

Summary
Yersinia pestis, the causative agent of plague, utilizes a type III secretion system (T3SS) to intoxicate host cells. The injection of T3SS substrates must be carefully controlled, and dysregulation leads to altered infection kinetics and early clearance of Y. pestis. While the sequence of events leading up to cell contact and initiation of translocation has received much attention, the regulatory events that take place after effector translocation is less understood. Here we show that the regulator YopK is required to maintain fidelity of substrate specificity, in addition to controlling translocation rate. YopK was found to interact with YopD within targeted cells during Y. pestis infection, suggesting that YopK’s regulatory mechanism involves a direct interaction with the translocation pore. In addition, we identified a single amino acid in YopK that is essential for translocation rate regulation but is dispensable for maintaining fidelity of translocation. Furthermore, we found that expression of YopK within host cells was sufficient to down-regulate translocation rate, but it did not affect translocation fidelity. Together, our data support a model in which YopK is a bifunctional protein whose activities are genetically and spatially dinstinct such that fidelity control occurs within bacteria and rate control occurs within host cells.
Introduction
Yersinia pestis, as well as many other Gram-negative pathogens, uses a type III secretion system (T3SS) as a major virulence factor during infection (Cornelis, 2002b, Cornelis et al., 1986, Straley & Bowmer, 1986). The T3SS, also termed injectisome, is composed of over 20 different proteins organized into a complex structure that transverses the inner and outer membranes of the bacterium and forms a conduit between the bacterium and eukaryotic cell (Cornelis, 2002a). Many of the proteins that comprise the T3SS basal body share homology to proteins of the bacterial flagellum, and like the flagellum, T3SS construction is precisely regulated (Erhardt et al., 2010). Once assembled, the T3SS injects an array of proteins, known as Yops (Yersinia outer proteins), into host cells with the net effect of immobilizing (Rosqvist et al., 1991, Rosqvist et al., 1990, Shao et al., 2003) and ultimately killing them (Park et al., 2007, Orth, 2002, Monack et al., 1997, Mills et al., 1997).
T3SS genes are located on a 70kb virulence plasmid, pCD1, and their expression is tightly regulated (Ben-Gurion & Shafferman, 1981, Straley & Bowmer, 1986). Temperature shift to 37°C activates transcription of T3SS genes by the global transcription factor VirF (Cornelis et al., 1986). When millimolar concentrations of calcium are present in the medium, the T3SS is inactive; however, the injectisome is built and Yops are expressed at low levels (Straley et al., 1993). Cell contact triggers polarized translocation of Yops from the bacterial cytosol into host cells (Pettersson et al., 1996, Rosqvist et al., 1994, Persson et al., 1995). In the absence of host cells, bacteria can be triggered to activate the injectisome and secrete Yops into the medium by chelating calcium (Straley & Bowmer, 1986, Michiels et al., 1990).
LcrQ (or the YscM1 and YscM2 homologs in Y. enterocolitica) is unique with no known homology outside of the Yersinia, which acts as a negative regulator of T3SS gene expression (Rimpiläinen et al., 1992, Allaoui et al., 1995, Cambronne et al., 2000, Wulff-Strobel et al., 2002). As a T3SS substrate, LcrQ (also YscM1 and YscM2) is translocated into host cells immediately after contact, thereby linking expression of effector Yops with cell contact (Pettersson et al., 1996, Rimpiläinen et al., 1992, Cambronne et al., 2000, Cambronne et al., 2004). It is suggested that LcrQ is injected into host cells to remove it from the bacterium, thus relieving transcriptional inhibition and allowing for robust expression of Yops (Pettersson et al., 1996, Cambronne et al., 2000, Cambronne et al., 2004).
Assembly of the injectisome begins with the formation of the basal body, which includes structural proteins that form a channel spanning the bacterial envelope, as well as a regulatory apparatus that controls export through the channel (Diepold et al., 2010, Diepold et al., 2011, Marlovits et al., 2004, Diepold et al., 2012). YscF is among the first substrates to be secreted through the channel, and it polymerizes into a needle that will span the gap between the bacterium and a host cell (Hoiczyk & Blobel, 2001). Upon completion of the needle, YscU in the export apparatus undergoes autocleavage (Edqvist et al., 2003), which results in a change of the substrate specificity from early Yops required for needle formation to middle Yops (such as LcrV, YopB and YopD) required for pore formation (Montagner et al., 2011). Upon secretion, LcrV forms a tip complex at the distal end of the YscF needle (Mueller et al., 2005). LcrV then aids in insertion of YopB and YopD into host cell membranes, which forms a translocon pore that completes the conduit between the bacterium and the target cell (Cornelis & Wolf-Watz, 1997, Goure et al., 2005).
Cell contact and completion of injectisome assembly triggers a second substrate specificity switch such that now only late Yops (Yops E, H, J, T, O, M, N, K, and LcrQ) are translocated, thereby ensuring that these proteins are delivered directly into host cells (Rosqvist et al., 1994, Cheng & Schneewind, 2000, Lee et al., 1998). Cell contact presumably triggers structural changes at the distal end of the injectisome, which are relayed to the basal body or the export apparatus to govern substrate specificity. The mechanism for assessing cell contact has not been determined; however, YopN is involved in preventing premature effector translocation (Forsberg et al., 1991, Lee et al., 2001, Cheng & Schneewind, 2000). TyeA binds to the C-terminal end of YopN and is proposed to tether YopN to the basal body of the injectisome (Iriarte et al., 1998, Cheng et al., 2001, Schubot et al., 2005). YscB and SycN bind to the N-terminal portion of YopN and aide in its secretion once its TyeA tether is released (Day & Plano, 1998, Jackson et al., 1998). Upon YopN translocation, the block on late Yops is relieved and these effectors are injected into host cells.
YopK is a Yersinia T3SS substrate (Garcia et al., 2006, Thorslund et al., 2011) and is a regulator of translocation (Holmstrom et al., 1997, Holmstrom et al., 1995a, Holmstrom et al., 1995b, Aili et al., 2008, Dewoody et al., 2011). YopK is a structural mystery. It shows no similarity to known proteins, apart from its homologs in Y. pseudotuberculosis (YopK) and Y. enterocolitica (YopQ), and there are no predicted functional domains. YopK is essential for virulence, as a Y. pestis yopK mutant triggers an early immune response, and colonization of the spleen and liver is impaired (Peters & Anderson, 2012, Straley & Bowmer, 1986, Straley & Cibull, 1989). Initial characterization of YopK function shows that a ΔyopK mutant injected higher levels of late Yops E and H into host cells while over expression of YopK results in less injection compared to wild type infection (Holmstrom et al., 1997). More recent work has shown that YopK specifically regulates the rate of late Yop injection into host cells (Dewoody et al., 2011). Furthermore, it was found that YopK exerts its regulatory activity inside host cells, indicating that YopK governs translocation from the distal end of the injectisome rather than at its base within the bacterium (Dewoody et al., 2011).
YopE has also been shown to play a role in translocation regulation (Aili et al., 2006, Aili et al., 2008, Dewoody et al., 2011). Using a Bla reporter assay, it was found that a ΔyopE mutant has a faster rate of Yop injection compared to wild type, but this phenotype is milder than that of a ΔyopK mutant (Dewoody et al., 2011). YopK was found to regulate translocation rate independently of YopE, and a double ΔyopEK mutant phenocopies the ΔyopK mutant, suggesting that YopK works upstream of YopE in T3SS regulation (Dewoody et al., 2011). YopE’s ability to regulate translocation is dependent on its GAP domain (GTPase activation protein) (Rosqvist et al., 1991, Dewoody et al., 2011). The GAP domain of YopE is also required for its ability to inactivate host Rho GTPases and disrupt actin polymerization (Black & Bliska, 2000, von Pawel-Rammingen et al., 2000). In addition, inhibitors of actin polymerization also inhibit effector translocation (Mejia et al., 2008). Together, these observations link translocation regulation by YopE with its ability to disrupt actin polymerization in the host cell. Distinguishing these two functions in order to understand their connection is troublesome, since both functions require the same conserved arginine finger motif in the GAP domain (Aili et al., 2002, Dewoody et al., 2011).
Our work indicated that while both YopE and YopK are required for controlling the rate of Yop translocation, they have distinct functions. Here we gain additional insight into how Yop translocation is controlled by further investigating the role of YopK. We find that ΔyopK leads to a loss of substrate specificity similar to a ΔyopN mutation, while a ΔyopE mutant maintains fidelity of translocation. Furthermore, we show that rate of translocation and fidelity of translocation are distinct functions, both of which can be attributed to YopK.
Results
Translocation phenotypes of multi-Yop mutants
In previous work, we found that YopK has a very strong negative effect on translocation while YopE has an intermediate effect (Dewoody et al., 2011). Because LcrQ is a negative regulator of Yop transcription, a ΔlcrQ mutation yields increased expression of Yops and a corresponding increase in Yop translocation (Rimpiläinen et al., 1992, Cambronne et al., 2000, Cambronne et al., 2004). Considering that LcrQ, YopE, and YopK all affect Yop injection, we wanted to better understand the relationship of these regulators. Toward that end, we created a series of single, double and triple mutants in Y. pestis KIM5. We then used a β-lactamase (Bla) reporter in combination with CCF2-AM dye to compare the injection phenotypes of each strain (Dewoody et al., 2011, Marketon et al., 2005, Charpentier & Oswald, 2004). A key feature of the CCF2-AM dye is a β-lactam ring linking coumarin and fluorescein groups, which leads to green fluorescence upon excitation with violet light. In the presence of Bla, the β-lactam ring is cleaved and fluorescence shifts from green to blue. When Bla fused to an effector Yop (Yop-Bla) is expressed by Y. pestis strains, Yop-Bla delivery into host cells can be easily quantified using flow cytometry to measure green vs. blue fluorescence.
To assess mutant phenotypes, CHO cells were infected with various Y. pestis strains carrying the YopJ-Bla reporter (Dewoody et al., 2011) at a multiplicity of infection (MOI) of 10 for 2 hours before cells were incubated with the dye. Figure 1 shows the average fluorescence of triplicate infections by strains carrying YopJ-Bla. The stacked bar graphs show uninjected cells (green fluorescence = white bar), cells with low-level injection (cells with both cleaved and uncleaved dye emit both blue and green fluorescence = grey bars), and cells with high-level injection (blue cells = black bars). A non-injectable Gst-Bla reporter was used as a negative control for all injection assays.
Figure 1. Translocation phenotypes of single, double and triple mutants.
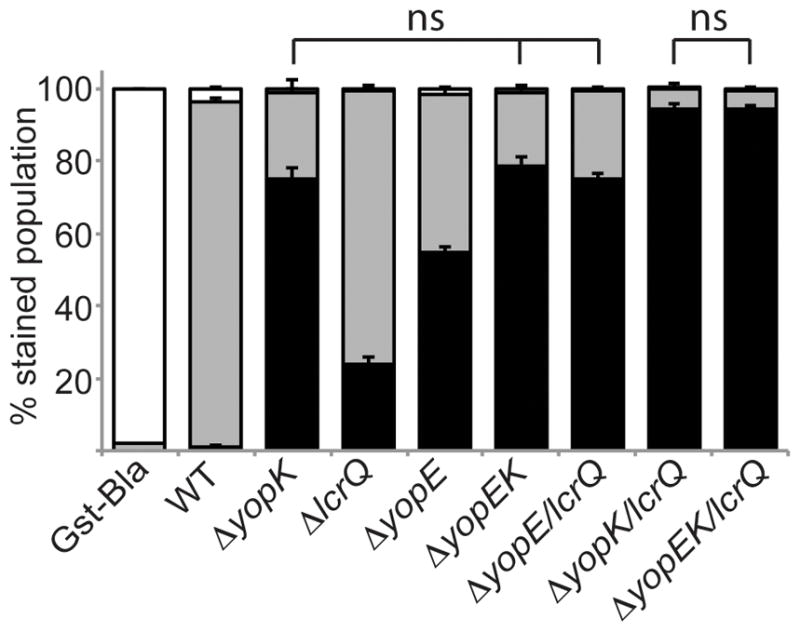
Y. pestis strains carrying the YopJ-Bla reporter or the Gst-Bla control were used to infect CHO cells at an MOI of 10 for 2 hours. Cells were then incubated with CCF2-AM and analyzed by flow cytometry. Each infection was performed in triplicate, samples were averaged, and standard deviation is shown. ANOVA followed by Tukey post-hoc test was done and n.s. indicates no significant difference among bracketed strains. The experiment was repeated at least twice. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection). Gst-Bla control plasmid was expressed in ΔyopK for this assay.
Wild type infection showed primarily low-level injection of the YopJ-Bla reporter (Fig. 1). In contrast, each single mutant showed increased injection, with the highest levels for the ΔyopK mutant followed by ΔyopE and ΔlcrQ mutants (P<0.001). The ΔyopK and ΔyopEK mutants were indistinguishable in phenotype, as previously reported (Fig. 1 and (Dewoody et al., 2011)). The addition of the ΔlcrQ mutation to all tested strains increased injection of the YopJ-Bla reporter. Thus, the ΔlcrQ mutation appeared to have an additive affect on injection by increasing the amplitude of effector translocation, consistent with previous findings (Cambronne et al., 2000, Cambronne et al., 2004). A ΔyopK mutation combined with either ΔlcrQ or ΔyopE/lcrQ mutations resulted in the highest level of injection, highlighting the importance of YopK as a regulator of translocation. Notably, combining a yopE deletion with either ΔyopK or ΔyopK/lcrQ did not have an additive effect. These results are consistent with a role for YopK upstream of YopE within host cells, while LcrQ works within bacteria to control the pool of Yops available for injection.
YopK ensures fidelity of injection
The above results, as well as previous data from us and others, demonstrate that a ΔyopK mutant over-injects effector Yops (late Yops). Furthermore, YopD translocation into host cells was observed in a Y. pseudotuberculosis yopK mutant (Francis & Wolf-Watz, 1998). We therefore hypothesized that the defective regulation of a yopK mutant extended to aberrant injection of other T3SS substrates that are not normally injected into host cells (early and middle Yops). In other words, do ΔyopK mutants lack control of substrate specificity (fidelity) as well as rate of translocation? To determine whether YopK contributes to T3SS fidelity, we employed digitonin fractionation to visualize the fate of representative Yops: YscF and YopR (early substrates), LcrV, YopD, and YopB (middle substrates), YopN, YopE, YopK, YopM, YopH (late substrates). CHO cells were infected with wild type and mutant Y. pestis strains and then lysed by digitonin. Digitonin selectively lyses eukaryotic membranes, allowing injected Yops (supernatant) to be separated from Yops associated with host membranes or attached bacteria (pellet) (Lee et al., 1998).
As seen in Figure 2, the ΔyopK mutant showed very high levels of late Yops in the digitonin supernatant (YopN, YopE, YopM and YopH) when compared to a wild type infection. Early substrates YscF and YopR were not injected into host cells by the ΔyopK mutant and were instead secreted into the culture medium in the same manner as wild type (data not shown). Interestingly, middle Yops LcrV and YopD were found in higher amounts in the digitonin supernatant fraction during infection with the ΔyopK mutant compared to wild type (Fig. 2). YopB was also found in the digitonin supernatant at higher levels than for wild type, though the increase was not as pronounced as for YopD and LcrV. This is not surprising since YopB has two transmembrane domains, and therefore is expected to partition with the membrane fraction even after translocation. Normally, middle Yops are translocated prior to cell contact, and late Yops are translocated after cell contact. Presumably, formation of the pore complex by the middle substrates completes injectisome assembly and triggers the second substrate specificity switch such that now late Yops travel through the needle. Thus, it appears that the ΔyopK mutant can still discriminate against early Yop injection but has lost the ability to reject middle Yops once cell contact is made.
Figure 2. A ΔyopK mutant lacks translocation fidelity.
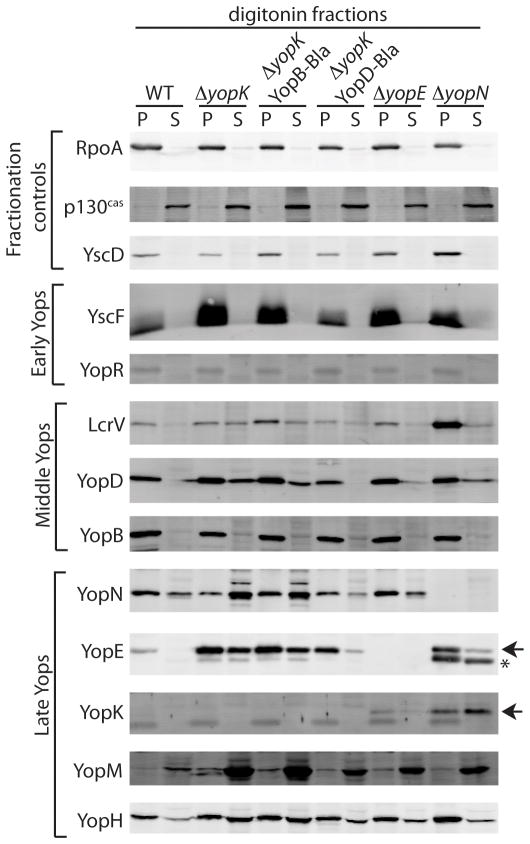
CHO cells were infected with Y. pestis strains for 3 hours before the media was removed and monolayer washed. Host cell membranes were lysed with digitonin and separated by centrifugation into supernatant (cytosolic content of host cells including injected Yops) and pellet (large membrane fragments and adherent bacteria). Samples were immunoblotted with RpoA (bacteria cytosolic protein), YscD (structural component of T3SS), and p130cas (eukaryotic cytosolic protein) serving as fractionation controls. * Denotes degradation products of YopE due to surface protease Pla (Sodeinde & Goguen, 1988, Sodeinde et al., 1988) while arrowheads indicate full-length proteins.
Because YopE also lacks proper control of translocation, we investigated whether a ΔyopE mutant lacks fidelity as well. We found that the ΔyopE mutant maintained a wild type phenotype with regard to early and middle substrate recognition, despite the fact that late Yops (YopM and YopH) were translocated at higher levels (Fig. 2). A ΔyopN mutant was included in the fractionation experiment, as it is known to lack fidelity during injectisome assembly such that middle and late Yops are both secreted into the medium and injected into host cells (Yother & Goguen, 1985, Day & Plano, 1998, Cheng & Schneewind, 2000). As expected, the ΔyopN strain injected both middle and late Yops, but not early Yops. Altogether, the data indicate that the substrate specificity switch responsible for rejecting early Yops is maintained within the ΔyopN and ΔyopK mutants. In contrast, the switch that is triggered by cell contact is compromised in both strains. Therefore, YopN and YopK both appear to participate in coordinating the delivery of late Yops into host cells.
To confirm the translocation fidelity phenotype of a ΔyopK mutant by an independent method and to quantify the defect, we generated two new Bla reporters: YopD-Bla and YopB-Bla as C-terminal fusions so as not to disturb their T3SS secretion signals. These reporter fusions were expressed and secreted at 37°C in the absence of calcium (Fig. S1 and data not shown). We attempted to evaluate an LcrV-Bla reporter, however, the protein fusion failed to secrete (data not shown). Expression of the YopD-Bla reporter caused a slight decrease in expression and injection of other Yops (Figs. S1 and 2), as expected since YopD is a negative regulator of Yop expression (Williams & Straley, 1998, Francis & Wolf-Watz, 1998, Anderson et al., 2002, Chen & Anderson, 2011). In addition, expression of YopD-Bla also led to slightly reduced levels of native YopD translocation (Fig. 2), which is likely due to competition for binding to the chaperone, LcrH, that is required for stability and efficient secretion of YopD (Francis et al., 2000, Wattiau et al., 1994, Neyt & Cornelis, 1999). It is important to note that strains expressing either YopD-Bla or YopB-Bla had abundant Yop secretion, demonstrating that the reporter fusions do not have a dominant negative impact on secretion of the late Yops (Fig. S1 and data not shown).
Although these are meant as middle Yop reporters, we tested whether YopD-Bla or YopB-Bla were functional and could complement a YopB or YopD mutant. To that end, we employed a GSK reporter fused to the C-terminus of YopJ (Garcia et al., 2006). The 13-residue GSK tag fused to YopJ is phosphorylated by host cell kinases only after injection (Garcia et al., 2006). A phospho-specific GSK antibody distinguishes between the phosphorylated (injected) and unphosphorylated (uninjected) YopJ-GSK. Arabinose was added to the bacterial cultures one hour before infection to induce expression of YopJ-GSK, while the Yop-Bla fusions were expressed upon shifting growth to 37°C. As shown in Figure S2, wild type and the ΔyopK mutant both gave rise to phospho-specific GSK bands only in the presence of arabinose. As expected there was more phosphorylated YopJ-GSK in cells infected with the ΔyopK mutant than in cells infected with wild type. Because WT and the ΔyopK mutant both carried either the YopB or YopD β-lactamase fusion when translocation took place, as shown by the Bla antibody, the data confirmed that expression of these reporters did not block translocation (Fig. S2). The ΔyopB mutant expressing YopB-Bla and the ΔyopD mutant expressing YopD-Bla did not show phospho-specific GSK bands when YopJ-GSK was induced with arabinose. This shows that the C-terminal Bla fusion disrupted the function of YopD and YopB in terms of translocation pore formation. However, the reporters were recognized as T3SS substrates since they were secreted (Fig. S1 and data not shown), and they did not prevent injection (Figs. 2 and S2). Therefore, despite their inability to participate in pore formation, the YopB- and YopD-Bla reporters are suitable middle Yop reporters.
Having confirmed the utility of the middle Yop reporters, CHO cells were infected with mutant Y. pestis strains carrying either YopD-Bla or YopB-Bla (Fig. 3A and 3B). As expected, wild type Y. pestis infection showed no injection of middle Yops into host cells. However, ~80% of host cells showed injection of YopD-Bla and ~20% injection of YopB-Bla during infection with the ΔyopK mutant. This is consistent with the digitonin fractionation experiment (Fig. 2), as well as previous work showing translocation of YopD into host cells by a Y. pseudotuberculosis yopK mutant (Francis & Wolf-Watz, 1998). Additionally, each ΔyopK double or triple mutant showed injection of middle Yops, again showing the importance of YopK in maintaining proper fidelity of the T3SS. Injection levels by ΔyopE, ΔlcrQ and ΔyopE/lcrQ mutants were not significantly different from wild type, indicating that these proteins do not play a direct role in maintaining fidelity. It is important to note that the ΔlcrQ mutation led to higher expression levels of all Yops, including the middle Yops, compared to the yopK mutant (Fig. S1), yet the single ΔlcrQ mutant (like WT) could not inject middle Yops (Fig. 3). Therefore, the differences in middle Yop injection cannot be explained simply by changes in Yop expression. However, when the ΔlcrQ mutation was combined with a ΔyopK or ΔyopEK mutation, there was an additive effect (P<0.001) to the reporter translocation level as shown previously (Fig. 1), consistent with a role for LcrQ in restricting the pool of potential T3SS substrates. In contrast, loss of yopE seems to counteract the fidelity phenotype, since the ΔyopEK and ΔyopEK/lcrQ strains both showed lower injection of the middle Yop reporters than ΔyopK or ΔyopK/lcrQ strains (P<0.001). This is in contrast to YopE’s role in translocation rate regulation, in which yopK appeared to be epistatic to yopE (Fig. 1 and (Dewoody et al., 2011)). Together, these data suggest that YopK plays an integral role in translocation fidelity and rate, while LcrQ influences the amount of proteins that are injected. It is unclear at this time whether YopE influences T3SS fidelity directly or indirectly.
Figure 3. YopK is essential for substrate specificity during Yop translocation.
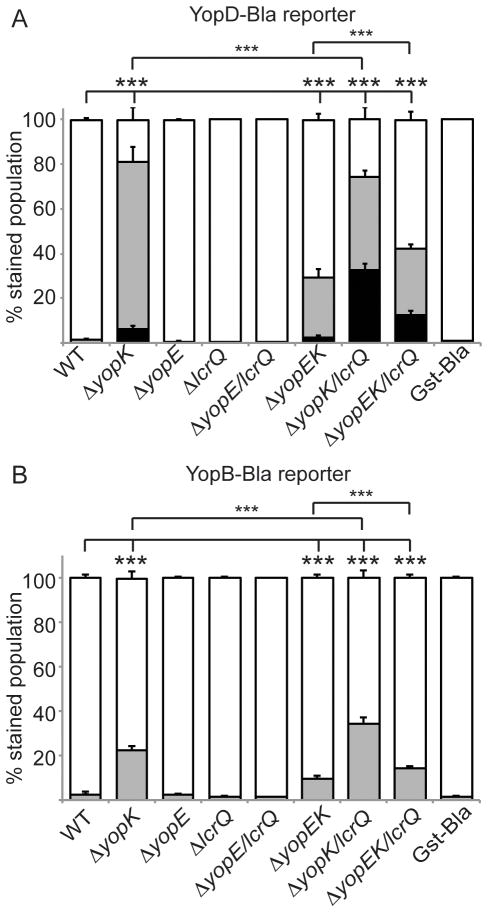
Y. pestis strains carrying the YopD-Bla (A) or YopB-Bla reporter (B)or the Gst-Bla control were used to infect CHO cells at an MOI of 10 for 5 hours. Cells were then incubated with CCF2-AM and analyzed by flow cytometry. Each infection was performed in triplicate, samples were averaged, and standard deviation shown. ANOVA followed by Tukey post-hoc test was done and *** indicates P<0.001. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection). Gst-Bla control plasmid was expressed in ΔyopK and ΔyopEK/lcrQ and averaged for this assay.
YopK residue D46A is essential to translocation regulation
It has been reported that YopK interacts with the host protein Rack1, and that a tandem mutation of residues threonine 45 (T45) and aspartic acid 46 (D46) of Y. pseudotuberculosis YopK abolished binding of Rack1 and reduced virulence in vivo (Thorslund et al., 2011). Y. pseudotuberculosis uses the adhesin invasin to bind to the extracellular face of host cell β-integrins, which in turn bind to Rack1 within the cytoplasm (Isberg & Barnes, 2001, Thorslund et al., 2011). Since Y. pestis lacks invasin and does not directly bind β-integrins, the relevance of these two residues or ability of YopK to bind Rack1 in this system is unknown. We therefore mutated the Y. pestis yopK open reading frame creating the double T45A/D46A mutation, as well as each single point mutation, and introduced them into WT and ΔyopK strains that also carried YopM-Bla. To verify that the YopK variants were expressed and secreted at similar levels to wild type YopK, we induced secretion by growing the strains in the absence of calcium at 37°C. Secreted proteins were separated by centrifugation from bacterial cells. Proteins were TCA precipitated and analyzed by immunoblotting. As seen in Figure 4A, expression and secretion of each YopK construct was at native YopK levels or higher. Additionally, expression of the YopK constructs did not alter secretion of other T3SS substrates.
Figure 4. Characterization of YopK point mutants.
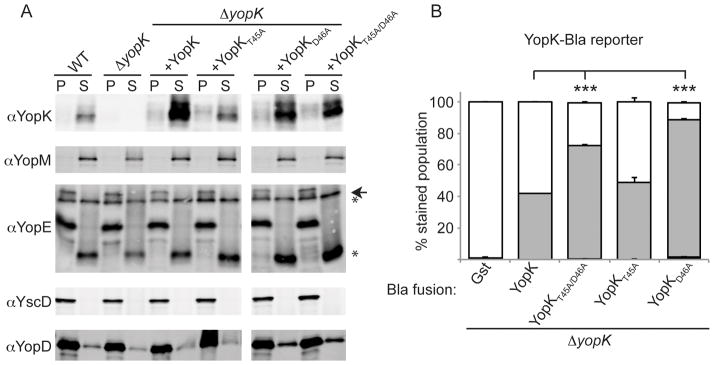
(A) ΔyopK Y. pestis carrying a YopK or YopK point mutant expression vector were induced to secrete using temperature switch to 37°C and low Ca2+. Separated by centrifugation, supernatants (S) contain secreted Yops while pellets (P) contain bacteria. YscD is a structural protein of the injectisome and a fractionation control. (arrowhead denotes full length protein while * indicates degradation products) (B) ΔyopK Y. pestis carrying YopK or YopK point mutants fused to Bla were used to infect CHO cells as in panel A. Triplicate samples were averaged and standard deviation is shown. ANOVA followed by Dunnett post-hoc test was done using the ΔyopK +native YopK infection as the control, and *** indicates P<0.001.
To eliminate the possibility that the point mutations in YopK abrogate its own injection, we introduced the same mutations into a YopK-Bla reporter. This allowed us to directly measure of translocation of YopK point mutants. CHO cells were infected with the ΔyopK mutant carrying only YopK-Bla or a YopK-Bla point mutant, and then injection was measured using CCF2-AM and flow cytometry (Fig. 4B). The experimental results are somewhat complex to interpret due to the fact that if the point mutant cannot regulate translocation (i.e. behaves like ΔyopK), the Bla reporter will be injected at higher levels along with the other Yops. However, we are primarily using these Bla fusions to determine whether their injection occurs, rather than to assess the function of individual point mutants. We observed ~40% injection of YopK-Bla, which was not statistically different from injection levels of YopKT45A-Bla. YopKT45A/D46A and YopKD46A were injected at slightly higher levels than native YopK. These results indicate that the point mutations do not prevent YopK translocation.
Having demonstrated their expression, secretion, and injection, we next tested the ability of each YopK variant to regulate injection by expressing them in the ΔyopK mutant carrying the YopM-Bla reporter. As expected, expression of native YopK reduced injection, whereas YopKT45A/D46A did not reduce injection levels by the ΔyopK mutant and instead gave results identical to the ΔyopK mutant alone (Fig. 5). To further distinguish which of these residues was important for function, we evaluated the ability of the single point mutants to complement the ΔyopK mutant. Expression of YopKT45A in the ΔyopK mutant resulted in injection levels similar to that of wild type Y. pestis, while YopKD46A expression did not complement the ΔyopK mutant and allowed high-level injection. This would suggest that aspartic acid 46 is essential for translocation regulation, but threonine 45 is dispensable.
Figure 5. Residues T45 and D46 of YopK are important for translocation regulation.
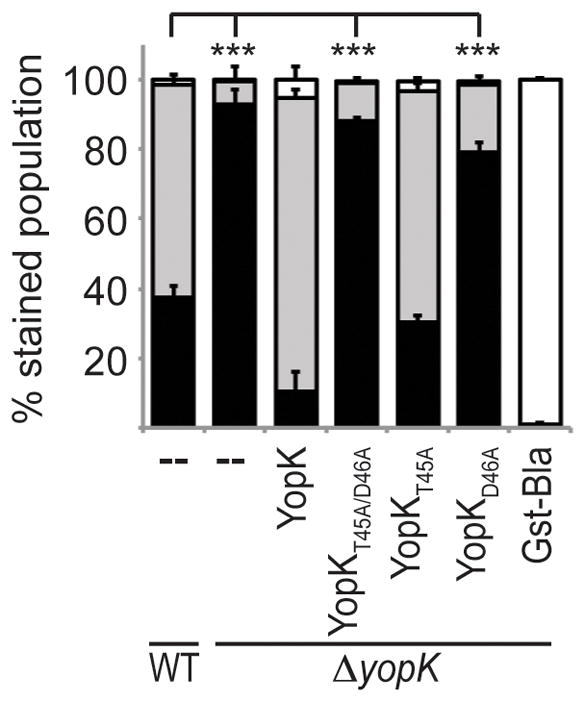
Y. pestis strains carrying the YopM-Bla reporter or the Gst-Bla control, along with either native YopK or YopK variant expression vectors, were used to infect CHO cells at an MOI of 10 for 3 hours. Cells were then incubated with CCF2-AM and analyzed by flow cytometry. Each infection was performed in triplicate, samples were averaged, and standard deviation shown. ANOVA followed by Dunnett post-hoc test was done using the WT infection as the control, and *** indicates P<0.001. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection).
YopK interacts with pore component YopD as well as host protein Rack1
Previous reports suggested that YopK interacts with the translocation pore components YopB and YopD. Purification of red blood cell membranes after Y. pseudotuberculosis infection revealed that YopK, along with pore components YopB and YopD, are present in the membrane fraction (Thorslund et al., 2011). Likewise, YopK co-immunoprecipitated with YopB in a different study (Brodsky et al., 2010). However, it remains to be determined whether the proposed interaction between YopK and the translocon components occurs within host cells, which is the site of YopK function. Toward this end, we performed a co-immunoprecipitation (co-IP) on cells infected with Y. pestis. CHO cells were transiently transfected with Keima-YopK, a eukaryotic expression vector which produces an N-terminal fusion of YopK to the red fluorescent protein, Keima (Dewoody et al., 2011). Transfected cells were infected with Y. pestis ΔyopK, then lysed and incubated with anti-YopK coated Dynabeads. Proteins isolated by the immunoprecipitation were analyzed by immunoblotting. Infection with a ΔyopK strain ensures that all precipitated proteins result from YopK interactions inside host cells.
As shown in Figure 6A, YopD was co-immunoprecipitated only when Keima-YopK was expressed in host cells and was absent from Keima-alone controls, despite the fact that equal amounts of YopD were present in each whole cell lysate (WCL), demonstrating that the interaction between YopK and YopD is specific. GRP78 is a chaperone of endoplasmic reticulum proteins found in the cytoplasm and perinuclear region of host cells (Reddy et al., 2003). This makes it a particularly good control for indiscriminant binding as the majority of Keima-YopK localized to the perinuclear region as viewed by fluorescence microscopy (data not shown). Probing WCL input and co-IP output samples with anti-GRP78 showed no indiscriminate binding to YopK. Interestingly, each of the YopK point mutations introduced into the Keima-YopK vector was also able to bind YopD, demonstrating that alanine substitutions of T45 and D46 do not inhibit YopD binding (Fig. 6A).
Figure 6. YopK interacts with YopD and Rack1 during infection.
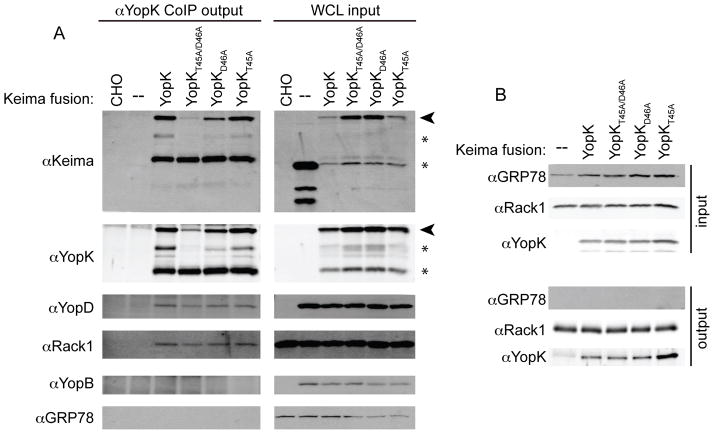
Co-immunoprecipitation with antibodies raised against YopK (A) or Rack1 (B). CHO cells were transiently transfected one day before assay. Cells were infected with ΔyopK Y. pestis for 3 hours at MOI 10 (A) or uninfected (B) before lysis. A sample of each whole cell lysates (WCL) was collected for analysis as input. Protein A Dynabeads bound to YopK antibody (A) or Rack1 antibody (B) were incubated with the WCL. Dynabeads were washed to remove non-specific proteins and bound proteins with removed with SDS buffer. Samples were boiled for 5 minutes and subjected to SDS-PAGE and Western blot. GRP78 antibody was used to probe both input and output samples to show specificity of co-IP and to act as a loading control for input samples. Arrowheads indicate full-length Keima-YopK fusions while * denote degradation products.
In addition to YopD, Keima-YopK pulled down the host protein Rack1 (Fig. 6A), which is not surprising due to the high homology between Y. pseudotuberculosis and Y. pestis YopK. In contrast to previously reported data (Thorslund et al., 2011), the Y. pestis YopKT45A/D46A mutant could pull down Rack1 (Fig. 6A). The single YopK point mutants were equally capable of binding Rack1 indicating that residues T45 and D46 of Y. pestis YopK are not required for Rack1 binding during infection, despite the fact that YopKD46A abrogated translocation rate regulation (Fig. 5).
The tandem T45A/D46A YopK mutation was shown to abolish Rack1 binding by yeast-2-hybrid analysis in a previous report (Thorslund et al., 2011), however, we showed in Figure 6A that YopKT45A/D46A pulls down Rack1 like native YopK. To confirm this observation and to determine whether formation of a translocation pore was required for the interaction, we performed a reciprocal co-IP using a commercially available Rack1 antibody bound to Protein A coated Dynabeads. CHO cells were transfected with Keima, Keima-YopK or the point mutant variants, lysed and then incubated with Rack1 coated Dynabeads. As seen in Figure 6B, Rack1 pulled down each of the Keima-YopK constructs. GRP78 was not present in the output fractions showing that the binding of Keima-YopK to Rack1 is specific. Furthermore, since the pull downs were performed on uninfected cells, there was no translocation pore, indicating that the YopK-Rack1 interaction is independent of pore formation. Our data show that YopK, YopKT45A/D46A, YopKT45A, and YopKD46A can all bind to Rack1 in host cells during infection. This means that the inactivation of YopK regulatory activity by aspartic acid 46 substitution is not due to loss of Rack1 binding (Fig. 5 and 6B).
YopK is a bifunctional regulator
We next assessed the role of residues 45 and 46 of YopK in the fidelity of Yop injection. We expressed either YopD-Bla (Fig. 7A) or YopB-Bla (Fig. 7B) in the ΔyopK mutant simultaneously with either native YopK or the YopK point mutants. Secretion assays were performed on each strain to ensure proper expression of the proteins of interest and as well as other Yops (Fig. S3 and S4). Y. pestis strains were then used to infect CHO cells followed by CCF2-AM staining and flow cytometry. Figure 7A shows that during a wild type infection YopD-Bla was not injected. It was, however, injected at moderate levels by the ΔyopK mutant. Interestingly, all YopK point mutants functioned as well as wild type in preventing YopD-Bla injection. Results for the YopB-Bla reporter were similar. As shown in Fig. 7B, the wild type infection showed no injection of YopB-Bla while a ΔyopK mutant led to injection of ~50% of host cells. The YopKT45A/D46A and YopKT45A point mutants completely rescued the ΔyopK mutant as well as native YopK, and YopKD46A was able to partially suppress the fidelity defect. Thus, these residues do not appear to be essential for YopK’s ability to regulate translocation fidelity. This is in contrast to the observation that D46A was essential to regulate the rate of translocation. This is the first evidence suggesting that YopK has two genetically distinct functions: one involved in regulating the rate of Yop translocation and one integral for substrate specificity of the injectisome.
Figure 7. YopK point mutants maintain fidelity of Yop injection.
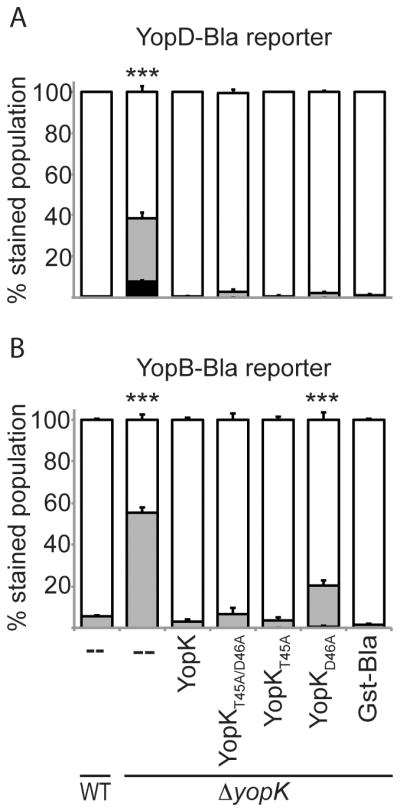
CHO cells were infected at MOI 10 for 5 hours with Y. pestis strains expressing YopD-Bla (A) or YopB-Bla (B) as well as a YopK or YopK point mutant complementation vector. ΔyopK Gst-Bla was included as a negative control. Cells were incubated with CCF2-AM and analyzed by flow cytometry. Each infection was performed in triplicate, samples were averaged, and standard deviation shown. ANOVA followed by Dunnett post-hoc test was done using the WT infection as the control, and *** indicates P<0.001. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection).
YopK functions in separate locations
After observing that the fidelity control and rate control aspects of YopK function could be separated, we investigated whether both phenomena occurred within host cells. We previously demonstrated that expression of Keima-YopK within host cells complements the defect of a ΔyopK mutant, as judged by the use of the YopM-Bla reporter, which is a measure of the rate control aspect (Dewoody et al., 2011). We therefore used the same approach, combined with the YopD-Bla reporter to assess fidelity. CHO cells were transiently transfected with either Keima or Keima-YopK and then infected, followed by CCF2-AM staining and flow cytometry. WT and ΔyopK Y. pestis strains expressing the YopM-Bla or YopD-Bla reporters were used for the infections. As shown in Figure 8, expression of Keima-YopK led to significantly lower injection levels of the YopM-Bla reporter during infection with either WT or ΔyopK Y. pestis. However results using the YopD-Bla reporter were very different. Expression of Keima-YopK during WT infection led to a very low, but significant, level of YopD-Bla injection. Importantly, there was no significant difference in YopD-Bla injection by the ΔyopK mutant regardless of Keima-YopK expression. These results indicate that while YopK must be injected into host cells in order to down-regulate translocation rate, the same is not true for fidelity control. Instead, it appears that YopK performs that function from the bacterial side of the injectisome.
Figure 8. YopK works within host cells and bacteria.
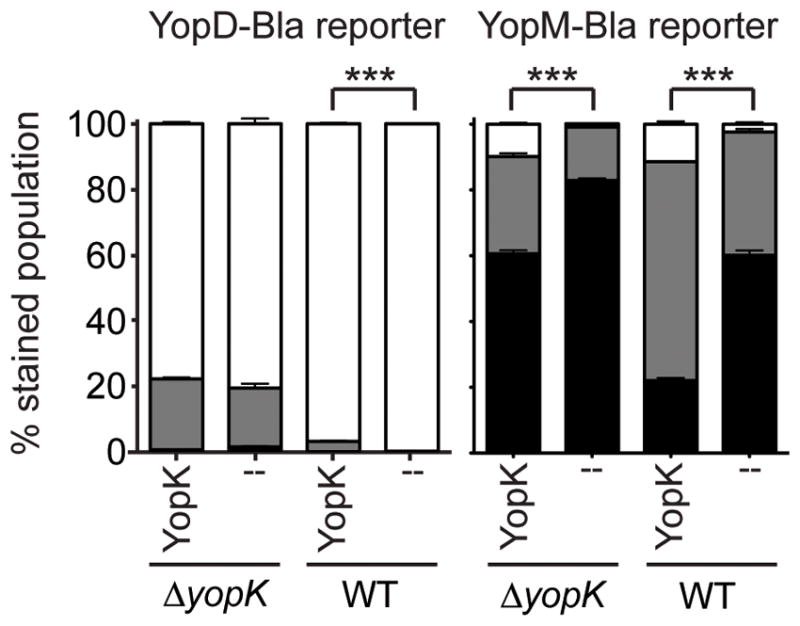
CHO cells were transfected and allowed to express either Keima (--) or Keima-YopK (YopK) for ~36 hours, followed by infection at MOI 10 for 4 hours with Y. pestis strains expressing either YopD-Bla or YopM-Bla. Infected cells were then stained with CCF2-AM and analyzed by flow cytometry. Each infection was performed in triplicate, samples were averaged, and standard deviation shown. Unpaired two-tailed t-tests were done and *** indicates P<0.001. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection).
Discussion
Herein we have investigated the roles of proteins involved in translocation regulation. We have found that YopK and YopE control rate of injection while LcrQ regulates the magnitude of the substrate pool (Fig. 1). The lcrQ mutation led to increased injection of reporter Yops (Figs. 1 and 3). This fits with the current understanding of the role of LcrQ as a negative regulator of Yop expression functioning to prevent the unnecessary production of Yops before cell contact is made. While the ΔyopE mutation led to increased injection compared to wild type, its effects when combined with ΔyopK mutation were not additive (Fig. 1). This corresponds with previous data suggesting that while both proteins play a role in translocation rate regulation, YopK functions upstream of YopE (Dewoody et al., 2011).
Fidelity of Yop translocation was assessed to determine if more than one facet of translocation was regulated from the host cell. Digitonin fractionation revealed that all strains tested maintained at least partial substrate specificity, as YscF and YopR were not injected into host cells (Fig. 2). Because the YscU autocleavage, which triggers the specificity switch from early to middle Yop secretion, is presumably irreversible, it is logical that a defect at the level of translocation into host cells would not allow a relapse into recognizing early T3SS substrates. Unlike wild type and the ΔyopE mutant, both ΔyopK and ΔyopN strains lacked fidelity of translocation, and could not prevent injection of LcrV, YopD and YopB into host cells (Fig. 2). Data presented here suggest a model in which YopK and YopN both control substrate specificity of the injectisome.
Interestingly, the yopE mutation in ΔyopK and ΔyopK/lcrQ backgrounds counteracted the loss of translocation fidelity (Fig. 3). Because YopE regulation of translocation and the actin cytoskeleton are tightly linked, it is possible that the difference in fidelity of the ΔyopEK and ΔyopEK/lcrQ mutants is due to the state of the host cell cytoskeleton. A ΔyopK mutant has active YopE during infection, which prevents actin polymerization. When YopE is absent, the ΔyopK infection takes place in the context of normal host cell cytoskeleton regulation. This would suggest that while YopE alone does not affect translocation fidelity, its disruption of the actin remodeling in response to infection makes the presence of YopK absolutely necessary to maintain fidelity. Based on our data in figure 8, YopK may perform that duty at the base of the injectisome in the bacterium. We would surmise that normal regulation of host actin cytoskeleton impacts translocation fidelity possibly by influencing the translocation pore conformation. More work is necessary to assess impact of the host cell cytoskeleton on the pore and translocation fidelity.
We have characterized two residues of YopK (T45 and D46) that have been implicated in binding to host cell protein Rack1 to prevent phagocytosis during Y. pseudotuberculosis infection (Thorslund et al., 2011). We found that residue D46 of YopK was essential for translocation rate regulation, while T45 was dispensable (Fig. 5). Interestingly, YopKD46A was able to maintain translocation fidelity (Fig. 7). These observations suggest that YopK regulates translocation rate and fidelity by separate mechanisms, and that D46 is only necessary for rate control. Furthermore, each activity of YopK appears to be be spatially separated (Fig. 8): YopK controls translocation rate within host cells while fidelity is maintained within bacteria.
Immunoprecipitation experiments revealed that Y. pestis YopK interacts with YopD in the host cell, and point mutations to YopK residues T45 and D46 do not disrupt this interaction (Fig. 6A). Previous work demonstrated YopK interactions with YopB (Brodsky et al., 2010) and YopD (Thorslund et al., 2011) in Y. pseudotuberculosis. Thus the interaction of Y. pestis YopK with YopD upon translocation into host cells is in agreement with prior findings. Unexpectedly, neither Keima-YopK nor any of the point mutants were able to pull down YopB in our experiments despite the presence of YopB in the whole cell lysate input. It could be that YopB was below the limit of detection for our YopB antibody in these experiments. Additionally, because YopB has two transmembrane domains, whereas YopD has only one, YopB may be extracted less efficiently from the plasma membrane in our attempts to capture YopK interactions.
An additional unexpected finding was that Y. pestis YopK point mutants readily bind Rack1 (Fig. 5 and Fig. 6A), which is in contrast to previous work showing that the Y. pseudotuberculosis YopKT45A/D46A mutant was not able to bind Rack1 (Thorslund et al., 2011). There are several possible explanations for the incongruity. In the Thorslund et al study, YopK-Rack1 binding was detected and characterized by Yeast-2-hybrid analysis using a HeLa cell library, and binding was not evaluated in target cells during a Yersinia infection, as we have done. In our work, we used CHO cells, so there could be differences associated with the relative affinities or binding characteristics of the HeLa vs. CHO Rack1 proteins. We previously attempted to screen a HeLa library by Yeast-2-hybrid analysis (unpublished data), and Rack1 was not identified using Y. pestis YopK as bait; however the negative result in this case could simply indicate that the screen was not saturated.
Given that in our experimental system, D46A abolished the rate control activity of YopK, without disrupting Rack1 binding, we cannot determine whether Rack1 participates in translocation regulation. It could be that there are separate pools of YopK within host cells: one pool of YopK is associated with translocation pores, while the other is associated with Rack1. Alternatively, YopK may be in a macromolecular complex that includes both YopD and Rack1, but the D46A mutation simply alters conformation of YopK and/or other proteins within the complex to exert the observed phenotype without abolishing the interactions altogether. Additionally, there may be other proteins involved in YopK binding, which participate in translocation regulation, and the D46A mutation abolishes the interaction.
Given our observations on the regulatory role of YopK, we propose a new model of translocation regulation, shown in Figure 9. During injectisome assembly, the apparatus goes through a series of assembly states corresponding to the types of substrates that are secreted at each step: early, middle, and late. In the early state, the substrates required for needle formation are secreted. Transition from early to middle occurs when needle assembly is finished, and then the middle substrates that are required for pore formation are secreted. During this stage, the YopN-TyeA complex associates with the injectisome to monitor fidelity of substrate recognition by preventing secretion of late Yops. Once cell contact is made, transition to the late state occurs along with release of YopN from the base of the injectisome. At this point, the injectisome is ON and YopN, YopK, and the other late Yops are injected into host cells. According to our data, YopK may interact with the base of the injectisome during the ON state to ensure that late Yops are recognized while middle Yops are rejected by the T3SS. During the ON state YopK would also accumulate within host cells and would interact with Rack1 and the translocation pore. Once inside the host, our model proposes that YopK shifts the injectisome to an OFF state to prevent over-injection of effectors. This would trigger the end of the infection and possibly release of the bacterium to intoxicate additional host cells.
Figure 9. Two roles for YopK in controlling the injectisome.
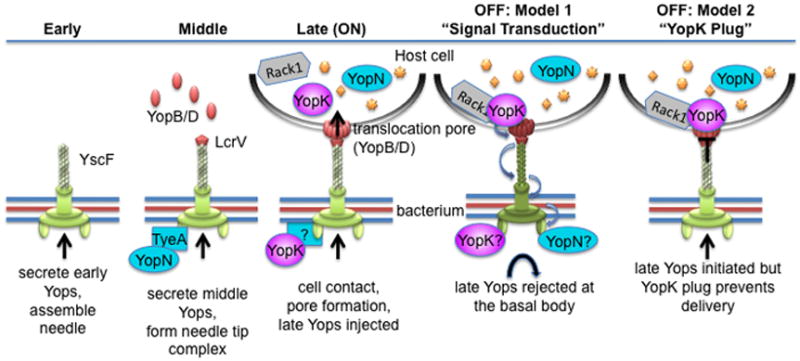
A model is shown to depict basic stages of injectisome assembly, activation, and deactivation. In the “early” stage, needle subunit (YscF) and other early substrates needed for needle assembly are secreted. Reaching optimal needle length triggers a substrate specificity switch and transition to the “middle” stage. This allows secretion of the translocators and assembly of the needle tip complex. Upon contact with a host cell, the translocators form a pore in the host membrane that completes the channel from the bacterium into the host. Channel completion coincides with transition to the “late” stage, in which the injectisome is “ON” and now injects effector Yops (late Yops) directly into host cells. During this time YopK accumulates in host cells and interacts with Rack1 and the translocation pore, and the end result is the down regulation of Yop injection (OFF state). In the first model, YopK would interact with the translocation pore, triggering a cascade of conformational changes throughout the injectisome, which ultimately prevent initiation of further Yop injection. In the second model, YopK would bind to the translocation pore and act as a cap that closes the channel to prevent further Yop injection.
In our model, we show two possible mechanisms for YopK-mediated down-regulation. In the first option, YopK binding to YopD triggers a conformational change in the pore that is transduced through the injectisome to retard or stop translocation. In the second option, YopK binding to YopD would essentially plug the channel to prevent further translocation. Currently we favor the first option for several reasons. First, in previous work expression of YopK correlated with the size of YopB/D-mediated pores (Holmstrom et al., 1997). In those experiments, over-expression of YopK led to smaller pores, while absence of YopK led to larger pores. Though these assays may not measure actively translocating pores, they may reflect the consequences of YopK binding to pore proteins. Perhaps YopK binding induces structural changes, which also stabilize the pore structure, and correspondingly, the absence of YopK results in a less stable pore structure that manifests as more leakiness (i.e.- a larger pore phenotype). An additional consideration is that a YopK point mutation (D46A) that abolished rate control did not abolish YopD binding, suggesting that the YopK variant may still interact with the pore proteins without inducing the proper conformational changes. If the second mechanism (the YopK plug model) were correct, then one would predict that mutations that abolish YopK activity would correspond with a lack of binding to the pore complex. We did not observe such an outcome with our YopKD46A mutant; however a more thorough analysis of the YopK open reading frame needs to be done to adequately test the prediction.
Regardless of the mechanism, one would predict that if YopK interacts with YopD to control injectisome function, then reciprocal mutations within YopD that are required for the interaction and subsequent translocation regulation should be identified. Though such YopD mutants have not been identified yet, previous work relied on deletion scanning mutagenesis(Francis & Wolf-Watz, 1998), rather than techniques involving generation of point mutants. Importantly, mutagenesis studies on YscF have revealed point mutations leading to distinct phenotypes for translocation, which presumably result from changes in overall needle conformations (Davis et al., 2010, Davis & Mecsas, 2007). Such an analysis on YopD may likewise reveal important residues associated with YopK binding and translocation regulation.
Finally, if YopK and YopN are both required for substrate specificity, they presumably interact with the T3SS basal body to influence substrate recognition. Determining if YopK associates with the base of the injectisome during translocation and defining the nature of the YopK and YopN interactions with the basal body will be an important future priority. YopN belongs to a family of regulators shared amongst T3SSs, which appear to regulate substrate specificity in response to external stimuli, such as changes in calcium levels, pH, or cell contact (Yu et al., 2010, Botteaux et al., 2009, Martinez-Argudo & Blocker, 2010, Crabill et al., 2012, Ferracci et al., 2005). In Yersinia, YopN functions within bacteria to prevent early secretion of late Yops, and this activity requires association with TyeA (Joseph & Plano, 2007, Ferracci et al., 2005, Sundberg & Forsberg, 2003, Iriarte et al., 1998, Day et al., 2003, Ferracci et al., 2004). It is thought that TyeA tethers YopN to the base of the injectisome, though the interacting proteins have not been identified. Dominant negative mutations in YopN, which inhibit dissociation of the YopN-TyeA complex also prevent YopN response to external stimuli (Ferracci et al., 2005). Therefore, in further support of Model 1 in Figure 9, it seems that external stimuli, such as cell contact, may be sensed by the distal portion of the injectisome and the information communicated to the base via conformational changes. That change likely stimulates release of the YopN-TyeA tether, allowing YopN to be injected into host cells (Day et al., 2003, Ferracci et al., 2004). Simultaneously, release of YopN may reveal a YopK binding site to enable YopK control of fidelity during injection. Upon its own translocation into host cells, YopK binding to the translocation pore may provide a final stimulus for structural changes in the injectisome to signal transition to the OFF state. Clearly, the events surrounding translocation regulation require further analysis with particular attention to the changes occurring at a molecular level in order to test the model predictions and resolve possible mechanisms. Future work also needs to include structural information on YopK to map important alleles onto possible functional domains.
Experimental procedures
Bacterial strains and media
Strains used in this study are shown in Table S2. Yersinia pestis KIM5, an attenuated variant of the Y. pestis mediaevalis strain KIM lacking the 102-kb pgm locus (Brubaker, 1969), was propagated on Heart Infusion Agar (HIA) plates at 26 °C for two days. Overnight cultures were grown in Heart Infusion Broth (HIB) at 26 °C. Antibiotics were added as appropriate to a final concentration of 20 μg/ml chloramphenicol (Cm), 50 μg/ml kanamycin (Km) or 50 μg/ml ampicillin (Ap). Escherichia coli strains were propagated in Luria-Bertani broth or agar, supplemented with 30 μg/ml Cm, 100 μg/ml Ap or 50 μg/ml Km, at 37 °C.
Strain and Plasmid creation
Plasmids used in this study are shown in Table S1. pRD12, pRD16, and pRD17 were created by quick change PCR using primers TD45-46AA for (GCTCATTTACTGAGAGCGGCTGTTTGTTCATTGGTC) and TD45-46AA rev (GACCAATGAACAAACAGCCGCTCTCAGTAAATGAGC) on plasmids pMM206 (YopK), pRD1 (Keima-YopK), and pMM84 (YopK-Bla) respectively. This introduced an alanine substitution for both T45 and D46 of YopK on each plasmid. pRD13, pRD18, pRD22 were created using the same primers on plasmids pMM206, pRD1, and pMM84 respectively which introduced T45A point mutations to the YopK ORF. pRD15, pRD19, and pRD24 were also created using primers TD45-46AA for and TD45-46AA rev on plasmids pMM206, pRD1, and pMM84 respectively that introduced D46A point mutations to YopK ORF on each vector. Each point mutation was confirmed by sequencing.
Bla-fusions to YopK (pMM84), YopB (pMM115), and YopD (pMM117) were created essentially as described previously for pMM83 (YopM-Bla) and pMM91 (Gst-Bla) (Marketon et al., 2005). YopK with its native promoter was amplified with the primers YopQpro-EcoRI (AAGAATTCCTGTTCATCTGTATAACC) and YopQORF-KpnI-2 (AAGGTACCTCCCATAATACATTCTTG) and cloned in front of Bla as an EcoRI-KpnI fragment. YopB and YopD were both expressed from the YopN promoter, which was amplified with the primers YopNpro-EcoRI (AAGAATTCACCCACCCCAACCTGAT) and YopNpro-Ndel (AACATATGAACTACTCCCTGAGATG) and cloned as an EcoRI-NdeI fragment in front of the YopB or YopD ORFs. The YopB ORF was amplified with the primers YopBORF-NdeI (AACATATGAGTGCGTTGATAACCCAT) and YopBORF-KpnI (AAGGTACCAACAGTATGGGGTCTGCC). The YopD ORF was amplified with the primers YopDORF-Ndel (AACATATGACAATAAATATCAAGAC) and YopDORF-KpnI (AAGGTACCGACAACACCAAAAGCGGC). Both ORFs were cloned between the YopN promoter fragment and Bla as NdeI-KpnI fragments.
PMY1, PMY2, PMY3 were created by inserting a stop-frameshift mutation of lcrQ into strains MEL18, DEW1, and MEL27 respectively. pMM54 (Sorg et al., 2005) was introduced into each stain and selected by plating with Cm. CmR colonies were cured of the insert and drug resistance marker by plating on 5% sucrose HIA plates. Colonies were screened by patch plating, PCR and sequencing.
PMY12 is a markerless, non-polar deletion of yopD in an otherwise wild type background. It was generated using a suicide construct on the SacB harboring plasmid pDS132 (Philippe et al., 2004). Two primer pairs were designed, ΔYopD 1 XbaI (TCTAGAGCGGCAGGATCTCAACTG)/ΔYopD 2 (CCAAGGTCATCAATGGTCAGACCATGGTTATTCCTCCTTAAACTTAAAC) and ΔYopD 3 (GTTTAAGTTTAAGGAGGAATAACCATGGTCTGACCATTGATGACCTTGG)/ΔYopD 4 SacI (GAGCTCCCATCAATAAGCACGGCACTAC), to amplify approximately 1kb fragments flanking the region to be deleted. Primers 2 and 3 overlap to facilitate joining of the upstream and downstream fragments by PCR-SOEing. The final PCR products were cloned into pDS132 via XbaI/SacI resulting in pMER9. pMER9 was moved into Y. pestis backgrounds by conjugal transfer. Matings were plated on Yersinia selective agar Cm plates and Campbell integrants (Cm-r, Suc-s) were identified by patching on HIA containing 5% sucrose. Merodiploids were resolved by overnight growth in the absence of Cm and subsequent plating on HIA with 5% Sucrose. Sucrose resistant isolates were patched onto HIA Cm to confirm excision of pMER9. ΔyopD mutants were confirmed by PCR using primers ΔYopD 5 (AAGGACAACAGCAAGAAGTCAC) and ΔYopD 6 (CTCCTCATGGCAACTCGATG), subsequent sequencing and western blot.
PMY4 was created with a stop-frameshift mutation of YopB. Primers YopB.KO-NotI (AAAAGCGGCCGCACCGCCGAATTAAAGATTTA)/YopB.KO-BglII-1 (AAAAAGATCTTCTACATGTTCCATCTCCTTTTTC) and YopB.KO-BglII-2 (AAAAAGATCTAGTGCGTTGATAACCCATGA)/YopB.KO-SacI (AAAAGAGCTCAATCAGCGTTATTATGTTGT) were used to amplify the regions upstream and downstream of YopB respectively. Each PCR fragment was inserted into the SmaI site of pBluescript creating pMER25 (yopB upstream with NotI/BglII sites) and pMER26 (yopB downstream with BglII/SacI sites). pMER25 was then cut with NotI/BglII while pMER26 was cut with BglII/SacI. These were introduced into pSR47s NotI/SacI by three-way ligation to generate pMER27. The mutant was generated by mating and selection as detailed above. The strain was confirmed by sequencing with primers YopB KO veri F (ACCGCCGAATTAAAGATTTATT) and YopB KO veri R (AACGGCTCCTACCCCTGA) and Western blot analysis.
Cell culture maintenance and transfections
Eukaryotic culture lines used in this study are listed in Table S2. CHO-K1 (ATCC) and CMV-Bla CHO-K1 (Invitrogen) were maintained in F12K (Cellgro) supplemented with 10% heat inactivated FBS (Cellgro). CMV-Bla CHO-K1 cells were supplemented with 1mg/mL geneticin (Gibco). Transfections were performed using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer. CHO cells were seeded at 6.5×105 cells per well into 6-well plates or 2.6×105 in 12-well plates 1 day prior to transfection. Transfection used 4μg of plasmid DNA and 10μL of Lipofectamine 2000 in 500mL of OptiMEM (Invitrogen) per well for a 6-well plate or 1.6μg of plasmid DNA and 4μL of Lipofectamine 2000 in 200mL of OptiMEM per well for a 12-well plate. Cells were incubated for 12–48 hours at 37 °C, 5% CO2 to allow expression.
Co-Immunoprecipitation
CHO cells were seeded at 2.6×105 in a 12 well plate one day prior to transfection. Cells were transfected with the 1.6μg of Keima or Keima-YopK DNA and Lipofectamine 2000 (Invitrogen) according to manufacture’s instructions. Cells were allowed to express vector fusions for 24 hours. For YopK co-IP, Y. pestis ΔyopK cultures were grown overnight in HIB to an OD600 between 0.4 and 0.8 and transferred to 37°C for 1 hour to pre-induce the T3SS. CHO were infected at an MOI of 10 for 3.5 hours, washed three times with PBS and incubated with lysis buffer (50mM NaCl, 0.5% NP-40, 1% TritonX-100, 10mM Tris, pH 7.4, 1mM EDTA, 1mM EGTA, pH 8, 0.2mM Na3VO4, 0.2mM PMSF, protease complete tab) at 4°C for 40 minutes. For Rack1 co-IP, CHO cells were not infected. Simultaneously, 50μL of Protein A coated Dynabeads (Invitrogen) per sample were incubated with 5% BSA in PBST (PBS with 0.02% Tween-20) for 1 hour at room temperature. Dynabeads were washed three times with PBST, then incubated with either 20μL YopK or 10μL Rack1 antibody per sample in 200μL PBST for 1 hour at room temperature. Dynabeads were washed three times in PBST and resuspended in 50μL PBST. CHO cell lysates were spun at 500g for 5 minutes to pellet unlysed cells, and lysate transferred to new tube where antibody coated beads were added. Dynabeads and lysate were incubated for 1 hour at room temperature before beads were washed three times with PBST and 25μL YSB (0.15M MgCl2, 4% SDS, 10% glycerol, 5% βME, 100mM Tris, pH 8, Bromophenol Blue) was added to Dynabeads. Samples were boiled for 5 minutes before immunoblotting.
Digitonin fractionation
Digitonin fractionation was completed by combining aspects of two protocols (Ramsby & Makowski, 2005, Lee et al., 1998). Digitonin extraction buffer was prepared in advance per Ramsby et al recipe and stored at −80°C until use. CHO cells were seeded a day in advance in T25 flasks to ~90% confluency. CHO cells were infected with various Y. pestis strains grown overnight in HIB at 26°C and pre-induced at 37°C for 1 hour. An MOI of 10 was used for the 3–4 hour infection. The monolayer was then washed three times with PBS to remove non-adherent bacteria and 5mL of digitonin extraction buffer was added to each flask for incubation at 4°C for 40 minutes with gentle agitation. The monolayer was scraped from the flask and transferred to a conical tube. Samples were spun at 10,000g for 30 minutes, and then the supernatant was transferred to a new tube for MeOH/CHCl3 extraction. The pellet was solubilized in 1% SDS in PBS then MeOH/CHCl3 extracted. All fractions were resuspended in 200μL YSB, boiled for 5 minutes and immunoblotted.
Injection assay
Injection assays were completed as previously reported with few modifications (Dewoody et al., 2011). CHO cells were seeded at 2.6×105 in 12-well plates 1 day prior to infection. CHO were infected with Y. pestis grown overnight in HIB with antibiotics to exponential phase and pre-induced at 37°C for 1 hour. Bacteria were added to CHO at an MOI of 10 and infection at 37°C occurred for 2–5 hours depending on the Bla reporter. YopJ-Bla reporter injection assays were performed with a 2 hour infection, YopM-Bla infections lasted for 3 hours, and infections with YopD-Bla and YopB-Bla took place for 5 hours. Samples with the Gst-Bla control were done for the same amount of time as the reporter infection for each experiment. Cells were washed with PBS and suspended in HBSSflow (1X HBSS, 0.5mM EDTA, 25mM HEPES, 2% BSA, pH 7.4) after infection. CHO cells were strained with 0.4μm filter and CCF2-AM was added to each sample per manufacturer directions. YopD and YopB-Bla injection assays were allowed to incubate with CCF2-AM for 1 hour at room temperature before transferring to ice and analyzing by flow cytometry while other reporters required only 30 minutes at room temperature. Flow cytometry analysis was performed on an LSRII in the Indiana University Flow Cytometry Core Facility. Flow cytometry data was analyzed using either FACSDiva (BD) or FlowJo (TreeStar) software as described previously (Dewoody et al., 2011). Statistical analysis was performed using GraphPad Prism. In the case of single pair-wise comparisons, two-tailed T-tests were done. In the case of multiples strains compared to a single control strain, one-way ANOVA followed by Dunnett post-hoc test was used. In the case of multiple pair-wise comparisons, one-way ANOVA followed by Tukey post-hoc test was used.
Secretion Assay
Secretion assays were performed as previously described (Marketon et al., 2005). Bacteria are grown overnight at 26°C in HIB with antibiotics as appropriate, then subcultured 1:20 into modified M9. They were grown an additional 2 hours at 26°C before transferring to 37°C for 3 hours to induce secretion. Samples were separated by centrifugation, then proteins in media supernatant and bacterial pellet fractions were TCA precipitated and visualized by immunoblotting.
Antibodies
Antibodies were purchased for Rack1 (B-3) (Santa Cruz Biotechnology), actin Ab5 (BD Transduction Laboratories), P-GSK 3β (Cell Signaling), β-lactamase (Millipore), Keima (MBL International Corp), GRP78 (BD Transduction Laboratories) and p130cas (BD Transduction Laboratories). YopK antibody (Dewoody et al., 2011) produced in the Marketon lab was affinity purified using Affi-Gel 15 activated affinity media (BioRad). Antibodies to RpoA, YscD, YopM, YscF and YopE were produced in the Marketon lab as previously reported (Dewoody et al., 2011, Houppert et al., 2012). YopD, YopN, YopR, YopH, and LcrV anitbodies were a gift from the laboratory of Olaf Schneewind and the YopB antibody a gift from the lab of Deborah Anderson.
GSK assay
This assay was done essentially as reported by Garcia et al. CHO cells were seeded at 6.5×105 in a 6-well plate one day prior to experiment. Y. pestis strains carrying either a YopB-Bla or YopD-Bla expression vector as well as the YopJ-GSK (Garcia et al., 2006) reporter were grown overnight at 26°C to exponential phase with appropriate antibiotics. Bacteria were transferred to 37°C to pre-induce the T3SS and 0.2% L-arabinose was added to induce YopJ-GSK expression. CHO cells were infected at MOI 10 for 3 hours before monolayers were washed with PBS and lysed with YSB supplemented with phosphatase inhibitor cocktail (Sigma), protease inhibitor cocktail (Sigma), and benzonase (Novagen) at 1:100. Samples were boiled for 5 minutes then immunoblotted with phospho-specific GSK antibody to test for phosphorylation of the GSK reporter.
Supplementary Material
Acknowledgments
Special thanks to Andrew Houppert and other members of the Marketon laboratory for helpful discussions on this work. This work was partially supported by NIH/NIAID award number R21AI083660 to Melanie Marketon and NIH Training Grant T32 GMOO7757 support for Rebecca Dewoody. Weacknowledge membership in and support provided by the RegionV “Great Lakes” RCE (NIH award 2-U54-AI-057153).
Citations
- Aili M, Hallberg B, Wolf-Watz H, Rosqvist R. GAP activity of Yersinia YopE. Methods Enzymol. 2002;358:359–370. doi: 10.1016/s0076-6879(02)58102-7. [DOI] [PubMed] [Google Scholar]
- Aili M, Isaksson EL, Carlsson SE, Wolf-Watz H, Rosqvist R, Francis MS. Regulation of Yersinia Yop-effector delivery by translocated YopE. Int J Med Microbiol. 2008;298:183–192. doi: 10.1016/j.ijmm.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Aili M, Isaksson EL, Hallberg B, Wolf-Watz H, Rosqvist R. Functional analysis of the YopE GTPase-activating protein (GAP) activity of Yersinia pseudotuberculosis. Cell Microbiol. 2006;8:1020–1033. doi: 10.1111/j.1462-5822.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- Allaoui A, Schulte R, Cornelis GR. Mutational analysis of the Yersinia enterocolitica virC operon: characterization of yscE, F, G, I, J, K required for Yop secretion and yscH encoding YopR. Molecular Microbiology. 1995;18:343–355. doi: 10.1111/j.1365-2958.1995.mmi_18020343.x. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Ramamurthi KS, Tam C, Schneewind O. YopD and LcrH regulate expression of Yersinia enterocolitica YopQ by a posttranscriptional mechanism and bind to yopQ RNA. J Bacteriol. 2002;184:1287–1295. doi: 10.1128/JB.184.5.1287-1295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Gurion R, Shafferman A. Essential virulence determinants of different Yersinia species are carried on a common plasmid. Plasmid. 1981;5:183–187. doi: 10.1016/0147-619x(81)90019-6. [DOI] [PubMed] [Google Scholar]
- Black DS, Bliska JB. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol. 2000;37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- Botteaux A, Sory MP, Biskri L, Parsot C, Allaoui A. MxiC is secreted by and controls the substrate specificity of the Shigella flexneri type III secretion apparatus. Mol Microbiol. 2009;71:449–460. doi: 10.1111/j.1365-2958.2008.06537.x. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker RR. Mutation rate to nonpigmentation in Pasteurella pestis. J Bacteriol. 1969;98:1404–1406. doi: 10.1128/jb.98.3.1404-1406.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne ED, Cheng LW, Schneewind O. LcrQ/YscM1, regulators of the Yersinia Yop virulon, are injected into host cells by a chaperone-dependent mechanism. Molecular Microbiology. 2000;37:263–273. doi: 10.1046/j.1365-2958.2000.01974.x. [DOI] [PubMed] [Google Scholar]
- Cambronne ED, Sorg JA, Schneewind O. Binding of SycH chaperone to YscM1 and YscM2 activates effector Yop expression in Yersinia enterocolitica. Journal of Bacteriology. 2004;186:829–841. doi: 10.1128/JB.186.3.829-841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier X, Oswald E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 β-Lactamase as a new fluorescence-based reporter. Journal of Bacteriology. 2004;186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Anderson DM. Expression hierarchy in the Yersinia type III secretion system established through YopD recognition of RNA. Mol Microbiol. 2011;80:966–980. doi: 10.1111/j.1365-2958.2011.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Schneewind O. Yersinia enterocolitica TyeA, an intracellular regulator of the type III machinery, is required for specific targeting of YopE, YopH, YopM, and YopN into the cytosol of eukaryotic cells. J Bacteriol. 2000;182:3183–3190. doi: 10.1128/jb.182.11.3183-3190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LW, Kay O, Schneewind O. Regulated secretion of YopN by the type III machinery of Yersinia enterocolitica. Journal of Bacteriology. 2001;183:5293–5301. doi: 10.1128/JB.183.18.5293-5301.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis G. The Yersinia Ysc-Yop virulence apparatus. Int J Med Microbiol. 2002a;291:455–462. doi: 10.1078/1438-4221-00153. [DOI] [PubMed] [Google Scholar]
- Cornelis G, Sory MP, Laroche Y, Derclaye I. Genetic analysis of the plasmid region controlling virulence in Yersinia enterocolitica 0:9 by Mini-Mu insertions and lac gene fusions. Microbial Pathogenesis. 1986;1:349–359. doi: 10.1016/0882-4010(86)90067-7. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. Yersinia type III secretion: send in the effectors. J Cell Biol. 2002b;158:401–408. doi: 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis GR, Wolf-Watz H. The Yersinia Yop virulon: a bacterial system for subverting eukaryotic cells. Mol Microbiol. 1997;23:861–867. doi: 10.1046/j.1365-2958.1997.2731623.x. [DOI] [PubMed] [Google Scholar]
- Crabill E, Karpisek A, Alfano JR. The Pseudomonas syringae HrpJ protein controls the secretion of type III translocator proteins and has a virulence role inside plant cells. Mol Microbiol. 2012;85:225–238. doi: 10.1111/j.1365-2958.2012.08097.x. [DOI] [PubMed] [Google Scholar]
- Davis AJ, De Jesús Díaz DA, Mecsas J. A dominant-negative needle mutant blocks type III secretion of early but not late substrates in Yersinia. Molecular Microbiology. 2010;76:236–259. doi: 10.1111/j.1365-2958.2010.07096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Mecsas J. Mutations in the Yersinia pseudotuberculosis type III secretion system needle protein, YscF, that specifically abrogate effector translocation into host cells. Journal of Bacteriology. 2007;189:83–97. doi: 10.1128/JB.01396-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JB, Ferracci F, Plano GV. Translocation of YopE and YopN into eukaryotic cells by Yersinia pestis yopN, tyeA, sycN, yscB and lcrG deletion mutants measured using a phosphorylatable peptide tag and phosphospecific antibodies. Molecular Microbiology. 2003;47:807–823. doi: 10.1046/j.1365-2958.2003.03343.x. [DOI] [PubMed] [Google Scholar]
- Day JB, Plano GV. A complex composed of SycN and YscB functions as a specific chaperone for YopN in Yersinia pestis. Molecular Microbiology. 1998;30:777–788. doi: 10.1046/j.1365-2958.1998.01110.x. [DOI] [PubMed] [Google Scholar]
- Dewoody R, Merritt PM, Houppert AS, Marketon MM. YopK regulates the Yersinia pestis type III secretion system from within host cells. Molecular Microbiology. 2011;79:1445–1461. doi: 10.1111/j.1365-2958.2011.07534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepold A, Amstutz M, Abel S, Sorg I, Jenal U, Cornelis GR. Deciphering the assembly of the Yersinia type III secretion injectisome. EMBO J. 2010 doi: 10.1038/emboj.2010.84. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepold A, Wiesand U, Amstutz M, Cornelis GR. Assembly of the Yersinia injectisome: the missing pieces. Molecular Microbiology. 2012 doi: 10.1111/j.1365-2958.2012.08146.x. no-no. [DOI] [PubMed] [Google Scholar]
- Diepold A, Wiesand U, Cornelis GR. The assembly of the export apparatus (YscR,S,T,U,V) of the Yersinia type III secretion apparatus occurs independently of other structural components and involves the formation of an YscV oligomer. Molecular Microbiology. 2011;82:502–514. doi: 10.1111/j.1365-2958.2011.07830.x. [DOI] [PubMed] [Google Scholar]
- Edqvist PJ, Olsson J, Lavander M, Sundberg L, Forsberg Å, Wolf-Watz H, Lloyd SA. YscP and YscU regulate substrate specificity of the Yersinia type III secretion system. Journal of Bacteriology. 2003;185:2259–2266. doi: 10.1128/JB.185.7.2259-2266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt M, Namba K, Hughes KT. Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harbor Perspectives in Biology. 2010:2. doi: 10.1101/cshperspect.a000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracci F, Day JB, Ezelle HJ, Plano GV. Expression of a functional secreted YopN-TyeA hybrid protein in Yersinia pestis is the result of a +1 translational frameshift event. Journal of Bacteriology. 2004;186:5160–5166. doi: 10.1128/JB.186.15.5160-5166.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracci F, Schubot FD, Waugh DS, Plano GV. Selection and characterization of Yersinia pestis YopN mutants that constitutively block Yop secretion. Molecular Microbiology. 2005;57:970–987. doi: 10.1111/j.1365-2958.2005.04738.x. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Viitanen AM, Skurnik M, Wolf-Watz H. The surface-located YopN protein is involved in calcium signal transduction in Yersinia pseudotuberculosis. Mol Microbiol. 1991;5:977–986. doi: 10.1111/j.1365-2958.1991.tb00773.x. [DOI] [PubMed] [Google Scholar]
- Francis MS, Aili M, Wiklund ML, Wolf-Watz H. A study of the YopD-lcrH interaction from Yersinia pseudotuberculosis reveals a role for hydrophobic residues within the amphipathic domain of YopD. Mol Microbiol. 2000;38:85–102. doi: 10.1046/j.1365-2958.2000.02112.x. [DOI] [PubMed] [Google Scholar]
- Francis MS, Wolf-Watz H. YopD of Yersinia pseudotuberculosis is translocated into the cytosol of HeLa epithelial cells: evidence of a structural domain necessary for translocation. Molecular Microbiology. 1998;29:799–813. doi: 10.1046/j.1365-2958.1998.00973.x. [DOI] [PubMed] [Google Scholar]
- Garcia JT, Ferracci F, Jackson MW, Joseph SS, Pattis I, Plano LRW, Fischer W, Plano GV. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect Immun. 2006;74:5645–5657. doi: 10.1128/IAI.00690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goure J, Broz P, Attree O, Cornelis GR, Attree I. Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. Journal of Infectious Diseases. 2005;192:218–225. doi: 10.1086/430932. [DOI] [PubMed] [Google Scholar]
- Hoiczyk E, Blobel G. Polymerization of a single protein of the pathogen Yersinia enterocolitica into needles punctures eukaryotic cells. Proceedings of the National Academy of Sciences. 2001;98:4669–4674. doi: 10.1073/pnas.071065798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom A, Pettersson J, Rosqvist R, Hakansson S, Tafazoli F, Fallman M, Magnusson KE, Wolf-Watz H, Forsberg A. YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol Microbiol. 1997;24:73–91. doi: 10.1046/j.1365-2958.1997.3211681.x. [DOI] [PubMed] [Google Scholar]
- Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A. Virulence plasmid-encoded YopK is essential for Yersinia pseudotuberculosis to cause systemic infection in mice. Infect Immun. 1995a;63:2269–2276. doi: 10.1128/iai.63.6.2269-2276.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A. YopK, a novel virulence determenant of Yersinia pseudotuberculosis. Contrib Microbiol Immunol. 1995b;13:239–243. [PubMed] [Google Scholar]
- Houppert AS, Kwiatkowski E, Glass EM, DeBord KL, Merritt PM, Schneewind O, Marketon MM. Identification of chromosomal genes in Yersinia pestis that influence type III secretion and delivery of Yops into target cells. PLoS ONE. 2012;7:e34039. doi: 10.1371/journal.pone.0034039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriarte M, Sory M-P, Boland A, Boyd AP, Mills SD, Lambermont I, Cornelis GR. TyeA, a protein involved in control of Yop release and in translocation of Yersinia Yop effectors. EMBO J. 1998;17:1907–1918. doi: 10.1093/emboj/17.7.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, Barnes P. Subversion of integrins by enteropathogenic. Yersinia Journal of Cell Science. 2001;114:21–28. doi: 10.1242/jcs.114.1.21. [DOI] [PubMed] [Google Scholar]
- Jackson MW, Day JB, Plano GV. YscB of Yersinia pestis functions as a specific chaperone for YopN. Journal of Bacteriology. 1998;180:4912–4921. doi: 10.1128/jb.180.18.4912-4921.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SS, Plano GV. Identification of TyeA residues required to interact with YopN and to regulate Yop secretion. Adv Exp Med Biol. 2007;603:235–245. doi: 10.1007/978-0-387-72124-8_21. [DOI] [PubMed] [Google Scholar]
- Lee VT, Anderson DM, Schneewind O. Targeting of Yersinia Yop proteins into the cytosol of HeLa cells: one-step translocation of YopE across bacterial and eukaryotic membranes is dependent on SycE chaperone. Mol Microbiol. 1998;28:593–601. doi: 10.1046/j.1365-2958.1998.00822.x. [DOI] [PubMed] [Google Scholar]
- Lee VT, Mazmanian SK, Schneewind O. A program of Yersinia enterocolitica type III secretion reactions is activated by specific signals. Journal of Bacteriology. 2001;183:4970–4978. doi: 10.1128/JB.183.17.4970-4978.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. Plague bacteria target immune cells during infecion. Science. 2005;309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlovits TC, Kubori T, Sukhan A, Thomas DR, Galán JE, Unger VM. Structural insights into the assembly of the type III secretion needle complex. Science. 2004;306:1040–1042. doi: 10.1126/science.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Argudo I, Blocker AJ. The Shigella T3SS needle transmits a signal for MxiC release, which controls secretion of effectors. Mol Microbiol. 2010;78:1365–1378. doi: 10.1111/j.1365-2958.2010.07413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia E, Bliska JB, Viboud GI. Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog. 2008;4:e3. doi: 10.1371/journal.ppat.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels T, Wattiau P, Brasseur R, Ruysschaert JM, Cornelis G. Secretion of Yop proteins by. Yersiniae Infection and Immunity. 1990;58:2840–2849. doi: 10.1128/iai.58.9.2840-2849.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills SD, Boland A, Sory MP, Van Der Smissen P, Kerbourch C, Finlay BB, Cornelis GR. YopP, a novel Yersinia effector protein, is delivered into macrophages to induce apoptosis. Proc Natl Acad Sci USA. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Mecsas J, Ghori N, Falkow S. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner C, Arquint C, Cornelis GR. Translocators YopB and YopD from Yersinia enterocolitica form a multimeric integral membrane complex in eukaryotic cell membranes. Journal of Bacteriology. 2011;193:6923–6928. doi: 10.1128/JB.05555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller CA, Broz P, Müller SA, Ringler P, Erne-Brand F, Sorg I, Kuhn M, Engel A, Cornelis GR. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science. 2005;310:674–676. doi: 10.1126/science.1118476. [DOI] [PubMed] [Google Scholar]
- Neyt C, Cornelis GR. Role of SycD, the chaperone of the Yersinia Yop translocators YopB and YopD. Mol Microbiol. 1999;31:143–156. doi: 10.1046/j.1365-2958.1999.01154.x. [DOI] [PubMed] [Google Scholar]
- Orth K. Function of the Yersinia effector YopJ. Curr Opin Microbiol. 2002;5:38–43. doi: 10.1016/s1369-5274(02)00283-7. [DOI] [PubMed] [Google Scholar]
- Park H, Teja K, O’Shea JJ, Siegel RM. The Yersinia Effector Protein YpkA Induces Apoptosis Independently of Actin Depolymerization. J Immunol. 2007;178:6426–6434. doi: 10.4049/jimmunol.178.10.6426. [DOI] [PubMed] [Google Scholar]
- Persson C, Nordfelth R, Holmstrom A, Hakansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995;18:135–150. doi: 10.1111/j.1365-2958.1995.mmi_18010135.x. [DOI] [PubMed] [Google Scholar]
- Peters KN, Anderson DM. Modulation of host cell death pathways by Yersinia species and the type III effector YopK. In: de Almeida AMP, Leal NC, editors. Advances in Yersinia Research. Springer; New York: 2012. pp. 229–236. [DOI] [PubMed] [Google Scholar]
- Pettersson J, Roland N, Dubinina E, Bergman T, Gustafsson M, Magnusson KE, Wolf-Watz H. Modulation of virulence factor expression by pathogen target cell contact. Science. 1996;273:1231–1233. doi: 10.1126/science.273.5279.1231. [DOI] [PubMed] [Google Scholar]
- Philippe N, Alcaraz J-P, Coursange E, Geiselmann J, Schneider D. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid. 2004;51:246–255. doi: 10.1016/j.plasmid.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Ramsby M, Makowski G. Differential Detergent Fractionation of eukaryotic cells. In: Walker Handbook J., editor. The Proteomics Protocols. Totowa, NJ: Humana Press Inc; 2005. [DOI] [PubMed] [Google Scholar]
- Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors. Journal of Biological Chemistry. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- Rimpiläinen M, Forsberg A, Wolf-Watz H. A novel protein, LcrQ, involved in the low-calcium response of Yersinia pseudotuberculosis shows extensive homology to YopH. Journal of Bacteriology. 1992;174:3355–3363. doi: 10.1128/jb.174.10.3355-3363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosqvist R, Forsberg A, Rimpilainen M, Bergman T, Wolf-Watz H. The cytotoxic protein YopE of Yersinia obstructs the primary host defence. Mol Microbiol. 1990;4:657–667. doi: 10.1111/j.1365-2958.1990.tb00635.x. [DOI] [PubMed] [Google Scholar]
- Rosqvist R, Forsberg A, Wolf-Watz H. Intracellular targeting of the Yersinia YopE cytotoxin in mammalian cells induces actin microfilament disruption. Infect Immun. 1991;59:4562–4569. doi: 10.1128/iai.59.12.4562-4569.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosqvist R, Magnusson KE, Wolf-Watz H. Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. EMBO J. 1994;13:964–972. doi: 10.1002/j.1460-2075.1994.tb06341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubot FD, Jackson MW, Penrose KJ, Cherry S, Tropea JE, Plano GV, Waugh DS. Three-dimensional structure of a macromolecular assembly that regulates type III secretion in. Yersinia pestis Journal of Molecular Biology. 2005;346:1147–1161. doi: 10.1016/j.jmb.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Shao F, Vacratsis PO, Bao Z, Bowers KE, Fierke CA, Dixon JE. Biochemical characterization of the Yersinia YopT protease: Cleavage site and recognition elements in Rho GTPases. Proceedings of the National Academy of Sciences. 2003;100:904–909. doi: 10.1073/pnas.252770599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde OA, Goguen JD. Genetic analysis of the 9.5-kilobase virulence plasmid of Yersinia pestis. Infect Immun. 1988;56:2743–2748. doi: 10.1128/iai.56.10.2743-2748.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde OA, Sample AK, Brubaker RR, Goguen JD. Plasminogen activator/coagulase gene of Yersinia pestis is responsible for degradation of plasmid-encoded outer membrane proteins. Infect Immun. 1988;56:2749–2752. doi: 10.1128/iai.56.10.2749-2752.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg JA, Miller NC, Marketon MM, Schneewind O. Rejection of impassable substrates by Yersinia type III secretion machines. Journal of Bacteriology. 2005;187:7090–7102. doi: 10.1128/JB.187.20.7090-7102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Bowmer WS. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect Immun. 1986;51:445–454. doi: 10.1128/iai.51.2.445-454.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Cibull ML. Differential clearance and host-pathogen interactions of YopE-and YopK-YopL- Yersinia pestis in BALB/c mice. Infect Immun. 1989;57:1200–1210. doi: 10.1128/iai.57.4.1200-1210.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Plano GV, Skrzypek E, Haddix PL, Fields KA. Regulation by Ca2+ in the Yersinia low-Ca2+ response. Molecular Microbiology. 1993;8:1005–1010. doi: 10.1111/j.1365-2958.1993.tb01644.x. [DOI] [PubMed] [Google Scholar]
- Sundberg L, Forsberg Å. TyeA of Yersinia pseudotuberculosis is involved in regulation of Yop expression and is required for polarized translocation of Yop effectors. Cellular Microbiology. 2003;5:187–202. doi: 10.1046/j.1462-5822.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- Thorslund SE, Edgren T, Pettersson J, Nordfelth R, Sellin ME, Ivanova E, Francis MS, Isaksson EL, Wolf-Watz H, Fallman M. The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS ONE. 2011;6:e16784. doi: 10.1371/journal.pone.0016784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Telepnev MV, Schmidt G, Aktories K, Wolf-Watz H, Rosqvist R. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol Microbiol. 2000;36:737–748. doi: 10.1046/j.1365-2958.2000.01898.x. [DOI] [PubMed] [Google Scholar]
- Wattiau P, Bernier B, Deslee P, Michiels T, Cornelis GR. Individual chaperones required for Yop secretion by Yersinia. Proc Natl Acad Sci USA. 1994;91:10493–10497. doi: 10.1073/pnas.91.22.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AW, Straley SC. YopD of Yersinia pestis plays a role in negative regulation of the low-calcium response in addition to its role in translocation of Yops. J Bacteriol. 1998;180:350–358. doi: 10.1128/jb.180.2.350-358.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff-Strobel CR, Williams AW, Straley SC. LcrQ and SycH function together at the Ysc type III secretion system in Yersinia pestis to impose a hierarchy of secretion. Molecular Microbiology. 2002;43:411–423. doi: 10.1046/j.1365-2958.2002.02752.x. [DOI] [PubMed] [Google Scholar]
- Yother J, Goguen JD. Isolation and characterization of Ca2+-blind mutants of. Yersinia pestis Journal of Bacteriology. 1985;164:704–711. doi: 10.1128/jb.164.2.704-711.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, McGourty JK, Liu M, Unsworth KE, Holden DW. pH sensing by intracellular Salmonella induces effector translocation. Science. 2010;328:1040–1043. doi: 10.1126/science.1189000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.