

Abstract
Over the first few postnatal weeks, extensive remodeling occurs at the developing murine retinogeniculate synapse, the connection between retinal ganglion cells (RGCs) and the visual thalamus. Although numerous studies have described the role of activity in the refinement of this connection, little is known about the mechanisms that regulate glutamate concentration at and around the synapse over development. Here we show that interactions between glutamate transporters and metabotropic glutamate receptors (mGluRs) dynamically control the peak and time course of the excitatory postsynaptic current (EPSC) at the immature synapse. Inhibiting glutamate transporters by bath application of TBOA (dl-threo-β-benzyloxyaspartic acid) prolonged the decay kinetics of both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and N-methyl-d-aspartate receptor (NMDAR) currents at all ages. Moreover, at the immature synapse, TBOA-induced increases in glutamate concentration led to the activation of group II/III mGluRs and a subsequent reduction in neurotransmitter release at RGC terminals. Inhibition of this negative-feedback mechanism resulted in a small but significant increase in peak NMDAR EPSCs during basal stimulation and a substantial increase in the peak with coapplication of TBOA. Activation of mGluRs also shaped the synaptic response during high-frequency trains of stimulation that mimic spontaneous RGC activity. At the mature synapse, however, the group II mGluRs and the group III mGluR7-mediated response are downregulated. Our results suggest that transporters reduce spillover of glutamate, shielding NMDARs and mGluRs from the neurotransmitter. Furthermore, mechanisms of glutamate clearance and release interact dynamically to control the glutamate transient at the developing retinogeniculate synapse.
Keywords: development, glutamate transporters, metabotropic glutamate receptors, synapses, visual system
fast excitatory neurotransmission in the central nervous system (CNS) is primarily mediated by the presynaptic release of glutamate and its clearance from the synaptic cleft. At the retinogeniculate synapse, glutamate is released from retinal ganglion cells (RGCs) onto thalamic relay neurons in the lateral geniculate nucleus (LGN). Over development, information is continuously relayed through this synapse to cortex (Akerman et al. 2002; Huttenlocher 1967; Krug et al. 2001; Moseley et al. 1988). Before eye opening, around postnatal day (P)12 in mice, information is encoded in correlated spontaneous retinal activity characterized by prolonged bursts of spiking reaching frequencies >20 Hz (Demas et al. 2003; Torborg et al. 2005). At this time, relay neurons receive synaptic contacts from more than 10 RGCs (Chen and Regehr 2000; Jaubert-Miazza et al. 2005). Consistent with this immature circuitry and patterns of retinal activity, relay neurons are exposed to episodic barrages of glutamate lasting seconds at a time (Mooney et al. 1996). Little is known about the mechanisms present at the immature synapse that handle potentially high concentrations of glutamate.
Two mechanisms of glutamate clearance are diffusion and removal or buffering of the neurotransmitter by high-affinity glutamate transporters (Diamond and Jahr 1997; Tzingounis and Wadiche 2007). Glutamate concentration at the cleft can also be reduced through a metabotropic glutamate receptor (mGluR)-mediated decrease in neurotransmitter release (Conn and Pin 1997; Min et al. 1998; Oliet et al. 2001; Renden et al. 2005; Scanziani et al. 1997). The relative roles of these three mechanisms have been shown to change over development at other CNS synapses (Cathala et al. 2005; Renden et al. 2005; Thomas et al. 2011). For example, in the hippocampus, increased neuropil density with age impedes diffusion and corresponds with a greater role of glutamate transporters (Diamond 2005; Thomas et al. 2011). At the calyx of Held, increased fenestration of the presynaptic terminal at older ages enhances diffusion and reduces the need for presynaptic mGluRs (Renden et al. 2005; Taschenberger et al. 2002). At the developing rodent retinogeniculate synapse, ultrastructural studies show extensive extracellular space before P4. By P8, this space is replaced by neuropil, and “coarse glial processes” are seen near RGC terminals (Aggelopoulos et al. 1989; Bickford et al. 2010). These structures are distinct from the adult synapse, where some glia form glomeruli around aggregates of presynaptic terminals (Budisantoso et al. 2012; Lieberman 1973; Rafols and Valverde 1973). At the mature synapse, diffusion is generally restricted to two dimensions because of the large contact size of the RGC bouton (Budisantoso et al. 2012). Although the structural detail of the immature synapse has been described, little is known concerning glutamate transporter function or how glutamate release is regulated during bursts of retinal activity.
Our results demonstrate that glutamate transporters actively shape synaptic currents at the retinogeniculate synapse. In addition, inhibiting transporters at the immature synapse leads to the activation of group II/III mGluRs that, in turn, decrease neurotransmitter release from RGC terminals. Frequencies that occur during retinal waves are sufficient to activate these mGluRs. Finally, we show downregulation of this mGluR-mediated response with age, suggesting neurotransmission is tightly controlled throughout this developmental period.
MATERIALS AND METHODS
Slice preparation and extracellular solutions.
Parasagittal slices (250 μm) that preserved optic tract and visual thalamus were prepared from P8–12 or P26–34 C57BL/6 or Black Swiss mice (Charles River, Wilmington, MA or Taconic Farms, Germantown, NY) of either sex, as described previously (Chen and Regehr 2000) and in accordance with federal guidelines and protocols approved by Children's Hospital Boston. Slices were prepared in ice-cold choline-based dissection solution containing the following (in mM): 130 choline chloride, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7.0 MgCl2, 0.5 CaCl2, and 25 glucose. Slices were incubated in this solution at 32°C for 15–20 min, followed by an additional 10 min in artificial cerebral spinal fluid (aCSF) containing (in mM) 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1.0 MgCl2, 2.0 CaCl2, and 25 glucose (Sigma, St. Louis, MO). aCSF was adjusted to 310–315 mosmol/l. Slices were then transferred into the recording chamber superfused with aCSF at 2–3 ml/min. All experiments were performed at 35 ± 1°C, unless otherwise indicated. aCSF and choline dissection solutions were saturated with 95% O2-5% CO2. Slices were used for up to 5 h after preparation. Experiments were performed with aCSF containing the γ-aminobutyric acid type A (GABAA) receptor antagonist bicuculline (20 μM) or picrotoxin (50 μM; Sigma), the GABAB-receptor antagonist 3-N-[1-(S)-3,4-(dichlorophenyl)ethyl]amino-2-(S)-hydroxypropyl-P-benzyl-phosphinic acid (CGP-55845; 2 μM), and the A1-adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; 10 μM). To isolate the N-methyl-d-aspartate receptor (NMDAR) excitatory postsynaptic current (EPSC), we included 2,3-dihydro-6-nitro-7-sulfamoyl-benzol[f]quinozaline-2,3-dione (NBQX; 5 μM) while holding the cell at +40 mV. To isolate the AMPA receptor (AMPAR) EPSC, we included 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid [(R)-CPP; 20 μM] and d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5; 50–100 μM) in the bath while holding the cell at −70 mV. For some experiments, AMPAR desensitization was inhibited by including 6-chloro-3,4-dihydro-3-(5-norbornen-2-yl)-2H-1,2,4-benzothiazidiazine-7-sulfonamide-1,1-dioxide (cyclothiazide; 50 μM) in the bath. To inhibit glutamate transport, we added 10–50 μM dl-threo-β-benzyloxyaspartic acid (TBOA) to the superfusion solution. The following agents were also added depending on the experiment: the antagonist to mGluRs, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY-341495; 50 μM); agonists to group I, II, and III mGluRs, respectively, (S)-3,5-dihydroxyphenylglycine [(S)-DHPG; 1–25 μM], (2R,4R)-4-aminopyrrolidine-2,4,-dicarboxylate (APDC; 1–30 μM), and l-(+)-2-amino-4-phosphonobutyric acid (l-AP4; 10–500 μM); and the low-affinity NMDAR antagonist l-(+)-2-amino-5-phosphonopentanoic acid (l-AP5; 1 mM). Stock solutions of pharmacological agents were stored at −20°C and diluted according to the final concentrations immediately before experiments. All pharmacological agents were purchased from Tocris Bioscience (Ellisville, MO) unless otherwise indicated.
Electrophysiology.
Whole cell recordings of thalamic relay neurons were acquired using glass patch pipettes (resistance 1.1–1.7 MΩ) filled with internal solution containing (in mM) 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and 0.1 methoxyverapamil hydrochloride (L-type Ca2+ channel antagonist; Sigma) and adjusted to 290–300 mosmol/l, pH 7.3. This solution is designed to minimize postsynaptic contributions to synaptic transmission. Cesium blocks K+ channels and thus optimizes voltage clamp, whereas EGTA and fluoride inactivate many second messenger systems. Relay neurons were visualized using differential interference contrast optics (Olympus). Voltage-clamp recordings were made using an Axopatch 200B or Multiclamp 700A amplifier (Axon Instruments, Foster City, CA), filtered at 1 kHz, and digitized at 4–50 kHz with an ITC-16 interface (InstruTECH, Port Washington, NY). EPSCs were evoked by current pulses delivered to the intact optic tract by a pair of aCSF-filled glass micropipettes (Drummond Scientific, Broomall, PA), connected to a stimulus isolator (World Precision Instruments, Sarasota, FL), with intensities that ranged from 10 to 150 μA. The membrane potential of the relay neuron was clamped at either −70 or +40 mV (for AMPAR-mediated EPSCs and NMDAR-mediated EPSCs, respectively) during stimulation of the optic tract and was held at 0 mV between stimulation trials. For baseline recordings, optic tract was stimulated at a frequency of 0.025 Hz for NMDAR EPSCs and 0.05 Hz for AMPA EPSCs. To ensure consistent access resistance of the recording electrode throughout the entire experiment, we monitored the peak amplitude of a brief (10 ms) hyperpolarizing test pulse (−5 mV) given before the optic tract stimulation. Access resistances of relay neurons were <15 MΩ.
Analysis.
Data acquisition and analysis was performed using custom software written in IgorPro (Wave-Metrics, Portland, OR), Prism (GraphPad Software, San Diego, CA), and Excel (Microsoft, Redmond, WA). EPSCs were analyzed as the average of 5–10 waves. The decay time course of the AMPAR EPSC is well fit with the double-exponential function f(x) = y0 + A1e(−x/τfast) + A2e(−x/τslow) and is quantified as the weighted tau: τ = [τfast × A1/(A1 + A2)] +[τslow × A2/(A1 + A2)]. At the immature synapse, the time constant of the slow component (τs) contributes to a significant component of the EPSC waveform (Liu and Chen 2008) and is the component most sensitive to TBOA. For experiments involving trains of stimuli, stimulus sets were performed using randomized, interleaved trains at frequencies of 10, 20, and 50 Hz. EPSC amplitudes following the first EPSC (EPSC2–5) were quantified as the difference between the peak EPSC and the baseline current immediately after the stimulus artifact. Data are summarized as means ± SE using the two-tailed paired t-test unless otherwise indicated.
RESULTS
Glutamate transporters shape the synaptic waveform at the immature retinogeniculate synapse.
To investigate the role of glutamate transporters at the immature retinogeniculate synapse, we tested the effects of the competitive nontransportable blocker of glutamate transporters, TBOA, on acute slices prepared from P8–12 mice. Whole cell recordings were made with patch electrodes in voltage-clamp mode from relay neurons in the LGN, and synaptic responses were evoked by stimulating optic tract fibers (Chen and Regehr 2000). Figure 1 illustrates the effects of bath application of TBOA (50 μM) while either NMDAR- or AMPAR-mediated EPSCs were recorded.
Fig. 1.
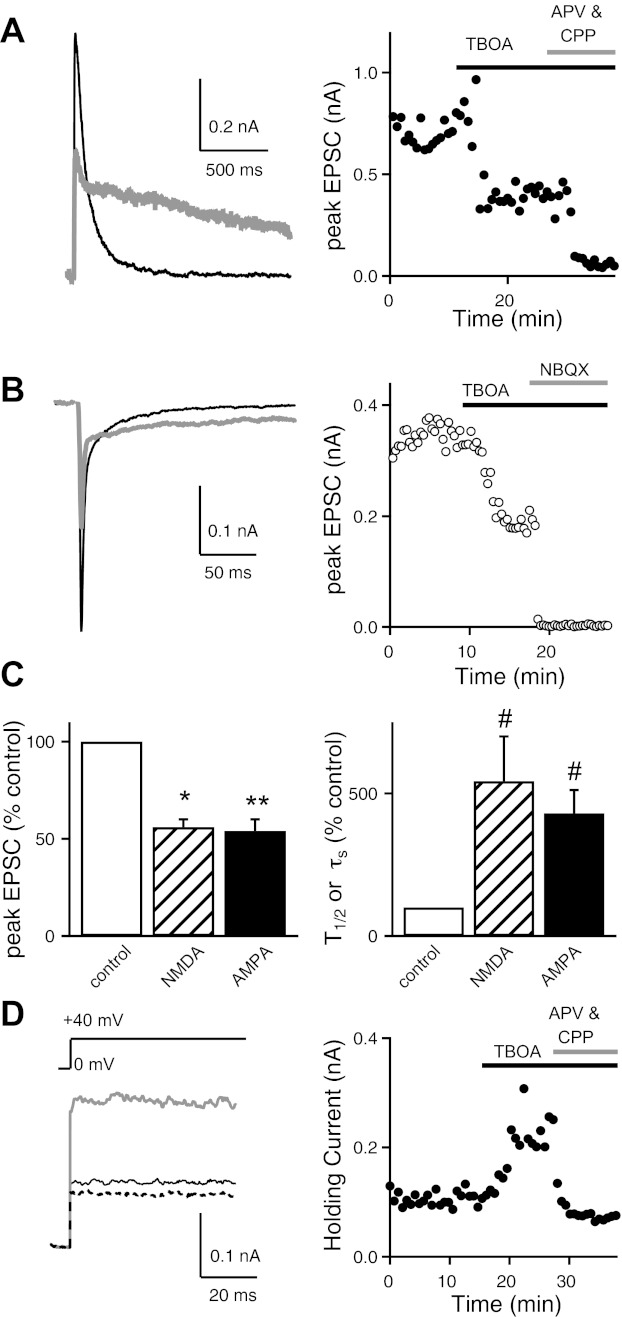
Effects of dl-threo-β-benzyloxyaspartic acid (TBOA) at the immature retinogeniculate synapse. Excitatory postsynaptic currents (EPSCs) were measured before and during bath application of 50 μM TBOA. A and B: traces from representative experiments are shown for N-methyl-d-aspartate receptor (NMDAR)-mediated (holding potential Vh, +40 mV; A, left) and AMPA receptor (AMPAR)-mediated EPSCs (Vh, −70 mV; B, left) before (control; black line) and during bath application of 50 μM TBOA (gray line). Graphs show time course of NMDAR (A, right) and AMPAR EPSC amplitudes (B, right) before and during bath application of 50 μM TBOA, followed by their respective antagonists, 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP) and 2-amino-5-phosphonopentanoic acid (APV; A, right) or 2,3-dihydro-6-nitro-7-sulfamoyl-benzol[f]quinozaline-2,3-dione (NBQX; B, right). C: summary graphs show the mean normalized amplitudes (±SE) of both NMDAR and AMPAR EPSCs (left) and the effects of TBOA on EPSC decay (T1/2 or τs) shown as a percentage of control (right).*P < 0.01; **P < 0.001; #P < 0.05. D: effects of TBOA on the holding current (Ihold) in response to a +40-mV step. Representative traces (left) and time course (right) are shown before (control; thin line) and during bath application of TBOA (gray line) and NMDAR antagonists (dotted line). Recordings were performed at 35 ± 1°C.
When we inhibited glutamate transport with TBOA, the NMDAR EPSC amplitude was reduced to 56 ± 4% of control (n = 5, P = 0.01) and the time course of decay was significantly prolonged [half-decay time (T1/2): 542.9 ± 156.9% of control, n = 5, P < 0.05; Fig. 1, A and C]. The remaining current in the presence of TBOA was blocked by NMDAR-specific antagonists (Fig. 1A, right). We found a similar effect of TBOA on the waveform of the AMPAR EPSC (Fig. 1, B and C). The peak amplitude of the AMPAR EPSC decreased to 54 ± 6% of control (n = 5, P = 0.001), and the time course of the current also slowed. The decay kinetics of the AMPAR EPSC at immature synapses is well described by a double-exponential relationship (Liu and Chen 2008). In the presence of TBOA, τs increased to 430 ± 82% of control (n = 5, P < 0.05) and the current could be completely blocked by AMPAR-specific antagonists (Fig. 1B, right). The average effects of TBOA on the peak EPSC and the decay time courses of both NMDAR and AMPAR currents are compared in Fig. 1C.
These results demonstrate that glutamate transporters are present at the immature retinogeniculate synapse and that they actively remove glutamate during synaptic transmission. Slowing of the EPSC decay kinetics implies that reduced glutamate clearance results in either extended activation of glutamatergic receptors in the synaptic cleft and/or spillover to receptors farther from the release sites. In addition, TBOA increased the relay neuron holding current (Ihold) elicited by a step depolarization from 0 to +40 mV to 200 ± 30% of control (n = 5, P < 0.05). The difference in Ihold, with and without TBOA, can be attributed to increased basal activation of NMDARs because it is reversed with bath application of NMDAR antagonists [100 μM dl-APV and 20 μM (R)-CPP; Fig. 1D]. Since NMDA antagonists do not block all of Ihold in control conditions, our results suggest that TBOA exposure increases ambient glutamate concentration by significantly more than 200%.
Biphasic synaptic response to glutamate transporter inhibition.
Although an increase in ambient glutamate or glutamate spillover could explain the slowing of NMDAR current decay kinetics and the change in Ihold in response to bath application of TBOA, it does not explain the decrease in EPSC peak amplitudes. However, we noticed in some experiments, such as in Fig. 1A, that there was often an initial transient increase followed by a reduction in the peak EPSC. Thus we asked whether the decrease in peak EPSC was a secondary effect of elevated glutamate concentrations. To test this possibility, we took advantage of the fact that the activity of glutamate receptors (both metabotropic and ionotropic) is more temperature dependent than glutamate diffusion. We reasoned that we might be able to better appreciate the initial transient increase in NMDAR EPSC peak amplitude by recording at room temperature (RT) and by sampling more frequently. Figure 2A shows that at RT the time course of the peak EPSC amplitude following inhibition of glutamate transport consisted of two distinct phases. Immediately following the addition of TBOA, evoked NMDAR currents increased in amplitude and duration. Over the ensuing 2–5 min, however, the current decreased in amplitude while maintaining slow decay kinetics. These observations could be explained by a mechanism wherein an increase in glutamate spillover and/or ambient glutamate concentration leads to a decrease in neurotransmitter release. Activation of mGluRs has been shown to decrease the probability of release at a number of synapses in the CNS (Baskys and Malenka 1991; Maki et al. 1994; Min et al. 1998; Oliet et al. 2001; Renden et al. 2005; Scanziani et al. 1997; von Gersdorff et al. 1997). However, little is known about the role of mGluRs at the immature retinogeniculate synapse.
Fig. 2.
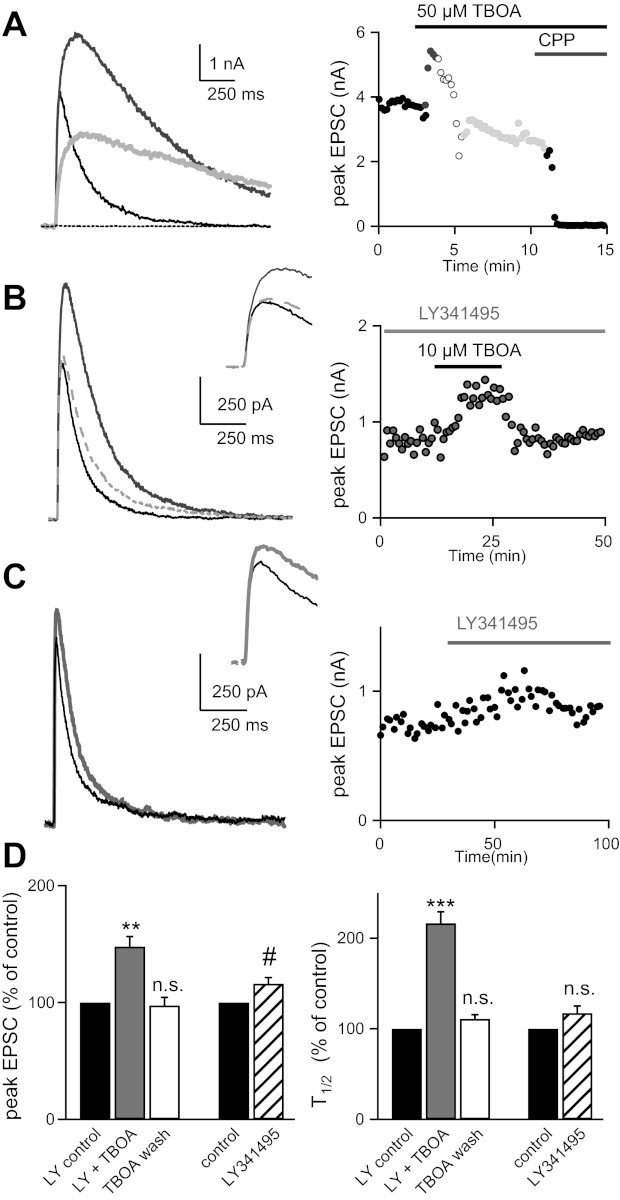
Inhibition of group II/III metabotropic glutamate receptors (mGluRs) prevents TBOA-induced reduction in EPSC amplitude at the immature synapse. A: traces (left) and time course (right) of NMDA EPSC responses recorded at room temperature before (black) and during (dark gray, minute 1; open circles, minutes 2–3, light gray: minutes 3–5) bath application of 50 μM TBOA, followed by the NMDAR antagonist CPP. Stimulation frequency was 0.1 Hz. B: representative traces (left) and time course (right) of NMDAR EPSC responses in the presence of LY-341495 (LY; 50 μM) before (black line), during (solid gray line), and after (dashed gray line) bath application of 10 μM TBOA. C: NMDAR EPSC traces (left) and time course (right) before (black line) and during (gray line) bath application of LY. D: summary of data (means ± SE). Average T1/2 is 87 ± 11 ms in LY vs. 185.4 ± 19.8 ms in LY and TBOA. #P < 0.05; **P < 0.01; ***P < 0.001 (n.s., not significant). Recordings in B and C were performed at 35 ± 1°C.
To test for the presence of mGluRs, we examined the effects of TBOA in the presence of an mGluR antagonist, LY-341495 (LY) (Kingston et al. 1998). LY is a very selective antagonist to group II mGluRs at low concentrations; however, at higher concentrations (50 μM), it has measureable effects on all mGluR groups (Linden et al. 2009). In the following experiments we used a lower concentration of TBOA, since we found that bath application of 50 μM TBOA often led to unstable recordings attributable to excessive changes in Ihold. As with 50 μM TBOA, 10 μM TBOA effectively reduced the NMDAR peak EPSC (to 75 ± 5% of control, n = 7, P < 0.05; data not shown); thus the previously described effect on the peak current is still present even with reduced TBOA concentration. In the presence of 50 μM LY we found that the inhibitory effect of TBOA on EPSC amplitude was prevented. Instead, there was a lasting increase in peak EPSC to nearly 150% of control (147 ± 8.6% of control; n = 6, P < 0.01; Fig. 2, B and D). Despite the fivefold reduction in TBOA concentration, we still observed a doubling in NMDAR EPSC T1/2 that was reversed with washout of the transporter inhibitor (Fig. 2, B and D). We also found smaller changes in Ihold in the presence of 10 μM TBOA compared with 50 μM TBOA, which were reversible (TBOA: 147.8 ± 12.6% of control n = 6, P = 0.01; wash: 112.9 ± 17.7% of control n = 5, P = 0.4; data not shown).
These results suggest that TBOA mediates the reduction in peak EPSC through accumulation of glutamate and subsequent activation of mGluRs. Application of LY alone had a small but significant effect on the NMDAR EPSC amplitude, but not T1/2, at baseline stimulation frequencies (peak increased to 116 ± 5% of control, n = 6, P < 0.05; 0.025-Hz stimulation, Fig. 2, C and D). Our baseline stimulation of the optic tract (0.025 Hz) is at a much lower frequency than the reported mean RGC firing rates of 0.3–0.45 Hz at ages P9–13 in mice (Demas et al. 2003; Torborg and Feller 2005). Thus we interpret these data to indicate that mGluRs can be activated by glutamate spillover and/or ambient glutamate and inhibit neurotransmitter release during physiological levels of RGC activity.
Activation of group II/III mGluRs regulates neurotransmitter release at the immature retinogeniculate synapse.
We next sought to identify the class of mGluRs responsible for the decrease in EPSC amplitude seen with application of TBOA. Our finding that both AMPAR and NMDAR peak currents decreased to a similar extent in the presence of TBOA favors a presynaptic mechanism. Moreover, our experimental conditions were designed to minimize known postsynaptic effects of mGluR signaling (see materials and methods). Thus we examined the effects of specific mGluR agonists on synaptic strength and the paired-pulse response (PPR) to address whether the probability of release was affected. At the retinogeniculate synapse, both pre- and postsynaptic mechanisms have been shown to contribute to PPR (Budisantoso et al. 2012; Chen et al. 2002). To accurately monitor a presynaptic process without contamination of postsynaptic AMPAR desensitization, we added 50 μM cyclothiazide (CTZ) to the bath. CTZ prevents desensitization of AMPARs and does not alter release probability at the retinogeniculate synapse (Chen and Regehr 2000). Figure 3, A–C, shows the effects of bath application of agonists to different classes of mGluRs on pairs of stimuli separated by an interstimulus interval of 50 ms. The time courses of the peak amplitude of the first (EPSC1) and second EPSCs (EPSC2) are plotted before and during agonist application (top right). The PPR values, calculated as EPSC2/EPSC1, are shown at bottom right. Application of group II (APDC) and group III (l-AP4) agonists led to a sustained reduction in EPSC1 amplitude (Fig. 3D). For example, 30 μM APDC inhibited the peak EPSC to 73 ± 4% of control (n = 5, P < 0.05) and increased PPR to 130 ± 4% of control (n = 5, P < 0.01), whereas 50 μM l-AP4 reduced the current to 68 ± 7% of control (n = 6, P < 0.05) and increased PPR to 150 ± 9% of control (n = 6, P < 0.05).
Fig. 3.

mGluR agonists modulate synaptic currents at the immature synapse. Representative traces (left) and time course (right) of AMPAR EPSC responses to pairs of pulses before (thin line) and during (thick line) bath application of the group II mGluR agonist (2R,4R)-4-aminopyrrolidine-2,4,-dicarboxylate (APDC; 30 μM; A), group III agonist l-(+)-2-amino-4- phosphonobutyric acid (l-AP4; 500 μM; B), and group I agonist (S)-3,5-dihydroxyphenylglycine (DHPG; 25 μM; C). Peak amplitude of first (EPSC1; circles) and second AMPAR EPSC (EPSC2; squares) and paired-pulse ratio (PPR; triangles) are plotted. D: summary data of peak EPSCs (top) and PPR (bottom) shown as a percentage of control in the presence of various concentrations of mGluR agonists. #P < 0.05; *P < 0.01. Recordings were performed at 25 ± 1°C.
The group III class of mGluRs includes mGluR4, -6, -7, and -8, which can all be activated by l-AP4. The EC50 or IC50 values of l-AP4 for mGluR4, -6, and -8 range from 0.4 to 1.2 μM (Conn and Pin 1997). However, mGluR7 has a lower affinity for the agonist and requires a much higher concentration than 50 μM for full activation (Conn and Pin 1997). Thus we tested the effects of higher concentrations of l-AP4. We found that 500 μM l-AP4 further reduced synaptic strength to 48.9 ± 4% of control (n = 5, P = 0.01; compared with 50 μM l-AP4: P < 0.05, unpaired t-test) and increased PPR to 196 ± 31% of control (n = 5, P < 0.01; Fig. 3, B and D). These data suggest that mGluR7 is also present at the immature retinogeniculate synapse.
In contrast to the group II and III agonists, the group I agonist DHPG (≥10 μM) did not significantly alter the average peak EPSC amplitude (110 ± 16% of control, n = 4, P = 0.7) or the PPR (110 ± 6% of control, n = 4, P = 0.4) (Fig. 3, C and D). The dose-dependent relationship of agonists to the three groups of mGluR are compared in Fig. 3D. These results demonstrate that agonists for group II and III, but not group I, mGluRs lead to a sustained reduction in release probability at the immature retinogeniculate synapse.
Role of glutamate transporters at the mature retinogeniculate synapse.
The functional properties of the rodent retinogeniculate synapse remodel dramatically over the first 3–4 postnatal weeks (Chen and Regehr 2000; Hooks and Chen 2006; Jaubert-Miazza et al. 2005). To test whether the role of glutamate transporters also changes over development at this visual synapse, we examined the effects of TBOA in LGN slices prepared from P27–34 mice. Bath application of 10 μM TBOA significantly alters the waveform of the mature NMDAR EPSC. In contrast to the immature synapse, where we found a decrease in NMDAR EPSC amplitude without LY (Fig. 1), TBOA caused a reversible increase in the EPSC amplitude to 163 ± 20.2% of control (n = 6, P < 0.05; Fig. 4A). The TBOA-mediated effects on EPSC kinetics and Ihold, however, were similar to those of the immature synapse, with a significant and reversible increase in the T1/2 (to 185 ± 23.9% of control, n = 6, P < 0.05; Fig. 4A) and Ihold (to 119 ± 3.9% of control, n = 6, P < 0.01; Fig. 4C).
Fig. 4.
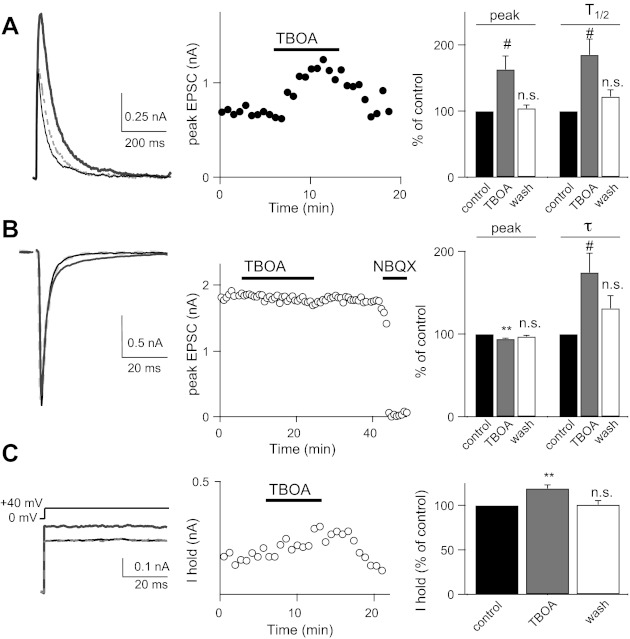
Effects of TBOA on EPSCs at the mature retinogeniculate synapse. A: representative traces (left) and time course (middle) of NMDAR EPSCs recorded from a mature (postnatal day 28) relay neuron before (black line), during (solid gray line), and after (dashed gray line) bath application of 10 μM TBOA; summary data (means ± SE) shown as a percentage of control (right). B: representative traces (left) and time course (middle) of AMPAR EPSCs recorded before (black line), during (dark gray line), and after (dashed gray line) application of TBOA, followed by receptor antagonist; summary data (means ± SE) shown as a percentage of control (right). C: Ihold in response to a +40-mV step. Average traces (left) and time course (middle) are shown before (black line), during (dark gray line), and after (dashed gray line) application of TBOA; summary Ihold data (means ± SE) shown as a percentage of control. Recordings were performed at 35 ± 1°C. #P < 0.05; *P < 0.01. **P < 0.001.
We also found a small but significant decrease in the mature AMPAR EPSC in the presence of 10 μM TBOA (to 94.2 ± 0.8% of control, n = 5, P < 0.01) and a slowing of the time constant of decay, τ (to 174 ± 23.2% of control, n = 5, P < 0.05; Fig. 4B). This reduction in peak amplitude is due to AMPAR desensitization, because in the presence of 50 μM CTZ, bath application of 10 μM TBOA had no significant effect on peak amplitude (105.5 ± 2.9% of control, n = 5, P = 0.12; data not shown). However, in the presence of CTZ, EPSC decay kinetics still increased in response to transporter inhibition (τ: 231 ± 3.8% of control, n = 5, P < 0.05; data not shown). AMPAR desensitization can occur at the mature retinogeniculate synapse as a result of glutamate spillover and/or increased ambient glutamate concentration (Budisantoso et al. 2012; Chen et al. 2002).
Downregulation of mGluRs at the mature retinogeniculate synapse.
The difference between the synaptic responses to TBOA at the mature and immature retinogeniculate synapse suggests a reduction over development in the negative feedback loop mediated through mGluRs. To confirm that there is a change in the role of mGluRs at the mature synapse, we tested for the presence of mGluRs by using specific agonists, as we did for the immature synapse. Figure 5A shows that bath application of 50 μM l-AP4 resulted in a small but persistent decrease in peak AMPAR EPSC (to 84.7 ± 5.6% of control, n = 5, P = 0.05), whereas PPR increased to 122.8 ± 5% of control (n = 5, P < 0.05). This suggests that group III mGluRs are present at the mature synapse. However, increasing the concentration of l-AP4 to 500 μM did not elicit further inhibition, as it did at the immature synapse (see Fig. 3). This is consistent with a loss of mGluR7 (500 μM l-AP4: EPSC1 amplitude, 82.7 ± 3.6% of control, n = 6, P < 0.05 and PPR, 122.4 ± 9% of control, n = 6, P < 0.05; but compared with 50 μM l-AP4: EPSC1 amplitude, P = 0.78, unpaired t-test; PPR, P = 0.97; Fig. 5, B and C). A summary of the dose dependence of l-AP4 on the mature AMPAR EPSC and PPR is shown in Fig. 5B. Thus, although some group III mGluRs remain at the synapse over development, activation of these receptors results in a smaller effect on synaptic strength and release probability in mature synapses compared with the immature synapses (compare Figs. 3 and 5).
Fig. 5.
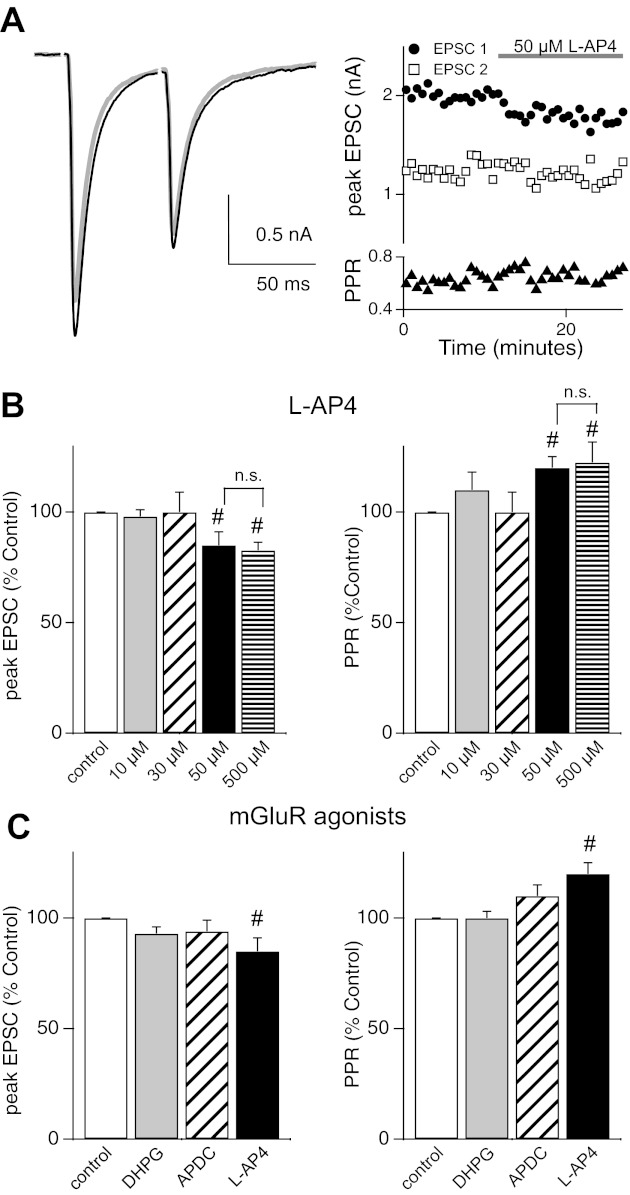
Downregulation of mGluRs at the mature retinogeniculate synapse. A: representative traces (left) and time course (right) of pairs of AMPAR EPSCs before (gray) and during (black) application of 50 μM l-AP4. Peak EPSC1 (circles), EPSC2 (squares), and PPR (triangles) are plotted. B: summary data (means ± SE) shown as a percentage of control in response to 4 concentrations of l-AP4 (10, 30, 50, and 500 μM). C: summary data (means ± SE) for different mGluR agonists: 10 μM DHPG (EPSC1: 93 ± 3% of control, n = 6, P = 0.16; PPR: 100 ± 3% of control, n = 6, P = 0.68) and 30 μM APDC (EPSC1: 94 ± 5% of control, n = 4, P = 0.24; PPR: 110 ± 5% of control, n = 4, P = 0.11). #P < 0.05. Recordings were performed at 25 ± 1°C.
In contrast to group III mGluRs, agonists of group I (10 μM DHPG) and group II (30 μM APDC) did not significantly alter the peak EPSC amplitude or the PPR at the mature retinogeniculate synapse (Fig. 5C). These results demonstrate that the role of group II mGluRs in mediating release probability is lost over development at the retinogeniculate synapse. Taken together, our data suggest that the mGluR negative feedback mechanism that is present at the immature retinogeniculate synapse is downregulated with age.
Activation of mGluRs during physiologically relevant stimulus trains.
Our findings demonstrate that inhibition of glutamate transporter activity can activate a group II/III mGluR-mediated reduction in release probability at the immature synapse. The data also show that there is activation of mGluRs during basal RGC activity (see Fig. 2C). We asked how mGluR activation alters the synaptic response during bursts of synchronous RGC activity that has been shown to occur during this developmental period. Action potential firing rates of RGCs during bursts in immature mice can increase to >20 Hz (Demas et al. 2003; Kerschensteiner and Wong 2008; Torborg and Feller 2005). Because the AMPAR current is relatively small compared with the NMDAR current early in development, we examined the response of NMDAR currents to trains of optic nerve stimulation. To accurately monitor changes in presynaptic neurotransmitter release using NMDAR currents as a reporter, we included the low-affinity NMDAR antagonist l-AP5 (1 mM) in the bath solution to reduce receptor saturation (Chen et al. 2002). We stimulated the optic tract with trains of 5 stimuli at frequencies of 10, 20, or 50 Hz. After a stable baseline was established, 50 μM LY was added to the solution. If tonic activation of group II/III mGluRs influence the synaptic response to trains of stimuli, we would predict that LY would antagonize the binding of glutamate to these receptors, resulting in an increase in the probability of neurotransmitter release.
Indeed, our results are consistent with this prediction. We found a significant increase in the strength of the EPSC1 and a decrease in PPR following relief of mGluR activation (Fig. 6). EPSC1 amplitude of each train increased to 126 ± 8% of control (n = 6, P < 0.05). Figure 6 (left) shows average traces in response to 5 stimuli at either 10 (A), 20 (B), or 50 Hz (C) before (black) and during (gray) bath application of 50 μM LY. A summary plot of the peak amplitudes of the subsequent EPSCs relative to EPSC1 is shown at right. The degree of synaptic depression in response to trains of stimuli was enhanced with higher frequencies in control conditions. Bath application of the mGluR antagonist reduced the peak amplitudes of subsequent EPSCs following EPSC1. Synaptic depression was significantly enhanced in the presence of LY (P < <0.002 for all frequencies tested, 1-way ANOVA). These results demonstrate that group II/III mGluR are activated during physiological RGC firing patterns and reduce release probability at the immature retinogeniculate synapse.
Fig. 6.
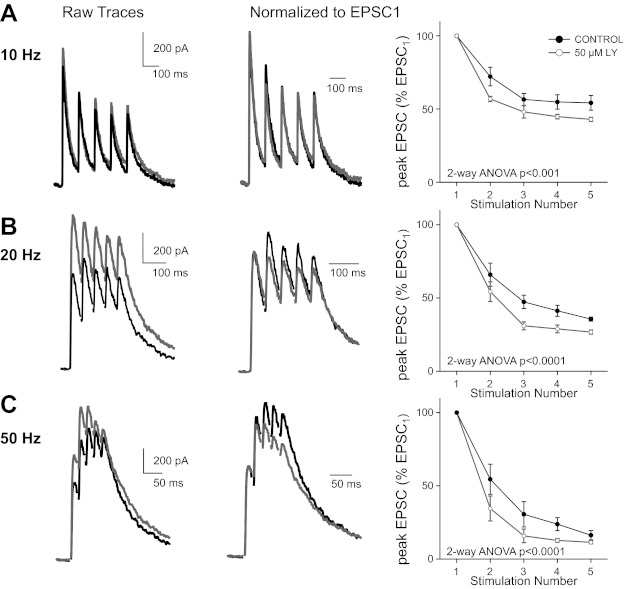
mGluRs are activated during physiologically relevant trains. A–C, left: representative traces of NMDAR EPSCs in response to trains of 5 stimuli at 10 (A), 20 (B), and 50 Hz (C) shown before (black) and during (gray) bath application of 50 μM LY. Middle: NMDAR responses normalized to EPSC1 before (black) and during (gray) application of 50 μM LY. Right: summary of the relative changes in EPSC amplitudes during trains of stimuli at frequencies before (black) and during (gray) application of LY (means ± SE). Peak amplitudes in a train are normalized to EPSC1. Synaptic depression in response to trains of stimuli was significantly increased in the presence of LY for 10, 20, and 50 Hz (P << 0.001; 2-way ANOVA). Recordings were performed at 35 ± 1°C.
DISCUSSION
Developmental regulation of synaptic glutamate concentration.
We examined mechanisms regulating glutamate release and clearance at the developing retinogeniculate synapse. First, we have shown that inhibiting glutamate transporters changes the shape of synaptic currents. This confirms an active role for transporters in removal of glutamate. Second, our experiments unveiled the presence of a negative feedback mechanism present at the immature synapse. When glutamate accumulates, both group II and III mGluRs are activated, resulting in a sustained reduction in neurotransmitter release. Moreover, our results demonstrate that these mGluRs are activated during presynaptic frequencies that mimic immature RGC firing patterns.
The mGluR-mediated negative feedback loop may play an important role during periods of high-frequency presynaptic activity. This mechanism could be advantageous early in development to prevent excess glutamate accumulation during correlated bursts of presynaptic activity. The inhibitory network is not fully matured in the LGN until after eye opening (P12–14) (Bickford et al.); thus the activation of group II/III mGluRs may be a major mechanism preventing excessive glutamate accumulation and excitotoxicity. Our data demonstrate that the mGluR-mediated feedback is downregulated at the mature synapse. This suggests that once connections between retina and thalamus have refined and stabilized, an mGluR type of autoregulation may no longer be needed to control glutamate release.
Glutamate transporters at the retinogeniculate synapse.
Glutamate clearance from the synaptic cleft is controlled by diffusion and by its active removal and buffering by transporters. In addition to removing synaptically released glutamate, transporters also maintain extracellular concentrations of the neurotransmitter (Herman and Jahr 2007). Consistent with previous studies (Tzingounis and Wadiche 2007), we have shown that glutamate transporters maintain ambient levels of extracellular glutamate, shape the synaptic waveform at the retinogeniculate synapse over development, and shield mGluRs from excessive activation at the immature synapse. However, what makes this visual synapse distinct from other CNS synapses is the sensitivity of both NMDAR and AMPAR currents to transporter inhibition.
At both the immature synapse when mGluRs are inhibited, and at the mature synapse, a low concentration of TBOA (10 μM) results in a robust increase in the NMDAR EPSC peak and a doubling of the decay kinetics (see Figs. 2 and 4). The degree of increase of the peak current is striking compared with the Schaffer collateral (SC)-CA1 synapse. At this hippocampal connection, TBOA did not alter the amplitude of the NMDAR EPSC at immature ages and had a small effect at older ages (Christie and Jahr 2006; Diamond 2005; Thomas et al. 2011). Here, at the retinogeniculate synapse, the peak of the NMDAR but not AMPAR EPSC increases in the presence of TBOA. This can be explained by the higher affinity of NMDARs for glutamate and the slower kinetics of the channel compared with the AMPAR. The increase in NMDAR EPSC peak is likely influenced by the extent of glutamate spillover and the activation of nearby extrasynaptic NMDARs. Moreover, our results also showed a significant increase in AMPAR EPSC decay kinetics throughout development consistent with spillover to neighboring synapses (Budisantoso et al. 2012). In contrast to our study, TBOA does not alter AMPAR kinetics at the SC-CA1 synapse (Christie and Jahr 2006). In addition, 200 μM TBOA has modest or no effects on AMPAR kinetics at the mossy fiber-granule cell synapse in the cerebellum and the calyx of Held, respectively (DiGregorio et al. 2002; Renden et al. 2005). Thus our findings suggest currents at this visual synapse rely heavily on glutamate transporters to shape the synaptic response. Interestingly, the effects of TBOA on this sensory synapse are similar to the effects seen on the primary afferent synapses in the mature spinal cord (Napier et al. 2012; Nie and Weng 2009).
It is still unclear which glutamate transporters are present at the immature retinogeniculate synapse. Five excitatory amino acid transporters (EAATs) comprise the family of glutamate transporters in the brain. EAAT1, EAAT2, and EAAT3, also known as GLAST, GLT-1, and EAAC1 in the rodent, are expressed throughout the nervous system (Arriza et al. 1997; Danbolt 2001; Fairman et al. 1995; Kanai and Hediger 1992; Pines et al. 1992; Storck et al. 1992). Given the IC50 values of dl-TBOA to GLAST, GLT-1, and EAAC1 (70, 6, and 6 μM, respectively), our results showing a dramatic effect of 10 μM TBOA suggest that GLT-1 and/or EAAC1 play a role in shaping the synaptic transient at the retinogeniculate synapse (Lebrun et al. 1997; Shimamoto et al. 2000). Our data are consistent with a previous report that localized both GLT-1 and GLAST to nearby glia at the mature synapse (Budisantoso et al. 2012). However, it remains unknown whether there are changes in transporter expression or their subcellular localization over development. It will be interesting in future studies to further investigate the distinct locations and roles of specific glutamate transporter subtypes at the immature synapse.
Autoregulation of neurotransmitter release by mGluRs over development.
We found that transporters at the immature retinogeniculate synapse prevented excessive glutamate binding to group II/III mGluRs. Consistent with reports from other synapses, activation of these metabotropic receptors reduced synaptic strength and increased PPR (Baskys and Malenka 1991; Conn and Pin 1997; Maki et al. 1994; Min et al. 1998; Oliet et al. 2001; Renden et al. 2005; Scanziani et al. 1997; von Gersdorff et al. 1997). The presence of these receptors was unexpected, because studies in the mature visual thalamus had shown that group II or III mGluRs regulate neurotransmitter release at corticothalamic projections but not at the retinogeniculate synapse (Alexander and Godwin 2005, 2006; Turner and Salt 1999). However, consistent with these previous studies at the mature synapse, we found that the group II/III mGluR-mediated responses are downregulated with age. Notably, the loss of mGluR function has been described over development at another sensory synapse, the calyx of Held. At this brain stem synapse, some presynaptic group II/III mGluRs are present in early development and then downregulate with age, although there is no evidence of presynaptic mGluR7-mediated function at this synapse (Renden et al. 2005). Interestingly, the spiral ganglion neurons in the cochlear hair cells that drive presynaptic input to the calyx of Held were recently shown to exhibit wavelike spontaneous activity, with prolonged bursts of spikes reaching 100 Hz, during a prehearing developmental period (Tritsch and Bergles 2010; Tritsch et al. 2010). The similarities in the two sensory synapses support the idea that mGluRs play an important role in regulating glutamate release during this period of activity-dependent synapse remodeling.
The rapid effects of group II/III mGluR agonists on release probability suggest that the receptors are located on the presynaptic RGC terminals as opposed to a neighboring cell. To date, no high-resolution immunoelectron microscopy study of the developing LGN has localized mGluRs. However, in another sensory nucleus of the developing thalamus, the ventral posterior nucleus of the somatosensory system, the group II class mGluR2/3 has been localized to 3% of presynaptic axon terminals of asymmetric synapses (Liu et al. 1998). This electron microscopy (EM) study also noted dense labeling of mGluR2 in glial processes near synapses. Our study cannot completely rule out that mGluR activation in glia could, through an indirect pathway, lead to the reduction of vesicular release at RGC axon terminals. An example of one such indirect pathway involves astrocyte-mediated accumulation of adenosine that binds to presynaptic A1 receptors and reduces release probability (Dittman and Regehr 1996; Pan et al. 1995; Pascual et al. 2005; Scanziani et al. 1992; Zhang and Schmidt 1999). However, we included the A1 receptor blocker DPCPX in all of our experiments. Thus, were astrocytes involved, a substance other than adenosine would be responsible for presynaptic modulation of release. Moreover, mGluR2 expression in astrocytes does not decrease over development in the thalamus (Liu et al. 1998). Thus we favor a model where mGluRs are transiently expressed in RGC terminals.
Our results demonstrate a dynamic range of mGluR-mediated regulation of glutamate release at the immature retinogeniculate synapse (see Figs. 1, 2, and 6). The negative feedback response appears to scale with the degree of glutamate accumulation and spillover. With low-frequency stimulation we see a small but significant mGluR-mediated effect, whereas a substantial decrease in synaptic strength is seen when transporters are inhibited. This could be explained by the subcellular localization of the different mGluRs and by their distinct affinities for glutamate. For example, EM studies in the hippocampus show immunoreactivity for the high-affinity group II mGluR2/3 to axon terminals outside of the synaptic cleft (Shigemoto et al. 1997; Tamaru et al. 2001). In contrast, low-affinity group III mGluR7 (Kd = 1 mM glutamate) is found in presynaptic active zones (Brandstatter et al. 1996; Conn and Pin 1997; Shigemoto et al. 1997). Interestingly, it has been shown that mGluR7s are activated during periods of robust presynaptic activity (Pelkey et al. 2005, 2007). Thus the varied location and glutamate affinities of different classes of mGluRs could contribute to a diverse repertoire of synaptic responses. Given the dynamic synaptic environment at the immature retinogeniculate synapse, it is possible that activation of mGluRs may play an important role in the process of developmental synaptic refinement.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants T32 EY007110 (to J. L. Hauser), F31 NS044793 (to E. B. Edson), F31 NS048630 (to B. M. Hooks), R01 EY013613 (to C. Chen), and Children's Hospital Boston Intellectual and Developmental Disabilities Research Center P01 HD18655.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.L.H., E.B.E., and C.C. conception and design of research; J.L.H., E.B.E., and B.M.H. performed experiments; J.L.H. and E.B.E. analyzed data; J.L.H., E.B.E., and C.C. interpreted results of experiments; J.L.H. and E.B.E. prepared figures; J.L.H. and C.C. drafted manuscript; J.L.H. and C.C. edited and revised manuscript; J.L.H. and C.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank A. Thompson, S. Park, J. Leffler, M. Do, T. Soulier, and X. Liu for helpful discussions of the experiments and manuscript.
REFERENCES
- Aggelopoulos N, Parnavelas JG, Edmunds S. Synaptogenesis in the dorsal lateral geniculate nucleus of the rat. Anat Embryol (Berl) 180: 243–257, 1989 [DOI] [PubMed] [Google Scholar]
- Akerman CJ, Smyth D, Thompson ID. Visual experience before eye-opening and the development of the retinogeniculate pathway. Neuron 36: 869–879, 2002 [DOI] [PubMed] [Google Scholar]
- Alexander GM, Godwin DW. Presynaptic inhibition of corticothalamic feedback by metabotropic glutamate receptors. J Neurophysiol 94: 163–175, 2005 [DOI] [PubMed] [Google Scholar]
- Alexander GM, Godwin DW. Unique presynaptic and postsynaptic roles of group II metabotropic glutamate receptors in the modulation of thalamic network activity. Neuroscience 141: 501–513, 2006 [DOI] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA 94: 4155–4160, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol 444: 687–701, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford ME, Slusarczyk A, Dilger EK, Krahe TE, Kucuk C, Guido W. Synaptic development of the mouse dorsal lateral geniculate nucleus. J Comp Neurol 518: 622–635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstatter JH, Koulen P, Kuhn R, van der Putten H, Wassle H. Compartmental localization of a metabotropic glutamate receptor (mGluR7): two different active sites at a retinal synapse. J Neurosci 16: 4749–4756, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budisantoso T, Matsui K, Kamasawa N, Fukazawa Y, Shigemoto R. Mechanisms underlying signal filtering at a multisynapse contact. J Neurosci 32: 2357–2376, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci 8: 1310–1318, 2005 [DOI] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron 33: 779–788, 2002 [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron 28: 955–966, 2000 [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci 26: 210–216, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237, 1997 [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol 65: 1–105, 2001 [DOI] [PubMed] [Google Scholar]
- Demas J, Eglen SJ, Wong RO. Developmental loss of synchronous spontaneous activity in the mouse retina is independent of visual experience. J Neurosci 23: 2851–2860, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci 25: 2906–2916, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17: 4672–4687, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron 35: 521–533, 2002 [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci 16: 1623–1633, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375: 599–603, 1995 [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci 27: 9736–9741, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron 52: 281–291, 2006 [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Development of cortical neuronal activity in the neonatal cat. Exp Neurol 17: 247–262, 1967 [DOI] [PubMed] [Google Scholar]
- Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci 22: 661–676, 2005 [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360: 467–471, 1992 [DOI] [PubMed] [Google Scholar]
- Kerschensteiner D, Wong RO. A precisely timed asynchronous pattern of ON and OFF retinal ganglion cell activity during propagation of retinal waves. Neuron 58: 851–858, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37: 1–12, 1998 [DOI] [PubMed] [Google Scholar]
- Krug K, Akerman CJ, Thompson ID. Responses of neurons in neonatal cortex and thalamus to patterned visual stimulation through the naturally closed lids. J Neurophysiol 85: 1436–1443, 2001 [DOI] [PubMed] [Google Scholar]
- Lebrun B, Sakaitani M, Shimamoto K, Yasuda-Kamatani Y, Nakajima T. New beta-hydroxyaspartate derivatives are competitive blockers for the bovine glutamate/aspartate transporter. J Biol Chem 272: 20336–20339, 1997 [DOI] [PubMed] [Google Scholar]
- Lieberman AR. Neurons with presynaptic perikarya and presynaptic dendrites in the rat lateral geniculate nucleus. Brain Res 59: 35–59, 1973 [DOI] [PubMed] [Google Scholar]
- Linden AM, Johnson BG, Trokovic N, Korpi ER, Schoepp DD. Use of MGLUR2 and MGLUR3 knockout mice to explore in vivo receptor specificity of the MGLUR2/3 selective antagonist LY341495. Neuropharmacology 57: 172–182, 2009 [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C. Different roles for AMPA and NMDA receptors in transmission at the immature retinogeniculate synapse. J Neurophysiol 99: 629–643, 2008 [DOI] [PubMed] [Google Scholar]
- Liu XB, Munoz A, Jones EG. Changes in subcellular localization of metabotropic glutamate receptor subtypes during postnatal development of mouse thalamus. J Comp Neurol 395: 450–465, 1998 [DOI] [PubMed] [Google Scholar]
- Maki R, Robinson MB, Dichter MA. The glutamate uptake inhibitor l-trans-pyrrolidine-2,4-dicarboxylate depresses excitatory synaptic transmission via a presynaptic mechanism in cultured hippocampal neurons. J Neurosci 14: 6754–6762, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron 21: 561–570, 1998 [DOI] [PubMed] [Google Scholar]
- Mooney R, Penn AA, Gallego R, Shatz CJ. Thalamic relay of spontaneous retinal activity prior to vision. Neuron 17: 863–874, 1996 [DOI] [PubMed] [Google Scholar]
- Moseley MJ, Bayliss SC, Fielder AR. Light transmission through the human eyelid: in vivo measurement. Ophthalmic Physiol Opt 8: 229–230, 1988 [DOI] [PubMed] [Google Scholar]
- Napier IA, Mohammadi SA, Christie MJ. Glutamate transporter dysfunction associated with nerve injury-induced pain in mice. J Neurophysiol 107: 649–657, 2012 [DOI] [PubMed] [Google Scholar]
- Nie H, Weng HR. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol 101: 2041–2051, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292: 923–926, 2001 [DOI] [PubMed] [Google Scholar]
- Pan WJ, Osmanovic SS, Shefner SA. Characterization of the adenosine A1 receptor-activated potassium current in rat locus ceruleus neurons. J Pharmacol Exp Ther 273: 537–544, 1995 [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science 310: 113–116, 2005 [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46: 89–102, 2005 [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Yuan X, Lavezzari G, Roche KW, McBain CJ. mGluR7 undergoes rapid internalization in response to activation by the allosteric agonist AMN082. Neuropharmacology 52: 108–117, 2007 [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain l-glutamate transporter. Nature 360: 464–467, 1992 [DOI] [PubMed] [Google Scholar]
- Rafols JA, Valverde F. The structure of the dorsal lateral geniculate nucleus in the mouse. A Golgi and electron microscopic study. J Comp Neurol 150: 303–332, 1973 [DOI] [PubMed] [Google Scholar]
- Renden R, Taschenberger H, Puente N, Rusakov DA, Duvoisin R, Wang LY, Lehre KP, von Gersdorff H. Glutamate transporter studies reveal the pruning of metabotropic glutamate receptors and absence of AMPA receptor desensitization at mature calyx of held synapses. J Neurosci 25: 8482–8497, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron 9: 919–927, 1992 [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 385: 630–634, 1997 [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17: 7503–7522, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, Lebrun B, Yumoto N, Nakajima T. Syntheses of optically pure beta-hydroxyaspartate derivatives as glutamate transporter blockers. Bioorg Med Chem Lett 10: 2407–2410, 2000 [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA 89: 10955–10959, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience 106: 481–503, 2001 [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leao RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36: 1127–1143, 2002 [DOI] [PubMed] [Google Scholar]
- Thomas CG, Tian H, Diamond JS. The relative roles of diffusion and uptake in clearing synaptically released glutamate change during early postnatal development. J Neurosci 31: 4743–4754, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg CL, Feller MB. Spontaneous patterned retinal activity and the refinement of retinal projections. Prog Neurobiol 76: 213–235, 2005 [DOI] [PubMed] [Google Scholar]
- Torborg CL, Hansen KA, Feller MB. High frequency, synchronized bursting drives eye-specific segregation of retinogeniculate projections. Nat Neurosci 8: 72–78, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Bergles DE. Developmental regulation of spontaneous activity in the Mammalian cochlea. J Neurosci 30: 1539–1550, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Rodriguez-Contreras A, Crins TT, Wang HC, Borst JG, Bergles DE. Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat Neurosci 13: 1050–1052, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol 519: 481–491, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8: 935–947, 2007 [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci 17: 8137–8146, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Schmidt JT. Adenosine A1 and class II metabotropic glutamate receptors mediate shared presynaptic inhibition of retinotectal transmission. J Neurophysiol 82: 2947–2955, 1999 [DOI] [PubMed] [Google Scholar]