

Summary
FMRP is an evolutionarily conserved protein that is highly expressed in neurons and its deficiency causes fragile X mental retardation syndrome. FMRP controls the translation of target mRNAs in part by promoting their dynamic transport in neuronal RNA granules. We have previously shown that high expression of mammalian FMRP induces formation of granules termed FMRP granules. These RNA granules are reminiscent of neuronal granules, of stress granules, as well as of the recently described in vitro-assembled granules. In contrast with mammalian FMRP, which has two paralog proteins, Drosophila FMRP (dFMRP) is encoded by a single gene that has no paralog. Using this genetically simple organism, we investigated formation and dynamics of FMRP granules. We found that increased expression of dFMRP in Drosophila cells induces the formation of dynamic dFMRP RNA granules. Mutagenesis studies identified the N-terminal protein–protein domain of dFMRP as a key determinant for FMRP granules formation. The RGG RNA binding motif of dFMRP is dispensable for dFMRP granules formation since its deletion does not prevent formation of those granules. Deletion of the RGG motif reduced, however, dFMRP trafficking between FMRP granules and the cytosol. Similarly, deletion of a large part of the KH RNA binding motif of dFMRP had no effect on formation of dFMRP-granules, but diminished the shuttling activity of dFMRP. Our results thus suggest that the mechanisms controlling formation of RNA granules and those promoting their dynamics are uncoupled. This study opens new avenues to further elucidate the molecular mechanisms controlling FMRP trafficking with its associated mRNAs in and out of RNA granules.
Key words: FMRP, RNA, Protein translation
Introduction
Via its control of mRNA expression, the formation of RNA granules is critical for an adequate cellular response to external stimuli. RNA granules are dynamic and can be reversibly associated and dissociated in vivo as well as in a cell-free system. In vitro assembly of RNA granules involves trapping of granules components through specific low complexity polypeptide sequences present within specific RNA binding proteins (Han et al., 2012; Kato et al., 2012). Whether similar mechanisms control formation and dynamics of RNA granules in vivo remain unknown.
The RNA-binding protein Fragile X Mental Retardation (FMRP) is an evolutionarily conserved RNA granule component that is particularly abundant in the brain due to its high expression in neurons (Ashley et al., 1993; Siomi et al., 1993; Khandjian et al., 1995). The absence of FMRP causes the development of fragile X syndrome, the most frequent form of hereditary mental retardation (McLennan et al., 2011; Santoro et al., 2012). FMRP is considered to be a nucleocytoplasmic shuttling protein (Eberhart et al., 1996; Siomi et al., 1996; Sittler et al., 1996; Tamanini et al., 1999). In the cytoplasm, the major fraction of FMRP is associated with mRNP complexes bound to polyribosomes (Corbin et al., 1997; Feng et al., 1997a; Feng et al., 1997b), in support of a translational role for FMRP (Bassell and Warren, 2008; Cheever and Ceman, 2009a; Cheever and Ceman, 2009b; Santoro et al., 2012). In neurons, FMRP may also act as a translational repressor by trapping mRNAs into neuronal RNA granules which are then transported out of the soma in a repressed state until they reach their destination in the neurites (Bassell and Warren, 2008). It was previously suggested that mammalian FMRP might also promote translation repression of its mRNA targets under stress conditions by trapping them into stress granules (Mazroui et al., 2002). SG are cytoplasmic bodies whose formation during stress correlates with the inhibition of translation initiation and might constitute the actual sites where stalled translation initiation complexes accumulate (Anderson and Kedersha, 2009; Balagopal and Parker, 2009). The formation of SG, which occurs under stress conditions, requires the phosphorylation of eIF2α (Kedersha et al., 1999), a key pathway known to induce translation initiation arrest upon stress (Holcik and Sonenberg, 2005). Phosphorylation of eIF2α has been also implicated in formation of RNA SG-like granules following overexpression of specific RNA granules (Solomon et al., 2007; Kedersha and Anderson, 2009; Reineke et al., 2012). Recent studies showed, however, that formation of RNA granules in vitro can occurs directly through aggregation of RNA-binding proteins (Han et al., 2012; Kato et al., 2012), suggesting that specific mechanisms may promote formation of RNA granules in vivo. Induction of SG-like granules has also been reported upon an increased expression of mammalian FMRP in the absence of external stress (Mazroui et al., 2002; Mazroui et al., 2003), to which we herein refer as FMRP granules. These FMRP granules are reminiscent of SG since the two types of RNA granules share common components such as translation initiation factors, mRNA, and various RNA-binding proteins, including FMRP (Mazroui et al., 2002; Mazroui et al., 2003). In addition to FMRP, mammalian genomes encode two other members of this family, namely FXR1 and FXR2 (Khandjian, 1999) whose overexpression also induces the formation of cytoplasmic granules in which they co-localize together with FMRP (Mazroui et al., 2002). These FMRP granules also resemble cytoplasmic granules found under conditions where endogenous FMRP and its homologue FXR1 are particularly abundant, such as in brain (Khandjian, 1999) and tumors (Baguet et al., 2007; Comtesse et al., 2007), raising the possibility that these FMRP-containing granules may fulfill similar functions, e.g. the regulation of translation of FMRP mRNA targets. However, the molecular mechanisms leading to the formation of these FMRP granules are still undefined. On the other hand, Drosophila encodes only one member of the FMRP family, i.e. dFMRP (Wan et al., 2000). dFMRP shares the basic molecular functional determinants with its mammalian homologues, implying a conservation of FMRP functions between flies and mammals (Wan et al., 2000; Zhang et al., 2001; Zhang and Broadie, 2005). These conserved domains include the N-terminal protein–protein domain which is known to promote FMRP dimerization and interactions with its partners, as well as KH and the RGG box, which act as RNA-binding motifs (Siomi et al., 1996).
In the present study, we investigated FMRP granules formation and dynamics in Drosophila. We found that increasing dFMRP level induces dFMRP granule formation, in constant equilibrium with free dFMRP. These dFMRP granules contain several canonical RNA granule markers including deIF4E, poly(A)-binding protein (dPABP) as well as poly(A)+ mRNA. The formation of dFMRP granules seems to be unrelated to a stress response, but involves the N-terminal protein–protein interaction domain of dFMRP. RNA-binding motifs of dFMRP are dispensable for dFMRP granules formation but contribute in dFMRP dynamics in and out dFMRP granules. The kinetics of the shuttling activity in FMRP granules are thus conserved between flies and mammals.
Results
GFP-tagged dFMRP expression induces the formation of dFMRP granules which requires its protein–protein interaction domain
We have previously shown that increased expression of mammalian FMRP or of its homologues FXR1 and FXR2 induces the formation of cytoplasmic granules resembling neuronal and stress granules, termed FMRP granules (Mazroui et al., 2002; Mazroui et al., 2003; Dolzhanskaya et al., 2006; Kim et al., 2006). We thus asked whether an increased expression of dFMRP might induce the formation of dFMRP granules in Drosophila cells. To address this question, we assessed the formation of RNA granules upon expression of the GFP-dFMRP fusion protein using different antibodies that detect dFMRP (Fig. 1A), deIF4E or dPABP (Fig. 2) as RNA granules markers. Poly(A)+ mRNA within these RNA granules was detected using a labeled oligo (dT) probe (Fig. 3). Expression of GFP alone did not induce the formation of FMRP granules, as evidenced by the normal diffuse distribution of the SG markers dFMRP, deIF4E, and dPABP (Fig. 1A, Fig. 2). We found that GFP-dFMRP overexpression induces the formation of large granules in >50% of transfected cells. These granules are positive for GFP-dFMRP fusion protein (Fig. 1A) and for both deIF4E and dPABP (Fig. 2). Using anti-dFMRP antibodies, we detected a clear fluorescence signal in those granules (Fig. 1A). However, we could not conclude from this experiment whether this signal corresponds to endogenous dFMRP since the dFMRP antibody used also recognizes GFP-dFMRP (see below). The size of these FMRP granules ranged from 2 to 4 µm, and contained poly(A)+ mRNA, as evidenced by the FISH signal detected using labeled oligo (dT) probes (Fig. 3). Quantification of the FISH signal shows that a large fraction of poly(A)+ mRNA (∼70%) is present in dFMRP-induced granules (Fig. 3). FISH experiments also revealed the presence of significant amounts (∼60%) of dFMR1 mRNA, a well-known FMRP-mRNA target, in those granules (Fig. 4). From these experiments we concluded that the expression of GFP-dFMRP induces the formation of large RNA cytoplasmic granules where specific RNA granule markers and poly(A)+ mRNA are entrapped. These results are consistent with in vitro data describing FMRP as a factor that may promote granules formation (Han et al., 2012; Kato et al., 2012). We thus used these granules as an in vivo model to investigate how dFMRP could induces RNA granules. For these experiments, we constructed several GFP-dFMRP versions in which each known conserved domain has been selectively deleted, leaving the rest of the protein intact (Fig. 1B). ΔKH lacks the conserved KH domain at positions 226–335, ΔRGG is a construct lacking the RGG box (470–507), and ΔPP refers to dFMRP lacking the protein–protein interaction domain (116–212). The formation of dFMRP granules upon expression of GFP fusion proteins was assessed using antibodies that detect the SG markers dFMRP (Fig. 1C), deIF4E and dPABP (Fig. 2), and labelled-oligo(dT) probes to visualise poly(A)+ mRNA (Fig. 3). We found that expression of both ΔKH and ΔRGG mutants induces the formation of dFMRP granules in >50% of transfected cells (Fig. 1C, Fig. 2). FISH experiments showed that these dFMRP granules contain poly(A)+ mRNA (Fig. 3A). Quantification of FISH signal showed that the amount of poly(A)+ mRNA present in dFMRP granules that are induced by either ΔKH or ΔRGG (∼70%) is similar to that induced by GFP-dFMRP (Fig. 3B). This accumulation of mRNA in dFMRP granules induced in the dFMRP mutants ΔKH or ΔRGG which are defective in RNA binding was unexpected. This might be due to indirect interactions with mRNA bound to endogenous dFMRP, which dimerizes with ΔKH and ΔRGG (see Discussion). Nevertheless, our results indicate that the expression of GFP-dFMRP and of its ΔKH and ΔRGG mutants induces the recruitment of a significant amount of mRNA in dFMRP granules. The size of these dFMRP granules is, however, different, ranging from 0.5–1 µm in cells expressing ΔKH, to 4 µm in cells expressing ΔRGG mutant protein. This indicates that the level of mRNA present in dFMRP granules is not dependent on the size of the mRNA-harboring granules. The FMRP epitope recognized by the anti-dFMRP antibody which was used in these experiments lies within the RGG region (Fig. 1D; data not shown). Since this antibody cannot recognize the ΔRGG mutant (Fig. 1D), the signal detected by this antibody in dFMRP granules induced by ΔRGG likely corresponds to endogenous dFMRP (Fig. 1C). Finally, expression of ΔPP resulted in a completely diffuse cytoplasmic distribution of the protein and the lack of detectable dFMRP granules (Fig. 1C, Fig. 2, Fig. 3A). The differential ability of dFMRP mutants to induce the formation of dFMRP granules is not due to their differential levels of protein expression as evidenced by western blot analysis using anti-GFP antibodies, which showed similar expression of either WT or any of the mutant proteins (Fig. 5A, top panel). Moreover, increasing either the amount of transfected DNA or transfection time did not result in the formation of dFMRP granules in the ΔPP mutant or affect the formation of RNA granules induced by GFP-dFMRP, ΔKH and ΔRGG mutants. We conclude that dFMRP clearly participates in the mechanism(s) leading to dFMRP granule induction when highly expressed. The mechanism of formation of dFMRP granules in Drosophila cells involves the protein–protein interaction domain of dFMRP, although we cannot exclude a contribution of its RNA-binding motifs (see Discussion).
Fig. 1. Expression of dFMRP induces dFMRP granules: role of the PP domain.
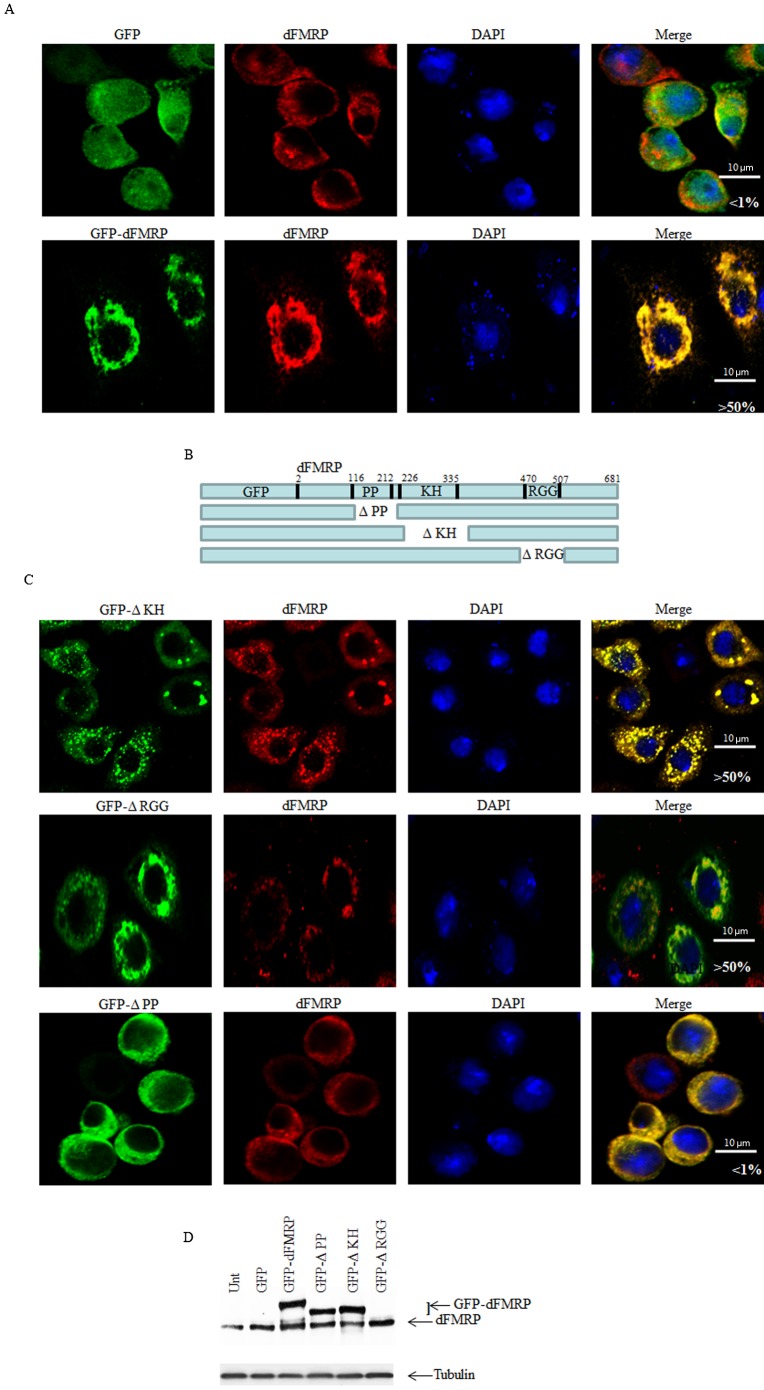
(A) Schneider cells were transfected with either GFP or GFP-dFMRP constructs for 48 h. Cells were then processed for immunofluorescence to detect GFP or GFP-dFMRP (green). The intracellular localization of endogenous dFMRP (red) is revealed using specific anti-dFMRP antibodies. Scale bar: 10 µm. (B) Schematic representation of GFP-dFMRP (top) and its deletion versions. (C) Cells were transfected with GFP-dFMRP mutants and then processed for immunofluorescence as in Fig. 1A. Note that while the ΔPP mutant fails to induce dFMRP granules, both ΔKH and ΔRGG mutants induce granules that are positive for the corresponding GFP-dFMRP mutants and for endogenous dFMRP as well. All pictures were taken using a 63× objective at 1.5 zoom. The percentage of cells harboring FMRP granules from 5 different fields and 5 different experiments containing a total of 1,000 cells is indicated at the bottom of merged images. Scale bar: 10 µm. (D) Cells expressing GFP-dFMRP were collected and protein extracts were next analyzed by immunoblotting for GFP-dFMRP expression using dFMRP-specific antibodies. Tubulin was used as a loading control.
Fig. 2. Co-localization of GFP-dFMRP mutants with the RNA granules markers deIF4E, dPABP and with poly(A)+ mRNA within dFMRP granules in Schneider cells.
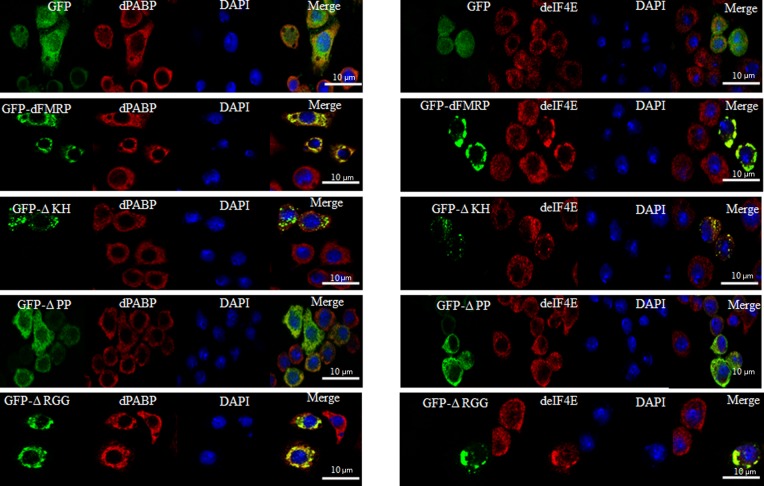
Following transfection with the indicated GFP polypeptides, cells were fixed, permeabilized, and then processed for immunofluorescence to detect GFP or GFP-dFMRP (green). The intracellular localization of dPABP and deIF4E (red) is revealed using the appropriate specific antibodies. Scale bar: 10 µm.
Fig. 3. Co-localization of GFP-dFMRP mutants with poly(A)+ mRNA within dFMRP granules in Schneider.
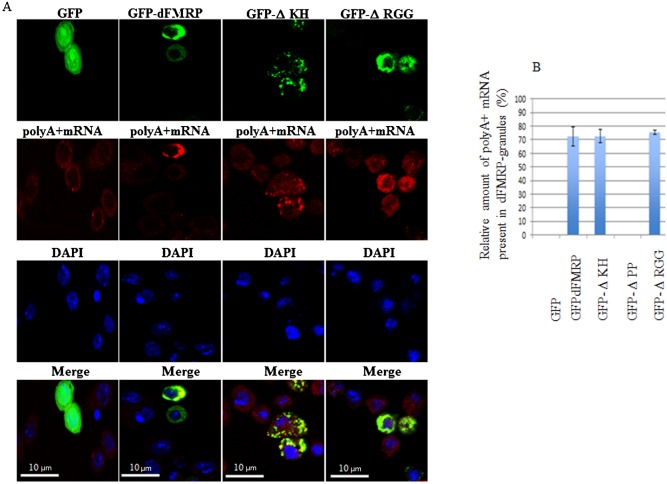
Following transfection with the indicated GFP polypeptides, cells were fixed, permeabilized, and then incubated with 0.2 µM of an Alexa Fluor 594-labeled oligo(dT) probe to detect poly(A)+ mRNA (red), as described in Materials and Methods. (A) dFMRP granules were visualized by confocal microscopy using anti-dFMRP antibodies (green). Scale bar: 10 µm. (B) Densitometry of FISH poly(A)+ mRNA signal with Adobe Photoshop software. The number of pixels and mean intensity were recorded for the selected regions (SG, cytoplasm and background). The mean intensity was multiplied by the number of pixels for the region selected in order to obtain the absolute intensity. The absolute intensity of the background region was subtracted from each region of interest. In order to compare the intensity of two given regions of interest, relative intensities were next calculated. The relative intensity corresponds to the absolute intensity normalized to the absolute intensity of the region of reference.
Fig. 4. Detection of dFMR1 mRNA in dFMRP granules by FISH.
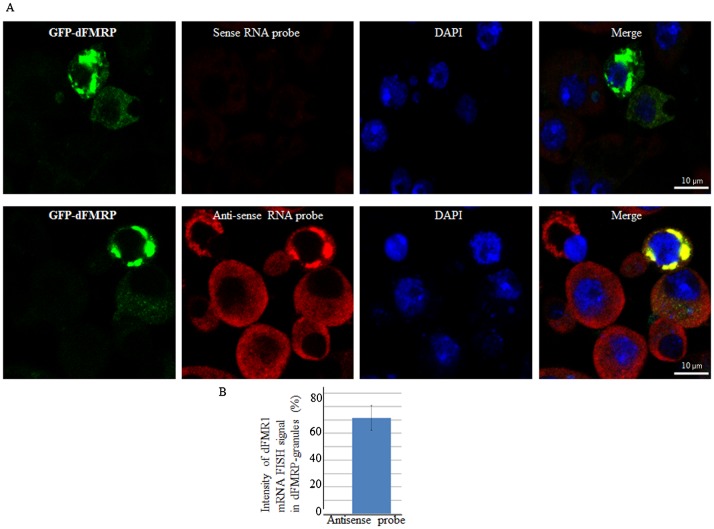
(A) Schneider cells were transfected with GFP-dFMRP. Cells were then fixed, permeabilized, and incubated with 3 nM of Alexa Fluor 488-labeled antisense RNA probe to detect dFMR1 mRNA (panels 6 and 8) or with Alexa Fluor 488-labeled sense probe (panels 2 and 4) as control. dFMRP granules were visualized as green fluorescence. Scale bar: 10 µm. (B) Densitometry quantification of FISH poly(A)+ mRNA signal was done with Photoshop software as above.
Fig. 5. Expression of either dFMRP or human FMRP does not induce eIF2α phosphorylation.
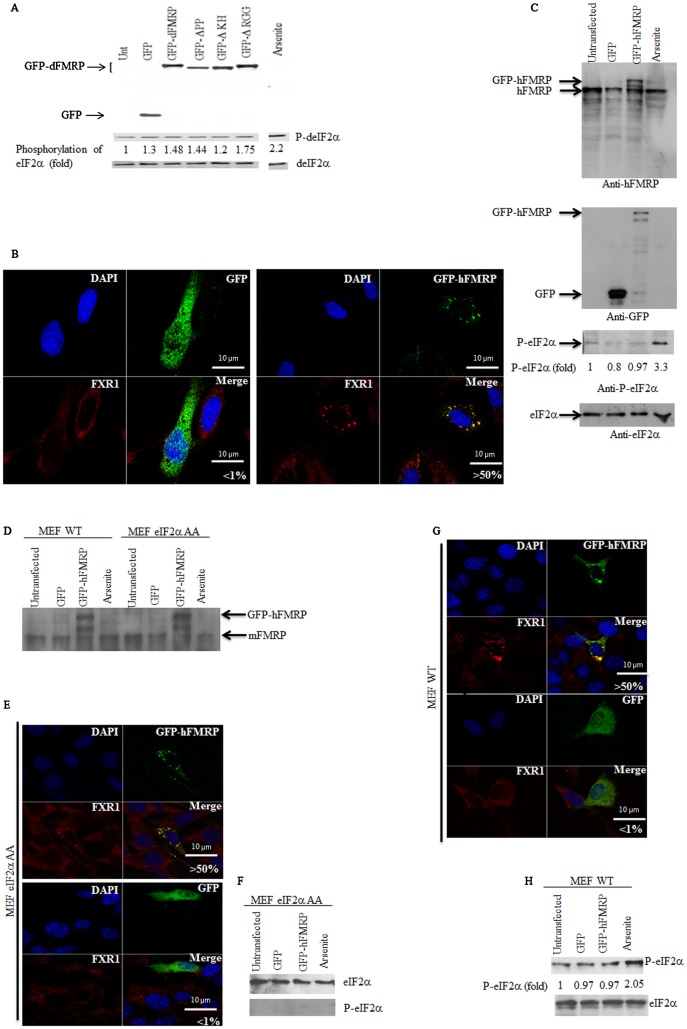
(A) Analysis of eIF2α phosphorylation upon dFMRP expression. Following transfection with GFP-dFMRP or its mutants, Schneider cells were lysed and protein content was analyzed by immunoblotting for GFP-dFMRP expression using anti-GFP antibodies. Phosphorylation of eIF2α was analyzed by western blot (P-eIF2α; center panel) using specific antibodies. An extract isolated from arsenite-treated Schneider cells was used as a positive control for eIF2α phosphorylation. Total eIF2α was analyzed using the pan-eIF2α antibodies and used as a loading control (bottom panel). The amount of phosphorylated eIF2α was determined by quantitation of the film signals by densitometry using the Adobe Photoshop and expressed as a percentage of total eIF2α. The results are representative of 5 different experiments. (B) Expression of GFP-hFMRP in HeLa cells induces FMRP granules. Cells expressing GFP-hFMRP or GFP alone were processed for fluorescence to detect GFP or GFP-hFMRP (green). The intracellular localization of the FMRP partner FXR1 in FMRP granules (red) was revealed by immunofluorescence using specific antibodies. The indicated percentage of FMRP granules that are induced by expressing GFP-hFMRP in HeLa cells is representative of 3 different experiments. (C) Analysis of eIF2α phosphorylation upon dFMRP expression. HeLa cells expressing GFP-hFMRP or GFP alone were collected and their protein extracts prepared for western blot analysis of eIF2α phosphorylation and of eIF2α (as loading control) using specific antibodies (bottom panels). GFP-hFMRP and endogenous FMRP were detected using anti-FMRP antibodies (top panel). GFP was detected using anti-GFP antibodies, which also detect the GFP-hFMRP fusion (center panel). The indicated amount of phosphorylated eIF2α was quantified as described in Fig. 2A. (D–H) GFP-hFMRP induces FMRP granules independently of eIF2α phosphorylation. (D) MEF-eIF2α A/A and its WT counterpart cell line were transfected with either GFP or GFP-hFMRP. Cells were then collected and their protein extracts were analyzed by western blotting for the expression of GFP-hFMRP and endogenous mFMRP using anti-FMRP antibodies. (E,G) Expression of GFP-hFMRP induces FMRP granules in both MEF eIF2α A/A (E) and its WT MEF counterparts (G). Cells expressing GFP or GFP-hFMRP were processed for confocal microscopy for GFP and GFP-hFMRP detection (green). The intracellular localization of the FMRP partner FXR1 in FMRP granules (red) is revealed by immunofluorescence using specific antibodies. The indicated percentage of FMRP granules that are induced by expressing GFP-hFMRP in MEF-eIF2a A/A and their WT MEF counterparts is representative of 3 different experiments. (F,H) The indicated cells expressing GFP or GFP-hFMRP were collected and their protein extracts prepared for western blot analysis of eIF2α phosphorylation and of eIF2α as a loading control using specific antibodies. Arsenite treatment was used as a positive control for eIF2α phosphorylation. The indicated percentage of phosphorylated eIF2α was calculated as described in Fig. 5A. Scale bars in B,E,G: 10 µm.
dFMRP granule formation occurs independently of eIF2α phosphorylation
Overexpression of several RNA granules components including TIA, Caprine and G3BP1, has been shown to induce formation of cytoplasmic SG-like granules through a mechanism involving the phosphorylation of eIF2α (Kedersha et al., 1999; Solomon et al., 2007; Reineke et al., 2012). We then assessed whether formation of dFMRP granules, which is as described above is mediated by the N-terminal protein–protein interaction domain, could also involve eIF2α phosphorylation. Following the expression of GFP-dFMRP protein in Schneider cells, total and phosphorylated eIF2α levels were assessed by western blot analysis using specific antibodies. No significant eIF2α phosphorylation could be detected in these experiments (Fig. 5A). These results suggest that dFMRP granule formation in Drosophila cells occurs independently of eIF2α phosphorylation. Moreover, the expression of GFP-dFMRP does not alter the phosphorylation of eIF2α observed during stress (data not shown), indicating that dFMRP-induced formation of dFMRP granules does not functionally affect the pathway of eIF2α phosphorylation. Our results (cf. Fig. 1, Fig. 5A) raise the possibility that overexpression of the mammalian homolog of dFMRP may induce FMRP granules in an eIF2α phosphorylation-independent fashion. We thus explored the possibility that the formation of both mammalian FMRP granules and dFMRP granules occurs independently of eIF2α phosphorylation. First, we found that expression of GFP-hFMRP in HeLa cells efficiently induced FMRP granules (Fig. 5B), as previously documented (Mazroui et al., 2002). Second, we found that this induction occurred without notable phosphorylation of eIF2α (Fig. 5C), suggesting that the formation of FMRP granules in these cells may not require eIF2α phosphorylation. This conclusion was further supported using MEFs expressing a nonphosphorylatable eIF2 mutant (eIF2αS51A/S51A), obtained from knockin mice (Scheuner et al., 2001). These cells are unable to phosphorylate eIF2α and do not form SG upon arsenite treatment (McEwen et al., 2005), which is known to induce SG in an eIF2α phosphorylation-dependent manner. We found that expression of GFP-hFMRP (Fig. 5D) induces FMRP granules in eIF2αS51A/S51A (Fig. 5E), as efficiently as in WT MEFs (Fig. 5G). As expected, GFP-hFMRP expression induced the formation of FMRP granules in WT MEFs without inducing eIF2α phosphorylation (Fig. 5G,H). Control experiments confirmed that no eIF2α phosphorylation is detectable in eIF2αS51A/S51A (Fig. 5F). We concluded that the formation of FMRP granules, in both Drosophila and mammalian cells, does not require the activation of the general eIF2α phosphorylation stress response, but involves dimerization of FMRP.
The formation of dFMRP granules involves dFMRP dimerization
Mammalian FMRP is known to homodimerize, to heterodimerize with its homologues FXR1 and FXR2, and to interact with other proteins via its N-terminal protein–protein interaction domain. Our above-described results showed that deleting the protein–protein interaction domain of dFMRP prevents the formation of dFMRP granules. This result also ruled out the possibility that dFMRP granule formation upon the expression of GFP-dFMRP might be due to GFP dimerization. The prevention of dFMRP granule formation upon deletion of the PP domain might be due to the lack of self-interaction of GFP-ΔPP or of its interaction with endogenous dFMRP or other protein partners. Our immunoprecipitation experiments using anti-GFP antibodies showed that GFP-ΔPP fails to associate efficiently with dFMRP in Schneider cells, compared to GFP-dFMRP, as assessed by western blot using anti-dFMRP antibodies (Fig. 6A). To validate the identity of the co-immunoprecipitated dFMRP as endogenous dFMRP, we relied upon depletion experiments. Depletion of dFMRP should result in the loss of endogenous dFMRP in GFP-dFMRP precipitates. For these experiments, we used two siRNAs targeting endogenous dFMR1 mRNA, but not GFP-dFMR1 mRNA (Fig. 6B; data not shown). As expected, treatment of Schneider cells with these siRNAs significantly (55%) suppressed endogenous dFMRP synthesis. As a result, the band corresponding to endogenous dFMRP was lost in GFP-dFMRP precipitates (Fig. 6C), thus validating the identity of the immunoprecipitated protein as endogenous dFMRP. We concluded that GFP-dFMRP, but not its GFP-ΔPP mutant, interacts with endogenous dFMRP. Altogether, these results suggest that the N-terminal domain of dFMRP is required for dFMRP granule formation, at least in part by promoting the dimerization of dFMRP.
Fig. 6. Formation of FMRP granules involves FMRP dimerization.

(A–C) Co-immunoprecipitation of GFP-dFMRP mutant polypeptides with endogenous dFMRP. (A) Schneider cells were transfected with GFP-dFMRP or GFP-ΔPP. 48 h post transfection, cells were lysed and their extracts used to immunoprecipitate dFMRP using anti-GFP antibodies. IgG were used as a control immunoprecipitation. IP: immunoprecipitate; total represents 5% of the input used for immunoprecipitation. Immunoprecipitated proteins were analyzed by western blot for dFMRP using anti-dFMRP antibodies. The positions of GFP-dFMRP, GFP-ΔPP, and endogenous dFMRP are indicated by arrows. (B,C) Validation of GFP-dFMRP interaction with endogenous dFMRP. Schneider cells were first treated with siRNAs specific to the 3′UTR of dFMR1 mRNA and were then transfected with GFP-dFMRP 48 h later. Twenty-four h later, cells were lysed and their extracts used to immunoprecipitate dFMRP using anti-GFP antibodies. (B) Total: 5% of the input used for immunoprecipitation. Proteins were analyzed by western blot for dFMRP and GFP-dFMRP expression using anti-dFMRP antibodies Tubulin serves as a loading control. (C) IP: immunoprecipitate. Immunoprecipitated proteins were analyzed by western blot for dFMRP using anti-dFMRP antibodies. The positions of GFP-dFMRP and endogenous dFMRP are indicated by arrows. (D–G) GFP-I304N is a weak inducer of FMRP granules in STEK cells. MEF and STEK cells were transfected with either GFP-hFMRP or GFP-I304N (D,F) for 48 h. Cells were then processed for immunofluorescence to detect GFP fusion proteins (green). The intracellular localization of the FMRP partner FXR1 in FMRP granules (red) was revealed using specific antibodies. The indicated percentage of FMRP granules induced by expressing either GFP-hFMRP (D,E) or GFP-I304N (F,G) in MEFs and STEK is representative of 3 different experiments. Scale bars in D,F: 10 µm.
We then evaluated whether the role of FMRP N-terminal dimerization domain in promoting FMRP granule formation is conserved in mammals. For this, we assessed the formation of FMRP granules in MEF cells derived from fmr1−/− knockout mice (termed STEK) (Mazroui et al., 2002) using GFP-hFMRP I304N. The missense mutation I304N found in the FMRP KH2 motif in a fragile X patient (De Boulle et al., 1993) prevented FMRP homodimerization and therefore prevented the association of FMRP with polysomes (Feng et al., 1997a). This mutant associates, however, with WT FMRP as well as with its partners FXR1 and FXR2 (Laggerbauer et al., 2001). If an interaction between FMRP monomers is required to induce the formation of FMRP granules, the expression of I304N mutant in STEK should prevent formation of the latter. In contrast, expression of the I304N mutant in WT MEFs should result in FMRP granule formation, at least in part via its interaction with endogenous FMRP. Finally, expression of GFP-hFMRP in both WT MEFs and STEK cells, should lead to the formation of FMRP granules in part due to GFP-hFMRP homodimerization. As expected, GFP-hFMRP induced FMRP granule formation with similar efficiency in both STEK and WT MEFs, as assessed by the localization of FMRP granule marker FXR1 (Mazroui et al., 2002) (Fig. 6D,E). Expression of GFP-I304N barely induced FMRP granules in STEK compared to WT MEFs (Fig. 5F,G). The lack of granules in I304N mutants is not due to low protein expression in the mutant as assessed by western blot analysis (data not shown). Altogether, these results suggest a conserved role for the dimerization domain of FMRP in inducing formation of FMRP RNA granule. These results are also consistent with recent data suggesting that formation of in vitro assembled RNA granules involves homotypic trapping of RNA granules components.
Characterization of FMRP granule dynamics
The above-described results showed that expression of GFP-dFMRP and its mutants ΔKH and ΔRGG induces the formation of RNA granules. However, the relative stability of these FMRP granules is as yet unknown. To gain insight on the kinetics of FMRP trafficking between RNA granules and the cytosol, and to determine the role of dFMRP functional domains in such trafficking, we relied upon fluorescence recovery after photobleaching (FRAP) experiments. Following the transfection of Schneider cells with various GFP-dFMRP constructs, individually formed dFMRP granules were bleached and allowed to recover over a period of 140 s. The intensity of recovering fluorescence was recorded every 5 s by confocal microscopy and plotted against time. The procedure was repeated twice to verify the reproducibility of recovery and the independence of percentage of recovery from the photobleached granules. With these experimental settings, GFP-dFMRP recovered up to 40±7% of original fluorescence (Fig. 7A,B), thus providing the first demonstration that FMRP granules induced by FMRP expression are dynamic, labile foci. Both GFP-ΔKH and GFP-ΔRGG appeared to recover to a lower extent than GFP-dFMRP, i.e. to 30±5% (Fig. 6A,B, P<0.004). We validated these results by measuring the mobile fraction (MF), which provides a measure of the concentration of free molecules within the bleached area. The data show that the percentage of the MF for each GFP-dFMRP species correlates with the percentage of recovery recorded (Fig. 7C, P<0.04). Taken together, our results show that dFMRP is in constant exchange between dFMRP granules and the cytosol. We then tested whether FMRP shuttling activity is conserved in mammals. Using FRAP experiments, single FMRP granules were photobleached following transfection of HeLa cells with GFP-hFMRP, and the fluorescence recovery was monitored for 140 s. FMRP was significantly mobile in FMRP granules, as ∼50% of fluorescence was recovered after 140 s (Fig. 8). Overall, these results are indicative of a conserved trafficking of a significant fraction of cellular FMRP in and out FMRP granules, which followed kinetics similar to that found for dFMRP in RNA neuronal granules. Trafficking of dFMRP in FMRP RNA granules is promoted by its RNA binding motifs.
Fig. 7. Analysis of dFMRP kinetics in dFMRP granules by FRAP.

Schneider cells were transfected with GFP-dFMRP and its mutant versions. (A–C) 48 h post transfection, a single dFMRP granule (red circle; indicated by arrow) was photobleached and fluorescence recovery was recorded over 140 s using a confocal microscope. The FRAP methodology is described in Materials and Methods. Scale bar: 10 µm. The images shown in (A) were selected for illustration and correspond to merged differential interference contrast (DIC) microscopy and fluorescence pictures. The recovery of dFMRP fluorescence in the photobleached area was quantified as described in Materials and Methods and plotted as a function of time as indicated in (B). Curves are representative of 3 independent experiments with a total of 100 photobleached granules for each indicated GFP fusion protein. (C) Bar graphs for the MF of each GFP fusion protein are shown with error bars corresponding to the SD of 3 independent experiments. The indicated P-values are calculated with unpaired Student's t-test (n = 3).
Fig. 8. Dynamics of GFP-hFMRP within FMRP granules in HeLa cells by FRAP.
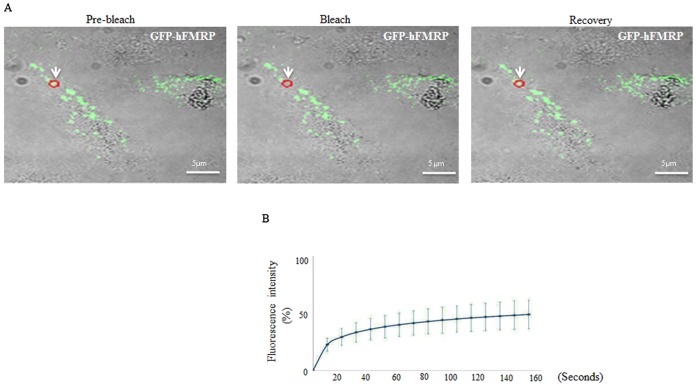
(A,B) Cells were transfected with GFP-hFMRP. Forty-eight h post transfection, a single FMRP-granule (red circle; indicated by arrow) was photobleached (A) and the fluorescence recovery (B) was recorded over 140 s using a confocal microscope as described in Fig. 7. Scale bar in A: 5 µm.
Discussion
RNA granules play diverse role in controlling the turnover and translation of specific mRNAs. FMRP is one of the major components of RNA granules including neuronal granules (Barbee et al., 2006), stress granules (Mazroui et al., 2002), and in vitro assembled granules (Han et al., 2012; Kato et al., 2012). Overexpression of FMRP induces formation of FMRP granules that are reminiscent of RNA granules. Here we used FMRP granules formed in Drosophila cells as a model to investigate how FMRP could promote formation of RNA granules and to define the determinants of FMRP involved in its trafficking in and out RNA granules.
During the past few years, several mammalian SG nucleating factors have been shown to induce the formation of SG-like RNA granules based on the effect of their overexpression. Formation of these SG-like granules was reported to occur through mechanisms which involve the phosphorylation of eIF2α, a key event essential for the triggering of SG formation upon stress (Kedersha et al., 1999; Kedersha et al., 2005). It has been thus assumed that formation of RNA granules via overexpression of SG nucleating factors requires eIF2α phosphorylation (Solomon et al., 2007; Kedersha and Anderson, 2009; Reineke et al., 2012). Our study indicates that this may not be always the case, although we cannot exclude the possibility that FMRP overexpression might induce other forms of stress that could contribute to FMRP granule formation. First, no increase in eIF2α phosphorylation was detected upon expression of GFP-dFMRP or its mutants in Drosophila cells (Fig. 5A), although dFMRP granules were formed (Figs 1–5). Second, the expression of GFP-hFMRP in human cells induces FMRP granules but fails to induce eIF2α phosphorylation (Fig. 5B,C). And lastly, GFP-hFMRP expression induced FMRP granules in eIF2αS51A/S51A as efficiently as in WT MEFs (Fig. 6), demonstrating that eIF2α phosphorylation is dispensable for dFMRP granule formation, which in this case likely involves specific mechanisms. This is also consistent with recent data describing formation of FMRP-containing RNA granules in lysates of unstressed cells (Han et al., 2012; Kato et al., 2012), indicating that RNA granules can form independently of stress-mediated phosphorylation of eIF2α.
We found that formation of dFMRP granules requires the N-terminal conserved PP domain of dFMRP, since the expression of a dFMRP mutant lacking this domain (ΔPP) fails to induce dFMRP granules (Fig. 1C, Figs 3, 4). This result suggests that dFMRP granule formation occurs via mechanisms that involve dFMRP-protein interactions. In addition, we found that expression of dFMRP mutants lacking RNA-binding motifs (ΔKH or ΔRGG) still induces dFMRP granules (Fig. 1C, Figs 3, 4), further indicating that dFMRP granule formation indeed requires the N-terminal PP domain of dFMRP which remains intact in these mutants. This conserved PP domain may promote both the dimerization of dFMRP and its interactions with other protein partners. Our results show that mutant ΔPP polypeptides fail to interact with endogenous dFMRP, suggesting that self-interactions of dFMRP and/or its interactions with endogenous dFMRP, are essential for granule formation. Self-interactions of specific RNA binding proteins have been recently shown to promote their trapping within in vitro assembled RNA granules (Han et al., 2012; Kato et al., 2012). These self-interactions are mediated by low complexity sequences (LC) characterized by repetitive amino acids. In these pioneer in vitro studies, the authors also reported a specific LC polypeptide sequence that promotes formation of RNA granules in a cell-free system. The reported LC sequence was shown to be both essential and sufficient for formation of RNA granules through trapping of RNA binding protein-containing LC sequences, including FMRP. The LC sequence of FMRP lies within its RGG motif, suggesting a key role of RGG in inducing FMRP granules through its LC sequence (Han et al., 2012; Kato et al., 2012). This is consistent with our previous study showing that the expression of mammalian FMRP mutants lacking the RGG motif fails to induce granules in fmr1−/− MEF cells (Mazroui et al., 2003). The LC-containing RGG motif of FMRP is conserved in Drosophila, yet expression of ΔRGG mutants still induce formation of dFMRP granules in Schneider cells. In the latter case, dFMRP granule formation induced by ΔRGG mutants in Schneider cells is likely due both to self-interactions of these mutants and to their interplay with endogenous FMRP (via the PP domain), which through its LC sequence likely contributes to FMRP granules formation. Expression of ΔKH mutant also induces dFMRP granules suggesting that binding of FMRP to RNA, which is mediated by the KH motif, is not required for formation of FMRP granules. This idea is consistent with in vitro data which have ruled out an essential role of RNA in mediating RNA granules formation in cell-free systems (Han et al., 2012; Kato et al., 2012). We cannot exclude, however, that indirect interactions of ΔKH mutant with RNA via its association with endogenous dFMRP or its partners, could contribute to formation of dFMRP granules observed in Schneider cells. In any case, our result demonstrates a key role of the N-terminal dimerization domain of dFMRP in inducing formation of dFMRP granules. Likewise, it is noteworthy that the expression of the naturally occurring mutant I304N fails to induce granules in MEFs lacking FMRP (Fig. 6) most likely as it cannot homodimerize (Feng et al., 1997b). Expression of this mutant does, however, induce FMRP granules in WT MEFs (Fig. 7), likely as a result of its direct interaction with endogenous FMRP (Laggerbauer et al., 2001). However, this result contrasts with previous studies showing that expression of I304N-EGFP (but not WT-FMRP-EGFP) from a tetracyclin-inducible promoter does not induce granules in PC12 neuron cells (De Diego Otero et al., 2002; Schrier et al., 2004). The latter lack of granule formation may nonetheless owe to an increased turnover of I304N in neuron cells, as recently suggested (Zang et al., 2009). Altogether, our results suggest that FMRP-protein interactions are key determinants of the induction of FMRP granule formation, which could further be promoted by FMRP-RNA interactions. FMRP is highly expressed in neurons where it localizes in RNA granules to regulate the expression of its mRNA targets. Whether FMRP regulates neuronal granules formation is currently unknown. Based on our results, we speculate a potential role of FMRP through its dimerization domain in regulating formation of these naturally occurring neuronal RNA granules, although we did not demonstrate this in the present work.
Although mammalian FMRP expression has been shown to induce FMRP granules years ago, the dynamic aspects of this phenomenon had not previously been investigated. Using FRAP, we demonstrate that dFMRP is in constant exchange between a “free” cytosolic pool and dFMRP granules (Fig. 7). This property is not specific to Drosophila since we observed similar dynamic changes with mammalian FMRP (Fig. 8). Shuttling of dFMRP between dFMRP granules and the cytosol was slightly but significantly reduced by deletion of the RNA-binding domains, suggesting that RNA-binding activity mediated by each RNA-binding domain of dFMRP may contribute, albeit to a limited extent, to its shuttling between dFMRP granules and cytosol (Fig. 7). Because expression of a dFMRP mutant lacking the PP domain does not induce dFMRP granules (Figs 1–3) we could not assess whether protein interactions are required for dFMRP shuttling between the two compartments. Ling and colleagues have previously reported that dFMRP-containing granules can move in the cytoplasm and that this movement is mediated by both kinesin-1 and dynein motor proteins (Ling et al., 2004). This is consistent with recent reports showing that movement of FMRP granules in neurons involves the interaction between FMRP and KIF-3 motor protein (Davidovic et al., 2007). It will be interesting to test if dFMRP shuttling between dFMRP-granules and the cytosol involves dFMRP interactions with motor proteins such as kinesin-1 and dynein. Estes and colleagues found that dFMRP rapidly shuttles between the naturally occurring neuronal RNA granules and the “free” cytosolic fraction (Estes et al., 2008). The shuttling kinetics of mRFP-dFMRP in endogenous neuronal granules are similar to those described here for GFP-dFMRP in dFMRP granules. It will be interesting to assess whether dFMRP shuttling to and from neuronal granules requires its RNA-binding domains. In their study, Estes et al. demonstrated that dFMRP shuttling promotes trafficking of its mRNA targets between RNA neuronal granules and the cytosolic fraction (Estes et al., 2008). The mechanisms governing the promotion of mRNA trafficking by dFMRP are still unknown, as well as the relative efficiency of the latter process. Our investigations using dFMRP granules predicts a working model in which FMRP dimerization induces the recruitment of FMRP with its bound mRNA targets into RNA granules where they are incorporated in complexes whose dynamics are maintained by RNA interactions. Future experiments using FMRP granules should contribute to dissect the mechanism(s) by which FMRP-RNA interaction might control its shuttling activity; as well as the actual trafficking pathway of its associated mRNAs between RNA granules and the “free” cytosolic fraction. Posttranslational modifications of FMRP, such as phosphorylation and methylation, are known to regulate FMRP interactions with RNA, polysomes and proteins (Siomi et al., 2002; Ceman et al., 2003; Narayanan et al., 2007; Narayanan et al., 2008; Cheever and Ceman, 2009a; Cheever and Ceman, 2009b; Coffee et al., 2012). We hypothesize that such modifications are likely to play a key role in modulating FMRP shuttling with its bound RNA between RNA granules and cytosol, thus ensuring tight regulation of the expression of FMRP-mRNA targets.
Materials and Methods
Cell lines and cultures
HeLa cervical cancer cells were obtained from the American Type Culture Collection (Manassas, VA; ATCC). Wild-type (WT) MEFs, fmr1−/− MEFs (termed STEK), and MEFs harboring the eIF2αS51A/S51A mutation (eIF2αAA) were described previously (Scheuner et al., 2001). Cells were cultured at 37°C in DMEM (Sigma–Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (all supplements from Sigma–Aldrich). Drosophila Schneider cells were obtained from Dr Robert Tanguay (Laval University) and were cultured at 25°C in Schneider medium (Sigma–Aldrich) supplemented with 10% FBS, penicillin and streptomycin.
Antibodies
Phospho-specific anti-eIF2α and the pan anti-eIF2 were purchased from Cell Signaling Technology (Beverly, MA). Anti-deIF2α (EIF2S1) was obtained from Abcam (Cambridge, MA). Anti-FMRP was previously described (Gareau et al., 2011) and anti-FXR1 was obtained from Dr Edward Khandjian (Laval University). Anti-deIF4E and anti-dPABP were kindly provided by Dr Nahum Sonenberg (McGill University). Anti-dFMRP hybridoma (anti-dFMRP, 5B6-f) was obtained from Developmental Studies Hybridoma Bank (Iowa City, IA) and cultured as recommended by the manufacturer to produce anti-dFMRP antibodies. Anti-GFP antibodies were purchased from Abcam.
Small-interfering RNA (siRNA) experiments
siRNA-dFMRP and non-targeting control siRNA were purchased from Dharmacon (Lafayette, CO). siRNA transfections were performed essentially as described (Gareau et al., 2011), using HiPerFect reagent (Qiagen) following the manufacturer's protocol. Twenty-four h before transfection, Schneider cells were plated on concanavalin A-treated coverslips 24 h before transfection at a density leading to 60–80% confluence at the moment of transfection. For a 6-well plate, annealed duplexes were used at a final concentration of 50 nM. Forty-eight h postransfection, cells were treated with siRNA (50 nM) for an additional 48 h. Cells were then either fixed and processed for immunofluorescence, or harvested for protein extraction. The sequences of the siRNAs used are:
siRNA-3′UTR-dFMRP-1: 5′-CAACACAACUCAACAACAA-3′
siRNA-3′UTR-dFMRP-2: 5′-UUGUUGUUGAGUUGUGUUG-3′.
Immunofluorescence and RNA FISH
Following fixation and permeabilization (20 min in 3.7% paraformaldehyde at room temperature followed by a 15-min immersion in MeOH at −20°C), cells were incubated with primary antibodies diluted in 0.1% (v/v) Tween-20/PBS (PBST) for 2 h at room temperature. After rinsing with PBST, cells were incubated with goat anti-mouse/rabbit IgG (H+L) secondary antibodies conjugated with the Alexa Fluor dye of the appropriate maximum absorption wavelength (405, 488 or 594 nm) for 1 h, washed, and then mounted.
For FISH experiments, cells were first fixed in 3.7% paraformaldehyde for 20 min at room temperature, then permeabilized by a 15-min immersion in 0.1% Triton X-100/PBS. Poly(A)+ mRNAs were detected using a custom made 5′-tagged Alexa Fluor® 594-oligo [dT]25 (Invitrogen, Burlington, ON, Canada) diluted in PBS to a final concentration of 0.2 µM. Hybridization was performed by modifying the method presented in (Chakraborty et al., 2006). Briefly, cells were incubated with the oligo (dT)/PBS for 30 minutes at 42°C, then overnight at 37°C. Cells were then washed twice with 2× SSC (20 min at 37°C) followed by one wash with 0.5× SSC (20 min at 37°C), and finally with PBS.
For specific FISH experiments, the dFMRP coding region was amplified by PCR using primers fused either with T3 (dFMRP-forward: 5′- AATTAACCCTCACTAAAGGGTTTGTGGATGTGGACGGCGT-3′) or T7 (dFMRP-reverse: 5′-TAATACGACTCACTATAGGGGGTGAGAGATTACGAAAATGC-3′) minimal promoter sequences. The amplified fragments were used as templates for in vitro transcription to produce either a dFMRP antisense RNA from the T7 promoter, or a dFMRP sense RNA from the T3 promoter, using the FISH Tag RNA Green Kit with Alexa Fluor 488 (Invitrogen). In the latter technique, in vitro transcription incorporates an amine-modified UTP into the probe template. The purified RNA is then incubated with an amine-reactive Alexa Fluor 488 dye (e.g. the succinimidyl ester of Alexa Fluor 488 carboxylic acid) which binds and reacts with the modified UTP. The conjugated probe is then purified, quantified, denatured, and incubated with cells. Before hybridization, cells were fixed and permeabilized as described above, and then prehybridized in 50% PBST/50% hybridization buffer (50% formamide, 5× SSC, 1 mM phosphate buffer, pH 7.4, 1× Denhardt's solution, and 160 ng/ml of denatured salmon sperm DNA) at room temperature for 10 min with gentle rocking. After two washes with fresh hybridization buffer for 30 min at 42°C, the probe was added to hybridization buffer and incubated with the cells for 16 h at 42°C. After hybridization, cells were processed for immunofluorescence as described above. RNA and proteins were visualized using the LSM 700 confocal laser scanning microscope (Zeiss), equipped with a ZEN 2009 software for image acquisition and analysis. Images were acquired using the following settings: 63× oil objective (zoom 1.0), 0.06 µm for pixel size, and 1.00 airy units as pinhole.
DNA manipulation
pAC-GFP-C2 vectors encoding GFP-hFMRP and GFP-I304N were produced as follows: pET21a-FMRP and pET21a-I304N (Mazroui et al., 2003) were first digested with EcoR1 and BamH1. The isolated FMRP fragments were then inserted into pAC-GFP-C2 previously digested with EcoR1 and BamH1. To generate the vector pAc5.1/V5-HisA (Invitrogen) encoding GFP-dFMRP, total RNA was extracted from Schneider cells with the Omniscript Reverse Transcription kit (Qiagen) and used in a reverse transcription reaction to make dFMRP cDNA using oligo(dT). The reverse transcription product was then subjected to a PCR reaction using dFMRI-XhoI-F (5′-GGCCTCGAGCTATGGAAGATCTCCTCGTG-3′) and dFMR1-EcoRI-Rend (5′ GGCGAATTCTTAGGACGTGCCATTGAC 3′) in order to amplify the dFMRP cDNA. Amplified dFMRP cDNA was digested, purified and then incorporated into the digested (XhoI/EcoRI) pAc-GFP-C1 vector (Invitrogen) by ligation to generate GFP-dFMRP construct. GFP-dFMRP was then amplified by PCR using GFP-EcoRI-F oligo (5′ GGCGAATTCCGCCACCATGGTGAGCAA 3′) and dFMR1-EcoRI-Rend (5′ GGCGAATTCTTAGGACGTGCCATTGAC 3′). The PCR product GFP-dFMR1 was then digested at both ends with EcoRI and purified for insertion into the pAc5.1/V5-HisA Drosophila vector previously digested with EcoRI. pAc5.1/V5-HisA vectors encoding the GFP-dFMRP ΔKH, ΔRGG, and ΔPP variants were generated by ligation of PCR products amplified from pAc5.1/V5-HisA-GFP-dFMRP. The PCR products were first digested with the corresponding restriction enzymes whose sites are present in the primers used for PCR amplification before ligation. For the GFP-dFMRP-ΔPP mutant, GFP-EcoRI F and dFMRP-BamHI R342 oligos were used to amplify the first PCR fragment. Oligos used to amplify the second fragment were dFMRP-BamHI F664 and dFMRP-XbaI Rend. Both fragments were digested and joined to pAc5.1/V5-HisA previously digested with EcoRI and XbaI. For GFP-dFMRP-ΔKH mutant, the first PCR fragment was amplified with the GFP-EcoRI F and dFMRP-BamHI R672 oligos, and the second PCR fragment amplified with the dFMRP-BamHI F1012 and dFMRP-XbaI Rend. Amplified fragments were digested and ligated into pAc5.1/V5-HisA previously digested with EcoRI and XbaI. For the GFP-dFMRP-ΔRGG mutant, we used the GFP-EcoRI F with dFMRP-BamHI R1413 primers to amplify the first PCR fragment and the dFMRP-BamHI F1519 with dFMRP-XbaI Rend primers to amplify the second PCR fragment. The PCR fragments were digested and ligated into pAc5.1/V5-HisA that was digested with EcoRI and XbaI. The following primers were used: GFP-EcoRI-F, 5′-GGCGAATTCCGCCACCATGGTGAGCAA-3′; dFMRP-BamHI R342, 5′-GGCGGATCCCAGACGACCCAATTCACA-3′; dFMRP-BamHI F654, 5′-GGCGGATCCTACGTTGAGGAGTTCCGT-3′; dFMRPI-XbaI Rend, 5′-GGCTCTAGATTAGGACGTGCCATTGAC-3′; dFMRP-BamHI R672, 5′-GGCGGATCCCTCAACGTAGTTTCCACG-3′; dFMRP-BamHI F1012, 5′-GGCGGATCCCTGGCGCATGTACCCTTT-3′; dFMRP-BamHI R1413, 5′-GGCGGATCCGTTGTAGCCACGCTGCTG-3′; dFMRP-BamHI F1519, 5′-GGCGGATCCAACGATCAGCAGAATGGC-3′.
DNA transfection and immunoprecipitation
For DNA transfection, Schneider, MEFs and HeLa cells were transfected with 0.5 µg of DNA in a 6-well plate using the Effectene transfection reagent kit (Qiagen). For immunoprecipitation, cells were collected and lysed at 4°C with lysis buffer (50 mM Tris-HCl, pH 7.4; 0.5% NP-40; 150 mM NaCl; 1 mM MgCl2; 0.25 mM phenylmethanesulfonylfluoride; 0.5 mM DTT) containing a cocktail of protease inhibitors (Roche, Laval, QC, Canada) and 40 U/µl RNase Inhibitor (Invitrogen). The extract was then incubated with protein A Sepharose CL-4B beads (GE Healthcare Life Sciences, QC, Canada) conjugated with the appropriate antibody. Following three washes with lysis buffer, proteins were eluted by resuspending the beads with an equal volume of loading dye buffer. Five percent of the suspension was used for immunoblot analysis of the immunoprecipitated proteins.
Imaging and FRAP
Images were acquired on a Zeiss LSM 700 confocal system (Zeiss). For live cell imaging and SG formation monitoring, acquisitions using the 488-nm line at 2% and differential interference contrast (DIC) mode were taken before and after arsenite treatment. Using the same parameters, videos were acquired by taking images every 3 min for 90 min and merging them side to side. For FRAP, a single GFP-labeled granule per cell was photobleached using the Photo Bleach function of the Zeiss LSM 700 imaging system with the diode laser 488-line set at 100%. The acquisition of recovery time-points was done using the laser 488-line set at 2%. A first picture was taken before FRAP and then, pictures were continuously taken during 30 cycles. Each picture shot required an average of 5 s, depending of the size of the photobleached region, for a total time of approximately 140 s. The FRAP analysis included the determination of the average fluorescence intensity of a region of interest containing an unbleached granule as well as an area of background fluorescence. To ensure that the bleaching laser did not damage the cell, the same granules were photobleached, and fluorescence recovery was recorded again. Measurements of fluorescence were done using imaging ZEN software (Zeiss). Briefly, background fluorescence was subtracted from the bleached and unbleached granules and recovery fluorescence values were normalized to a percentage of original fluorescence. The bleached granule was then corrected to the fluorescence of the unbleached granule to adjust for slight changes in focus or slight time-dependent bleaching. Recovery could then be compared in multiple granules of different sizes and from different cells across multiple experimental sessions. Mobile fraction (MF) measurements (i.e. the percentage of fluorescence proteins capable of diffusing into a bleached region of interest during the time-course of the experiment) were determined using ZEN software (Zeiss).
Acknowledgments
We are grateful to Dr Nahum Sonenberg for his gift of anti-deIF4E and anti-dPABP antibodies, and to Dr Edward Khandjian for providing the anti-FXR1 ML13 antibodies. We would also like to thank Christine Filion for her technical help and Drs Yves Labelle and Richard Poulin for editing the manuscript and for stimulating discussions. This work was supported by the Natural Sciences and Engineering Research Council of Canada (MOP-CG095386) and by a Canadian Foundation for Innovation grant (MOP-GF091050) to R.M. C.G. is a recipient of a scholarship from the Faculty of Medicine at Laval University. R.M. is a recipient of a CIHR New Investigator Scholarship award.
References
- Anderson P., Kedersha N. (2009). RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 10, 430–436 10.1038/nrm2694 [DOI] [PubMed] [Google Scholar]
- Ashley C. T., Sutcliffe J. S., Kunst C. B., Leiner H. A., Eichler E. E., Nelson D. L., Warren S. T. (1993). Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat. Genet. 4, 244–251 10.1038/ng0793-244 [DOI] [PubMed] [Google Scholar]
- Baguet A., Degot S., Cougot N., Bertrand E., Chenard M. P., Wendling C., Kessler P., Le Hir H., Rio M. C., Tomasetto C. (2007). The exon-junction-complex-component metastatic lymph node 51 functions in stress-granule assembly. J. Cell Sci. 120, 2774–2784 10.1242/jcs.009225 [DOI] [PubMed] [Google Scholar]
- Balagopal V., Parker R. (2009). Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr. Opin. Cell Biol. 21, 403–408 10.1016/j.ceb.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbee S. A., Estes P. S., Cziko A. M., Hillebrand J., Luedeman R. A., Coller J. M., Johnson N., Howlett I. C., Geng C., Ueda R.et al. (2006). Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 52, 997–1009 10.1016/j.neuron.2006.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell G. J., Warren S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214 10.1016/j.neuron.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S., O'Donnell W. T., Reed M., Patton S., Pohl J., Warren S. T. (2003). Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum. Mol. Genet. 12, 3295–3305 10.1093/hmg/ddg350 [DOI] [PubMed] [Google Scholar]
- Chakraborty P., Satterly N., Fontoura B. M. (2006). Nuclear export assays for poly(A) RNAs. Methods 39, 363–369 10.1016/j.ymeth.2006.07.002 [DOI] [PubMed] [Google Scholar]
- Cheever A., Ceman S. (2009a). Phosphorylation of FMRP inhibits association with Dicer. RNA 15, 362–366 10.1261/rna.1500809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever A., Ceman S. (2009b). Translation regulation of mRNAs by the fragile X family of proteins through the microRNA pathway. RNA Biol. 6, 175–178 10.4161/rna.6.2.8196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee R. L., Jr, Williamson A. J. A. d. k. i. n. s., M C., Gray M. C., Page T. L., Broadie K. (2012). In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum. Mol. Genet. 21, 900–915 10.1093/hmg/ddr527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comtesse N., Keller A., Diesinger I., Bauer C., Kayser K., Huwer H., Lenhof H.–P., Meese E. (2007). Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 120, 2538–2544 10.1002/ijc.22585 [DOI] [PubMed] [Google Scholar]
- Corbin F., Bouillon M., Fortin A., Morin S., Rousseau F., Khandjian E. W. (1997). The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum. Mol. Genet. 6, 1465–1472 10.1093/hmg/6.9.1465 [DOI] [PubMed] [Google Scholar]
- Davidovic L., Jaglin X. H., Lepagnol–Bestel A. M., Tremblay S., Simonneau M., Bardoni B., Khandjian E. W. (2007). The fragile X mental retardation protein is a molecular adaptor between the neurospecific KIF3C kinesin and dendritic RNA granules. Hum. Mol. Genet. 16, 3047–3058 10.1093/hmg/ddm263 [DOI] [PubMed] [Google Scholar]
- De Boulle K., Verkerk A. J., Reyniers E., Vits L., Hendrickx J., Van Roy B., Van den Bos F., de Graaff E., Oostra B. A., Willems P. J. (1993). A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 3, 31–35 10.1038/ng0193-31 [DOI] [PubMed] [Google Scholar]
- De Diego Otero Y., Severijnen L. A., van Cappellen G., Schrier M., Oostra B., Willemsen R. (2002). Transport of fragile X mental retardation protein via granules in neurites of PC12 cells. Mol. Cell. Biol. 22, 8332–8341 10.1128/MCB.22.23.8332-8341.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhanskaya N., Merz G., Aletta J. M., Denman R. B. (2006). Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J. Cell Sci. 119, 1933–1946 10.1242/jcs.02882 [DOI] [PubMed] [Google Scholar]
- Eberhart D. E., Malter H. E., Feng Y., Warren S. T. (1996). The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 5, 1083–1091 10.1093/hmg/5.8.1083 [DOI] [PubMed] [Google Scholar]
- Estes P. S., O'Shea M., Clasen S., Zarnescu D. C. (2008). Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol. Cell. Neurosci. 39, 170–179 10.1016/j.mcn.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Feng Y., Gutekunst C.–A., Eberhart D. E., Yi H., Warren S. T., Hersch S. M. (1997a). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neuroscience 17, 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Absher D., Eberhart D. E., Brown V., Malter H. E., Warren S. T. (1997b). FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol. Cell 1, 109–118 10.1016/S1097-2765(00)80012-X [DOI] [PubMed] [Google Scholar]
- Gareau C., Fournier M. J., Filion C., Coudert L., Martel D., Labelle Y., Mazroui R. (2011). p21WAF1/CIP1 upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS ONE 6, e20254 10.1371/journal.pone.0020254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T. W., Kato M., Xie S., Wu L. C., Mirzaei H., Pei J., Chen M., Xie Y., Allen J., Xiao G.et al. (2012). Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 10.1016/j.cell.2012.04.016 [DOI] [PubMed] [Google Scholar]
- Holcik M., Sonenberg N. (2005). Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6, 318–327 10.1038/nrm1618 [DOI] [PubMed] [Google Scholar]
- Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J.et al. (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N., Anderson P. (2009). Regulation of translation by stress granules and processing bodies. Prog. Mol. Biol. Transl. Sci. 90, 155–185 10.1016/S1877-1173(09)90004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N. L., Gupta M., Li W., Miller I., Anderson P. (1999). RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 147, 1431–1442 10.1083/jcb.147.7.1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke–Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., Anderson P. (2005). Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 10.1083/jcb.200502088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandjian E. W. (1999). Biology of the fragile X mental retardation protein, an RNA-binding protein. Biochem. Cell Biol. 77, 331–342 10.1139/o99-035 [DOI] [PubMed] [Google Scholar]
- Khandjian E. W., Fortin A., Thibodeau A., Tremblay S., Côté F., Devys D., Mandel J. L., Rousseau F. (1995). A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell culture. Hum. Mol. Genet. 4, 783–789 10.1093/hmg/4.5.783 [DOI] [PubMed] [Google Scholar]
- Kim S. H., Dong W. K., Weiler I. J., Greenough W. T. (2006). Fragile X mental retardation protein shifts between polyribosomes and stress granules after neuronal injury by arsenite stress or in vivo hippocampal electrode insertion. J. Neurosci. 26, 2413–2418 10.1523/JNEUROSCI.3680-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B., Ostareck D., Keidel E. M., Ostareck–Lederer A., Fischer U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338 10.1093/hmg/10.4.329 [DOI] [PubMed] [Google Scholar]
- Ling S. C., Fahrner P. S., Greenough W. T., Gelfand V. I. (2004). Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc. Natl. Acad. Sci. USA 101, 17428–17433 10.1073/pnas.0408114101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazroui R., Huot M. E., Tremblay S., Filion C., Labelle Y., Khandjian E. W. (2002). Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Hum. Mol. Genet. 11, 3007–3017 10.1093/hmg/11.24.3007 [DOI] [PubMed] [Google Scholar]
- Mazroui R., Huot M. E., Tremblay S., Boilard N., Labelle Y., Khandjian E. W. (2003). Fragile X Mental Retardation protein determinants required for its association with polyribosomal mRNPs. Hum. Mol. Genet. 12, 3087–3096 10.1093/hmg/ddg335 [DOI] [PubMed] [Google Scholar]
- McEwen E., Kedersha N., Song B., Scheuner D., Gilks N., Han A., Chen J. J., Anderson P., Kaufman R. J. (2005). Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933 10.1074/jbc.M412882200 [DOI] [PubMed] [Google Scholar]
- McLennan Y., Polussa J., Tassone F., Hagerman R. (2011). Fragile X syndrome. Curr. Genomics 12, 216–224 10.2174/138920211795677886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U., Nalavadi V., Nakamoto M., Pallas D. C., Ceman S., Bassell G. J., Warren S. T. (2007). FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neuroscience 27, 14349–14357 10.1523/JNEUROSCI.2969-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U., Nalavadi V., Nakamoto M., Thomas G., Ceman S., Bassell G. J., Warren S. T. (2008). S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482 10.1074/jbc.C800055200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke L. C., Dougherty J. D., Pierre P., Lloyd R. E. (2012). Large G3BP-induced granules trigger eIF2α phosphorylation. Mol. Biol. Cell 23, 3499–3510 10.1091/mbc.E12-05-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M. R., Bray S. M., Warren S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245 10.1146/annurev-pathol-011811-132457 [DOI] [PubMed] [Google Scholar]
- Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner–Weir S., Kaufman R. J. (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 10.1016/S1097-2765(01)00265-9 [DOI] [PubMed] [Google Scholar]
- Schrier M., Severijnen L. A., Reis S., Rife M., van't Padje S., van Cappellen G., Oostra B. A., Willemsen R. (2004). Transport kinetics of FMRP containing the I304N mutation of severe fragile X syndrome in neurites of living rat PC12 cells. Exp. Neurol. 189, 343–353 10.1016/j.expneurol.2004.05.039 [DOI] [PubMed] [Google Scholar]
- Siomi H., Siomi M. C., Nussbaum R. L., Dreyfuss G. (1993). The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298 10.1016/0092-8674(93)90420-U [DOI] [PubMed] [Google Scholar]
- Siomi M. C., Zhang Y., Siomi H., Dreyfuss G. (1996). Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol. Cell. Biol. 16, 3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi M. C., Higashijima K., Ishizuka A., Siomi H. (2002). Casein kinase II phosphorylates the fragile X mental retardation protein and modulates its biological properties. Mol. Cell. Biol. 22, 8438–8447 10.1128/MCB.22.24.8438-8447.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittler A., Devys D., Weber C., Mandel J. L. (1996). Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of FMR1 protein isoforms. Hum. Mol. Genet. 5, 95–102 10.1093/hmg/5.1.95 [DOI] [PubMed] [Google Scholar]
- Solomon S., Xu Y., Wang B., David M. D., Schubert P., Kennedy D., Schrader J. W. (2007). Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2α, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol. 27, 2324–2342 10.1128/MCB.02300-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanini F., Bontekoe C., Bakker C. E., van Unen L., Anar B., Willemsen R., Yoshida M., Galjaard H., Oostra B. A., Hoogeveen A. T. (1999). Different targets for the fragile X-related proteins revealed by their distinct nuclear localizations. Hum. Mol. Genet. 8, 863–869 10.1093/hmg/8.5.863 [DOI] [PubMed] [Google Scholar]
- Wan L., Dockendorff T. C., Jongens T. A., Dreyfuss G. (2000). Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20, 8536–8547 10.1128/MCB.20.22.8536-8547.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang J. B., Nosyreva E. D., Spencer C. M., Volk L. J., Musunuru K., Zhong R., Stone E. F., Yuva–Paylor L. A., Huber K. M., Paylor R.et al. (2009). A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 5, e1000758 10.1371/journal.pgen.1000758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Q., Broadie K. (2005). Fathoming fragile X in fruit flies. Trends Genet. 21, 37–45 10.1016/j.tig.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Zhang Y. Q., Bailey A. M., Matthies H. J., Renden R. B., Smith M. A., Speese S. D., Rubin G. M., Broadie K. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603 10.1016/S0092-8674(01)00589-X [DOI] [PubMed] [Google Scholar]