

Abstract
A hydride generation (HG) procedure has been described for determination of Pb by ICP-MS using potassium hexacyanomanganate(III), K3Mn(CN)6, as an additive to facilitate the generation of plumbane (PbH4). Potassium hexacyanomanganate(III) was prepared in acidic medium as it was unstable in water. The stability of hexacyanomanganate(III) was examined in dilute solutions of HCl, HNO3 and H2SO4. The solutions prepared in 1% v/v/ H2SO4 were found to be stable for over a period of 24 h. The least suitable medium was 1% v/v HNO3. For generation of plumbane, acidic hexacyanomanganate(III) and sample solutions were mixed online along a 5-cm long tygon tubing (1.14 mm i.d.) and then reacted with 2% m/v sodium borohydride (NaBH4). A concentration of 0.5% m/v K3Mn(CN)6 facilitated the generation of PbH4 remarkably. In comparison to H2SO4, HCl provided broader working range for which optimum concentration was 1% v/v. No significant interferences were noted from transition metals and hydride forming elements, up to 0.5 μg mL−1 levels, except Cu which depressed the signals severely. The depressive effects in the presence of 0.1 μg mL−1 Cu were alleviated by increasing the concentration of K3Mn(CN)6 to 2% m/v. Under these conditions, the sensitivity was enhanced by a factor of at least 42 to 48. The detection limit (3s) was 0.008 μg L−1 for 208Pb isotope. Average signal-to-noise ratio (S/N) ranged between 18 and 20 for 1.0 μg mL−1 Pb solution. The accuracy of the method was verified by analysis of several certified reference materials, including Nearshore seawater (CASS-4), Bone ash (SRM 1400), and Mussel tissue (SRM 2976). The procedure was also successfully applied to the determination of Pb in coastal seawater samples by ICP-MS.
Keywords: Lead, Hydride generation, Hexacyanomanganate(III), ICP-MS
1. Introduction
Hydride generation (HG) has been realized as one the most powerful analytical tools in atomic spectrometry despite the fact it is applicable to the determination of a small number of elements, including As, Bi, Pb, Sb, Se, Sn, and Te that form covalent hydrides at ambient conditions [1–9]. The advantages of the HG are mainly due to the more efficient transport of gaseous sample to the excitation or atomization source. In comparison to conventional solution nebulization, sensitivity could be boosted as high as two orders of magnitude for certain hydride forming elements, such as As, Se and Te that exhibit poor atomization and excitation.
Generation of lead hydride (PbH4, plumbane) was reported first by Thompson and Thomerson [10] about four decades ago. Numerous papers have been published since then to improve the analytical figures of merit (selectivity, sensitivity, detection limit and precision) for determination of Pb by hydride generation [11–26]. It has been well documented now that generation of plumbane is more sensitive to acidity of the medium than hydrides of other elements. In addition to relatively narrow range of acidity, difficulties are associated with, efficiency of oxidizing agents, instability of plumbane, and interferences of transition metals and hydride forming elements. Plumbane is assumed to be generated from the Pb(IV) oxidation state of Pb. Therefore, various oxidizing agents have been utilized in most studies to oxidize Pb(II) to Pb(IV) in acidic solutions. These include hydrogen peroxide [18–22], ammonium or sodium persulfate [19,23,24], potassium dichromate [19,21], ammonium cerium (IV) nitrate [25,26] The use of chelating agents is rather scarce. Chen at al. [27] reported that a number of chelating agents, including nitroso R salt, Bromo pyrogallol red, Pyrocatechol violet, Alizarine red-S, 5-Br-PADAP and PAN-S enhanced signals in determination of Pb by hydride generation. Contrary to the scenario with oxidizing agents, their results suggested that plumbane could also be generated directly from the chelated Pb(II).
Potassium hexacyanoferrate(III) (K3Fe(CN)6) was first used in determination of Pb by hydride generation atomic spectrometry in 1985 [28]. It is by far the most effective reagent to date in generation of plumbane in acidic solutions; consequently, it has become inevitable for determination of Pb by hydride generation using various spectrometric techniques [12–17, 29–34]. As a mild oxidizing agent, its action in plumbane generation was attributed to the oxidation of Pb(II) to Pb(IV) although the reduction potential (E° = 0.36 V) of [Fe(CN)6]3− to [Fe(CN)6]4− is much lower than that required for oxidation of Pb2+ to Pb4+ (E° = −1.69 V). This assumption has been disproved recently that plumbane was generated as efficiently as even if [Fe(CN)6]3− was not interacted directly with Pb(II) in the solution prior to reaction with NaBH4 [31]. In another study, generation of plumbane was not affected from the reduction of [Fe(CN)6]3− to [Fe(CN)6]4− such that trace amounts of [Fe(CN)6]3− was sufficient for formation of PbH4 [17]. The effectiveness of hexacyanoferrate(III) was explained by the formation of reactive intermediates, such as hydroboranes, facilitating the generation of PbH4.
Transition metals form complex cyanides that possess different thermodynamic and catalytic properties. Only a few of these cyanide complexes are stable (e.g., Fe, Ag and Au), and hence, they have not been considered as an additive in hydride and vapor generation approaches apart from the most popular hexacyanoferrate(III). In a recent paper, we have demonstrated that hexacyanochromate(III) could facilitate the generation of Cd vapor allowing better sensitivity and selectivity to be achieved in determination of Cd [35]. In this paper, we have investigated the performance of a new transition metal cyanide complex, potassium hexacyanomanganate(III) (K3Mn(CN)6), for generation of plumbane and described a highly sensitive and robust method for determination of Pb by hydride generation. Plumbane was generated by interacting Pb(II) in acidic solution with K3MnCN)6 followed by reaction with sodium borohydride (NaBH4). The gaseous hydride was transported rapidly to argon plasma of ICP-MS through a cooled spray chamber. The effects of solution acidity, concentrations of K3Mn(CN)6 and NaBH4, and the manifold parameters were optimized. The tolerance of the method to chemical interferences of transition metals and hydride forming elements was investigated. The method was applied to the determination of Pb in various certified reference materials and samples of coastal seawater by ICP-MS.
2. Experimental
2.1. Materials and solutions
All solutions were prepared with double deionized water (18 MΩ cm resistivity) obtained from a Barnstead E-Pure system fed by a reverse-osmosis system (SpectraPure). A 1.0 μg mL−1 Pb standard solution was prepared from a 1000 μg mL−1 stock solution (SPEX Certiprep, Metuchen, NJ) and stored in 1% v/v HNO3 (Trace metal grade, BDH Chemicals). All experimental solutions and calibration standards were prepared from this solution. Solutions of the other trace elements, including As, Bi, Ca, Cd, Co, Cr, Cu, Fe, K, Mg, Na, Ni, Sb, Se and Zn, were either prepared from high-purity salts or from 1000 μg mL−1 stock solutions (Spex Certiprep). Sodium borohydride (NaBH4, ≥98%) was purchased from Sigma Aldrich, St. Louis, MO. Sodium borohydride (NaBH4) solution was prepared daily in 0.1% m/v sodium hydroxide (NaOH, Fisher Scientific). Hydrogen peroxide (H2O2, 99.999%, Sigma Aldrich) was used along with HNO3 in digestion of tissue samples.
Potassium hexacyanomanganate(III) (K3Mn(CN)6, reagent grade) was obtained from MP Biomedicals, Solon, OH (Lot No: 41218). It was prepared in 1% v/v H2SO4 (trace metal grade, Fisher Scientific). Lead impurities were removed from K3Mn(CN)6 solutions by co-precipitation of Pb(II) with calcium fluoride (CaF2). Detailed information about the cleaning procedure is provided below using ammonium fluoride (NH4F) and calcium nitrate solutions. Ammonium fluoride (NH4F, electronics grade, 40% m/v, ~12 mol L−1) was purchased from Sigma Aldrich. Calcium nitrate solution was prepared by dissolving 2.5 g of ultrapure CaCO3 (99.99%, Alfa Aesar) slowly with 20% v/v HNO3 and completed to 20 mL with water yielding 50 mg mL−1 Ca(II) (as nitrate). The HNO3 solution was added incrementally during dissolution of CaCO3 to obtain a neutral to slightly acidic Ca(NO3)2 solution. The solution was heated briefly to convert carbonates to carbon dioxide.
2.2. Instrumentation and hydride generation manifold
Measurements were performed by using a Varian 820MS ICP-MS instrument (Varian, Australia). The instrument was equipped with a peltier-cooled double-pass glass spray chamber, standard one-piece low flow ball-and-socket connection quartz torch, standard Ni sampler and skimmer cones, and all-digital discrete dynode electron multiplier detector. The instrument was optimized daily in nebulization mode for sensitivity, doubly charged ions (<2%) and oxides (<3%) with 5 μg L−1 solution of 138Ba, 25Mg, 115In, 140Ce, 208Pb. Samples were introduced manually to the instrument. Data were collected by ICP-MS Expert software package (version 2.2 b126) at peak hopping mode using the 206Pb, 207Pb and 208Pb. The operating parameters of the instrument are summarized in Table 1.
Table 1.
Operating conditions for Varian 820MS ICP–MS instrument and hydride generation system
|
| |
| ICP–MS | |
|
| |
| RF Power (kW) | 1.4 |
| Plasma Ar flow (L min−1) | 18 |
| Auxiliary Ar flow (L min−1) | 1.8 |
| Nebulizer Ar flow (L min−1) | 1.2 |
| Sheath Ar flow (L min−1) | 0.1 |
| Sampling depth (mm) | 7 |
| Pump rate (mL min−1) | 1.0 |
| Stabilization time (sec) | 40 |
| Spray chamber temperature (°C) | 2 |
| Scan mode | Peak hopping |
| Dwell time (ms) | 50 |
| Points/peak | 1 |
| Scans/peak | 6 |
| Scans/replicate | 10 |
| Isotopes measured | 206Pb, 207Pb and 208Pb |
|
| |
| Hydride generation | |
|
| |
| Sample acidity − flow rate | 1% v/v HCl − 1.0 mL min−1 |
| K3Mn(CN)6 concentration − flow rate | 2% m/v − 0.5 mL min−1 |
| NaBH4 concentration − flow rate | 2% m/v − 1.0 mL min−1 |
Schematic diagram of the hydride generation manifold is illustrated in Fig. 1. The stand-alone spray chamber with 100 mL inner volume was used as gas-liquid separator. The pneumatic nebulizer (Ari-mist) was removed and a polypropylene T-piece (4.0 mm id) was installed as shown in Fig. 1. Carrier argon was introduced by means of the T-piece inserted to the inlet of the spray chamber. Its flow rate was controlled by tuning the flow rate of the nebulizer argon gas. Tygon peristaltic pump tubings were red/red stop (1.14 mm i.d.) for sample and NaBH4, and black/black stop (0.76 mm i.d.) for K3Mn(CN)6. The waste line was on a separate peristaltic pump utilizing a purple/white stop tygon tubing (2.79 mm i.d.). The reaction coil was 5.0 cm long tygon tubing (1.14 mm i.d.). The transfer line was 20 cm long PTFE tubing (1.6 mm i.d.) which was extended into the spray chamber through the T-piece (Fig. 1). Other lines used for connections were made of 0.8 mm i.d. PTFE tubing. Sample solutions in 1% v/v HCl were pumped at 1.0 mL min−1 and mixed on-line with 2% m/v K3Mn(CN)6 solution in 1% v/v H2SO4 running at 0.5 mL min−1. This mixture was then reacted with 2% m/v NaBH4 pumped at a flow rate of 1.0 mL min−1.
Fig. 1.
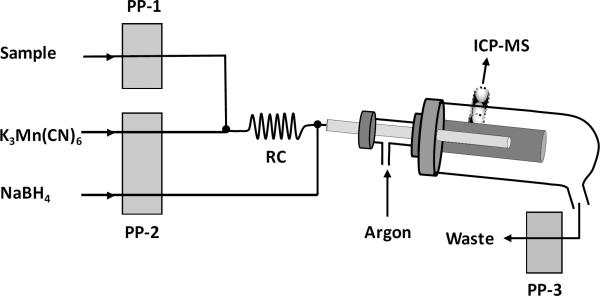
Schematic illustration of the hydride generation manifold. The gas–liquid separator is the stand–alone glass double pass spray chamber with 100 mL inner volume. The spray chamber is peltier cooled to 2 °C for removal excess water vapor. Reaction coil (RC) is 5.0 cm long tygon tubing (1.14 mm i.d.).
2.3. Preparation and cleaning of potassium hexacyanomanganate(III) solution
Potassium hexacyanomanganate(III) was unstable in water, therefore, the experimental solutions could not be prepared in water. Further, the reagent grade chemical was found to contain significant Pb impurity. A cleaning procedure was developed based on the co-precipitation of Pb impurities with CaF2 according to the procedures described elsewhere [36]. A 6% m/v K3Mn(CN)6 solution was prepared in 2% v/v H2SO4 by dissolving 0.6 g of the reagent in 10 mL of 2% v/v H2SO4. The reagent was placed into a dry 15-mL tube and dissolved by adding 2% v/v H2SO4 solution (ca. 8 mL) directly onto the reagent. Dissolution was relatively slow. Therefore, the contents were vortexed repeatedly yielding a deep-red solution in about 4–5 min. The volume was completed to 10 mL with 2% v/v H2SO4 solution. Care should be given to avoid contact with water as it results in loss of reagent even after the solution is acidified. It should be noted that contact with water does not deteriorate the stability of the solution once the solid reagent is successfully dissolved in acidic solution.
The 6% m/v K3Mn(CN)6 stock solution was pipetted as 1.0 mL portions into 2-mL micro-centrifuge tubes. To each tube, first 0.1 mL of 12 mol L–1 NH4F solution was added. Then, 0.2 mL of 50 mg mL−1 Ca(II) was added resulting in intense precipitation of CaF2 in acidic solution. The contents were allowed to precipitate a few minutes and then centrifuged for 30 min at 12,000 rpm by Eppendorf micro-centrifuge (Model 5415D). The supernatant in each tube was decanted rapidly to another 2-mL micro-centrifuge tube for a second co-precipitation by adding NH4F and Ca(II) as described above. At the conclusion of the second centrifugation, the liquid phase in each tube was collected and diluted to 30 mL with 1% v/v H2SO4 yielding 2% m/v K3Mn(CN)6 in 1% v/v H2SO4. This solution was stable for at least 48 h after preparation. Solution-based ICP-MS analysis of the diluted solutions of cleaned K3Mn(CN)6 indicated that Pb impurity was reduced to below 0.02 μg L−1.
2.4. Samples and preparation
The accuracy of the procedure was verified by determination of Pb concentration in different certified reference materials (CRM)s), such as Nearshore seawater (CASS-4), Mussel tissue (SRM 2976) and Bone ash (SRM 1400). Coastal seawater samples were also analyzed by the procedure. CASS-4 was obtained from the National Research Council Canada, Ottawa, ON. The coastal seawater samples were collected at about 1 m depth from Galveston Bay, Galveston, TX into acid-cleaned polypropylene bottles and acidified to 0.1% (v/v) HNO3 at the sampling site. At the laboratory, they were filtered through 0.45 μm membrane filters and stored in 0.1% v/v HNO3 until analysis. Sub-samples (5.0 mL) of CASS-4 and coastal seawater were taken and acidified to 1% v/v HCl for analysis. Another set of sub-samples were spiked with 0.02 and 0.5 μg L−1 Pb for CASS-4 and coastal seawater, respectively, and analyzed simultaneously.
Bone ash (SRM 1400) was obtained from the National Institutes of Standards and Technology, Gaithersburg, MD. It is produced from high temperature calcinations of bone and thus is predominantly calcium phosphate matrix with μg g−1 levels of levels of Al, F, Fe, Sr and Zn. Sub-samples of about 25 mg were dissolved in 1.0 mL concentrated HNO3 in 4-mL PTFE tubes (Savillex) at 120 °C using Digiprep Cube digestion system (SCP Science, Champlain, NY) as described elsewhere [17,33]. The solutions were heated to dryness. The final residue was dissolved and diluted to 10 mL with 1% v/v HNO3. For HG-ICP-MS determination, 1.0 mL was taken and diluted to 20 mL with 1% v/v HCl.
Mussel tissue (SRM 2976) was also obtained from the National Institutes of Standards and Technology, Gaithersburg, MD. This material was characterized with As, Cu, Fe and Zn at high μg g−1 levels. Approximately, 50 mg sub-samples were digested with 2.0 mL concentrated HNO3 and 1.0 mL H2O2 in 60-mL screw-capped PTFE tubes (Savillex) at 140 °C for 2 h. At the conclusion, caps were opened and additional 1.0 mL H2O2 was added to hot solution to oxidize the organics. The contents were heated to dryness, and then redissolved with 2 mL water and heated again to remove all residual H2O2. The contents were then dissolved in 1% v/v HNO3 and completed to 10 mL. Determinations were made in 10-fold diluted solutions in 1% v/v HCl.
2.5. Examining the stability K3Mn(CN)6
Preliminary experiments showed that potassium hexacyanomanganate(III), (K3Mn(CN)6), was highly unstable in water. Contact with water yielded insoluble black precipitate due to the formation of oxide or hydroxides. On the other hand, it dissolved readily and was stable to some extent in dilute acids, such as HCl and H2SO4. Therefore, 0.5% m/v K3Mn(CN)6 was prepared from reagent grade chemical directly (e.g., without any cleaning) in 1% v/v HCl, 1% v/v HNO3 and 1% v/v H2SO4 to determine its stability in each acid solution. In this experiment, measurements were made with freshly prepared and aged K3Mn(CN)6 solution for 10 μg L−1 Pb solutions acidified from 0 to 7% v/v HCl. Each Pb solution was mixed on-line with 0.5% m/v K3Mn(CN)6 solution and then reacted with 1% m/v NaBH4.
2.6. Optimization of hydride generation conditions
Once the optimum medium for stabilization of K3Mn(CN)6 was identified, the effect of solution acidity was examined for 1.0 μg L−1 Pb solutions prepared in HCl and H2SO4. The acidity was varied from 0 to 7% v/v for each acid. In this experiment, 0.5% m/v K3Mn(CN)6 solutions were prepared in 1% v/v H2SO4 from the cleaned stock solution (2% m/v) and mixed on-line similarly with the acidified sample solution.
The concentration of K3Mn(CN)6 solution was examined between 0 and 2.5% m/v K3Mn(CN)6 in 1% v/v H2SO4. The length of reaction coil (1.14 mm i.d. tygon tubing) was varied from 2 to 20 cm to affect the signals. Then, the flow rates for the sample and the nebulizer argon gas were optimized. The former was examined between 0.5 and 2.0 mL min−1 and the latter was varied from 0.8 to 1.4 L min−1. The sodium borohydride concentration was optimized by varying its concentration from 0 to 3% m/v for the highest Pb signals. The flow rates for K3Mn(CN)6 and NaBH4 solutions were not varied and kept at 0.5 mL min−1 and 1.0 mL min−1, respectively, since all other variables were optimized for these settings.
2.7. Examination of matrix effects
As for most hydride forming elements, determination of Pb by hydride generation is vulnerable to the interferences of transition metals (Co, Cu and Ni). In general, effects are manifested by depression or enhancement of the signals depending on the nature of acid-oxidant system and catalytic properties of the interfering element. Thus, the effects of a number of matrix elements, including As(III), Bi(III), Cd(II) Co(II), Cu(II), Fe(III), Mn(II), Ni(II), Sb(III), Se(IV), Sn(II) and Zn(II) were investigated for 1.0 μg L−1 Pb(II) in the presence of 0.1 and 0.5 μg mL−1 of each element. Effects for Ca(II), K(I), Mg(II), and Na(I) were tested for 1000 μg mL−1 solutions containing 1.0 μg L−1 Pb(II).
3. Results and discussion
3.1. Stability of K3Mn(CN)6solution
Manganese forms a number of complexes with cyanide. The standard reduction potentials of hexacyanomanganate complexes are given below [37].
| (1) |
| (2) |
| (3) |
The data show that cyanide complexes of Mn(II) and Mn(I), [Mn(CN)6]4− and [Mn(CN)6]5−, respectively, are stabilized best at Mn(III) oxidation state. In other words, both [Mn(CN)6]5− and [Mn(CN)6]4− ions are easily oxidized to [Mn(CN)6]3−. The reduction of potential for Mn3+ = Mn2+ system is +1.5 V which is relatively high in comparison to −0.24 V for [Mn(CN)6]3− = Mn(CN)6]4−. This information also substantiates that Mn(III) is stabilized significantly by complexing with cyanide.
Despite its stability, hexacyanomanganate(III) hydrolyzes in water as shown in the reaction (4) yielding insoluble Mn(OH)3 [37].
| (4) |
The value of Kdiss/Ksp (e.g., the ratio of dissociation constant to the solubility product constant of Mn(OH)3) was estimated to be 2 × 104, indicating that [Mn(CN)6]3− is thermodynamically unstable in water [37]. In acidic solution, however, [Mn(CN)6]3− undergoes disproportionation and decomposition reactions producing hexacyanomanganate(IV), [Mn(CN)6]2−, as shown in reactions (5) and (6) [38].
| (5) |
| (6) |
It was reported that the disproportionation reaction (5) was very fast and quantitative at room temperature [38] yielding [Mn(CN)6]2− ion. Although this complex is more stable in acidic medium, it was also found to slowly decompose producing Mn(II) (e.g., Mn(H2O)62+) and cyanogen gas, (CN)2 according to the reaction (6) [38].
3.2. Effects of acids on stability of K3Mn(CN)6
The evidence from the literature [37, 38] clearly indicated that it would not be feasible to utilize K3Mn(CN)3 robustly in generation of PbH4 for quantitative determination of Pb unless the reagent is stabilized sufficiently. In initial attempts of PbH4 generation, about 0.5% m/v K3Mn(CN)6 was prepared in very dilute solutions of HCl, such as 0.2% and 0.5% v/v HCl since Pb standard solutions were also prepared in the same acid. Evidently, it was feasible to prepare K3Mn(CN)6 solutions that enhanced the signals. However, temporal stability was poor. Visible precipitation or deposition was noted within an hour of preparation in some solutions, especially with lower acid concentration. This result was consistent with the literature and was indicative of the slow degradation of the reagent in dilute acid solutions. To overcome this hurdle, additional experiments were performed by increasing HCl concentration as well as affecting the stability with dilute solutions of HNO3 and H2SO4. The results of the experiments are illustrated in Fig. 2 for 1% v/v solutions of HCl, HNO3 and H2SO4. Signals for 10 μg L−1 Pb solutions were almost identical when measurements were made with freshly prepared K3Mn(CN)6 solutions regardless of the acid used for dissolution. On the other hand, extended or long–term stability of the reagent varied remarkably depending on the acid. Apparently, neither 1% v/v HCl nor 1% v/v HNO3 medium provided adequate stabilization (Fig. 2A and 2B). For K3Mn(CN)6 solution prepared in 1% v/v HCl, signals decreased significantly to about 52% of the initial signals (e.g., 48% lower) when measurements were repeated 6 h later (see Fig. 2A). According to the reactions 5 and 6, this decrease was due to the instability of the acid dissolution product, viz. [Mn(CN)6]2− in 1% v/v HCl. Higher concentrations of HCl (e.g., 5% v/v) provided better stability. Yet, signals did drop by 30 to 35% within 6 h. Degradation of K3Mn(CN)6 in 1% v/v HNO3 was even faster such that 3 h later the sensitivity dropped by 72% in comparison to that for the fresh solution (Fig. 2B). This was attributed to the oxidizing nature of HNO3, viz. decomposition of [Mn(CN)6]2− to Mn(IV). Further, a brown–black precipitate formed as the solution was allowed to wait longer. This precipitate was thought to be manganese(IV) oxide as a result of decomposition of [Mn(CN)6]2− in HNO3 medium.
Fig. 2.
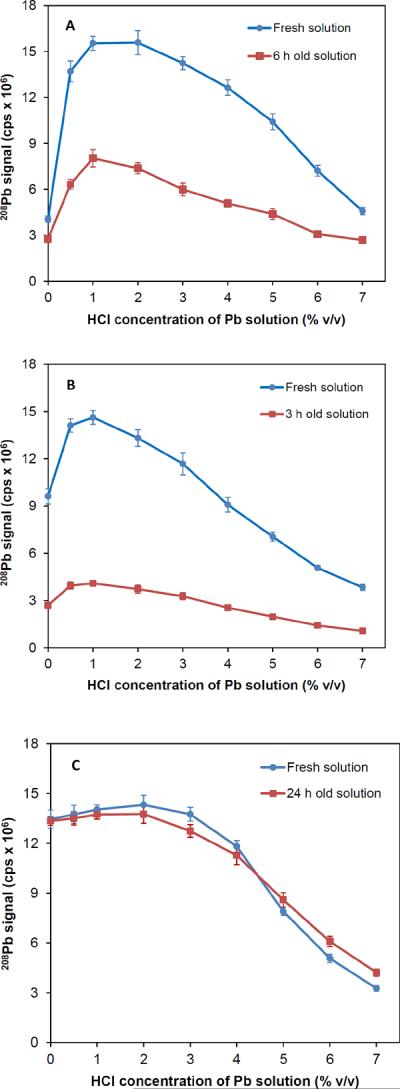
Temporal stability of 0.5% m/v K3Mn(CN)6 solutions prepared in (A) 1% v/v HCl, (B) 1% v/v HNO3, (C) 1% v/v H2SO4. Signals were collected for 10 μg L−1 Pb solution acidified from 0 to 7% v/v HCl.
Unlike those in HCl and HNO3, K3Mn(CN)6 was found to be stable in 1% v/v H2SO4 medium (Fig. 2C). The signals for 10 μg L−1 Pb solutions were almost identical for fresh and aged solutions of 0.5% m/v K3Mn(CN)6. No significant changes occurred in signals when measurements were performed 24 h later with the same K3Mn(CN)6 solution. Sensitivity varied only about 3 to 5%. Stability was not examined instrumentally beyond 24–h period. However, no significant degradation or precipitation was noted for at least 48 h when K3Mn(CN)6 solutions were inspected visually.
Higher concentrations of H2SO4 (ca. 5 to 6% v/v) did not deteriorate its stability, instead facilitated the dissolution of the reagent, which was advantageous for preparing concentrated stock solutions. Saturation data for K3Mn(CN)6 in dilute H2SO4 is not available; however, our results suggested that up to 8% m/v solutions could be prepared in 2 to 3% v/v H2SO4. Consequently, experimental solutions of K3Mn(CN)6 were prepared in dilute H2SO4 and cleaned according to the procedure described in section 2.3 in subsequent experiments.
3.3. Optimization of solution acidity for generation of plumbane
Hydrochloric acid is often the desired reagent for acidification of solutions for generation of plumbane (PbH4) in the presence of potassium hexacyanoferrate(III), K3Fe(CN)6 [12–17, 27–33]. In this study, however, K3Mn(CN)6 was best stabilized in dilute H2SO4, therefore, we attempted to examine the performances of HCl and H2SO4 on generation of plumbane within a concentration gradient of 0 to 7% v/v. The results are illustrated in Fig. 3. The optimum acidity was between 0.5 to 2% v/v for HCl, which was consistent with that in Fig. 2C. Unlike that for HCl, the optimum range for H2SO4 was significantly narrow (0 to 0.5% v/v) exhibiting an optimum concentration around 0.25% v/v H2SO4. The sensitivity was relatively better (e.g., 15 to 20%) in H2SO4 medium. Nevertheless, HCl appeared to be more robust because of the broader working range of sample acidity. Thus, all solutions in subsequent studies were prepared in 1% v/v HCl and mixed on–line with K3Mn(CN)6 solutions in 1% v/v H2SO4.
Fig. 3.
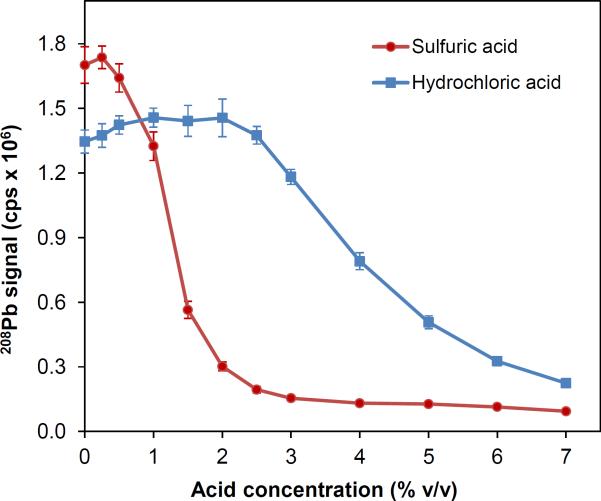
Effect of solution acidity adjusted by HCl or H2SO4 on the signals profiles for 1.0 μg L−1 Pb. K3Mn(CN)6 = 0.5% m/v in 1% v/v H2SO4, NaBH4 = 1% m/v in 0.1% m/v NaOH.
3.4. Effect of K3Mn(CN)6 concentration on PbH4 generation
The effect of K3Mn(CN)6 concentration on generation of PbH4 was studied between 0.1 and 2.5% m/v levels. Signals for 1.0 μg L−1 Pb solutions increased steeply with 0.1% m/v K3Mn(CN)6 indicating that very small concentration of K3Mn(CN)6 was sufficient for the formation of PbH4 from acidic sample solutions. This pattern was consistent with that for K3Fe(CN)6 that also boosted Pb signals at relatively small levels [12,17,31]. Optimum concentration was around 0.5% m/v K3Mn(CN)6 to attain maximum signals. However, it should be noted that this concentration reflects the performance of K3Mn(CN)6 in the absence of matrix or any interferences. Further, Pb is a highly ubiquitous element. Consequently, its determination suffers from contamination from the reagents that hampers the limit of detection (LOD), especially when utilizing highly sensitive approaches, such as hydride generation. Thus, the concentration of K3Mn(CN)6 was tentatively kept at 0.5% m/v until further examination under matrix conditions.
The generation of PbH4 is often assumed to take place via oxidation of Pb(II) to Pb(IV) with the aid of the additives in acidic solution. The enhancement in the case of K3Mn(CN)6, however, cannot be attributed to the oxidation of Pb2+ to Pb4+ since hexacyanomanganate(III) ([Mn(CN)6]3−) is oxidized to hexacynomanganate(IV) ([Mn(CN)6]2−) in dilute H2SO4 solution as mentioned above [37, 38], viz. reduction of [Mn(CN)6]2− to [Mn(CN)6]3− is not favored. It is therefore likely that K3Mn(CN)6, albeit [Mn(CN)6]2− complex, affected the formation of PbH4 by the same mechanism involving the formation of reactive hydroborane intermediates facilitating the generation of PbH4 as proposed in the presence of K3Fe(CN)6 in that the generation of PbH4 was found to be independent of the sequence whether or not K3Fe(CN)6 was mixed with acidic sample solution or NaBH4 solution first [31]. This phenomenon was ascribed to the formation of reactive “borane complex intermediates” in the presence of K3Fe(CN)6 affecting the generation of PbH4 from Pb(II).
3.5. Optimization of NaBH4 concentration and flow rates
The influence of NaBH4 concentration on signals is shown in Fig. 4A for Pb (1.0 μg L−1) and 0.5% m/v K3Mn(CN)6 setup. NaBH4 was highly influential on the generation of plumbane. In comparison to those with 1% m/v NaBH4, the signals constantly increased up to 1.5% m/v and exhibited a plateau with further increase up to 3% m/v. Apparently, a concentration of 2% m/v was optimum to maintain the sensitivity. This result was also consistent with previous results reported for generation of PbH4 [17]. Water vapor is a major by-product of the reaction of NaBH4 with HCl, which could induce instability in plasma at higher concentrations of NaBH4 as reported elsewhere [17]. No significant instability was noted in this study from continuous introduction of 2% m/v NaBH4 since the cooled–spray chamber (2 °C) afforded efficient removal of water vapor before reaching to plasma.
Fig. 4.
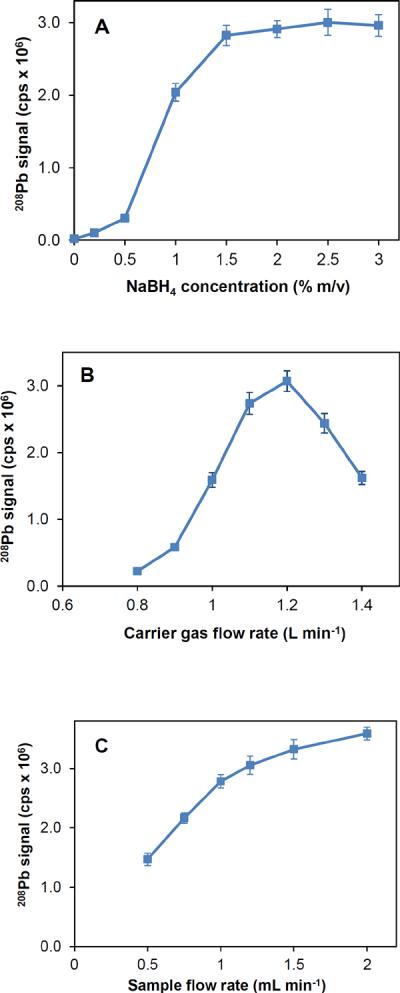
Effects of various experimental parameters on signals during generation of PbH4 in 1.0 μg L−1 Pb using 0.5% m/v K3Mn(CN)6. (A) NaBH4 concentration, (B) Carrier (nebulizer) argon flow rate, and (C) flow rate of sample solution.
The effect of the nebulizer flow rate on signals is shown in Fig. 4B. The sensitivity was at maximum between 1.1 and 1.2 L min−1 when the flow rate of nebulizer argon was varied from 0.8 to 1.4 L min−1. Signals dropped significantly at lower and higher flow rates due to the alterations in the sampling depth in the plasma. The flow rate for the sample solution was around 1.0 mL min−1 to achieve adequate sensitivity (Fig. 4C). Signals increased with increasing flow rates up to 1.5 to 2.0 mL min−1, but at the cost of increased sample consumption. Thus, the flow rate of the sample solution was kept constant at 1.0 mL min−1.
3.6. Analytical figures of merit
The detection limits obtained for 1% v/v HCl blanks with 2% m/v K3Mn(CN)6 were 0.007, 0.007 and 0.008 μg L−1 for 206Pb, 207Pb and 208Pb isotopes. These detection limits reflect the analyte concentration equivalent to three times the standard deviation of the blanks signals (n=13). Calibration curves were linear within 0.02 to 5.0 μg L−1 (r2 = 0.998 – 1.0). The detection limits for traditional ICP-MS measurements were 0.078, 0.075 and 0.090 μg L−1 for 206Pb, 207Pb and 208Pb isotopes, respectively. In comparison to the traditional ICP–MS, the sensitivity was enhanced by a factor of 42 to 48 based on the slopes of the calibration curves for direct ICP–MS and HG–ICP–MS. However, the enhancement in the detection limits were about 10-fold because of the higher background signals at HG mode. The average signal–to–ratio (S/N) at 1.0 μg L−1 was, however, about 18 to 20 which was mainly because of the residual Pb impurities in K3Mn(CN)6 solution despite efficient cleaning by coprecipitation. Relative standard deviations (RSD) varied between 0.4% and 6.3% for replicate measurements (n = 6) of the standard solutions (0.02 to 2 μg L−1). No memory effects were noted for replicate measurements even with 10 μg L−1 Pb solution that was continuously introduced during method optimization. A washout of 20 s with 1% v/v HCl (at 4 mL min−1) was sufficient for cleaning up the system between samples.
3.7. Tolerance to chemical interferences
Hydride generation (HG) offers the capability to alleviate the matrix interferences and better sensitivity for accurate determination by ICP-MS. Nevertheless, the chemical interferences from transition metals are still important setbacks in HG approaches [1,2,4,15,16,26]. The interferences in solution are mainly mediated by the transition metals, while the hydride forming elements cause interferences in vapor phase. Further, the interfering elements vary greatly with the hydride generation medium. Wang et al. [18], for instance, reported depressive effects from Cu(II), Fe(III), Mn(II) and Ni(II) on plumbane generation in HCl-H2O2 medium, while the same elements as well as Co(II) and Zn(II) enhanced the signals in HNO3-H2O2 medium [39,40]. In contrast, Co(II) induced interferences in determination of Pb by hydride generation in K3Fe(CN)6 medium [33]. The depressive effect was attributed to the reduction of Fe(III) and Pb(II) through cobalt borides. Among the transitions metals, Cu(II) has been the most reported for its interferences in the presence of oxidizing agents, such as hexacyanoferrate(III) [17], hydrogen peroxide [18] and cerium(IV) ammonium nitrate [26].
The tolerance of K3Mn(CN)6 to the effects of alkali, alkaline and transition metals and hydride forming elements are summarized in Table 2. The values reflect the relative responses obtained from 1.0 μg L−1 Pb solution with 500 μg L−1 of the interfering element to that of single element Pb solution using 0.5% m/v K3Mn(CN)6. No significant interferences were observed from any of the transition metals and hydride forming elements, excluding Cu(II), when test concentration was varied from 100 to 500 μg L−1. Signals varied from 94% for Ni(II) to 106% for Zn(II). The test concentrations for the alkali (Na and K) and alkaline earth elements (Ca and Mg) were 1000 μg mL−1. The relative responses in the solutions of these elements varied between 98% (Mg) and 106% (K), indicating that they were highly benign in the generation of PbH4.
Table 2.
Effects of matrix elements on relative signals in 1.0 μg L−1 Pb solution using 0.5% m/v K3Mn(CN)6
| Element | Concentration (μg L−1) | Relative signal (%) |
|---|---|---|
| As(III) | 500 | 96 ± 2 |
| Bi(III) | 500 | 102 ± 2 |
| Co(II) | 500 | 98 ± 5 |
| Cr(III) | 500 | 102 ± 5 |
| Cu(II) | 100 | 42 ± 3 |
| 500 | 12 ± 2 | |
| Fe(III) | 500 | 103 ± 2 |
| Mn(II) | 500 | 98 ± 4 |
| Ni(II) | 500 | 94 ± 7 |
| Sb(III) | 500 | 100 ± 2 |
| Se(IV) | 500 | 98 ± 3 |
| Sn(II) | 500 | 99 ± 2 |
| Zn(II) | 500 | 106 ± 4 |
Catalytic decomposition of [Fe(CN)6]3− was reported in the presence Cu(II) [41,42]. In this study, Cu(II) induced severe depression in K3Mn(CN)6 medium, suggesting that interferences of Cu(II) were due to the decomposition of Mn(CN)6]3− (e.g., [Mn(CN)6]2−). The instability of the reagent could have also contributed to the severity of the interferences such that the signals were suppressed by more than 50% even in the presence of 0.1 μg mL−1 Cu(II). To overcome this hurdle, the concentration of K3Mn(CN)6 was re-examined along different Cu(II) concentrations. The results are shown in Fig. 5 for 0.5, 1 and 2% m/v K3Mn(CN)6. It is clear that 0.5% m/v K3Mn(CN)6 was not adequate to abate the depressive effects of Cu(II), though it performed successfully in the presence of other interfering elements, such as Ni(II) and Co(II). The signals, however, improved significantly by increasing K3Mn(CN)6 concentration to 2% m/v for which percent relative response was about 92% in the presence of 0.1 μg mL−1 Cu(II) matrix.
Fig. 5.
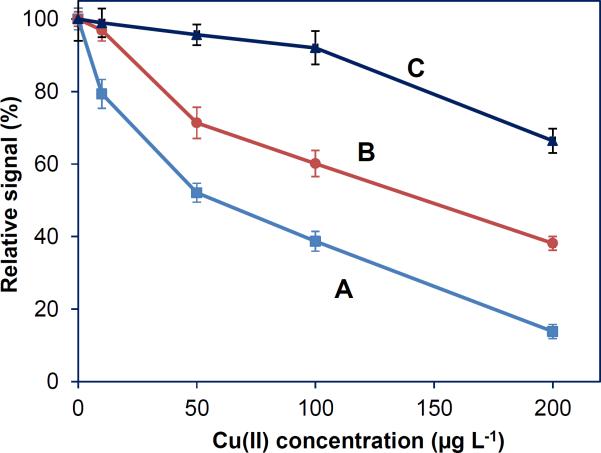
The effect of Cu(II) concentration on signals for 1.0 μg L−1 Pb solution as a function of K3Mn(CN)6 concentration. (A) 0.5% m/v K3Mn(CN)6, (B) 1.0% m/v K3Mn(CN)6, (C) 2% m/v K3Mn(CN)6.
Table 3 summarizes the performance of the method in the presence of 2% m/v K3Mn(CN)6 with those reported earlier. It is clear that Cu(II) was the only single element that interfered with the formation of PbH4 in any medium, even in the presence of potassium hexacyanoferrate(III) medium [15,17,43]. The effects were severest in both HNO3-H2O2 [16] and HCl-H2O2 [16, 18] such that accuracy was low even in freshwater samples containing part per billion levels of transitions metal ions [16]. In HNO3-H2O2, isotope dilution (ID) approach provided better detection limits and accuracy [16]. The detection limits obtained by using oxalic acid-ammonium cerium(IV) nitrate were similar to those in this study, but (NH4)2Ce(NO3)6 medium appears to be highly susceptible to the depressive effects of Cu(II) despite the masking ability of oxalate ion [26].
Table 3.
Comparison of the performance of the procedure with other methods applied to the determination of Pb by hydride generation
| Technique | Medium | Matrix | Concentration (μ g mL−1) | R(%) | LOD (μg L−1) | Ref.# |
|---|---|---|---|---|---|---|
| HCl | This work | |||||
| HG–ICPMS | K3Mn(CN)6 | Cu(II) | 0.1 | 92 | 0.008 | |
| HG–ICPMS | Cu(II) | 0.5 | 90 | 0.003 | 15 | |
| HCl–H2C2O4 | Co(II) | 0.5 | 91 | |||
| k3Fe(CN)6 | Ni(II) | 0.5 | 91 | |||
| HG–ICPMS | HNO3–H2O2 | Freshwaters | – | 130 | 0.2 | 16 |
| Seawater | 89 | |||||
| 20% NaCl | 41 | |||||
| HG–ID–ICPMS | HNO3–H2O2 | Freshwatera | – | 100 | 0.02 | 16 |
| Seawater | 99 | |||||
| 20% NaCl | 102 | |||||
| HCl | ||||||
| HG–ICPAES | K3Fe(CN)6 | Cu(II) | 1.0 | 72 | 0.1 | 17 |
| FI–HG–ICPMS | HCl–H2O2 | Cu(II) | 0.1 | 36 | 0.04 | 18 |
| Fe(III) | 0.1 | 41 | ||||
| Ni(II) | 0.5 | 93 | ||||
| H2C2O4 | 0.02 | 99 | 0.007 | 26 | ||
| HG–ETV–ICPMS | (NH4)2Ce(NO3)6 | Cu(II) | 0.1 | 64 | ||
| HG–ETV–ICPMS | HCl | Lakewaterb | – | 98 | 0.07 | 32 |
| K3Fe(CN)6 | ||||||
| HG–ICPAES | HCl | Cu(II) | 1.0 | 50 | 0.7 | 43 |
| K3Fe(CN)6 | Fe(III) | 1.0 | 92 |
SRM 1623, NIST Freshwater certified reference material.
NWRI TM-28.3 certified reference material (trace elements fortified water from Lake Ontario), (5-fold diluted).
3.7. Application to real samples
Calibration was made with single element external standards for the analysis of samples by the optimized HG-ICP-MS procedure. The standard solutions ranged from 0, 0.02, 0.05, 0.1, 0.2, 0.5, 1.0 to 2.0 μg L−1 Pb in 1% v/v HCl. The signals from five standard solutions bracketing that of the sample were used to calculate the Pb concentration in the samples. The Nearshore certified reference seawater (CASS-4) and the coastal seawater were analyzed by adjusting the acidity of sub-samples to 1% v/v HCl. Because the certified value of Pb in CASS-4 was within the vicinity of the detection limits of the method, accuracy was further verified with spiked solutions for both water samples. The results are summarized in Table 4 for 208Pb isotope. The experimental results for CASS-4 were consistent with the certified value of Pb in CASS-4 at 95% confidence level. The concentration of Pb in the coastal seawater was much higher which was attributed to the contributions from local sources (creeks and rivers) as well as leaching from the surface and bottom sediments. Similarly, the concentrations obtained from the unspiked samples were consistent with that for the spiked seawater samples. The concentration of Cu(II) in CASS-4 was 0.592 ± 0.055 μg L−1 and 0.920 ± 0.06 μg L−1 in coastal seawater. These levels were well below the threshold value of Cu(II) (100 μg L−1) to affect the accuracy.
Table 4.
Elemental concentrations for 208Pb from the analysis of Nearshore certified seawater reference material (CASS–4), coastal seawater, bone ash (SRM 1400) and mussel tissue (SRM 2976) certified reference materials by HG–ICP–MS procedure. Results are given as mean ± standard deviation of five replicate analyses for each sample. Spike concentrations are 0.02 and 0.05 μg L−1 for CASS–4 and coastal seawater, respectively
| Sample | Determined |
Certified value | |
|---|---|---|---|
| Unspiked sample | Spiked sample | ||
| Nearshore seawater (CASS–4)(μg L−1) | 0.0013 ± 0.004 | 0.030 ± 0.006 | 0.0098 ± 0.0036 |
| Coastal seawater(μg L−1) | 0.580 ± 0.020 | 1.18 ± 0.06 | – |
| Bone ash (SRM 1400) (μg g −1) | 8.93 ± 0.34 | – | 9.07 ± 0.12 |
| Mussel tissue (SRM 2976) (μg g−1) | 1.33 ± 0.22 | – | 1.19 ± 0.18 |
The results for Bone ash (SRM 1400) and Mussel tissue (SRM 2976) are also summarized in Table 4. The bone ash samples were rich in calcium and phosphate along with aluminum, iron, fluoride and magnesium at μg mL−1 levels. The mussel tissue (SRM 2976) was composed of organic matrix with high levels of Fe and Zn. Determinations were performed in 20− and 10-fold diluted solutions for SRM 1400 and SRM 2976, respectively, to bring the concentration of Pb within the calibration range. Accurate results were obtained for Pb in both reference materials. The experimental concentrations were in agreement with the certified values within 95% confidence level (Table 4).
4. Conclusions
Efficiency of potassium hexacyanoferrate(III) for generation of lead hydride has been known for decades now. In this study, we have utilized another transition metal cyanide complex, potassium hexacyanomanganate(III), for the first time for generation of plumbane and investigated its performance in determination of Pb by HG-ICP-MS. Potassium hexacyanomanganate(III) is very insoluble and unstable in water that renders this catalytically active reagent unusable for hydride generation. On the other hand, the acidic conditions provided by H2SO4 were conducive to achieve better stabilization. The results confirmed that K3Mn(CN)6 was uniquely suited for effective generation of plumbane from acidic sample solutions of Pb(II) when interacted with NaBH4. It is assumed that that hexacyanomanganate(III) mediates the generation of PbH4 by similar mechanism as hexacyanoferrate(III) does. Future studies are expected to shed light onto the mechanism of this process.
The method described here is highly sensitive and robust for quantitative determination of Pb accurately in various samples by ICP-MS and other atomic spectroscopy techniques. It is also not affected from the presence of common transition metals (Co, Cr, Fe, Ni,) and hydride forming elements. Cu(II) impacts the stability and hence performance of K3Mn(CN)6 adversely at sub-ppm levels. Nevertheless, the effects could easily be abated using on-line approach and higher concentrations of K3Mn(CN)6. Because of the simplicity, the method could easily be interfaced to inductively coupled plasma atomic emission spectroscopy (ICP-AES) for sub-ppb determination of Pb. It is envisioned that this catalytically active reagent could also offer the capability of hydride generation for multiple element determination.
Highlights
Potassium hexacyanomanganate(III), (K3Mn(CN)6, was utilized first time for hydride generation (HG).
Hexacyanomanganate(III) promoted generation of lead hydride (PbH4) remarkably.
The HG method using K3Mn(CN)6 enhanced sensitivity by at least 40-fold.
The method detection limits for Pb were as low as 7 ng L−1 by ICP-MS.
The method is highly suitable for quantitative determination of Pb in various samples and salt matrices by ICP-MS.
Acknowledgements
This project was supported by grants from the National Center for Research Resources (5 G12 RR013459-15), the National Institute on Minority Health and Health Disparities (8 G12 MD007581-15) and the Research Initiative for Scientific Enhancement (2 R25 GM067122) from the National Institutes of Health. The views expressed herein are those of authors and do not necessarily represent the official views of the NIH and any of its sub-agencies. The authors also acknowledge financial support from the Turkish Higher Education Council to Dr. Vedat Yilmaz during the course of this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Tsalev DL. J. Anal. At. Spectrom. 1999;14:147–162. [Google Scholar]
- [2].Sturgeon RE, Mester Z. Appl. Spectrosc. 2002;56:202A–213A. [Google Scholar]
- [3].Pohl P. Trends Anal. Chem. 2004;23:87–101. [Google Scholar]
- [4].Pohl P, Zyrnicki W. Anal. Chim. Acta. 2002;468:71–79. [Google Scholar]
- [5].McLaughlin RLJ, Brindle ID. J. Anal. At. Spectrom. 2002;17:1540–1548. [Google Scholar]
- [6].Farias S, Rodriguez RE, Ledesma A, Batistoni DA, Smichowski P. Microchem. J. 2002;73:79–88. [Google Scholar]
- [7].Salgado SG, Nieto MAQ, Simon MMB. J. Chromatogr. Part A. 2006;1129:54–60. [Google Scholar]
- [8].Koh J, Kwon Y, Pak Y-N. Microchem. J. 2005;80:195–199. [Google Scholar]
- [9].Rojas I, Murillo M, Carrión,·J N. Chirinos, Anal. Bioanal. Chem. 2003;376:110–117. doi: 10.1007/s00216-003-1856-7. [DOI] [PubMed] [Google Scholar]
- [10].Thompson KC, Thomerson DR. Analyst. 1974;99:595–601. [Google Scholar]
- [11].Mena CM, Cabrera C, Lorenzo ML, Lopez MC, Agric J. Food Chem. 1997;45:1812–1815. [Google Scholar]
- [12].Ertas N, Arslan Z, Tyson JF. J. Anal. At. Spectrom. 2008;23:223–228. [Google Scholar]
- [13].Elci L, Arslan Z, Tyson JF. J. Hazard. Mat. 2009;162:880–885. doi: 10.1016/j.jhazmat.2008.05.113. [DOI] [PubMed] [Google Scholar]
- [14].Chuachuad W, Tyson JF. J. Anal. At. Spectrom. 2005;20:282–288. [Google Scholar]
- [15].Chen S, Zhang Z, Yu H, Liu W, Sun M. Anal. Chim. Acta. 2002;463:177–188. [Google Scholar]
- [16].Petrov PK, Wibetoe G, Tsalev DL. Spectrochim. Acta Part B. 2006;61:50–57. [Google Scholar]
- [17].Afonso DD, Arslan Z, Baytak S. J. Anal. At. Spectrom. 2010;25:726–729. doi: 10.1039/b920280c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang XR, Viczian M, Lasztity A, Barnes RM. J. Anal. Atom. Spectrom. 1988;3:821–827. [Google Scholar]
- [19].Castillo JR, Mir JM, Martinez C. J. Vol, P. Colon, Mikrochim. Acta. 1985;85:253–263. [Google Scholar]
- [20].Hon PK, Lau OW, Cheung WC, Wong MC. Anal. Chim. Acta. 1980;115:355–359. [Google Scholar]
- [21].Jin K, Taga M. Anal. Chim. Acta. 1982;143:229–236. [Google Scholar]
- [22].Mena CM, Cabrera CM, Lorenzo L, Lopez MC. J. Agric. Food Chem. 1997;45:1812–1815. [Google Scholar]
- [23].Maleki N, Safavi A, Ramezani Z. J. Anal. At. Spectrom. 1999;14:1227–1230. [Google Scholar]
- [24].dos Santos EJ, Herrmann AB, Frescura VLA, Welz B, Curtius AJ. Anal. Bioanal. Chem. 2007;388:863–868. doi: 10.1007/s00216-006-1081-2. [DOI] [PubMed] [Google Scholar]
- [25].Li J, Liu Y, Lin T. Anal. Chim. Acta. 1990;231:151–155. [Google Scholar]
- [26].Li J, Lu F, Umemura T, Tsunoda K–I. Anal. Chim. Acta. 2000;419:65–72. [Google Scholar]
- [27].Chen H, Tang F, Gu C, Brindle ID. Talanta. 1993;40:1147–1155. doi: 10.1016/0039-9140(93)80180-y. [DOI] [PubMed] [Google Scholar]
- [28].Thao R, Zhou H. Fenxi Huaxue. 1985;13:283–285. [Google Scholar]
- [29].Cankur O, Korkmaz D, Ataman OY. Talanta. 2005;66:789–793. doi: 10.1016/j.talanta.2004.12.035. [DOI] [PubMed] [Google Scholar]
- [30].Tyson JF, Ellis RI, Carnrick G, Fernandez F. Talanta. 2000;52:403–410. doi: 10.1016/s0039-9140(00)00341-6. [DOI] [PubMed] [Google Scholar]
- [31].D'Ulivo A, Onor M, Spiniello R, Pitzalis E. Spectrochim. Acta Part B. 2008;63:835–842. [Google Scholar]
- [32].Gil S, de Loos–Vollebregt MTC, Bendicho C. Spectrochim. Acta Part B. 2009;64:208–214. [Google Scholar]
- [33].Zheng L, Lin L, Zhu L, Jiang M. Spectrochim. Acta Part B. 2009;64:222–228. [Google Scholar]
- [34].Li R, Yan H, Yang X, Li Z, Guo Y. J. Anal. At. Spectrom. 2011;26:1488–1493. [Google Scholar]
- [35].Yilmaz V, Rose L, Arslan Z, Little MD. J. Anal. At. Spectrom. 2012;27:1895–1902. doi: 10.1039/C2JA30195D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Arslan Z. Talanta. 2005;65:1326–1334. doi: 10.1016/j.talanta.2004.09.009. [DOI] [PubMed] [Google Scholar]
- [37].Chadwick BM, Sharpe AG. In: Advances in Inorganic Chemistry and Radiochemistry. Emeleus HJ, Sharpe AG, editors. Vol. 8. Academic Press; New York: 1966. pp. 103–109. [Google Scholar]
- [38].Lopes–Cueto G, Ubide C. Can. J. Chem. 1986;64:2301–2304. [Google Scholar]
- [39].Bonilla M, Rodriguez L, Camara C. J. Anal. At. Spectrom. 1987;2:157–161. [Google Scholar]
- [40].Aroza I, Bonilla M, Madrid Y, Camara C. J. Anal. At. Spectrom. 1989;4:163–166. [Google Scholar]
- [41].López–Cueto L, Casado–Riobó JA. Talanta. 1979;26:127–132. doi: 10.1016/0039-9140(79)80228-3. [DOI] [PubMed] [Google Scholar]
- [42].López–Cueto G, Casado–Riobó JA. Talanta. 1979;26:151–153. doi: 10.1016/0039-9140(79)80236-2. [DOI] [PubMed] [Google Scholar]
- [43].Brindle ID, McLaughlin R, Tangtreamjitmun N. Spectrochim. Acta Part B. 1998;53:1121–1129. [Google Scholar]