

Abstract
High-density lipoprotein receptors scavenger receptor class B type I [HDLR-SR-B1 (SR-B1)] is a key player in reverse cholesterol transport and maintaining blood cholesterol. We demonstrated that human SR-B1 is transcriptionally activated by 17β-estradiol (E2) in HEPG2 and JAR cells. SR-B1 promoter contains multiple estrogen response elements (ERE half-sites) along with some Sp1 binding sites. Knockdown of estrogen receptor (ER)α and ERβ down-regulated E2-induced SR-B1 expression. ERs were bound to SR-B1 promoter EREs in an E2-dependent manner. Along with ERs, mixed-lineage leukemia (MLL) histone methylases, especially MLL1 and MLL2, play key roles in E2-mediated SR-B1 activation. MLL1 and MLL2 bind to SR-B1 promoter in an E2-dependent manner and control the assembly of transcription pre-initiation complex and RNA polymerase II (RNAPII) recruitment. ERs and MLLs play critical roles in determining the cholesterol uptake by steroidogenic tissues/cells, and their knockdown suppressed the E2-induced cholesterol uptake efficiencies of the cells. Intriguingly, MLL2 knockdown in mice resulted in a 33% increase in plasma cholesterol level and also reduced SR-B1 expression in mice liver, demonstrating its crucial functions in controlling plasma cholesterol in vivo.
Cholesterol trafficking between lipoproteins and cells is a fundamental biological process that ultimately determines plasma cholesterol level (1, 2). Blood cholesterol is a major factor in the development of atherosclerotic cardiovascular disease. There are five major lipoprotein classes: high-density lipoproteins (HDLs), low-density lipoproteins (LDLs), and others. LDL delivers cholesterol to peripheral tissues that requires cholesterol for membrane formation or steroid hormone biosynthesis (3, 4). In contrast, HDLs, which are rich in cholesterol but poor in triglycerides, carry cholesterol from peripheral tissues to liver, where cholesterol is excreted in bile as free cholesterol or converted into bile salts (5–7). HDL and LDL are major players in trafficking of cholesterol between liver and peripheral tissues (5, 8–10). HDL receptors scavenger receptor class B type I [HDLR-SR-B1 (SR-B1)] is a cell surface receptor for HDL and is expressed at high levels in the liver and other steroidogenic tissues where it mediates the selective uptake of cholesteryl ester (CE) from cholesterol-rich HDL (5–7). Alteration in SR-B1 expression profoundly influences diverse physiological functions including female fertility, atherosclerosis, abnormal red blood cell development, and development of coronary heart diseases (11, 12). Although SR-B1 is critical for cholesterol homeostasis and cardiovascular diseases, little is known about its transcriptional regulation. Herein, we examined the roles of steroid hormone such as estrogen in transcriptional regulation of SR-B1 and its impact on cholesterol homeostasis.
Estrogen signaling may follow distinct mechanisms (13, 14). In a classical mechanism, upon binding to estrogen (ligand), estrogen receptors (ERs) get activated, migrate to nucleus, bind to the estrogen response elements (EREs) present in the promoter of estrogen-responsive genes leading to target gene activation. ERα and ERβ are two major ERs that mediate transcriptional activation of estrogen-responsive genes. Both ERα and ERβ have high affinity for 17β-estradiol (E2) and bind to EREs. Along with ERs, estrogen-mediated gene activation requires various ER coregulators that include the steroid receptor coactivator (SRC) family of coactivators, cAMP response element-binding protein binding protein (CBP)/p300 histone acetyltransferases (HATs), histone methyltransferases (HMTs), etc. (15–22). Coactivators facilitate chromatin modification and remodeling of the target gene promoter leading to transcriptional activation.
Mixed-lineage leukemias (MLLs) are human histone H3 lysine-4 (H3K4)-specific HMTs that play key roles in gene activation (23). In humans, there are several MLLs such as MLL1, MLL2, MLL3, MLL4, etc. MLLs are master regulators of HOX genes. MLLs are often rearranged and misregulated in cancer (24). MLLs exist as multiprotein complexes inside the cell, methylate histones, and play crucial roles in gene activation, cell cycle progression, and stress response (25–30). Recent studies demonstrate that MLLs participate in nuclear receptor (NR)-mediated gene activation. MLLs interact with NRs, get recruited to the promoters of target genes, modify chromatins, and modulate gene expression (31–36). Various ER coactivators and HATs are linked with cardiovascular diseases (37–39). However, the role of histone-methylating enzymes in cardiovascular function and disease remains mostly unexplored.
Herein, we investigated potential roles of MLL HMTs in transcriptional regulation of SR-B1 expression. We demonstrated that SR-B1 is an estrogen (E2)-responsive gene. MLLs coordinate with ERs and regulate E2-dependent SR-B1 expression. Importantly, MLLs also play critical roles in regulation of cholesterol uptake by the liver and controlling plasma cholesterol level in vivo.
Materials and Methods
Cell culture and estrogen treatment
Human hepatocellular carcinoma (HEPG2; American Type Culture Collection, Manassas, VA) and placental choriocarcinoma (JAR; American Type Culture Collection) cells were grown in DMEM (Sigma Chemical Co., St. Louis, MO) supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, and penicillin/streptomycin (100 U and 0.1 mg/ml, respectively) in the presence of 5% CO2 at 37 C in a humidified incubator (28). For E2 treatment, HEPG2/JAR cells were grown in phenol-red-free DMEM-F12 containing charcoal-stripped FBS for three generations and then treated with E2. Cells were harvested for RNA and protein extraction or fixed in 4% formaldehyde as needed.
For knockdown experiments, cells were grown up to 60% confluency and transfected with MLLs, ERs, and scrambled antisense oligonucleotides separately in FBS-free media using iFECT transfection reagent (MoleculA) as instructed by the manufacturer and described by us previously (28). Respective knockdown was confirmed at both mRNA and protein levels using RT-PCR and Western blotting.
Extraction of RNA, RT-PCR, and real-time-PCR analysis
Cells were harvested, lysed in diethylpyrocarbonate-treated buffer A [20 mm Tris-HCl (pH 8), 1 mm EDTA, 10 mm KCl, 1.5 mm MgCl2] for 10 min in ice, and centrifuged (3500 × g) for 5 min. The aqueous phase containing the cytoplasmic RNA was subjected to phenol-chloroform extraction. The RNA was precipitated by LiCl by incubating at −80 C for 1 h, quantified, and subjected to RT-PCR. Reverse transcription reactions were performed in a total volume of 25 μl containing 1 μg total RNA or 500 ng mRNA, 2.4 μm oligo-deoxythymidine, 100 U Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI), 1× first-strand buffer (Promega), 100 μm dNTPs mix (dATP, dGTP, dCTP, and dTTP), 1 mm dithiothreitol, and 20 U RNaseOut (Invitrogen, Carlsbad, CA). This cDNA (1 μl) was used for semiquantitative PCR (RT-PCR) with primer pairs listed in Table 1.
Table 1.
Primers used for RT-PCR, ChIP, and antisense experiments
| Primers | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| PCR primers | ||
| β-Actin | AGAGCTACGAGCTGCCTGAC | GTACTTGCGCTCAGGAGGAG |
| SR-B1 | CTGTGGGTGAGATCATGTGG | GCCAGAAGTCAACCTTGCTC |
| SR-B1-ERE1 | GCTGGCGAAGTCCTACTCAC | CTTCCTTTTCCCTCCTCCTC |
| SR-B1-ERE2 | CTGCCAGGCCACGTTCTA | GCCACCATCACCCTATGACT |
| SR-B1-ERE3 | CCACTACGCCTGGCTAATTT | GAGGTCGGGAGTTGGAGAC |
| MLL1 | GAGGACCCCGGATTAAACAT | GGAGCAAGAGGTTCAGCATC |
| MLL2 | GTGCAGCAGAAGATGGTGAA | GCACAATGCTGTCAGGAGAA |
| MLL3 | AAGCAAACGGACTCAGAGGA | ACAAGCCATAGGAGGTGGTG |
| MLL4 | CCCTCCTACCTCAGTCGTCA | CAGCGGCTACAATCTCTTCC |
| ERα | AGCACCCTGAAGTCTCTGGA | GATGTGGGAGAGGATGAGGA |
| ERβ | AAGAAGATTCCCGGCTTTGT | TCTACGCATTTCCCCTCATC |
| Sp1RR | ACGAGGCCTCCTCAGCTC | GCCTTGGGCTTCAGGATT |
| Cloning primersa | ||
| SR-B1-ERE1 | CGGATACCTGGGAGAACAGA | CCACCTATGTCTCCCCTCCT |
| SR-B1-ERE2 | CTCCCAAAGTGCTGGGATTA | GCCACCATCACCCTATGACT |
| SR-B1-ERE3 | AGCTTCCCTGTGCATCAGTT | GAGACCAGCCTGACCAACAT |
| SR-B1-non ERE | ATGGGGGTGTGGGTTATTTT | TCACGAAAGGACTCTGAGCA |
| Antisense oligonucleotidesb | ||
| MLL1 antisense | TGCCAGTCGTTCCTCTCCAC | |
| MLL2 antisense | ACTCTGCCACTTCCCGCTCA | |
| MLL3 antisense | CCATCTGTTCCTTCCACTCCC | |
| MLL4 antisense | CCTTCTCTTCTCCCTCCTTGT | |
| ERα antisense | CATGGTCATGGTCAG | |
| ERβ antisense | GAATGTCATAGCTGA | |
| Scramble antisense | CGTTTGTCCCTCCAGCATCT |
Flanked by specific restriction sites.
Phosphorothioate antisense oligonucleotide.
For real-time PCR, cDNA was amplified using SsoFast EvaGreen supermix (Bio-Rad, Hercules, CA) and primers as described in Table 1, using CFX96 real-time PCR detection system, and results were analyzed using the CFX Manager.
Protein extraction and Western blotting
Cells were incubated in whole-cell protein extraction buffer [50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 0.05% Nonidet P-40, 0.2 mm phenylmethylsulfonyl fluoride, and 1× protease inhibitors cocktail] for 20 min on ice, centrifuged (13,000 rpm for 10 min) and the supernatant containing the protein extract was separated and subjected to Western blotting using anti-MLL1 (Lifespan Bioscience, Seattle, WA), anti-MLL2 (Lifespan Bioscience), anti-MLL3 (Abgent, San Diego, CA), anti-MLL4 (Sigma), anti-ERα (Santa Cruz Biotechnology, Santa Cruz, CA), anti-ERβ (Santa Cruz), anti-SR-B1 (Abcam, Cambridge, MA), and anti-β-actin (Sigma) antibodies. Western blots were developed using alkaline phosphatase method.
Dual luciferase reporter assay
The EREs of SR-B1 promoter along with their flanking regions (250–400 bp) were inserted upstream of the promoter of a firefly luciferase gene in pGL3 promoter vector (Promega) (primers are in Table 1) (31, 32). HEPG2 cells (4 × 105 in six-well plate) were cotransfected with 1500 ng of these ERE-containing luciferase reporter constructs along with 150 ng of a reporter plasmid containing renilla luciferase (pRLTk; Promega) as an internal transfection control using FuGENE6 transfection reagent. Control transfections were done using pGL3 promoter vector without any ERE insertion or with a luciferase construct-containing segment of SR-B1 promoter containing no ERE (nonspecific control, non-ERE). At 24 h after transfection, cells were treated with 100 nm E2, incubated for 6 h, and subjected to luciferase assay using dual luciferase reporter assay kit (Promega). Firefly luciferase activities were assayed and normalized to those of renilla luciferase. Each treatment was done in four replicates, and the experiment was repeated at least twice.
Transient expression of ERα
To analyze the effect of transient expression of exogenous ERα in SR-B1 in HEPG2 cells, ERα expression construct pEGFP-C1-ERα was obtained from Addgene (Cambridge, MA). For transient transfection, the cells were grown overnight in phenol-red-free media (up to 60% confluency) and treated with varying concentrations of pEGFP-C1-ERα expression construct using Lipofectamine 2000 transfection reagent (Invitrogen). At 24 h after transfection, cells were treated with 100 nm E2, incubated for 6 h, and subjected to RNA and protein extraction. Each treatment was done in three replicates, and the experiment was repeated at least twice.
Chromatin immunoprecipitation (ChIP) experiment
For the ChIP assay, E2-treated and control cells were fixed in 4% formaldehyde, sonicated to shear the chromatin (∼300 nucleotides), and subjected to ChIP analysis as described by us previously (31, 32). ChIP assay was performed using antibodies against MLL1 (Lifespan Bioscience), MLL2 (Lifespan Bioscience), MLL3 (Abgent), MLL4 (Sigma), ERα (Santa Cruz), (Santa Cruz), H3K4 trimethyl (Upstate Biotechnology, Lake Placid, NY), RNA polymerase II (RNAPII) (Abcam), transcription factor IIB (TFIIB) (Abgent), TFIIF (Abgent), TATA box binding protein associated factor 250 kD (Upstate), human capping enzyme (hCE) (kind gift from Reinberg's laboratory), SRC1 (Santa Cruz), p300 (Santa Cruz), CBP (Santa Cruz), Sp1 (Upstate), and β-actin (Sigma). Immunoprecipitated DNA was PCR amplified using primers specific to different EREs of SR-B1 promoter. The real-time PCR analysis of the ChIP DNA was done using SsoFast EvaGreen supermix (Bio-Rad). Each PCR was done in triplicate.
Cholesterol uptake assay
JAR cells were grown up to 60% confluency and treated with phosphorothioate antisense oligonucleotides specific to MLL1, MLL2, MLL3, MLL4, and ERα by using iFECT transfection reagent for 48 h. The knocked-down cells were treated with 100 nm E2 for an additional 4 h before addition of 200 μg HDL packaged with 1.5 μCi [3H]cholesterol for an additional 6 h (40). Cells were washed with phenol-red-free DMEM four times followed by PBS twice. Cells were then lysed, and an equal amount of cell lysate from different samples was subjected to liquid scintillation counting for detection of tritium-labeled cholesterol.
Animal experiments
Experimentally naive 6-wk-old, male athymic nude mice (nu/nu) (Harlan, Indianapolis, IN) were housed three per cage with same-sex cage mates in a temperature- and humidity-controlled environment under a 12-h light, 12-h dark cycle. All animals had free access to food and water throughout the study and were maintained and cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at the University of Texas at Arlington approved the experimental protocol. Every care and precaution was taken to minimize the pain/stress on the animals. One group of three mice was injected with MLL2 antisense (300 μg/20 g body weight, ip) in PBS (41). Control mice (group of three) were injected with an equal volume of the diluent (PBS) or scramble antisense alone. Three injections of antisense were administered at 24-h intervals. The mice were either perfused with PBS followed by 4% paraformaldehyde in PBS or euthanized via cervical dislocation at 48 and 96 h after final antisense administration. The experiment was repeated at least thrice.
Immunohistological analysis
For immunohistochemical staining, the liver tissue of the sequentially perfused mice was sectioned and subjected to immunohistochemical staining with SR-B1 antibody (42). In brief, the liver sections were incubated in blocking buffer (containing donkey serum) and then with primary antibodies specific to SR-B1. Sections were then incubated with biotinylated donkey secondary antibody followed by avidin-biotin complex followed by peroxidase labeling using a 3,3′-diaminobenzidine substrate assay kit (Vector Laboratories, Burlingame, CA). Sections were examined under light microscope.
Determination of plasma cholesterol level in mice
Athymic nude mice (nu/nu) (groups of three in each treatment) were ip administered with MLL2 antisense (300 μg/20 g body weight) in PBS. Control mice were injected with an equal volume of the diluent (PBS) or scramble antisense alone. Three shots of antisense were administered at 24-h intervals. The mice were euthanized at 48 and 96 h after final antisense administration, and the blood samples were collected. The blood was analyzed to detect level of cholesterol by using a commercial cholesterol assay kit (Amplex Red cholesterol assay kit; Invitrogen). The experiment was repeated at least thrice.
Statistical analysis
Each experiment was done in two to three replicates, and then cells were pooled (and treated as one sample), subjected to RNA extraction, RT-PCR, and ChIP analysis, and each experiment was repeated at least thrice (n = 3). The real-time PCR analysis of such samples were done in three replicate reactions and repeated in all three independent experiments (n = 3). For luciferase assay and cholesterol uptake assays, each treatment was done in four replicates and the experiment was repeated at least twice (n = 2). Normally distributed data were analyzed by ANOVA, and nonnormally distributed data were analyzed using Student's t tests (SPSS Inc., Chicago, IL) to determine the level of significance between individual treatments. The treatments were considered significantly different at P ≤ 0.05.
Results
Human HDLR-SR-B1 (SR-B1) is transcriptionally regulated by estrogen
To assess the effect of estrogen in transcriptional regulation of SR-B1, we treated HEPG2 cells (hepatocytes) with varying concentrations of E2 and examined the level of SR-B1 expression in the absence and presence of E2. RNA was isolated from the E2-treated and control cells, reverse transcribed into cDNA, and analyzed by real-time-PCR using primers specific to SR-B1 pre-mRNA. Our results demonstrated that SR-B1 was overexpressed upon treatment with E2 in a concentration-dependent manner (Fig. 1A). The level of SR-B1 mRNA was increased by 3- and 8-fold in presence of 10 and 100 nm E2, respectively (Fig. 1A, qPCR data are in the right panel). The SR-B1 expression was also increased in protein level upon treatment with E2 (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Notably, similar E2-dependent SR-B1 expression was also observed in other steroidogenic JAR cells (placenta choriocarcinoma) and application of E2 antagonist tamoxifen inhibited the E2-induced expression of SR-B1 (Supplemental Fig. 2). Temporal studies demonstrated that E2-mediated stimulation of SR-B1 was initiated as early as 1 h and continued to increase up to 6 h and then plateaued (Fig. 1B, right panel shows the real-time PCR quantification). These results demonstrated that SR-B1 is an E2-responsive gene.
Fig. 1.
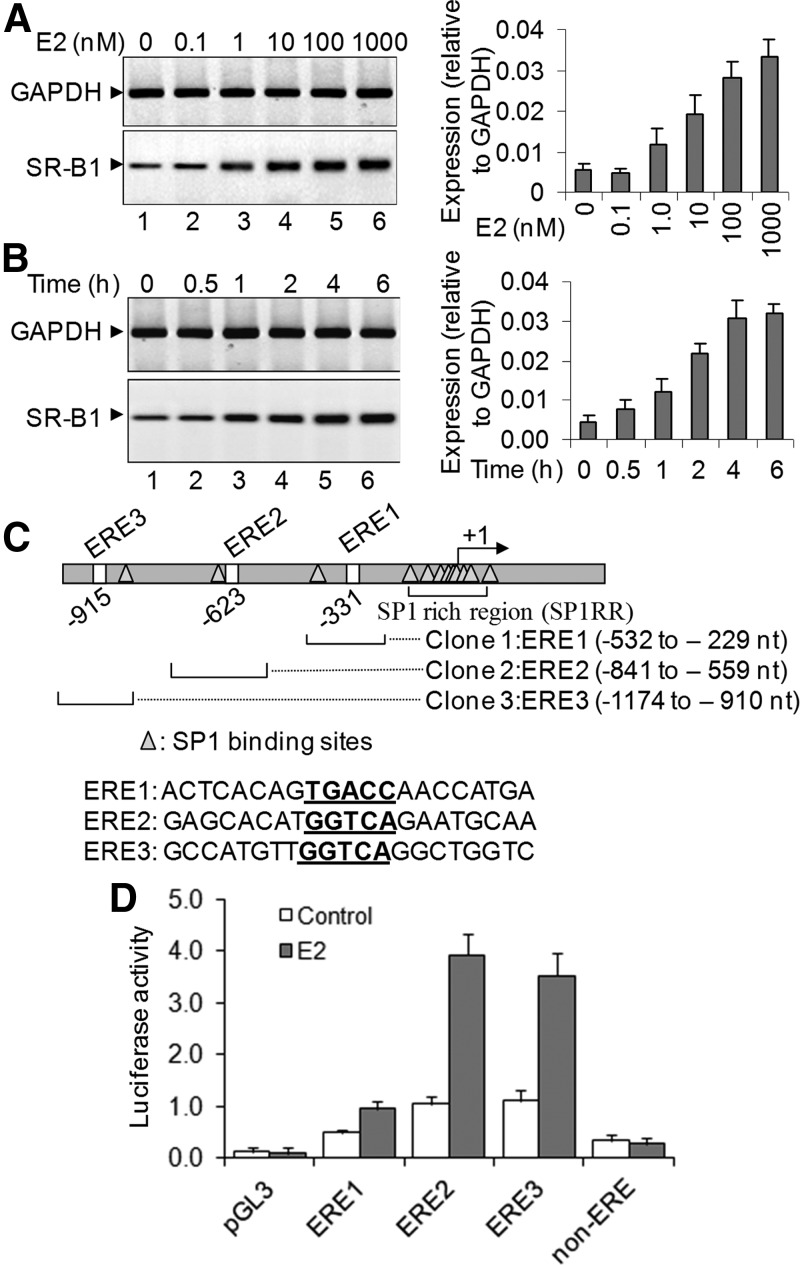
Effects of E2 on SR-B1 gene expression and promoter activity. A, Effect of E2 on SR-B1 expression. HEPG2 cells, grown in phenol-red-free media, were treated with varying concentrations of E2 for 6 h. RNA was isolated and analyzed by RT-PCR using primers specific to SR-B1. GAPDH was used as quantitative control. The real-time-PCR quantification is shown in the right panel. Bars indicate se (n = 3; P ≤ 0.05). B, Temporal effect of E2 exposure on SR-B1 expression. HEPG2 cells were treated with 100 nm E2 for varying time periods. RNA was analyzed by RT-PCR. The real-time PCR quantification is shown in the right panel (n = 3). C, SR-B1 promoter showing different EREs (ERE half-sites) and Sp1 binding sites. ERE1–3 and a non-ERE region were cloned into luciferase-based reporter construct pGL3. D, Luciferase assay. Each ERE-pGL3 construct, along with renilla luciferase construct, were cotransfected into HEPG2 cells for 24 h. Control cells were treated with empty pGL3 vector or non-ERE-pGL3. Cells were then treated with 100 nm E2 for an additional 6 h and then subjected to luciferase assay by using Dual-Glo luciferase assay system. The luciferase activities in the presence of E2 (over untreated controls and normalized against renilla luciferase expression) were plotted. The experiment with four replicate treatments was repeated at least twice (n = 8). Bars indicate se (P ≤ 0.05).
To further understand the mechanism of transcriptional regulation of SR-B1, we analyzed the promoter sequences of SR-B1 and found that SR-B1 promoter contains three putative EREs (ERE half-sites, GGTCA or TGACC) within 1000 nucleotides upstream of the transcription start site (Fig. 1C). We cloned each ERE region separately in a luciferase-based human expression construct pGL3 and analyzed their E2 response. ERE-pGL3 constructs were transfected in HEPG2 cells, exposed to E2, and then subjected to luciferase activity analysis using a commercial luciferase detection kit. We also cotransfected a renilla luciferase construct (pRLTk) as an internal transfection control, and luciferase activities from the ERE-pGL3 constructs were normalized to renilla luciferase activity. Transfection with either control pGL3 plasmid (with no ERE) or non-ERE-pGL3 did not show any E2-induced luciferase activity (Fig. 1D). However, transfection with different ERE(1–3)-pGL3 constructs resulted in differential E2-dependent luciferase induction (Fig. 1D). ERE2- and ERE3-pGL3 showed a more robust E2 response in comparison with ERE1 (Fig. 1D). These results indicated that ERE2 and ERE3 regions of SR-B1 promoter are potentially involved in E2-mediated SR-B1 activation.
ERs are key players in E2-induced SR-B1 expression
ERs are major players in estrogen-induced regulation of estrogen-responsive genes. There are two major types of ERs, ERα and ERβ, that play major roles in E2-induced gene activation. Notably, HEPG2 cells are known to express a relatively lesser amount of ERs in comparison with other steroidogenic cells such as human breast cancer cells (MCF7, see Western blot analysis in Supplemental Fig. 3A). To examine potential roles of endogenous ERs in E2-induced SR-B1 expression, we knocked down ERα and ERβ independently (or in combination), using specific antisense oligonucleotide (knockdown efficiency of ER antisense oligonucleotide in protein level is shown in Supplemental Fig. 4), in HEPG2 cells, exposed the cells to E2, and examined its impact on SR-B1 expression. Our results demonstrated that knockdown of ERα suppressed significantly the E2-induced expression of SR-B1 (Fig. 2A, lane 4, quantification on the bottom panel). Independent knockdown of ERβ also decreased E2-induced expression of SR-B1 (Fig. 2A, lane 5). The combined knockdown of ERα and ERβ showed similar level of decreased expression of SR-B1 mRNA as observed during their independent knockdowns (Fig. 2A, lane 6). Scramble antisense (which has no homology to ERs) showed no significant effect on E2-induced SR-B1 expression (Fig. 2A, lane 3). These results demonstrated that SR-B1 is an E2-responsive gene, and both ERα and ERβ play key roles during E2-induced SR-B1 expression. Notably, there is some basal of level of SR-B1 expression still retained even after knockdown of both ERs, which may indicate the presence of some ER-independent mechanism of SR-B1 expression.
Fig. 2.
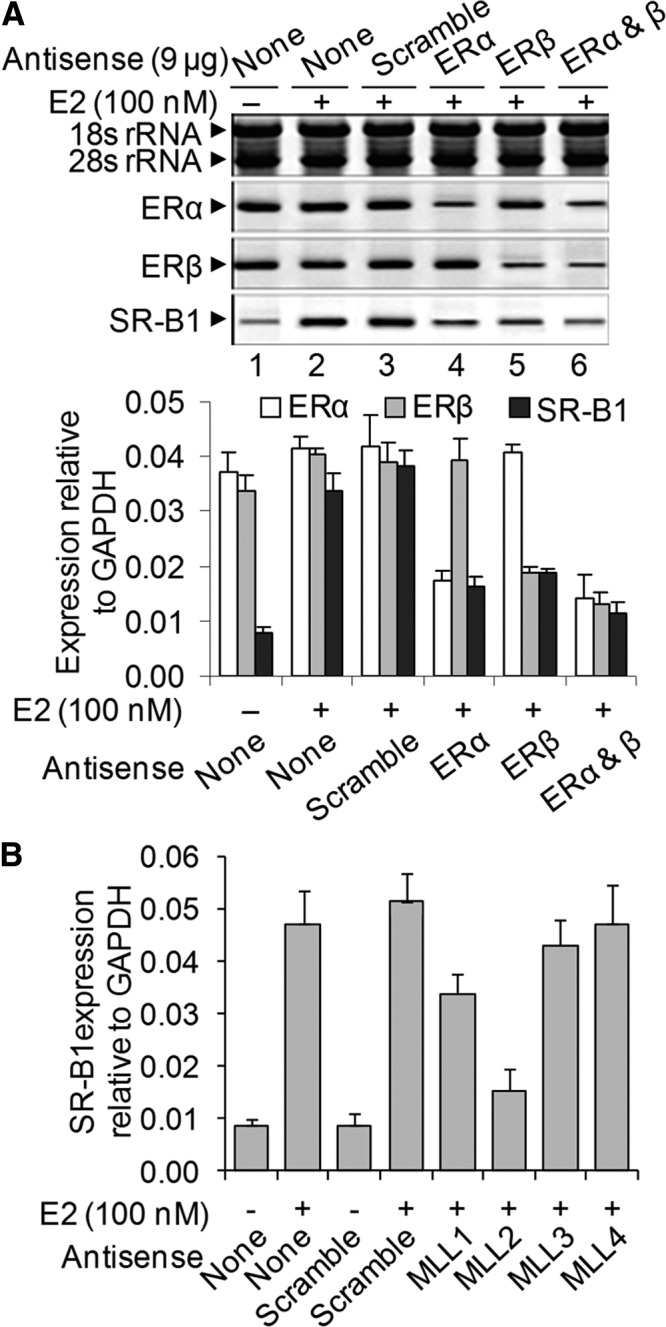
Effect of knockdown of ERs and MLLs on E2-dependent SR-B1 expression. A, Effects of ER knockdown. HEPG2 cells were transfected with ERα and ERβ antisense oligonucleotides separately or in combination for 48 h and then treated with 100 nm E2 for 6 h. A scramble antisense was also transfected for negative control. RNA from the control and antisense-treated cells were subjected to RT-PCR analysis using primers specific to SR-B1 and ERs. rRNA was used as loading control. Real time quantification of gene expression relative to GAPDH is shown in the bottom panel. Bars indicate standard errors (n = 3; P ≤ 0.05). B, Effects of MLLs knockdown: HEPG2 cells were transfected with MLL1, MLL2 MLL3, MLL4, and scramble antisense oligonucleotides separately for 48 h and then treated with 100 nm E2 for 6 h. RNA was reverse-transcribed and subjected to real-time PCR analysis for the expression of SR-B1. The expression of SR-B1 relative to GAPDH is plotted. Bars indicate standard errors (n = 3; P ≤ 0.05).
To examine further whether endogenous ER is sufficient for E2-induced SR-B1 expression, we transfected HEPG2 cells with an ERα expression construct for exogenous overexpression of ERα and then exposed the cells to E2 followed by analysis of SR-B1 expression. Our analysis showed that overexpression (confirmed by Western blot, Supplemental Fig. 3A) of exogenous ERα has a very subtle effect on E2-induced SR-B1 expression in HEPG2 cells (Supplemental Fig. 3B). This observation indicated that endogenous ERs of HEPG2 cells are sufficient for E2-induced SR-B1 expression.
MLL histone methylases act as ER coregulators during E2-induced SR-B1 expression
Along with ERs, various ER coregulators including HATs and HMTs play crucial roles in transcription of E2-responsive genes. Recent studies demonstrated that MLL histone methylases act as ER coactivators in NR-mediated gene expression. Herein, we examined potential involvement of MLL histone methylases in E2-induced SR-B1 expression. We knocked down different MLLs (MLL1–4) independently using respective antisense oligonucleotide, exposed the cells to E2, and then analyzed its effects on E2-induced SR-B1 expression (Fig. 2B and Supplemental Fig. 5). Scramble antisense (without any homology to MLL) was used in parallel as negative control. The specific MLL knockdowns were confirmed by analyzing their respective gene expression (Supplemental Fig. 5, Western blot analysis of MLL knockdowns are shown in Supplemental Fig. 6). The MLL knocked-down and E2-treated RNA was converted into cDNA and subjected to real-time-PCR analysis for the SR-B1 expression (Fig. 2B). Interestingly, the level of E2-induced SR-B1 expression was affected by knockdown of MLL1 or MLL2, although the MLL2 knockdown affected the most (more than 80%) (Fig. 2B). However, MLL3, and MLL4 knockdowns showed minor or no effect on E2-induced SR-B1 expression (Fig. 2B). The target specificity of the MLL2 was further confirmed by using real-time PCR (Supplemental Fig. 7), and the off-target effect of MLL2 was alleviated using a second MLL2 antisense (MLL2-2, Supplemental Fig. 8). These analyses demonstrated that histone methylases MLL1 and MLL2 play crucial roles during E2-induced SR-B1 expression.
ERs and MLLs bind to SR-B1 promoter in an E2-dependent manner
To investigate further roles of MLLs and ERs in E2-dependent SR-B1 expression, we examined their binding in the SR-B1 promoter in the presence of E2 using ChIP assay. Briefly, chromatins isolated from E2-treated and control cells were subjected to immunoprecipitation using ER- and MLL-specific antibodies, and the immunoprecipitated DNA fragments were reverse cross-linked and PCR amplified using primers specific to different ERE regions of SR-B1 promoter. ChIP analysis demonstrated that the binding of ERα was enriched in the ERE2 and ERE3 regions of SR-B1 promoter in the presence of E2 (lanes 3–6, Fig. 3A, real-time PCR quantifications are shown in the right panels). The E2-dependent binding of ERβ was also observed in those ERE regions (Fig. 3A, lanes 3–6). These observations further demonstrated that both ERα and ERβ are involved in E2-dependent activation of SR-B1, likely via binding through ERE2 and ERE3 regions of SR-B1 promoter.
Fig. 3.
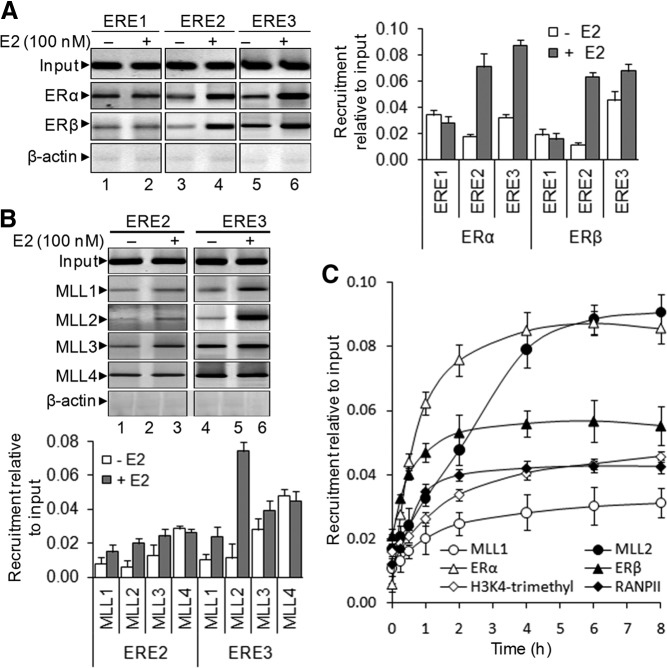
E2-dependent recruitment of ERs and MLLs (MLL1–4), level of H3K4 trimethylation, and RNAPII recruitment onto SR-B1 promoter (ChIP assay). A, Recruitment of ERα and ERβ. HEPG2 cells were treated with 100 nm E2 for 6 h, fixed with formaldehyde, sonicated to shear the chromatin, and subjected to ChIP assay using antibodies against ERα and ERβ separately. Antibody specific to β-actin was used as IgG control. The ChIP DNA was PCR amplified using primers specific to ERE1–3 of SR-B1 promoter. Real-time quantification of recruitment level of ERs in the absence and presence of E2, relative to input, are shown in the right panel. Bars indicate se (n = 3; P ≤ 0.05). B, Recruitment of MLL1–4. E2-treated and control HEPG2 cells were subjected to ChIP assay using antibodies against ML1–4 separately. The immunoprecipitated DNA fragments were PCR amplified using primers specific to ERE2 and ERE3 regions of SR-B1 promoter. Real-time quantification of level of recruitment relative to input is shown in the bottom panel. Bars indicate se (n = 3; P ≤ 0.05). C, Kinetics of recruitment of MLLs, ERs, and RNAPII and enrichments of H3K4 trimethylation level. HEPG2 cells were treated with 100 nm E2 for varying time periods and subjected to ChIP assay using antibodies against ERα, ERβ, MLL1, MLL2, H3K4 trimethyl, and RNAPII separately. The immunoprecipitated DNA fragments were subjected to real-time PCR analysis using primers specific to the ERE3 region of SR-B1 promoter. The recruitment levels relative to input were plotted. Bars indicate se (n = 3; P ≤ 0.05).
Because MLLs were found to be critical for E2-induced expression of SR-B1, we also examined the binding of MLLs into the SR-B1 promoter using ChIP assay. We limited our ChIP analysis to ERE2 and ERE3 regions because these EREs were more responsive to E2 exposure (Fig. 1, C and D) and ER binding (Fig. 3A). ChIP analysis demonstrated that binding of MLL2 was significantly enhanced in the ERE3 region (Fig. 3B, real-time-quantification in the bottom panel). Slight enrichment of MLL1 was observed in the ERE3 region in the presence of E2 (Fig. 3B). A subtle increase of MLL2 recruitment was also observed in the ERE2 region (Fig. 3B). Notably, the binding of MLL3 and MLL4 was mostly not sensitive to E2 exposure, and these results are in agreement with the respective knockdown analysis presented in Fig. 2B. These observations further demonstrated that MLL2 and to some extent MLL1 are associated with E2-induced SR-B1 expression.
Because ERα, ERβ, MLL1, and MLL2 are found to be essential for E2-induced SR-B1 expression, we analyzed their recruitment kinetics in ERE3 regions (the most responsive ERE) after treatment with E2 (Fig. 3C). Because MLLs are histone H3K4-specific methyltransferases and H3K4 trimethylation is associated with transcriptional activation, we examined the H3K4 trimethylation level in the SR-B1 promoter as a function of time. Our results demonstrated that, along with ERα, ERβ, MLL1, and MLL2, the level of H3K4 trimethylation and recruitment of RNAPII were increased in the SR-B1 promoter. This association of H3K4 trimethylation and RNAPII followed a similar pattern as the binding of MLLs and ERs, indicating their coordination during E2-induced transcription of SR-B1 (Fig. 3C).
MLL2 regulates assembly of transcription pre-initiation complex (PIC)
To explore more details about the mechanism of E2-induced SR-B1 transactivation, we examined the recruitment of various general transcription factors and other ER coactivators using ChIP assay in the absence and presence of E2. Our results demonstrated that transcription factors TAF250 (component of TFIID), TFIIB, TFIIF, and hCE were enriched in the SR-B1 promoter (ERE3 region) upon treatment with E2 (Fig. 4A). Similarly, recruitment of ER coactivators such as SRC1, p300, and CBP were also enhanced upon addition of E2 (Fig. 4A). These observations demonstrated that estrogen stimulated the formation of transcription PIC as well as recruitment of transcription coactivators that helps in histone acetylation/methylation and chromatin remodeling leading to transcriptional activation.
Fig. 4.
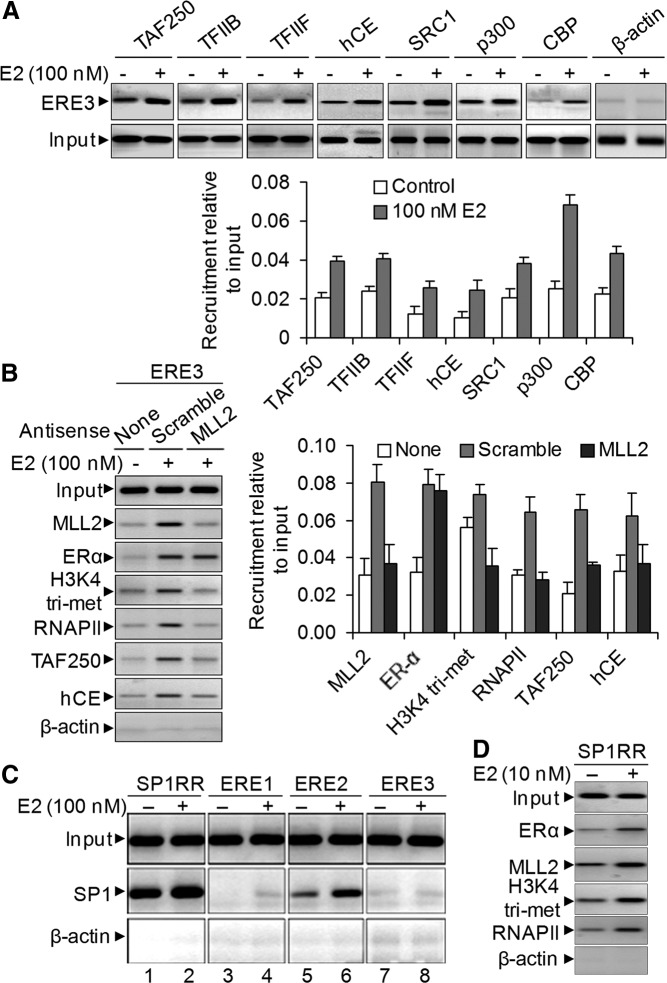
E2-induced PIC formation and recruitment of ER coactivator on the SR-B1 promoter. A, HEPG2 cells were treated with 100 nm E2 for 6 h and subjected to ChIP assay using antibodies specific to transcription factors TFIIB, TFIIF, TAF250, and hCE and ER coactivators SRC-1, p300, and CBP. The immunoprecipitated DNA fragments were PCR amplified using primers specific to the ERE3 region of SR-B1 promoter. Real-time quantification of recruitment level relative to input is shown in the bottom panel. Bars indicate se (n = 3; P ≤ 0.05). B, Roles of MLL2 on PIC assembly. HEPG2 cells were transfected with MLL2 antisense (and scramble) for 48 h and then treated with 100 nm E2 for 6 h and subjected to ChIP assay using antibodies specific to MLL2, ERα, H3K4 trimethyl, RNAPII, TAF250, and hCE. The ChIP DNA fragments were PCR amplified using primers specific to the ERE3 region of SR-B1 promoter. Real-time quantification of level of recruitment relative to input is shown in the right panel. Bars indicate se (n = 3; P ≤ 0.05). C, E2-induced recruitment of Sp1 in SR-B1 promoter. HEPG2 cells were treated with 100 nm E2 for 6 h and subjected to ChIP assay using antibodies specific to transcription factor Sp1. The immunoprecipitated DNA fragments were PCR amplified using primers specific to ERE1, ERE2, ERE3, and Sp1RR of SR-B1 promoter. D, E2-induced enrichment of ERα, MLL2, and RNAPII and E2-induced increase of H3K4 trimethylation level in Sp1RR.
To examine the potential roles of MLL histone methylases in PIC assembly, we knocked down MLL2 (the most responsive) and then analyzed the E2-dependent binding of general transcription factor (GTF) and coactivators using ChIP assay (Fig. 4B). Our results demonstrated that MLL2 knockdown does not affect the E2-dependent binding of ERα but reduced the level of TAF250 and hCE on the ERE3 region of the SR-B1 promoter (Fig. 4B, quantification on the right panel). MLL2 knockdown also reduced the level of H3K4 trimethylation and RNAPII recruitment in SR-B1 promoter (Fig. 4B). These observations suggested that MLL2 plays critical roles in recruitment of other ER coactivators as well as assembly of GTFs forming PIC before transcription activation.
Analysis of the SR-B1 promoter showed the presence of several Sp1 binding sites clustered around the +167- to −128-bp region (Supplemental Fig. 9A). To examine whether Sp1 is involved in E2-induced SR-B1 expression, we examined the binding of the Sp1 and also analyzed the enrichment of RNAPII, H3K4 trimethylation, histone acetylation, and histone methylase MLL2 in the Sp1 binding sites in the absence of presence of E2 using ChIP assay. These analyses demonstrated that Sp1 binding is enriched in the Sp1 binding [Sp1-rich region (Sp1RR)] sites in the presence of E2, although a significant amount of constitutive binding of Sp1 was observed in the Sp1RR site in the absence of E2 (Fig. 4C). E2-dependent enrichment of Sp1 was also observed in the ERE2 region, which may be likely due the presence of an Sp1 binding site in the close proximity of the ERE2 region (Fig. 4C). No significant binding of Sp1 was observed in either ERE1 or ERE3 regions in both the absence and presence of E2. The E2-dependent enrichment of RNAPII, H3K4 trimethylation, histone acetylation, and MLL2 was also observed in the Sp1 binding sites (Sp1RR, Fig. 4D). The E2-dependent enrichment of Sp1 along with other gene activation markers and ER coregulators in the Sp1 binding sites indicated involvement of an Sp1-mediated pathway during E2-induced SR-B1 gene activation, and notably, this is likely in combination with an ERE-dependent pathway.
ERs and MLLs play critical roles in cholesterol uptake by steroidogenic JAR cells
Because SR-B1 is a well-known player in reverse cholesterol transport and cholesterol uptake by liver and other steroidogenic tissues, we investigated any potential role of MLLs and ERs in controlling the cholesterol uptake efficiencies. For the cholesterol uptake assays, we used JAR cells, because the E2-induced SR-B1 expression was more robust in JAR cells. Also, it has been shown that the capacity of selective cholesteryl esters (CE) uptake by JAR cell lines is closely related to the expression level of SR-BI. In addition, a high level of adenoviral overexpression of SR-BI resulted in an enhanced capacity for selective CE uptake by JAR cells (40). Thus, JAR cells serve as suitable in vitro models to study selective CE uptake experiments (40).
We knocked down different MLLs (MLL1–4) and ERs separately using respective antisense oligonucleotides in JAR cells and then analyzed cholesterol uptake efficiencies of these knocked-down cells in the presence and absence of E2 following a previously described procedure (40). In brief, we packaged the 3H-labeled cholesterol oleate (CE) into human HDL. Then cells were incubated with these radiolabeled CEs containing HDL for 6 h at 37 C and washed thoroughly with PBS, and then the cells were lyse, and equivalent amounts of cell lysates were analyzed by using a scintillation counter to measure the entrapped cholesterol inside the JAR cells. Our results demonstrated that the cells that were treated with [3H]CE-labeled HDL but not with E2 has entrapped some amount of cholesterol (compared with control) (Fig. 5, compare lanes 1–3 with lane 4). However, the level of this entrapped [3H]CE was increased by 3.3-fold upon addition of E2, likely due to the induction of SR-B1 expression (Fig. 5, compare lanes 4 and 5). Interestingly, independent knockdown of MLL1, MLL2, MLL3, ERα, and ERβ resulted in a reduced level of entrapped cholesterol. MLL4 knockdown had no significant effect. Knockdown of MLL2 showed the most significant effect and suppressed the [3H]CE uptake almost to the basal level (2.5-fold less compared with control, in lane 5 Fig. 5). Knockdown of MLL1, MLL3, or ERα also decreased the [3H]CE uptake significantly (1.3-, 1.2-, and 1.7-fold less, respectively, Fig. 5). In addition, ERβ has down-regulated the CE uptake, although to a lesser extent in comparison with ERα (Fig. 5). These results demonstrated that along with ERs, histone methylase, especially MLL2, plays critical roles in cholesterol uptake by JAR cells in the presence of E2. Notably, MLL2 was also the most responsive MLL for E2-induced SR-B1 expression in HEPG2 cells (Figs. 2 and 3) and JAR cells (data not shown).
Fig. 5.

Role of ERs and MLLs (MLL1–4) on cholesterol uptake in JAR cells. JAR cells were grown up to 60% confluency before transfection with MLL1, MLL2, MLL3, MLL4, ERα, ERβ, and scramble antisense oligonucleotides separately for 48 h. The knocked-down cells were then treated with 100 nm E2 for an additional 4 h and then incubated with 200 μg HDL packaged with tritium-labeled CE for an additional 6 h. Cells were washed with phenol-red-free DMEM four times followed by PBS twice. Cells were then lysed and subjected to liquid scintillation counting for detection of entrapped tritium-labeled cholesterol and plotted (radioactivity uptake). Bars indicate se (n = 4; P ≤ 0.05).
Knockdown of MLL2 results in elevation of plasma cholesterol in mice
To further examine potential roles of MLLs on SR-B1 gene expression and cholesterol uptake in vivo, we administered MLL2-specific antisense in mice and then analyzed its effect on blood cholesterol level. In brief, athymic nude (nu/nu) mice were administrated with 300 μg phosphorothioate MLL2 antisense ip three times at 24-h intervals. Notably, this dose of antisense was selected because this is not toxic to animals (data not shown) and effective in knocking down target genes in mice. Mice were euthanized 48 and 96 h after the antisense administration, and the blood samples were collected. Control mice were administered with either PBS (vehicle) or a scramble antisense with no homology to MLL2. Our results demonstrated that knockdown of MLL2 increased the level of mice blood cholesterol from 138 mg/dl (control) to 180 mg/dl at 96 h after MLL2 antisense administration (Fig. 6A). The cholesterol level was increased in a time-dependent manner. These results demonstrated that MLL2 plays critical roles in maintaining plasma cholesterol levels.
Fig. 6.
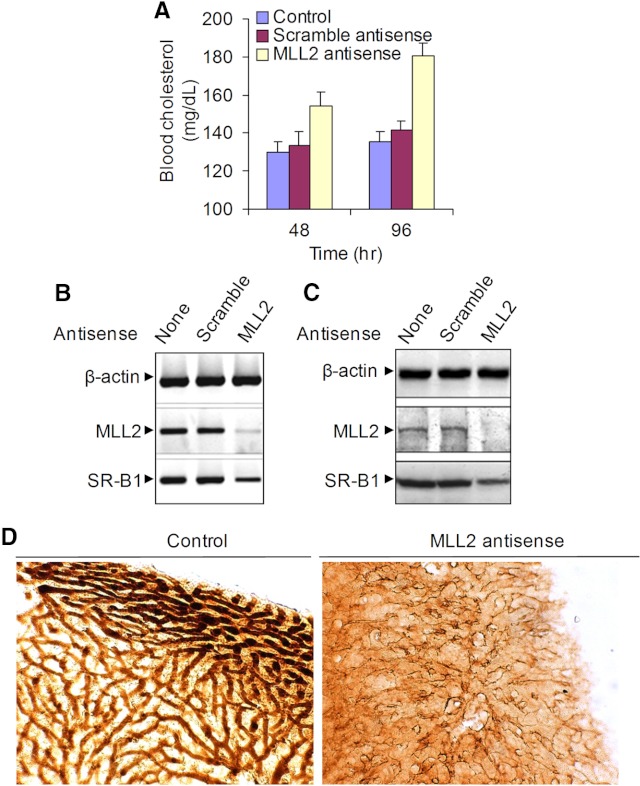
Effect of MLL2 knockdown on SR-B1 expression and plasma cholesterol level in mice. Athymic nude (nu/nu) mice were ip administered 300 μg phosphorothioate MLL2 antisense three times at 24-h intervals. Mice were euthanized or sequentially perfused with 4% formaldehyde at 48 and 96 h after final administration. Control mice were administered either PBS or a scramble antisense with no homology with MLL2. A, Plasma cholesterol level. Blood from MLL2 antisense-treated and control mice were collected at 48 and 96 h and subjected to total cholesterol quantification using a commercial cholesterol assay kit. Cholesterol levels in control, scramble, or antisense-treated mice were plotted. Bars indicate se (n = 3, P ≤ 0.05). B and C, Levels of MLL2 and SR-B1 expression in the control and MLL2 antisense-treated mouse liver. The liver tissues of the euthanized mice were subjected to RNA and protein. B, RNA was analyzed by RT-PCR with primers specific to MLL2 and SR-B1. C, The protein was analyzed by Western blotting using antibodies specific to MLL2 and SR-B1. D, Immunohistological staining of mouse liver showing the SR-B1 expression. The liver tissue of the sequentially perfused MLL2 antisense-treated and control mice was sectioned and subjected to immunohistological (diaminobenzidine) staining with SR-B1 antibody and examined under a light microscope.
Because SR-B1 plays a critical role in maintaining blood cholesterol levels, we analyzed whether MLL2 antisense treatment affected the expression of SR-B1 in mouse liver tissue. RT-PCR and Western blot analysis demonstrated that application of scramble antisense had no effect on MLL2 expression in the mouse liver. However, MLL2 antisense application knocked down MLL2 both in mRNA and protein levels in mouse liver (Fig. 6, B and C). In addition, MLL2 antisense treatment also down-regulated the expression of SR-B1 at the mRNA and protein level in the mouse liver, indicating critical roles of MLL2 in controlling SR-B1 expression in vivo. The level of SR-B1 expression in the mouse liver was further confirmed by immunohistological staining. Our analysis demonstrated that SR-B1 protein is highly expressed in the cell membranes of the control mouse liver and the level of SR-B1 expression is dramatically reduced in the MLL2 antisense-treated mouse liver, further demonstrating critical roles of MLL2 in SR-B1 expression in vivo (Fig. 6D). ChIP analysis showed that MLL2 knockdown reduced the recruitment of RNAPII and level of H3K4 trimethylation in the SR-B1 promoter in the mouse liver tissue, indicating critical roles of MLL2 in transcriptional activation of SR-B1 in vivo (Supplemental Fig. 10). Our results identify an important role for MLL2 in cholesterol homeostasis. Because SR-B1 is a critical player in plasma cholesterol trafficking to liver, the regulation of its expression by MLL2 is likely a key factor in the overall effects of this transcriptional coregulator on cholesterol control.
Discussion
Cardiovascular disease is one of the major causes of deaths all over the world. In general, women show less cardiovascular disease than age-matched men in the premenopausal stage. However, the incidence of cardiovascular diseases in women increases in the postmenopausal periods. Estrogen is well recognized to have beneficial effects in premenopausal women by preventing cardiovascular diseases by keeping low plasma cholesterol levels, and this protection is reduced in the postmenopausal state when the estrogen level goes down (43, 44). Estrogen replacement therapy in postmenopausal women reduces the risk of cardiovascular disease, although it may significantly increase the risk of other diseases including cancer (44). Oral estrogen administration lowers plasma LDL and raises HDL levels (45). Notably, significant amounts of estrogen are also produced in men by local tissue aromatization of androgenic precursors from the testes and adrenal glands. Although it is not very clear what roles this endogenous estrogen plays in men, it appears to play crucial roles in lipoproteins and glucose metabolism (44). Reduction in estrogen levels in males has been linked with a decrease in HDL levels. The estradiol level is negatively correlated to LDL cholesterol and fasting blood glucose levels in healthy men, indicating crucial roles of estrogen in maintaining a desirable profile of lipid and glucose metabolism (46, 47). Hypocholesterolemic effects of estradiol has also been demonstrated using animal models including male and female rats and ovariectomized animals (43). Overall, estrogen appears to play crucial roles in controlling plasma lipoprotein levels and cholesterol metabolism in both female and males (45, 48–50). In addition to cardiovascular function, estrogen also plays major roles in various other aspects of mammalian physiology including reproduction, development of secondary sexual characteristics, and stress response (48, 51, 52).
LDL receptor (LDLR) and SR-B1 are two major players in receptor-mediated cholesterol trafficking between lipoprotein to various tissues and maintenance of plasma cholesterol (1). LDLR primarily recognizes the LDL particle that helps delivering cholesterol into peripheral tissues and thus a major player in forward cholesterol transport. In contrast, SR-B1, which is expressed on the liver cell surface, recognizes the HDL particles rich in cholesterol and facilitates in delivering cholesterol into liver for further metabolism. The mechanism underlying these lipid transfers is distinct from classic receptor-mediated endocytosis, but it remains poorly understood (10). SR-BI-knockout mice and SR-BI-overexpressing mice have significant impacts on HDL metabolism (11, 12). Although many studies have been done in an effort to understand the roles of LDLR and SR-B1 in cholesterol homeostasis, which is well known to be influenced by steroid hormones, little is known about the transcriptional regulation of SR-B1 and LDLR, especially in the presence of hormones. Herein, we examined the epigenetic mechanism of transcriptional regulation of SR-B1 under an estrogen environment.
Our studies demonstrated that human SR-B1 is transcriptionally regulated by E2. Because steroids are well known to play critical roles in controlling blood cholesterol level and cardiovascular disease, our studies showing the E2-mediated transcriptional activation of SR-B1 provides a novel link between estrogen signaling and cholesterol homeostasis. Sequence analysis showed that the SR-B1 promoter contains three ERE half-sites, and a luciferase-based reporter assay demonstrated that ERE2 and ERE3, which are located −623 and −915 nucleotides upstream of transcriptional start sites are responsive to E2 exposure. Antisense-mediated knockdown of ERα and ERβ suppressed the E2-mediated activation of SR-B1. ChIP analysis demonstrated that both ERα and ERβ were primarily enriched to ERE2 and ERE3 regions of the SR-B1 promoter in an E2-dependent manner, further supporting our previous observations that these EREs are primarily involved in E2-induced SR-B1 expression. Notably, our studies demonstrated that both ERα and ERβ are involved in E2-mediated activation of SR-B1. Knockdown of either ERα or ERβ significantly inhibited the expression of SR-B1. These observations indicate that ERα and ERβ may form a heterodimer that recognizes the EREs in the SR-B1 promoter followed by recruitment of other ER coregulators and PIC assembly. Notably, the ERs are known to form the homo- and heterodimers in recognition of EREs during transcriptional regulation of estrogen-responsive genes.
Similar to ERs, knockdown of the MLL family of histone methylases also suppressed E2-induced SR-B1 expression. Our ChIP analysis showed that E2 treatment led to a significant increase in the recruitment of MLL2 in the ERE2 and ERE3 regions of the SR-B1 promoter. Some amount of E2-dependent enrichment of MLL1 was also observed in the ERE3 region. These observations indicated that MLL2 and MLL1 coordinate with ERs and act as coregulators for SR-B1 expression under an E2 environment (31–35). MLLs contain one or more LXXLL domains (NR box) (53). Proteins containing NR boxes are likely to interact with NRs. MLL1 contains one LXXLL domain, whereas MLL2, MLL3, and MLL4 contain five, three, and four LXXLL domains, respectively (53). NR coregulators interact with NRs through their NR boxes and get recruited to the promoter of target genes (54, 55). Thus presence of one or more LXXLL domain makes the MLLs as potential coregulators for E2-mediated activation of SR-B1. However, our studies demonstrated that among all the MLLs, knockdown of MLL2 exhibited the strongest effect on E2-indcued SR-B1 expression.
Our studies also demonstrated that along with increased recruitment of ERs and MLL2, E2 treatment also induced the recruitment of general transcription factors such as TFIID subunit TAF250, TFIIB, TFIIF, and hCEs and stimulated the pretranscription initiation complex assembly (56, 57). E2 also induced the recruitment of other ER coactivators (54, 55, 58) such as SRC1, CBP, and p300. SRC1, CBP, and p300 are well-known HATs, and thus, their recruitment likely induces histone acetylation and, hence, chromatin remodeling and transcription activation of SR-B1. Importantly, knockdown of MLL2 impaired the E2-dependent recruitment of GTFs and ER coactivators in the SR-B1 promoter, although binding of ERs was unaffected. These observations indicated that the recruitment of MLL2 takes place after ER recruitment, and MLL2 aids the recruitment of other ER coactivators, GTFs, and RNAPII. MLL2 recruitment also increased the H3K4 trimethylation level in the SR-B1 promoter, indicating potential roles of MLL2-mediated histone H3K4 trimethylation in E2-induced SR-B1 expression. Notably, in the cholesterol uptake experiments, MLL2 knockdown had a stronger impact than individual ER knockdowns. This observation indicates that E2-mediated transcription activation of SR-B1 may follow an ER-dependent as well as to some extent an ER-independent pathway, and MLL2 may participate in both an ER-dependent as well as an ER-independent pathway. Similarly, the knockdown of MLL2 decreased H3K4 trimethylation even below the basal level in HEPG2 (Fig. 4B), which indicated that MLL2 is potentially involved in basal as well as activated transcription of SR-B1.
SR-B1 is a critical player in cholesterol uptake by the liver and other steroidogenic tissues. Because our studies showed ERs and MLL2 as important regulators of SR-B1, we examined their potential roles in cholesterol uptake by the liver cells. Our studies demonstrated that the cholesterol uptake efficiency of HEPG2 cells was increased upon treatment with E2. Importantly, knockdown of ERs and MLLs (MLL1–4) affected differentially the E2-dependent cholesterol uptake efficiency of HEPG2 cells. Knockdown of either ERα or ERβ reduced the cholesterol uptake efficiency (up to 46 and 26%, respectively) of the HEPG2 cells. Interestingly, knockdown of MLL2 abolished the cholesterol uptake efficiency of the HEPG2 cell almost to the basal level, demonstrating critical roles of MLL2 in this process.
Intriguingly, antisense-mediated knockdown of MLL2 in mice down-regulated SR-B1 expression in the liver tissue both at the mRNA and protein levels and resulted in 33% increase in the level of blood cholesterol within 96 h after MLL2 antisense treatment. Immunohistological analysis also demonstrated that SR-B1 is expressed in the membrane of mouse liver cells, and this expression was significantly reduced upon MLL2 knockdown. ChIP analysis of mouse liver tissue showed that MLL2 binds to SR-B1 promoter and induces histone H3K4 trimethylation and RNAPII recruitment, indicating critical roles of MLL2 in transcriptional activation of SR-B1 in vivo that is reflected in the level of SR-B1 expression in mouse liver cells. These studies demonstrated that MLL2 is a key player in transcriptional activation of SR-B1 and controlling blood cholesterol both in vitro and in vivo. Notably, similar to human SR-B1 promoter, sequence analysis showed that mouse SR-B1 promoter also contains several putative EREs and Sp1 binding sites near the transcription start site (Supplemental Fig. 9B), although sequences around the ERE regions between mouse and human SR-B1 promoter are not conserved. The presence of these EREs in both mouse and human SR-B1 indicate that both are potentially regulated by estrogen.
In summary, our studies demonstrate that histone methylase MLL2, in coordination with ERs, plays critical roles in E2-induced expression of SR-B1. They also regulate the cholesterol uptake efficiency of the steroidogenic tissues and influences plasma cholesterol levels. Notably, the levels of cholesterol in vivo very much depend on various other hormone levels as well as male/female sex and age of animals, and further investigation is needed to address more detail on the roles of MLLs in sex-specific regulation of plasma cholesterol. Furthermore, histone deacetylases and various other epigenetic regulators have been previously implicated in regulation of blood cholesterol (37–39). Herein, we demonstrate that HMTs, especially MLLs, are critical players in cholesterol homeostasis and maintenance of blood cholesterol. Our studies revealed novel epigenetic regulators associated with estrogen signaling and maintenance of plasma cholesterol levels. Mutation or malfunction in MLL-ER interaction may increase the cardiovascular risk, and therefore, stabilizing the interaction between MLL and ERs may be beneficial in developing novel cardiovascular therapy.
Supplementary Material
Acknowledgments
We thank Bogala M. Reddy and other lab members for helpful discussions.
Research in the Mandal laboratory is supported by grants from the National Institutes of Health (1R15 ES019129-01) and the American Heart Association (0765160Y).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CBP
- cAMP response element-binding protein-binding protein
- CE
- cholesteryl ester
- ChIP
- chromatin immunoprecipitation
- E2
- 17β- estradiol
- ER
- estrogen receptor
- ERE
- estrogen response element
- FBS
- fetal bovine serum
- GTF
- general transcription factor
- HAT
- histone acetyltransferase
- hCE
- human capping enzyme
- HDL
- high-density lipoproteins
- HDLR-SR-B1 (SR-B1)
- HDL receptors scavenger receptor class B type I
- H3K4
- histone H3 lysine-4
- HMT
- histone methyltransferase
- LDL
- low-density lipoproteins
- LDLR
- LDL receptor
- MLL
- mixed-lineage leukemia
- NR
- nuclear receptor
- PIC
- pre-initiation complex
- RNAPII
- RNA polymerase II
- SRC
- steroid receptor coactivator
- TFIIB
- transcription factor IIB.
References
- 1. Brown MS, Goldstein JL. 1983. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J Clin Invest 72:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grummer RR, Carroll DJ. 1988. A review of lipoprotein cholesterol metabolism: importance to ovarian function. J Anim Sci 66:3160–3173 [DOI] [PubMed] [Google Scholar]
- 3. Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. 2003. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11:25–33 [DOI] [PubMed] [Google Scholar]
- 4. Osono Y, Woollett LA, Herz J, Dietschy JM. 1995. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J Clin Invest 95:1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams DL, Wong JS, Hamilton RL. 2002. SR-BI is required for microvillar channel formation and the localization of HDL particles to the surface of adrenocortical cells in vivo. J Lipid Res 43:544–549 [PubMed] [Google Scholar]
- 6. Fukasawa M, Adachi H, Hirota K, Tsujimoto M, Arai H, Inoue K. 1996. SRB1, a class B scavenger receptor, recognizes both negatively charged liposomes and apoptotic cells. Exp Cell Res 222:246–250 [DOI] [PubMed] [Google Scholar]
- 7. Krieger M. 1999. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem 68:523–558 [DOI] [PubMed] [Google Scholar]
- 8. Matsui M, Sakurai F, Elbashir S, Foster DJ, Manoharan M, Corey DR. 2010. Activation of LDL receptor expression by small RNAs complementary to a noncoding transcript that overlaps the LDLR promoter. Chem Biol 17:1344–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. 1998. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem 273:5599–5606 [DOI] [PubMed] [Google Scholar]
- 10. Nieland TJ, Penman M, Dori L, Krieger M, Kirchhausen T. 2002. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci USA 99:15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Silver DL, Jiang XC, Arai T, Bruce C, Tall AR. 2000. Receptors and lipid transfer proteins in HDL metabolism. Ann NY Acad Sci 902:103–111; discussion 111–112 [DOI] [PubMed] [Google Scholar]
- 12. Ueda Y, Gong E, Royer L, Cooper PN, Francone OL, Rubin EM. 2000. Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J Biol Chem 275:20368–20373 [DOI] [PubMed] [Google Scholar]
- 13. Nilsson S, Gustafsson JA. 2002. Estrogen receptor action. Crit Rev Eukaryot Gene Expr 12:237–257 [DOI] [PubMed] [Google Scholar]
- 14. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. 1995. The nuclear receptor superfamily: the second decade. Cell 83:835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moggs JG, Orphanides G. 2001. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep 2:775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barish GD, Downes M, Alaynick WA, Yu RT, Ocampo CB, Bookout AL, Mangelsdorf DJ, Evans RM. 2005. A nuclear receptor atlas: macrophage activation. Mol Endocrinol 19:2466–2477 [DOI] [PubMed] [Google Scholar]
- 17. Lonard DM, O'Malley BW. 2005. Expanding functional diversity of the coactivators. Trends Biochem Sci 30:126–132 [DOI] [PubMed] [Google Scholar]
- 18. Tremblay GB, Giguère V. 2002. Coregulators of estrogen receptor action. Crit Rev Eukaryot Gene Expr 12:1–22 [DOI] [PubMed] [Google Scholar]
- 19. Lee JS, Kim KI, Baek SH. 2008. Nuclear receptors and coregulators in inflammation and cancer. Cancer Lett 267:189–196 [DOI] [PubMed] [Google Scholar]
- 20. Carlberg C, Seuter S. 2010. Dynamics of nuclear receptor target gene regulation. Chromosoma 119:479–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim SW, Park K, Kwak E, Choi E, Lee S, Ham J, Kang H, Kim JM, Hwang SY, Kong YY, Lee K, Lee JW. 2003. Activating signal cointegrator 2 required for liver lipid metabolism mediated by liver X receptors in mice. Mol Cell Biol 23:3583–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim GH, Park K, Yeom SY, Lee KJ, Kim G, Ko J, Rhee DK, Kim YH, Lee HK, Kim HW, Oh GT, Lee KU, Lee JW, Kim SW. 2009. Characterization of ASC-2 as an antiatherogenic transcriptional coactivator of liver X receptors in macrophages. Mol Endocrinol 23:966–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bannister AJ, Schneider R, Kouzarides T. 2002. Histone methylation: dynamic or static? Cell 109:801–806 [DOI] [PubMed] [Google Scholar]
- 24. Hess JL. 2004. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 10:500–507 [DOI] [PubMed] [Google Scholar]
- 25. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13:713–719 [DOI] [PubMed] [Google Scholar]
- 26. Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. 2006. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 13:852–854 [DOI] [PubMed] [Google Scholar]
- 27. Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. 2004. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24:5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mishra BP, Ansari KI, Mandal SS. 2009. Dynamic association of MLL1, H3K4 trimethylation with chromatin and Hox gene expression during the cell cycle. FEBS J 276:1629–1640 [DOI] [PubMed] [Google Scholar]
- 29. Ansari KI, Kasiri S, Mandal SS. 27 August 2012. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene 10.1038/onc.2012.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ansari KI, Kasiri S, Mishra BP, Mandal SS. 2012. Mixed lineage leukaemia-4 regulates cell-cycle progression and cell viability and its depletion suppresses growth of xenografted tumour in vivo. Br J Cancer 107:315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ansari KI, Hussain I, Shrestha B, Kasiri S, Mandal SS. 2011. HOXC6 Is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J Mol Biol 411:334–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ansari KI, Shrestha B, Hussain I, Kasiri S, Mandal SS. 2011. Histone methylases MLL1 and MLL3 coordinate with estrogen receptors in estrogen-mediated HOXB9 expression. Biochemistry 50:3517–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dreijerink KM, Mulder KW, Winkler GS, Höppener JW, Lips CJ, Timmers HT. 2006. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res 66:4929–4935 [DOI] [PubMed] [Google Scholar]
- 34. Lee S, Lee DK, Dou Y, Lee J, Lee B, Kwak E, Kong YY, Lee SK, Roeder RG, Lee JW. 2006. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc Natl Acad Sci USA 103:15392–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mo R, Rao SM, Zhu YJ. 2006. Identification of the MLL2 complex as a coactivator for estrogen receptor α. J Biol Chem 281:15714–15720 [DOI] [PubMed] [Google Scholar]
- 36. Ansari KI, Hussain I, Kasiri S, Mandal SS. 2012. HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol 48:61–75 [DOI] [PubMed] [Google Scholar]
- 37. Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. 2011. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 183:1402–1410 [DOI] [PubMed] [Google Scholar]
- 38. Hamamori Y, Schneider MD. 2003. HATs off to Hop: recruitment of a class I histone deacetylase incriminates a novel transcriptional pathway that opposes cardiac hypertrophy. J Clin Invest 112:824–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, Geschwind DH, Gottesfeld JM, Pandolfo M. 2008. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One 3:e1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wadsack C, Hrzenjak A, Hammer A, Hirschmugl B, Levak-Frank S, Desoye G, Sattler W, Malle E. 2003. Trophoblast-like human choriocarcinoma cells serve as a suitable in vitro model for selective cholesteryl ester uptake from high density lipoproteins. Eur J Biochem 270:451–462 [DOI] [PubMed] [Google Scholar]
- 41. Singhal SS, Singhal J, Yadav S, Dwivedi S, Boor PJ, Awasthi YC, Awasthi S. 2007. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (Ral-binding protein 1). Cancer Res 67:4382–4389 [DOI] [PubMed] [Google Scholar]
- 42. Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ. 2009. Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. J Neurosci 29:3529–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu D, Bachmann KA. 1998. An investigation of the relationship between estrogen, estrogen metabolites and blood cholesterol levels in ovariectomized rats. J Pharmacol Exp Ther 286:561–568 [PubMed] [Google Scholar]
- 44. Sudhir K, Komesaroff PA. 1999. Clinical review 110: Cardiovascular actions of estrogens in men. J Clin Endocrinol Metab 84:3411–3415 [DOI] [PubMed] [Google Scholar]
- 45. Walsh BW, Schiff I, Rosner B, Greenberg L, Ravnikar V, Sacks FM. 1991. Effects of postmenopausal estrogen replacement on the concentrations and metabolism of plasma lipoproteins. N Engl J Med 325:1196–1204 [DOI] [PubMed] [Google Scholar]
- 46. Bagatell CJ, Knopp RH, Rivier JE, Bremner WJ. 1994. Physiological levels of estradiol stimulate plasma high density lipoprotein2 cholesterol levels in normal men. J Clin Endocrinol Metab 78:855–861 [DOI] [PubMed] [Google Scholar]
- 47. Shono N, Kumagai S, Higaki Y, Nishizumi M, Sasaki H. 1996. The relationships of testosterone, estradiol, dehydroepiandrosterone-sulfate and sex hormone-binding globulin to lipid and glucose metabolism in healthy men. J Atheroscler Thromb 3:45–51 [DOI] [PubMed] [Google Scholar]
- 48. Eacker SM, Agrawal N, Qian K, Dichek HL, Gong EY, Lee K, Braun RE. 2008. Hormonal regulation of testicular steroid and cholesterol homeostasis. Mol Endocrinol 22:623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. 2001. Requirement of Sp1 and estrogen receptor α interaction in 17β-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology 142:1546–1553 [DOI] [PubMed] [Google Scholar]
- 50. Lopez D, Sanchez MD, Shea-Eaton W, McLean MP. 2002. Estrogen activates the high-density lipoprotein receptor gene via binding to estrogen response elements and interaction with sterol regulatory element binding protein-1A. Endocrinology 143:2155–2168 [DOI] [PubMed] [Google Scholar]
- 51. Lindholm J, Winkel P, Brodthagen U, Gyntelberg F. 1982. Coronary risk factors and plasma sex hormones. Am J Med 73:648–651 [DOI] [PubMed] [Google Scholar]
- 52. Semmens J, Rouse I, Beilin LJ, Masarei JR. 1983. Relationship of plasma HDL-cholesterol to testosterone, estradiol, and sex-hormone-binding globulin levels in men and women. Metabolism 32:428–432 [DOI] [PubMed] [Google Scholar]
- 53. Ansari KI, Mandal SS. 2010. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J 277:1790–1804 [DOI] [PubMed] [Google Scholar]
- 54. McKenna NJ, Lanz RB, O'Malley BW. 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344 [DOI] [PubMed] [Google Scholar]
- 55. Hermanson O, Glass CK, Rosenfeld MG. 2002. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab 13:55–60 [DOI] [PubMed] [Google Scholar]
- 56. Mandal SS, Chu C, Wada T, Handa H, Shatkin AJ, Reinberg D. 2004. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc Natl Acad Sci USA 101:7572–7577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sims RJ, 3rd, Mandal SS, Reinberg D. 2004. Recent highlights of RNA-polymerase-II-mediated transcription. Curr Opin Cell Biol 16:263–271 [DOI] [PubMed] [Google Scholar]
- 58. Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. 1996. Nuclear receptor coactivators and corepressors. Mol Endocrinol 10:1167–1177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.