

Abstract
Although thymic involution has been linked to the increased testosterone in males after puberty, its detailed mechanism and clinical application related to T-cell reconstitution in bone marrow transplantation (BMT) remain unclear. By performing studies with reciprocal BMT and cell-specific androgen receptor (AR) knockout mice, we found that AR in thymic epithelial cells, but not thymocytes or fibroblasts, played a more critical role to determine thymic cellularity. Further dissecting the mechanism using cell-specific thymic epithelial cell-AR knockout mice bearing T-cell receptor transgene revealed that elevating thymocyte survival was due to the enhancement of positive selection resulting in increased positively selected T-cells in both male and female mice. Targeting AR, instead of androgens, either via genetic knockout of thymic epithelial AR or using an AR-degradation enhancer (ASC-J9®), led to increased BMT grafting efficacy, which may provide a new therapeutic approach to boost T-cell reconstitution in the future.
Thymic atrophy is associated with aging and accompanied by decreased thymic cellularity, disrupted thymic architecture, and diminished emigrating naïve T-cells into peripheral pools (1, 2). The reduction of exported T-cells from thymus sequentially causes the expansion of existing memory T-cells (3), limits the diversity of T-cell receptor (TCR) repertoire (4), and dampens the immune response when encountering new foreign antigens.
The interactions between thymocytes and lymphostromal cells in thymus are critical to maintain functional T-cells exportation (5). Cortical thymic epithelial cells (cTECs) play crucial roles to mediate positive selection by physical engagement of TCR in thymocytes with major histocompatibility complex (MHC)-self-peptides complexes (6). After positive selection, CD4+CD8+ double positive (DP) thymocytes encounter other thymic stromal cells (TSCs) such as macrophages, dendritic cells, and medullary TECs (mTECs) in the medulla for negative selection to eliminate auto-reactive thymocytes (7).
To determine the target cells of androgen/androgen receptor (AR) effects on thymopoiesis, the expression of AR in thymocytes and TSCs has been extensively analyzed (8). Although some early ligand-binding studies indicated that thymocytes had no AR expression (9), later studies have shown AR protein detection in thymocytes by flow cytometry, immunoblotting in mice, and ligand-binding assays in humans (10, 11). AR expression in thymic stroma was also substantially characterized in the early studies and demonstrated that AR was predominantly expressed in TECs by histochemical study of thymic tissue and analysis of cultured cell lines (8).
By using reciprocal bone marrow transplantation (BMT) strategy together with newly established specific AR knockout (ARKO) mice, we clearly demonstrated that AR in TECs, but not in thymocytes or fibroblasts, play a more critical role to determine thymic cellularity.
Conversely, targeting AR either by genetic knockout of AR in TECs or using the AR-degradation enhancer, ASC-J9® (12), to predominantly degrade AR in TECs leading to increased BMT efficacy, might be applicable in the clinical setting to boost T-cell reconstitution in patients.
Materials and Methods
Mice, cells, and reagents
The floxed AR mice (C57BL/6) were described previously (13) and ACTB cre transgenic (tg) mice (FVB) and lck cre tg mice (C57BL/6) were purchased from The Jackson Laboratory (Bar Harbor, ME). The bovine cytokeratin 5 cre tg mice (FVB) were kindly provided by Dr. D.G Johnson (University of Texas MD Anderson Cancer Center, Smithville, TX) (14). Fsp1 cre mice (C57BL/6) were a gift from Dr. N.A. Bhowmick (Vanderbilt University, Nashville, TN) (15). The AND-TCR/Rag2 KO mice originally in C57BL/10 background were a gift from Dr. R.N. Germain (National Institutes of Health/National Institute of Allergy and Infectious Diseases, Bethesda, MD). HY-TCR tg mice (C57BL/6) were purchased from Taconic Farms, Inc. (Germantown, NY). For the positive and negative selection study, mice were maintained in C57BL/6 background by backcrossing more than seven to eight generations. Animal study protocols were approved by the University of Rochester Committee on Animal Resources, and the mice were kept in a specific pathogen-free environment. Primary fluorochrome or biotinylated-conjugated antibodies, CD45.1(A20), CD45.2 (104), CD3 (145–2C11), CD4 (GK1.5), CD8a (53–6.7), CD44 (IM7), CD25 (3C7), CD69 (H1.2F3), T-cell receptor-β (H57–597), Thy 1.2 (53–2.1), CD45R/B220 (RA3–6B2), bromodeoxyuridine (BrdU) (PRB-1), MHC II I-A/I-E (M5/114.15.2), EpCAM (G8.8), HY-TCR (T3.70), and anti-murine FcR block (2.4G2) were purchased from e-Bioscience. Biotinylated UEA-1 was purchased from Vector Laboratories, Inc. (Burlingame, CA). ASC-J9® compound was obtained from AndroScience Corp. (San Diego, CA).
Flow cytometry
Bone marrow cells, thymocytes, and splenocytes were washed in fluorescence-activated cell sorting (FACS) buffer (PBS + 1% heat-inactivated fetal bovine serum and 0.1% sodium azide) and 1–3 × 106 cells were incubated with primary antibodies at 4 C for 30 min, and then washed twice with FACS buffer. If necessary, cells were incubated with Streptavidin-conjugated fluorochrome for another 30 min at 4 C. The stained cells were resuspended in FACS buffer and analyzed by FACS CantoII Flow Cytometry (Becton Dickinson and Co., Franklin Lakes, NJ). The data analysis was conducted by using FlowJo V7.5.5 (Tree Star, Inc., Ashland, OR). For the BrdU incorporation assay, mice were IP injected four times with 10 mg/kg BrdU at 6-h intervals. The final injection was finished 1 h before animals were killed. The isolated thymocytes were stained with surface markers first and then fixed with 2% paraformaldehyde. The BrdU staining protocol was according to manufacturer's instructions (Becton Dickinson). For thymocytes apoptosis assays, freshly isolated thymocytes were stained with Annexin-V and 7-AAD (eBioscience) and analyzed by FACS.
Histological and immunohistochemical analysis
Freshly dissected thymi were immediately immersed in 10% neutral formalin for overnight incubation. The tissues were paraffin embedded, sectioned (5 μm), and stained by hematoxylin and eosin. For the AR expression determination, anti-AR antibody (C-19, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was incubated at 4 C overnight and then labeled with appropriate secondary antibody, ABC kit (Vector Laboratories), and diaminobenzidene substrate detection were used. For double immunofluorescent staining, the thymus sections were colabeled with antibodies against pan-CK (PCK26, Sigma-Aldrich, St. Louis, MO) and AR (C19) at 4 C overnight. The binding of the antibody was detected by Alexa Fluor 488 goat antirabbit IgG or 594 goat antimouse IgG (Molecular Probes, Inc., Eugene, OR). The images were acquired using an E800 microscope (Nikon, Melville, NY) and a SPOT camera (Diagnostic Instruments, Arnold, MD).
Thymus stromal cells isolation and phenotypic characterization
This procedure was modified slightly from the previously published protocol (16). Each thymus was washed with PBS three times and cleaned of fat and connective tissue. The thymus was then minced into approximately 1-mm3 pieces with fine scissors, resuspended in 10% fetal bovine serum (FBS) RPMI 1640 medium with agitation for 5 min several times after which the pellet was allowed to settle. The supernatants were removed for collection. Thymic pellets were then incubated with 0.125% (wt/vol) Collagenase D and 0.1% (wt/vol) deoxyribonuclease I (Roche, Indianapolis, IN) in 10% FBS RPMI 1640 medium at 37 C for 10 min with gentle agitation. After each digestion, the supernatant was collected and digestion was repeated another three times. The remaining aggregates were incubated with 0.125% (wt/vol) Collagenase/Dispase (Roche) and 0.1% (wt/vol) deoxyribonuclease I in RPMI 1640 medium for another 10 min. The remaining thymic aggregates were retriturated by 1 ml blue tip to segregate macroscopic fragments several times. Cells from all supernatant fractions were then centrifuged at 450 × g for 5 min, pooled, and resuspended in cold FACS buffer. Cells were filtered through a 70-μm strainer and counted. For phenotypic characterization of thymic stromal cells, 1–2 × 106 cells were stained with appropriate primary fluorochrome-conjugated antibodies.
Mice treatment, γ-irradiation, and bone marrow transplantation
Recipient male mice at 5–6 wk of age (FVB/C57BL/6, CD45.1+CD45.2+) were injected ip daily with vehicle-dimethylsulfoxide (DMSO) or 50 mg/kg ASC-J9® (AndroScience Corp.). After 2 wk injection, T-cells-depleted (TCD) BM cells were transplanted iv into recipients that had previously received two doses of 5 Gy total body irradiation (4 h apart) from a Cesium source 24 h before BMT. This dose effectively destroys the hematopoietic cells of recipient mice. Donor BM cells were removed aseptically from femurs and tibias of F1 (CD45.1+CD45.2−) progeny mice from B6.SJL-Ptprca Pep3b/BoyJ (CD45.1+, The Jackson Laboratory) and FVB (CD45.1+). Donor BM cells were depleted of T-cells by incubation with anti-Thy 1.2 (30-H-12, Becton Dickinson) for 40 min at 4 C and subsequently incubated with Low-TOX-M rabbit complement (Cedarlane Laboratories, Burlington, Ontario, Canada) for 60 min at 37 C. BM cells were resuspended in 2% FBS RPMI medium and transplanted into the recipient mice by iv injection (17). For the cell proliferation assay, 4 × 105 splenocytes isolated from reconstituted recipients 6 wk after BMT were plated into 96-well plates, which were precoated with plate-bound anti-CD3e and CD28 antibody (eBioscience, San Diego, CA). After 2 d culture, 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma-Aldrich) was added to the wells 3 h before harvest and then subjected to OD measurement by ELISA. For the intracellular IFN-γ detection, 1–2 × 106 splenocytes isolated from reconstituted recipients 6 wk after BMT were stimulated with 50 ng/ml phorbol-12-myristate 13-acetate and 1 μm ionomycin in the presence of 10 μg/ml Brefeldin A for 4 h. After incubation, surface markers of CD45.1, CD45.2, and CD8 were stained and fixed by paraformaldehyde. The cells were permeabilized by Cytofix/Cytoperm kit (BD Bioscience, Palo Alto, CA) and stained with anti-IFN-γ antibody (Ab) (eBioscience). Flow cytometer and software were used as described above.
Statistics
The data are presented as mean ± sem and the two-tailed Student's t test or one-way ANOVA were used for the parametric analyses. The following P values were considered as statistically significant: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Results
Enlarged thymus with increased thymic cellularity in mice lacking whole-body AR
To unravel the AR functions in thymus development, we established a mouse model that lacks AR expression in the whole-body (G-AR−/y) (13). Evidence from thymus genotyping together with AR mRNA and protein detection confirmed AR disruption in this male G-AR−/y mouse (Supplemental Fig. 1, A–C, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). For the phenotypic characterization, we found the thymus was significantly enlarged in G-AR−/y mice compared with wild-type (WT) littermates (AR+/y) at the age of 8–12 wk (Fig. 1A). The enlarged thymus was correlated with increased thymic cellularity in G-AR−/y mice (Fig. 1B). To determine the thymocyte differentiation after loss of AR, we analyzed the CD4−CD8− (double negative, DN), CD4+CD8+ (DP), and CD4+ or CD8+ (single positive, SP) populations by flow cytometry. We found that the populations of DN, DP, and SP were similar in both genotypes of mice (Fig. 1C). In addition, using surface markers of CD44 and CD25 to define the subpopulations of DN: CD44+CD25− (DN1), CD44+CD25+ (DN2), CD44−CD25+ (DN3), and CD44−CD25− (DN4) in both genotypes of mice, the results also showed no noticeable differences (Fig. 1D). As expected, we found the cellularity of thymocyte subsets was proportionally increased in G-AR−/y mice as compared with WT control (Fig. 1, E and F). The triple-negative CD3−CD4−CD8− (TN) thymocytes were gated from Thy 1.2+ and CD3− population (Fig. 1F). Our findings from G-AR−/y mice also confirmed the previous reports using castration (18) or naturally occurring AR mutant testicular–feminized (Tfm) mice (8) showing similar results.
Fig. 1.
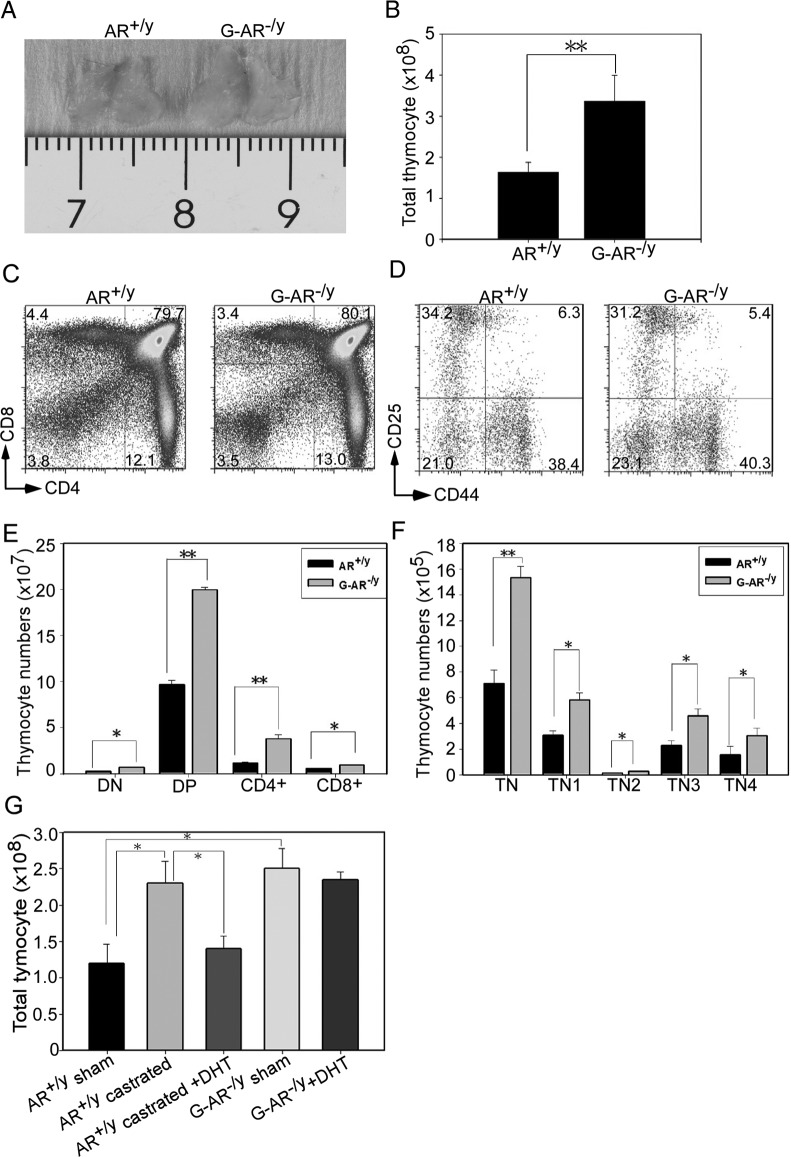
Loss of universal AR in male G-AR−/y mice leads to enlarged thymus and increased thymocyte cellularity. A, Enlarged thymus was observed in male G-AR−/y mice at 8–12 wk of age. The representative photograph is shown from 8–10 mice per group. B, Total thymocyte numbers were increased in G-AR−/y mice compared with AR+/y littermate control mice. C, The populations of CD4+, CD8+, DP, and DN thymocytes did not change in G-AR−/y mice. Expression of CD4 and CD8 from AR+/y and G-AR−/y mouse thymocytes were characterized by flow cytometry. The numbers in the corners of each quadrant represent the percentage of cells. D, Immature subsets of thymocyte population defined by CD25 and CD44 did not alter in G-AR−/y mice. E, Cellularity of DN, DP, CD4+ SP, and CD8+ SP was elevated in G-AR−/y mice. F, Cell numbers of TN (CD3−CD4−CD8−) subsets were counted and showed an increase in G-AR−/y mice. The TN populations were gated from Thy1.2+. The data of Fig. 1, B–F were shown from four independent experiments with 8–10 mice per group, and each bar represents the mean ± sem. *, P < 0.05; **, P < 0.01 compared with AR+/y mice. G, AR+/y male mice were subjected to sham or surgical castration to remove systemic androgens. The DHT pellets or placebo was implanted into castrated AR+/y mice and G-AR−/y mice. The thymus from each group was harvested, and thymocyte numbers were determined after 2 wk of DHT pellet implantation (n = 6–8; *, P < 0.05).
We have previously shown that serum testosterone levels were dramatically reduced in male G-AR−/y mice (13) and some studies also suggested that androgens can function in non-AR-dependent pathways to alter intracellular signaling for cell behavior changes (19). We therefore asked whether the enlarged thymus in G-AR−/y mice was due to the decreased androgens or deficiency of AR. By using castration and dihydrotestosterone (DHT) pellet replacement approaches, we failed to see the reversed phenotype of enlarged thymus in G-AR−/y mice when supplemented with DHT (Fig. 1G). This result suggested that AR-dependent pathways are more critical, whereas AR-independent pathways are dispensable in mediating this phenotype.
Together, results from Fig. 1, A–G, suggested that knockout of AR in the whole body results in thymus enlargement and increased thymic cellularity. Importantly, the AR-dependent regulations are more critical than AR-independent pathways to control thymus development.
Lacking AR in non-bone marrow-derived stromal cells is responsible for thymus enlargement
The thymic stromal cells are comprised of BM-derived and thymus residential cells. BM-derived stromal cells such as macrophages and dendritic cells are more sensitive to γ-irradiation (20), yet thymus residential cells such as cTECs, mTECs, fibroblasts, and endothelial cells are more resistant (21). To dissect AR functions in specific cellular compartments, we performed reciprocal BMT between AR+/y and G-AR−/y mice by transplanting BM cells reciprocally into γ-irradiated G-AR−/y and AR+/y recipients. By comparing the reconstituted thymus from BM chimera mice, we found that BM cells transplanted from either AR+/y or G-AR−/y mice into the irradiated G-AR−/y recipients resulted in enlarged thymus compared with thymus of AR+/y mice receiving the same BMT (Fig. 2A). In contrast, AR+/y or G-AR−/y BM cells transplanted into AR+/y recipient mice consistently showed no enlargement in thymus (Fig. 2A). This conclusion is consistent with the previous report using similar reciprocal BMT approaches between WT and Tfm (AR−/y) mice (8).
Fig. 2.
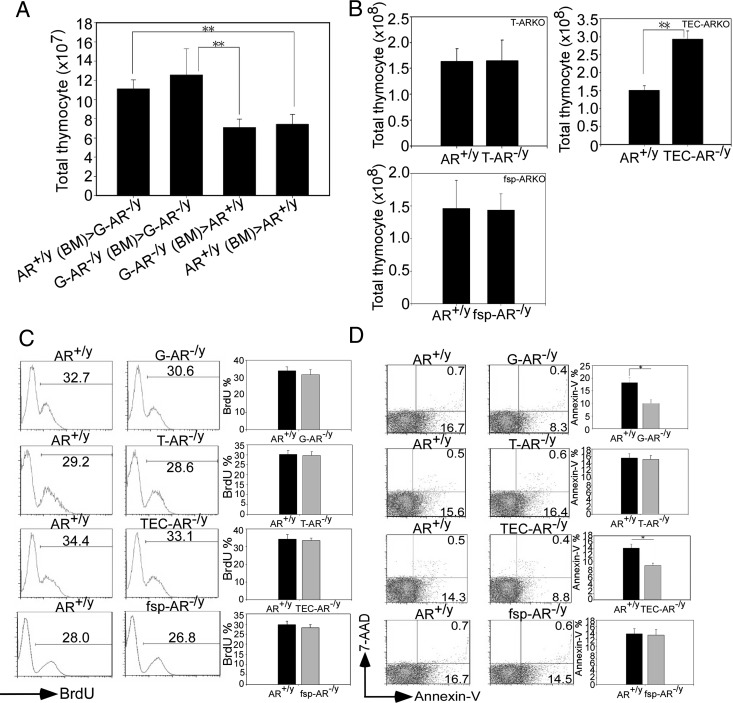
AR in TECs is crucial for maintaining the thymocyte cellularity. A, γ-Irradiated 6 wk-old AR+/y and G-AR−/y recipient, and mice were adoptively transferred with AR+/y or G-AR−/y BM cells. AR+/y (BM)>G-AR−/y means that AR+/y BM cells were transferred into G-AR−/y recipient, vice versa. After BM engraftment, thymi from individual BM chimera mice were harvested and the numbers of repopulated thymocytes were counted. Each bar represents the mean ± sem from three independent experiments with 6 to 8 mice per group (**, P < 0.01). B, Thymocyte numbers were increased in the TEC-AR−/y mice, but not in the T-AR−/y or fsp-AR−/y mice. The cellularity was determined from 8- to 12-wk old male T-AR−/y, TEC-AR−/y, fsp-AR−/y, and AR+/y littermate control mice. C, The percentage of BrdU+ DP thymocytes was assayed by flow cytometry (left panels), and the results showed comparable % between AR+/y and AR-deficient mice. The quantification data are provided in the right panels. D, G-AR−/y and TEC-AR−/y mice showed decreased apoptotic thymocytes compared with AR+/y littermate control. The apoptotic thymocytes were characterized by labeling with Annex-V and 7-AAD for flow cytometric analysis, and the quantification data are provided in the right panels. The representative and quantitative data shown in Fig. 2, B–D. were obtained from three independent experiments with 6 to 8 mice per group, and each bar represents mean ± sem. (**, P < 0.01; *, P < 0.05 as compared with AR+/y littermate control mice).
Collectively, results obtained from reciprocal BMT demonstrated that AR in the thymus residential stromal cells, but not the hematopoietic derived cells, plays more critical roles to determine T-cell repopulation.
Specific disruption of AR in thymic epithelium leads to thymus enlargement
To further delineate which stromal cells play more dominant roles in thymus development, we applied the cre/loxP system to generate cell-specific ARKO mice, including T-cell-specific ARKO (T-AR−/y, cre was driven by proximal lymphocyte-specific protein tyrosine kinase, lck promoter) (22), thymic epithelial cell-specific ARKO (TEC-AR−/y, cre was driven by bovine cytokeratin 5 promoter) (14), and fibroblast-specific ARKO (fsp-AR−/y, cre was driven by fsp1 promoter) (15) to address this question. The genotyping results of thymus from individual AR knockout mice are provided in Supplemental Fig. 1D, and the tissue-selective AR deletion was further confirmed using tissue genotyping from TEC-AR−/y and T-AR−/y mice (Supplemental Fig. 1, E and F). The three important androgen-targeted organs, brain, pituitary, and testis, were concurrently examined using tissue genotyping and AR quantitative PCR analysis to show AR intact among those organs in AR+/y and three specific AR−/y mice (Supplemental Fig. 1, G and H). For the serum testosterone level detection in these specific ARKO mice, we have provided the data for T-AR−/y (Supplemental Fig. 1I) and TEC-AR−/y mice (23). The tissue genotyping and serum testosterone level measurement in fsp-AR−/y mice have been documented in our previous reports (17, 24). By comparing these three specific ARKO mice and their WT littermate controls, we found that increased thymocyte numbers only appeared in TEC-AR−/y mice (Fig. 2B), indicating that loss of thymic epithelial AR is critical to mediate thymus enlargement. This observation is consistent with the previous report (8) showing that ARs in TECs, but not in the thymocytes, are more important to mediate the thymocyte development. We then determined thymocyte differentiation in these three specific ARKO mice and found the subpopulations were comparable (data not shown). The cellularity of thymocyte subsets were proportionally increased in the TEC-AR−/y mice compared with AR+/y mice (Supplemental Fig. 2, A and B). Furthermore, the enhanced positive selection was evidenced by showing elevation of DP CD69hi population in TEC-AR−/y mice compared with AR+/y controls (Supplemental Fig. 2, A and B). Thus, the proliferative potentials of splenic T-lymphocytes did not change substantially after plate-bound anti-CD3e/CD28 stimulation in both genotypes of mice (Supplemental Fig. 2D).
To explore the potential mechanisms resulting in thymus enlargement, we used BrdU incorporation assay to measure proliferative thymocytes and found BrdU+ DP thymocytes were comparable in these ARKO mice and their AR+/y littermate controls (Fig. 2C). However, we did observe less apoptotic thymocytes in the G-AR−/y and TEC-AR−/y mice (not in T-AR−/y and fsp-AR−/y mice) compared with their AR+/y controls (Fig. 2D).
Together, results from these cell specific ARKO mice indicated that loss of AR in TECs leads to thymus enlargement and increased thymocyte cellularity. The reduction of apoptotic cells (increasing cell survival) may contribute to the increased cellularity in G-AR−/y and TEC-AR−/y mice.
Deletion of AR in cTECs increases thymocyte survival through enhancing positive selection in the AND-TCR system
Because TECs have been known to play important roles for the positive/negative selection processes (6), we studied whether AR in TECs could modulate positive and/or negative selection processes. We first bred cytokeratin 5 (K5) cre mice with Rosa26R-LacZ reporter mice to determine cre expression. It has been documented that bipotent thymic epithelial progenitor cells can differentiate from K5+/K8+ progenitors to K5−/K8+ cTECs and K5+/K8− mTECs (25). By using β-gal staining, we were able to detect cre expression in both cTECs and mTECs (Supplemental Fig. 3, upper panel arrows), which was further confirmed by using anti-β-gal IHC staining (Supplemental Fig. 3, lower panel arrows). By crossing K5 cre mice with floxed AR mice, we can simultaneously ablate AR in cTECs and mTECs, which provide a proper in vivo mouse model to manifest thymic epithelial AR functions.
To study the AR role in TECs during positive selection process, we generated male AR+/y and TEC-AR−/y mice bearing AND-TCR tg (expressed TCR V α 11/V β 3 specific for a carboxy-terminal fragment of pigeon cytochrome c) expressed exclusively in CD4+ T-cells in the context of I-Ab. Evidences of establishing these compound TCR tg mice are shown (Supplemental Fig. 4A). We found the cellularity of total thymocytes, CD4+ thymocytes, and CD4+ splenic T-cells was increased in TEC-AR−/y mice (Fig. 3A) as well as in the population of positively-selected CD4+ cells (Fig. 3B).
Fig. 3.
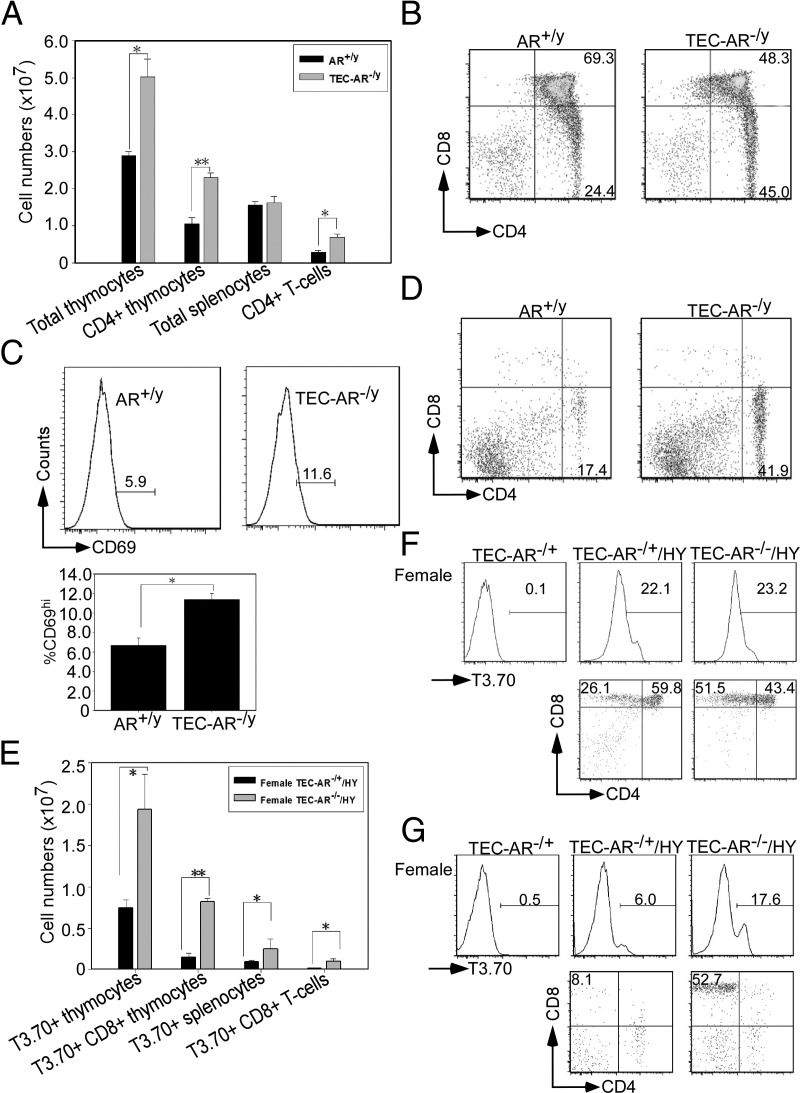
Lack of AR in cTECs promotes thymocytes survival through alteration of positive selection. The male mice used in Fig. 3, A–D, contained AND-TCR tg, and the female mice in Fig. 3, E–G, carried HY-TCR tg. All these mice are of the Rag2 null background. A, Increased positively selected CD4+ T-cells in TEC-AR−/y mice. 10–12 wk of age male AR+/y and TEC-AR−/y mice were killed and thymocytes/splenocytes collected for characterization. The total numbers of thymocytes, CD4+ thymocytes, splenocytes, and CD4+ T-cells were graphed. B, Thymocytes were stained with anti-CD4 and CD8 Abs for flow cytometric analysis. The numbers indicate the percentage of cells in each quadrant. C (upper panels), The representative histogram was gated from DP populations and the expression of CD69hi was analyzed. The quantification data are provided in the lower panel. D, Splenocytes were stained with anti-CD4 and CD8 Abs for flow cytometric analysis. E, Female TEC-AR−/+ and TEC-AR−/− mice 10–12 wk of age were killed, and thymocytes/splenocytes were harvested for characterization. The cell numbers of clonotypic T3.70 gated thymocytes, CD8+ thymocytes, splenocytes, and CD8+ T-cells were counted. F, Thymocytes were stained with anti-T3.70, CD4, and CD8 Abs for flow cytometric analysis. The mice without HY-TCR tg served as negative control. The lower plots were shown from T3.70 gated population, and the numbers indicate the percentage of cells in each quadrant. G, Splenocytes were stained with anti-T3.70, CD4, and CD8 Abs for flow cytometric analysis. The lower plots were shown from T3.70 gated population. The representative and quantitative data collected from Fig. 3 were from three independent experiments with 6 to 8 mice per group. (*, P < 0.05; **, P < 0.01 compared with male AR+/y control mice or female TEC-AR−/+ mice).
During the positive selection process, up-regulation of surface TCR precedes the step in which DP thymocytes will differentiate into SP. DP thymocytes also up-regulate the expression of early activation antigen CD69 during positive selection (26). As expected, the populations of CD69hi DP thymocytes were higher in TEC-AR−/y mice (AR+/y 5.9% vs. TEC-AR−/y 11.6%), suggesting that more active positive selection occurred in male TEC-AR−/y mice (Fig. 3C). Furthermore, the similar results were observed in spleen, in which elevation of CD4+ population was displayed in the TEC-AR−/y mice (Fig. 3D). Interestingly, we also examined the proliferation potential of positively-selected CD4+ T-cells from AR+/y/AND-TCR and TEC-AR−/y/AND-TCR mice using BrdU incorporation assay. The proliferative ability showed comparable between these two genotypes of mice (data not shown), implicating selected CD4+ T-cells are functional competent in TEC-AR−/y mice and the cellularity of selected CD4+ T-cells increased in TEC-AR−/y mice may not be due to the more active proliferation of CD4+ T-cells in TEC-AR−/y mice.
Collectively, these data clearly indicated that DP thymocytes from TEC-AR−/y mice received more potent TCR-mediated signals and therefore displayed increased positive selection.
We also extended the above findings to female mice by generating TEC-AR−/+ (knockout of only one allele of Ar gene in TECs) and TEC-AR−/ − mice (knockout of both Ar alleles) in HY-TCR tg mice as evidenced in Supplemental Fig. 4B. Thymocytes in HY-TCR tg mice (express TCR V α 17/V β 8.2 specific for a mouse male antigen SMCY 738–764) in the context of H-2-D-b will be positively selected in female mice, but negatively selected in male mice (27). The cellularity of transgenic Vα chain T3.70+ thymocytes (recognizing male antigen HY in the context of H2-D-b) and CD8+ thymocytes were increased in female TEC-AR−/− mice as compared with TEC-AR−/+ mice (Fig. 3E) as well as the T3.70+CD8+ population (Fig. 3F). The similar results were consistently observed in the T3.70+ gated CD8+ splenocytes (Figs. 3, E and G).
Taken together, we used male mice (AND-TCR system) and female mice (HY-TCR system) to demonstrate that AR in cTECs could suppress positive selection in these two TCR tg mouse models. The elevation of positively selected thymocytes may account for the increased thymocyte survival, leading to increased thymic cellularity.
Inactivation of AR in mTECs does not alter negative selection in the HY-TCR system
As described, a majority of thymocytes in HY-TCR male mice will be negatively selected in the context of H-2-D-b. We subsequently generated male AR+/y and TEC-AR−/y mice bearing HY-TCR tg for negative selection study as evidenced in Supplemental Fig. 4C. The cellularity of T3.70+ thymocytes and T3.70+ CD8+ thymocytes showed comparable amounts in both genotypes of mice (Supplemental Fig. 5A). Furthermore, the population of T3.70+ DP and CD8+ thymocytes also showed no evident difference in both genotypes of mice (Supplemental Fig. 5B). In the spleen, we consistently observed similar results (Supplemental Fig. 5, A and C).
Mice lacking thymic epithelial AR have increased epithelial cellularity and proliferative potential
Next, we wanted to explore whether AR was involved in the growth and differentiation of TECs. First, thymi from AR+/y and TEC-AR−/y mice were subjected to TSCs isolation and we found nonhematopoietic CD45− TSCs constituted about 1.5% of total thymic cells in AR+/y (Fig. 4, A and B) and TEC-AR−/y mice (data not shown). The compositions of TEC subsets were defined by analyzing surface expression of MHC II, mTECs marker UEA-1, and general TEC marker EpCAM. EpCAM and UEA-1 double staining was used to distinguish the subsets of cTECs (EpCAM+/UEA-1−) and mTECs (EpCAM+/UEA-1+), and we found both genotypes of mice had comparable TEC subsets containing 20–23% cTECs and 50–55% of mTECs (Fig. 4C). This conclusion was further strengthened by using MHC II and UEA-1 colabeling to distinguish cTECs (MHC II+/UEA-1−), mTECslo (MHC IIlo/UEA-1+), and mTECshi (MHC IIhi/UEA-1+) subsets. We found that the populations of these three subsets (cTECs 18–19%, mTECslo 43–46%, and mTECshi 21–24%) in both genotypes of mice also showed no noticeable difference (Fig. 4D). Furthermore, we found the cellularity of TEC subsets was proportionally increased in the TEC-AR−/y mice (Fig. 4E) concurrent with higher proliferative potential of TECs in TEC-AR−/y mice (AR+/y vs. TEC-AR−/y, 2.2 to 4.3% in TECs, 1.9 to 3.2% in cTECs, and 7.1 to 14.2% in mTECs) (Fig. 4, F and G).
Fig. 4.
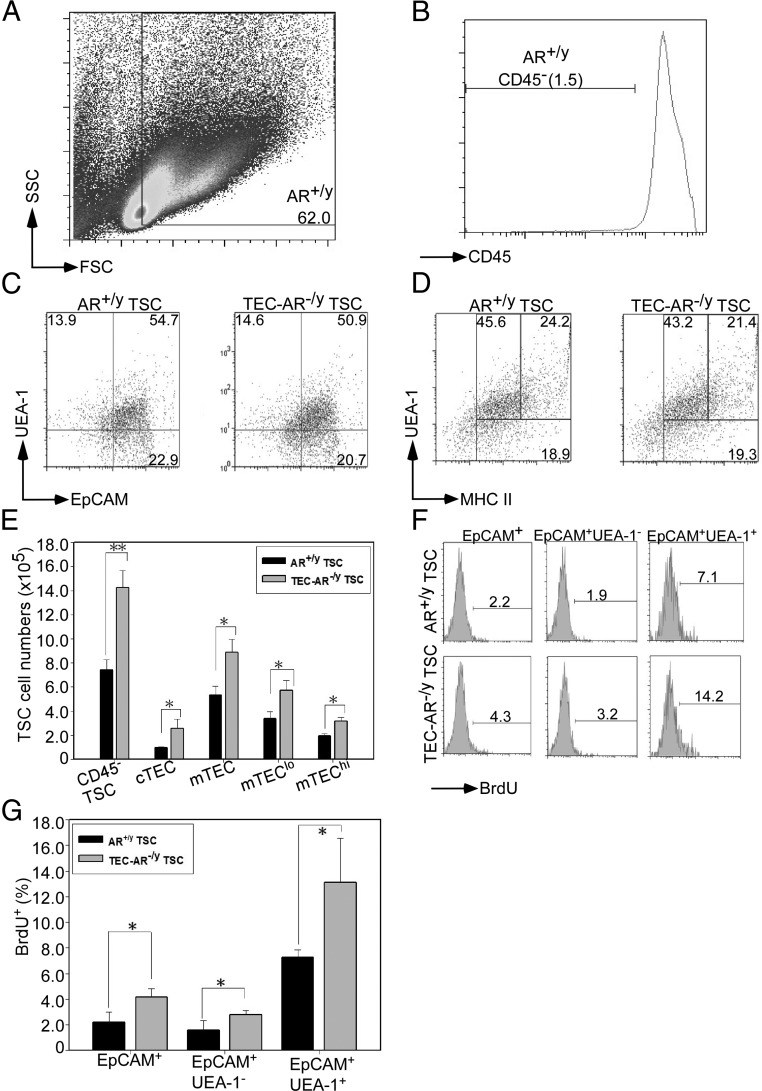
Mice lacking thymic epithelial AR have increased epithelial cellularity and enhanced proliferative potential. A, The representative dot plot showed gating of FSC/SSC in enzyme-digested thymus from 8- to 12-wk old AR+/y male mice. The number denotes the percentage of cells in gated regions. B, Histogram shows gating of CD45− TSCs from AR+/y male mice. The histogram was gated from Fig. 4A 62% population, and the number denotes the percentage of cells in gated regions. C, Flow cytometric analysis of discrete subsets of thymic epithelial cells was employed by staining anti-CD45, biotinylated UEA-1, and EpCAM Abs in AR+/y and TEC-AR−/y TSCs. A subset of EpCAM+UEA-1− cells (gated from CD45− cells) was defined as cTECs, and a subset of EpCAM+UEA-1+ cells (gated from CD45− cells) was defined as mTEC. D, Flow cytometric analysis of mTEC subsets by determining the expression of MHC II and UEA-1. A subset of MHC II+UEA-1− cells, MHC II hi in UEA-1+ cells, and MHC IIlo in UEA-1+ cells was determined. These subsets were gated from CD45− cells. E, Cellularity of different TEC subsets was determined. TSCs (CD45−), cTECs (CD45−MHC II+UEA-1−), mTECs (CD45− MHC II+UEA-1+), mTECslo (CD45−MHC IIloUEA-1+), and mTECshi (CD45−MHC IIhi UEA-1+) were analyzed. F, The percentage of BrdU+ TSC subsets, TECs (CD45−EpCAM+), cTECs (CD45−EpCAM+UEA-1−), and mTECs (CD45−EpCAM+ UEA-1−), and mTECs (CD45−EpCAM+UEA-1+) were determined. G, The quantitative data of BrdU+ TEC subsets were determined. The representative flow plot and quantitative data in Fig. 4 were from three independent experiments with 6 to 8 mice per group (*, P < 0.05; **, P < 0.01 compared with AR+/y littermate control mice).
Mechanistically, we used primary culture of TSCs to characterize the AR-regulated genes and confirmed by using flow cytometry to detect EpCAM expression (Supplemental Fig. 6, A and B). The potential AR-regulated gene screening used quantitative PCR, and the results showed that costimulatory molecules (CD40, CD80, and CD86) and thymopoiesis-suppressive cytokines (TGF-β1and IL-6) were decreased in TEC-AR−/y TECs. In contrast, the potent thymopoietic cytokine IL-7 and T precursor cells recruitment chemokine CCL21 were increased in TEC-AR−/y TECs (Supplemental Fig. 6C). Notably, a previous report has documented that CD28KO or B7 (CD80/CD86) double KO mice constituently showed increased selection of mature CD4 and CD8 SP T-cells, indicating that CD28-B7 engagement plays a negative regulatory role for positive selection (28). Interestingly, we also found that CD80 was positively regulated by AR in TE71.1 murine TECs (Supplemental Fig. 7, A–D), suggesting that AR in TECs may control the positive selection process via CD80 regulation. The underlying mechanism governing the CD80 expression by which AR may go through the direct promoter binding was demonstrated using the promoter luciferase assay (Supplemental Fig. 7, E and F). The results showed that the potential AREI (−1350 to −1336) but not the AREII (−203 to −189) could be the AR binding site to mediate AR effects on the CD80 expression (Supplemental Fig. 7F). To further confirm the AR binding site located at AREI of CD80 promoter regions, we conducted the AR chromatin immunoprecipitation assay (Supplemental Fig. 7G). Other altered growth factors/cytokines/chemokines (Supplemental Fig. 6C) in the thymic selection functions are still unresolved at this point.
To clarify whether the increased epithelial cell proliferation in TEC-AR−/y mice is due to the intrinsic effects of AR loss or needs the support from positively selected thymocytes, we used AR small interfering RNA (siRNA) to knock down AR in TE71.1 and examined the cell growth by MTT assay. We demonstrated that silencing of AR in TE71.1 did not alter the cell growth (Supplemental Fig. 8A) and confirmed with BrdU incorporation assay (Supplemental Fig. 8B). For the in vivo evidence, we dissected the thymi from AR+/y/Rag2 KO and TEC-AR−/y/Rag2 KO male mice and stained with proliferation marker proliferating cell nuclear antigen. The results consistently showed there is no evident difference of epithelial proliferating cell nuclear antigen staining in both genotypes of mice (Supplemental Fig. 8C). This conclusion was further supported by the previous report indicating that positively selected thymocytes were able to regulate thymic epithelial cells via RANK-RANKL and CD40 signaling (29).
Together, results from Fig. 4 and Supplemental Figs. 6–8 showed that TECs did not undergo extensive compositional changes of subpopulations in TEC-AR−/y mice. Instead, we found the cellularity of three subsets of TECs was proportionally increased in TEC-AR−/y mice. Conversely, thymic epithelial AR may utilize multiple mechanisms such as regulation of costimulatory molecules, such as CD80 and thymopoietic related cytokines/chemokines, to keep thymic cellularity under control. The increased cellularity of thymic epithelial cells after AR deletion appears to rely on the thymocytes' support.
Ablation of AR in TECs enhances BMT grafting efficacy
Previously described findings led us to believe that suppression of AR expression in TECs by genetic knockout in recipients may be beneficial to increase grafting efficacy, especially T-cell reconstitution. To mimic human autologous BMT, we performed BMT between two congenic mice, FVB/C57BL/6 (CD45.1+CD45.2+) as recipients and FVB/C57BL/6.SJL (CD45.1+CD45.2−) as donors, which differ only at one CD45.2 gene allele. To prove this supposition, we first conducted congenic BMT on lethally γ-irradiated recipient AR+/y and TEC-AR−/y mice. After BMT, we harvested the BM, thymocytes, and splenocytes from recipients and found donor-derived thymocyte (3 and 6 wk after BMT) and splenocyte numbers (6 wk after BMT) were substantially increased in TEC-AR−/y recipients (Fig. 5A). The better transplantation efficacy was also demonstrated by detecting increased amounts of donor-derived DP, CD4+ thymocytes (3 wk after BMT) and DP, CD4+, and CD8+ thymocytes (6 wk after BMT) (Fig. 5B). Similar results were also seen in donor-derived splenic CD4+ and CD8+ T-cells in TEC-AR−/y recipients 6 wk after BMT (Fig. 5C).
Fig. 5.
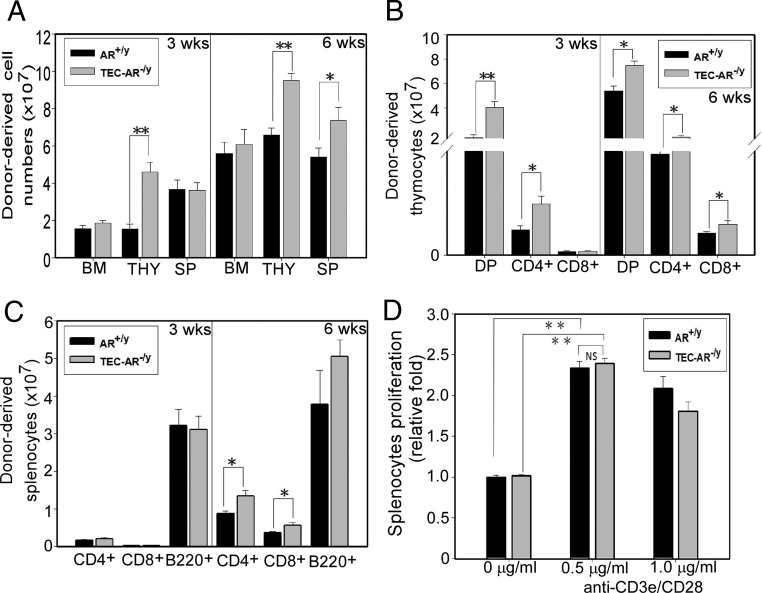
Ablation of AR in TECs enhances BMT grafting efficacy. A, Male AR+/y or TEC-AR−/y mice, 8–10 wk of age, in C57BL/6/FVB (CD45.1+CD45.2+) background were irradiated with a BM lethal dose of γ-irradiation; 5 × 106 B6-SJL/FVB (CD45.1+CD45.2−) donor-derived TCD BM cells were transferred into recipient mice. After 3 and 6 wk of BMT, the cellularity of BM, thymus, and spleen was determined. B, Donor-derived thymic cellularity was determined by using congenic (anti-CD45.1 and CD45.2) and T-cell markers (CD4 and CD8). The donor-derived DP thymocytes (CD45.1+CD45.2−CD4+CD8+), CD4+ SP (CD45.1+CD45.2−CD4+CD8−), and CD8+ SP (CD45.1+CD45.2−CD4−CD8+) were characterized after 3 and 6 wk of BMT. C, Donor-derived splenic cellularity was determined from donor-derived CD4+ T-cells, CD8+ T-cells, and B-cells (B220+) after 3 and 6 wk of BMT. D, Splenocytes were harvested from BM chimera mice either from AR+/y or TEC-AR−/y at 6 wk after BMT and stimulated with 0.5 or 1 μg/ml of plate-bound anti-CD3e and CD28 Abs for 2 d. The cell proliferation was analyzed using MTT assay. All data collected from Fig. 5 represent mean ± sem from four independent experiments with 8–10 mice per group (*, P < 0.05; **, P < 0.01; NS, no significance compared with AR+/y recipients, P > 0.05).
To further assess the function of repopulated peripheral T-cells 6 wk after BMT (donor-derived cell populations are more than 80% of total splenic cells; data not shown), the proliferative potential of splenic T-cells was determined using anti-CD3e/CD28 Abs cross-linking stimulation, and followed with MTT cell growth assay. We found the T-cell proliferation isolated from these two BM chimera mice showed no significant difference (Fig. 5D).
Together, results from Fig. 5, A–D, clearly demonstrated that targeting thymic epithelial AR, using TEC-AR−/y mice as recipients, created a better transplantation efficacy as evidenced by earlier T-cells reconstitution after congenic grafting.
Targeting AR via ASC-J9® leads to the acceleration of T-cell reconstitution after BMT
To validate the potentials of targeting thymic epithelial AR to increase grafting efficacy in the clinical application, a strategy that could mimic genetic knockout of thymic epithelial AR, with minimal side-effects, is required. So far, two potential approaches that could target AR are AR-siRNA (30, 31) or the chemical compound ASC-J9® (12, 17, 31). The technology using siRNA has been known to show difficulty of in vivo delivery and toxicity (30), whereas ASC-J9® has been shown to exert in vivo receptor degradation activity (12, 17, 31); therefore, we decided to use ASC-J9® here to test its potentials in improving BMT efficacy.
First, we treated male C57BL/6 (B6) mice with ASC-J9® via daily ip injections at 50 mg/kg and found 2 wk of ASC-J9® treatment substantially increased the cellularity of thymocytes and splenocytes (Fig. 6A). The relative cell populations in thymus and spleen were similar in mice either receiving vehicle-DMSO or ASC-J9® (Supplemental Fig. 9, A–D). To confirm AR degradation in the ASC-J9®-treated group, we harvested the thymus after treatment and performed AR IHC and pan-CK/AR double immunofluorescent (IF) staining. As expected, AR expression in TECs was significantly reduced in mice receiving ASC-J9® (Fig. 6B, arrows).
Fig. 6.
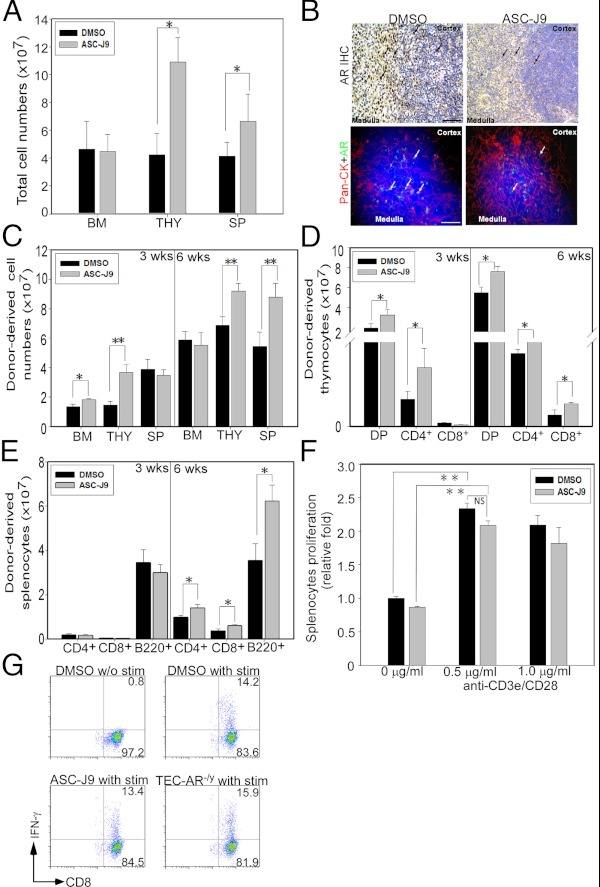
Targeting AR via ASC-J9® in recipients leads to enhanced T-cell reconstitution after BMT. A, Total BM cells, thymocytes, and splenocytes from 8-wk-old male C57BL/6 mice were harvested 2 wk after vehicle-DMSO control or ASC-J9® daily injection. Results are presented as mean ± sem of six to eight mice per group (*, P < 0.05). B, Thymi from male C57BL/6 mice receiving 2 wk of vehicle-DMSO control or ASC-J9® injection were harvested for histological examination. Paraffin-embedded thymus was stained with anti-AR Ab, and arrows indicate AR expression cells (upper panel). Double immunofluorescent staining was used to determine thymic epithelial AR expression (lower panel). Arrows indicate epithelial AR expression. Blue indicates DAPI; red indicates pan-CK; and green indicates AR. Scale bar, 50 μm. C, The cellularity of donor-derived BM, thymocytes, and splenocytes was determined at 3 and 6 wk after BMT. Male WT recipients, 5–6 wk of age (C57BL/6/FVB, CD45.1+CD45.2+), were treated with vehicle-DMSO or 50 mg/kg ASC-J9® for 2 wk after a lethal dose of γ-irradiation. B6-SJL/FVB (CD45.1+CD45.2−) TCD BM cells (5 × 106) were then transferred into recipients and harvested 3 and 6 wk after BMT. D, Donor-derived thymic cellularity was determined from BM chimera mice at 3 and 6 wk after BMT using congenic and T-cell marker immunostaining. The donor-derived DP, CD4+ SP, and CD8+ SP thymocytes were characterized. E, Three and 6 wk after BMT, splenic cellularity was determined from donor-derived CD4+ T-cells, CD8+ T-cells, and B220+ B-cells. F, Splenocytes were harvested from BM chimera mice (recipients received either vehicle-DMSO or ASC-J9® before BMT) at 6 wk after BMT and stimulated with plate-bound anti-CD3e and CD28 Abs (0.5 and 1 μg/ml) for 2 d. The cell proliferation was analyzed by MTT assay. G, The intracellular IFN-γ expression from donor-derived splenic CD8+ T-cells at 6 wk after BMT was determined by using congenic markers CD45.1, CD45.2, CD8, and IFN-γ immunostaining. Six weeks after BMT, donor-derived splenic T-cells were stimulated with phorbol-12-myristate 13-acetate (PMA) and ionomycin in the presence of brefeldin (10 ug/ml) for 4 h for flow cytometric analysis. Representative plots were gated from CD45.1+CD45.2−CD8+, and the numbers at the upper right corner represent the percentage of donor-derived splenic CD8+ T cells that expressed IFN-γ. The representative and quantitative data collected from Fig. 6 were from four independent experiments with 8–10 mice per group. Each bar represents mean ± sem. *, P < 0.05; **, P < 0.01; NS, no significance compared with vehicle-DMSO group, P > 0.05. IHC, Immunohistochemistry.
To test whether degradation of AR protein by ASC-J9® could enhance BMT grafting efficacy, we treated recipients (FVB/C57BL/6, CD45.1+CD45.2+) with ASC-J9® for 2 wk before congenic BMT (donor: FVB/C57BL/6.SJL, CD45.1+CD45.2−). After treatment, mice were subjected to γ-irradiation and BMT was performed within 24 h (mice did not continue to receive ASC-J9® treatment during reconstitution). We then harvested the BM, thymocytes, and splenocytes for characterization and found donor-derived BM cells and thymocytes were increased in ASC-J9®-treated group at 3 wk after BMT (Fig. 6C). The increased amounts of donor-derived thymocytes and splencoytes were also observed in ASC-J9®-treated recipients 6 wk after grafting (Fig. 6C). Consistently, DP and SP thymocyte numbers in ASC-J9®-treated recipients also showed a significant increase at 3 wk and 6 wk after BMT (Fig. 6D). The increased cellularity of donor-derived splenic T- and B-cells was also observed 6 wk after BMT (Fig. 6E), indicating that pretreatment with ASC-J9® in recipients can boost the BMT grafting efficacy.
To assess the functional activity of peripheral T-cells in recipients receiving ASC-J9® treatment before congenic BMT, we tested the proliferative ability of splenic T-cells 6 wk after BMT and ASC-J9® treatment did not affect the cell proliferation (Fig. 6F). This result was further supported by measuring IFN-γ production (Fig. 6G).
Collectively, pretreatment with ASC-J9® in recipients before BMT leads to enhanced T-cell reconstitution with little impact on T-cell functions, most likely through predominant degradation of AR in TECs similar to the observation in which recipients are from G-AR−/y or TEC-AR−/y mice.
Discussion
Here, we demonstrated that AR in TECs is critical to determine thymic cellularity through multiple mechanisms such as modulation of positive selection, thymopoiesis-related cytokines/chemokines, and costimulatory molecules. Early studies (32–34) indicated that growth factors/cytokines related to thymopoiesis such as TGF-β, IL-6, IL-7, and chemokines CCL21/CCL25 might contribute to this process. For example, TGF-β was suggested to use suppressive effects either directly and/or indirectly to up-regulate cytokines, such as IL-6 and leukemia inhibitory factor, to impede thymopoiesis (35). Consistently, we found that the expressions of TGF-β1 and IL-6 were decreased in the TEC-AR−/y TECs (Supplemental Fig. 6C). Furthermore, we found IL-7 and CCL21 expressions were increased in TEC-AR−/y mice (Supplemental Fig. 6C), which are in agreement with early reports showing their effects to promote thymopoiesis (32). Collectively, these results suggested that increased thymic cellularity and outputs in TEC-AR−/y mice may be due to the consequence of increased positively selected T-cells together with the above possible mechanisms.
In this present study, we used male HY-TCR tg mouse system to study negative selection and indicated that deletion of thymic epithelial AR did not perturb negative selection in this unique model. Although male HY-TCR tg mice have been known to exhibit the early deletion of thymocytes in DN or early DP stage, alternative more physiological mouse models such as superantigen-mediated deletion of Vβ subsets or other available TCR tg systems, such as P14, AND-TCR in H2s background, or CD4+ HY-TCR, are also applicable to cross with ARKO mice in the future (36, 37). However, our data (Supplemental Fig. 10, A–D) showed that aged TEC-AR−/y male mice still displayed thymic involution similar to AR+/y mice without obvious immune cells infiltration, making it unlikely that negative selection defects such as autoimmunity would occur in this TEC-AR−/y mouse. Although previous studies have demonstrated that castrated mice or rats are more prone to autoimmune disease development such as experimental autoimmune encephalomyelitis (38), the systemic androgen depletion is not equal to loss of thymic epithelial AR. Importantly, AR in peripheral T-cells can inhibit the proliferation and modulate Th1-Th2 balance (39), yet AR in thymocytes is dispensable for development, indicating castration effects may not be equal to thymic epithelial AR deletion.
Androgen deprivation therapy via reduction or prevention of androgens binding to AR, instead of targeting AR directly, has been used as the standard treatment for prostate cancer since its discovery by Huggins and Hodges (40). However, systemic suppression of circulating androgens not only failed to cure prostate cancer, but also yield many unpleasant side-effects such as impotence, loss of libido, osteoporosis, and fatigue (41). Based on the above concerns, although androgen deprivation therapy (surgical or chemical castration, LHRH agonist) has been shown to increase BMT efficacy (42), its similar side effects and largely unknown mechanisms may impede its further application. The major difference between this report and previous findings is that we clearly proved here that it is AR, not androgens, that play a more critical role in determining thymic cellularity as evidenced using castration and DHT restoration to conclude that AR-dependent signaling is more critical (Fig. 1G). This conclusion was further strengthened by the characterization of male TEC-AR−/y mice showing thymus enlargement with normal serum testosterone and female mice (in HY-TCR system) with almost nondetectable androgens, but still expressed AR proteins (43). We suggest that AR, not androgens, in female mice also plays critical roles in maintaining thymic cellularity via modulation of positive selection process to increase thymocyte survival (Fig. 3E).
In summary, all the above evidence support the supposition that targeting AR by ASC-J9®, but not androgens, represents a more promising therapeutic approach to enhance the grafting efficacy for human autologous BMT with much less side-effects and could be very applicable for other androgen/AR-related diseases.
Supplementary Material
Acknowledgments
We thank Karen Wolf (George H. Whipple Laboratory, University of Rochester Medical Center, Rochester, NY) and Charles Shih (AndroScience Corp., San Diego, CA) for manuscript preparation and critical review. ASC-J9® was a gift from AndroScience Corp.
This work was supported by National Institutes of Health Grant DK73414, George H. Whipple Professorship Endowment, Taiwan Department of Health Clinical Trial and Research Center of Excellence grant DOH99-TD-B-111-004 (China Medical University, Taichung, Taiwan).
Disclosure Summary: ASC-J9® was patented by the University of Rochester, the University of North Carolina, and AndroScience Corp. and then licensed to AndroScience Corp. Both the University of Rochester and C. Chang own royalties and equity in AndroScience Corp. The remaining authors declare no competing financial interests.
Footnotes
- Ab
- Antibody
- AR
- androgen receptor
- ARKO
- AR knockout
- BM
- bone marrow
- BMT
- bone marrow transplantation
- BrdU
- bromodeoxyuridine
- cTEC
- cortical TEC
- DMSO
- dimethylsulfoxide
- DN
- double negative
- DP
- double positive
- FACS
- fluorescence-activated cell sorting
- FBS
- fetal bovine serum
- MHC
- major histocompatibility complex
- mTEC
- medullary TEC
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- siRNA
- small interfering RNA
- TCD
- T-cell depleted
- TCR
- T-cell receptor
- TEC
- thymic epithelial cell
- tg
- transgenic
- TSC
- thymic stromal cell
- WT
- wild type.
References
- 1. Hince M, Sakkal S, Vlahos K, Dudakov J, Boyd R, Chidgey A. 2008. The role of sex steroids and gonadectomy in the control of thymic involution. Cell Immunol 252:122–138 [DOI] [PubMed] [Google Scholar]
- 2. Taub DD, Longo DL. 2005. Insights into thymic aging and regeneration. Immunol Rev 205:72–93 [DOI] [PubMed] [Google Scholar]
- 3. Globerson A, Effros RB. 2000. Ageing of lymphocytes and lymphocytes in the aged. Immunol Today 21:515–521 [DOI] [PubMed] [Google Scholar]
- 4. Kilpatrick RD, Rickabaugh T, Hultin LE, Hultin P, Hausner MA, Detels R, Phair J, Jamieson BD. 2008. Homeostasis of the naive CD4+ T cell compartment during aging. J Immunol 180:1499–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson G, Harman BC, Hare KJ, Jenkinson EJ. 2000. Microenvironmental regulation of T cell development in the thymus. Semin Immunol 12:457–464 [DOI] [PubMed] [Google Scholar]
- 6. Starr TK, Jameson SC, Hogquist KA. 2003. Positive and negative selection of T cells. Annu Rev Immunol 21:139–176 [DOI] [PubMed] [Google Scholar]
- 7. Kyewski B, Klein L. 2006. A central role for central tolerance. Annu Rev Immunol 24:571–606 [DOI] [PubMed] [Google Scholar]
- 8. Olsen NJ, Olson G, Viselli SM, Gu X, Kovacs WJ. 2001. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology 142:1278–1283 [DOI] [PubMed] [Google Scholar]
- 9. McCruden AB, Stimson WH. 1984. Androgen receptor in the human thymus. Immunol Lett 8:49–53 [DOI] [PubMed] [Google Scholar]
- 10. Viselli SM, Olsen NJ, Shults K, Steizer G, Kovacs WJ. 1995. Immunochemical and flow cytometric analysis of androgen receptor expression in thymocytes. Mol Cell Endocrinol 109:19–26 [DOI] [PubMed] [Google Scholar]
- 11. Viselli SM, Stanziale S, Shults K, Kovacs WJ, Olsen NJ. 1995. Castration alters peripheral immune function in normal male mice. Immunology 84:337–342 [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, Merry DE, Sobue G, Chen LM, Chang SS, Chang C. 2007. ASC-J9® ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med 13:348–353 [DOI] [PubMed] [Google Scholar]
- 13. Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. 2002. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA 99:13498–13503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berton TR, Matsumoto T, Page A, Conti CJ, Deng CX, Jorcano JL, Johnson DG. 2003. Tumor formation in mice with conditional inactivation of Brca1 in epithelial tissues. Oncogene 22:5415–5426 [DOI] [PubMed] [Google Scholar]
- 15. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. 2004. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303:848–851 [DOI] [PubMed] [Google Scholar]
- 16. Gray DH, Fletcher AL, Hammett M, Seach N, Ueno T, Young LF, Barbuto J, Boyd RL, Chidgey AP. 2008. Unbiased analysis, enrichment and purification of thymic stromal cells. J Immunol Methods 329:56–66 [DOI] [PubMed] [Google Scholar]
- 17. Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ, Chang C. 2009. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-α expression. J Clin Invest 119:3739–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olsen NJ, Watson MB, Henderson GS, Kovacs WJ. 1991. Androgen deprivation induces phenotypic and functional changes in the thymus of adult male mice. Endocrinology 129:2471–2476 [DOI] [PubMed] [Google Scholar]
- 19. Rahman F, Christian HC. 2007. Non-classical actions of testosterone: an update. Trends Endocrinol Metab 18:371–378 [DOI] [PubMed] [Google Scholar]
- 20. Frasca D, Guidi F, Arbitrio M, Pioli C, Poccia F, Cicconi R, Doria G. 2000. Hematopoietic reconstitution after lethal irradiation and bone marrow transplantation: effects of different hematopoietic cytokines on the recovery of thymus, spleen and blood cells. Bone Marrow Transplant 25:427–433 [DOI] [PubMed] [Google Scholar]
- 21. Zinkernagel RM, Althage A. 1999. On the role of thymic epithelium vs. bone marrow-derived cells in repertoire selection of T cells. Proc Natl Acad Sci USA 96:8092–8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hennet T, Hagen FK, Tabak LA, Marth JD. 1995. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proc Natl Acad Sci USA 92:12070–12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee SO, Tian J, Huang CK, Ma Z, Lai KP, Hsiao H, Jiang M, Yeh S, Chang C. 2012. Suppressor role of androgen receptor in proliferation of prostate basal epithelial and progenitor cells. J Endocrinol 213:173–182 [DOI] [PubMed] [Google Scholar]
- 24. Yu S, Yeh CR, Niu Y, Chang HC, Tsai YC, Moses HL, Shyr CR, Chang C, Yeh S. 2012. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 72:437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossi SW, Chidgey AP, Parnell SM, Jenkinson WE, Scott HS, Boyd RL, Jenkinson EJ, Anderson G. 2007. Redefining epithelial progenitor potential in the developing thymus. Eur J Immunol 37:2411–2418 [DOI] [PubMed] [Google Scholar]
- 26. Swat W, Dessing M, von Boehmer H, Kisielow P. 1993. CD69 expression during selection and maturation of CD4+8+ thymocytes. Eur J Immunol 23:739–746 [DOI] [PubMed] [Google Scholar]
- 27. Van Ewijk W, Kisielow P, Von Boehmer H. 1990. Immunohistology of T cell differentiation in the thymus of H-Y-specific T cell receptor α/β transgenic mice. Eur J Immunol 20:129–137 [DOI] [PubMed] [Google Scholar]
- 28. Vacchio MS, Williams JA, Hodes RJ. 2005. A novel role for CD28 in thymic selection: elimination of CD28/B7 interactions increases positive selection. Eur J Immunol 35:418–427 [DOI] [PubMed] [Google Scholar]
- 29. Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, Matsumoto M, Matsuo K, Penninger JM, Takayanagi H, Yokota Y, Yamada H, Yoshikai Y, Inoue J, Akiyama T, Takahama Y. 2008. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity 29:438–450 [DOI] [PubMed] [Google Scholar]
- 30. Sun A, Tang J, Terranova PF, Zhang X, Thrasher JB, Li B. 2010. Adeno-associated virus-delivered short hairpin-structured RNA for androgen receptor gene silencing induces tumor eradication of prostate cancer xenografts in nude mice: a preclinical study. Int J Cancer 126:764–774 [DOI] [PubMed] [Google Scholar]
- 31. Miyamoto H, Yang Z, Chen YT, Ishiguro H, Uemura H, Kubota Y, Nagashima Y, Chang YJ, Hu YC, Tsai MY, Yeh S, Messing EM, Chang C. 2007. Promotion of bladder cancer development and progression by androgen receptor signals. J Natl Cancer Inst 99:558–568 [DOI] [PubMed] [Google Scholar]
- 32. Liu C, Ueno T, Kuse S, Saito F, Nitta T, Piali L, Nakano H, Kakiuchi T, Lipp M, Hollander GA, Takahama Y. 2005. The role of CCL21 in recruitment of T-precursor cells to fetal thymi. Blood 105:31–39 [DOI] [PubMed] [Google Scholar]
- 33. Alves NL, Richard-Le Goff O, Huntington ND, Sousa AP, Ribeiro VS, Bordack A, Vives FL, Peduto L, Chidgey A, Cumano A, Boyd R, Eberl G, Di Santo JP. 2009. Characterization of the thymic IL-7 niche in vivo. Proc Natl Acad Sci USA 106:1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldberg GL, Sutherland JS, Hammet MV, Milton MK, Heng TS, Chidgey AP, Boyd RL. 2005. Sex steroid ablation enhances lymphoid recovery following autologous hematopoietic stem cell transplantation. Transplantation 80:1604–1613 [DOI] [PubMed] [Google Scholar]
- 35. Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, Patel DD, Haynes BF. 2000. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol 164:2180–2187 [DOI] [PubMed] [Google Scholar]
- 36. Baldwin TA, Sandau MM, Jameson SC, Hogquist KA. 2005. The timing of TCR α expression critically influences T cell development and selection. J Exp Med 202:111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrero I, Anjuère F, MacDonald HR, Ardavín C. 1997. In vitro negative selection of viral superantigen-reactive thymocytes by thymic dendritic cells. Blood 90:1943–1951 [PubMed] [Google Scholar]
- 38. Dalal M, Kim S, Voskuhl RR. 1997. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J Immunol 159:3–6 [PubMed] [Google Scholar]
- 39. Roden AC, Moser MT, Tri SD, Mercader M, Kuntz SM, Dong H, Hurwitz AA, McKean DJ, Celis E, Leibovich BC, Allison JP, Kwon ED. 2004. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol 173:6098–6108 [DOI] [PubMed] [Google Scholar]
- 40. Huggins C, Hodges CV. 1972. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin 22:232–240 [DOI] [PubMed] [Google Scholar]
- 41. Berges R, Bello U. 2006. Effect of a new leuprorelin formulation on testosterone levels in patients with advanced prostate cancer. Curr Med Res Opin 22:649–655 [DOI] [PubMed] [Google Scholar]
- 42. Goldberg GL, King CG, Nejat RA, Suh DY, Smith OM, Bretz JC, Samstein RM, Dudakov JA, Chidgey AP, Chen-Kiang S, Boyd RL, van den Brink MR. 2009. Luteinizing hormone-releasing hormone enhances T cell recovery following allogeneic bone marrow transplantation. J Immunol 182:5846–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sajjad Y, Quenby S, Nickson P, Lewis-Jones DI, Vince G. 2007. Androgen receptors are expressed in a variety of human fetal extragenital tissues: an immunohistochemical study. Asian J Androl 9:751–759 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.