgl II-digested pCAGGS expression vector overnight at 14°C. The ligated products were transformed into competent DH5α, and selected on ampicillin plates. DNA was isolated from overnight cultures using the Qiagen miniprep kit, digested to confirm the presence of insert and sequenced in both directions by the Northwestern Genomic Core Facility. Large scale DNA preparations were isolated using Qiagen Endo-Free Plasmid maxiprep kit and used in subsequent experiments. EBV gL in pCAGGS was previously described (Haan et al., 2001).
HSV sgH was generated by PCR amplification from wild type gH (Pertel et al., 2001) in pCAGGS using the N-terminal primer 5′-GGCAGATCTGCCACCATGGGGAATGGTTTATGGTTC-3′ and the C-terminal stop containing primer 5′-GCGAGATCTTCATTAGGGCGCAATTGCGGCCACGGGCTGC-3′.
The PCR products were purified by Qiagen PCR purification kit, digested with Bgl II, purified by Qiagen gel extraction kit and ligated into the gel purified Bgl II-digested pCAGGS expression vector overnight at 14°C. The ligated products were transformed into competent DH5α, and selected on ampicillin plates. DNA was isolated from overnight cultures using the Qiagen miniprep kit, digested to confirm the presence of insert. Clones were sequenced to confirm directionality by the Northwestern Genomic Core Facility. HSV gL in pCAGGS was previously described (Pertel et al., 2001).
HLA-DRα and HLA-DRβ were cloned into pSG5 (Stratagene) as previously described (Haan et al., 2000).
Transfection
CHO-K1 cells were transfected in Opti-Mem (Gibco) medium using Lipofectamine 2000 (Invitrogen) according to the manufactures directions. Briefly, 24 hrs after cell plating in a six well dish, various combinations of expression vectors were transfected with lipofectamine in Opti-Mem overnight.
Expression (Fig. 2A): pCAGGS (vector), wtgp42 2μg, DI 0.5μg, gL 0.5μg; wtgp42 2μg, DI-DII .5μg, gL 0.5μg; wtgp42 2μg, DI-DII-III 0.5μg, gL 0.5μg; wtgp42 2μg, sgH .5μg, gL 0.5μg; (Fig. 2B): pCAGGS (vector) 4μg; DI 2μg, gL 2μg; DI-DII 2μg, gL 2μg; DI-DII-III 2μg, gL 2μg; sgH 2μg, gL 2μg
Endogenous Binding (Fig. 3A): pCAGGS (vector) 4μg; sgH 0.5μg, gL 0.5ug; gB 0.5μg, pCAGGS 2.5μg; sgH 0.5μg, gL 0.5ug; gp42 2μg, pCAGGS 1μg; sgH 0.5μg, gL 1ug; wtgH 0.5μg, pCAGGS 2μg
Exogenous binding (Fig. 3B): pCAGGS 4μg; wtgp42 4μg
Fusion (Fig. 4): Target cells: (all lanes) T7 polymerase 0.5μg; (lane 1 and 2) pCAGGS 4μg; (lane 3, 4, 5) pCAGGS 2μg; HLA-DRα 1μg, HLA-DRβ 1μg; Effector cells: (all lanes) T7 luciferase 0.8μg; (lane 1) pCAGGS 3μg; (lane 2, 3 and 5) gp42 2μg, gH 0.5μg, gL 0.5μg, gB 0.5μg; (lane 4) gp42 2μg, sgH 0.5μg, gL 0.5μg, gB 0.5μg
Fusion and expression (Fig. 5):Target cells: (all lanes) T7 polymerase 0.5μg; (lane 1 and 2) pCAGGS 4μg; (lane 3, 4, 5) pCAGGS 2μg; HLA-DRα 1μg, HLA-DRβ 1μg; Effector cells: (all lanes) T7 luciferase 0.8μg; gp42 2μg, gL 2.5μg, gB 0.5μg; (lane 2) gH 0.5μg; (lane 3) gH 1μg; (lane 4) gH 1.5μg; (lane 5) gH 2μg; (lane 6) gH 0.5μg, sgH 0.5μg; (lane 7) gH 0.5μg, sgH 1μg; (lane 8) gH 0.5μg, sgH 1.5μg
Fusion and expression (Fig. 6): Target cells: (all lanes) T7 polymerase 0.5μg; (lane 1 and 2) pCAGGS 4μg; (lane 3, 4, 5) pCAGGS 2μg; HLA-DRα 1μg, HLA-DRβ 1μg; Effector cells: (all lanes): T7 luciferase 0.8μg; gp42 2μg, gL 2.5μg, gB 0.5μg; (lane 2–7) gH 0.5μg; (lane 3) gH 1μg; (lane 4) HSV sgH 1μg; HSV gL 1μg; (lane 5) DI 1μg; (lane 6) DI/II 1μg; (lane 7) DI/II/III 1μg; (lane 8) sgH 1μg
gp42 expression (Fig. 7): (all lanes) gp42 2μg; (lane 1) pCAGGS 1μg; (lane 2) DI 0.5μg, gL 0.5μg; (lane 3) DI/II 0.5μg, gL 0.5μg; (lane 4) DI/II/III 0.5μg; gL 0.5μg; (lane 5) sgH 0.5μg gL 0.5μg (lane 6) wt gH 0.5μg, gL 0.5μg
Western Blotting
CHO-K1 cells were transfected as described above. The medium was changed 16 hrs post transfection to complete Ham’s F12 medium and cells and culture supernatants were collected at 48 h post transfection. Cells were detached with Versene, washed with PBS, and lysed using a 1% Triton x-100 buffer containing protease inhibitors (One milliliter of lysis buffer/10 million cells). Culture supernatants were collected prior to cell detachment and spun down to pellet detached cells. Supernatants and lysates were run on 4–20% Bio-Rad Criterion gels (Figure 2B) and 12% Bio-Rad Criterion gels (Figure 2A and 7) in sodium dodecyl sulphate (SDS) sample buffer at 90V for l.5 hrs. Proteins were transferred to Whatman Optitran 0.45um nitrocellulose membrane in transfer buffer at 100V for 90 min. Blots were blocked in Tris-buffered saline with 5% milk for 1 hr at room temperature or overnight at 4°C and then incubated for 2 hrs at room temperature with polyclonal anti-FLAG antibody (Sigma, F7425) 1:1000 (Figure 2) or polyclonal anti-gp42 antibody serum (PB114) 1:2000 (Figure 7) in blocking solution, as previously described (Fan and Longnecker, 2010; McShane et al., 2003). Blots were washed and IRDye® 800CW conjugated goat (polyclonal) anti-rabbit IgG (H+L) (Li-Cor biosciences, 926-32211) 1:10,000 in blocking solution was applied for 1hr at room temperature with an aluminium foil cover. The blots were washed and analyzed with Li-Cor Biosciences Odyssey® infared imaging studio software.
Cell enzyme-linked immunosorbent Assay (cELISA)
cELISA was used to determine sgH binding (Figure 3A). Briefly, CHO-K1 cells were co-transfected in a 6 well dish with each of the FLAG tagged mutants and wild-type gp42, gB or wild type gH. The medium was changed 16 hours post transfection and one hour later the cells were detached with Versene, counted using a Beckman Coulter Z1 particle counter, 37,500 cells were transferred to a 96 well plate and the total volume was adjusted to 150 ul with complete Ham’s F12 medium. Twenty-four hours later cells were washed once with phosphate-buffered saline (PBS), and cELISA was performed using the monoclonal antibody anti-FLAG-M2 (F1804; Sigma). After incubation with antibody, the cells were washed, fixed, and incubated with biotinylated goat anti-mouseIgG (Sigma), followed by streptavidin-horseradish peroxidase (HRP) (GE Healthcare) and TMB one component HRP substrate (BioFX). Absorbance readings were taken at 380 nm using a Wallac-Victor luminometer (Perkin-Elmer).
cELISA and Fusion Assay
CHO-K1 cells were transiently transfected as described above. The medium was changed 16 hrs post transfection and the cells were detached with Versene and 37,500 cells were transferred to duplicate 96-well plates, one plate was used for cELISA with monoclonal anti-gp42 antibody (3H3) and the other plate was overlaid with equal numbers of CHO-K1 target cells transfected with T7 polymerase and HLA-DRα 1μg, HLA-DRβ. The total volume was adjusted to 150 μl with complete Ham’s F12 medium. Eighteen to twenty hours after overlay, cells were washed with PBS and lysed for 10 min with 50 μl passive lysis buffer (Promega) per well. Luciferase activity was measured with a Perkin-Elmer Victor plate reader immediately after addition of 50 μl/well of luciferase reagent (Promega).
Monolayer Binding Assay
A “monolayer binding assay” was performed as previously described (Fan and Longnecker, 2010) to assess the abilities of the secreted gH mutants to bind gB, gp42 or gH (Figure 3B). CHO-K1 cells seeded in six well plates for 24 hrs were transfected with wild type gp42 using 6 μl of Lipofectamine 2000. After 24 h of incubation, the cells were transferred to 96 well dishes in Ham’s F12 supplemented with 10%FBS. Twenty four hours later the cells were washed twice with cold PBS and overlayed with each of the gH FLAG mutant protein supernatants (isolated following 48 hr transfection and normalized to each other by Western blot, data not shown) in PBS-ABC for 1 h at 4°C. As a negative control, each of the FLAG mutant protein supernatants was also overlayed onto untransfected cells. The cells were then washed with cold PBS four times and soluble gH binding was detected by cELISA using the monoclonal antibody anti-FLAG-M2 (F1804; Sigma) for one hour at room temperature. After incubation with primary antibody, the cells were washed, fixed, and incubated with biotinylated goat anti-mouseIgG (Sigma) for 30 minutes at room temperature, followed by streptavidin-horseradish peroxidase (HRP) (GE Healthcare) for 30 minutes at room temperature and TMB one component HRP substrate (BioFX). Absorbance readings were taken at 380 nm using a Wallac-Victor luminometer (Perkin-Elmer).
Epstein-Barr virus (EBV) soluble gH/gL (sgH/gL) inhibits EBV-induced membrane fusion and does not function in fusion.
sgH/gL stably binds gp42, but not gB nor gH/gL.
Domain IV of gH/gL is required for proper folding and the transmembrane domain and cytoplasmic tail of EBV gH/gL are required for the most efficient fusion.
Acknowledgments
We thank the members of the Longnecker laboratory for their help and support. We thank Qing Fan for critical reading of this manuscript and Nanette Susmarski for excellent technical support. This research was supported by AI076183 (R.L. and T.J.) from National Institute of Allergy and Infectious Diseases by CA117794 (R.L. and T.J.) and CA133063 (R.L. and C.L.R) from the National Cancer Institute and by 12POST9380013 (J.C.) from the American Heart Association.
Abbreviations
- EBV
Epstein Barr Virus
- cELISA
cell enzyme-linked immunosorbent assay
- a.a
amino acid
- PCR
polymerase chain reaction
- ug
microgram
- V
volts
- hr
hour
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CLR, SAC, and JC were responsible for the experimental work, intellectual design, and drafting the manuscript. RL and TSJ participated in the intellectual design of this study and the final manuscript. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. Journal of virology. 2010;84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A. 2007;104:18718–1872. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Swanson-Mungerson M, Amick AC, Longnecker R. Epstein-Barr virus in Burkitt’s lymphoma: a role for latent membrane protein 2A. Cell Cycle. 2010;9:901–908. doi: 10.4161/cc.9.5.10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med. 2002;8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- Browne HM, Bruun BC, Minson AC. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J Gen Virol. 1996;77 (Pt 10):2569–2573. doi: 10.1099/0022-1317-77-10-2569. [DOI] [PubMed] [Google Scholar]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- Chen J, Rowe CL, Jardetzky TS, Longnecker R. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. MBio. 2012;3 doi: 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova LS, Hutt-Fletcher LM. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. Journal of virology. 2011;85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A. 2009;106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol. 2010;17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011;7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, Longnecker R. The Ig-like v-type domain of paired Ig-like type 2 receptor alpha is critical for herpes simplex virus type 1-mediated membrane fusion. J Virol. 2010;84:8664–8672. doi: 10.1128/JVI.01039-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan KM, Kwok WW, Longnecker R, Speck P. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J Virol. 2000;74:2451–2454. doi: 10.1128/jvi.74.5.2451-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan KM, Lee SK, Longnecker R. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology. 2001;290:106–114. doi: 10.1006/viro.2001.1141. [DOI] [PubMed] [Google Scholar]
- Harman A, Browne H, Minson T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. Journal of virology. 2002;76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JO, Lin E, Spear PG, Longnecker R. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J Virol. 2010;84:2038–2046. doi: 10.1128/JVI.02215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NA, Geraghty RJ. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology. 2004;324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J Virol. 2006;80:9444–9454. doi: 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure. 2009;17:223–233. doi: 10.1016/j.str.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. Embo J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient To Activate NF-kappaB. Journal of virology. 2012;86:6555–6562. doi: 10.1128/JVI.00295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol. 1997;71:4657–4662. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longnecker R. Molecular biology of Epstein-Barr virus. In: McCance DJ, editor. Human tumor viruses. American Society for Virology; Washington, D.C: 1998. pp. 133–174. [Google Scholar]
- Matsuura HAKRLTJ. The crystal structure of Epstein-Barr Virus gH and gL complex. Presented orally at the International Herpesvirus Workshop; July 2010.2010. [Google Scholar]
- McShane MP, Mullen MM, Haan KM, Jardetzky TS, Longnecker R. Mutational analysis of the HLA class II interaction with Epstein-Barr virus glycoprotein 42. J Virol. 2003;77:7655–7662. doi: 10.1128/JVI.77.13.7655-7662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen MM, Haan KM, Longnecker R, Jardetzky TS. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell. 2002;9:375–385. doi: 10.1016/s1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- Pertel PE, Fridberg A, Parish ML, Spear PG. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology. 2001;279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. Journal of virology. 2009;83:7678–7689. doi: 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulford DJ, Lowrey P, Morgan AJ. Co-expression of the Epstein-Barr virus BXLF2 and BKRF2 genes with a recombinant baculovirus produces gp85 on the cell surface with antigenic similarity to the native protein. J Gen Virol. 1995;76 (Pt 12):3145–3152. doi: 10.1099/0022-1317-76-12-3145. [DOI] [PubMed] [Google Scholar]
- Rickinson A, Kieff E. Epstein-Barr virus. In: Fields BN, DMK, Howley PM, editors. Fields’ Virology. Lippincott Williams& Wilkins; Philadelphia: 2007. pp. 2656–2700. [Google Scholar]
- Ryckman BJ, Chase MC, Johnson DC. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorem J, Jardetzky TS, Longnecker R. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J Virol. 2009;83:6664–6672. doi: 10.1128/JVI.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs MK, Armitage RJ, Comeau MR, Strockbine L, Farrah T, Macduff B, Ulrich D, Alderson MR, Mullberg J, Cohen JI. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J Virol. 1996;70:5557–5563. doi: 10.1128/jvi.70.8.5557-5563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K. Role of Epstein-Barr virus in Burkitt’s lymphoma. Curr Top Microbiol Immunol. 2001;258:141–151. doi: 10.1007/978-3-642-56515-1_9. [DOI] [PubMed] [Google Scholar]
- Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin Cancer Res. 2004;10:803–821. doi: 10.1158/1078-0432.ccr-0670-3. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Chase MC, Johnson DC. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. Journal of virology. 2011;85:11638–11645. doi: 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. Journal of virology. 2008;82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WI, Sham JS. Nasopharyngeal carcinoma. Lancet. 2005;65:2041–2054. doi: 10.1016/S0140-6736(05)66698-6. [DOI] [PubMed] [Google Scholar]
- Wilson DW, Davis-Poynter N, Minson AC. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J Virol. 1994;68:6985–6993. doi: 10.1128/jvi.68.11.6985-6993.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hutt-Fletcher LM. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology. 2007;363:148–155. doi: 10.1016/j.virol.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen LR, Stephens EB, Davenport LC, Hutt-Fletcher LM. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology. 1993;195:387–396. doi: 10.1006/viro.1993.1388. [DOI] [PubMed] [Google Scholar]