

Abstract
Cholinergic neurons in the laterodorsal tegmental (LDT) and peduncolopontine tegmental (PPT) nuclei regulate reward, arousal, and sensory gating via major projections to midbrain dopamine regions, the thalamus, and pontine targets. Muscarinic acetylcholine receptors (mAChRs) on LDT neurons produce a membrane hyperpolarization and inhibit spike-evoked Ca2+ transients. Pharmacological studies suggest M2 mAChRs are involved, but the role of these and other localized mAChRs (M1--M4) has not been definitively tested. To identify the underlying receptors and to circumvent the limited receptor selectivity of available mAChR ligands, we used light- and electron-immunomicroscopy and whole cell recording with Ca2+ imaging in brain slices from knockout mice constitutively lacking either M2, M4, or both mAChRs. Immunomicroscopy findings support a role for M2 mAChRs, since cholinergic and noncholinergic LDT and pedunculopontine tegmental neurons contain M2-specific immunoreactivity. However, whole cell recording revealed that the presence of either M2 or M4 mAChRs was sufficient, and that the presence of at least one of these receptors was required for these carbachol actions. Moreover, in the absence of M2 and M4 mAChRs, carbachol elicited both direct excitation and barrages of spontaneous excitatory postsynaptic potentials (sEPSPs) in cholinergic LDT neurons mediated by M1 and/or M3 mAChRs. Focal carbachol application to surgically reduced slices suggest that local glutamatergic neurons are a source of these sEPSPs. Finally, neither direct nor indirect excitation were knockout artifacts, since each was detected in wild-type slices, although sEPSP barrages were delayed, suggesting M2 and M4 receptors normally delay excitation of glutamatergic inputs. Collectively, our findings indicate that multiple mAChRs coordinate cholinergic outflow from the LDT in an unexpectedly complex manner. An intriguing possibility is that a local circuit transforms LDT muscarinic inputs from a negative feedback signal for transient inputs into positive feedback for persistent inputs to facilitate different firing patterns across behavioral states.
Keywords: muscarinic receptors, GIRK channels, R-type Ca2+ channels, cholinergic neural pathways, brain slice, sleep, arousal, motivation, ascending arousal system
neurons of the laterodorsal tegmental (LDT) and pedunculopontine tegmental (PPT) nuclei have been implicated in numerous functions, including the regulation of motivation and reward (Blaha et al. 1996; Forster and Blaha 2000; Lodge and Grace 2006), arousal and rapid eye movement (REM) sleep (for review, see Jones 2005) and sensory gating (Bosch and Schmid 2008). These functions are mediated, in part, by cholinergic LDT and PPT neurons which project extensively to midbrain dopamine areas (Beninato and Spencer 1988; Oakman et al. 1995; Omelchenko and Sesack 2006; Woolf and Butcher 1986), to brain stem nuclei, including the reticular formation (Satoh and Fibiger 1986; Schofield et al. 2011; Semba and Fibiger 1992; Semba et al. 1990) and to the thalamus (Hallanger and Wainer 1988; Pare et al. 1988; Steriade et al. 1988).
These cholinergic neurons appear regulated by cholinergic afferents (Semba and Fibiger, 1992; Surkis et al., 1996) which may provide feedback signals supporting their firing patterns across behavioral state (see discussion). These signals are likely to be complex, since cholinergic analogs activate both ionotropic nicotinic acetylcholine (ACh) receptors (nAChRs) and G protein-coupled muscarinic ACh receptors (mAChRs) to produce a myriad of pre- and postsynaptic actions in the LDT and PPT (Good et al. 2007; Ishibashi et al. 2009; Kohlmeier and Leonard 2006; Leonard and Llinás 1994; Luebke et al. 1993; Ye et al. 2010), as in basal forebrain cholinergic neurons (Khateb et al. 1997). In LDT cholinergic neurons, muscarinic receptors mediate at least two distinct postsynaptic actions, a membrane hyperpolarization via activation of G protein-coupled inwardly rectifying potassium (GIRK) channels (Kohlmeier and Leonard 2006; Leonard and Llinás 1994; Luebke et al. 1993) and a reduction in spike-evoked Ca2+ transients (SpECTs) via inhibition of R-type Ca2+-channels (Kohlmeier and Leonard 2006). Such complexity underscores the necessity to identify the underlying receptors and interconnections to better understand how these signals interact.
Five mammalian mAChR genes (M1-M5) have been identified that give rise to receptors forming two classes based on G protein coupling: M2 and M4 mAChRs have selectivity for Gi proteins, while M1, M3, and M5 mAChRs have selectivity for Gq proteins (for review, see Caulfield 1993; Wess 1996). These receptors are distributed throughout the brain and body (for review, see Levey 1993; Nathanson 2008) where they mediate a vast number of functions. In situ hybridization indicates M2, M3, and M4 mAChR mRNAs codistribute with cholinergic neurons in the LDT and PPT (Sugaya et al. 1997; Vilaro et al. 1994; Vilaro et al. 1992). Receptor autoradiography suggests M1, M2, and M3 mAChRs are present in the LDT, but only M2 and M3 sites were detected in the PPT (Baghdoyan 1997). Studies using subtype-specific antibodies have noted the presence of M2 immunoreactivity in these regions (Brischoux et al. 2008; Levey 1993; Levey et al. 1991), but definitive double-labeling studies are lacking.
Pharmacological studies using brain slices are consistent with M2 mAChRs mediating the membrane hyperpolarization in LDT and PPT neurons (Leonard and Llinás 1994; Luebke et al. 1993; Ye et al. 2010), while the mAChRs mediating inhibition of the SpECTs have not been studied. Moreover, in vivo measures of ACh release in pontine targets of LDT and PPT are consistent with M2 and/or M4 mAChRs acting as inhibitory presynaptic autoreceptors (Baghdoyan et al. 1998; Coleman et al. 2004a; Roth et al. 1996). Nevertheless, it is well recognized that pharmacological approaches are hampered by available ligands showing only limited receptor subtype selectivity (for review, see Caulfield and Birdsall 1998; Eglen et al. 2001) and the presence of multiple mAChR subtypes in most brain regions. A more promising approach has been to utilize mice engineered to be lacking one or more mAChRs (for review, see Wess 2004; Wess et al. 2007). Accordingly, we utilized light and electron microscopy immunocytochemistry and whole cell recordings in brain slices from wild-type (WT) mice and mice constitutively lacking M2, M4, or both M2 and M4 mAChRs to definitively test the role of these receptors in mediating the muscarinic membrane hyperpolarization and inhibition of SpECTs in LDT neurons.
METHODS
Ethical approval.
All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of New York Medical College and were compliant with National Institutes of Health guidelines for ethical treatment of animals.
Mice.
Mice constitutively lacking either the M2 (M2−/− mice), M4 (M4−/− mice), or both mAChRs (M2M4−/− mice) were used in this study. Generation of these mice has been described previously (Duttaroy et al. 2002; Gomeza et al. 1999a; Gomeza et al. 1999b), and all strains were backcrossed for >10 generations onto the C57BL/6NTac background (Taconic Farms, Germantown, NY). WT mice used for control were obtained from Taconic Farms and were of the same C57BL/6NTac genetic background.
Immunocytochemistry for light and electron microscopy.
WT (n = 8) mice and M2−/− (n = 2) mice were perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer (PB). The brain was then removed, and 50-μm-thick sections were cut using a vibratome. Adjacent sections of WT and M2−/− mice were incubated for 30 min in 10% normal goat serum (NGS) or normal rabbit serum (NRS) in phosphate-buffered saline (PBS) and then transferred into solutions of the following antibodies diluted in 1% normal goat serum or normal rabbit serum (in PBS) for overnight incubation: goat-anti-choline acetyltransferase (ChAT) (Chemicon AB144P; 1:300), rabbit-anti-neuronal nitric oxide synthase (nNOS) (Diasorin 24287; 1:8,000), mouse-anti-nNOS (Sigma N2280; 1:2,000), rat-anti-M2 (Chemicon MAB367; 1:1,000), or rabbit-anti-M1 (Sigma M9808; 1:500). The following day the sections were rinsed in PB and incubated for 1 h in a 1:100 dilution of one of the following antibodies (all from Vector Laboratories): biotinylated-goat-anti-mouse, biotinylated-goat-anti-rabbit, biotinylated-goat-anti-rat, or biotinylated-rabbit-anti-goat. The sections were then rinsed in PB and incubated for 1 h in a 1:100 dilution of avidin and biotinylated horseradish peroxidase (ABC kit, Vector), rinsed in PB, and reacted with a nickel-enhanced diaminobenzidine solution. Sections were subsequently mounted on slides and coverslipped for light microscopy, or embedded for electron microscopy as described below.
For electron microscopy, sections were postfixed in osmium tetroxide, dehydrated with an alcohol series, and flat embedded in Durcupan resin between two sheets of Aclar plastic. Embedded sections were examined using a light microscope, and selected areas of the LDT were mounted on resin blocks. Thin sections (∼70-nm thick) were cut using an ultramicrotome, collected on Formvar-coated slot grids, and stained with uranyl acetate. Sections were examined using an electron microscope, and images were captured using a digital camera.
Immunocytochemistry for confocal microscopy.
Sections from WT mice were incubated in the following combinations of antibodies: rat-anti-M2 (1:1,000), and goat-anti-ChAT (1:300), rat-anti-M2 (1:1,000), and rabbit-anti-nNOS (1:5,000), or rabbit-anti-M1 (1:250) and mouse-anti-nNOS (1:1,000). The following day the sections were rinsed in PB and incubated for 1 h in 1:100 dilutions of rabbit-anti-rat-Alexa 488 and rabbit-anti-goat-Alexa-546, goat-anti-rat-Alexa-488 and goat-anti-rabbit-Alexa-546 or goat-anti-rabbit-Alexa 488 and goat-anti-mouse Alexa-546 (all from Molecular Probes). The sections were then rinsed in PB, mounted on slides, and coverslipped for viewing using a confocal microscope.
Slice preparation and artificial cerebrospinal fluid.
Brain slices were prepared from 12- to 30-day-old mice. Slices were typically prepared from a single mouse each recording day, and recordings from each genotype were interleaved to reduce any effect of drift in experimental conditions over time. Animals were decapitated following induction of deep anesthesia with isofluorane. A block of the brain containing the LDT was rapidly removed and incubated in ice-cold artificial cerebrospinal fluid (ACSF), which contained (in mM): NaCl, 121; KCl, 5; NaH2PO4, 1.2; CaCl2, 2.7; MgSO4, 1.2; NaHCO3, 26; dextrose, 20; and oxygenated by bubbling with carbon (95% O2, 5% CO2). The brain stem was then blocked in a coronal plane and sectioned at 250 μm on a Leica vibratome (VT1000S). Slices containing the LDT were incubated at 35°C for 15 min and then stored at room temperature. Recordings were obtained from slices submerged in a recording chamber, which was perfused at 2–5 ml/min with continuously oxygenated ACSF at room temperature.
Drugs.
Carbachol (CCh; Sigma) was dissolved in water in 10 mM aliquots and dissolved in ACSF the day of the experiment to a final concentration of 10 μm, unless otherwise noted. CCh was bath applied in the majority of experiments; however, in localization studies, it was “puffed” with a brief pressure pulses (100 ms; Picospritzer II, General Valve) from a patch pipette positioned just above the slice but downstream from the recorded cell at a concentration of 10 mM. This method allowed very local application of drug with a quick wash-in and wash-out time. 5-HT and atropine (Sigma), an irreversible muscarinic receptor antagonist, were applied at a final concentration of 50 μM and 5 μM, respectively. Tertiapin (Peptide Institute) and SNX-482 (Peptides International) were dissolved in ACSF at a stock concentration and dissolved in ACSF the day of the experiment and applied at 10 nM and 100 nM, respectively. The M1 mAChR preferring antagonist pirenzepine (10 μM) and the M4 antagonist tropicamide (1 μM) were obtained from Sigma and diluted in water at 1,000 times the final concentration. The M2 preferring antagonist AF-DX 116 (1 μM; Tocris Bioscience) was dissolved in DMSO at a stock solution 105 times final concentration. Tetrodotoxin (TTX) (Alomone) was dissolved in ACSF to a final concentration of 500 nM to block voltage-gated sodium channels. The ionotropic receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX) (15 μM; Sigma), (2R)-amino-5-phosphonovaleric acid (APV) (50 μM; Sigma), bicuculline (10 μM; Sigma), and strychnine (2.5 μM; Sigma) were added to ACSF in some recordings. For low calcium solution recordings, the Ca2+ concentration of the ACSF was buffered to <20 μM by the addition of 2.7 mM EGTA (calculated with Patcher's Power Tools XOP for Igor Pro).
Whole cell electrophysiological recording and calcium imaging.
Micropipettes (2–4 MΩ) used for patch clamp recordings (Borosilicate, cat. number 8050, AM systems) were pulled on a horizontal puller (Sutter Instruments, P87). Pipettes were filled with a recording solution of either the potassium salt of bis-fura 2 (50 μM; Molecular Probes) dissolved in the recording solution (in mM), 144 potassium-gluconate, 3 MgCl2, 10 HEPES, 0.3 NaGTP, and 4 Na2ATP, or, in those cases where calcium imaging was not being conducted, pipettes were filled with a recording solution containing (in mM) 144 potassium-gluconate, 0.2 EGTA, 3 MgCl2, 10 HEPES, 0.3 NaGTP, and 4 Na2ATP (osmolarity = 310). Inhibitory postsynaptic currents (IPSCs) were recorded with a pipette solution containing high chloride and ACSF with blockers of glutamatergic synaptic transmission to reverse the chloride reversal potential and optimize detection of inhibitory events (in mM): 128.4 KCl, 3 MgCl2, 10 HEPES, 0.3 NaGTP, 4 Na2ATP, 0.2 EGTA.
In all cases, biotinylated Alexa-594 (25 μM; Molecular Probes) was also included in the patch solution so cells could be recovered following slice fixation and histochemically identified. Neurons were visualized for whole cell recordings at ×160 magnification with visible-light, differential interference contrast optics, using a Nuvicon tube camera (Dage VE-1000) mounted on a fixed-stage microscope (Olympus BX50WI). Cells for recording were chosen within the boundary of the LDT nuclei which were identified using a ×4 objective.
Gigaseals were obtained under visual control using an Axopatch 200B amplifier (Molecular Devices) operated in voltage clamp mode and filtered at 2 or 5 kHz with a four-pole Bessel filter at the amplifier output and sampled at 4, 10, or 20 kHz. After establishing the whole cell recording configuration, cells were filled with both the calcium indicator and biotinylated Alexa-594 for Ca2+ imaging by either passive diffusion or brief hyperpolarizing pulses. Data were not collected until at least 10–20 min had passed following break-in to allow dye equilibration. Neurons were imaged through a ×40 water immersion objective (0.8 numerical aperture) using a cooled, charge-coupled device camera equipped with a back illuminated EEV 57 frame transfer chip having an imaging area of 512 × 512 pixels (field size = 160 μm/side; MicroMax, Roper Scientific). Bis-fura 2 was excited at 380 nm with light from a 75-W Xenon lamp that was shuttered to reduce light exposure to the tissue in between data acquisition. Recordings were either conducted in voltage clamp or “I-clamp fast” current clamp mode, following appropriate compensation for the pipette capacitance; quality of the cells was assayed by monitoring the holding current and the input resistance as determined by the voltage or current response to a brief, negative going step. Recordings were uncorrected for liquid junction potentials which were calculated to be ∼12 mV. Current and voltage traces were digitized and command pulses were generated by custom-designed software (TIWB; Inoue et al. 1998) run on a Mac OS computer which controlled an ITC-18 interface (HEKA Instruments). TIWB software also controlled the camera and shutter which allowed precise synchronization between electrophysiological and optical signals. The camera was read out through a 1-MHz, 14-bit A/D converter. Images were binned on the chip at 4 × 4 and acquired every 50 ms, a rate fast enough to monitor changes in calcium accompanying rapid alterations of the membrane potential. Changes in fluorescence (dF/F) were quantified by the average pixel values within regions of interest (ROI) that were positioned on background- and baseline-subtracted fluorescent images. The background was determined from a ROI that was positioned at a location remote from the filled cell or its processes. Baseline fluorescence was determined as the fluorescence measured in the first few frames of each sequence before stimulation. Bis-fura 2 fluorescence decreases when calcium increases and is bound by the dye; however, for purposes of clarity, dF/F responses have been inverted in figures such that positive deflections of the dF/F traces indicate rises in calcium. To measure spike-evoked calcium transients, we measured the dF/F produced by five action potentials triggered by brief current pulse (2 ms) delivered at 8 Hz. dF/F was computed from a ROI positioned over the soma.
Electrophysiological analysis.
All recorded neurons used for further analysis had stable resting potentials and overshooting action potentials when recorded in current clamp mode. When recorded in voltage-clamp mode, neurons were held at −60 mV, and recordings were discarded if more than 50 pA were required to hold cells at this membrane potential. Current and voltage waveforms were analyzed using Igor Pro Software (Wavemetrics). Holding current was measured from 1-s averages of the holding current at −60 mV. Detection of postsynaptic currents (PSCs) and some analyses were done using Mini Analysis software (Synaptosoft, Decatur, GA). To compare the effects of CCh on the frequency and amplitude distributions of PSCs, epochs of at least 30 s were used before and after superfusion of CCh. Cumulative distributions were then compared using Kolmogorov-Smirnov statistics (K-S test) with the level of significance set at values below 0.05.
Additional analysis and figure preparation were conducted using Igor Pro software. Differences between means were compared using a two-sample, two-tailed t-test corrected for multiple comparisons (when necessary) and repeated-measures ANOVA using DataDesk 6 software (Data Description, Ithaca, NY). Numerical results are reported as means ± SE.
Identification of recorded neurons.
For immunoidentification of recorded neurons, slices were fixed overnight in 4% paraformaldehyde at 4°C and then stored in 0.01 M PBS and 30% sucrose for cryoprotection. Slices were then resectioned at 40 μm and incubated with gentle shaking at 4°C for 24 h in the dark with a nNOS antibody (Sigma, catalog no. N7280, rabbit polyclonal 1:400) which is an excellent marker for cholinergic neurons in the LDT (Vincent et al. 1983) followed by incubation with an Alexa 488-conjugated secondary antibody (A11008, goat anti-rabbit, Molecular Probes). Further histochemical processing was not necessary to visualize the Alexa-594 filled cell which was imaged using a Texas-red filter-cube set.
RESULTS
M2 mAChRs are present in cholinergic and noncholinergic LDT neurons.
To clarify whether M2 mAChRs are localized to both cholinergic and noncholinergic LDT and PPT neurons, we utilized light and electron microscopic immunocytochemical methods. Initially, we stained adjacent sections from an M2−/− mouse with antibodies directed against nNOS and ChAT to verify that the expression pattern of ChAT/nNOS remained grossly normal in the LDT and PPT of M2−/− mice. An example of ChAT labeling in the LDT region from an M2−/− mouse is illustrated in Fig. 1A. Staining with an anti-M2 antibody failed to stain LDT or PPT neurons or their processes in tissue obtained from M2−/− mice (Fig. 1C) but revealed distinct somatic and dendritic labeling often associated with the plasma membrane in sections from a WT mouse in the LDT (Fig. 1, B and D) and PPT (not shown). The absence of staining in tissue from the M2−/− mouse and presence of staining in WT tissue indicates that our conditions resulted in specific M2 mAChR labeling.
Fig. 1.
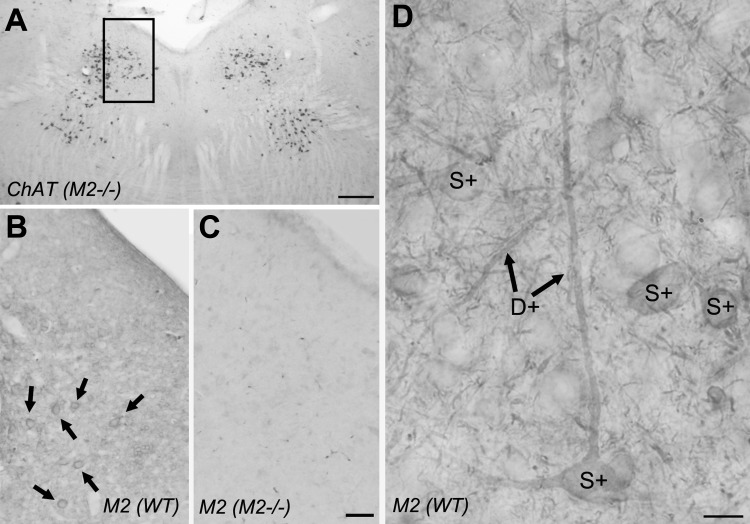
Neurons in the laterodorsal tegmental (LDT) express M2 muscarinic acetylcholine receptors (mAChRs). A: a coronal section through the mouse brain stem of an M2−/− mouse stained with an antibody against choline acetyltransferase (ChAT) reveals a grossly normal distribution of cholinergic neurons in the LDT region. The box indicates the approximate area stained with an antibody against the M2 receptor in sections from wild-type (WT; B) and M2−/− mice (C). In WT mice, the M2 mAChR antibody outlines somata (examples indicated by arrows in B and by “S+” in D) and dendrites (D+). Scale in A = 250 μm, scale in C = 50 μm and also applies to B. Scale in D = 10 μm.
We then examined the colocalization of M2 mAChR immunoreactivity with nNOS immunoreactivity in the LDT from WT mice using confocal microscopy (Fig. 2). This revealed that M2 mAChR immunofluorescence was associated with somatic and dendritic membranes of both nNOS+ (arrows) and nNOS-negative (asterisks) neurons. A similar pattern was seen in tissue stained with antibodies against ChAT and M2 mAChRs (not shown). Thus M2 mAChR are somatodendritic autoreceptors in cholinergic LDT neurons and are also hetero-receptors in noncholinergic LDT neurons.
Fig. 2.
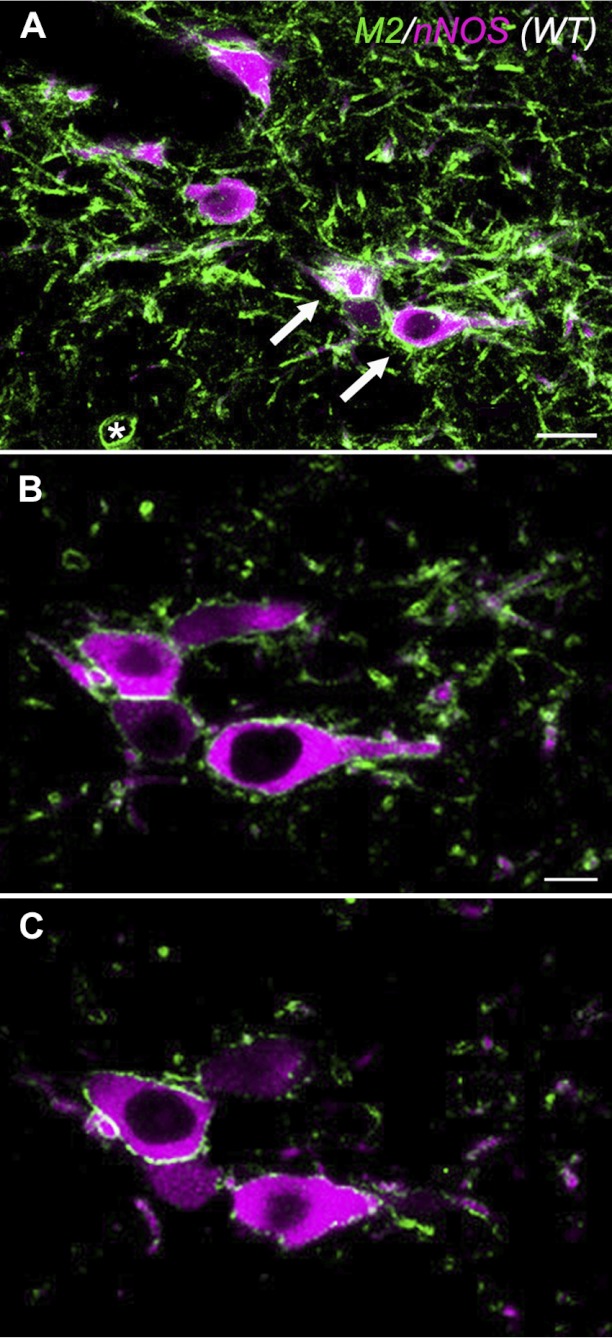
Neuronal nitric oxide synthase (nNOS) neurons in the LDT express M2 mAChRs. Confocal images illustrate LDT cells labeled with antibodies against the M2 mAChR (green) and nNOS (purple). A: a merged stack of optical sections. B and C: single 1-μm optical sections within the stack illustrated in A. M2 mAChRs are expressed on the membranes of nNOS+ cells (arrows) and nNOS− cells (asterisk). Scale bar in A = 20 μm. Scale bar in B = 10 μm and also applies to C.
M2 mAChRs are distributed in pre- and postsynaptic profiles in the LDT.
To confirm the postsynaptic membrane localization of M2 mAChR immunoreactivity, we examined the distribution of label at the ultrastructural level (Fig. 3). This revealed that M2 immunolabel was distributed near the plasma membrane of somata and proximal dendrites (Fig. 3, A and B) as expected from the pattern observed by confocal microscopy and throughout the profiles of smaller dendrites (Fig. 3, C and D). At higher magnification, it was also apparent that M2 immunolabel was present in presynaptic terminals contacting both unlabeled (Fig. 3, E and F) and labeled postsynaptic dendritic profiles (Fig. 3, G and H). Thus M2 mAChRs are also expressed by presynaptic terminals where they could influence the release of neurotransmitter in the LDT.
Fig. 3.
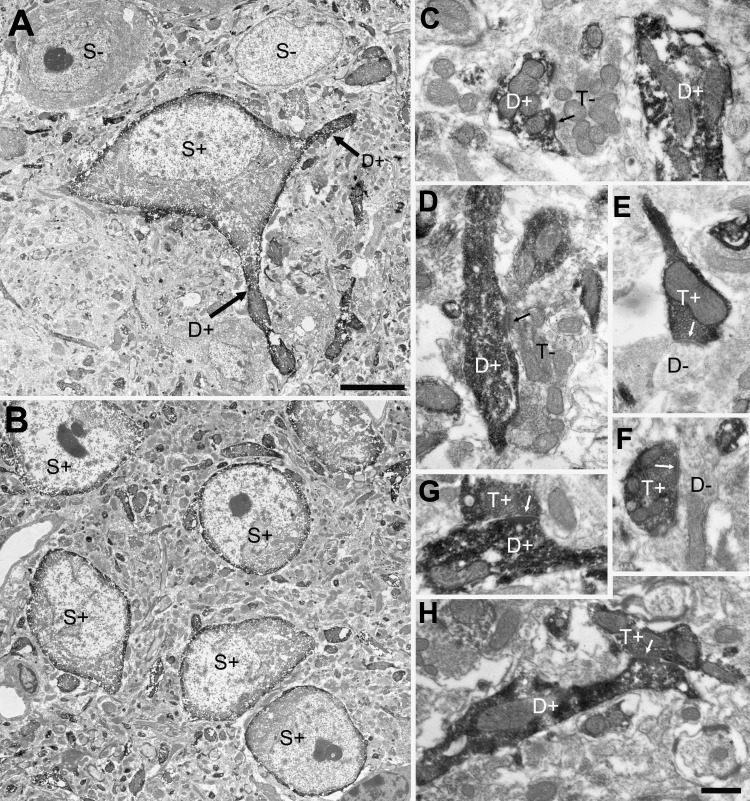
M2 mAChRs are distributed in pre- and postsynaptic profiles in the LDT. Electron micrographs illustrate the distribution of M2 mAChR staining in the LDT. A and B: M2 mAChR staining is distributed near the outer membranes of somata (S+) and proximal dendrites (D+). C and D: M2 mAChR staining is distributed throughout smaller dendrites (D+) that are contacted (arrows) by unlabeled synaptic terminals (T−). E and F: M2 mAChR staining is distributed in synaptic terminals (T+) that contact (arrows) unlabeled dendrites (D−). G and H: M2 mAChR staining is distributed in synaptic terminals (T+) that contact (arrows) labeled dendrites (D+). Scale in A = 5 μm (also applies to B). Scale in H = 0.5 μm (applies to C–G).
Neither the CCh-induced membrane hyperpolarization nor inhibition of SpECTs were blocked in slices from M2−/− or M4−/− mice.
To directly test the possibility that M2 mAChRs mediate the membrane hyperpolarization and inhibition of SpECTs in LDT neurons, we compared the effect of CCh on LDT neurons recorded in brain slices obtained from WT, M2−/−, and M4−/− mice. In slices from WT mice, the average membrane potential of LDT neurons was −62.3 ± 0.4 mV (n = 24). CCh (10 μM) hyperpolarized 71% of the cells by an average of 3.8 ± 0.62 mV with an average input resistance decrease of 55.2 ± 2.3% (n = 17/24; Fig. 4, A1 and D1). SpECTs were decreased in the same cells by 39.8 ± 2.9% (Fig. 4, A2 and D2). These CCh-induced changes were similar to those reported in a previous study using brain slices from a different cohort of C57BL6 mice (Kohlmeier and Leonard 2006). In about 47% of LDT neurons, we found that the hyperpolarization was followed by a depolarization (n = 8/17) which has not been previously reported for LDT neurons. In the remainder of cells, CCh elicited just a depolarization (25%, n = 6/24) or failed to elicit any response (n = 1/24).
Fig. 4.
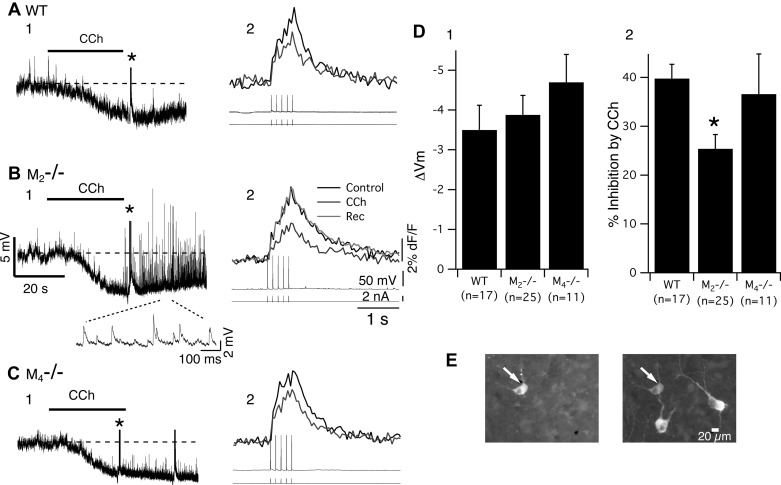
Neither M2 nor M4 mAChRs are necessary for the carbachol (CCh)-mediated membrane hyperpolarization or inhibition of spike-evoked Ca2+ transients (SpECTs) in LDT neurons. A: CCh (10 μM; 90 s) elicited a membrane hyperpolarization, as well as a decrease in SpECTs in cholinergic LDT neurons from WT mice. Similar inhibitory actions were elicited in slices from either M2−/− (B) or M4−/− (C) mice, indicating that neither M2 nor M4 mAChRs alone are necessary for these actions. Asterisks in A1, B1, and C1 mark the 8-Hz train of five spikes producing the SpECTs in A2, B2, and C2. D: histograms detailing CCh-induced hyperpolarization (D1) and decrease in SpECTs (D2) in normal C57, M2−/−, and M4−/− mice. *P < 0.05. E: a representative example of a LDT cell from which recordings in this report were obtained. Copresence of Alexa-594 (arrow, left) with Alexa-488 immunofluorescence for nNOS (arrow, right) indicate that this recorded cell is cholinergic.
In slices from M2−/− mice, the average membrane potential was −61.6 ± 0.6 mV (n = 42) which was not different from that seen in WT slices. CCh hyperpolarized 60% of these cells (n = 25/42) by an average of 3.5 ± 0.49 mV with a concurrent decrease in membrane resistance of 49.8 ± 4.6% (n = 25; Fig. 4, B1 and D1) which was also not different from that seen in WT slices. In cells that hyperpolarized to CCh, SpECTs were inhibited by 25.4 ± 2.9% dF/F (n = 25), which was smaller, on average, than the change seen in cells from WT mice (P < 0.05; Fig. 4D2), but was, nevertheless, still clearly present. Thus, in spite of M2 mAChRs receptors being present in the LDT from WT mice, their absence in the LDT from M2−/− mice did not affect membrane potential, and they were not necessary for either inhibitory action of CCh. Similar to findings utilizing WT slices, 48% of the cells which initially hyperpolarized to CCh also showed a depolarization of 5.1 ± 0.8 mV (12/25) in slices from M2−/− mice. Moreover, a striking barrage of synaptic activity was observed in many cells from these mice, a point examined in more detail later (Fig. 4B1, inset). In the remaining cells, CCh produced only a depolarization (n = 16/42; 38%), while one cell failed to show a membrane potential change.
We next examined the effect of CCh on LDT neurons recorded in slices from M4−/− mice. We found that, in these cells, the average membrane potential was not different from that in WTs (−61.3 ± 1.1 mV; n = 14). When CCh was applied, 78% of these cells hyperpolarized by an average of −4.7 ± 0.65 mV, with a concurrent input resistance decrease of 61.1 ± 6.6% (n = 11/14; Fig. 4, C1 and D1) which was also not different from that seen in WT slices. Similarly, SpECTs decreased in conjunction with the hyperpolarization (reduction of SpECTs in CCh: 36.6 ± 4.5%, n = 11/14, Fig. 4, C2 and D2) to the same extent seen in WT slices. Thus M4 mAChRs are also not necessary for either inhibitory action of CCh in LDT neurons. Additionally, in the absence of M4 mAChRs, one of the responding LDT neurons showed a hyperpolarization followed by depolarization which was a lower percentage than that seen in slices from WT (47%) and M2−/− mice (48%). Moreover, there was an increase in synaptic activity (Fig. 4C1) in some LDT neurons, but it did not appear as pronounced as that in neurons from M2−/− mice. In 21% of the cells, CCh elicited a membrane depolarization (n = 3/14) which was similar to the number encountered in slices from WT and M2−/− mice.
Although only about one-half of the recorded neurons were recovered for histology from these recordings, many were immunoreactive for nNOS (Fig. 4E). Twenty-six cells across the three genotypes were identified as nNOS+, indicating that CCh inhibition, which was not blocked in M2−/− and M4−/− mice, was also not blocked in cholinergic LDT neurons from M2−/− and M4−/− mice.
Effectors in the M2 or M4 single knockouts.
As noted in the Introduction, CCh hyperpolarizes LDT neurons via GIRK channel activation and reduces SpECTs by inhibiting Ca2+ influx through R-type calcium channels. Although CCh responses were qualitatively similar in the single receptor knockouts, we investigated whether the same effectors as observed in WT slices are involved in each knockout. We first confirmed that these CCh actions were mediated by muscarinic receptors. As expected, atropine (5 μM) completely blocked the hyperpolarization, the change in input resistance, and the decrease in SpECTs produced by CCh in both genotypes (Table 1, M2−/−, n = 4; M4−/−, n = 7, P < 0.05).
Table 1.
Antagonist effects on CCh actions in single mAChR knockouts
| Condition | n | Effect on Vm, mV | Effect on Rin, %control | Effect on Spike-Evoked dF/F, %control |
|---|---|---|---|---|
| M2−/− | ||||
| Control | 4 | −2.5 ± 0.8* | −40.7 ± 14.2* | −11.2 ± 5.7* |
| + Atropine | −0.8 ± 0.2 | +2.3 ± 4.1 | −1.0 ± 2.2 | |
| Control | 4 | −2.1 ± 1.4* | −32.3 ± 10.6* | −13.9 ± 5.8* |
| + Tertiapin | +2.0 ± 1.5* | +1.4 ± 12.6 | −19.1 ± 16.9* | |
| Control | 4 | −2.5 ± 0.5* | −47.2 ± 10.8* | −16.3 ± 5.1* |
| + SNX-482 | −2.9 ± 0.9* | −44.6 ± 15.0* | −5.6 ± 3.4 | |
| M4−/− | ||||
| Control | 7 | −5.3 ± 0.9* | −51.9 ± 8.3* | −46.2 ± 1.1* |
| + Atropine | +3.5 ± 3.4 | +3.2 ± 17.2 | −0.8 ± 1.4 | |
| Control | 3 | −5.8 ± 0.7* | −54.8 ± 21.6* | −47.6 ± 2.9* |
| + Tertiapin | −0.8 ± 0.2 | −25.1 ± 7.3* | −33.0 ± 2.6* | |
| Control | 4 | −2.7 ± 0.8* | −47.7 ± 15.2* | −44.9 ± 4.5* |
| + SNX-482 | −2.8 ± 0.7* | −55.2 ± 18.7* | −10.6 ± 4.4 | |
Values are means ± SE; n, no. of neurons. CCh, carbachol; mAChR, Muscarinic acetylcholine receptors; Vm, membrane potential; Rin, input resistance; dF/F, change in fluorescence.
P < 0.05.
Inhibition of GIRK channels by tertiapin (10 nM) significantly reduced membrane hyperpolarization and input resistance changes induced by CCh (Table 1, M2−/−, n = 4; M4−/−, n = 3; P < 0.05 for both), but did not alter the inhibition of SpECTs in slices from either genotype, as expected. Moreover, although significantly reduced from control, a significant decrease in input resistance also remained in M4−/− slices (n = 3, P < 0.05). Often, this decrease in input resistance was followed by a depolarization. Indeed, in slices from both genotypes, CCh resulted in a significant depolarization (Table 1, n = 4, P < 0.05) in ACSF containing tertiapin. The effectiveness of tertiapin indicates that GIRK channels contribute to the membrane hyperpolarization produced by CCh in LDT neurons from M2−/− and M4−/− mice.
As expected from studies in slices from WT mice, SNX-482 significantly reduced CCh inhibition of SpECTs in both genotypes (Table 1; M2−/−, n = 4; M4−/−, n = 4; P < 0.05 for both), but did not prevent the membrane hyperpolarization. This indicates that R-channels are involved with CCh-evoked inhibition of SpECTs in LDT neurons from M2−/− and M4−/− mice.
Collectively, these data indicate that neither the M2 nor M4 mAChR are necessary to produce the GIRK channel-mediated membrane hyperpolarization or inhibition of R-channel-mediated SpECT produced by CCh. Since inhibition of SpECTs was smaller but not abolished in LDT neurons from M2−/− mice, part of this M2 mAChR-mediated inhibition cannot be complemented by remaining mAChRs.
Presence of either M2 or M4 mAChRs is sufficient and necessary to mediate the CCh-induced membrane hyperpolarization and inhibition of SpECTs in LDT neurons.
To determine whether receptors other than M2 and M4 mAChRs can mediate these inhibitory CCh responses, we examined the actions of CCh in slices from M2M4−/− mice. In these slices, CCh failed to elicit a membrane hyperpolarization (n = 17/17; Fig. 5, A1 and C1), a reduction in SpECTs (n = 17/17; Fig. 5, A2 and C2), or when recorded in voltage-clamp mode, an outward current (n = 62/62) in all LDT cells examined, including nNOS+ neurons (Fig. 5B). Moreover, in the absence of these inhibitory actions, CCh elicited a depolarization (3.43 ± 0.6 mV, n = 17/17) or an inward current (−17.27 ± 4.75 pA, n = 58/62; 4/62 failed to respond) in the overwhelming majority of these recordings (Fig. 5C1). CCh also produced an increase in synaptic activity in a majority of these recordings. Thus either M2 or M4 receptors can mediate these inhibitory actions of CCh.
Fig. 5.
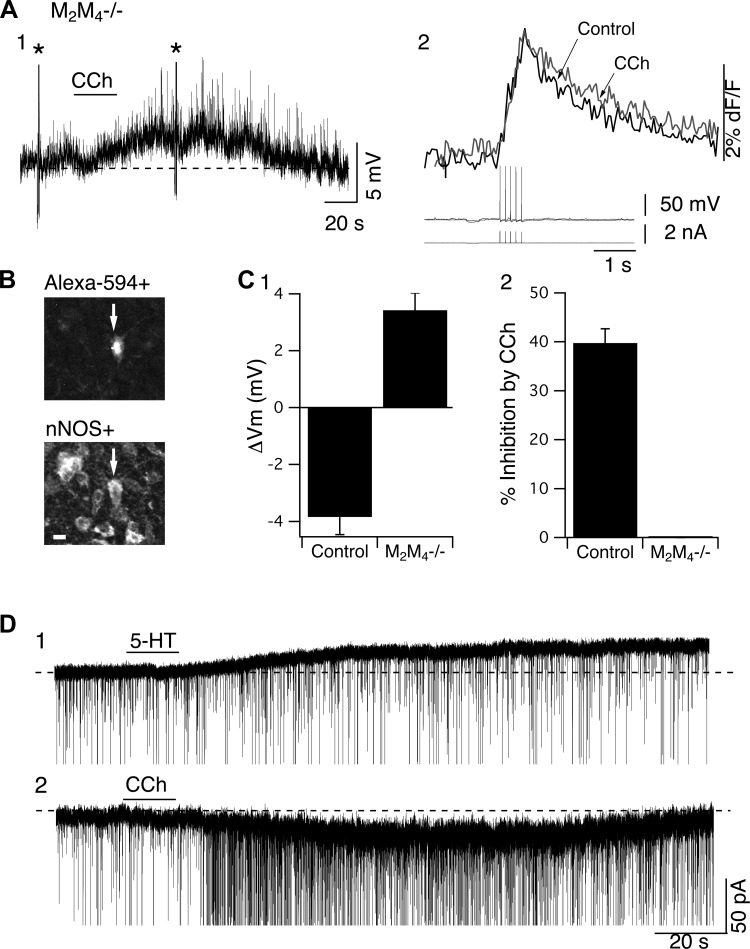
Only M2 and M4 mAChRs mediate the CCh-induced membrane hyperpolarization and inhibition of SpECTs in LDT neurons. In slices from M2M4−/− mice, CCh elicited a depolarization rather than a hyperpolarization (A1) and did not alter SpECTs (A2). Asterisk in A1 marks the 8-Hz train of five spikes producing the SpECTs in A2. B: presence of Alexa 594+ (top) and nNOS+ (bottom) in the cell from which electrophysiological recordings were obtained, as shown in A, indicate that these CCh-induced effects are elicited in cholinergic neurons. Scale: 20 μm. C: bar graphs summarizing the depolarizing action of CCh and the failure of CCh to inhibit SpECTs in neurons recorded in slices from M2M4−/− mice. ΔVm, change in membrane potential. Nevertheless, serotonin (5-HT) elicited a hyperpolarizing current (D1) in the same cells in which CCh was found to subsequently induce a depolarizing current (D2). These data indicate failure of CCh to induce hyperpolarization is not due to alterations in the ability of G protein-coupled inwardly rectifying potassium (GIRK) channels to be activated.
An alternate possibility is that the loss of both M2 and M4 mAChRs impairs the function of the effectors, even if other remaining mAChRs might be competent to activate them. To confirm that this was not the case, we exploited a previous finding from occlusion experiments indicating that CCh and serotonin produce membrane hyperpolarization by action on an overlapping pool of K+ channels in LDT neurons (Leonard and Llinás 1994). We, therefore, tested whether serotonin could still elicit hyperpolarization in LDT neurons from M2M4−/− mice. Serotonin (10 μM) reliably elicited an outward current of 20.1 ± 4.9 pA (n = 3/4; Fig. 5D1) in LDT neurons for which CCh subsequently elicited an inward current (−28.9 ± 9.7 pA, n = 4/4; Fig. 5D2). We conclude that the hyperpolarizing mechanism is still intact in neurons from M2M4−/− mice, but that remaining CCh-sensitive receptors cannot activate these effectors. Thus M2 and M4 mAChRs are necessary and sufficient to mediate membrane hyperpolarization and inhibition of SpECTs in LDT neurons.
CCh stimulates glutamatergic but not GABAergic/glycinergic afferents to LDT neurons in slices from M2M4−/− mice.
The robust barrages of synaptic potentials evoked by CCh in slices from M2M4−/− mice were surprising to us since no such responses were noted in three prior slice studies of CCh actions on LDT neurons (Kohlmeier and Leonard 2006; Leonard and Llinás 1994; Luebke et al. 1993). We, therefore, thought it was important to study the nature of these synaptic potentials. We first applied antagonists of ionotropic GABA/glycine receptors or ionotropic glutamate receptors to block spontaneous IPSCs (sIPSCs) or spontaneous excitatory postsynaptic currents (sEPSCs) respectively, under voltage clamp conditions. For cells in which CCh elicited synaptic activity in addition to inward current, CCh was reapplied following superfusion with ACSF containing either bicuculline and strychnine, or APV and DNQX. In blockers of GABA/glycine transmission, the slow inward current evoked by CCh was not changed (control: −21.3 ± 8.9 pA; strychnine and bicuculline: −19.3 ± 7.8 pA, P > 0.05, n = 7), and the increase in synaptic activity appeared similar to that in control conditions (Fig. 6A1). However, application of APV and DNQX essentially abolished the remaining synaptic activity and its increase to CCh while the amplitude of the slow inward current was not affected (control: −25.6 ± 9.8 pA, DNQX and APV: −21.3 ± 9.6 pA, P > 0.05, n = 10 Fig. 6, A2 and A3). We quantified the effect of CCh on these sEPSCs in seven cells (30-s epochs selected before and after CCh application) in which CCh elicited both inward current and synaptic activity in ACSF containing bicuculline and strychnine (Fig. 6B). Under these conditions, CCh increased the number of events and shifted the interval distribution to shorter values in each cell (K-S test, P < 0.05, n = 7), and the mean interval across cells was significantly shortened (control: 217.9 ± 59.9 ms; CCh: 46.5 ± 11.6 ms; Fig. 6B). CCh also slightly increased the amplitude of sEPSCs in just over one-half of these cells, (K-S test, P < 0.05 n = 4/7; Fig. 6B).
Fig. 6.
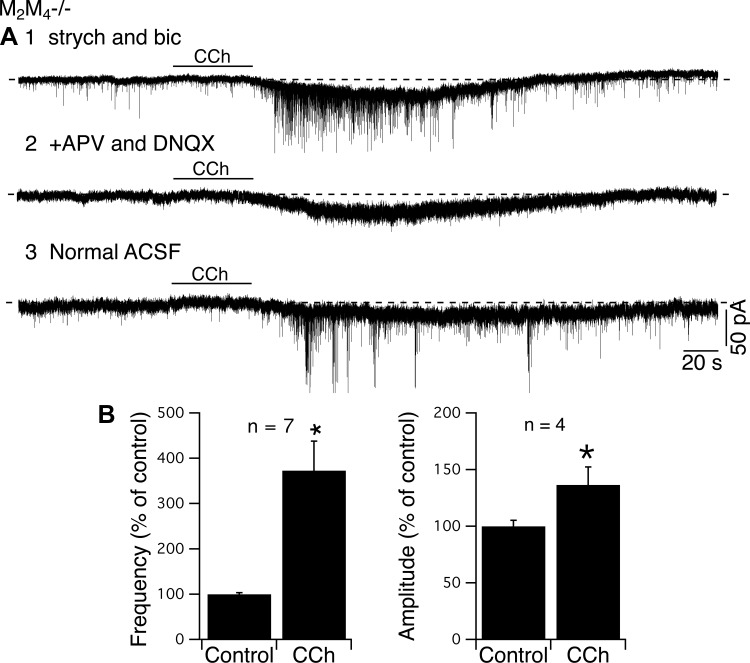
CCh induces excitatory synaptic activity [spontaneous excitatory postsynaptic currents (sEPSCs)] and does not induce inhibitory [spontaneous inhibitory postsynaptic currents (sIPSCs)] synaptic activity in slices from M2M4−/− mice. A1: application of strychnine and bicuculline did not block CCh-induced inward currents or postsynaptic currecnts (PSCs). A2: however, adding (2R)-amino-5-phosphonovaleric acid (APV) and 6,7-dinitroquinoxaline-2,3-dione (DNQX) in the artificial cerebrospinal fluid (ACSF) blocked CCh-induced PSC activity. A3: partial recovery following washout of APV and DNQX. B: summary graph of effects of CCh on frequency and amplitude of excitatory postsynaptic currents (EPSCs). *P < 0.05.
To better examine possible actions of CCh on sIPSCs, we also isolated them by blocking excitatory postsynaptic currents (EPSCs) with APV and DNQX and by using a high Cl− internal solution to make the IPSCs large and inward. Under these conditions, CCh induced a slow inward current but produced only nonsignificant changes (P > 0.05, n = 6) in the mean number (−4.2 ± 6.9%), interval (9.2 ± 25.7%), and amplitude (−5.7 ± 8.7%) of sIPSCs. Collectively, these data indicate that the slow inward current in LDT neurons was not mediated by ionotropic GABA, glycine, or glutamate receptors, and that glutamatergic, but not GABAergic or glycinergic, afferents to LDT neurons were stimulated by CCh.
TTX blocked the increase in EPSCs but not the slow inward current evoked by CCh in slices from M2M4−/− mice.
To determine whether the CCh-evoked increase in sEPSC frequency and inward current depend on action potentials, we applied TTX (Fig. 7A). For a group of cells in which CCh decreased the mean interval between sEPSCs from 267.4 ± 145 ms to 56 ± 19.5 ms and increased the number of events by 378 ± 108% under control conditions, neither the interval (K-S test, P > 0.05, n = 5) nor the number of muscarinic EPSCs were influenced by CCh following application of TTX (K-S test, P > 0.05 n = 5/5). The mean interval was 488 ± 149.0 ms in control conditions and 618 ± 26.0 ms in CCh. In the same group of cells, TTX did not affect the CCh-induced inward current (control inward current: −29.3 ± 10.0 pA vs. inward current in TTX: −28.2 ± 11.7 pA, n = 5, P > 0.05; Fig. 7A3). As TTX blocked CCh's effect on EPSC frequency in all neurons tested, it is likely that the receptors mediating the increase in EPSCs are located at locations remote from the terminals of presynaptic glutamatergic neurons (e.g., the cell body). Since TTX had no effect on the slow inward current/depolarization, it is likely that the mediating receptors are located on the recorded LDT neurons.
Fig. 7.
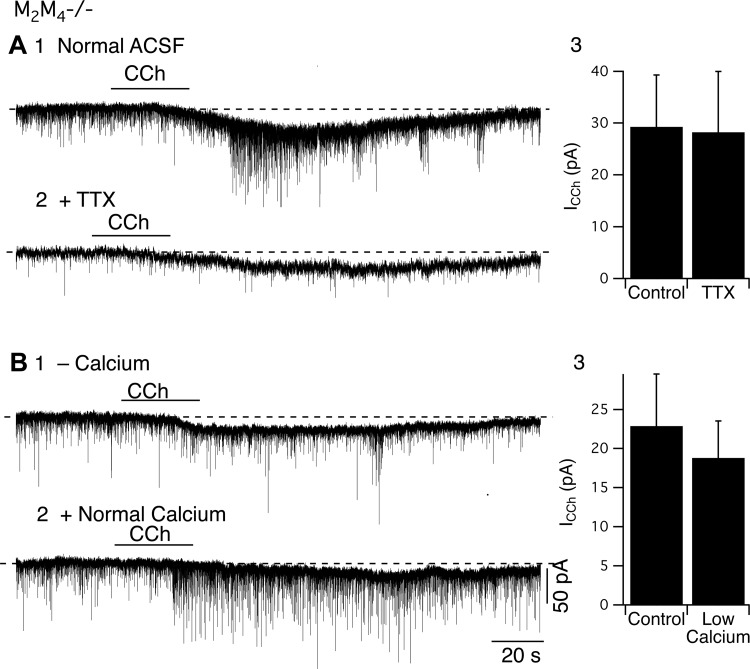
The CCh-mediated inward current (ICCh) results from a direct postsynaptic action, while the increase in sEPSCs results from action potentials in presynaptic glutamatergic neurons. A: in slices from M2M4−/− mice, the ICCh was not attenuated by application of tetrodotoxin (TTX) (A2 and A3), while the increase in EPSC frequency was abolished. B: application of low-calcium ACSF did not attenuate the ICCh, but prevented the increase in EPSC frequency (B1 and B3). B2: reapplication of normal calcium ACSF reinstated the increase in EPSC frequency produced by CCh.
To further test these interpretations, we examined the effect of CCh in low-calcium ACSF (2.7 mM Ca, 2.7 mM EGTA), which blocks Ca2+-dependent transmitter release (Fig. 7B). We found that the amplitude of the slow inward current measured under this condition was not different from that measured following replacement with normal ACSF (normal ACSF: −22.9 ± 6.6 pA, low Ca: −18.8 ± 4.7 pA, P > 0.05, n = 5, Fig. 7B3). In contrast, CCh failed to elicit an increase in sEPSC frequency in low-calcium ACSF (Fig. 7B1), an effect that was reversed following replacement with normal calcium ACSF (n = 5, Fig. 7B2). Taken together, these data indicate that CCh has dual-excitatory actions in LDT neurons from M2M4−/− mice: it directly activates postsynaptic receptors located on LDT neurons to produce the slow inward current, and it activates presynaptic glutamatergic afferents to fire action potentials which increases the sEPSC frequency.
Both direct and indirect CCh excitation of LDT neurons in slices from M2M4−/− mice are mediated by residual muscarinic receptors.
As CCh is a mixed nicotinic and muscarinic agonist, we first tested whether these effects were blocked by atropine. In four cells in which bath-applied CCh induced a slow inward current (−23.4 ± 7.2 pA) and barrages of EPSPs, atropine (5 μM) significantly reduced the slow current (−3.9 ± 3.6 pA, P < 0.05) and blocked the barrage of sEPSPs, as expected from experiments in the single knockouts, indicating that bath-applied CCh excites LDT neurons from the M2M4−/− slices primarily, if not exclusively, via muscarinic receptors. We also tested the M1-preferring receptor antagonist pirenzepine on these two excitatory actions in slices from M2M4−/− mice at a concentration (10 μM) sometimes used in brain slice experiments. This concentration blocked the inward current elicited by CCh (control: −27.3 ± 5.9 pA, pirenzepine: −1.2 ± 5.2, n = 4, P < 0.05, Fig. 8, A and B1) and completely blocked the increase in sEPSC frequency (Fig. 8, A and B2). In two cells, the experiment was conducted in ACSF containing TTX to better isolate the inward current. In these cases, pirenzepine reduced the inward current from −22.5 ± 7.9 pA to −4.0 ± 5.2 pA. To investigate whether M1 mAChRs could plausibly mediate these responses, we also performed immunocytochemistry using antibodies against M1 mAChRs. We found extensive M1 immunoreactivity in the LDT (Fig. 8C) which was codistributed with nNOS+ neurons (Fig. 8D) and was localized to both nNOS+ and nNOS− neurons (Fig. 8E). Taken together, these data indicate it is likely that residual M1 and possibly M3 mAChRs mediate the direct and indirect cholinergic excitation of LDT neurons, including neurons verified to be cholinergic, as indicated by presence of nNOS (n = 2). Nevertheless, this interpretation is necessarily tentative since specificity of both the pharmacology and this antibody need to be confirmed in the corresponding receptor knockouts.
Fig. 8.
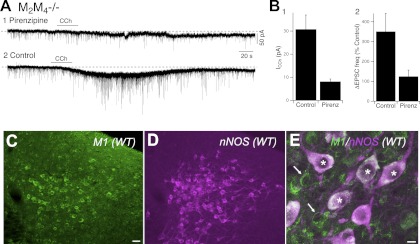
M1 and/or M3 mAChRs mediate pre- and postsynaptic actions of CCh in slices from M2M4−/− mice. A: pirenzepine greatly attenuated the CCh-mediated inward current and excitatory synaptic activity. In this neuron, the direct and indirect excitatory actions were reinstated following the wash-out of pirenzepine. B: bar graph summarizing the pirenzepine attenuation of the CCh-induced inward currents (B1; n = 5) and increase in sEPSC frequency (B2; n = 5). C, D, and E: confocal images of a section through the LDT illustrates immunocytochemical staining for M1 mAChRs (C), nNOS (D), and both M1 receptors and nNOS (E). M1 receptors are expressed by nNOS+ cells (asterisks) and nNOS− cells (arrows). Scale bar in C = 20 μm and applies to D. Scale bar in E = 5 μm.
Indirect CCh excitation of LDT neurons may arise from local glutamatergic neurons.
The sites at which CCh causes TTX-sensitive action potentials in glutamatergic afferents are likely to be relatively close to cholinergic LDT neurons since long-distance connections are disrupted in brain slices. To determine whether they are within the LDT, we surgically isolated the LDT region in slices from M2M4−/− mice and focally applied CCh at different locations within the LDT. Cuts were made at the lateral and ventral borders of the LDT. The slices were then positioned to ensure ACSF flowed in a dorsoventral direction. CCh was then applied via a puffer pipette positioned above the slice and approaching the recorded cell from the downstream side of the ACSF flow (Fig. 9A). In spite of this restricted connectivity, CCh still evoked EPSCs in LDT neurons. In 56% of the cells tested, CCh elicited sEPSCs and inward currents (n = 9/16), while, in 37.5%, CCh just elicited inward currents (n = 6/16, 1/16 failed to respond). To verify that these effects resulted from local actions of CCh, we moved the puff-pipette in increments downstream from the recorded cell (Fig. 9, A and B). In all cases, we found positions ventral to the LDT in which puffer application failed to elicit EPSCs or inward currents (n = 6/6). This failure was not due to a loss of puffer patency, since EPSCs and inward current could be re-elicited by returning the puffer to the original position. Results from such an experiment are illustrated for a neuron subsequently shown to contain nNOS in Fig. 9C.
Fig. 9.
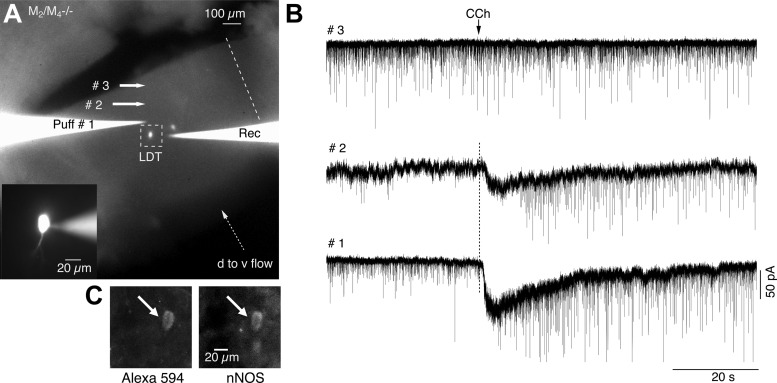
CCh activates local glutamatergic neurons. A: low power fluorescence image of the LDT which has been surgically isolated from more ventral portions of the slice. Midline is indicated by the dotted line, and the dorsal-to-ventral flow of ACSF is indicated by the dotted arrow (d to v flow). Both the recording pipette (right) and puff pipette (Puff #1) contain Alexa 594 and are fluorescent, as are two recorded and filled neurons which are visible. Inset shows the leftmost neuron at higher magnification during the recording (pipette seen on right side). Responses shown in B were obtained from this neuron, with the puff pipette located at the corresponding positions. B: the increase in sEPSC frequency and postsynaptic inward current decreased as the puff location was moved out of the LDT. C: immunocytochemistry following fixation revealed the recorded cell was nNOS+ and hence cholinergic.
In three cells, we confirmed that this locally evoked increase in sEPSC frequency was blocked by atropine (5 μM), indicating that mAChRs within the LDT elicit barrages of EPSCs in cholinergic LDT neurons in M2M4R−/− mice. Collectively, these data suggest that CCh elicits TTX-sensitive spiking in local glutamatergic neurons that innervate LDT cholinergic neurons (see discussion).
CCh increases the frequency of EPSPs in slices from WT mice but with a longer latency than in slices from M2M4−/− mice.
To determine whether the indirect excitation induced by CCh was an adaptation to the receptor knockouts, we measured the prevalence and latency of CCh-evoked sEPSCs in LDT neurons recorded in slices from WT mice. In a total of 36 LDT neurons, bath-applied CCh elicited depolarization/inward currents in 31% (n = 11/36), hyperpolarization/outward currents in 67% (n = 24/36), and no response in one cell. Of the 67% showing an inhibitory response, 50% (n = 12/24) also showed a late increase in sEPSC/spontaneous excitatory postsynaptic potentials (sEPSPs). In some cases, this excitatory synaptic activity accompanied a late depolarization/inward current (n = 7/12; Fig. 10A), while, in others, it occurred without an underlying depolarization/inward current (5/12). In one case, a hyperpolarization was followed by a depolarization that was not accompanied by excitatory synaptic activity (1/24). The remainder of cells showed a pure hyperpolarizing response not accompanied by any excitation (n = 11/24). Thus a CCh-mediated increase in sEPSC/sEPSP frequency was prevalent in slices from WT mice.
Fig. 10.
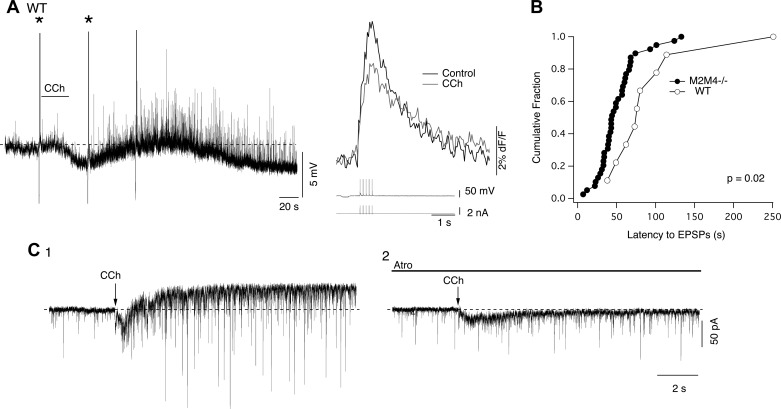
Recordings from LDT neurons obtained from WT slices reveal that CCh can induce an inward current and barrage of EPSPs in cholinergic neurons. A: current clamp recording from a cell in the LDT that responded to bath-applied CCh with an initial hyperpolarization and a decrease in the SpECT (right). This hyperpolarization was followed by depolarization and a barrage of EPSP activity. Asterisks (left) mark the 8-Hz train of five spikes producing the SpECTs shown at higher temporal resolution to the right. B: cumulative distribution of excitatory postsynaptic potential (EPSP) barrage latency recorded in slices from M2M4−/− and WT mice. The barrage latency was shifted to longer times in WT mice. C: voltage clamp recordings from an identified LDT cholinergic neuron from a WT slice following puffer application of CCh before (C1) and after the application of atropine (C2). CCh evoked an increase in EPSC frequency and an outward current that were blocked following application of atropine. Note the early atropine-resistant inward current which is attributable to activation of nicotinic acetylcholine receptors by the rapid application method.
To test our impression that the barrage of sEPSPs might be delayed in slices from WT mice, we compared the barrage onset latencies measured from WT and M2M4−/− slices. Indeed, the latency distribution of this barrage was shifted to longer times in neurons from WT slices (Fig. 10B, K-S test, P = 0.02) with the mean onset latency shifting from 51.3 ± 4.3 s (n = 39) in slices from M2M4−/− mice to 93.8 ± 21.1 s (n = 9) in slices from WT mice. The difference between these distributions is consistent with an early CCh-mediated inhibition of presynaptic glutamatergic neurons via M2 and M4 mAChRs in slices from WT mice.
To determine whether mAChRs within the LDT trigger the sEPSCs in slices from WT mice, we again puff-applied CCh within the LDT of WT slices while recording from LDT neurons. Under these conditions, CCh elicited an outward current in 4/4 neurons (27.1 ± 6.2 pA, paired t-test, P < 0.05) and 2 of these neurons were subsequently identified as nNOS+. In 4/4 of these cells, CCh also induced a significant increase in sEPSC frequency (268.8 ± 40.4% of control; paired t-test, P < 0.05; Fig. 10C1). To verify that this effect was due to activation of muscarinic receptors in these WT slices, we puffed CCh again after applying atropine (5 μM), which fully blocked the outward current and the increase in sEPSC frequency (Fig. 10C2). This revealed that CCh also activated a transient inward current whose onset preceded the outward current. The early peak of this inward current was unaffected by atropine (n = 4/4, paired t-test, P = 0.81, Fig. 10C2), suggesting that brief puffer-applications of CCh can activate desensitizing nicotinic receptors, as we previously demonstrated (Ishibashi et al. 2009). Nevertheless, these data clearly indicate that, in WT mice, activation of mAChRs in the LDT stimulates excitatory synaptic input to cholinergic LDT neurons, and that this effect does not simply arise from the constitutive loss of M2/M4 mAChRs.
M2 and M4 mAChR antagonists block the CCh-evoked membrane hyperpolarization in slices from WT mice but have nonspecific actions on the mAChRs mediating the depolarization and sEPSC barrage.
In the final series of experiments, we sought confirmation in slices from WT mice that M2/M4 mAChRs activated an outward current, and that the inward current and barrage of sEPSCs are blocked by pirenzepine. After determining that CCh elicited an outward current and sEPSPs, CCh was then reapplied in ACSF containing AF-DX 116 (1 μM) and tropicamide (1 μM) to block M2 and M4 mAChRs, respectively. Under these conditions, CCh failed to elicit an outward current, as expected for M2/M4 antagonism (n = 6/6); however, only little residual inward current and no barrage of sEPSCs (n = 6/6) remained. A subsequent application of pirenzepine (10 μM) did not further attenuate these responses (n = 6/6). This could indicate that pirenzepine-insensitive mAChRs mediate the depolarization and sEPSCs in WTs, unlike in slices from M2M4−/− mice, or that these drugs are also blocking pirenzepine-sensitive receptors. We, therefore, examined the effect of these antagonists in slices from M2M4−/− mice. CCh alone evoked inward current and sEPSCs as expected, but these concentrations of AF-DX 116 and tropicamide also blocked CCh-evoked sEPSCs and significantly attenuated the inward current (P < 0.05, n = 4/4). Subsequent application of pirenzepine failed to further attenuate the remaining depolarization, indicating nonspecific actions at these concentrations. Collectively, these data support a role for WT M2/M4 mAChRs in the CCh-mediated membrane hyperpolarization, but demonstrate that these antagonists, at least at the concentrations we used, also block non-M2/M4 mAChRs.
DISCUSSION
The major findings of this study are as follows: 1) that M1- and M2-specific immunoreactivity is present at the soma-dendritic membranes of cholinergic and noncholinergic neurons of the LDT and PPT; 2) that expression of either M2 or M4 mAChRs is necessary and sufficient to mediate two previously described muscarinic actions in LDT neurons: membrane hyperpolarization and inhibition of SpECTs; and 3) that non-M2/M4 mAChRs elicit dual excitatory actions consisting of direct depolarization and barrages of glutamatergic sEPSPs. Since each finding was verified in cholinergic neurons, they indicate that M2, M4, and one or more residual mAChRs are soma-dendritic autoreceptors in cholinergic LDT neurons. Finally, by using surgically reduced slices, we found that mAChRs within, or very close, to the LDT stimulates a TTX-sensitive barrage of sEPSPs in cholinergic LDT neurons, revealing, for the first time, a local excitatory input that is regulated by mAChRs. Neither the direct nor indirect excitation was a compensatory artifact from the knockouts, since both were detected in slices from WT mice. Interestingly, we found that the onset of the CCh-evoked sEPSP barrage was delayed in slices from WT mice, suggesting that M2 and M4 receptors function to delay the depolarizing effect of cholinergic input to local glutamatergic neurons that are presynaptic to LDT cholinergic neurons. Collectively, these findings suggest that mAChRs within the LDT coordinate cholinergic outflow in a time-dependent manner. One possible function of these circuit elements is to transform cholinergic input from a negative feedback signal for transient inputs to positive feedback for persistent inputs to promote low or high firing rates associated with different behavioral states.
Anatomical localization of M2 mAChRs in the LDT and PPT.
Our light and electron microscopy results establish for the first time that M2 mAChRs are localized to both cholinergic and noncholinergic LDT neurons. The absence of immunoreactivity in tissue sections from M2−/− mice confirmed the specificity of the antibody and indicates that tissue immunoreactivity reflects receptor localization. In confocal images, immunostaining was closely associated with soma-dendritic membranes, and this localization was confirmed by electron microscopy (Fig. 3), indicating that M2 mAChRs are membrane-associated and are likely to be functional. These findings extend prior in situ hybridization studies from rat (Vilaro et al. 1994; Vilaro et al. 1992) and mouse (Lein et al. 2007), showing that M2 mRNA codistributes with cholinergic neurons in the LDT and PPT. Our findings also extend prior immunocytochemistry studies which noted regional M2 mAChR immunostaining, but did not colocalize with a cholinergic marker (Brischoux et al. 2008; Levey 1993; Levey et al. 1991), and are consistent with evidence for M2 mAChR binding sites found in the rat LDT and PPT (Baghdoyan 1997; Gill and Gallagher 1998).
M2 and M4 mAChRs function as autoreceptors and heteroreceptors in the LDT.
Our physiological studies using slices from single- and double-receptor knockouts establish that M2 mAChRs are sufficient to activate the membrane hyperpolarization, since it was abolished in slices from M2M4−/− mice, but was unaltered in slices from M4−/− mice. This substantiates prior suggestions that M2 mAChRs mediate this hyperpolarization (Leonard and Llinás 1994; Luebke et al. 1993) which were based on weak antagonism by pirenzepine, as argued for neighboring parabrachial neurons (Egan and North 1986) and by the finding that methoctramine (10 μM) fully or partially antagonized the hyperpolarization in PPT neurons (Ye et al. 2010). Nevertheless, the presence of multiple mAChRs and overlapping antagonist selectivity make these pharmacological experiments inconclusive. This selectivity problem was well illustrated by the role of M4 mAChRs revealed in our studies (see below) and by our observation that M2/M4-preferring antagonists (AF-DX 116, 1 μM; tropicamide, 1 μM) also effectively blocked the pirenzepine-sensitive depolarization in slices from both WT and M2M4−/− mice. Thus, by using mAChR knockouts, we circumvented these limitations and were able to definitively assign a role to M2 mAChRs in LDT neurons. We also established that M2 mAChRs are sufficient to mediate inhibition of SpECTs in the same neurons, although the pharmacology of this action had not been previously studied in the LDT.
Our findings also clearly establish a role for M4−/− mAChRs since they are sufficient to mediate both of these responses: in slices from M2−/− mice, the CCh-evoked membrane hyperpolarization was unaltered, and inhibition of SpECTs was still present, even though both responses were absent in slices from M2M4−/− mice. Consistent with these findings, M4 mRNA is normally expressed in cholinergic and noncholinergic neurons of the LDT and PPT in rat (Sugaya et al. 1997; Vilaro et al. 1994) and M4 mRNA codistributes with ChAT mRNA in a mouse expression atlas (Lein et al. 2007). Thus hyperpolarization and inhibition of SpECTs are likely to be normal functions of M4 mAChRs in LDT neurons. However, since the CCh-evoked inhibition of SpECTs was somewhat attenuated in M2−/− slices, M2 mAChRs may normally play a more important role in mediating this response. To our knowledge, no immunohistochemical or receptor autoradiography studies of M4 mAChR in the LDT or PPT have been published, although immunoprecipitation studies indicate modest levels of M4 mAChR protein in the midbrain and pons (Gomeza et al. 1999b; Yasuda et al. 1993). In preliminary experiments using a commercial M4 mAChR antibody (Chemicon MAB1576; 1:1,000), we were unable to detect specific staining in the LDT and PPT or neighboring brain stem regions, despite specific staining in the striatum (M. E. Bickford, unpublished observation), suggesting that brain stem M4 protein levels are below our detection limit.
Collectively our physiological findings suggest that both M2 and M4 mAChRs mediate the same dual actions since each receptor activated GIRK channels and inhibited R-type Ca2+ channels within the same LDT neurons. While expression system studies indicate that both receptors can mediate both these actions, it is interesting that studies of natively expressed receptors suggest that each receptor principally targets either GIRK or Ca2+ channels (Fernandez-Fernandez et al. 1999; Shapiro et al. 1999). For example, in rat sympathetic neurons, M2 mAChRs activate GIRK channels (when present), and M4 mAChRs inhibit voltage-dependent Ca2+ channels in the same cells, leading to the suggestion that there must be spatial segregation of receptors, G proteins, and channels in these neurons to prevent redundant actions (Brown 2010). Our findings suggest that such a segregation is not necessary, at least when considering the entire soma-dendritic compartment.
Given the potential heterogeneity of LDT neurons, it is worth considering whether some cells might express only M2 or M4 mAChRs. This, however, seems unlikely, since the fraction of LDT neurons showing a CCh-evoked hyperpolarization was not reduced in slices from single-receptor knockouts. Alternatively, our findings might reflect a compensatory upregulation of the remaining receptor following the constitutive loss of the other. On the other hand, previous studies have found no evidence for such a compensatory upregulation (Gomeza et al. 1999a; Gomeza et al. 1999b), and, in mouse sympathetic neurons, knockout of M2 mAChRs cleanly abolishes fast inhibition of the Ca2+ current, while knockout of M4 mAChRs had no effect (Shapiro et al. 1999). Moreover, we found that the effectors in single-receptor knockouts were tertiapin- and SNX-482-sensitive as in WT slices, suggesting that the remaining receptors coupled to the same effectors. Even in slices from M2M4−/− mice, a membrane hyperpolarization was still activated by 5-HT, as expected for WT animals (Leonard and Llinás 1994; Leonard et al. 2000). Taken together, it seems likely that both receptors activate GIRK channels and inhibit Ca2+ influx through R-type Ca2+ channels in WT LDT neurons. Experiments using more localized application of agonist or the synaptic release of ACh will be necessary to see if these functions are segregated over smaller, perhaps even synaptic, domains within the soma-dendritic compartment of these neurons.
Our findings also fit well with previous studies indicating M4, and perhaps M2, mAChRs function as inhibitory autoreceptors at cholinergic terminals from LDT and PPT. At mouse pontine reticular targets, mAChR antagonists enhance the release of ACh with an order of potency suggesting terminal M2 and/or M4 mAChRs (Coleman et al. 2004b). In the ventral tegmental area (VTA), baseline levels of ACh were elevated in M4−/− but not in M2−/− mice, while the ability of locally infused scopolamine to enhance ACh efflux was greatly attenuated in M4−/− mice, but not in M2−/− mice (Tzavara et al. 2004). Our findings show that both M4 and M2 mAChRs couple to effectors appropriate to inhibit terminal ACh release from these neurons. Since inhibition of transmitter release appears mainly mediated by inhibition of Ca2+ influx (Brown and Sihra 2008), it is possible that M4 mAChRs exclusively couple to voltage-dependent Ca2+ channels in the VTA terminals of cholinergic LDT neurons.
Non-M2/M4 mAChRs mediate direct and indirect excitation of LDT neurons.
In slices from M2M4−/− mice, CCh evoked a depolarization/inward current in almost all LDT neurons and increased sEPSC frequency in the majority of these neurons. Since we found ∼30% of the cells from WT mice showed only a hyperpolarization to CCh, our findings imply that M2/M4 receptor signaling normally suppresses detection of the direct and indirect muscarinic excitation of these neurons. Nevertheless, we did find evidence for both direct and indirect muscarinic excitation in slices from WT mice, and both of these effects were blocked by pirenzepine, suggesting that M1 and/or M3 mAChRs mediate these actions. Since some of these cells were nNOS+, our findings suggest that M1 and/or M3 mAChRs function as excitatory autoreceptors as well as heteroreceptors in the LDT. This fits with prior receptor autoradiography evidence for M1 and M3 mAChRs in the LDT (Baghdoyan 1997), with M3 mAChR mRNA expression in rat LDT (Vilaro et al. 1994), and LDT expression of M1 and M3 mAChR mRNA in a mouse expression atlas (Lein et al. 2007). Furthermore, our immunocytochemical results suggest that M1 receptors are expressed by both nNOS+ and nNOS− cells in the LDT. These findings are also consistent with a recent study of thalamic-projecting PPT neurons in rat (Ye et al. 2010), which found that some neurons were directly and indirectly excited by CCh effects that were blocked by 10 μM pirenzepine.
Indirect muscarinic excitation of LDT cholinergic neurons may arise from local glutamatergic neurons.
An unanticipated finding was that CCh evokes a barrage of TTX-sensitive glutamatergic postsynaptic currents/potentials in cholinergic LDT neurons in slices from M2M4−/−, M2−/−, and less commonly from M4−/− mice. Although not previously detected, we reexamined this issue in slices from WT mice and found that CCh indeed evokes such sEPSC/sEPSPs, but the barrage onsets are delayed compared with those in slices from M2M4−/− mice (see Fig. 10). By surgically reducing the slice and using focal CCh application, we determined that these barrages were activated by local mAChRs, perhaps expressed in local glutamatergic neurons. An obvious candidate population mediating these EPSC/EPSPs are the numerous VGluT2 neurons recently identified within the LDT (Wang and Morales 2009). Since the barrage of EPSC/EPSPs was shifted to earlier times in slices lacking M2/M4 mAChRs, we hypothesize these local glutamatergic neurons were among those nNOS-negative LDT neurons showing an M2/M4 mAChR-mediated hyperpolarization followed by a depolarization. Although their local connections have not been studied, these LDT glutamatergic neurons project to the VTA (Wang et al. 2010) like cholinergic LDT neurons (Oakman et al. 1995) and may be functionally related, since their firing also appears to depend on behavioral state (Boucetta and Jones 2009). Another possibility is that the EPSP barrage results from cholinergic neurons that corelease glutamate (Allen et al. 2006; Ren et al. 2011), as previously suggested for LDT neurons (Clements and Grant 1990). This, however, seems unlikely since neither VGluT1 or VGluT3 message was detected in the LDT, and only ∼1% of VGluT2 neurons were immunopositive for ChAT (Wang and Morales 2009). Yet another possibility is that mAChRs depolarize LDT glutamatergic axons sufficiently to fire TTX-sensitive action potentials, but insufficiently to directly cause transmitter release. While theoretically possible, we are not aware of any such examples in the literature. Thus, although further clarification is necessary, we hypothesize that some of these VGluT2 neurons receive cholinergic inputs and provide glutamatergic collaterals to LDT cholinergic and noncholinergic neurons. Such a local glutamatergic circuit could also explain how orexin peptides evoke barrages of TTX-sensitive EPSCs in cholinergic and noncholinergic LDT neurons (Burlet et al. 2002) and how noradrenalin evokes TTX-sensitive barrages of EPSPs in noncholinergic LDT neurons (Kohlmeier and Reiner 1999). Indeed, just such pools of local glutamatergic neurons regulate output from cholinergic and noncholinergic neurons in the medial septum (Manseau et al. 2005) and have been proposed to regulate VTA dopamine neurons (Dobi et al. 2010).
Functional implications.
Our findings indicate that it will be important to clarify the details of LDT neuronal interconnectivity in future studies since its effect on cholinergic and glutamatergic outflow to target regions will critically depend on these details. One intriguing possibility is that cholinergic afferents to the LDT provide synaptic feedback from functionally related mesopontine cholinergic neurons (Semba and Fibiger 1992; Surkis et al. 1996). Based on our present findings, we would expect this feedback to be time dependent with transient or low levels of activity producing negative feedback, but sustained or high levels of activity producing positive feedback due to the build-up of direct and indirect excitation. Such a mechanism could promote bistable firing patterns and could help sustain cholinergic outflow during periods of high cholinergic activity expected during cortical activation of arousal and REM sleep (Boucetta and Jones 2009; Kayama et al. 1992). Indeed, positive feedback is a necessary component for the well-known reciprocal interaction model (Pace-Schott and Hobson 2002), proposed to drive REM and non-REM sleep alternation, and is thought to be a general feature of reticulo-reticulo interconnections (Ropert and Steriade 1981).
At a systems level, cholinergic outflow from mesopontine cholinergic neurons is implicated in numerous functions, including motivation and reward (Blaha et al. 1996; Forster and Blaha 2000; Lodge and Grace 2006), arousal and sleep (Jones 2005), and sensory gating (Bosch and Schmid 2008; Schofield et al. 2011). Related behavioral phenotypes might therefore be expected in M2 and/or M4 knockouts mice. For example, LDT/PPT cholinergic output to the rodent pontine reticular formation is thought to be important for the regulation of REM sleep via activation of M2/M4 mAChRs (Coleman et al. 2004b; Lydic et al. 2002; Shuman et al. 1995), while cholinergic output to the thalamus is thought to be important for activation of the cortical EEG (Dossi et al. 1991; Steriade et al. 1991; Williams et al. 1994). However, M2M4−/− mice are reported to have normal baseline REM sleep with only subtle alterations in EEG activity (Goutagny et al., 2005). Moreover, baseline sleep appears normal in single receptor knockouts with only modest alteration in slow-wave sleep rebound following sleep deprivation in M2−/− mice (Turner et al. 2010). Similarly, mesopontine cholinergic neurons are implicated in mediating prepulse inhibition of acoustic startle via M2/M4 mAChRs in the nucleus pontis caudalis (Bosch and Schmid 2006; 2008). However, prepulse inhibition was not different in M2−/− or M4−/− mice, even though these mice show increased acoustic startle (Turner et al. 2010). Nevertheless, the lack of expected phenotypes from constitutive knockouts does not mean that signaling via these receptors is unimportant, especially in light of their widespread expression. Future studies with conditional knockouts, and methods to interrogate these specific circuits, should provide stronger tests for the behavioral functions of these circuits.
GRANTS
Research was supported by National Institutes of Health grants NS27881 (C. S. Leonard), HL64150 (C. S. Leonard), and NS35377 (M. E. Bickford).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.A.K., M.E.B., and C.S.L. conception and design of research; K.A.K., M.I., and M.E.B. performed experiments; K.A.K., M.I., M.E.B., and C.S.L. analyzed data; K.A.K., M.I., J.W., M.E.B., and C.S.L. interpreted results of experiments; K.A.K., M.I., M.E.B., and C.S.L. prepared figures; K.A.K. and C.S.L. drafted manuscript; K.A.K., M.I., J.W., M.E.B., and C.S.L. edited and revised manuscript; K.A.K., M.I., J.W., M.E.B., and C.S.L. approved final version of manuscript.
REFERENCES
- Allen TG, Abogadie FC, Brown DA. Simultaneous release of glutamate and acetylcholine from single magnocellular “cholinergic” basal forebrain neurons. J Neurosci 26: 1588–1595, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdoyan HA. Location and quantification of muscarinic receptor subtypes in rat pons: implications for REM sleep generation. Am J Physiol Regul Integr Comp Physiol 273: R896–R904, 1997 [DOI] [PubMed] [Google Scholar]
- Baghdoyan HA, Lydic R, Fleegal MA. M2 muscarinic autoreceptors modulate acetylcholine release in the medial pontine reticular formation. J Pharmacol Exp Ther 286: 1446–1452, 1998 [PubMed] [Google Scholar]
- Beninato M, Spencer RF. The cholinergic innervation of the rat substantia nigra: a light and electron microscopic immunohistochemical study. Exp Brain Res 72: 178–184, 1988 [DOI] [PubMed] [Google Scholar]
- Blaha CD, Allen LF, Das S, Inglis DW, Latimer MP, Vincent SR, Winn P. Modulation of dopamine efflux in the nucleus accumbens after cholinergic stimulation of the ventral tegmental area in intact, pedunculopontine nucleus-lesioned, and laterodorsal tegmental nucleus-lesioned rats. J Neurol 16: 714–722, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch D, Schmid S. Activation of muscarinic cholinergic receptors inhibits giant neurones in the caudal pontine reticular nucleus. Eur J Neurosci 24: 1967–1975, 2006 [DOI] [PubMed] [Google Scholar]
- Bosch D, Schmid S. Cholinergic mechanism underlying prepulse inhibition of the startle response in rats. Neuroscience 155: 326–335, 2008 [DOI] [PubMed] [Google Scholar]
- Boucetta S, Jones BE. Activity profiles of cholinergic and intermingled GABAergic and putative glutamatergic neurons in the pontomesencephalic tegmentum of urethane-anesthetized rats. J Neurosci 29: 4664–4674, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brischoux F, Mainville L, Jones BE. Muscarinic-2 and orexin-2 receptors on GABAergic and other neurons in the rat mesopontine tegmentum and their potential role in sleep-wake state control. J Comp Neurol 510: 607–630, 2008 [DOI] [PubMed] [Google Scholar]
- Brown DA. Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. J Mol Neurosci 41: 340–346, 2010 [DOI] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol 184: 207–260, 2008 [DOI] [PubMed] [Google Scholar]
- Burlet S, Tyler CJ, Leonard CS. Direct and indirect excitation of laterodorsal tegmental neurons by Hypocretin/Orexin peptides: implications for wakefulness and narcolepsy. J Neurosci 22: 2862–2872, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP. Muscarinic receptors–characterization, coupling and function. Pharmacol Ther 58: 319–379, 1993 [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJM. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50: 279–290, 1998 [PubMed] [Google Scholar]
- Clements JR, Grant S. Glutamate-like immunoreactivity in neurons of the laterodorsal tegmental and pedunculopontine nuclei in the rat. Neurosci Lett 120: 70–73, 1990 [DOI] [PubMed] [Google Scholar]
- Coleman CG, Lydic R, Baghdoyan HA. Acetylcholine release in the pontine reticular formation of C57BL/6J mouse is modulated by non-M1 muscarinic receptors. Neuroscience 126: 831–838, 2004a [DOI] [PubMed] [Google Scholar]
- Coleman CG, Lydic R, Baghdoyan HA. M2 muscarinic receptors in pontine reticular formation of C57BL/6J mouse contribute to rapid eye movement sleep generation. Neuroscience 126: 821–830, 2004b [DOI] [PubMed] [Google Scholar]
- Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci 30: 218–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossi RC, Pare D, Steriade M. Short-lasting nicotinic and long-lasting muscarinic depolarizing responses of thalamocortical neurons to stimulation of mesopontine cholinergic nuclei. J Neurophysiol 65: 393–406, 1991 [DOI] [PubMed] [Google Scholar]
- Duttaroy A, Gomeza J, Gan JW, Siddiqui N, Basile AS, Harman WD, Smith PL, Felder CC, Levey AI, Wess J. Evaluation of muscarinic agonist-induced analgesia in muscarinic acetylcholine receptor knockout mice. Mol Pharmacol 62: 1084–1093, 2002 [DOI] [PubMed] [Google Scholar]
- Egan TM, North RA. Acetylcholine hyperpolarizes central neurones by acting on an M2 muscarinic receptor. Nature 319: 405–407, 1986 [DOI] [PubMed] [Google Scholar]
- Eglen RM, Choppin A, Watson N. Therapeutic opportunities from muscarinic receptor research. Trends Pharmacol Sci 22: 409–414, 2001 [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Wanaverbecq N, Halley P, Caulfield MP, Brown DA. Selective activation of heterologously expressed G protein-gated K+ channels by M2 muscarinic receptors in rat sympathetic neurones. J Physiol 515: 631–637, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster GL, Blaha CD. Laterodorsal tegmental stimulation elicits dopamine efflux in the rat nucleus accumbens by activation of acetylcholine and glutamate receptors in the ventral tegmental area. Eur J Neurosci 12: 3596–3604, 2000 [DOI] [PubMed] [Google Scholar]
- Gill TM, Gallagher M. Evaluation of muscarinic M2 receptor sites in basal forebrain and brainstem cholinergic systems of behaviorally characterized young and aged Long-Evans rats. Neurobiol Aging 19: 217–225, 1998 [DOI] [PubMed] [Google Scholar]
- Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H, Wess J. Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A 96: 1692–1697, 1999a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A 96: 10483–10488, 1999b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CH, Bay KD, Buchanan R, Skinner RD, Garcia-Rill E. Muscarinic and nicotinic responses in the developing pedunculopontine nucleus (PPN). Brain Res 1129: 147–155, 2007 [DOI] [PubMed] [Google Scholar]
- Hallanger AE, Wainer BH. Ascending projections from the pedunculopontine tegmental nucleus and the adjacent mesopontine tegmentum in the rat. J Comp Neurol 274: 483–515, 1988 [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18: 5366–5373, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M, Leonard CS, Kohlmeier KA. Nicotinic activation of laterodorsal tegmental neurons: implications for addiction to nicotine. Neuropsychopharmacology 34: 2529–2547, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci 26: 578–586, 2005 [DOI] [PubMed] [Google Scholar]
- Kayama Y, Ohta M, Jodo E. Firing of “possibly” cholinergic neurons in the rat laterodorsal tegmental nucleus during sleep and wakefulness. Brain Res 569: 210–220, 1992 [DOI] [PubMed] [Google Scholar]
- Khateb A, Fort P, Williams S, Serafin M, Jones BE, Muhlethaler M. Modulation of cholinergic nucleus basalis neurons by acetylcholine and N-methyl-d-aspartate. Neuroscience 81: 47–55, 1997 [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Leonard CS. Transmitter modulation of spike-evoked calcium transients in arousal related neurons: muscarinic inhibition of SNX-482-sensitive calcium influx. Eur J Neurosci 23: 1151–1162, 2006 [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Reiner PB. Noradrenaline excites non-cholinergic laterodorsal tegmental neurons via two distinct mechanisms. Neuroscience 93: 619–630, 1999 [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176, 2007 [DOI] [PubMed] [Google Scholar]
- Leonard CS, Llinás R. Serotonergic and cholinergic inhibition of mesopontine cholinergic neurons controlling REM sleep: an in vitro electrophysiological study. Neuroscience 59: 309–330, 1994 [DOI] [PubMed] [Google Scholar]
- Leonard CS, Rao SR, Inoue T. Serotonergic inhibition of action potential evoked calcium transients in NOS-containing mesopontine cholinergic neurons. J Neurophysiol 84: 1558–1572, 2000 [DOI] [PubMed] [Google Scholar]
- Levey AI. Immunological Localization of m1-m5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci 52: 441–448, 1993 [DOI] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11: 3218–3228, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc Natl Acad Sci U S A 103: 5167–5172, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, McCarley RW, Greene RW. Inhibitory action of muscarine agonists on neurons in the rat laterodorsal tegmental nucleus in vitro. Journal of Neurophysiology 70: 2128–2135, 1993 [DOI] [PubMed] [Google Scholar]
- Lydic R, Douglas CL, Baghdoyan HA. Microinjection of neostigmine into the pontine reticular formation of C57BL/6J mouse enhances rapid eye movement sleep and depresses breathing. Sleep 25: 835–841, 2002 [DOI] [PubMed] [Google Scholar]
- Manseau F, Danik M, Williams S. A functional glutamatergic neurone network in the medial septum and diagonal band area. J Physiol 566: 865–884, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson NM. Synthesis, trafficking, and localization of muscarinic acetylcholine receptors. Pharmacol Ther 119: 33–43, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakman SA, Faris PL, Kerr PE, Cozzari C, Hartman BK. Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J Neurosci 15: 5859–5869, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Cholinergic axons in the rat ventral tegmental area synapse preferentially onto mesoaccumbens dopamine neurons. J Comp Neurol 494: 863–875, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci 3: 591–605, 2002 [DOI] [PubMed] [Google Scholar]
- Pare D, Smith Y, Parent A, Steriade M. Projections of brainstem core cholinergic and non-cholinergic neurons of cat to intralaminar and reticular thalamic nuclei. Neuroscience 25: 69–86, 1988 [DOI] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, Luo M. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron 69: 445–452, 2011 [DOI] [PubMed] [Google Scholar]
- Ropert N, Steriade M. Input-output organization of midbrain reticular core. J Neurophysiol 46: 17–31, 1981 [DOI] [PubMed] [Google Scholar]
- Roth MT, Fleegal MA, Lydic R, Baghdoyan HA. Pontine acetylcholine release is regulated by muscarinic autoreceptors. Neuroreport 7: 3069–3072, 1996 [DOI] [PubMed] [Google Scholar]
- Satoh K, Fibiger HC. Cholinergic neurons of the laterodorsal tegmental nucleus: Efferent and afferent connections. J Comp Neurol 253: 277–302, 1986 [DOI] [PubMed] [Google Scholar]
- Schofield BR, Motts SD, Mellott JG. Cholinergic cells of the pontomesencephalic tegmentum: Connections with auditory structures from cochlear nucleus to cortex. Hear Res 279: 85–95, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba K, Fibiger HC. Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and antero-grade transport and immunohistochemical study. J Comp Neurol 323: 387–410, 1992 [DOI] [PubMed] [Google Scholar]
- Semba K, Reiner PB, Fibiger HC. Single cholinergic mesopontine tegmental neurons project to both the pontine reticular formation and the thalamus in the rat. Neuroscience 38: 643–654, 1990 [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca(2+) channels by using knockout mice. Proc Natl Acad Sci U S A 96: 10899–10904, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman SL, Capece ML, Baghdoyan HA, Lydic R. Pertussis toxin-sensitive G proteins mediate carbachol-induced REM sleep and respiratory depression. Am J Physiol Regul Integr Comp Physiol 269: R308–R317, 1995 [DOI] [PubMed] [Google Scholar]
- Steriade M, Dossi R, Pare D, Oakson G. Fast oscillations (20–40 Hz) in thalamocortical systems and their potentiation by mesopontine cholinergic nuclei in the cat. Proc Natl Acad Sci U S A 88: 4396–4400, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Pare D, Parent A, Smith Y. Projections of cholinergic and non-cholinergic neurons of the brainstem core to relay and associational thalamic nuclei in the cat and macaque monkey. Neuroscience 25: 47–67, 1988 [DOI] [PubMed] [Google Scholar]
- Sugaya K, Clamp C, Bryan D, McKinney M. mRNA for the m4 muscarinic receptor subtype is expressed in adult rat brain cholinergic neurons. Brain Res Mol Brain Res 50: 305–313, 1997 [DOI] [PubMed] [Google Scholar]
- Surkis A, Taylor B, Peskin CS, Leonard CS. Quantitative morphology of physiologically identified and intracellularly labeled neurons from the guinea pig laterodorsal tegmental nucleus in vitro. Neuroscience 74: 375–392, 1996 [DOI] [PubMed] [Google Scholar]
- Turner J, Hughes LF, Toth LA. Sleep, activity, temperature and arousal responses of mice deficient for muscarinic receptor M2 or M4. Life Sci 86: 158–169, 2010 [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, McKinzie DL, Felder C, Nomikos GG. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J 18: 1410–1412, 2004 [DOI] [PubMed] [Google Scholar]
- Vilaro MT, Palacios JM, Mengod G. Multiplicity of muscarinic autoreceptor subtypes? Comparison of the distribution of cholinergic cells and cells containing mRNA for five subtypes of muscarinic receptors in the rat brain. Brain Res Mol Brain Res 21: 30–46, 1994 [DOI] [PubMed] [Google Scholar]
- Vilaro MT, Wiederhold KH, Palacios JM, Mengod G. Muscarinic M2 receptor mRNA expression and receptor binding in cholinergic and non-cholinergic cells in the rat brain: a correlative study using in situ hybridization histochemistry and receptor autoradiography. Neuroscience 47: 367–393, 1992 [DOI] [PubMed] [Google Scholar]
- Vincent SR, Satoh K, Armstrong DM, Fibiger HC. NADPH-Diaphorase: A selective histochemical marker for the cholinergic neurons of the pontine reticular formation. Neurosci Lett 43: 31–36, 1983 [DOI] [PubMed] [Google Scholar]
- Wang H, Chakraborti A, Ng T, Yamaguchi T, Morales M. Ventral tegmental input from the pedunculopontine and laterodorsal tegmental nuclei is dominated by glutamatergic and GABAergic, rather than cholinergic neurons. In: Program Num 366.4 Neuroscience Meeting Planner (Online) San Diego, CA: Society for Neuroscience, 2010 [Google Scholar]
- Wang HL, Morales M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci 29: 340–358, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol 10: 69–99, 1996 [DOI] [PubMed] [Google Scholar]
- Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol 44: 423–450, 2004 [DOI] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov 6: 721–733, 2007 [DOI] [PubMed] [Google Scholar]
- Williams JA, Comisarow J, Day J, Fibiger H, Reiner PB. State-dependent release of acetylcholine in rat thalamus measured by in vivo microdialysis. J Neurosci 9: 5236–5242, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic systems in the rat brain. III. Projections from the pontomesencephalic tegmentum to the thalamus, tectum, basal ganglia and basal forebrain. Brain Res Bull 16: 603–637, 1986 [DOI] [PubMed] [Google Scholar]
- Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Development of antisera selective for m4 and m5 muscarinic cholinergic receptors: distribution of m4 and m5 receptors in rat brain. Mol Pharmacol 43: 149–157, 1993 [PubMed] [Google Scholar]
- Ye M, Hayar A, Strotman B, Garcia-Rill E. Cholinergic modulation of fast inhibitory and excitatory transmission to pedunculopontine thalamic projecting neurons. J Neurophysiol 103: 2417–2432, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]