

Abstract
To account for benzodiazepine-induced spinal analgesia observed in association with an inflammation-induced shift in the influence of the GABAA receptor antagonist gabazine on nociceptive threshold, the present study was designed to determine whether persistent inflammation is associated with the upregulation of high-affinity GABAA receptors in primary afferents. The cell bodies of afferents innervating the glabrous skin of the rat hind paw were retrogradely labeled, acutely dissociated, and studied before and after the induction of persistent inflammation. A time-dependent increase in GABAA current density was observed that was more than twofold by 72 h after the initiation of inflammation. This increase in current density included both high- and low-affinity currents and was restricted to neurons in which GABA increased intracellular Ca2+. No increases in GABAA receptor subunit mRNA or protein were detected in whole ganglia. In contrast, the increased current density was completely reversed by 20-min preincubation with the tyrosine kinase inhibitor genistein and partially reversed with the Src kinase inhibitor PP2. Genistein reversal was partially blocked by the dynamin inhibitor peptide P4. Changes in nociceptive threshold following spinal administration of genistein and muscimol to inflamed rats indicated that the pronociceptive actions of muscimol observed in the presence of inflammation were reversed by genistein. These results suggest that persistent changes in relative levels of tyrosine kinase activity following inflammation provide not only a sensitive way to dynamically regulate spinal nociceptive signaling but a viable target for the development of novel therapeutic interventions for the treatment of inflammatory pain.
Keywords: dorsal root ganglion, nociceptor, patch clamp, persistent pain
spinal γ-aminobutyric acid A-type (GABAA) receptor signaling in the adult is normally inhibitory in the absence of tissue injury such that spinal administration of GABAA receptor agonists increases nociceptive threshold (Anseloni and Gold 2008; Yamamoto and Yaksh 1991) while administration of antagonists results in hypersensitivity (Anseloni and Gold 2008; Yamamoto and Yaksh 1993). However, we have recently obtained evidence suggesting that in the presence of persistent inflammation there is a shift in spinal GABAA receptor signaling such that the agonist muscimol, at least at low doses, facilitates inflammatory hypersensitivity, while the antagonist gabazine is analgesic (Anseloni and Gold 2008). Interestingly, in contrast to results obtained with the full agonist, we (Anseloni and Gold 2008) and others (Knabl et al. 2008) have demonstrated that spinal benzodiazepine administration is analgesic in both the presence and the absence of tissue injury. While an inflammation-induced depolarizing shift in Cl− equilibrium potential (EGABA), the prevailing hypothesis regarding the basis for injury-induced changes in GABAA receptor signaling (Price et al. 2009), could account for the results obtained with muscimol and gabazine, the benzodiazepine results suggest that an additional mechanism(s) must also be present. Furthermore, we have recently obtained evidence indicating that mechanisms in addition to a depolarizing shift in EGABA must contribute to the persistent inflammation-induced shift in GABA signaling (Zhu et al. 2012). As one of these mechanisms was an increase in GABAA current, we hypothesized that the emergence of a benzodiazepine-resistant, high-affinity extrasynaptic GABAA receptor contributes to the persistent inflammation-induced shift in spinal GABAA signaling.
While persistent inflammation-induced changes in spinal GABAA receptor signaling may reflect presynaptic changes in the central terminals of primary afferents and/or postsynaptic changes in dorsal horn neurons, the available evidence supports a presynaptic site. For example, in an acute inflammation model, GABAA receptor antagonist administration results in a decrease in transmitter release from the central terminals of primary afferents (Sluka et al. 1994). The generation of antidromically conducting action potentials in the central terminals of primary afferents, referred to as the dorsal root reflex observed in acute inflammation models, is also blocked by spinal GABAA receptor antagonists (Lin et al. 1999). More recently, we obtained data indicating that persistent inflammation results in the emergence of excitatory GABAA receptor signaling that is detectable in isolated dorsal root ganglion (DRG) neurons (Zhu et al. 2012). Thus we hypothesize that a high-affinity extrasynaptic GABAA receptor that contributes to the inflammation-induced shift in spinal GABAA receptor signaling is increased on the primary afferent neurons. The present study was designed to begin to test this hypothesis.
Results of the present study partially supported our central hypothesis with evidence that in the presence of persistent inflammation there is an increase in both high- and low-affinity GABAA currents. Strikingly, this increase in current does not appear to be due to an increase in subunit transcription or translation but rather a persistent increase in the relative activity of a tyrosine kinase. Furthermore, behavioral data indicate that this increase in tyrosine kinase activity is sufficient to account for the inflammation-induced shift in spinal GABAA signaling from inhibition to excitation.
MATERIALS AND METHODS
Animals.
Adult male Sprague-Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) weighing between 250 and 350 g were used for all experiments. Rats were housed two per cage in the University of Pittsburgh Association for Assessment and Accreditation of Laboratory Animal Care-approved animal facility on a 12:12-h light-dark schedule with food and water available. All procedures involving animals were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Labeling and inflammation.
DRG neurons that innervate the glabrous skin of rat hind paw were retrogradely labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; Invitrogen, Carlsbad, CA), which was freshly dissolved in DMSO (170 mg/ml) and subsequently diluted 1:10 in saline. DiI was injected (3–5 sites at 3–2 μl/site) with a 30-gauge needle 14–17 days prior to electrophysiological recording. Complete Freund's adjuvant (CFA; Sigma-Aldrich, St. Louis, MO; mixed 1:1 with saline) was injected (100 μl) into the site previously injected with DiI. Unless otherwise noted, “inflamed” DRG neurons were studied 72 h after CFA injection. Both DiI and CFA were injected under isoflurane-induced anesthesia.
Preparation of isolated DRG neurons.
Prior to tissue harvest, rats were deeply anesthetized with a subcutaneous injection (1 ml/kg) of a cocktail containing ketamine (55 mg/ml), xylazine (20 mg/ml), and acepromazine (5.5 mg/ml). L4 and L5 DRG were harvested bilaterally and either pooled for noninflamed animals or processed in parallel for inflamed animals so that neurons ipsilateral and contralateral to the site of inflammation could be studied. DRG were cleaned of connective tissue, enzymatically treated, and mechanically dispersed as previously described (Lu et al. 2006). Isolated neurons were plated on poly-l-lysine-coated coverslips, and electrophysiology experiments were performed 2–8 h after plating.
Electrophysiology.
Voltage-clamp recordings were performed with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) controlled with pCLAMP software (Molecular Devices) via a Digidata 1320A A/D converter. Data were low-pass filtered at 5 kHz and digitally sampled at 2 kHz. For voltage-clamp protocols, capacity transients were canceled via amplifier circuitry. Borosilicate glass (WPI, Sarasota Springs, FL) patch electrodes were pulled on a Sutter P-2000 horizontal puller (Sutter Instruments, Novato CA) and were ∼1.5–2.5 MΩ in resistance when filled with electrode solution of the following composition (in mM): 140 CsCl, 1 MgCl2·6H2O, 1 CaCl2, 11 EGTA, 10 HEPES, 2 ATP, and 1 GTP; pH was adjusted to 7.2 with Tris base, and osmolality was adjusted to 317 mmol/kgH2O with sucrose. The bath solution used to record GABA currents in relative isolation was composed of (in mM) 140 choline-Cl, 2.5 CaCl2, 1.2 MgCl2, 5 HEPES, and 10 glucose; pH was adjusted to 7.4 with Tris base, and osmolality was adjusted to 325 mmol/kgH2O with sucrose. All salts were obtained from Sigma-Aldrich.
Test compounds were applied with a piezo-driven perfusion system (SF-77B Perfusion Fast-Step; Warner Instruments, Hamden, CT), that enables on- and off-rates of under 10 ms. To minimize the potential impact of GABAB receptor activation, all recording was performed in the presence of the GABAB receptor antagonist CGP 55845 (1 μM; Tocris Biosciences, via R & D Systems, Minneapolis, MN).
For all the concentration-response, pharmacological, and kinase studies, GABA (with CGP 55845) was applied at 3-min intervals. For the concentration-response analysis, GABA was applied from lowest to highest concentration and the amplitude of GABA current plotted as a function of concentration was fitted with a Hill equation to generate estimates of peak current (efficacy), the concentration for 50% of peak (EC50, potency), as well as the Hill coefficient. For pharmacological studies, we used both within-cell and between-cell designs. For the within-cell design, test compounds were applied after a stable baseline of GABAA current amplitude was established, and the change in GABAA current observed in the presence of the test compound was analyzed relative to baseline. At least a 5-min intertest interval was used in these experiments. For the between-cell design, neurons were preincubated in either the test compound or its vehicle control, and comparisons were made between groups.
Calcium imaging.
Fura-2 was used to assess changes in the concentration of intracellular Ca2+, as previously described (Zhu et al. 2012). Briefly, DRG neurons were loaded with fura-2 AM ester with Pluronic F-127 (TEF Labs, Austin, TX) for 20 min at room temperature. Imaging was performed on an inverted microscope (Nikon Eclipse TE2000-U) with neurons continuously superfused with bath solution of the following composition (in mM): 130 NaCl, 2.5 CaCl2, 0.6 MgCl2, 10 HEPES, and 10 glucose; pH was adjusted to 7.4 with Tris base, and osmolality was adjusted to 325 mmol/kgH2O with sucrose. Excitation of fura-2 (340/380 nm) was controlled by a DG-4 filter changer (Sutter Instrument). Fura-2 emission (510 nm) data were acquired with Metafluor software (Molecular Devices) via a charge-coupled device (CCD) camera (Quant-EM 512sc; Photometrics, Tucson, AZ) at 1 Hz during drug application. A neuron was considered a “responder” if GABA application resulted in an increase in fluorescence ratio >10% over baseline. This number is three standard deviations greater than the average baseline fluctuation.
PCR.
Rats from inflamed and naive groups were anesthetized, and L4/L5 DRG were harvested. Total RNA was extracted from DRG with TRIzol (Invitrogen). cDNA synthesis was carried out with 2 μg of total RNA with SuperScript II reverse transcriptase (Invitrogen), 0.1 M DTT, RNaseOUT, dNTP mix, 5× first-strand buffer, and random primer (Invitrogen) at 42°C for 50 min, as described by the manufacturer's protocol. cDNA was then kept at −20°C for future use.
For conventional PCR, we used hot-start Taq DNA polymerase; all reactions were denatured at 95°C, annealed at 58°C, and extended at 72°C. A gradient of ∼25–40 cycles was executed, and the product was separated on a 2% agarose gel. The gel was then stained with 0.5 μg/ml ethidium bromide and imaged with an LAS3000 imager (Fujifilm). The optical density of the bands of PCR product of specific genes was then plotted against the number of the cycles, and a cycle number that was in the rising phase of the amplification curve was chosen for the specific genes. The abundance of the target message RNA was estimated based on the optical density of the PCR product (normalized to GAPDH), and comparisons were made between naive and inflamed rats.
For real-time PCR, SYBR Green PCR CORE reagent (Applied Biosystems, Life Technologies, Carlsbad, CA) was used, with a PCR protocol that started with 50°C for 2 min followed by 95°C for 12 min prior to 40 cycles of 95°C for 15 s and 60°C for 60 s. The reaction was run on a thermal cycler (Applied Bioscience) and then analyzed with Prism 7000 SDS software. Amplification efficiency of primers was evaluated, and conditions were optimized so that the efficiency of amplification of target gene and internal comparator were comparable. The ΔΔCT method (where CT is threshold cycle) was used to compare transcriptional levels of target genes between inflamed and naive rat DRG. Primers for the gene products of interest were designed to span at least one intron. The primer sequences used are available upon request.
Western blot.
L4 and L5 DRG were homogenized with a Teflon tube and mortar for <10 strokes in ice-cold RIPA buffer supplied with protease inhibitors as described previously (Zhu et al. 2012). Lysates were collected in 0.5-ml tubes. Teflon tubes were rinsed with RIPA buffer, and the solutions were combined with the lysates previously collected. Lysates were centrifuged for 5 min at 10,000 rpm and 4°C. Protein concentration was determined via BCA protein assay with a BCA assay kit (Thermo-Fisher, Pittsburgh, PA); lysates were then mixed with Laemmli buffer (2×, 400 μl + 100 μl β-ME) and boiled for 5 min before loading. Protein (30 μg) from one animal was then loaded per lane, separated on a 7% SDS-PAGE gel, and transferred to nitrocellulose membrane. Membranes were blocked with 5% milk for 1 h at room temperature and then incubated with primary antibody at 4°C overnight [1:200 for GABAA receptor antibodies, 1:1,000 for GAPDH, diluted with 5% milk-Tris-buffered saline-Tween 20 (TBST)]. The blots were washed and then incubated with peroxidase-conjugated secondary antibody (1:3,000 in 5% milk-TBST; Jackson ImmunoResearch Laboratories, West Grove, PA) for an hour at room temperature. An ECL kit (Amersham Biosciences, Piscataway, NJ) was used for detection of immunoreactivity, where luminescence data were collected on an LAS3000 imager (Fujifilm). The sources of GABAA receptor subunit antibodies were as follows: δ, Santa Cruz Biotechnology (sc-31438; Santa Cruz, CA); β2/3, Millipore (05-474; Billerica, MA); and γ2, Millipore (AB 5954).
Behavioral experiments.
Intrathecal catheters were placed via methods modified from those previously described (Yaksh and Rudy 1976). Rats were anesthetized with rat cocktail, and the subarachnoid space was cannulated with a 32-gauge polyethylene tube (0041, ReCathCo, Pittsburgh, PA) through the atlantooccipital membrane. The tip of the catheter was advanced 8 cm so as to correspond with the lumbar enlargement; the other end was attached to PE-10 tubing, which was fixed to the subcutaneous tissue to avoid movement of the catheter. The rats were allowed to recover for 6 days before testing. Rats showing symptoms of infection, motor dysfunction, or a mistargeted catheter (determined at the end of testing) were excluded from further analysis. CFA was injected into the glabrous skin of rat hind paws as described above for rats in inflamed groups. As with the majority of electrophysiological experiments, behavioral experiments were performed on inflamed rats 72 h after the injection of CFA.
Six days after intrathecal catheterization, the rats were divided into four groups defined by the presence of inflammation (inflamed vs. naive) and the administration of a tyrosine kinase inhibitor (genistein or its inactive isoform, genistin). Mechanical nociceptive threshold was assessed with an electronic von Frey device (IITC Life Science, Woodland Hills, CA) fitted with a ridged tip (∼1 mm in diameter) applied to the dorsolateral surface of the hind paw of rats gently restrained in the toe of a sock. The average of three responses obtained with an interstimulus interval of at least 15 s was used as threshold. Threshold data were collected before and 20 min after the administration of genistein or genistin (both at 0.1 mg/10 μl dissolved in DMSO), where 10 μl of drug was followed by 10 μl of saline administered at a rate of 20 μl/min via a perfusion pump (WPI). After the second determination of threshold, all rats received a second intrathecal injection of muscimol (0.1 μg/10 μl dissolved in saline), which was followed 10 min later by a third determination of mechanical threshold. After the testing, animals were deeply anesthetized and subsequently euthanized after Trypan blue was injected through the catheter. The location of the catheter was determined via visual inspection. The experimenter collecting behavioral data was blinded to whether rats received genistein or genistin. All behavioral data were collected in the University of Pittsburgh Rodent Behavior Analysis Core Facility.
Test compounds.
Capsaicin, diazepam, 3α,21-dihydroxy-5α-pregnan-20-one (THDOC), forskolin, γ-aminobutyric acid (GABA), genistein, genistin, muscimol, phorbol 12,13-dibutyrate (PDBu), picrotoxin, PP2, and ZnCl2 were from Sigma-Aldrich. Bicuculline methiodide, CGP 55845, and dynamin inhibitory peptide (P4 peptide) were from Tocris Bioscience. GABA, ZnCl2, muscimol, and P4 peptide were dissolved in water, diazepam, forskolin, genistein, genistin, PDBu, picrotoxin, PP2, and THDOC in DMSO, and capsaicin in ethanol to make stock solutions. Stock solutions were then kept in a −20°C freezer and diluted with bath solution in ratios of 1:1,000 to their working concentration before use.
Statistics.
All pooled data are presented as means ± SE. A Student's t-test was used for two-group comparisons (i.e., naive vs. inflamed), a Mann-Whitney rank sum test was used for two group comparisons that failed normality test (i.e., cell body size), a one-way ANOVA was used for multiple group comparisons (i.e., current amplitude vs. day after CFA injection), and a two-way ANOVA was used to assess the presence of a significant interaction between inflammation and tyrosine kinase inhibitor on GABA current amplitude or muscimol effect. The Tukey test was used for post hoc pairwise multiple comparisons. P < 0.05 was considered statistically significant. Pearson correlation was used to test the correlation between peak current density and cell size. Cumulative distribution plots of GABA current density before and after inflammation were fitted with a single or double Boltzmann equation.
RESULTS
GABA currents in cutaneous DRG neurons.
Electrophysiological data were collected from 203 and 228 cutaneous DRG neurons from 28 naive and 32 inflamed rats, respectively. The initial set of experiments was performed to assess the impact of retrograde labeling and persistent inflammation on the biophysical properties of GABAA current in cutaneous DRG neurons. As previously described for unlabeled DRG neurons (Lee et al. 2012; White 1990), as well as DRG neurons retrogradely labeled from the glabrous skin (Zhu et al. 2012), 100% of cutaneous DRG neurons studied were responsive to GABA, with a rapidly activating inward current when evoked from a holding potential of −60 mV (Fig. 1A). This was true of neurons from both naive and inflamed rats. This current was completely blocked by the competitive GABAA receptor antagonist bicuculline (50 μM, n = 5) and suppressed by the noncompetitive GABAA receptor antagonist picrotoxin (100 μM, n = 5). There was a weak (r2 = 0.36) but significant (P < 0.01) correlation between peak current amplitude (in response to 1 mM GABA) and cell body capacitance (Fig. 1B). Current density was relatively constant between neurons, even those with putatively distinct functional properties such as small-diameter neurons responsive to capsaicin (a subpopulation of nociceptive afferents) and those with a large cell body diameter unresponsive to capsaicin (putative nonnociceptive afferents) (Fig. 1C). Also consistent with previous results (Flake et al. 2005; Zhu et al. 2012), the membrane capacitance of cutaneous neurons from inflamed rats (42.03 pF; 25th percentile: 34.93 pF, 75th percentile: 49.62 pF) was significantly larger than that of neurons from naive rats (34.15 pF; 25th percentile: 27.80 pF, 75th percentile: 40.28 pF; P < 0.01). This was clearly illustrated with a histogram of cell body capacitance plotted as a fraction of the total number of neurons studied (Fig. 1D). Because of the correlation between cell body size and current amplitude and the impact of inflammation on cell body diameter, current density (peak current/membrane capacitance) was used for all subsequent analyses to enable comparisons between groups of neurons.
Fig. 1.
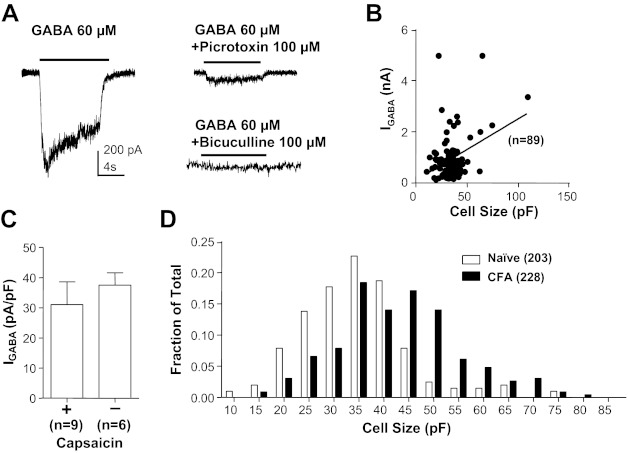
GABA current in acutely dissociated cutaneous dorsal root ganglion (DRG) neurons from adult rats. A, left: typical current evoked from a holding potential of −60 mV in response to an 8-s application of GABA (60 μM) to a small-diameter cutaneous neuron from a naive rat. Right: GABA current was almost (to 78%) completely blocked by the noncompetitive antagonist picrotoxin (100 μM) and completely blocked by the competitive antagonist bicuculline (50 μM). Scale bar on left also applies to traces on right. Data with picrotoxin were from the same neuron on left, while those with bicuculline were from another neuron with comparable baseline current. B: peak current evoked in response to 1 mM GABA (IGABA) is plotted as a function of the membrane capacitance for cutaneous neurons from naive rats. There is a small (r2 = 0.36) but significant (P < 0.01) correlation between current amplitude and membrane capacitance. C: GABA current density (current divided by membrane capacitance) is comparable (P > 0.05) even in distinct subpopulations of cutaneous neurons such as small diameter capsaicin responsive (+) and large diameter capsaicin nonresponsive (−) neurons. D: histogram of cell body size, as determined by membrane capacitance, for cutaneous neurons from naive and inflamed [Complete Freund's adjuvant (CFA)] rats illustrates the significant (P < 0.01) inflammation-induced increase in cell body size. In this and subsequent figures, data are plotted as means ± SE, where the number of neurons studied is indicated in parentheses.
Impact of inflammation on GABA currents in cutaneous DRG neurons.
Consistent with our previous results obtained with perforated-patch recording (Zhu et al. 2012), GABA currents were much larger in cutaneous neurons from inflamed rats compared with those from naive rats (Fig. 2A). This difference was associated with an increase in current density, which was significant (P < 0.01), despite the inflammation-induced increase in membrane capacitance (Fig. 2B), and only found in labeled DRG neurons. Current density in nonlabeled neurons from inflamed animals (27.44 ± 3.26 pA/pF, n = 10) was not significantly different (P > 0.05) from that in cutaneous neurons from naive rats (Fig. 2B) or unlabeled neurons from naive rats (25.45 ± 5.40 pA/pF, n = 22).
Fig. 2.
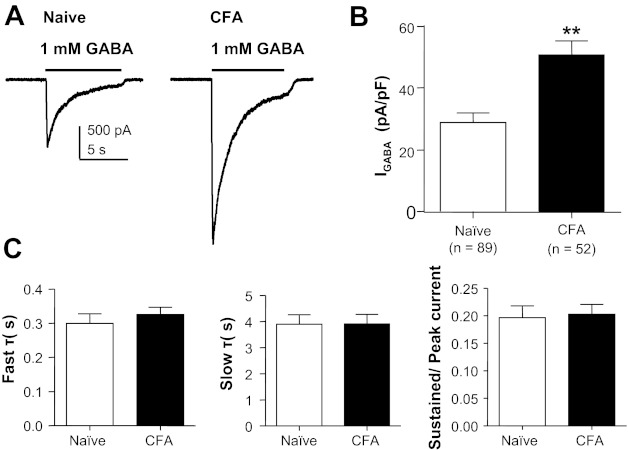
Persistent inflammation-induced increase in GABA current density. A: representative current evoked in response to 1 mM GABA applied to cutaneous neurons from a naive rat (left) and a rat 72 h after the induction of inflammation (CFA, right). B: pooled data indicated that the current density in cutaneous neurons from inflamed rats was almost double that in neurons from naive rats. C: there was no detectable difference (P > 0.05) between cutaneous neurons from naive and inflamed rats with respect to the rate of GABA current inactivation, which was defined by both fast (left) and slow (center) time constants (τ). There was also no difference (P > 0.05) between these groups with respect to the ratio of peak to sustained currents (right). **P < 0.01.
Despite the dramatic increase in current density, there was no detectable influence of inflammation on other biophysical properties of the GABA current as quantified, for example, by the rates of current decay or the ratio of “sustained” to “peak” current observed during sustained application of GABA (Fig. 2C). The current decay was well fitted with a double-exponential equation, yielding both fast and slow time constants of decay that were comparable in currents evoked from naive and inflamed cutaneous neurons.
Pharmacological properties of GABAA receptor in DRG neurons did not change after inflammation.
A key prediction of our central hypothesis was that persistent inflammation would result in an increase in high-affinity current in cutaneous neurons as we have recently described in DRG neurons with time in culture (Lee et al. 2012) as well as in response to the acute application of inflammatory mediators (Lee and Gold 2012). Because an increase in high-affinity current should result in a leftward shift in the GABA concentration-response curve, we assessed the impact of a range of GABA concentrations on cutaneous neurons from naive and inflamed rats. GABA was applied at an interapplication interval no shorter than 3 min based on pilot data indicating that the current evoked with 3 mM GABA was stable over at least five applications (data not shown). There was a significant (P < 0.01) increase in efficacy (Fig. 3), as measured by peak current density (naive 29.53 ± 4.52 pA/pF vs. inflamed 70.14 ± 7.97 pA/pF). However, there was no detectable influence of inflammation on potency, as measured by EC50. EC50 values were 140.61 ± 77.27 μM (n = 23) and 121.83 ± 36.06 μM (n = 14) in neurons from naive and inflamed rats, respectively (Fig. 3B). A concentration-response curve of sustained current, which was measured at the end of an 8-s GABA application, was also plotted, and the results showed a similar change: maximal current density increased (from 5.50 ± 1.06 pA/pF to 12.01 ± 1.40 pA/pF, P < 0.01), with no significant change in EC50 (naive 18.74 ± 1.625, inflamed 16.78 ± 2.369 μM) (Fig. 3C).
Fig. 3.
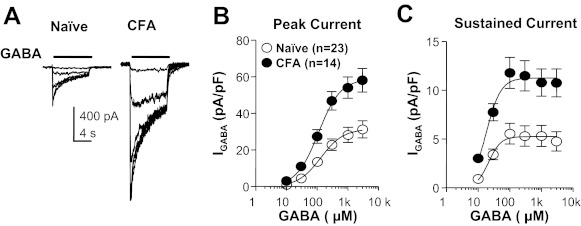
Inflammation-induced increase in efficacy but not potency of GABA current activation. A: representative currents evoked in response to increasing concentrations of GABA applied to a cutaneous neuron from a naive (left) and an inflamed (CFA, right) rat. B: pooled concentration-response curves of peak GABA current in cutaneous neurons from naive and inflamed rats: there was a significant increase in the maximum current in the inflamed group (P < 0.01) compared with naive rats but no change in EC50. C: pooled concentration-response curve of sustained GABA current: there was also a significant increase of sustained current following inflammation (P < 0.01) and no change in EC50.
The absence of a detectable change in biophysical properties or shift in the GABA concentration-response curve suggested that the inflammation-induced increase in current density reflected a generalized increase in all GABAA receptor subtypes already present in the absence of inflammation. To confirm this suggestion, we employed three pharmacological tools that have relative selectivity for subtypes of GABAA receptors defined by their subunit composition. The presence of the δ-subunit results in a high-affinity receptor resistant to benzodiazepines but sensitive to ZnCl2 and the neurosteroid THDOC (Saxena and Macdonald 1994; Wohlfarth et al. 2002). In contrast, the presence of the γ2-subunit results in a low-affinity receptor that can be facilitated by benzodiazepines such as diazepam but is resistant to the facilitatory effect of THDOC and the blocking effect of ZnCl2. Consistent with the suggestion that there are at least two types of GABAA receptors present in sensory neurons (Lee et al. 2012), there were neurons in which GABA currents were potentiated by THDOC (Fig. 4, A and B) and inhibited by ZnCl2 (Fig. 4, C and D) as well as facilitated by diazepam (Fig. 4, E and F). THDOC- and ZnCl2-sensitive GABA currents were present in every neuron tested from both naive and inflamed animals. However, this was not the case for diazepam. Only 34.4% (10 of 29) of the neurons from naive animals were responsive to diazepam as determined by a 20% increase in evoked current. In contrast, the percentage of diazepam-responsive neurons from inflamed rats increased to 70% (14 of 20, P < 0.05, χ2-test). Despite the heterogeneity among neurons with respect to the presence of different current types, the densities of THDOC-, ZnCl2-, and diazepam-sensitive current were all significantly increased in neurons from inflamed rats.
Fig. 4.
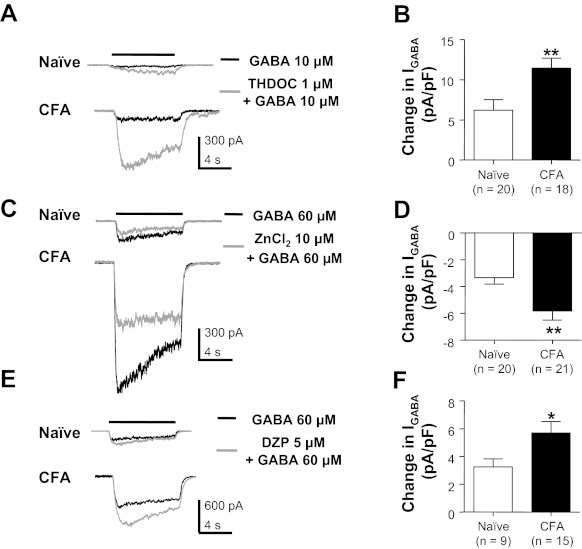
Pharmacological analysis of GABA currents in cutaneous neurons from naive and inflamed rats. GABA was applied before (black traces) and after (gray traces) the application of compounds used to facilitate or block specific GABAA receptor subtypes. 3α,21-Dihydroxy-5α-pregnan-20-one (THDOC) potentiated GABA currents in cutaneous neurons from both naive and inflamed (CFA) rats (A), where pooled data (B) indicated that the increase in current was significantly larger in neurons from inflamed rats. In contrast, Zn2+ attenuated GABA currents in DRG neurons from both naive and inflamed rats (C). Again, the magnitude of the Zn2+-sensitive current was significantly larger in neurons from inflamed rats (D). Diazepam (DZP) potentiated GABA-evoked current in cutaneous neurons from both naive and inflamed rats (E); data pooled from neurons responsive to DZP (F) indicated that DZP-induced increase in current was significantly larger in neurons from inflamed rats. *P < 0.05, **P < 0.01.
GABA “excites” a subpopulation of cutaneous neurons from inflamed rats.
Cumulative distributions of GABA current density in cutaneous neurons from naive and inflamed rats suggested the emergence of a subpopulation of neurons in the presence of inflammation in which GABA current density was selectively increased. That is, the frequency distribution for naive neurons was well fitted with a single-exponential equation, whereas the distribution for inflamed neurons required a double-exponential equation. More importantly, the median of the frequency distribution for naive neurons (23.2 pA/pF) was almost identical to that for the lower-density group of inflamed neurons (23.1 pA/pF), while that of the higher-density group of inflamed neurons was 69.0 pA/pF (Fig. 5A). This shift in distribution suggested that the inflammation-induced increase in current was restricted to a subpopulation of cutaneous neurons. This observation is consistent with our recent results suggesting that persistent inflammation results in a shift in GABA signaling in a subpopulation of cutaneous DRG neurons (Zhu et al. 2012). As with our previous study, Ca2+ imaging was used to screen cutaneous neurons from naive and inflamed rats for the presence of an excitatory response. While we acknowledge that an evoked Ca2+ transient is an indirect measure of an excitatory response, our previous data indicate that the Ca2+ transient is due to Ca2+ influx following a GABAA receptor-mediated depolarization-induced activation of low-voltage-activated Ca2+ channels (Zhu et al. 2012). Consistent with these previous results, none of the 12 neurons from naive rats responded to GABA (3 mM) with an increase in intracellular Ca2+. In contrast, 8 of the 15 neurons from inflamed rats responded to GABA with a reproducible increase in intracellular Ca2+ (Fig. 5, B and C). All of these neurons (8 of 8) were responsive to capsaicin and had a small-diameter cell body size, suggesting that they were nociceptive and could account for the observation that a shift in GABA signaling contributes to inflammatory hyperalgesia.
Fig. 5.
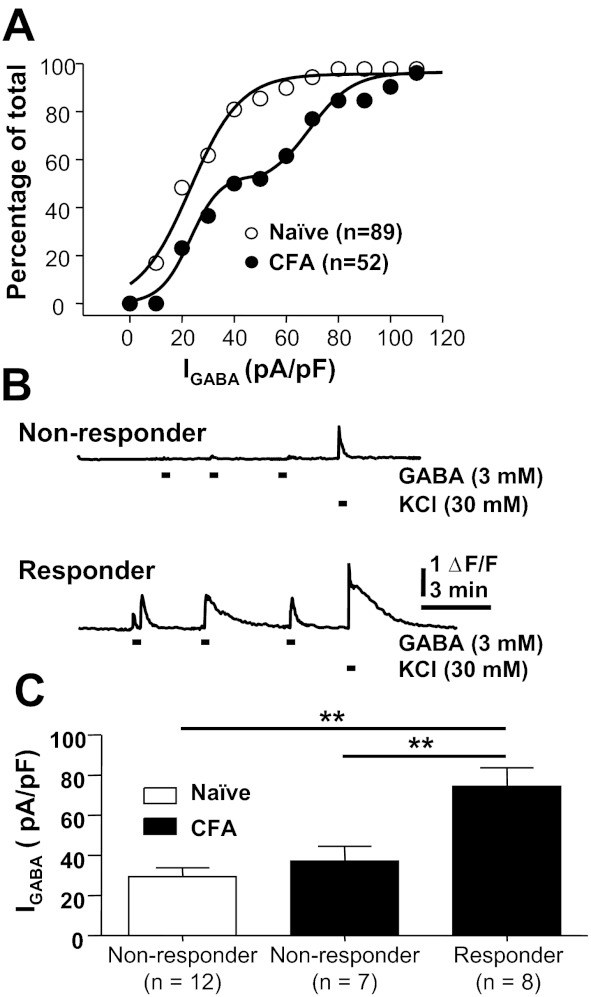
Inflammation-induced increase in GABA current density in a subpopulation of cutaneous neurons. A: cumulative distribution of peak GABA current density in cutaneous neurons from naive and inflamed (CFA) rats. Both curves are fitted with logistic equations used to estimate the median current density. The biexponential rise of the cumulative distribution curve for neurons from inflamed rats suggested that the increase in current density was restricted to a subpopulation of cutaneous neurons. B: fura-2-based Ca2+ imaging was used to screen for the presence of GABA-mediated “excitation” of cutaneous neurons. Neurons in which GABA (3 mM) failed to drive an increase in intracellular Ca2+ were considered nonresponders (top), while those in which GABA evoked an increase in Ca2+ were considered responders (bottom). Data from both neurons in B were from inflamed rats. ΔF/F, increase in fluorescence ratio. C: GABA (1 mM) current density was assessed in neurons from naive and inflamed rats prescreened with Ca2+ imaging. All 12 cutaneous neurons from naive rats were nonresponders, while over half of the neurons from inflamed rats were responders. Pooled current density measurements for the neurons in each group indicated that the increase in current density was restricted to the responder neurons from the inflamed group. **P < 0.01.
GABA current density was assessed after each neuron was screened with Ca2+ imaging. Pooled data (Fig. 5C) from the 27 neurons studied in this way indicated that the current density in GABA responders (74.35 ± 9.23, n = 8) was significantly greater than that in nonresponders from inflamed rats (37.12 ± 7.42, n = 7) as well as those from naive rats (29.46 ± 4.38, n = 12; P < 0.01). While an increase in current density was observed in only ∼50% of cutaneous neurons from inflamed rats, prescreening was not used in subsequent experiments, in order to avoid potential confounds associated with fura-2 imaging.
Time course of inflammation-induced increase in GABA current.
All previous data from inflamed rats were collected from cutaneous neurons harvested 72 h after the induction of inflammation with CFA. In an effort to implicate mechanisms that may contribute to the inflammation-induced increase in GABA current in cutaneous neurons, we assessed the time course for the development of the increase in current density. Neurons were harvested for subsequent study 24, 48, and 120 h after CFA injection. The increase in current density took time to develop, as it was not significant until 3 days after the induction of inflammation (Fig. 6). Importantly, with respect to the potential role of the increase in GABA current density in inflammatory hyperalgesia, an increase in current density was no longer significant by 5 days after inflammation, a time point at which rats are well on their way to recovery from the inflammatory hyperalgesia (Boegel et al. 2011).
Fig. 6.
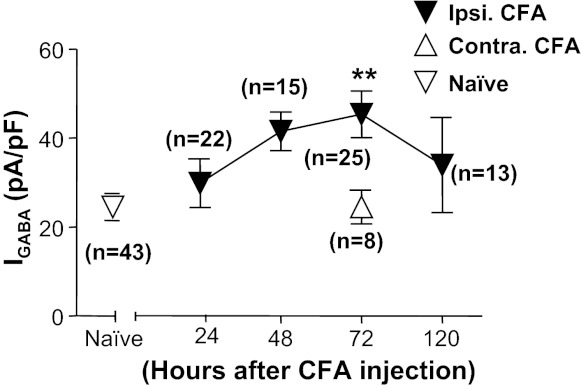
Time course of CFA-induced increase of GABA current density in cutaneous neurons. Data were collected from cutaneous neurons from naive rats and from rats 24, 48, 72, and 120 h after the induction of inflammation (ipsi CFA). A group of neurons was also studied from the side contralateral (contra) to the site of inflammation at the 72 h time point. There was a significant effect of time after inflammation relative to control, where post hoc analysis indicated that the current density at 72 h after inflammation was significantly greater than that in neurons from naive rats and in neurons contralateral to the site of inflammation. **P < 0.01.
Inflammation was associated with no detectable changes in GABAA receptor subunit mRNA or protein levels.
The delayed increase in GABA current density in the presence of persistent inflammation suggested that the increase may be due to increase in receptor subunit expression and/or transcriptional modulation. Semiquantitative, conventional PCR was initially used to screen mRNA extracted from L4 and L5 ganglia for inflammation-induced changes in the expression of 19 GABAA receptor subunits. While the results of this analysis indicated that mRNAs encoding all 19 subunits were detected in mRNA extracted from whole DRG, there were no detectable differences between naive and inflamed ganglia with respect to the mRNA levels of any of the subunits assessed (n = 3 per group; Fig. 7). To confirm the negative results obtained with the semiquantitative approach employed, real-time PCR analysis was used to assess levels of δ- and α1-subunits, where the δ-subunit appeared to contribute to the inflammation-induced increase in current density, according to our pharmacological analysis. The β2-subunit was included as an additional control. Despite the increased sensitivity of the real-time PCR analysis, no differences between naive and inflamed ganglia were detected with respect to the expression of any of these three subunits (n = 3 per group; Fig. 7, inset).
Fig. 7.
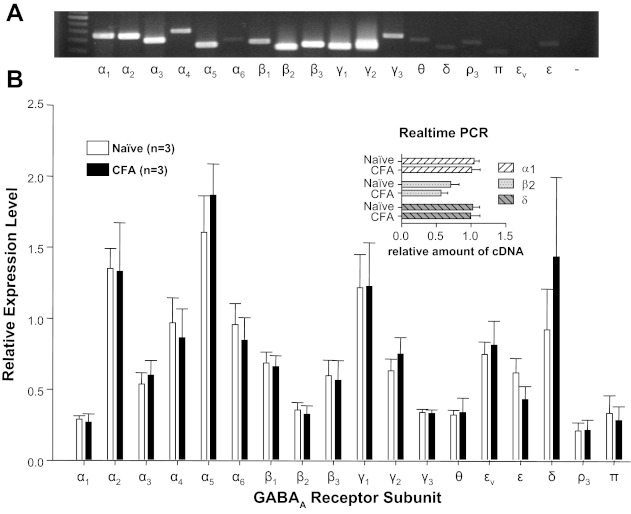
GABAA receptor subunit expression in L4-L5 DRG from naive and inflamed rats. A: example of an ethidium bromide-stained gel loaded with the PCR product of each of the GABAA receptor subunits. A size marker was loaded in the 1st lane, and the subunit amplified is indicated below each subsequent lane; εv is a splice variant of the ε-subunit, and “−” is from a control reaction in which the “template” was generated from a reverse transcription reaction in which no reverse transcriptase was added to the reaction mixture. B: pooled data from mRNA harvested from naive and inflamed (CFA) rats amplified with conventional PCR were normalized to expression levels of GAPDH. All subunits were detectable with <35 cycles of amplification, yet there were no detectable differences in relative expression levels between naive and inflamed rats. Real-time PCR (inset) was used to confirm the absence of a detectable change in the expression levels of α1-, β2-, and δ-subunits in mRNA extracted from whole DRG from naive and inflamed rats.
Given the evidence that inflammation is associated with increases in protein levels that appear to be due to posttranscriptional processing (Ji et al. 2002), we assessed total protein levels of the δ-, γ2-, and β2/3-subunits in L4 and L5 DRG from naive and inflamed rats by Western blot. Consistent with the results of our mRNA analysis, there was no detectable increase in subunit protein levels in DRG from inflamed rats relative to that in naive rats (n = 4 per group; Fig. 8).
Fig. 8.
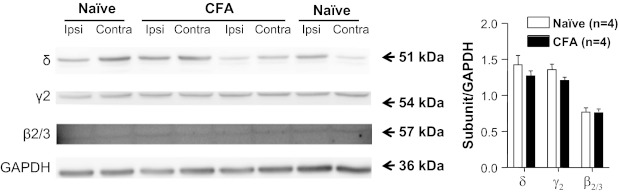
GABAA receptor subunit protein levels in DRG from naive and inflamed (CFA) rats. Levels of δ-, γ2-, and β2/3-subunit protein were assessed in the total protein extracted from L4-L5 ganglia ipsilateral (Ipsi) or contralateral (Contra) to the site of inflammation (or left side), harvested from 4 naive rats and 4 inflamed rats. GAPDH was used as a loading control. Pooled data are plotted on right. There was no apparent inflammation-induced change in any of these 3 subunits.
Tyrosine kinase inhibitor genistein abolished the increase of GABA current in inflamed DRG neurons.
While the failure to detect inflammation-induced changes in mRNA or protein levels for GABAA receptor subunits should be viewed with caution given the relatively small subpopulation of neurons in which these changes were manifest, these negative results suggested that the increase in current density may be due to a persistent change in posttranslational receptor regulation. GABAA receptors are substrates of multiple kinases, and phosphorylation of GABAA receptors may have a profound influence on current amplitude, kinetics, and trafficking (Kittler and Moss 2003). Therefore, we next assessed the potential contribution of changes in relative levels of kinase activity to the inflammation-induced increase in GABA current.
Despite evidence that GABA currents may be increased or decreased by increases in kinase activity, where the direction of the change in current amplitude depends on both the specific kinase and the subunit composition, we started with the nonselective protein kinase inhibitor K252a, because there is evidence that the activity of multiple kinases is upregulated during inflammation, or at least in response to an array of inflammatory mediators (Gold and Gebhart 2010). The impact of a 6-min application of K252a (1 μM) on GABA-evoked currents was assessed relative to baseline, where the stability of baseline currents was determined with at least three consecutive GABA applications. Current density was significantly (P < 0.05) reduced (relative to control) in neurons from both naive and inflamed rats by K252a. However, the observation that the decrease in current density was significantly larger in neurons from naive rats (Fig. 9A) suggested that if the relative level of kinase activity was responsible for the inflammation-induced increase in current, it would likely reflect a specific class of kinase.
Fig. 9.
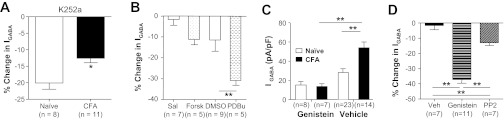
Influence of kinase activity on GABA current density in cutaneous neurons from naive and inflamed (CFA) rats. A: GABA current density was significantly reduced during a 6-min incubation with general kinase inhibitor K252a (1 μM). However, the decrease in current was significantly greater in neurons from naive rats than those from inflamed rats. B: there was no significant influence of a 6-min incubation with forskolin (Forsk, 10 μM) on GABA current density in neurons from naive rats. In contrast, 6 min with phorbol 12,13-dibutyrate (PDBu) resulted in a significant decrease in GABA current density. Each comparison was made to control neurons incubated with vehicle [saline (Sal) for forskolin and DMSO for PdBu] specific to each compound. C: cutaneous neurons from naive and inflamed rats were preincubated (20 min) with genistein (10 μM, which was sufficient to totally abolish the inflammation-induced increase in GABA current density). D: GABA current density in neurons from inflamed rats was significantly attenuated after a 6-min incubation with genistein (10 μM) or PP2 (10 μM) relative to vehicle (Veh)-treated control neurons. **P < 0.01.
To begin to explore the contribution of specific kinases to GABA current density in cutaneous DRG neurons, we next assessed the impact of increases in PKA and PKC activity on DRG neurons from naive animals, because of evidence that both are involved in inflammatory hyperalgesia (Khasar et al. 1999; Taiwo and Levine 1991). We included the study of PKC in this series of experiments because of evidence that PKC activation can increase GABAA currents (Poisbeau et al. 1999; Weiner et al. 1994), despite results from many studies suggesting that PKC activation drives a decrease in GABAA current (Kellenberger et al. 1992; Kittler et al. 2000; Kumar et al. 2005; Ragozzino and Eusebi 1993). GABA currents were evoked before and 5 min after the application of the adenylate cyclase activator forskolin (10 μM; Gold et al. 1998), as an indirect way to stimulate PKA activity, and the PKC activator PDBu (1 μM; Gold et al. 1998). Only PDBu produced significant decrease in current density relative to vehicle-treated control neurons (Fig. 9B), as the forskolin-induced decrease in current was not significant. These results argue against a role for an increase in PKA activity as a mechanism underlying the inflammation-induced increase in current density and suggest that for PKC to contribute to the inflammation-induced increase in GABA current density there would have to be a relative decrease in PKC activity.
Because of compelling data indicating that tyrosine kinase-dependent phosphorylation of GABAA receptors results in a decrease in receptor endocytosis and consequently an increase in current density (Michels and Moss 2007), we next assessed the impact of the general tyrosine kinase inhibitor genistein on GABA current in DRG neurons. Preincubating cutaneous neurons from naive rats with genistein (10 μM) for 20 min had no significant influence on current density (Fig. 9C). However, the same treatment of neurons from inflamed rats totally abolished the inflammation-induced increase of GABA current density (Fig. 9C).
Given evidence that Src kinase is activated in DRG neurons in the presence of persistent inflammation (Li et al. 2006) and the availability of relatively specific inhibitors of this kinase such as PP2 (Li et al. 2006), we next sought to determine the extent to which Src was the target for genistein in cutaneous neurons. Cutaneous neurons from inflamed rats were incubated with PP2 (10 μM) or genistein (10 μM) for 6 min after assessment of baseline GABA current density. As with genistein, GABA current density was significantly (P < 0.05) reduced in neurons incubated with PP2 compared with those incubated in vehicle control (Fig. 9D). However, the magnitude of PP2-induced suppression of GABA current density was significantly less than that obtained with genistein (Fig. 9D), suggesting that protein tyrosine kinase(s) other than Src may also contribute to the increase of GABA current following inflammation.
Role of endocytosis in genistein-induced inhibition of GABA current.
Because tyrosine phosphorylation of the γ2-subunit blocks dynamin-dependent endocytosis of GABAA receptors, resulting in an increase in GABA current (Kittler et al. 2008; Kittler and Moss 2003), the decoy inhibitory peptide, P4, was used to determine the contribution of endocytosis to the inflammation-induced changes in GABAA current density. A scrambled peptide was used as a control for the actions of the P4 peptide. In the presence of the P4 peptide, a 20.3 ± 6.6% (n = 11) increase in GABAA current was observed over 6 min. This increase was not significantly (P > 0.05) different from the increase observed in the presence of the control peptide over the same time period 3.8 ± 5.2% (n = 9). However, the genistein-induced suppression of GABA current was significantly attenuated in neurons treated with the P4 peptide (Fig. 10).
Fig. 10.
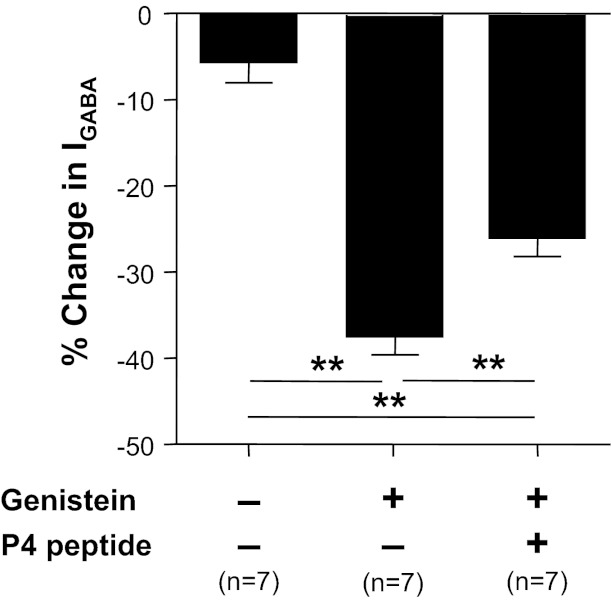
Dynamin-dependent receptor cycling contributes to the regulation of GABA current density in cutaneous neurons. Cutaneous neurons from inflamed rats were incubated with vehicle control, genistein (10 μM), or the combination of dynamin inhibitory peptide (P4, 20 μM) and genistein (10 μM) for 6 min. Genistein-induced suppression of GABA current was significantly attenuated in the presence of P4 peptide. **P < 0.01.
Intrathecal injection of tyrosine kinase inhibitor genistein reversed the inflammation-induced switch in GABAA signaling.
To begin to assess the functional consequences of an inflammation-induced increase in the relative level of tyrosine kinase activity on spinal GABAA receptor signaling, we assessed the impact of tyrosine kinase inhibition on muscimol-induced changes in nociceptive threshold. Naive or inflamed rats previously fitted with intrathecal catheters were treated with genistein or its inactive isoform, genistin (each at 0.1 mg/10 μl), 20 min prior to the intrathecal administration of muscimol (0.2 μg /10 μl). Nociceptive threshold was determined before each intrathecal injection and then again 10 min after muscimol. Doses and timing of genistein and muscimol were based on results from previous studies (Anseloni and Gold 2008; Guo et al. 2002). The impact of muscimol on mechanical threshold was calculated as percent change from the mechanical threshold obtained after the first intrathecal injection. As expected, inflammation was associated with a significant decrease in mechanical threshold from 67.94 ± 6.05 g (n = 17) to 34.87 ± 5.60 g (n = 13) (Fig. 11A). There was no significant influence of genistein treatment on mechanical threshold in either naive or inflamed rats. However, there was a significant interaction between inflammation and treatment prior to the administration of muscimol (P < 0.01, 2-way ANOVA), where post hoc analysis confirmed that this interaction was due to a contrasting influence of muscimol on nociceptive threshold in the presence of inflammation that depended on whether or not animals had received the active tyrosine kinase inhibitor: in control-treated animals muscimol exacerbated inflammatory hyperalgesia, further decreasing the mechanical threshold by 43.7 ± 4.0% (n = 5), while in genistein-treated animals muscimol attenuated inflammatory hyperalgesia, increasing the mechanical threshold by 32.1 ± 18.4% (n = 8; Fig. 11B).
Fig. 11.
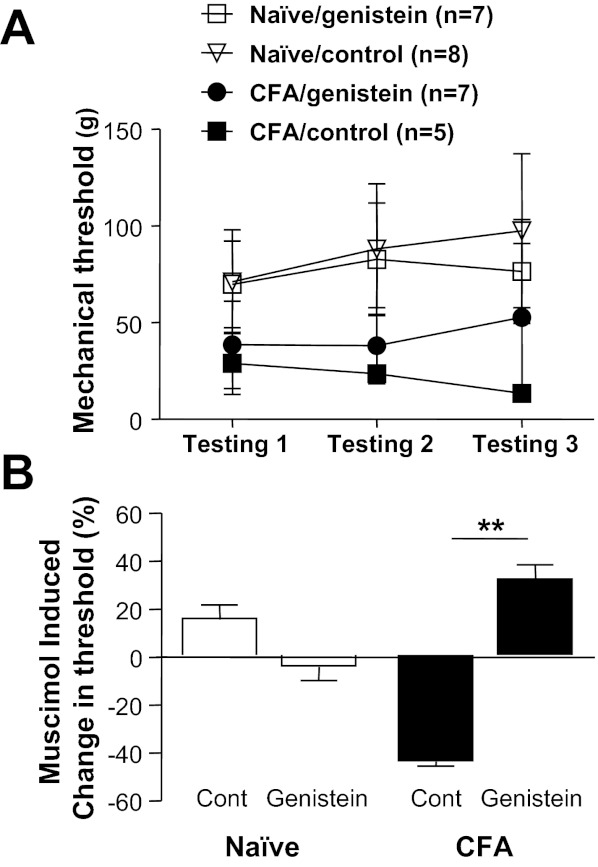
The inflammation-induced shift in spinal GABAA signaling is reversed by intrathecal (i.t.) administration of genistein. Test agents were administered via chronically implanted i.t. catheters to naive and inflamed (CFA) rats. A: mechanical nociceptive threshold was determined before i.t. injection of test agents (testing 1), 20 min after the injection of genistein or genistin (0.1 mg/10 μl, testing 2), and then again 10 min after the injection of muscimol (0.1 μg/10 μl, testing 3). As expected, inflammation was associated with a significant decrease in mechanical threshold. B: a 2-way ANOVA was used to assess the impact of inflammation and tyrosine kinase inhibitor on muscimol-induced changes in nociceptive threshold. The interaction between these 2 factors was significant, with post hoc analysis indicating that this was due to the differential response to muscimol in inflamed rats, which depended on whether or not rats had received an active tyrosine kinase inhibitor. **P < 0.01.
DISCUSSION
The initial goal of this study was to test the hypothesis that a high-affinity extrasynaptic GABAA receptor that contributes to the inflammation-induced shift in spinal GABAA signaling is increased in primary afferent neurons. While patch-clamp data only provided partial support for this hypothesis, they led to a number of novel observations related to GABAA receptor signaling in primary afferent neurons as well as mechanisms of inflammatory hyperalgesia. First, consistent with our initial hypothesis, we observed an increase in GABAA current with pharmacological properties of extrasynaptic receptors. Second, this increase in current was restricted to a subpopulation of neurons in which GABA was able to evoke an excitatory response. Third, the increase in GABA current was due to an increase in both low-affinity and high-affinity currents. Fourth, while the time course for the development of the inflammation-induced increase in GABA current density was relatively slow, there was no evidence of an increase in GABAA receptor subunit mRNA or protein. Fifth, and potentially most striking, acute inhibition of tyrosine kinase by genistein completely abolished the persistent inflammation-induced increase of GABA current. Sixth, the actions of genistein were partially blocked by a dynamin inhibitory peptide. Seventh, spinal administration of genistein was able to acutely reverse the persistent inflammation-induced switch in spinal GABAA signaling. A model summarizing these main observations is illustrated in Fig. 12.
Fig. 12.
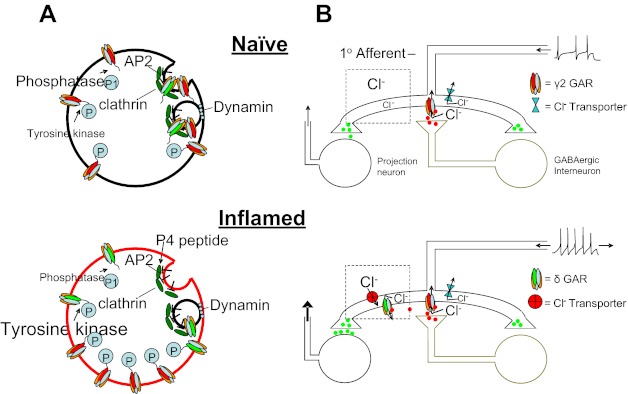
Model of inflammation-induced changes in GABAA receptor (GAR) signaling. A: at a cellular level, in the absence of inflammation (top), the relative level of tyrosine kinase activity is low. Consequently, turnover of GABAA receptors is high, with many receptors internalized in clathrin-coated pits via a dynamin-dependent process. In the presence of inflammation (bottom), there is an increase in the relative level of tyrosine kinase activity and consequently an increase in the phosphorylation of GABAA receptors. As this phosphorylation of receptors can mask an AP2 binding site, internalization of GABAA receptors is decreased and current density is increased. Application of genistein reduces tyrosine kinase activity, resulting in a decrease in phosphorylation of GABAA receptors, an increase in receptor internalization, and a decrease in current. The P4 peptide is designed to block the binding of dynamin to amphiphysin and thereby prevent endocytosis, attenuating the decrease in GABAA current associated with genistein. B: GABAA receptor activation in the spinal cord in normally inhibitory, where a presynaptic site of action is the primary target illustrated. In the presence of inflammation, there is an increase in both synaptic and extrasynaptic GABAA receptors. However, to account for our previous observation (Anseloni and Gold 2008) as well as those of others (Knabl et al. 2008; Witschi et al. 2011) that benzodiazepines retain their analgesic efficacy in the presence of inflammation, we hypothesize that the pronociceptive actions of GABA are due to activity at extrasynaptic GABAA receptors that are coupled to a relatively depolarized Cl− equilibrium potential. These receptors are targeted by the low dose of muscimol used in the present study. A decrease in GABAA current density via genistein eliminates the pronociceptive actions of muscimol, leaving only a residual antinociceptive activity as synaptic receptors.
Extrasynaptic receptors composed of α4/α6- and δ-subunits are blocked by Zn2+ and potentiated by THDOC. Thus, while not highly specific for receptors with this subunit composition, results with these compounds suggest that extrasynaptic receptors are present in cutaneous neurons from naive rats and that this receptor density is increased with inflammation. Consistent with the presence of these receptors in naive rats, mRNA encoding these subunits was detected in whole ganglia, as was δ-subunit protein. Recent results from unlabeled DRG neurons suggest that the majority of δ-subunit-containing receptors are normally trafficked out of the DRG neuron cell body (Lee et al. 2012), suggesting that high-affinity currents detected in the present study are an underrepresentation of relative levels of GABAA receptor subunits produced in sensory neurons. That additional GABAA receptor subtypes contribute to the total current density in cutaneous neurons is suggested by the observations that Zn2+ blocked <50% of the current in neurons from naive and inflamed rats. The presence of mRNA encoding ε- and ρ-subunits suggests that at least some of the additional subtypes in cutaneous neurons will also be extrasynaptic.
The inflammation-induced increase in low-affinity GABA current in addition to the increase in proportion of neurons with benzodiazepine-sensitive current was not originally anticipated because of evidence that there is a decrease in the analgesic potency of the benzodiazepine agonist midazolam in the presence of inflammation (Anseloni and Gold 2008). Nevertheless, with recent data confirming that at least some of the analgesic efficacy of spinal benzodiazepines is due to an action on primary afferent neurons (Witschi et al. 2011), the increase in low-affinity current suggests that at least some of the increase in GABA current may serve to counter pronociceptive processes upregulated in the presence of inflammation, including excitatory GABAA signaling (Fig. 12).
One of the most striking observations in this study, and potentially most important from the perspective of novel therapeutic approaches for the treatment of pain, was that indicating that persistent inflammation is associated with a relative increase in a tyrosine kinase activity. While an increase in kinase activity has long been appreciated as a mechanism of inflammatory hypersensitivity (Khasar et al. 1999; Taiwo and Levine 1991), the observation that inhibitors of prostaglandin production retain their analgesic efficacy in the face of persistent inflammation suggests that an increase in kinase activity is only maintained by elevated levels of agonist. On the other hand, more recent evidence has highlighted distinct pathways resulting in an increase in kinase activity in the afferent cell body. These include activity-dependent increases in MAP kinase (MAPK)-ERK activation (Mizushima et al. 2007) and trophic factor-dependent increases in phosphatidylinositol 3-kinase (PI3K) and MAPK (Patapoutian and Reichardt 2001). However, these increases in kinase activity are either transient (Mizushima et al. 2007) or studied in the context of pathways, such as CREB or JUNK, involved in the regulation of gene expression (Doya et al. 2005; Qiao and Vizzard 2004). Negative results with mRNA and Western blot analysis do not rule out the possibility that there are changes in the expression of GABAA receptor subunits that contributed to the inflammation-induced increase in GABAA current density. However, results of the present study clearly indicate that altered expression and translation are not the only ways in which persistent changes in afferent phenotype are maintained. In addition to these more traditional mechanisms, a relative increase in tyrosine kinase activity also plays a critical role in the maintenance of an “injury” phenotype.
As a general tyrosine kinase inhibitor, genistein can inhibit receptor tyrosine kinase (RTK) and protein tyrosine kinase (PTK). There is even evidence that genistein can directly inhibit GABAA currents, at least α1β2-subunit-containing receptors in a heterologous expression system (Huang et al. 1999). With respect to the latter mechanism of action, we suggest that a direct influence of genistein in GABAA currents in the present study is likely to be minimal, as no more than a 15% block of currents was reported in this previous study with a genistein concentration five times higher than that used in the present study. Furthermore, genistein increased current desensitization in this previous study, an effect not observed in DRG neurons. With respect to the former target of genistein action, nerve growth factor (NGF), a prototypical ligand for RTKs, plays critical role in the initiation and maintenance of inflammatory hyperalgesia (McMahon 1996). The retrograde transport of an activated RTK could account for the delayed increase in GABAA current observed in the present study. However, the available evidence also argues against this specific RTK in the increase in GABA currents in cutaneous neurons, as even prolonged application of NGF was shown to influence the kinetics but not the amplitude of GABA current in DRG neurons (Fabbro and Nistri 2004). In contrast, previous evidence indicates that there is a persistent increase in src activity in DRG neurons in the presence of inflammation (Li et al. 2006). src phosphorylates β- and γ2-subunits of the GABAA receptor, resulting in an increase of GABA current (Brandon et al. 2001; Moss et al. 1995; Wan et al. 1997), and the src kinase-selective inhibitor PP2 resulted in a significant reduction in the inflammation-induced increase in GABA current in cutaneous neurons. Because the concentration of PP2 employed should have been sufficient for complete block of src (Tong and Gibb 2008), the observation that the influence of PP2 was less than that of genistein raises the possibility that other tyrosine kinases such as fyn and hck (Jeong et al. 2008; Jurd et al. 2010) contributed to the inflammation-induced increase in GABA current.
Tyrosine phosphorylation can increase GABA current in two different ways: changes in channel gating and changes in channel recycling. With respect to gating, an increase in receptor phosphorylation results in an increase in open channel probability associated with an increase in mean channel open time (Moss et al. 1995). With respect to channel recycling, there is a relatively high level of receptor turnover, where clathrin-dependent endocytosis mediates receptor internalization (Kittler et al. 2008). Tyrosine kinase-dependent phosphorylation of at least the γ2-subunit prevents the association between the receptor and internalization machinery, resulting in an increase in GABAA receptor density. The observation that the P4 peptide partially blocked the genistein-induced decrease in current density suggests that at least some of the inflammation-induced increase in current density is due to a decrease in GABAA receptor endocytosis and the subsequent accumulation of receptors on the plasma membrane (Fig. 12).
Because of the complexities of tyrosine kinase signaling in nociceptive processing, the impact of genistein on the behavioral changes in response to muscimol was the critical test of the functional impact of the changes in GABAA signaling we had described in vitro. In this regard, the minimal influence of muscimol in naive rats pretreated with genistein was consistent with the results from our in vitro experiments, which suggested that there may be a resting level of tyrosine kinase activity that maintains even baseline levels of GABAA receptors in the plasma membrane: a decrease in spinal GABAA receptors in naive rats would attenuate the antinociceptive effects of muscimol. The situation is more complex in the presence of inflammation, where genistein not only attenuated the pronociceptive actions of muscimol but conferred antinociceptive activity. This occurred despite the fact that genistein reduced GABA current density in inflamed neurons to a level that was comparable to that in naive neurons. The implication of these observations is that there is an interaction between the tyrosine kinase-dependent increase in GABA current density and other tyrosine kinase-independent changes in nociceptive processing. For example, the increase in GABAA current density may be associated with changes in voltage-gated Na+ channel properties, where the latter are tyrosine kinase independent. If such changes in Na+ channel properties result in an appropriate shift in the balance of channel activation and inactivation, then they may confer an antinociceptive influence to muscimol despite the decrease in GABA current density.
The use of muscimol was a critical test of our in vitro results, in part because we had anticipated an analgesic effect of genistein alone, both because of the attenuation of the pronociceptive phosphorylation of the NR2B subunit of the NMDA receptor (Guo et al. 2002) and because of evidence that in the presence of inflammation endogenous GABAA receptor activation contributes to inflammatory hyperalgesia (Anseloni and Gold 2008). Thus the absence of an effect of genistein alone raises the possibility that there are antinociceptive processes in the spinal cord dependent on tyrosine kinase activity that serve to counter pronociceptive changes such as the increase in synaptic GABAA receptors (Fig. 12). Along these lines, while the dose of muscimol used in the present study was chosen to bias activity at high-affinity receptors, given evidence of the upregulation of receptors composed of several different subunit compositions, it will be of interest in future studies to employ additional pharmacological tools in vivo to begin to tease out the relative contribution of GABAA receptor types to the observed changes in behavior.
In summary, the results of the present study have revealed a novel mechanism underlying the regulation of GABAA receptors in DRG neurons. This mechanism, an increase in the relative activity of tyrosine kinase, contributes to a shift in spinal GABAA signaling from inhibition to excitation. While the hypothesis that a shift in GABAA signaling is mediated by a Na+-K+-Cl−-dependent depolarizing shift in the Cl− equilibrium potential is appealing in both its simplicity and its ability to account for many of the changes observed in association with relatively acute noxious stimuli (Cervero et al. 2003; Price et al. 2009; Willis 1999), results of the present study suggest that such a mechanism is insufficient to account for the changes in GABAA signaling observed in the presence of persistent inflammation. This added level of complexity serves as yet another reminder of why drug discovery in pain research has been so difficult.
GRANTS
This work was supported in part by National Institute of Neurological Disorders and Stroke Grant NS-063010-01 (M. S. Gold) and Foundation for Anesthesia Education and Research (FAER) MSARF 01/01/2011 (S. Dua).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z. and M.S.G. conception and design of research; Y.Z. and S.D. performed experiments; Y.Z., S.D., and M.S.G. analyzed data; Y.Z. and M.S.G. interpreted results of experiments; Y.Z. and M.S.G. prepared figures; Y.Z. drafted manuscript; Y.Z., S.D., and M.S.G. approved final version of manuscript; M.S.G. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Ronald Dubner, Man-Kyo Chung, and Steven Prescott for helpful feedback on the preparation of this manuscript.
REFERENCES
- Anseloni VC, Gold MS. Inflammation-induced shift in the valence of spinal GABA-A receptor-mediated modulation of nociception in the adult rat. J Pain 9: 732–738, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegel K, Gyulai FE, Moore KK, Gold MS. Deleterious impact of a gamma-aminobutyric acid type A receptor preferring general anesthetic when used in the presence of persistent inflammation. Anesthesiology 115: 782–790, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ. Constitutive tyrosine phosphorylation of the GABAA receptor gamma2 subunit in rat brain. Neuropharmacology 41: 745–752, 2001 [DOI] [PubMed] [Google Scholar]
- Cervero F, Laird JM, Garcia-Nicas E. Secondary hyperalgesia and presynaptic inhibition: an update. Eur J Pain 7: 345–351, 2003 [DOI] [PubMed] [Google Scholar]
- Doya H, Ohtori S, Fujitani M, Saito T, Hata K, Ino H, Takahashi K, Moriya H, Yamashita T. c-Jun N-terminal kinase activation in dorsal root ganglion contributes to pain hypersensitivity. Biochem Biophys Res Commun 335: 132–138, 2005 [DOI] [PubMed] [Google Scholar]
- Fabbro A, Nistri A. Chronic NGF treatment of rat nociceptive DRG neurons in culture facilitates desensitization and deactivation of GABAA receptor-mediated currents. Br J Pharmacol 142: 425–434, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flake NM, Bonebreak DB, Gold MS. Estrogen and inflammation increase the excitability of rat temporomandibular joint afferent neurons. J Neurophysiol 93: 1585–1597, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med 16: 1248–1257, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci 18: 10345–10355, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci 22: 6208–6317, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RQ, Fang MJ, Dillon GH. The tyrosine kinase inhibitor genistein directly inhibits GABAA receptors. Brain Res Mol Brain Res 67: 177–183, 1999 [DOI] [PubMed] [Google Scholar]
- Jeong DG, Park WK, Park S. Artemin activates axonal growth via SFK and ERK-dependent signalling pathways in mature dorsal root ganglia neurons. Cell Biochem Funct 26: 210–220, 2008 [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36: 57–68, 2002 [DOI] [PubMed] [Google Scholar]
- Jurd R, Tretter V, Walker J, Brandon NJ, Moss SJ. Fyn kinase contributes to tyrosine phosphorylation of the GABAA receptor gamma2 subunit. Mol Cell Neurosci 44: 129–134, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Malherbe P, Sigel E. Function of the alpha1beta2gamma2S gamma-aminobutyric acid type A receptor is modulated by protein kinase C via multiple phosphorylation sites. J Biol Chem 267: 25660–25663, 1992 [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron 24: 253–260, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, Smith KR, McAinsh K, Arancibia-Carcamo IL, Saenger W, Haucke V, Yan Z, Moss SJ. Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc Natl Acad Sci USA 105: 3616–3621, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ. Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci 20: 7972–7977, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol 13: 341–347, 2003 [DOI] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451: 330–334, 2008 [DOI] [PubMed] [Google Scholar]
- Kumar S, Khisti RT, Morrow AL. Regulation of native GABAA receptors by PKC and protein phosphatase activity. Psychopharmacology (Berl) 183: 241–247, 2005 [DOI] [PubMed] [Google Scholar]
- Lee KY, Charbonnet M, Gold MS. Upregulation of high-affinity GABAA receptors in cultured rat dorsal root ganglion neurons. Neuroscience 208: 133–142, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Gold MS. Inflammatory mediators potentiate high affinity GABAA currents in rat dorsal root ganglion neurons. Neurosci Lett 518: 128–132, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Ennes HS, Trevisani M, Nicoletti P, Mittal Y, Mayer EA. Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol 291: G219–G228, 2006 [DOI] [PubMed] [Google Scholar]
- Lin Q, Wu J, Willis WD. Dorsal root reflexes and cutaneous neurogenic inflammation after intradermal injection of capsaicin in rats. J Neurophysiol 82: 2602–2611, 1999 [DOI] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol 577: 169–190, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB. NGF as a mediator of inflammatory pain. Philos Trans R Soc Lond B Biol Sci 351: 431–440, 1996 [DOI] [PubMed] [Google Scholar]
- Michels G, Moss SJ. GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol 42: 3–14, 2007 [DOI] [PubMed] [Google Scholar]
- Mizushima T, Obata K, Katsura H, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Mashimo T, Noguchi K. Intensity-dependent activation of extracellular signal-regulated protein kinase 5 in sensory neurons contributes to pain hypersensitivity. J Pharmacol Exp Ther 321: 28–34, 2007 [DOI] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature 377: 344–348, 1995 [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272–280, 2001 [DOI] [PubMed] [Google Scholar]
- Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci 19: 674–683, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev 60: 149–170, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao LY, Vizzard MA. Up-regulation of phosphorylated CREB but not c-Jun in bladder afferent neurons in dorsal root ganglia after cystitis. J Comp Neurol 469: 262–274, 2004 [DOI] [PubMed] [Google Scholar]
- Ragozzino D, Eusebi F. Inhibition of GABA and glycine responses by glutamate in rat hippocampal neurons. Brain Res 628: 115–120, 1993 [DOI] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Assembly of GABAA receptor subunits: role of the delta subunit. J Neurosci 14: 7077–7086, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA, Willis WD, Westlund KN. Inflammation-induced release of excitatory amino acids is prevented by spinal administration of a GABAA but not by a GABAB receptor antagonist in rats. J Pharmacol Exp Ther 271: 76–82, 1994 [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience 44: 131–135, 1991 [DOI] [PubMed] [Google Scholar]
- Tong H, Gibb AJ. Dopamine D1 receptor inhibition of NMDA receptor currents mediated by tyrosine kinase-dependent receptor trafficking in neonatal rat striatum. J Physiol 586: 4693–4707, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Man HY, Braunton J, Wang W, Salter MW, Becker L, Wang YT. Modulation of GABAA receptor function by tyrosine phosphorylation of beta subunits. J Neurosci 17: 5062–5069, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Zhang L, Carlen PL. Potentiation of GABAA-mediated synaptic current by ethanol in hippocampal CA1 neurons: possible role of protein kinase C. J Pharmacol Exp Ther 268: 1388–1395, 1994 [PubMed] [Google Scholar]
- White G. GABAA-receptor-activated current in dorsal root ganglion neurons freshly isolated from adult rats. J Neurophysiol 64: 57–63, 1990 [DOI] [PubMed] [Google Scholar]
- Willis WD., Jr Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res 124: 395–421, 1999 [DOI] [PubMed] [Google Scholar]
- Witschi R, Punnakkal P, Paul J, Walczak JS, Cervero F, Fritschy JM, Kuner R, Keist R, Rudolph U, Zeilhofer HU. Presynaptic alpha2-GABAA receptors in primary afferent depolarization and spinal pain control. J Neurosci 31: 8134–8142, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT, Macdonald RL. Enhanced neurosteroid potentiation of ternary GABAA receptors containing the delta subunit. J Neurosci 22: 1541–1549, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17: 1031–1036, 1976 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL. Effects of intrathecal strychnine and bicuculline on nerve compression-induced thermal hyperalgesia and selective antagonism by MK-801. Pain 54: 79–84, 1993 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL. Spinal pharmacology of thermal hyperesthesia induced by incomplete ligation of sciatic nerve. I. Opioid and nonopioid receptors. Anesthesiology 75: 817–826, 1991 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Lu S, Gold MS. Persistent inflammation increases GABA-induced depolarization of rat cutaneous dorsal root ganglion neurons in vitro. Neuroscience 220: 330–340, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]