

Abstract
Atrial fibrillation (AF) is the most commonly encountered cardiac arrhythmia, and is a significant source of healthcare expenditures throughout the world. It is an arrhythmia with a very clearly defined predisposition for individuals of advanced age, and this fact has led to intense study of the mechanistic links between aging and AF. By promoting oxidative damage to multiple subcellular and cellular structures, reactive oxygen species (ROS) have been shown to induce the intra- and extra-cellular changes necessary to promote the pathogenesis of AF. In addition, the generation and accumulation of ROS have been intimately linked to the cellular processes which underlie aging. This review begins with an overview of AF pathophysiology, and introduces the critical structures which, when damaged, predispose an otherwise healthy atrium to AF. The available evidence that ROS can lead to damage of these critical structures is then reviewed. Finally, the evidence linking the process of aging to the pathogenesis of AF is discussed.
Keywords: Arrhythmia, Cardiac, Atrial fibrillation, Aging, Redox, Free radicals, Oxidative stress
1. Introduction
Atrial fibrillation (AF) is the most commonly encountered clinical cardiac arrhythmia, and recent data suggests the prevalence of this disease in Western societies is 1%-3% of adults over the age of 20.[1],[2] Individuals with AF are at increased risk of cardiovascular and cerebrovascular morbidity and mortality compared with individuals without this condition.[2],[3] As a result, AF contributes significantly to healthcare costs worldwide. The estimated yearly cost of treating patients with AF in the United States alone is estimated to be over six billion USD.[3] In addition, population studies of AF have consistently shown AF is skewed towards individuals over the age of 70.[4] In one study conducted in a random male population from the United Kingdom, AF prevalence was approximately 2.3% in individuals aged 65–74, but 8.3% in individuals aged 75–84.[2] This consistently observed correlation between age and AF prevalence has led some researchers to look for a common link between the mechanisms of aging and the mechanisms underlying the pathophysiology of AF. One likely link is the formation of reactive oxygen species (ROS) and the subsequent disruption of cellular redox balance, as increased tissue ROS appear to be positively correlated with increasing age.[5]
This review begins with a description of AF pathophysiology and describes the critical cellular and subcellular components involved in this disease. It then discusses current evidence regarding the susceptibility of these components to oxidative damage mediated by ROS, and the role this process may play in AF. Finally, the limited available evidence that age alone predisposes individuals to AF through ROS-mediated mechanisms will be discussed.
2. AF pathophysiology
The pathophysiology of AF is complex and, in the most general terms, involves three distinct mechanisms. These mechanisms are ectopic firing, atrial electrical remodeling and atrial structural remodeling.[6]–[8]
Ectopic firing, or ectopic depolarization of tissue, is the process underlying the initiation of AF “triggers”. Ectopic firing may be caused by proarrhythmic mechanisms such as enhanced automaticity, early after-depolarizations (EADs) or delayed after-depolarizations (DADs) (Figure 1).[9] Enhanced automaticity occurs in cardiomyocytes when the inward rectifier potassium current in these cells is reduced, usually through compromised structure or function of the membrane-bound voltage-independent inward-rectifier potassium channel. This unmasks pre-existing pacemaker currents in cardiomyocytes, allowing sodium channel-mediated membrane depolarization to occur during diastole.[10],[11] EADs, similarly, may be caused by prolongation of action potential duration (APD) that occurs as a result of reduced voltage-dependent delayed rectifier potassium channel function or reactivation of voltage-dependent sodium or L-type calcium channels during diastole.[12] Finally, DADs are caused by diastolic cytoplasmic calcium leak due to dysfunction in such mediators of intracellular calcium homeostasis as the sarcoplasmic reticulum calcium-release channel (ryanodine receptor) or the sarcoplasmic reticulum calcium-uptake pump (SERCA).[13],[14] Intracellular calcium leak induces a transient inward current that activates diastolic sodium channel-mediated membrane depolarization. Despite mechanistic differences, enhanced automaticity, EADs, and DADs all induce premature, unexpected diastolic membrane depolarization which causes AF when coupled to a vulnerable atrial substrate.
Figure 1. Mechanisms of ectopic firing in atrial fibrillation.
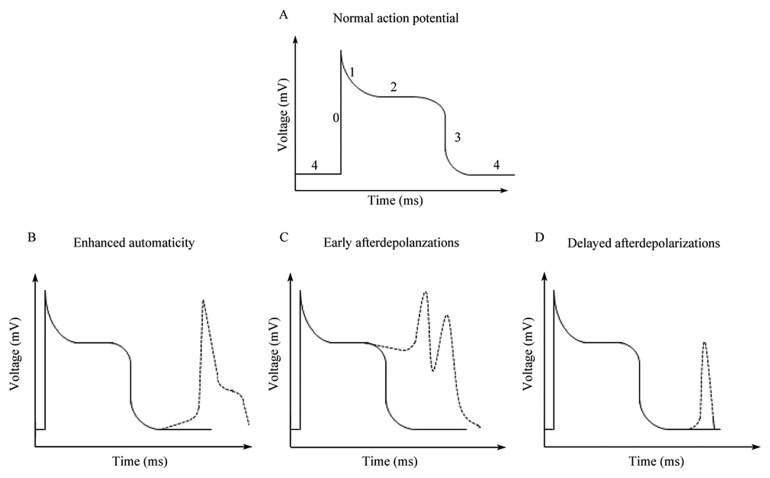
(A): A normal atrial myocyte action potential has 5 phases, labeled 0–4; (B): Enhanced automaticity occurs when unmasked pacemaker currents lead to spontaneous depolarization in phase 4; (C): Early afterdepolarizations are associated with reactivation of voltage-dependent sodium channels or L-type calcium channels in phase 2 of the action potential; (D): Late afterdepolarizations are caused by dysfunctional intracellular calcium handling that results in depolarization during phase 4 of the action potential.
As important as the mechanisms underlying AF triggers are the cells that have been implicated as the source of ectopic firing in patients with AF. Recent studies suggest several unique cell populations exist in the atrium and pulmonary vein sleeves that may contribute to AF triggers. These different cells types include periodic acid-schiff (PAS) positive sinus node-like cells,[15],[16] interstitial Cajal-like cells or cardiac telocytes,[17],[18] pulmonary vein cardiomyocytes (PVCMs),[19]–[21] and cardiac melanocyte-like cells (CMLCs) (Table 1).[22]
Table 1. Cell-types implicated in promoting atrial fibrillation triggers (references in brackets).
| Pulmonary vein cardiomyocytes | Myocardial telocytes | Sinus node-like PAS-positive cells | Cardiac melanocyte-like cells | |
| Anatomic location | Pulmonary vein sleeves | Greater number in atrium vs. ventricle; subepicardial and mid-myocardial (28 ) | Pulmonary vein sleeves; Pulmonary vein-LA junction; right atrial appendage (16) | Pulmonary vein os; posterior atrial wall; mitral and tricuspid annulus; intra-atrial septum (22) |
| Histology | Striated cells that are rod-like or bifurcated (20) | Oval somas with multiple, long, interstitial dendrites; c-kit+ (28) | Pale cytoplasm with PAS-positive staining (15) | Bipolar dendritic morphology; pigmented in murine hearts; c-kit+ (22) |
| Cellular EP characteristics | APD shorter than atrial myocytes; spontaneous and inducible automaticity and triggered activity (19) | Not directly assessed | Not directly assessed | Atrial myocyte-like action potentials; spontaneous and inducible afterdepolarizations (22) |
| Stimuli that promote ectopy | Ryanodine; muscarinic and beta-adrenergic agonists (19); ouabain (21) | Not assessed | Ryanodine and beta-adrenergic agonists (16) | Muscarinic and beta-adrenergic agonists (22) |
APD: action potential duration; EP: electrophysiology; PAS: Periodic acid-Schiff; LA: left atrium.
The pulmonary venous myocardium (PVM) creates a sleeve of developmentally distinct muscle fibers which project from the left atrium to cover the distal portion of the pulmonary veins.[23] Electrical activity originating in the PVM was shown to play an important role in human AF in seminal work conducted and published by Haissaguerre and co-workers in 1998.[24] While heterogenous in terms of its cellular composition, the predominant cell-type in this tissue is the pulmonary vein (PV) cardiomyocyte. Because of their postulated role in AF, PV cardiomyocytes have been extensively studied, and it is clear from these investigations that the electrophysiological properties of PVCMs differ markedly from those of left atrial cardiomyocytes. In 2002, Chen et al.[25] demonstrated the presence of cardiomyocytes with spontaneous pacemaker activity, or enhanced automaticity, in rabbit PVM. This automatic activity was subsequently correlated with reduced inward rectifier potassium currents in these cells.[25] Chen et al.[25] also demonstrated isoproterenol-induced after-depolarizations linked to intracellular calcium flux in isolated rabbit PVM preparations. In a separate set of experiments, Honjo et al.[26] demonstrated that pretreatment of isolated rabbit PV cardiomyocytes with ryanodine, followed by rapid pacing, was sufficient to induce spontaneous membrane depolarizations in these cells. Given the role of ryanodine in increasing the cytoplasmic pool of calcium, these results implicate the induction of depolarizing plasma membrane currents, likely through activation of the sodium-calcium exchanger, in the pathogenesis of spontaneous electrical activity in PVCMs. More recently, a direct comparison between the electrophysiology of PVCMs and left atrial cardiomyocytes was performed by Ehrlich et al.[27] These studies demonstrated that PVCMs have a more positive resting membrane potential, shorter APD, and smaller voltage-independent inward rectifier potassium current when compared to cardiomyocytes from the left atrium.[27] Taken as a whole, results from these groups support the presence of an ionic channel complement capable of serving as a substrate for ectopic activity from the cardiomyocytes of the PVM.
With regards to the existence of other cell types that may contribute to AF triggers, Perez-Lugones et al.[15] identified sinus node-like cells within human pulmonary veins that were associated with AF. These cells were identified by their pale cytoplasm and positive response to PAS staining within human cadaveric PV samples. Based upon the attached clinical histories for each sample, the authors found that samples from four patients with a history of AF had sinus node-like cells in the pulmonary veins. However, no such cells could be detected in the pulmonary veins of five hearts from patients without AF following transplantation. While the authors of this study were clear that the connection of sinus-node like cells to clinical atrial arrhythmias required further investingation, studies in canine models have provided some evidence for the contribution of PAS-positive cells in the PVs to atrial arrhythmias. In a study by Chuo et al.,[16] the authors induced voltage-independent triggered ectopy, by applying ryanodine and isoproterenol, which induced atrial arrhythmias in Langendorf-perfused canine hearts. The authors then identified PAS-positive cells clustered in the endocardium of the PV muscular sleeves from one preparation near the region from which they mapped frequent focal discharges, while few PAS-positive cells could be found in another preparation from a heart with no focal discharges.
More recently, a few studies have demonstrated the presence of cardiac telocytes within the muscular sleeves of PVs.[17],[18],[28] Morel et al.[17] described a rough correlation between cardiac telocytes in the PVs and clinical AF, where two of three patients with detectable PV cardiac telocytes had documented AF. The report by Gherghiceanu et al.[18] described the presence of cardaic telocytes in human PVs with no analysis of their connection to arrhythmia. However, neither of these studies characterized the electrophysiological properties of cardiac telocytes or explored mechanisms by which they may contribute to atrial arrhythmias. This is partially due to the fact Interstitial Cells of Cajal (ICCs) are well characterized within enteric smooth muscle and known to mediate gut motility in response to autonomic efflux.[29]–[31] Therefore, the assumption that cardiac telocytes within human PVs probably exhibit similar electrophysiologic behavior leading to PV ectopy is reasonable. However, in the gut, ICCs mediate very slow electrical responses (on the order of seconds), which are too sluggish to explain the rapid response time of PV ectopy that triggers AF (which is on the scale of milliseconds). While the contribution of cardiac telocytes to atrial arrhythmias will require further validation, it is intriguing to consider the possibility that cardiac telocytes in the heart may contribute to atrial arrhythmias either as a source of triggered activity (as they appear to in the intestines), or that they may increase arrhythmogenicity by contributing to zones of slowed conduction and promoting local reentry. In either case, these reports highlight the potential contribution of non-cardiomyocytes to atrial arrhythmogenesis, and open the way for exploring the contribution of other cell populations to atrial arrhythmias.
Figure 2. Role of ROS in the pathogenesis of atrial fibrillation(references in brackets).
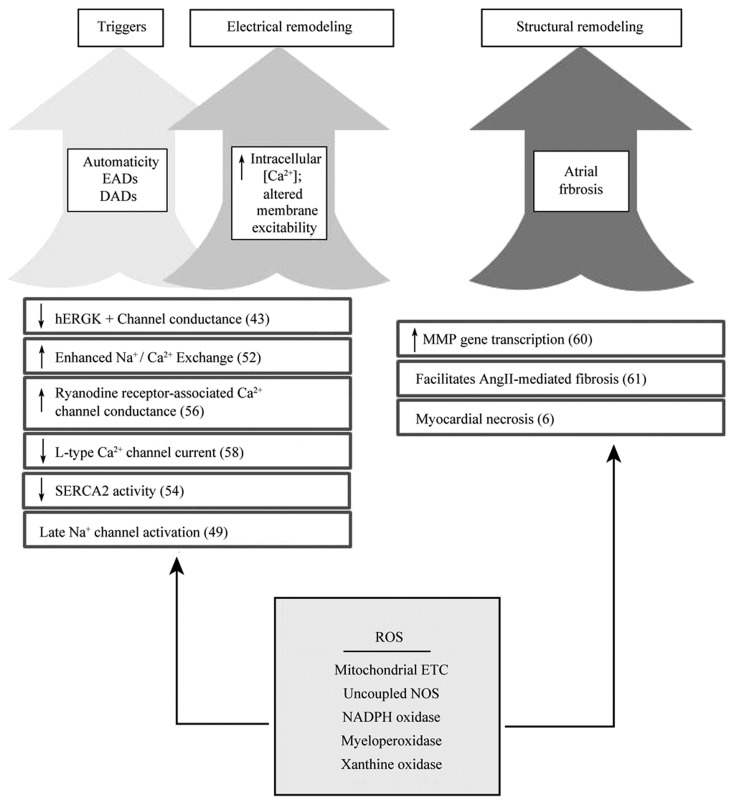
ROS-mediated damage impacts multiple cellular and subcellular structures which promotes AF pathogenesis. References are in parenthesis. DADs: delayed afterdepolarizations; EADs: early afterdepolarizations; ETC: electron transport chain; hERG; human ether-a-go-go related gene; MMP: matrix metalloproteinases; NOS: nitric oxide synthase; ROS: reactive oxygen species; SERCA2: Sarcoplasmic reticulum calcium-ATPase, type2.
Less extensively studied, but potentially as interesting as these other cell populations, are the CMLCs. These cells, first described by our laboratory in mice and later shown to be present in human hearts, are found near the pulmonary vein ostia and in the posterior walls of both cardiac atria. CMLCs express markers of the melanocyte lineage but are not derived from neural crest, making their origin unclear at this time. They also express adrenergic and muscarinic receptors, and are usually found in close proximity to autonomic nerve terminals. The electrophysiologic properties of CMLCs are of particular interest, and we have shown these cells display cardiomyocyte-like excitability, and the ability to electrically couple with cardiomyocytes. The proximity of these cells to autonomic innervation in the heart which corresponds to the anatomic sites that most often give rise to clinical AF triggers, their complement of adrenergic and muscarinic receptors, and their electrical excitability make them excellent candidates for sources of ectopic firing in AF.[22]
While AF triggers play an integral role in arrhythmia initiation, a right and left atrial substrate capable of supporting AF wavefronts is also essential to promote atrial fibrilation.[6],[7],[9] One process involved in the generation of this substrate is called “electrical remodeling” and involves alterations in ion channel currents on a subcellular scale. Electrical remodeling involves the activation of compensatory mechanisms designed to protect atrial cardiomyocytes from the increased cytoplasmic calcium concentrations that occur as a result of tachyarrhythmias.[32] Part of the electrical remodeling process provoked by AF includes augmenting voltage-dependent inward rectifier potassium currents and the inactivation of calcium currents to reduce atrial cardiomyocyte APD. As a consequence of these ionic changes, the shortening of atrial cardiomyocyte APD may facilitate micro-reentry necessary to perpetuate AF.[33] Simultaneously, changes in global calcium handling predispose atrial cardiomyocytes to DADs as described above. Together, the reduced APDs and propensity for calcium-induced ectopic firing provide a substrate which supports and perpetuates AF wavefronts.
Working in concert with atrial electrical remodeling to produce a vulnerable substrate for AF, is the process of “structural remodeling”. In contrast to electrical remodeling, structural remodeling occurs on a macroscopic scale and largely involves interstitial fibrosis.[34] The intercalation of collagen bundles between atrial cardiomyocytes impairs electrical continuity between these cells, and creates regions of slow and discoordinated conduction. At the same time, the fibroblasts which synthesize this collagen matrix can electrically interact with atrial cardiomyocytes to produce ectopic electrical activity.[35] The end result of these changes is an alteration in atrial tissue composition such that the functional reentry necessary to support AF can now occur.[34]
3. ROS and AF
Given its high metabolic profile, the heart bears a large burden of exposure to ROS. Sources of ROS in the atrium include the byproducts of endogenous and exogenous redox systems such as NADPH oxidase, xanthine oxidase, the mitochondrial electron transport chain, myeloperoxidase (MPO), and uncoupled nitric oxide synthase.[36]
NADPH oxidase and the mitochondrial electron transport chain (mETC) are major sources of superoxide anion in the heart, a highly reactive and unstable ROS. Both NADPH oxidase and the mETC have been shown in humans and animal models to play a role in AF pathophysiology. Using a tachypacing goat model, Reilly et al.[37] demonstrated an NADPH oxidase-dependent increase in atrial superoxide content after only two weeks of persistent AF. After 6 months of persistent AF, this increase in tissue superoxide anion was still present, but was shown to now be dependent on the mETC. Human studies conducted by Kim et al.[38] demonstrated a significant increase in superoxide production in tissue samples taken from the right atrium of patients with AF compared to those taken from patients in sinus rhythm. Importantly, these increases in superoxide anion were correlated with increased NADPH oxidase function. Taken together, these results suggest an important role for endogenous superoxide anion production in the pathophysiology of AF.
Uncoupled nitric oxide synthase (NOS), and the reactive species nitric oxide (NO) which is a byproduct of NOS activity, have also been implicated in AF pathogenesis. Three isoforms of NOS exist that are distributed in varying proportions within the cells of the atrium including atrial cardiomyocytes, macrophages, neutrophils, and endothelial cells.[36] In an uncoupled state, NOS enzymatic activity produces superoxide anion. As with NADPH oxidase, uncoupled NOS-mediated production of superoxide anion has been implicated in AF pathophysiology. Induction of NOS isoform 2 (NOS2) has been reported in the atrium of a canine tachypacing heart failure model. These changes in NOS2 expression correlated with decreased atrial effective refractory periods and an increase in inducible AF.[39] In a similar fashion, increases in atrial NOS expression were observed in a canine model of AF induced by seven days of tachypacing.[40] While these experiments suggest an important correlation exists between increased NOS2 expression and AF, they did not establish a causative link or identify the actual cell-type responsible for increased atrial NO production.
MPO is a unique source of ROS in the heart, as it is a bactericidal enzyme released from activated polymorphonuclear leukocytes (PMNs). Extracellular MPO generates hypochlorous acid, a ROS which activates multiple intracellular signaling cascades and matrix metalloproteinases (MMPs). Functional MPO was shown to be necessary for generation of an angiotensin II-mediated mouse model of AF.[41] MPO has also been implicated in human AF, as a correlation was found between early recurrence of AF following radiofrequency ablation and increased plasma MPO levels.[42] Increased plasma levels of MPO have also been described in patients with paroxysmal AF.[41]
With regard to the mechanisms by which ROS may cause AF, a number of experiments have demonstrated the critical subcellular and cellular components of the atria and PVM which promote AF are susceptible to ROS-mediated damage. For instance, ROS is known to affect the function of both potassium and calcium channels.[43]–[46] Exposure of isolated cells to oxidative stress has also been shown to induce ectopic firing due to enhanced automaticity, EADs, and DADs.[47]–[50] In an attempt to correlate ROS exposure to PV triggers, Lin et al.[51] exposed isolated rabbit PVs to hydrogen peroxide and recorded action potentials from this tissue. As expected, with ROS exposure a significant increase in spontaneous depolarizations and EADs was observed from the PVs. Similarly, our lab has observed increases in CMLC APD with exposure of these cells to hydrogen peroxide in vitro (data not published). We believe that this increase in CMLC APD in response to ROS exposure predisposes these cells to EADs and DADs. Taken as a whole, these results from both groups strongly suggest a role for ROS-mediated ionic dysfunction as a cause of enhanced automaticity, EADs, and DADs that generate the ectopic firing associated with AF triggers.
ROS are likely to play an integral role in atrial electrical and structural remodeling that underlie AF as well. In experimental systems, exposure to ROS significantly alters calcium handling in cardiomyoctyes through effects on the L-type calcium current, ryanodine receptor, the cardiac sarcoplasmic reticulum calcium-uptake pump (SERCA2), and the Na/Ca exchanger.[52]–[59] In addition, exposure of isolated rabbit left atrium to hydrogen peroxide has been shown to decrease left atrial APD.[51] This shortening in left atrial APD, which may be related to alterations in calcium handling, is the hallmark of atrial electrical remodeling in AF.
ROS are also likely to exert a direct influence on the expression and secretion of MMPs in the heart.[35] In vitro, elevated superoxide production in cardiomyocytes has been shown to result in increased MMP gene transcription through mitogen-activated protein kinase pathways.[60] Furthermore, in vivo studies of reactive species in cardiac fibrosis have confirmed these in vitro findings. Zhao et al.[61] have demonstrated that the deposition of collagen in an angiotensin II-induced rat model of cardiac fibrosis was dependent on superoxide anion production. Perhaps more interestingly, in vivo mouse studies conducted by Rudolph et al.[41] demonstrated a requirement for functional MPO in the generation of angiotensin II-mediated atrial fibrosis. This unique finding implicated PMN-derived ROS as a facilitator of cardiac fibrosis. These observations have added to a growing body of evidence linking ROS to not only atrial electrical remodeling but also atrial structural remodeling in AF.
4. Aging, ROS and AF
Aging is a complex multi-factorial process characterized by a progressive decline in mitochondrial function. On a cellular level, mitochondrial dysfunction results in the accumulation of ROS, as the respiratory and electron transport capacity of this organelle is compromised.[62] Invariably, accumulation of reactive species leads to redox imbalance, and an oxidative environment which causes the functional decline of both cells and tissues.[63] Evidence of oxidative damage to critical cellular structures as a result of redox imbalance has been demonstrated in multiple different tissues including the heart, and it is conceivable that ROS-mediated damage in the PVM and atrium may serve as a mechanism which promotes AF in the elderly.[64]
In an attempt to elucidate a role for aging in the development of AF triggers, Wongcharoen et al.[65] evaluated the electrophysiologic properties of PVs isolated from both aged (3 year-old) and young (3 month-old) rabbits. Pulmonary vein preparations from aged rabbits consistently demonstrated elevated resting membrane potentials and larger amplitude afterdepolarizations when compared to PV preparations from young rabbits. These differences in afterdepolarization amplitude were linked to abnormal intracellular calcium handling, as aged PVs had significantly increased expression of ryanodine receptor and Na/Ca exchanger protein.[65] These results suggest a relationship between the aging process and the pathogenesis of AF triggers, but they do not establish a direct mechanistic link between ROS generated through the aging process and the observed ionic alterations.
Antioxidant therapies, employed specifically to reduce ROS effects on the atria, have been studied in both humans and animal models. In a Wistar rat model of aging, Xu et al.[66] demonstrated a significant reduction in age-related atrial fibrosis and inducible AF through treatment with the PPAR-gamma activator pioglitazone. Furthermore, these results supported the notion that blocking age-induced ROS could prevent atrial changes supporting AF, as treatment with pioglitazone was shown to reverse an aging-induced decrease in tissue free radical reduction in this model. While encouraging, these results must be interpreted carefully. An important limitation of this animal model is the fact that the type of atrial fibrosis observed in aged Wistar rats is not seen in the atria of aging humans devoid of other co-morbidities.[67] Still, human studies of antioxidant therapy for AF have shown some promise. A recent meta-analysis of studies examining prophylactic administration of the antioxidant N-acetylcysteine, to cardiac surgery patients revealed that pretreatment with this compound could significantly decrease the incidence of post-operative AF.[68] Similar results have been obtained with 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors such as atorvastatin, and abrogation of inflammatory pathways leading to ROS generation have been postulated to be the way in which these drugs reduce post-operative AF.[69] As with results from animal models, these studies warrant careful interpretation. While they appear to support the hypothesis that restoring tissue redox balance can help prevent AF, the studies look at redox imbalance that involves a large component of inflammatory mediators present because of the post-operative state. One would not expect such a large concentration of tissue ROS through the normal process of aging.
With respect to atrial remodeling, very little experimental evidence exists linking the process of aging to the electrical and structural remodeling that generates a vulnerable atrial substrate for AF. Results from animal models have suggested that aging alone is associated with the left atrial cardio myocyte APD shortening commonly associated with atrial electrical remodeling seen in AF.[70] However, no evidence currently exists to suggest that a change in APD in humans occurs with aging. Quite to the contrary, by studying right atrial monophasic action potentials and effective refractory periods in patients from four separate age groups (25 to 64 years-old), Brorson et al.[71] demonstrated no correlation between age and APD. Similarly, with respect to atrial structural remodeling, current evidence suggests human atrial fibrosis does not occur in the absence of other precipitating factors, such as AF. An evaluation of post-mortem histology from human atria performed by Platonov et al.[67] demonstrated a lack of any correlation between age and the presence of atrial fibrosis. Taken as a whole, these results suggest that significant atrial remodeling necessary to generate a cardiac substrate capable of supporting AF is unlikely to be a consequence of aging alone. Rather, these changes are likely to occur secondary to other insults to the atria, such as volume overload from mitral valve regurgitation or as a consequence of AF itself.
5. Conclusions
As our current understanding of the mechanisms that promote and support AF continue to develop, this cardiac arrhythmia appears to more closely resemble a disorder acquired through a combination of exposures and comorbidities, rather than through purely genetic disposition. To be certain, through serving as agents of oxidative cellular damage, ROS are likely to be a major causative factor in the development of AF. As described above, the cellular changes brought about by ROS-mediated damage are sufficient to promote tissue changes consistent with AF triggers, and atrial electrical and structural remodeling. However, what is not immediately apparent is the threshold above which the heart is no longer capable of withstanding oxidative insult, or the extent to which the normal process of aging can generate oxidative damage that exceeds this limit. The role of genetics in this process must also be considered as there is a 2-3 fold increase in the risk of developing AF when at least one parent is afflicted with the disease.[72],[73] Furthermore, recent Genome-Wide Association Studies (GWAS) have demonstrated strong associations between human genomic loci and a predisposition for the development of AF.[74] Whether the genes which reside at these loci are important for modulating the effects of ROS on the heart is currently unknown, but will likely be determined in the future.
Determining what impact, if any, aging has on the pathogenesis of AF also has implications for the treatment of this disease. Appropriately, current AHA/ACCF/HRS guidelines do not address the use of antioxidants for treating AF in a preventative fashion.[75] It goes without saying that a well-designed prospective study of the effectiveness of prophylactic antioxidant therapy in preventing AF will be required prior to any formal recommendations for their use in this capacity. However, even prior to clinical trials, a much better understanding of the intersection between AF pathophysiology and aging mechanisms will need to be outlined. Only then will logical therapeutic targets, with the potential to provide more benefit than detriment throughout the life of an individual, actually be defined.
References
- 1.Naccarelli GV, Varker H, Lin J, et al. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol. 2009;104:1534–1539. doi: 10.1016/j.amjcard.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Davis RC, Hobbs FDR, Kenkre JE, et al. Prevalence of atrial fibrillation in the general population and in high-risk groups: the ECHOES study. Europace. 2012 doi: 10.1093/europace/eus087. [Epub ahead of print]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22490371. [DOI] [PubMed] [Google Scholar]
- 3.Kim MH, Johnston SS, Chu BC, et al. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovasc Qual Outcomes. 2011;4:313–320. doi: 10.1161/CIRCOUTCOMES.110.958165. [DOI] [PubMed] [Google Scholar]
- 4.Kannel WB, Wolf PA, Benjamin EJ, et al. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82(8A):2N–9N. doi: 10.1016/s0002-9149(98)00583-9. [DOI] [PubMed] [Google Scholar]
- 5.Jackson MJ, McArdle A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol (Lond) 2011;589(Pt 9):2139–2145. doi: 10.1113/jphysiol.2011.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 7.Patel VV. Novel insights into the cellular basis of atrial fibrilation. Expert Rev Cardiovasc Ther. 2010;8:907–916. doi: 10.1586/erc.10.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 9.Iwasaki Y, Nishida K, Kato T, et al. Atrial fibrillation pathophysiology: implications for management. Circulation. 2011;124:2264–2274. doi: 10.1161/CIRCULATIONAHA.111.019893. [DOI] [PubMed] [Google Scholar]
- 10.Stillitano F, Lonardo G, Zicha S, et al. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Dhamoon AS, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. 2005;2:316–324. doi: 10.1016/j.hrthm.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Z, Xie Y, Wen H, et al. Role of the transient outward potassium current in the genesis of early afterdepolarizations in cardiac cells. Cardiovasc Res. 2012;95:308–316. doi: 10.1093/cvr/cvs183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voigt N, Li N, Wang Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fink M, Noble PJ, Noble D. Ca2+-induced delayed afterdepolarizations are triggered by dyadic subspace Ca22+ affirming that increasing SERCA reduces aftercontractions. Am J Physiol Heart Circ Physiol. 2011;301:921–935. doi: 10.1152/ajpheart.01055.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Lugones A, McMahon JT, Ratliff NB, et al. Evidence of specialized conduction cells in human pulmonary veins of patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:803–809. doi: 10.1046/j.1540-8167.2003.03075.x. [DOI] [PubMed] [Google Scholar]
- 16.Chou CC, Nihei M, Zhou S, et al. Intracellular calcium dynamics and anisotropic reentry in isolated canine pulmonary veins and left atrium. Circulation. 2005;111:2889–2897. doi: 10.1161/CIRCULATIONAHA.104.498758. [DOI] [PubMed] [Google Scholar]
- 17.Morel E, Meyronet D, Thivolet-Bejuy F, et al. Identification and distribution of interstitial Cajal cells in human pulmonary veins. Heart Rhythm. 2008;5:1063–1067. doi: 10.1016/j.hrthm.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 18.Gherghiceanu M, Hinescu ME, Andrei F, et al. Interstitial Cajal-like cells (ICLC) in myocardial sleeves of human pulmonary veins. J Cell Mol Med. 2008;12:1777–1781. doi: 10.1111/j.1582-4934.2008.00444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YJ, Chen SA. Electrophysiology of pulmonary veins. J Cardiovasc Electrophysiol. 2006;17:220–224. doi: 10.1111/j.1540-8167.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 20.Yu MC, Huang CF, Chang CM, et al. Diverse cell morphology and intracellular calcium dynamics in pulmonary vein cardiomyocytes. Heart Vessels. 2011;26:101–110. doi: 10.1007/s00380-010-0035-y. [DOI] [PubMed] [Google Scholar]
- 21.Wongcharoen W, Chen YC, Chen YJ, et al. Effects of a Na+/Ca2+ exchanger inhibitor on pulmonary vein electrical activity and ouabain-induced arrhythmogenicity. Cardiovasc Res. 2006;70:497–508. doi: 10.1016/j.cardiores.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 22.Levin MD, Lu MM, Petrenko NB, et al. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest. 2009;119:3420–3436. doi: 10.1172/JCI39109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mommersteeg MTM, Christoffels VM, Anderson RH, et al. Atrial fibrillation: a developmental point of view. Heart Rhythm. 2009;6:1818–1824. doi: 10.1016/j.hrthm.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Haïssaguerre M, Jaïs P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 25.Chen YJ, Chen SA, Chen YC, et al. Electrophysiology of single cardiomyocytes isolated from rabbit pulmonary veins: implication in initiation of focal atrial fibrillation. Basic Res Cardiol. 2002;97:26–34. doi: 10.1007/s395-002-8384-6. [DOI] [PubMed] [Google Scholar]
- 26.Honjo H, Boyett MR, Niwa R, et al. Pacing-induced spontaneous activity in myocardial sleeves of pulmonary veins after treatment with ryanodine. Circulation. 2003;107:1937–1943. doi: 10.1161/01.CIR.0000062645.38670.BD. [DOI] [PubMed] [Google Scholar]
- 27.Ehrlich JR, Cha TJ, Zhang L, et al. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol (Lond) 2003;551(Pt 3):801–813. doi: 10.1113/jphysiol.2003.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostin S. Myocardial telocytes: a specific new cellular entity. J Cell Mol Med. 2010;14:1917–1921. doi: 10.1111/j.1582-4934.2010.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillock RJ, Kalman JM, Roberts-Thomson KC, et al. Multiple focal atrial tachycardias in a healthy adult population: characterization and description of successful radiofrequency ablation. Heart Rhythm. 2007;4:435–438. doi: 10.1016/j.hrthm.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 30.Kistler PM, Sanders P, Hussin A, et al. Focal atrial tachycardia arising from the mitral annulus: electrocardiographic and electrophysiologic characterization. J Am Coll Cardiol. 2003;41:2212–2219. doi: 10.1016/s0735-1097(03)00484-4. [DOI] [PubMed] [Google Scholar]
- 31.Hirst GDS, Edwards FR. Electrical events underlying organized myogenic contractions of the guinea pig stomach. J Physiol (Lond) 2006;576(Pt 3):659–665. doi: 10.1113/jphysiol.2006.116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greiser M, Lederer WJ, Schotten U. Alterations of atrial Ca (2+) handling as cause and consequence of atrial fibrillation. Cardiovasc Res. 2011;89:722–733. doi: 10.1093/cvr/cvq389. [DOI] [PubMed] [Google Scholar]
- 33.Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–1877. doi: 10.1093/eurheartj/ehs079. [DOI] [PubMed] [Google Scholar]
- 34.Goudis CA, Kallergis EM, Vardas PE. Extracellular matrix alterations in the atria: insights into the mechanisms and perpetuation of atrial fibrillation. Europace. 2012;14:623–630. doi: 10.1093/europace/eur398. [DOI] [PubMed] [Google Scholar]
- 35.Friedrichs K, Baldus S, Klinke A. Fibrosis in atrial fibrillation-Role of reactive species and MPO. Front Physiol. 2012;3:214. doi: 10.3389/fphys.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonilla IM, Sridhar A, Györke S, et al. Nitric oxide synthases and atrial fibrillation. Front Physiol. 2012;3:105. doi: 10.3389/fphys.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reilly SN, Jayaram R, Nahar K, et al. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2011;124:1107–1117. doi: 10.1161/CIRCULATIONAHA.111.029223. [DOI] [PubMed] [Google Scholar]
- 38.Kim YM, Guzik TJ, Zhang YH, et al. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–636. doi: 10.1161/01.RES.0000183735.09871.61. [DOI] [PubMed] [Google Scholar]
- 39.Nishijima Y, Sridhar A, Bonilla I, et al. Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc Res. 2011;91:71–79. doi: 10.1093/cvr/cvr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiroshita-Takeshita A, Brundel BJJM, Lavoie J, et al. Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc Res. 2006;69:865–875. doi: 10.1016/j.cardiores.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 41.Rudolph V, Andrié RP, Rudolph TK, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med. 2010;16:470–474. doi: 10.1038/nm.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter B, Gwechenberger M, Socas A, et al. Markers of oxidative stress after ablation of atrial fibrillation are associated with inflammation, delivered radiofrequency energy and early recurrence of atrial fibrillation. Clin Res Cardiol. 2012;101:217–225. doi: 10.1007/s00392-011-0383-3. [DOI] [PubMed] [Google Scholar]
- 43.Kolbe K, Schönherr R, Gessner G, et al. Cysteine 723 in the C-linker segment confers oxidative inhibition of hERG1 potassium channels. J Physiol (Lond) 2010;588(Pt 16):2999–3009. doi: 10.1113/jphysiol.2010.192468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Takeda S, Mochizuki S, et al. Mechanisms of hydrogen peroxide-induced increase in intracellular calcium in cardiomyocytes. J Cardiovasc Pharmacol Ther. 1999;4:41–48. doi: 10.1177/107424849900400107. [DOI] [PubMed] [Google Scholar]
- 45.Van Wagoner DR. Redox modulation of cardiac electrical activity. J Cardiovasc Electrophysiol. 2001;12:183–184. doi: 10.1046/j.1540-8167.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- 46.Bérubé J, Caouette D, Daleau P. Hydrogen peroxide modifies the kinetics of HERG channel expressed in a mammalian cell line. J Pharmacol Exp Ther. 2001;297:96–102. [PubMed] [Google Scholar]
- 47.Corretti MC, Koretsune Y, Kusuoka H, et al. Glycolytic inhibition and calcium overload as consequences of exogenously generated free radicals in rabbit hearts. J Clin Invest. 1991;88:1014–1025. doi: 10.1172/JCI115361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol (Lond) 1997;500(Pt 3):631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song Y, Shryock JC, Wagner S, et al. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 50.Barrington PL, Meier CF, Jr, Weglicki WB. Abnormal electrical activity induced by H2O2 in isolated canine myocytes. Basic Life Sci. 1988;49:927–932. doi: 10.1007/978-1-4684-5568-7_151. [DOI] [PubMed] [Google Scholar]
- 51.Lin YK, Lin FZ, Chen YC, et al. Oxidative stress on pulmonary vein and left atrium arrhythmogenesis. Circ J. 2010;74:1547–1556. doi: 10.1253/circj.cj-09-0999. [DOI] [PubMed] [Google Scholar]
- 52.Goldhaber JI. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol. 1996;271(3 Pt):H823–H833. doi: 10.1152/ajpheart.1996.271.3.H823. [DOI] [PubMed] [Google Scholar]
- 53.Hinata M, Matsuoka I, Iwamoto T, et al. Mechanism of Na+/Ca2+ exchanger activation by hydrogen peroxide in guineapig ventricular myocytes. J Pharmacol Sci. 2007;103:283–292. doi: 10.1254/jphs.fp0060015. [DOI] [PubMed] [Google Scholar]
- 54.Morris TE, Sulakhe PV. Sarcoplasmic reticulum Ca(2+)-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. . Free Radic. Biol. Med. 1997;22(1-2):37–47. doi: 10.1016/s0891-5849(96)00238-9. [DOI] [PubMed] [Google Scholar]
- 55.Zissimopoulos S, Docrat N, Lai FA. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem. 2007;282:6976–6983. doi: 10.1074/jbc.M607590200. [DOI] [PubMed] [Google Scholar]
- 56.Anzai K, Ogawa K, Ozawa T, et al. Oxidative modification of ion channel activity of ryanodine receptor. Antioxid. Redox Signal. 2000;2:35–40. doi: 10.1089/ars.2000.2.1-35. [DOI] [PubMed] [Google Scholar]
- 57.Goldhaber JI, Liu E. Excitation-contraction coupling in single guinea-pig ventricular myocytes exposed to hydrogen peroxide. J Physiol (Lond) 1994;477(Pt 1):135–147. doi: 10.1113/jphysiol.1994.sp020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudasek K, Brown ST, Fearon IM. H2O2 regulates recombinant Ca2+ channel alpha1C subunits but does not mediate their sensitivity to acute hypoxia. Biochem Biophys Res Commun. 2004;318:135–141. doi: 10.1016/j.bbrc.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 59.Guo J, Giles WR, Ward CA. Effect of hydrogen peroxide on the membrane currents of sinoatrial node cells from rabbit heart. Am J Physiol Heart Circ Physiol. 2000;279:H992–H999. doi: 10.1152/ajpheart.2000.279.3.H992. [DOI] [PubMed] [Google Scholar]
- 60.Spallarossa P, Altieri P, Garibaldi S, et al. Matrix metalloproteinase-2 and-9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc Res. 2006;69:736–745. doi: 10.1016/j.cardiores.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 61.Zhao W, Zhao T, Chen Y, et al. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 62.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 63.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 64.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wongcharoen W, Chen YC, Chen YJ, et al. Aging increases pulmonary veins arrhythmogenesis and susceptibility to calcium regulation agents. Heart Rhythm. 2007;4:1338–1349. doi: 10.1016/j.hrthm.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 66.Xu D, Murakoshi N, Igarashi M, et al. PPAR-γ activator pioglitazone prevents age-related atrial fibrillation susceptibility by improving antioxidant capacity and reducing apoptosis in a rat model. J Cardiovasc Electrophysiol. 2012;23:209–217. doi: 10.1111/j.1540-8167.2011.02186.x. [DOI] [PubMed] [Google Scholar]
- 67.Platonov PG, Mitrofanova LB, Orshanskaya V, et al. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J Am Coll Cardiol. 2011;58:2225–2232. doi: 10.1016/j.jacc.2011.05.061. [DOI] [PubMed] [Google Scholar]
- 68.Gu WJ, Wu ZJ, Wang PF, et al. N-Acetylcysteine supplementation for the prevention of atrial fibrillation after cardiac surgery: a meta-analysis of eight randomized controlled trials. BMC Cardiovasc Disord. 2012;12:10. doi: 10.1186/1471-2261-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karimi A, Bidhendi LM, Rezvanfard M, et al. The effect of a high dose of atorvastatin on the occurrence of atrial fibrillation after coronary artery bypass grafting. Ann Thorac Surg. 2012;94:8–14. doi: 10.1016/j.athoracsur.2012.01.054. [DOI] [PubMed] [Google Scholar]
- 70.Wongcharoen W, Chen YC, Chen YJ, et al. Effects of aging and ouabain on left atrial arrhythmogenicity. J Cardiovasc Electrophysiol. 2007;18:526–531. doi: 10.1111/j.1540-8167.2007.00781.x. [DOI] [PubMed] [Google Scholar]
- 71.Brorson L, Olsson SB. Right atrial monophasic action potential in healthy males. Studies during spontaneous sinus rhythm and atrial pacing. Acta Med Scand. 1976;199:433–446. doi: 10.1111/j.0954-6820.1976.tb06761.x. [DOI] [PubMed] [Google Scholar]
- 72.Fox CS, Parise H, D'Agostino RB, Sr, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 73.Arnar DO, Thorvaldsson S, Manolio TA. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–712. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 74.Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/ HRS focused update on the management of patients with atrial fibrillation (Updating the 2006 Guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Heart Rhythm. 2011;8:157–176. doi: 10.1016/j.hrthm.2010.11.047. [DOI] [PubMed] [Google Scholar]