

Abstract
Mutations in TRPM1 are found in humans with an autosomal recessive form of complete congenital stationary night blindness (cCSNB). The Trpm1−/− mouse has been an important animal model for this condition. Here we report a new mouse mutant, tvrm27, identified in a chemical mutagenesis screen. Genetic mapping of the no b-wave electroretinogram (ERG) phenotype of tvrm27 localized the mutation to a chromosomal region that included Trpm1. Complementation testing with Trpm1−/− mice confirmed a mutation in Trpm1. Sequencing identified a nucleotide change in exon 23, converting a highly conserved alanine within the pore domain to threonine (p.A1068T). Consistent with prior studies of Trpm1−/− mice, no anatomical changes were noted in the Trpm1tvrm27/tvrm27 retina. The Trpm1tvrm27/tvrm27 phenotype is distinguished from that of Trpm1−/− by the retention of TRPM1 expression on the dendritic tips of depolarizing bipolar cells (DBCs). While ERG b-wave amplitudes of Trpm1+/− heterozygotes are comparable to wild type, those of Trpm1+/tvrm27 mice are reduced by 32%. A similar reduction in the response of Trpm1+/tvrm27 DBCs to LY341495 or capsaicin is evident in whole cell recordings. These data indicate that the p.A1068T mutant TRPM1 acts as a dominant negative with respect to TRPM1 channel function. Furthermore, these data indicate that the number of functional TRPM1 channels at the DBC dendritic tips is a key factor in defining DBC response amplitude. The Trpm1tvrm27/tvrm27 mutant will be useful for elucidating the role of TRPM1 in DBC signal transduction, for determining how Trpm1 mutations impact central visual processing, and for evaluating experimental therapies for cCSNB.
Keywords: TRP channel, electroretinogram, congenital stationary night blindness, depolarizing bipolar cell
light-induced hyperpolarization of photoreceptors results in a decline in presynaptic glutamate release onto the dendrites of bipolar and horizontal cells. Hyperpolarization is conserved in horizontal and hyperpolarizing bipolar cells as a result of postsynaptic ionotropic glutamate receptors. In contrast, photoreceptor hyperpolarization is inverted in depolarizing bipolar cells (DBCs) as a result of a decrease in activation of postsynaptic metabotropic glutamate receptor 6 (GRM6) and subsequent opening of a transient receptor potential melastatin 1 protein (TRPM1)-containing cation-selective channel (Koike et al. 2010a, 2010b; Morgans et al. 2009, 2010; Shen et al. 2009; Shiells et al. 1981; Slaughter and Miller 1981).
Mutations in several genes disrupt photoreceptor-to-DBC synaptic transmission. Most encode proteins that control glutamate release from photoreceptor terminals or play a role in DBC signal transduction. Several are implicated in the Schubert-Bornschein (1952) class of congenital stationary night blindness (CSNB). Miyake et al. (1986) divided Schubert-Bornschein CSNB into “incomplete” (iCSNB) and “complete” (cCSNB) forms on the basis of clinical examination and functional characteristics. In this classification, the electroretinogram (ERG) is a critical test: while both iCSNB and cCSNB have normal a-waves, indicating preserved photoreceptor function (Penn and Hagins 1969), they differ with respect to the extent of involvement of postreceptoral ERG components (b-waves and oscillatory potentials). In patients with iCSNB, b-waves and oscillatory potentials are present but significantly reduced in amplitude, while in patients with cCSNB these response components are absent.
Mutations in CACNA1F and in CABP4 have been implicated in iCSNB (Bech-Hansen 1998; Strom et al. 1998; Zeitz et al. 2006), and both mediate photoreceptor glutamate release. In comparison, all four genes associated with human cCSNB (NYX: Bech-Hansen 2000; Pusch et al. 2000; GRM6: Dryja et al. 2005; Zeitz et al. 2005; TRPM1: Audo et al. 2009; Li et al. 2009; Nakamura et al. 2010; van Genderen et al. 2009; GPR179: Audo et al. 2012; Peachey et al. 2012) are expressed by DBCs, and ERG analyses show a comparable no b-wave (nob) phenotype observed in mouse mutants for Nyx (Gregg et al. 2007; Pardue et al. 1998), Grm6 (Maddox et al. 2008; Masu et al. 1995; Pinto et al. 2007), Gpr179 (Peachey et al. 2012), and Trpm1 (Koike et al. 2010a; Morgans et al. 2009; Shen et al. 2009). The availability of these models provides important opportunities to understand cCSNB and to evaluate experimental treatments for this disorder.
In this study, we report a new mouse model identified through an ERG screen of chemically mutagenized mice in the Translational Vision Research Models (TVRM) program of The Jackson Laboratory (Won et al. 2011). The nob phenotype we document in homozygous tvrm27 mutant mice is caused by a point mutation in the Trpm1 gene that affects the pore domain of the protein. The Trpm1tvrm27/tvrm27 mutant will be a valuable model for human cCSNB due to TRPM1 point mutations and will be useful in furthering our understanding of how TRPM1 channels function, how TRPM1 interacts with other proteins required for normal DBC signal transduction, and the role of TRPM1 with respect to trafficking and localization of the various components of the DBC signal transduction cascade.
METHODS
Mice and mapping.
All procedures used in animal experiments were approved by the Institutional Animal Care and Use Committees of the institutions involved. Homozygous tvrm27 mice were identified from a mutagenesis program (Won et al. 2011). Male C57BL/6J mice were mutagenized with ethylnitrosourea that was administered in intraperitoneal injections of 80 mg/kg for 3 wk (Justice et al. 2000). G3 offspring, generated by using a three-generation backcross mating scheme to identify recessive mutations (Herron et al. 2002), were screened by an ERG protocol (Hawes et al. 2000) to identify mice with abnormal ERGs.
Trpm1tm1Lex/tm1Lex, hereafter referred to as Trpm1−/− mice, were generated by Lexicon Genetics and acquired from the European Mouse Mutant Archive (www.emmanet.org). DBA/2J mice were obtained from The Jackson Laboratory. Grm6nob3 (Maddox et al. 2008) mice were obtained from The Jackson Laboratory. Nyxnob (Pardue et al. 1998) and Gpr179nob5 (Peachey et al. 2012) mutant mice were obtained from our local breeding colonies.
DNA was prepared from tail biopsies by proteinase K digestion and isopropyl alcohol extraction. DNA samples isolated from 261 tvrm27 F2 progeny were genotyped with 103 simple sequence length polymorphic markers distributed throughout the genome (Taylor et al. 1994).
Sequencing.
Total RNA was isolated from retinas with TRIzol reagent per the manufacturer's protocol (Life Technologies, Carlsbad, CA). cDNA was synthesized from total RNA with SuperScript II reverse transcriptase per the manufacturer's protocol (GE Biosystems).The entire coding region of Trpm1 was amplified with primers [Trpm1–001 (5′-ATGGGGTCCATGAGGAA-3′) and Trpm1–003 (5′-TCAGCACTCAGTTTCCGCGC-3′)], cloned, and sequenced on an Applied Biosystems (Carlsbad, CA) 3130XL Sequencer (using a 50-cm array and POP7 polymer). The causative mutation was confirmed to be present in genomic DNA.
Electroretinography.
After overnight dark adaptation, mice were anesthetized with ketamine (80 mg/kg) and xylazine (16 mg/kg) and their pupils were dilated with 1% tropicamide and 2.5% phenylephrine HCl eye drops. ERGs were recorded with a stainless steel wire contacting the anesthetized (1% proparacaine HCl) corneal surface that was referenced to a needle electrode placed in the cheek; a second needle electrode placed in the tail served as ground. Responses were amplified (0.03–1,000 Hz) and stored with an LKC (Gaithersburg, MD) UTAS E-3000 signal averaging system. For screening, a single flash luminance was used (1.4 log cd s/m2). ERGs were also recorded under stimulus conditions that allow the response properties of rod- and cone-driven components to be defined with published protocols (Chang et al. 2006; Gregg et al. 2007).
Electron and confocal microscopy: immunohistochemistry.
Eyes enucleated from mice killed by anesthetic overdose were placed in fresh fixative (2.5% glutaraldehyde-2% paraformaldehyde, 100 mM cacodylate, pH 7.4) for 30 min. Tissues were postfixed with 1% OsO4 for 1 h, en bloc stained with 2% uranyl acetate for 30 min, dehydrated in a graded ethanol series, and infiltrated and embedded in medium (Polybed 812; Polysciences, Warrington, PA). Thin sections were cut with an ultramicrotome (MT 7000; Ventana, Tucson, AZ), collected onto nickel grids, stained with 2% uranyl acetate and lead citrate, and imaged in a transmission electron microscope (Morgagni; FEI, Hillsboro, OR) at 80 kV. Additional details regarding these procedures are provided by Goldberg et al. (2007). Retinal structure and ribbon synapse ultrastructure were examined in both eyes of two wild-type (WT) and two Trpm1tvrm27/tvrm27 mice.
For confocal microscopy, dissected retinas were immersion fixed for 15 min in 4% (wt/vol) paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 (PB), then washed in PB, cryoprotected through a graded sucrose series, and frozen in OCT (Sakura Finetek, Torrence, CA)-20% sucrose (2:1; Barthel and Raymond 1990). Sixteen-micrometer sections were cut on a cryostat, mounted onto SuperFrost glass slides, air-dried, and stored at −80°C.
Sections were brought to room temperature, washed in PBS for 5 min and in PBS containing 0.5% (vol/vol) Triton X-100 (PBX) for 5 min and then incubated in blocking solution [PBX containing 5% (vol/vol) normal goat serum] for 1 h. Primary antibodies were diluted in blocking solution and incubated on retinal sections at room temperature overnight. Primary antibodies and dilutions used were anti-GRM6 (1:1,000; Koike et al. 2010a), anti-TRPM1 (1:100; Koike et al. 2010a), and anti-RIBEYE (1:1,000; BD Biosciences). After incubation with the primary antibody, sections were washed three times in PBS for 5 min each and subsequently incubated with fluorescently labeled secondary antibodies (1:1,000 in blocking solution) at room temperature for 1 h. Secondary antibodies were Alexa 488 goat anti-rabbit and Alexa 555 goat anti-mouse (Invitrogen, Carlsbad, CA). Slides were then washed three times in PBS and coverslipped with Immunomount (Thermo Shandon, Pittsburgh, PA). Sections were imaged on an Olympus (Center Valley, PA) FV1000 confocal microscope with a ×60 oil objective (1.45 NA). Images shown are maximum projections of confocal stacks, adjusted for contrast and brightness with FluoView software.
Bipolar cell patch-clamp electrophysiology.
After death, whole retinas were isolated from 4- to 6-wk-old mice and placed on a 0.65-μm cellulose acetate-nitrate membrane filter (Millipore, Billerica, MA) secured with vacuum grease to a glass slide adjacent to the recording chamber. Retinal slices (100 μm) were cut with a tissue slicer (Stoelting, Wood Dale, IL), transferred to the recording chamber while remaining submerged, and viewed with a Nikon (Tokyo, Japan) E600FN upright microscope equipped with a water-immersion ×40 objective and DIC optics. Slices were continuously perfused with Ames medium bubbled with 95% O2-5% CO2. In all experiments, inhibitory conductances were blocked with 100 μM picrotoxin (Sigma-Aldrich, St. Louis, MO), 10 μM strychnine (Sigma-Aldrich), and 50 μM (1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid (Sigma-Aldrich). Patch pipettes of resistance 7–9 MΩ were fabricated from borosilicate glass (WPI, Sarasota, Fl) with a two-stage vertical puller (Narishige, Tokyo, Japan) and filled with a K+ gluconate-based solution that also contained (in mM) 0.5 EGTA, 10 HEPES, 4 ATP, and 1 GTP (pH 7.4 by CsOH) and 14 μg/ml Alexa 488 (Invitrogen). The mGluR6 agonist l-AP4 (4 μM; Tocris Bioscience, Ellisville, MO) was added to the bath, and pharmacological agents [metabotropic receptor antagonist LY341495 (Tocris Bioscience), TRP channel agonist capsaicin (Sigma-Aldrich)] were delivered to the retina from a pipette with positive pressure (2–4 psi) with a computer-controlled solenoid valve (Picospritzer; General Valve, Fairfield, NJ). Retinal dissection, manipulations, and recordings were made in room light.
RESULTS
The nob ERG phenotype in homozygous tvrm27 mice is caused by a missense mutation in Trpm1.
To map the tvrm27 locus, affected mice were crossed to WT DBA/2J mice and the resulting F1 mice were intercrossed. F2 progeny homozygous for the tvrm27 locus were identified by ERG and their DNA genotyped with microsatellite markers across the genome. The tvrm27 phenotype was mapped to chromosome 7, between markers D7MIT230 and D7MIT195. This region contained Trpm1, which we considered an excellent candidate gene because Trpm1−/− mice are reported to have a similar ERG phenotype (Koike et al. 2010a; Morgans et al. 2009; Shen et al. 2009). To test whether tvrm27 was a new allele of Trpm1, we crossed homozygous tvrm27 mice to Trpm1−/− mice. All F1 offspring from this complementation cross lacked the ERG b-wave, indicating that tvrm27 is an allele of Trpm1, henceforth referred to as Trpm1tvrm27. The responsible mutation was identified by sequencing cDNA clones representing the Trpm1tvrm27 allele. A single nucleotide change c.G3399A (accession no. AY180104) resulted in an alanine to threonine missense mutation at position 1068 (p.A1068T; Fig. 1A). A1068 lies in the TRPM1 channel pore region (Fig. 1B) and is invariant in all mammals sequenced to date as well as in other vertebrates including platypus, zebra finch, and pufferfish (Fig. 1C).
Fig. 1.

A: sequences from control and Trpm1tvrm27/tvrm27 mice. B: schematic denoting the location of the p.A1068T mutation. C: alignment of murine TRPM1 amino acid sequence (1051–1087, accession no. NP_001034193.2) with the orthologous region of TRPM1 in 17 other vertebrate species. This region, including the A1068 residue (yellow), is highly conserved.
Retinal anatomy is normal in Trpm1tvrm27/tvrm27 mice.
We examined retinal anatomy by light and electron microscopy (Fig. 2). Examination of retinal cross sections of control and Trpm1tvrm27/tvrm27 mice showed normal cellular and synaptic layers (Fig. 2A). Furthermore, ultrastructural analyses indicated that Trpm1tvrm27/tvrm27 rod (Fig. 2B) and cone (Fig. 2C) photoreceptor terminals have normal ribbon synapses (Fig. 2B). Thus the abnormal ERG phenotype of Trpm1tvrm27/tvrm27 mice cannot be attributed to a loss of DBCs or a gross disruption of synaptic structure.
Fig. 2.

Comparison of retinal anatomy of control and Trpm1tvrm27/tvrm27 mice. A: by light microscopy, no difference between control and Trpm1tvrm27/tvrm27 retinas was observed. Scale bar, 15 μm. B and C: electron micrographs show that rod (B) and cone (C) ribbon synapses in Trpm1tvrm27/tvrm27 mice are present and have a morphology similar to those in wild-type (WT) mice. Scale bars, 500 nm. B, bipolar cell; H, horizontal cell.
Trpm1 expression is preserved in Trpm1tvrm27/tvrm27 retina.
Because Trpm1tvrm27/tvrm27 results from a missense mutation, we examined whether the mutant protein was expressed and localized similarly to WT. We double-labeled WT, Trpm1tvrm27/tvrm27, and Trpm1−/− retina sections with antibodies to TRPM1 and GRM6 (Fig. 3). Consistent with published data (Koike et al. 2010a; Pearring et al. 2011), WT retina had TRPM1 puncta on the tips of DBC dendrites and also diffuse label throughout their cell body (Fig. 3A). GRM6 label (Fig. 3B) colocalized with the TRPM1-positive puncta (Fig. 3C). The expression patterns of both TRPM1 and GRM6 were similar in the Trpm1tvrm27/tvrm27 retina (Fig. 3, D–F). In contrast, the Trpm1−/− retina lacked TRPM1 staining and maintained the WT distribution for GRM6 (Fig. 3, G–I). These data show that the mutant TRPM1 p.A1068T protein is correctly localized, indicating that the nob ERG phenotype in the Trpm1tvrm27/tvrm27 mutant is caused by a loss of channel function.
Fig. 3.

Immunohistochemistry for GRM6 (red) and TRPM1 (green) in WT (A–C), Trpm1tvrm27/tvrm27 (D–F), and Trpm1−/− (G–I) retinas. Note that TRPM1 expression is comparable in WT and Trpm1tvrm27/tvrm27 but absent in Trpm1−/− retina. Scale bars, 5 μm.
To further characterize GRM6 expression in the Trpm1tvrm27/tvrm27 retina, we incubated sections with antibodies to GRM6 and RIBEYE, a component of the photoreceptor ribbon complex. As can be seen in Fig. 4, TRPM1 expression on WT (Fig. 4A) and Trpm1tvrm27/tvrm27 (Fig. 4B) dendritic tips is surrounded by RIBEYE expression. Taken together, the data in Figs. 3 and 4 show that the expression of GRM6 and TRPM1 is comparable in WT and Trpm1tvrm27/tvrm27 DBCs.
Fig. 4.
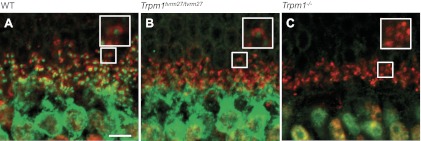
Immunohistochemistry for TRPM1 (green) and RIBEYE (red) in WT (A), Trpm1tvrm27/tvrm27 (B), and Trpm1−/− (C) retinas. Note that the apposition of pre- and postsynaptic elements are comparable to WT in Trpm1tvrm27/tvrm27 retina. Scale bar, 5 μm. Larger box displays the field contained in the small box at ×2 magnification.
Trpm1tvrm27 mice lack an ERG b-wave.
To further characterize the Trpm1tvrm27 mutant, we recorded ERGs under dark- and light-adapted conditions (Fig. 5). Dark-adapted ERGs obtained from representative WT and Trpm1tvrm27/tvrm27 mice are shown in Fig. 5A. Throughout the luminance range examined, WT ERGs are dominated by the positive-polarity b-wave, which increases in amplitude with increasing luminance and reflects light-induced activity of DBCs (Kofuji et al. 2000). At higher luminances, the b-wave is preceded by the negative-polarity a-wave, reflecting the light-induced closure of cation channels along rod photoreceptor outer segments (Penn and Hagins 1969). In comparison, the b-wave component is absent from responses of Trpm1tvrm27/tvrm27 mice. The net result is that the a-wave is followed by slow PIII, a negative-polarity component generated by Kir4.1 channel activity in Müller glial cells (Kofuji et al. 2000; Samuels et al. 2010) that is normally masked by the larger-amplitude b-wave. This nob ERG phenotype, in which the b-wave is absent while the a-wave is preserved, indicates that rod phototransduction is unaffected by the mutation, while DBC activity is grossly abnormal (cf. McCall and Gregg 2008).
Fig. 5.

Comparison of electroretinogram (ERG) waveforms of WT (black traces) and affected Trpm1tvrm27/tvrm27 (blue traces) mice. A: dark-adapted ERGs. The b-wave response present in WT ERGs is missing in Trpm1tvrm27/tvrm27 mice, revealing the electronegative slow PIII component. B: light-adapted ERG waveforms of Trpm1tvrm27/tvrm27 mice are abnormal. Values indicate flash luminance in log cd s/m2.
Figure 5B compares light-adapted ERGs obtained from representative WT and Trpm1tvrm27/tvrm27 mice. Under these conditions the ERG represents activity of the cone pathway. The WT cone ERG is dominated by the positive-polarity b-wave and high-frequency oscillatory potentials, which reflect activity through the DBC pathway (Sharma et al. 2005; Shirato et al. 2008). In contrast, the Trpm1tvrm27/tvrm27 cone ERGs are electronegative and rarely rise above the prestimulus baseline. This abnormal cone ERG waveform closely matches that of Trpm1−/− (Koike et al. 2010a; Morgans et al. 2009) as well as other DBC mutants (Nyx: Pardue et al. 1998; Grm6: Maddox et al. 2008; Pinto et al. 2007; Gpr179: Peachey et al. 2012).
DBC function is reduced in Trpm1+/tvrm27 heterozygotes.
Mutant TRPM1tvrm27 protein is correctly localized in Trpm1tvrm27/tvrm27 retina (Fig. 3); therefore the molecular composition of TRPM1 channels in Trpm1+/tvrm27 heterozygotes should be heterogeneous. Since TRP channels are tetramers (Clapham 2003; Moiseenkova-Bell et al. 2008; Stewart et al. 2010), individual channels can incorporate 0–4 Trpm1tvrm27 subunits. If WT and mutant (M) proteins are expressed at equal levels and participate equally in channel formation, the expected distribution of channels across the five possible stoichiometries is: 6.25% WT(4)M(0); 25% WT(3):M(1); 37.5% WT(2):M(2); 25% WT(1):M(3) and 6.25% WT(0)M(4). To investigate whether p.A1068T mutant subunits can coassemble with WT to generate a dominant-negative effect, we compared DBC function of WT mice to Trpm1+/tvrm27 and Trpm1+/− heterozygotes, using dark-adapted ERGs and patch-clamp recordings in retinal slices.
Representative WT, Trpm1+/tvrm27, and Trpm1+/− dark-adapted ERGs and summary data from multiple animals are shown in Fig. 6. These results show that the a-waves in all three genotypes are indistinguishable. Furthermore, the b-waves of WT and Trpm1+/− mice are also comparable. In contrast, Trpm1+/tvrm27 b-waves are reduced across an ∼5-log unit range of flash luminance (Fig. 6, A and B). Across flash luminance, the average Trpm1+/tvrm27 b-wave amplitude was 32% less than the WT response. These results indicate that the p.A1068T mutant subunit integrates into TRPM1 tetramers. The extent of the b-wave reduction is consistent with a simple model in which channels containing 0–2 mutant subunits (68.75% of all possible combinations) retain function while those containing either 3 or 4 mutant subunits (31.25%) are dysfunctional.
Fig. 6.

Comparison of dark-adapted ERGs recorded from Trpm1 heterozygotes. A: representative waveforms obtained from WT, Trpm1+/tvrm27, and Trpm1+/− mice. The amplitude of the b-wave is selectively reduced in Trpm1+/tvrm27 mice. Values indicate flash luminance in log cd s/m2. B: luminance-response functions for a-wave and b-wave amplitude in WT, Trpm1+/tvrm27, and Trpm1+/− mice. Data points indicate the average ± SE of 7 WT and Trpm1+/tvrm27 and 3 Trpm1+/− mice. C: luminance-response functions for WT, Grm6+/nob3/Gpr179+/nob5, and Nyx+/nob mice. For each mouse tested, the amplitude of the b-wave amplitude is expressed relative to the amplitude of the maximum a-wave. Data points indicate the average ± SE of 19 WT, 8 Grm6+/nob3Gpr179+/nob5, and 5 Nyx+/nob mice.
To determine whether heterozygous mouse models for other cCSNB genes (Nyx, Grm6, Gpr179) exhibit a similar selective ERG b-wave reduction, we also recorded dark-adapted ERGs from Grm6+/nob3Gpr179+/nob5 compound heterozygous mice and Nyx+/nob female mice and compared their results to WT littermates. To minimize variability across groups of mice due to factors such as age and genetic background, responses of each mouse were normalized to the maximum a-wave amplitude. Figure 6C presents luminance-response functions for Grm6+/nob3Gpr179+/nob5 and Nyx+/nob mice and their combined WT littermates. The b-wave response functions of Grm6+/nob3Gpr179+/nob5 double heterozygotes and WT mice superimpose. In comparison, Nyx+/nob heterozygote b-waves are consistently reduced in amplitude, consistent with X inactivation and DBCs expressing either Nyx+ or Nyxnob.
We used whole cell patch-clamp electrophysiology to directly examine the function of TRPM1 channels in rod DBCs. Application of either the mGluR6 antagonist LY341495 or the TRP agonist capsaicin (Caterina et al. 1997; Shen et al. 2009) evokes robust responses from WT DBCs (Fig. 7). Under the same conditions, the current-voltage relationships for LY341495 and capsaicin are similar, and both responses are blocked by capsazepine (Shen et al. 2009). The waveforms of the Trpm1+/tvrm27 DBC responses were similar to WT but were reduced in amplitude (Fig. 7). The variability in response waveform is reflective of our experience across multiple studies (Rampino et al. 2011; Shen et al. 2009, 2012) and may reflect variability in diffusion characteristics of individual puffs as well as the location of the puffer pipette with respect to the membrane patch being analyzed. In comparison, responses of Trpm1tvrm27/tvrm27 DBCs under these conditions were indistinguishable from background noise (Fig. 7). Figure 7B summarizes response amplitudes to LY34195 and capsaicin application from WT, Trpm1+/tvrm27, and Trpm1tvrm27/tvrm27 DBCs. These results show that the presence of the p.A1068T protein in Trpm1+/tvrm27 DBCs decreases their response amplitude by 49% for LY341495 and 42% for capsaicin, compared with WT. This decrease is comparable to the decrease (32%) seen in the amplitude of the ERG b-wave.
Fig. 7.

Patch-clamp responses of rod depolarizing bipolar cells (DBCs). A: representative responses of WT, Trpm1+/tvrm27, and Trpm1tvrm27/tvrm27 littermates to the mGluR6 agonist LY341495 or the TRP channel antagonist capsaicin. B: average peak response amplitude for WT (n = 7), Trpm1+/tvrm27 (n = 4), and Trpm1tvrm27/tvrm27 (n = 5) rod DBCs with a holding potential of +40 mV. Error bars indicate SE. **P = 0.002; *P = 0.01.
DISCUSSION
TRPM1 was identified as a potential cause of night blindness in Appaloosa horses (Bellone et al. 2008) some 30 years after Witzel et al. (1978) documented that they were night blind and had a nob ERG phenotype. Subsequently, several independent groups reported that Trpm1−/− mice have a nob ERG phenotype (Koike et al. 2010a; Morgans et al. 2009; Shen et al. 2009) and lack a DBC response to light (Koike et al. 2010a). In human studies, TRPM1 mutations have been identified in a subset of patients with cCSNB (Audo et al. 2009; Li et al. 2009; Nakamura et al. 2010; van Genderen et al. 2009) and a nob ERG phenotype was reported in a patient with a homozygous microdeletion of 15q13.3 that includes TRPM1 (Lepichon et al. 2010; Spielmann et al. 2011). Serum antibodies against TRPM1 have been identified in patients with melanoma-associated retinopathy, an acquired form of DBC dysfunction (Dhingra et al. 2011; Kondo et al. 2011). In CHO cells transfected with TRPM1, GRM6, and GNA0, TRPM1 channel activity is modulated by GRM6 activation via GNA0 (Koike et al. 2010a). Together, these results provide strong evidence that TRPM1 is the DBC transduction channel.
In this report, we describe a new mutant allele of Trpm1 that was generated as part of a large-scale mutagenesis program (Won et al. 2011). The Trpm1tvrm27 allele results from a missense mutation, p.A1068T, in TRPM1. Ala1068 is located in the predicted pore region of the channel (Xu et al. 2001) and is highly conserved in vertebrates, including marsupials, other mammals, birds, fish and amphibians. This level of conservation and the absence of any other detectable mutation suggest that Ala1068 is critical for channel function and that p.A1068T is the mutation responsible for the abnormalities observed in Trpm1tmrm27/tvrm27 mice.
Trpm1tvrm27/tvrm27 retinal anatomy at the light and electron microscopic level is normal. This is similar to descriptions of retinal anatomy in other mouse DBC signal transduction mutants (Grm6, Maddox et al. 2008; Masu et al. 1995; Pinto et al. 2007; Nyx, Ball et al. 2003; Pardue et al. 1998, 2001; Gpr179, Peachey et al. 2012), although some components of DBC ribbon synapses are mislocalized in Grm6−/− mice (Ishii et al. 2009). This is in contrast to mouse models for iCSNB genes and related proteins in which the outer plexiform layer collapses, ribbon synapses are malformed, and second-order neurons elaborate ectopic neurites (Cacna1f, Bayley and Morgans 2007; Chang et al. 2006; Mansergh et al. 2005; Cacnb2, Ball et al. 2002; Cacna2d4, Ruether et al. 2000; Wycisk et al. 2006; Cabp4, Haeseleer et al. 2004).
TRPM1 channel density sets the maximum response amplitude of DBCs.
To date, all studies of cCSNB mouse models have involved mutants with null alleles. Our data indicate that Trpm1tvrm27 is not a null allele but is instead a nonfunctional allele that is correctly localized to the DBC tips. We have used this mutation to begin to identify the rate-limiting steps in DBC signal transduction. Mice heterozygous for the Trpm1tvrm27 allele show decreased ERG b-wave and DBC response amplitudes to light or the activation of TRPM1 by capsaicin or of GRM6 by LY34195, respectively. In contrast, heterozygotes for the TRPM1 knockout allele have a normal ERG. DBC function is also normal in mice heterozygous for null alleles of Grm6 (Fig. 5C), Gpr179 (Peachey et al. 2012) or Gna0 (Okawa et al. 2010). These results indicate that none of these proteins is rate-limiting, even when expressed at half the WT level. This is not unexpected given their roles in the early stages of DBC signal transduction. In contrast, the incorporation of mutant TRPM1tvrm27 subunits disrupts a substantial fraction of TRPM1 channels, leading to reduced DBC function.
We propose that b-wave amplitude is dependent on the number of functional TRPM1 channels at the tips of the DBCs. This predicts reduced b-wave amplitudes in other mutants that impair normal TRPM1 expression, such as the Nyx+/nob mouse, because TRPM1 expression at the tips of DBCs depends on Nyx expression (Pearring et al. 2011). In Nyx+/nob female heterozygotes, because of X inactivation, approximately half of the DBCs express the WT allele and the other half express the Nyxnob mutant allele. Consistent with our proposal, the Nyx+/nob ERG b-wave amplitude is decreased. These data also predict that ERG b-wave reductions will be observed in female carriers of NYX mutations. Finally, b-wave reductions would also be expected in individuals heterozygous for TRPM1 mutations that are nonfunctional but trafficked normally. It will be important for future studies to fully characterize these individuals to determine whether they have previously unappreciated visual abnormalities.
GRANTS
This research was supported by grants from the National Institutes of Health (R21-EY-021852 to N. S. Peachey and R. G. Gregg; R01-EY-12354 to R. G. Gregg; R01-EY-014701 to M. A. McCall; RR-017890 to A. F. X. Goldberg; R01-EY-016501 to P. M. Nishina; R01-EY-010254 to S. Nawy; P30-CA-34196, The Jackson Laboratory Cancer Center Core Grant), the Department of Veterans Affairs Medical Research Service, Hope for Vision, a Foundation Fighting Blindness Center Grant to the Cole Eye Institute, Cleveland Clinic, and unrestricted awards from Research to Prevent Blindness to the Department of Ophthalmology, Cleveland Clinic Lerner College of Medicine and to the Department of Ophthalmology and Visual Sciences, University of Louisville.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.S.P., S.N., P.M.N., and R.G.G. conception and design of research; N.S.P., J.N.P., P.B., M.E.H., G.S.-S., T.A.R., Y.S., and P.M.N. performed experiments; N.S.P., J.N.P., P.B., A.F.G., Y.S., S.N., P.M.N., and R.G.G. analyzed data; N.S.P., T.F., C.K., A.F.G., M.A.M., P.M.N., and R.G.G. interpreted results of experiments; N.S.P., T.A.R., Y.S., S.N., and R.G.G. prepared figures; N.S.P., A.F.G., P.M.N., and R.G.G. drafted manuscript; N.S.P., J.N.P., P.B., T.F., C.K., S.N., P.M.N., and R.G.G. edited and revised manuscript; N.S.P., J.N.P., P.B., M.E.H., G.S.-S., T.A.R., T.F., C.K., A.F.G., Y.S., M.A.M., S.N., P.M.N., and R.G.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Doug Howell for running the initial ERG screens associated with this project, to Loan Dang for her assistance with ultrastructural analyses, and to Dr. Vera Moiseenkova-Bell for helpful comments on the manuscript.
Present addresses: J. N. Pearring, Dept. of Ophthalmology, Duke University, Durham, NC 27710; C. Koike, College of Pharmaceutical Sciences, Ritsumeikan University, 1-1-1 Noji-Higashi, Kusatsu, Shiga 525-8577, Japan; Y. Shen, Eye Center of Wuhan University Renmin Hospital, Eye Institute of Wuhan University, Wuhan, China.
REFERENCES
- Audo I, Kohl S, Leroy BP, Munier FL, Guillonneau X, Mohand-Saïd S, Bujakowska K, Nandrot EF, Lorenz B, Preising M, Kellner U, Renner AB, Bernd A, Antonio A, Moskova-Doumanova V, Lancelot ME, Poloschek CM, Drumare I, Defoort-Dhellemmes S, Wissinger B, Léveillard T, Hamel CP, Schorderet DF, De Baere E, Berger W, Jacobson SG, Zrenner E, Sahel JA, Bhattacharya SS, Zeitz C. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 85: 720–729, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audo I, Bujakowska K, Orhan E, Poloschek CM, Defoort-Dhellemmes S, Drumare I, Kohl S, Luu TD, Lecompte O, Zrenner E, Lancelot ME, Antonio A, Germain A, Michiels C, Audier C, Letexier M, Saraiva JP, Leroy BP, Munier FL, Mohand-Saïd S, Lorenz B, Friedburg C, Preising M, Kellner U, Renner AB, Moskova-Doumanova V, Berger W, Wissinger B, Hamel CP, Schorderet DF, De Baere E, Sharon D, Banin E, Jacobson SG, Bonneau D, Zanlonghi X, Le Meur G, Casteels I, Koenekoop R, Long VW, Meire F, Prescott K, de Ravel T, Simmons I, Nguyen H, Dollfus H, Poch O, Léveillard T, Nguyen-Ba-Charvet K, Sahel JA, Bhattacharya SS, Zeitz C. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 90: 321–330, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball SL, Pardue MT, McCall MA, Gregg RG, Peachey NS. Immunohistochemical analysis of the outer plexiform layer in the nob mouse shows no abnormalities. Vis Neurosci 20: 267–272, 2003 [DOI] [PubMed] [Google Scholar]
- Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG. Role of the β2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci 43: 1595–1603, 2002 [PubMed] [Google Scholar]
- Barthel LK, Raymond PA. Improved method for obtaining 3-microns cryosections for immunocytochemistry. J Histochem Cytochem 38: 1383–1388, 1990 [DOI] [PubMed] [Google Scholar]
- Bayley PR, Morgans CW. Rod bipolar cells and horizontal cells form displaced synaptic contacts with rods in the outer nuclear layer of the nob2 retina. J Comp Neurol 500: 286–298, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet 19: 264–267, 1998 [DOI] [PubMed] [Google Scholar]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Sparkes RL, Koop B, Birch DG, Bergen AA, Prinsen CF, Polomeno RC, Gal A, Drack AV, Musarella MA, Jacobson SG, Young RS, Weleber RG. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet 26: 319–323, 2000 [DOI] [PubMed] [Google Scholar]
- Bellone RR, Brooks SA, Sandmeyer L, Murphy BA, Forsyth G, Archer S, Bailey E, Grahn B. Differential gene expression of TRPM1, the potential cause of congenital stationary night blindness and coat spotting patterns (LP) in the Appaloosa horse (Equus caballus). Genetics 179: 1861–1870, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824, 1997 [DOI] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci 23: 11–24, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature 426: 517–524, 2003 [DOI] [PubMed] [Google Scholar]
- Dhingra A, Fina ME, Neinstein A, Ramsey DJ, Xu Y, Fishman GA, Alexander KR, Qian H, Peachey NS, Gregg RG, Vardi N. Autoantibodies in melanoma-associated retinopathy target TRPM1 cation channels of retinal ON bipolar cells. J Neurosci 31: 3962–3967, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA, Alexander KR, Derlacki DJ, Rajagopalan AS. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci USA 102: 4884–4889, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AF, Ritter LM, Khattree N, Peachey NS, Fariss RN, Dang L, Yu M, Bottrell AR. An intramembrane glutamic acid governs peripherin/rds function for photoreceptor disk morphogenesis. Invest Ophthalmol Vis Sci 48: 2975–2986, 2007 [DOI] [PubMed] [Google Scholar]
- Gregg RG, Kamermans M, Klooster J, Lukasiewicz PD, Peachey NS, Vessey KA, McCall MA. Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J Neurophysiol 98: 3023–3033, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci 7: 1079–1087, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes NL, Chang B, Hageman GS, Nusinowitz S, Nishina PM, Schneider BS, Smith RS, Roderick TH, Davisson MT, Heckenlively JR. Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens. Invest Ophthalmol Vis Sci 41: 3149–3157, 2000 [PubMed] [Google Scholar]
- Herron BJ, Lu W, Rao C, Liu S, Peters H, Bronson RT, Justice MJ, McDonald JD, Beier DR. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat Genet 30: 185–189, 2002 [DOI] [PubMed] [Google Scholar]
- Ishii M, Morigiwa K, Takao M, Nakanishi S, Fukuda Y, Mimura O, Tsukamoto Y. Ectopic synaptic ribbons in dendrites of mouse retinal ON- and OFF-bipolar cells. Cell Tissue Res 338: 355–375, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice MJ, Carpenter DA, Favor J, Neuhauser-Klaus A, Hrabé de Angelis M, Soewarto D, Moser A, Cordes S, Miller D, Chapman V, Weber JS, Rinchik EM, Hunsicker PR, Russell WL, Bode VC. Effects of ENU dosage on mouse strains. Mamm Genome 11: 484–8, 2000 [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20: 5733–5740, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike C, Obara T, Uriu Y, Numata T, Sanuki R, Miyata K, Koyasu T, Ueno S, Funabiki K, Tani A, Ueda H, Kondo M, Mori Y, Tachibana M, Furukawa T. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc Natl Acad Sci USA 107: 332–337, 2010a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike C, Numata T, Ueda H, Mori Y, Furukawa T. TRPM1: A vertebrate TRP channel responsible for retinal ON bipolar function. Cell Calcium 48: 95–101, 2010b [DOI] [PubMed] [Google Scholar]
- Kondo M, Sanuki R, Ueno S, Nishizawa Y, Hashimoto N, Ohguro H, Yamamoto S, Machida S, Terasaki H, Adamus G, Furukawa T. Identification of autoantibodies against TRPM1 in patients with paraneoplastic retinopathy associated with ON bipolar cell dysfunction. PLoS One 6: e19911, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepichon JB, Bittel DC, Graf WD, Yu S. A 15q13.3 homozygous microdeletion associated with a severe neurodevelopmental disorder suggests putative functions of the TRPM1, CHRNA7, and other homozygously deleted genes. Am J Med Genet A 152A: 1300–1304, 2010 [DOI] [PubMed] [Google Scholar]
- Li Z, Sergouniotis PI, Michaelides M, Mackay DS, Wright GA, Devery S, Moore AT, Holder GE, Robson AG, Webster AR. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet 85: 711–719, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox DM, Vessey KA, Yarbrough GL, Invergo BM, Cantrell DR, Inayat S, Balannik V, Hicks WL, Hawes NL, Byers S, Smith RS, Hurd R, Howell D, Gregg RG, Chang B, Naggert JK, Troy JB, Pinto LH, Nishina PM, McCall MA. Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J Physiol 586: 4409–4424, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet 14: 3035–3046, 2005 [DOI] [PubMed] [Google Scholar]
- Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R, Takada M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Specific deficit of the ON response in visual transmission by targeted disruption of the mGIuR6 gene. Cell 80: 757–765, 1995 [DOI] [PubMed] [Google Scholar]
- McCall MA, Gregg RG. Comparison of structural and functional abnormalities in mouse b-wave mutants. J Physiol 586: 4385–4392, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch Ophthalmol 104: 1013–1020, 1986 [DOI] [PubMed] [Google Scholar]
- Moiseenkova-Bell VY, Stanciu LA, Serysheva, Tobe BJ, Wensel TG. Structure of the TRPV1 channel revealed by electron cryomicroscopy. Proc Natl Acad Sci USA 105: 7451–7455, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW, Brown RL, Duvoisin RM. TRPM1: the endpoint of the mGluR6 signal transduction cascade in retinal ON bipolar cells. Bioessays 32: 609–614, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW, Zhang J, Jeffrey BG, Nelson SM, Burke NS, Duvoisin RM, Brown RL. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc Natl Acad Sci USA 106: 19174–19178, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Sanuki R, Yasuma TR, Onishi A, Nishiguchi KM, Koike C, Kadowaki M, Kondo M, Miyake Y, Furukawa T. TRPM1 mutations are associated with the complete form of congenital stationary night blindness. Mol Vis 16: 425–37, 2010 [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Pahlberg J, Rieke F, Birnbaumer L, Sampath AP. Coordinated control of sensitivity by two splice variants of Gαo in retinal ON bipolar cells. J Gen Physiol 136: 443–454, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue MT, McCall MA, LaVail MM, Gregg RG, Peachey NS. A naturally-occurring mouse model of X-linked congenital stationary night blindness. Invest Ophthalmol Vis Sci 39: 2443–2449, 1998 [PubMed] [Google Scholar]
- Pardue MT, Ball SL, Candille SI, McCall MA, Gregg RG, Peachey NS. nob: A mouse model of CSNB1. In: New Insights into Retinal Degenerative Diseases, edited by Hollyfield JG, Anderson RE, LaVail MM. New York: Kluwer/Plenum, 2001, p. 319–328 [Google Scholar]
- Peachey NS, Ray TA, Florijn R, Rowe LB, Sjoerdsma T, Contreras-Alcantara S, Baba K, Tosini G, Pozdeyev N, Iuvone PM, Bojang P, Jr, Pearring JN, Simonsz HJ, van Genderen M, Birch DG, Traboulsi EI, Dorfman A, Lopez I, Ren H, Goldberg AF, Nishina PM, Lachapelle P, McCall MA, Koenekoop RK, Bergen AA, Kamermans M, Gregg RG. GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 90: 331–339, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearring JN, Bojang P, Jr, Shen Y, Koike C, Furukawa T, Nawy S, Gregg RG. A role for nyctalopin, a small leucine rich repeat protein, in localizing the TRPM1 channel to retinal depolarizing bipolar cell dendrites. J Neurosci 31: 10060–10066, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn RD, Hagins WA. Signal transmission along retinal rods and the origin of the electroretinographic a-wave. Nature 223: 201–204, 1969 [DOI] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, Balannik V, McDearmon EL, Omura C, Lumayag S, Invergo BM, Glawe B, Cantrell DR, Inayat S, Olvera MA, Vessey KA, McCall MA, Maddox D, Morgans CW, Young B, Pletcher MT, Mullins RF, Troy JB, Takahashi JS. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis Neurosci 24: 111–123, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, Maurer J, Jacobi FK, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, Berger W, Meindl A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet 26: 324–327, 2000 [DOI] [PubMed] [Google Scholar]
- Rampino MA, Nawy SA. Relief of Mg2+-dependent inhibition of TRPM1 by PKCα at the rod bipolar cell synapse. J Neurosci 31: 13596–13603, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruether K, Grosse J, Matthiessen E, Hoffmann K, Hartmann C. Abnormalities of the photoreceptor-bipolar cell synapse in a substrain of C57BL/10 mice. Invest Ophthalmol Vis Sci 41: 4039–4047, 2000 [PubMed] [Google Scholar]
- Samuels IS, Sturgill GM, Grossman GH, Rayborn ME, Hollyfield JG, Peachey NS. Light-evoked responses of the retinal pigment epithelium: changes accompanying photoreceptor loss in the mouse. J Neurophysiol 104: 391–402, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert G, Bornschein H. Beitrag zur analyse des menschlichen Elektroretinogramms. Ophthalmologica 123: 396–412, 1952 [DOI] [PubMed] [Google Scholar]
- Sharma S, Ball SL, Peachey NS. Pharmacological studies of the mouse cone electroretinogram. Vis Neurosci 22: 631–636, 2005 [DOI] [PubMed] [Google Scholar]
- Shen Y, Heimel JA, Kammermans M, Peachey NS, Gregg RG, Nawy S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J Neurosci 29: 6088–6093, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rampino MA, Carroll RC, Nawy S. G-protein-mediated inhibition of the Trp channel TRPM1 requires the Gβγ dimer. Proc Natl Acad Sci USA 109: 8752–8757, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiells RA, Falk G, Naghshineh S. Action of glutamate and aspartate analogues on rod horizontal and bipolar cells. Nature 294: 592–594, 1981 [DOI] [PubMed] [Google Scholar]
- Shirato S, Maeda H, Miura G, Frishman LJ. Postreceptoral contributions to the light-adapted ERG of mice lacking b-waves. Exp Eye Res 86: 914–928, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter MM, Miller RF. 2-Amino-4-phosphonobutyric acid: a new pharmacological tool for retina research. Science 211: 182–185, 1981 [DOI] [PubMed] [Google Scholar]
- Spielmann M, Reichelt G, Hertzberg C, Trimborn M, Mundlos S, Horn D, Klopocki E. Homozygous deletion of chromosome 15q13.3 including CHRNA7 causes severe mental retardation, seizures, muscular hypotonia, and the loss of KLF12 and TRPM1 potentially causing macrocytosis and congenital retinal dysfunction in siblings. Eur J Med Genet 54: e441–e445, 2011 [DOI] [PubMed] [Google Scholar]
- Stewart AP, Egressy K, Lim A, Edwardson JM. AFM imaging reveals the tetrameric structure of the TRPM8 channel. Biochem Biophys Res Commun 394: 383–386, 2010 [DOI] [PubMed] [Google Scholar]
- Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Rüther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet 19: 260–263, 1998 [DOI] [PubMed] [Google Scholar]
- Taylor BA, Navin A, Phillips SJ. PCR-amplification of simple sequence repeat variants from pooled DNA samples for rapidly mapping new mutations of the mouse. Genomics 21: 626–632, 1994 [DOI] [PubMed] [Google Scholar]
- van Genderen MM, Bijveld MM, Claassen YB, Florijn RJ, Pearring JN, Meire FM, McCall MA, Riemslag FC, Gregg RG, Bergen AA, Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet 85: 730–736, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzel DA, Smith EL, Wilson RD, Aguirre GD. Congenital stationary night blindness: an animal model. Invest Ophthalmol Vis Sci 17: 788–795, 1978 [PubMed] [Google Scholar]
- Won J, Shi LY, Hicks W, Wang J, Hurd R, Naggert JK, Chang B, Nishina PM. Mouse model resources for vision research. J Ophthalmol 2011: 391384, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wycisk KA, Budde B, Feil S, Skosyrski S, Buzzi F, Neidhardt J, Glaus E, Nürnberg P, Ruether K, Berger W. Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci 47: 3523–3530, 2006 [DOI] [PubMed] [Google Scholar]
- Xu XZ, Moebius F, Gill DL, Montell C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc Natl Acad Sci USA 98: 10692–10697, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz C, van Genderen M, Neidhardt J, Luhmann UF, Hoeben F, Forster U, Wycisk K, Mátyás G, Hoyng CB, Riemslag F, Meire F, Cremers FP, Berger W. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci 46: 4328–4335, 2005 [DOI] [PubMed] [Google Scholar]
- Zeitz C, Kloeckener-Gruissem B, Forster U, Kohl S, Magyar I, Wissinger B, Mátyás G, Borruat FX, Schorderet DF, Zrenner E, Munier FL, Berger W. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet 79: 657–667, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]