

SUMMARY
Obesity is a highly heritable disease driven by complex interactions between genetic and environmental factors. Human genome-wide association studies (GWAS) have identified a number of loci contributing to obesity; however, a major limitation of these studies is the inability to assess environmental interactions common to obesity. Using a systems genetics approach, we measured obesity traits, global gene expression, and gut microbiota composition in response to a high-fat/high-sucrose (HF/HS) diet of more than 100 inbred strains of mice. Here we show that HF/HS feeding promotes robust, strain-specific changes in obesity that is not accounted for by food intake and provide evidence for a genetically determined set-point for obesity. GWAS analysis identified 11 genome-wide significant loci associated with obesity traits, several of which overlap with loci identified in human studies. We also show strong relationships between genotype and gut microbiota plasticity during HF/HS feeding and identify gut microbial phylotypes associated with obesity.
INTRODUCTION
The dramatic increase in obesity during the past few decades is tightly associated with the increase in obesity-related complications, such as type 2 diabetes, heart disease, and cancer. Energy-rich diets containing high levels of fat and refined carbohydrates along with sedentary lifestyles are believed to be the most significant environmental factors contributing to this epidemic (Finucane et al., 2011; Malik et al., 2010). Understanding the genetic and environmental interactions contributing to obesity is thus crucial for developing novel therapies and preventive strategies. Human genome-wide association studies (GWAS) and studies of rare monogenic forms of obesity, as well as biochemical studies with cells and animal models, have identified relevant genes and pathways important in obesity; however, given the complexity of the obesity phenotype and the small amount of variance that can be explained by known obesity alleles, it is clear that much remains to be discovered (Attie and Scherer, 2009; Bouchard et al., 1990; Knights et al., 2011; Sandholt et al., 2010; Speakman et al., 2011).
Obesity is strongly heritable in humans, with estimates ranging from 50 to 90% (Barsh et al., 2000; Stunkard et al., 1986). Large human GWAS explain less than 3% of this heritable component and environmental interactions with diet composition likely add significant complexity to such studies (Kilpelainen et al., 2011; Sonestedt et al., 2009; Speliotes et al., 2010). Indeed, long-term overfeeding in monozygotic twins promotes striking within-pair similarities in fat mass gain, demonstrating that gene-by-diet interactions may be highly heritable and have a large impact on obesity (Bouchard et al., 1990). In addition to host genetic contributions, the microbial community within the gut has been shown to influence obesity in humans and mice (Turnbaugh et al., 2009a; Turnbaugh et al., 2006). The gut microbiota is a transmissable trait and can undergo dynamic population shifts with varied dietary composition (Benson et al., 2010; Turnbaugh et al., 2009b; Yatsunenko et al., 2012). Obese subjects have an altered gut microbiota compared to lean individuals, which may be an important contributing factor to the obesity epidemic (Turnbaugh et al., 2009a). To date, very little is known about the genetic basis of gene-by-diet and gut microbiota-diet interactions to common obesogenic factors, such as the consumption of energy-rich diets.
Identification of genes contributing to complex traits, such as obesity, in mice has been hampered by the poor mapping resolution of traditional genetic crosses (Bhatnagar et al., 2011; Burrage et al., 2010; Dokmanovic-Chouinard et al., 2008; Ehrich et al., 2005b; Flint et al., 2005; Lawson et al., 2011; York et al., 1999; Zhang et al., 1994). Based on the genome sequencing of many mouse strains, the identification of millions of single nucleotide polymorphisms (SNPs) (Keane et al., 2011), and the development of an algorithm that corrects for population structure in association analysis (Kang et al., 2008), we recently developed a systems genetics resource in the mouse capable of high resolution genome-wide association mapping (Bennett et al., 2010). This resource, termed the Hybrid Mouse Diversity Panel (HMDP) is composed of more than 100 commercially available mouse strains and is ideal for systems-level analyses of gene-by-environment interactions. Association-based mapping approaches in rodents have recently been reviewed (Flint and Eskin, 2012).
Employing a systems genetics approach in the mouse, we integrated physical, molecular traits, and gut microbiota composition data in response to an energy-rich diet. Using GWAS rather than quantitative trait locus (QTL) analyses, we obtained biologically meaningful genetic mapping, such that several of the genetic loci identified contained between 1-3 genes, comparable to human GWAS. These genes were prioritized using expression quantitative trait locus (eQTL) analysis and we were able to show a significant overlap between mouse and human GWAS loci. We measured the change in fat dynamically, at five different points following high-fat/high-sucrose (HF/HS) feeding, providing strong evidence for a genetically controlled body fat set-point. Our use of inbred mice strains also enabled detailed analysis of the relationship between gut microbiota composition, obesity traits, and diet. Overall, gene-by-diet interactions were highly reproducible and pervasive, providing a partial explanation for the failure of human studies to explain a larger fraction of the genetic basis of obesity. Our results indicate that mouse GWAS and systems genetics analyses provide a powerful method to complement human studies and to address factors, such as gene-by-diet interactions, that would be difficult to study directly in humans.
RESULTS
Robust Variation in Gene-by-Diet Interactions
To assess gene-by-diet interactions common to obesity, male mice were fed ad libitum a HF/HS diet that represents a typical fast food diet, in terms of fat and refined carbohydrates (32% kcal from fat and 25% kcal from sucrose). Mice were maintained on a chow diet (6% kcal from fat) until 8 weeks of age and subsequently placed on a HF/HS diet for 8 weeks. Body fat percentage was assessed by magnetic resonance imaging (MRI) every two weeks and food intake was monitored for a period of one week at the middle of the study timeline (study schematic shown in Figure 1A). Altogether, about 100 inbred strains of male mice were studied, with an average of 6 mice of each strain (Table S1).
Figure 1. Natural Variation in Gene-by-Diet Interactions.
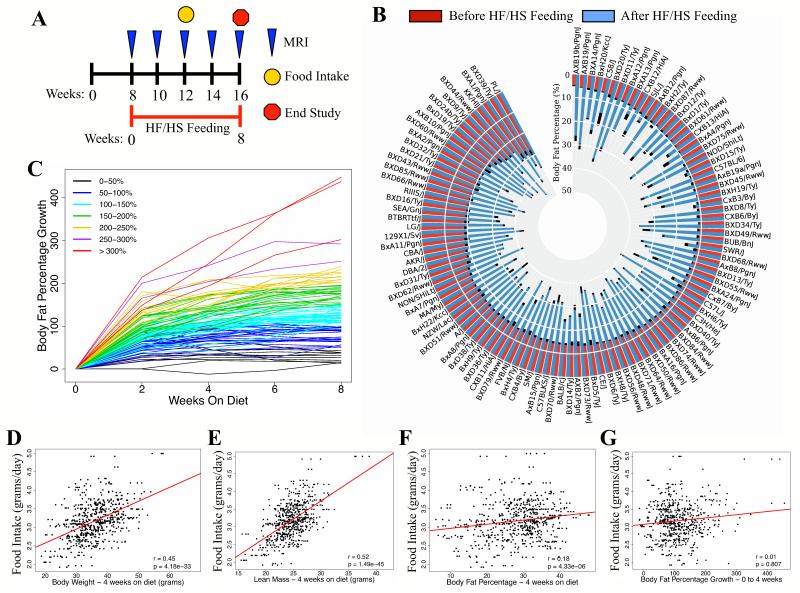
(A) Schematic of study design with indicated time points for HF/HS feeding (red), MRI (blue), food intake monitoring (yellow), and end of study (red). MRI, magnetic resonance imaging.
(B) Body fat percentage in male mice (108 strains) before (red) and after (blue) 8 weeks of HF/HS feeding. Error bars (black) represent standard error of the mean (SEM)
(C) Bi-weekly percent body fat percentage increase in male mice with indicated body fat percentage increase after 8 weeks of HF/HS feeding.
(D-G) Correlation of food intake (grams/day/mouse) with body weight (D), lean mass (E), body fat percentage – 4 weeks on HF/HS diet (F), and body fat percentage growth – 0 to 4 weeks (G), regression line (red). r, biweight midcorrelation, p, p-value.
A wide distribution in body fat percentage was observed in male mice before HF/HS feeding (Figures 1B). Dietary responses, as assessed by the body fat percentage increase during HF/HS feeding, varied widely among the strains (Figures 1C and S1). Although many strains exhibited a significant increase in body fat percentage throughout the study timeline, their individual responses differed significantly, from no change to a 200% increase in body fat percentage within the first two weeks (Figures 1C). Most strains responded during the first 4 weeks of HF/HS feeding and did not accumulate additional fat during the remainder of the study, suggesting an upper set-point whereby continued body fat percentage growth is resisted (Figure 1C) (Speakman et al., 2011). The large effect of the HF/HS feeding on fat accumulation was confirmed with age-matched (16 weeks old) male mice (Table S2) fed a chow diet, which displayed similar body fat percentage to male mice before the HF/HS diet intervention (Figure S1A). Additionally, comparison of individual male strains maintained on a chow diet or fed a HF/HS diet for 8 weeks showed an average increase in body fat percentage from 0 to more than 600% (Figure S1B).
We observed high heritability of about 80% for body fat percentage across the study timeline (Table 1). Changes in body fat percentage after HF/HS feeding were also highly heritable (>70%), suggesting that dietary responses are strongly controlled by genetics. Our results are consistent with the heritability estimates for body mass index (BMI) and obesity in humans (Barsh et al., 2000; Stunkard et al., 1986) and emphasize the importance of genetics in controlling obesity traits, such as gene-by-diet interactions.
Table 1. Heritability Estimates for Obesity and Dietary Responsiveness.
| Trait | Heritability (%) |
|---|---|
| Body Fat Percentage – 0 weeks on HF/HS diet | 80 |
| Body Fat Percentage – 2 weeks on HF/HS diet | 82 |
| Body Fat Percentage – 4 weeks on HF/HS diet | 83 |
| Body Fat Percentage – 6 weeks on HF/HS diet | 83 |
| Body Fat Percentage – 8 weeks on HF/HS diet | 85 |
| Body Fat Percentage growth - 0 to 2 weeks | |
| Body Fat Percentage growth - 0 to 4 weeks | 63 |
| Body Fat Percentage growth - 0 to 6 weeks | 67 |
| Body Fat Percentage growth - 0 to 8 weeks | 73 |
Body fat percentage growth calculated by quantifying the percentage increase of body fat after beginning HF/HS diet. Heritability calculated as described in experimental procedures.
Other Factors Contributing to Dietary Responsiveness
Over-consumption of high-calorie, energy-rich foods is a key environmental factor contributing to the global obesity epidemic (McCaffery et al., 2012). To understand the relationship between food intake and obesity, food intake was monitored and found to range from 2-5 grams per mouse per day. Total food intake per day was significantly correlated with body weight and lean mass (Figures 1D and E). In contrast, body fat percentage and body fat percentage change after 4 weeks of HF/HS feeding showed little to no correlation with food intake (Figures 1F and G). This suggests that factors outside of food intake largely underlie the variation of obesity and fat mass gain between the strains in response to HF/HS feeding.
To further define the contribution of energy consumption to the differences in fat accumulation, we performed in vivo metabolic chamber analyses of five inbred strains that are the progenitors of the recombinant inbred strains (and therefore contribute importantly to the overall genetic component) on chow diet. Significant strain differences were observed in total food intake, activity, heat production, and utilization of different energy substrates, as indicated by respiratory exchange ratio (RER) (Figures S2A, B, C, and D), all of which can influence dietary responses and subsequent fat accumulation.
GWAS and Systems Genetics Analysis
Association analysis was performed using about 100,000 informative SNPs, spaced throughout the genome, with efficient mixed model association (EMMA) adjusting for population structure (Kang et al., 2008). The threshold for genome-wide significance was based on simulation and permutations, as previously described (Farber et al., 2011). This approach has been validated using transgenic analyses and by comparison with linkage analysis (Bennett et al., 2010). Altogether, 11 genome-wide significant loci were found to be associated with obesity traits (Table 2). Loci averaged 500 kb to 2 Mb in size and in most cases contained 1 to 20 genes within a linkage disequilibrium (LD) block, an improvement of more than an order of magnitude as compared to traditional linkage analysis in mice which has a resolution of 10 to 20 Mb (Flint et al., 2005).
Table 2. Genome-wide Significant Loci For Obesity and Dietary Responsiveness.
| Trait | Chromosome | Peak SNP | Position (Mb) |
P-value | MAF | LD (Mb) | No. of Genes |
|---|---|---|---|---|---|---|---|
| Body Fat % increase – 0 to 8 wks | 1 | rs31849980 | 183730026 | 1.4E-08 | 15 | 178.3-184.5 | 60 |
| Body Fat % increase – 0 to 8 wks | 18 | rs30078681 | 9731125 | 4.3E-08 | 8 | 8.5-12.5 | 26 |
| Body Fat % increase – 0 to 8 wks | 16 | rs3148854 | 93933923 | 9.0E-08 | 40 | 93.0-94.0 | 8 |
| Body Fat % increase – 0 to 8 wks | 5 | rs13478388 | 91288973 | 1.5E-07 | 37 | 90.9-92.2 | 17 |
| Body Fat % increase – 0 to 8 wks | 18 | rs29628302 | 5395236 | 1.9E-07 | 11 | 4.6-5.4 | 3 |
| Body Fat % increase – 0 to 8 wks | 18 | rs13483184 | 3796540 | 2.6E-07 | 13 | 3.8-4.6 | 3 |
| Body Fat % increase – 0 to 8 wks | 11 | rs29417268 | 67079767 | 2.8E-07 | 12 | 65.8-67.4 | 14 |
| Body Fat % increase – 0 to 2 wks | 6 | rs13478690 | 30872499 | 2.8E-07 | 7 | 30.5-31.5 | 11 |
| Body Fat % increase – 0 to 8 wks | 3 | rs29982345 | 111983084 | 9.9E-07 | 14 | 110.2-113.4 | 7 |
| Body Fat % - 8 wks | 7 | rs13479513 | 134251677 | 6.7E-07 | 21 | 133.0-136.0 | 113 |
| Body Fat % increase – 0 to 2 wks | 2 | rs13476804 | 139322068 | 2.95E-06 | 10 | 138.9-139.4 | 1 |
MAF, Minor Allele Frequency; LD, Linkage Disequilibrium
Indicated genome-wide significant loci for obesity traits with indicated location, position, P-value, MAF, LD, and number of genes with in LD block (described in experimental procedures)
In order to help identify candidate genes at loci, global expression analyses of epididymal adipose tissue in male mice (16 weeks old) fed a chow diet were carried out to determine genetic regulation and correlation between gene expression and body fat percentage. The loci controlling transcript levels in adipose tissue were mapped using EMMA and are referred to as expression Quantitative Trait Loci (eQTL). Loci are termed “cis” if the locus maps within 1 Mb of the gene encoding the transcript and “trans” if the locus is outside 1 Mb. Overall, 3,960 cis and 4,496 trans eQTL were identified to have a genome-wide significance threshold (cis threshold: p < 1.4 × 10−3 and trans threshold: p < 6.1 × 10−6) (Figure S3A). Cis regulation indicates a potential functional genomic variation within or near a gene that significantly influences gene expression of a given gene. For example, Fto, the most widely replicated gene in human GWAS for obesity shows a strong cis eQTL in the adipose tissue of mice (Figure S5D), indicating genetic variation of this gene.
Global gene expression in epididymal adipose tissue was correlated with body fat percentage in chow fed mice (top 50 genes shown in Table S3). Many genes known to play a vital role in adipose biology showed significant correlations with body fat percentage. Leptin is a key adipose-derived hormone correlating with adipose tissue mass (Ioffe et al., 1998) and is strongly correlated (r = 0.75; p < 2.2 × 10−16) with body fat percentage (Figure 2A). Other important adipose tissue genes, such as Sfrp5 (Ouchi et al., 2010) (r = 0.76; p < 2.2 × 10−16), Chrebp (Herman et al., 2012) (r = −0.57; p = 2.28 × 10−9) and Tmem160 (r = 0.71; p = 4.21 × 10−16), a recently identified gene from a human GWAS for body mass index (BMI) (Speliotes et al., 2010), were also found to be highly correlated with body fat percentage (Figure 2B, S3B and S3C).
Figure 2. Genetic Control of Dietary Responses to HF/HS Feeding.
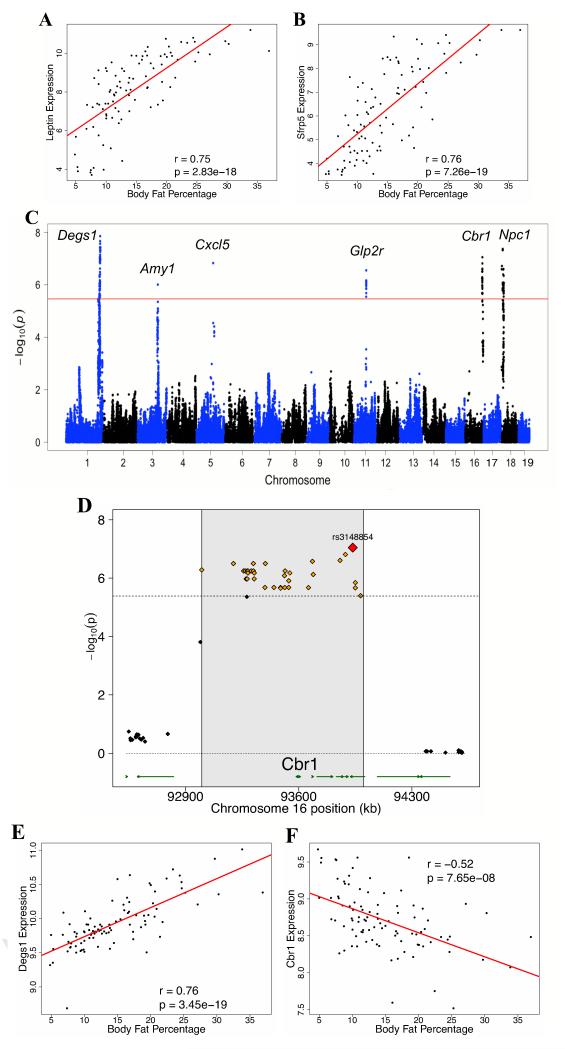
(A-B) Correlation of epididymal adipose gene expression of Leptin (Lep) (A) and Sfrp5 (B) with body fat percentage in male HMDP mice fed a chow diet, regression line (red). r, biweight midcorrelation, p, p-value.
(C) Manhattan plot showing the significance (−log10 of p) of all SNPs and percent body fat percentage increase after 8 weeks of HF/HS feeding in male HMDP mice. Candidate genes for genome-wide significant loci are indicated above genome-wide significant loci. Genome-wide significant threshold (red) of p=4.1 × 10−06 is indicated.
(D) Locus plot for genome-wide significant association at chromosome 16qC4 with approximate LD block (shaded in grey) and genome-wide significant SNPs (yellow) with the peak SNP (red).
(E-F) Correlation of epididymal adipose gene expression of Degs1 (E) and Cbr1 (F) with body fat percentage in male HMDP mice fed a chow diet, regression line (red). r, biweight midcorrelation, p, p-value.
Genetic Control of Obesity and Dietary Responsiveness
Most strains in the study showed a striking increase in body fat percentage within the first two weeks of HF/HS feeding (Figure 1C). Association analysis with body fat percent increase after 2 weeks of HF/HS feeding identified genome-wide significant loci on Chromosomes 2 and 6 (Table 2 and Figure S4A). The chromosome 2 locus (rs13476804; p = 2.95 × 10−6) contains one gene within the LD block, Sptlc3, which has been implicated in biogenesis of sphingolipids (Demirkan et al., 2012; Hornemann et al., 2009). The locus on chromosome 6 contains 11 genes within LD and the peak single nucleotide polymorphism (SNP), (rs13478690; p = 2.8 × 10−7) is 33.5kb upstream of Klf14, a primary candidate causal gene at this locus (Table 2 and Figure S4B). Klf14 has previously been identified in human GWAS for type 2 diabetes (Voight et al., 2010) and has recently been shown to be a master regulator of gene expression in adipose tissue (Small et al., 2011). Our results support a role of Klf14 in regulating changes in adipose tissue and indicate that Klf14 may also regulate dietary interactions.
Eight genome-wide significant loci were associated with body fat percentage growth after 8 weeks of HF/HS feeding (Table 2 and Figure 2C). The most significant signal (rs31849980; p = 1.4 × 10−8) maps to chromosome 1 and has genome-wide significant SNPs spanning a 5 Mb region with 60 genes within LD (Table 2). A primary candidate gene within this locus is Degs1, a fatty acid desaturase involved in the metabolism of important bioactive sphingolipids (Ternes et al., 2002). Degs1 expression in adipose tissue of chow fed male mice is strongly correlated (within top 10 genes) with body fat percentage (r = 0.7; p = 1.3 × 10−15) (Figure 3E). Previous linkage studies in mice have identified distal chromosome 1 as contributing importantly to obesity (Chen et al., 2008) and our results greatly refine this region and suggest Degs1 as a high-confidence candidate gene in the locus, although given the size of the locus multiple genes may be contributing to the signal.
Figure 3. Robust Shifts in Gut Microbiota Composition After HF/HS Feeding.
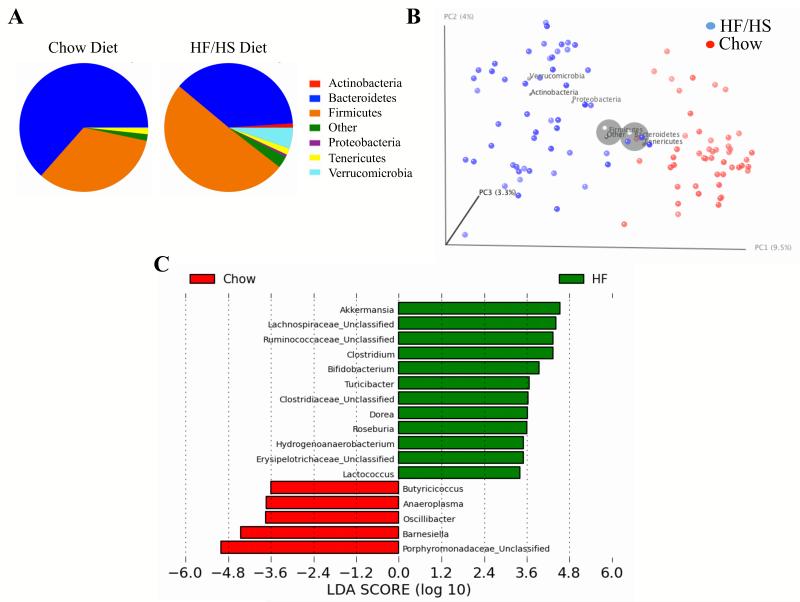
(A) Relative abundances of the different phyla after chow diet and HF/HS feeding (average among 52 matched strains).
(B) Principal Coordinates Analysis (PCoA) plot of the unweighted UniFrac distances. Each circle representing a different mice strain is colored according to the dietary conditions. PC1, PC2 and PC3 values for each mouse sample are plotted; percent variation explained by each PC is shown in parentheses.
(C) Linear Discriminant Analysis (LDA) coupled with effect size measurements identifies the most differentially abundant taxons between chow and HF/HS diets. HF/HS diet enriched taxa are indicated with a positive LDA score (green), and taxa enriched in normal chow diet have a negative score (red). Only taxa meeting an LDA significant threshold >2 are shown.
Of the eight loci associated with body fat percent growth after 8 weeks of HF/HS feeding both loci on chromosomes 16 and 18 contained genes with genome-wide cis eQTL and strong expression correlation with body fat percentage in epididymal adipose tissue (Figure 3C). The peak SNP at chromosome 18 (rs30078681; p = 4.3 × 10−8) contained 26 genes within LD and one gene, Npc1 was previously identified in a human GWAS for obesity (Meyre et al., 2009). Gene expression analysis of Npc1 indicated a strong negative correlation with body fat percentage (r = −0.4; p = 1.5 × 10−5) (Figure S3D) and the presence of a genome-wide significant cis eQTL (Figure S5A). Genomic sequence analysis indicated multiple non-synonymous coding variations within the Npc1 gene (Keane et al., 2011). Furthermore, Npc1 heterozygous knockout mice exhibit increased obesity on a high fat diet, but not on a chow diet (Jelinek et al., 2010), confirming the importance of Npc1 in regulating dietary interactions. The chromosome 16 locus (rs3148854; p = 9.0 × 10−8) has 8 genes within LD of the genome-wide significant SNPs and one gene in this region, Cbr1 (Figure 3D) is highly expressed in adipose tissue and is negatively correlated (r = −0.5; p = 1.6 × 10−7) with obesity (Figure 2F). Furthermore, Cbr1 has a cis eQTL (p = 1.1 × 10−4) (Figure 5B) and contains multiple non-synonymous coding variants (Keane et al., 2011). Cbr1 reduces carbonyl compounds via NADPH-dependent oxidoreductase activity and through its glutathione (GSH)-binding site Cbr1 can act on GSH-conjugated molecules and play a role in controlling oxidative stress (Bateman et al., 2008), an important component of obesity (Furukawa et al., 2004).
The loci for body fat growth following 8 weeks of HF/HS feeding also contained genes with known links to adipose biology and metabolism. The locus on chromosome 5 (rs13478388; p = 1.5 × 10−7) contains several C-X-C motif chemokine genes within LD. Cxcl5 is one of the genes in this cluster (Figure S4C), and expression of Cxcl5 is increased in adipose tissue of obese human subjects (Chavey et al., 2009). Cxcl5 contains non-synonymous coding polymorphisms and an alternative splice site among multiple mouse strains (Keane et al., 2011), making Cxcl5 a strong candidate at the chromosome 5 locus. Similarly, the significant SNP at chromosome 3 (rs29982345; p = 9.9 × 10−7) is within 1 Mb of three amylase genes, Amy2b, Amy2a5, and Amy1 (Figure S4D). Amylases play a critical role in the breakdown of polysaccharides to sugars and targeting amylases has been suggested as a potential method for treating obesity (Barrett and Udani, 2011). The significant peak on chromosome 11 (rs29417268; p = 2.8 × 10−7) has 11 genes within LD and one gene, Glp2r, plays an important role in intestinal homeostasis and feeding behavior (Guan et al., 2012). We also observed genome-wide significant loci for dietary response at two positions on chromosome 18 (rs13483184; p = 2.6 × 10−07 and rs29628302; p = 2.6 × 10−7) with no obvious candidate genes (Table 2).
In addition to loci influencing dietary response, there was one genome-wide significant locus at chromosome 7 associated with body fat percentage after 8 weeks of HF/HS feeding. This locus was gene-rich and contained 113 genes within LD of the peak SNP (rs13479513; p = 6.7 × 10−7). Two genes at this locus, Atp2a1 and Apob48r have previously been identified in human GWAS for BMI (Speliotes et al., 2010; Thorleifsson et al., 2009). Furthermore, this locus contains Sephs2, a strong candidate gene with both a significant cis eQTL (p = 1.4 × 10−4) and an exceptionally strong negative correlation with body fat percentage (r = 0.68; p = 2.0 × 10−14) (Figures S3E and S5C). Sephs2 is an evolutionarily conserved protein that is essential for production of selenocysteine (Xu et al., 2007), a key amino acid found in a diverse group of proteins with roles in the pathology of metabolic syndrome and diabetes (Rayman, 2012).
Conservation of Human and Mouse Metabolic Loci
A comparison of the genes/loci identified in our study with GWAS genes for human obesity and BMI revealed significant overlap (p = 4.0 × 10−4) (described in Experimental Procedures). In addition to genome-wide significant loci, we analyzed significant human GWAS genes in our association results and identified suggestive associations near Opcml and Sorl1 (Table 3). Both Opcml and Sorl1 have been found in large human GWAS for visceral adipose/subcutaneous adipose ratio and waist circumference, respectively (Fox et al., 2012; Smith et al., 2010). Our results demonstrate that genes associated with obesity and BMI in humans show functional variation in mice and are associated with dietary interactions and obesity.
Table 3. Significant Overlap of Mouse and Human GWAS.
| Mouse Trait | Chromosome | Peak SNP | P-value | Candidate Gene |
Corresponding Human GWAS Trait |
|---|---|---|---|---|---|
| Body Fat % increase – 0 to 8 weeks on HF/HS diet |
18 | rs30078681 | 4.33E-08 | Npcl | Obesity |
| Body Fat % increase – 0 to 2 weeks on HF/HS diet |
6 | rs13478690 | 2.75E-07 | Klf14 | Type 2 Diabetes, HDL Cholesterol |
| Body Fat % - 8 weeks on HF/HS diet | 7 | rs13479513 | 6.7E-07 | Atp2a1 | BMI, Weight |
| Body Fat % - 8 weeks on HF/HS diet | 7 | rs13479513 | 6.7E-07 | Apob48r | BMI |
| Body Fat % increase – 0 to 2 weeks on HF/HS diet |
9 | rs13459107 | 1.01E-05 | Opcml | Visceral Adipose/Subcutaneous Adipose Ratio |
| Body Fat % - 0 weeks on HF/HS diet | 9 | rs30236502 | 4.90E-05 | Sorll | Waist Circumference |
Identified mouse genetic loci containing genes previously identified in human GWAS studies for obesity and related traits. The corresponding mouse trait, location, P-value, candidate gene, and human GWAS Trait is indicated.
Contributions of Gut Microbiota to Obesity
A number of recent studies have implicated gut microbiota in metabolic and cardiovascular diseases (Turnbaugh et al., 2006; Vijay-Kumar et al., 2010; Wang et al., 2011). In order to investigate the genetic contributions to the gut microbiome and their relationship to obesity and dietary responses we examined the gut microbiota composition and distribution in strains after chow and HF/HS feeding. Since the microbiome is highly affected by environmental factors, all mice in the study were bred in the same facility and each strain was maintained in separate cages. We observed significant phylum-level shifts in the gut microbiota composition after HF/HS feeding (Figure 3A). The shifts between the two dominant phyla Bacteroidetes and Firmicutes after HF/HS feeding (Figure 3A and Table S4), are consistent with previous reports (Turnbaugh et al., 2009b). Principal Coordinates Analysis (PCoA) of unweighted UniFrac (similar results for weighted UniFrac shown in Figure S6B) (Lozupone and Knight, 2005) distances detected two clear clusters corresponding to the two dietary conditions (Figure 3B), suggesting that HF/HS feeding dramatically alters gut microbial communities across a variety of genetic backgrounds. Compared to mice on a chow diet the HF/HS fed mice had greater abundance of several genera classified to order Clostridales in Phyla Firmicutes and lower abundances of Bacteroidetes, classified to family Porphyromonadaceae (41% chow vs 18% HF/HS, p=4.36×10−11). In total we identified 17 genera whose abundance was significantly changed by diet (p<0.01) (Figure 3C, and Table S4). Interestingly, the abundance of Akkermansia genera of the Verrucomicrobia phylum explains much of the unweighted UniFrac distance clusters on PCoA plots in HF/HS diet (Figures S6C and S6D).
Robust effects of diet on the gut microbiota are well documented and recent evidence indicates that the gut microbiota constitute a complex polygenic trait under genetic regulation (Benson et al., 2010; McKnite et al., 2012; Qin et al., 2012). Consistent with the idea that the gut microbiota is influenced by genetic factors, we observed a strong effect of genetic background on the composition and plasticity of the gut microbiota after HF/HS feeding. Some strains showed large shifts in all major phyla, while other strains had little fluctuation in their gut microbiota after HF/HS feeding (Figure 4A). Additionally, Procrustes analysis was used to compare the relative orientation of matched strains after HF/HS and chow feeding and confirmed that the plasticity of gut microbial community is highly individualized (Figure 4B). Taken together, these results demonstrate a profound effect of HF/HS feeding on gut microbiota and show the strong influences of host genetics on influencing the plasticity of the gut microbiota in response to altered dietary compositions.
Figure 4. Plasticity of Gut Microbiota is Strain-Specific.
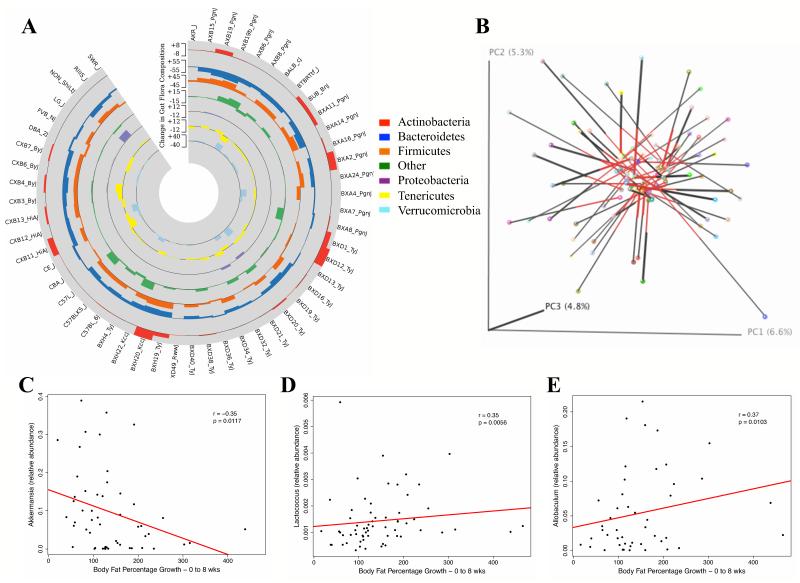
(A) Gut Microbiota phyla shift in 52 strains after HF/HS feeding for 8 weeks indicated by shift in percent composition of indicated phylum.
(B) Procrustes analysis of the same strains on chow diet and the HF/HS diet are linked with a bar. The unweighted UniFrac distances between the diets varies across strains, but are in general long, highlighting the shifts in microbial composition in response to diet.
(C-E) Correlation of relative abundance of Akkermansia (A), Lactococcus (B), and Allobaculum (C) with body fat percentage increase from 0 to 8 weeks in male mice fed a HF/HS diet, regression line (red), r, biweight midcorrelation, p, p-value.
To test the relationship between obesity, dietary responsiveness, and gut microbiota composition we analyzed all 11 genome-wide significant loci with the abundances of specific gut phylotypes. Only the chromosome 3 locus (rs29982345), which is associated with body fat growth after 8 weeks of HF/HS feeding showed a significant enrichment (p = 0.0033) of the genera Enterobacteriaceae (phylum Proteobacteria). This region of chromosome 3, which contains three amylase genes, may therefore contribute to both dietary responsiveness and gut microbiota composition. We also examined the relationship between specific gut phylotypes and obesity traits and observed a modest but statistically significant negative correlation with the abundance of the genera Akkermansia (phylum Verrucomicrobia) and body fat percentage growth after 8 weeks of HF/HS feeding (Figure 4C). Furthermore, body fat percentage growth after HF/HS feeding was positively correlated with the relative abundances of Lactococcus from phylum Firmicutes (Figure 4D) and with the genera Allobaculum (phylum Bacteroidetes) (Figure 4E). Additional studies are warranted to validate the connections of these specific gut microbiota and dietary interactions.
DISCUSSION
Our results have led to several findings about the genetic control of responses to energy-rich diets consumed by an increasing fraction of the world’s population. Although biochemical and genetic studies have revealed important regulators of body fat, including feedback pathways that modulate food intake and energy expenditure, these studies have not revealed how common genetic variation controls these processes. Human GWAS studies have been successful at identifying many loci controlling measures such as body mass index, but they have been unable to examine gene-by-environment interactions. Classic linkage studies in mice, on the other hand, have been able to partially address issues such as dietary responsiveness (Ehrich et al., 2005b), but have been hindered by poor mapping resolution, and, thus, only a handful of genes have been identified (Buchner et al., 2012; Dokmanovic-Chouinard et al., 2008; Ehrich et al., 2005a; York et al., 1999; Zhang et al., 1994). Our results add to this body of knowledge in several important ways. First, the improved mapping resolution enabled us to identify several candidate genes for gene-by-environment interactions in obesity. Second, because we examined obesity traits in a dynamic way, studying the mice following dietary challenge at multiple time points, we were able to provide strong evidence for the concept of a biologic set-point. Third, we have integrated the obesity traits with global adipose transcript levels and gut microbiota composition. Fourth, our results emphasize the importance of gene-by-environment interactions, with important implications for an understanding of the overall genetic architecture of obesity. Finally, our results provide the basis of a systems genetics resource for obesity traits that can be expanded to include multiple biologic scales using metabalomics, proteomics, and epigenetics. In particular it will be of interest to examine behavioral and neurological differences among the strains as they relate to obesity traits.
Using association rather than linkage, we were able to obtain biologically significant mapping data, as several of the loci identified for the response to a HF/HS diet contained between 1-3 genes, comparable to human GWAS results. Altogether, we examined only about 800 mice, much fewer than the tens of thousands examined in human GWAS. It is likely that power is greatly increased in mice, as compared to humans, because of the ability to accurately monitor the phenotypes (body fat, food intake) and control the environment. Also, for the loci containing larger numbers of candidates, examination of transcript levels in adipose tissue and access to complete DNA sequence information, allowed us to prioritize candidates. Indeed, several of the genes at such loci (Npc1 and Glp2r) have already been studied using transgenic approaches with results consistent with our findings (Guan et al., 2012; Jelinek et al., 2010).
Several different models have been developed to understand how genes and environment combine to regulate body fat. However, only limited studies have addressed the genetic basis of gene-by-environment interactions in obesity. The dual intervention point model postulates an upper and lower limit where genetic factors become dominant, and between which there is only weak genetic regulation (Speakman et al., 2011). Our results strongly support this concept, because after 4 weeks of HF/HS feeding most mouse strains reach an upper limit where there is no further increase in body fat (Figure 1). Thus, there must be strong genetic factors that resist continued fat accumulation beyond a certain point. Clearly, this set-point varies widely in the population; some strains have already achieved the set-point level on a chow diet whereas a few strains continue to accumulate fat throughout the 8 week feeding period, suggesting that the set-point mechanism is disrupted. It is noteworthy that obesity prevalence appears to be leveling off in developed countries, which is also consistent with the idea of a set-point (Flegal et al., 2012). Based on our data (Table 1) and a monozygotic twin study (Bouchard et al., 1990), acute body fat changes in response to an obesogenic environment are under strong genetic regulation, although this may be part of the same pathway that determines the set-point. Our data also show that food intake is only modestly correlated with obesity and dietary responsiveness, and indicates energy expenditure may be a likely mechanism contributing to the obesity set-point. Further metabolic studies of strains at the extremes will be required to confirm the effect of energy expenditure on obesity set-points.
The experimental design of our study, involving inbred strains of mice, allowed us to study the gut microbiota composition of the mice both on a chow and HF/HS diet. The HF/HS diet clearly influenced gut microbiota composition in all of the 52 strains examined on both chow and the HF/HS diet (Figure 3). Shifts in gut microbiota composition and diversity in response to dietary changes are similar to those previously noted in both human (Ley et al., 2006; Turnbaugh et al., 2009b) and mouse studies (Ley et al., 2005; Turnbaugh et al., 2006). Our data indicate that host genetic factors influence gut microbiota plasticity in response to diet; however, since the gut microbiota can vary significantly even in well-controlled cohorts we cannot exclude the possibility that other factors could partly explain the observed differences. We observed significant relationships between gut microbiota and metabolic traits (Figure 4C, 4D, 4E). The results are consistent with the concept that gut microbiota contribute to systemic functions and common diseases (Clemente et al., 2012). Future studies in the HMDP have the potential to test the roles of specific gut microbiota using transplantation and cross-fostering to systematically examine the relationship between genetic background, diet, and gut microbiota composition.
Recent successes in human GWAS for common diseases have yielded many genetic loci, but in most instances they have revealed only a very small fraction of the estimated heritability (Lander, 2011). While it is likely that some of the “missing heritability” will be found in rare variants in the population, our results, and those from other studies in animal models (Burrage et al., 2010; Lawson et al., 2011; Shao et al., 2008) suggest that genetic interactions are likely to have important contributions. Clearly, human studies have limited ability to identify gene-by-environment or gene-by-gene interactions (Zuk et al., 2012), and our results suggest that GWAS in mice will provide a powerful complement to human studies. A key feature of our experimental design is that genetically identical mice can be studied under multiple environmental conditions. Also, the greatly improved resolution of association as compared to classical linkage studies in mice greatly facilitates gene identification and mechanistic understanding. The substantial overlap of our study with human GWAS for obesity traits suggests that at least for metabolic disorders, the mouse is highly relevant and that natural variations of the two species affect shared pathways.
Because the inbred strains of the HMDP are permanent, data obtained about the strains is cumulative, allowing integration with previous results. The data compiled here provides a basis for further genetic studies of obesity-related traits in mice. It will be of interest, for example, to examine additional intermediate phenotypes, such as protein and metabolite levels, in the same set of strains, and to relate these to the obesity traits using association mapping, correlation, and modeling. In this study, we examined transcript levels in adipose tissue, and the composition of gut microbiota and in the future it would be valuable to examine transcript levels in other relevant tissues, such as the hypothalamus, liver, and muscle, as well as protein, metabolite levels, and epigenetic markers. Moreover, traits such as epigenetic variation and gut microbiota composition can be tested for effects on obesity-related traits in this set of strains.
EXPERIMENTAL PROCEDURES
Animals
All mice were obtained from The Jackson Laboratory and were bred at University of California, Los Angeles to generate mice used in this study. Mice were maintained on a chow diet (Ralston Purina Company) until 8 weeks of age when they were given a high-fat, high-sucrose diet (Research Diets-D12266B) with the following composition, 16.8 % kcal protein, 51.4 % kcal carbohydrate, 31.8 % kcal fat. Age-matched control male mice were fed a chow diet (18% kcal fat) for 16 weeks. A complete list of the strains included in our study is included in Supplemental Tables 1 and 2. The animal protocol for the study was approved by the Institutional Care and Use Committee (IACUC) at University of California, Los Angeles.
Association Analysis and Heritability Calculations
We performed the association testing of each SNP using a linear mixed model, which accounts for the population structure among the n animals using the following model (Kang et al., 2008):
,where μ is the mean, β is the allele effect of the SNP, x is the (nx1) vector of observed genotypes of the SNP, u is the random effects due to genetic relatedness with and e is the random noise with . K denotes the identity-by-state (IBS) kinship matrix estimated from all the SNPs, I denotes the (nxn) identity matrix and 1n is the (nx1) vector of ones. We estimated and using restricted maximum likelihood (REML), and computed p-values using the standard F-test to test the null hypothesis β = 0. Genome-wide significance threshold and genome-wide association mapping are determined as the family-wise error rate as the probability of observing one or more false positives across all SNPs for phenotype. We ran 100 different sets of permutation tests and parametric boot strapping of size 1,000, and observed that the mean and standard error of the genome-wide significance threshold at the family-wise error rate of 0.05 were 3.9 × 10−6 ± 0.3 × 10−6, and 4.0 × 10−6 ± 0.3 × 10−6, respectively. A detailed explanation of the analyses is provided in Bennett et. al. (Bennett et al., 2010). Linkage disequilibrium (LD) was determined by calculated pairwise r2 SNP correlations for each chromosome. Approximate LD boundaries were determined by visualizing r2 > 0.8 correlations in MATLAB (MathWorks)
Heritability (in the narrow-sense) is defined as the phenotypic variance explained by additive genetic effects and is computed using the following model (Yang et al., 2010):
We estimated and using REML and calculated the heritability (h2) for each trait as follows:
where tr(.) is the matrix trace and .
Body Composition Analysis
Animals were measured for total body fat mass and lean mass by magnetic resonance imaging (MRI) using Bruker Minispec with software from Eco Medical Systems, Houston, TX (Taicher et al., 2003). All animals in the study were measured at 0, 2, 4, 6, and 8 weeks of HF/HS feeding.
Assessment of food intake
After 4 weeks of HF/HS feeding, food intake was monitored for 5 days. Ninety to 100 grams of food was weighed and put in the food hopper, after 24 hours food was weighed, and then weighed again after 24 hours. The procedure was repeated again with fresh food, and a total of 4 × 24 hour period total food weight consumed was calculated by subtracting food weights; grams per mouse was then calculated by dividing average total food weight consumed in 4 × 24 hour periods by number of mice in cage.
In vivo metabolism assessment
Metabolic rate, activity, food and water consumption were assessed using a Columbus Instruments Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) equipped with sub-systems for open circuit indirect calorimetry, and monitoring of activity, feeding, and drinking. Detailed methods for in vivo metabolism assessment provided in Supplementary Experimental Procedures.
Gut Microbiota Analysis
Microbial community composition was assessed by pyrosequencing 16S rRNA genes derived from the cecal samples of chow diet and HF/HS diet animals. All mice used in the study were bred at UCLA to ensure uniform environmental conditions. Furthermore, strains were individually housed within the same vivarium throughout the duration of the study. To ensure comprehensive analysis we sequenced biological replicates on all mice included in the study. Biological replicates were carried out on all mice included in the study. For mice fed a HF/HS diet we sequenced a range of 2 to 5 mice per strain, with an average of 3 mice per strain (n=339 male mice from 110 strains). Chow diet mice were sequenced by pooling 2 to 4 mice per strain, with an average of 2 per strain (n=108 male mice from 52 strains). We also sequenced technical replicates on 8 mouse strains (5 strains on chow diet and 3 on HF/HS diet) and observed high reproducibility of UniFrac analysis between technical replicates (Figure S6A). Detailed methods for gut microbiota analysis provided in Supplementary Experimental Procedures.
Adipose RNA isolation and global gene expression analysis
Flash frozen epididymal adipose samples from male mice (16 weeks old) maintained on a chow diet were weighed and homogenized in Qiazol (Qiagen) and RNA isolated according to the manufacturer’s protocol using RNeasy columns (Qiagen). Isolated RNA (2 mice per strain as indicated in Supplementary Table 2) was analyzed for global gene expression using Affymetrix HT_MG430A arrays and was filtered as described (Bennett et al., 2010).
Expression eQTL analysis
GWAS for gene expression was performed using EMMA and defined as cis if peak SNP mapped within 1 Mb of gene position and trans if mapped outside (cis threshold: p < 1.4 × 10−3 and trans threshold: p < 6.1 × 10−6).
Statistics
Correlations were calculated using the biweight midcorrelation, which is robust to outliers (Wilcox, 2005). Overlap p-value between mouse and human GWAS genes was calculating using hypergeometric cumulative distribution function in MATLAB (MathWorks) using pval = 1 - hygecdf(x,M,K,N), where x = total number of genes in genome, M = total number of genes identified in mouse and humans, K = number of genes identified in obesity and BMI human GWAS, N = number of genes identified in mouse obesity traits.
Supplementary Material
HIGHLIGHTS.
Detailed analysis of diet-induced obesity in more than 100 inbred mouse strains
Identification of 11 genetic loci associated with obesity and dietary responsiveness
Significant overlap between mouse loci with human GWAS loci for obesity
Strain-specific shifts in gut microbiota composition in response to dietary intervention
ACKNOWLEDGEMENTS
We thank Hannah Qi, Zhiqiang Zhou, Judy Wu, and Tieyan Han for expert assistance with mouse experiments. We thank Raffi Hagopian for help with preparing figures and Luz Orozco for statistical advice. This work was supported by National Institutes of Health (NIH) grant HL028481 to A.J.L and by a Howard Hughes Medical Institute Early Career Scientist award to R.K. B.W.P. was supported by a NIH training grant T32-HD07228. E.O. was supported by MOBILITAS Postdoctoral Research Grant (MJD252). C.D.R. was supported by a NIH training grant T32-HL69766. M.C. was supported by American Heart Association postdoctoral fellowship (10POST3660048). L.K.U. was supported by NIH training grant T32-GM08759. A.H., B.Z., M.K., P.G., and T.K. are employees and shareholders of Bristol-Myers Squibb
Footnotes
Accession Numbers
All microarray data from this study are deposited in the NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) under the accession number XXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Attie AD, Scherer PE. Adipocyte metabolism and obesity. J Lipid Res. 2009;50(Suppl):S395–399. doi: 10.1194/jlr.R800057-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett ML, Udani JK. A proprietary alpha-amylase inhibitor from white bean (Phaseolus vulgaris): a review of clinical studies on weight loss and glycemic control. Nutr J. 2011;10:24. doi: 10.1186/1475-2891-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh GS, Farooqi IS, O’Rahilly S. Genetics of body-weight regulation. Nature. 2000;404:644–651. doi: 10.1038/35007519. [DOI] [PubMed] [Google Scholar]
- Bateman RL, Rauh D, Tavshanjian B, Shokat KM. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem. 2008;283:35756–35762. doi: 10.1074/jbc.M807125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Oler AT, Rabaglia ME, Stapleton DS, Schueler KL, Truchan NA, Worzella SL, Stoehr JP, Clee SM, Yandell BS, et al. Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet. 2011;7:e1002323. doi: 10.1371/journal.pgen.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G, Dussault J, Moorjani S, Pinault S, Fournier G. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. doi: 10.1056/NEJM199005243222101. [DOI] [PubMed] [Google Scholar]
- Buchner DA, Geisinger JM, Glazebrook PA, Morgan MG, Spiezio SH, Kaiyala KJ, Schwartz MW, Sakurai T, Furley AJ, Kunze DL, et al. The juxtaparanodal proteins CNTNAP2 and TAG1 regulate diet-induced obesity. Mamm Genome. 2012 doi: 10.1007/s00335-012-9400-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrage LC, Baskin-Hill AE, Sinasac DS, Singer JB, Croniger CM, Kirby A, Kulbokas EJ, Daly MJ, Lander ES, Broman KW, et al. Genetic resistance to diet-induced obesity in chromosome substitution strains of mice. Mamm Genome. 2010;21:115–129. doi: 10.1007/s00335-010-9247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavey C, Lazennec G, Lagarrigue S, Clape C, Iankova I, Teyssier J, Annicotte JS, Schmidt J, Mataki C, Yamamoto H, et al. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab. 2009;9:339–349. doi: 10.1016/j.cmet.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirkan A, van Duijn CM, Ugocsai P, Isaacs A, Pramstaller PP, Liebisch G, Wilson JF, Johansson A, Rudan I, Aulchenko YS, et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012;8:e1002490. doi: 10.1371/journal.pgen.1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokmanovic-Chouinard M, Chung WK, Chevre JC, Watson E, Yonan J, Wiegand B, Bromberg Y, Wakae N, Wright CV, Overton J, et al. Positional cloning of “Lisch-Like”, a candidate modifier of susceptibility to type 2 diabetes in mice. PLoS Genet. 2008;4:e1000137. doi: 10.1371/journal.pgen.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich TH, Hrbek T, Kenney-Hunt JP, Pletscher LS, Wang B, Semenkovich CF, Cheverud JM. Fine-mapping gene-by-diet interactions on chromosome 13 in a LG/J × SM/J murine model of obesity. Diabetes. 2005a;54:1863–1872. doi: 10.2337/diabetes.54.6.1863. [DOI] [PubMed] [Google Scholar]
- Ehrich TH, Kenney-Hunt JP, Pletscher LS, Cheverud JM. Genetic variation and correlation of dietary response in an advanced intercross mouse line produced from two divergent growth lines. Genet Res. 2005b;85:211–222. doi: 10.1017/S0016672305007603. [DOI] [PubMed] [Google Scholar]
- Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E, Kang HM, Furlotte N, Berberyan A, Ghazalpour A, et al. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 2011;7:e1002038. doi: 10.1371/journal.pgen.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Flint J, Eskin E. Genome-wide association studies in mice. Nat Rev Genet. 2012;13:807–817. doi: 10.1038/nrg3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N, Lohman K, Johnson AD, Foster MC, Greenawalt DM, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8:e1002695. doi: 10.1371/journal.pgen.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Shi X, Li X, Chang B, Wang Y, Li D, Chan L. GLP-2 receptor in POMC neurons suppresses feeding behavior and gastric motility. Am J Physiol Endocrinol Metab. 2012;303:E853–864. doi: 10.1152/ajpendo.00245.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann T, Penno A, Rutti MF, Ernst D, Kivrak-Pfiffner F, Rohrer L, von Eckardstein A. The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. J Biol Chem. 2009;284:26322–26330. doi: 10.1074/jbc.M109.023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe E, Moon B, Connolly E, Friedman JM. Abnormal regulation of the leptin gene in the pathogenesis of obesity. Proc Natl Acad Sci U S A. 1998;95:11852–11857. doi: 10.1073/pnas.95.20.11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek D, Heidenreich RA, Erickson RP, Garver WS. Decreased Npc1 gene dosage in mice is associated with weight gain. Obesity (Silver Spring) 2010;18:1457–1459. doi: 10.1038/oby.2009.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpelainen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, Demerath E, Ahmad T, Mora S, Kaakinen M, Sandholt CH, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011;8:e1001116. doi: 10.1371/journal.pmed.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R. Human-associated microbial signatures: examining their predictive value. Cell Host Microbe. 2011;10:292–296. doi: 10.1016/j.chom.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Lawson HA, Cady JE, Partridge C, Wolf JB, Semenkovich CF, Cheverud JM. Genetic effects at pleiotropic loci are context-dependent with consequences for the maintenance of genetic variation in populations. PLoS Genet. 2011;7:e1002256. doi: 10.1371/journal.pgen.1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik VS, Popkin BM, Bray GA, Despres JP, Hu FB. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffery JM, Papandonatos GD, Peter I, Huggins GS, Raynor HA, Delahanty LM, Cheskin LJ, Balasubramanyam A, Wagenknecht LE, Wing RR. Obesity susceptibility loci and dietary intake in the Look AHEAD Trial. Am J Clin Nutr. 2012 doi: 10.3945/ajcn.111.026955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, Andreux PA, Bastiaansen JW, Wang X, Kachman SD, Auwerx J, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One. 2012;7:e39191. doi: 10.1371/journal.pone.0039191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyre D, Delplanque J, Chevre JC, Lecoeur C, Lobbens S, Gallina S, Durand E, Vatin V, Degraeve F, Proenca C, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, Akasaki Y, Shimono A, Walsh K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010;329:454–457. doi: 10.1126/science.1188280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Rayman MP. Selenium and human health. Lancet. 2012;379:1256–1268. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- Sandholt CH, Sparso T, Grarup N, Albrechtsen A, Almind K, Hansen L, Toft U, Jorgensen T, Hansen T, Pedersen O. Combined analyses of 20 common obesity susceptibility variants. Diabetes. 2010;59:1667–1673. doi: 10.2337/db09-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Burrage LC, Sinasac DS, Hill AE, Ernest SR, O’Brien W, Courtland HW, Jepsen KJ, Kirby A, Kulbokas EJ, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci U S A. 2008;105:19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EN, Chen W, Kahonen M, Kettunen J, Lehtimaki T, Peltonen L, Raitakari OT, Salem RM, Schork NJ, Shaw M, et al. Longitudinal genome-wide association of cardiovascular disease risk factors in the Bogalusa heart study. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonestedt E, Roos C, Gullberg B, Ericson U, Wirfalt E, Orho-Melander M. Fat and carbohydrate intake modify the association between genetic variation in the FTO genotype and obesity. Am J Clin Nutr. 2009;90:1418–1425. doi: 10.3945/ajcn.2009.27958. [DOI] [PubMed] [Google Scholar]
- Speakman JR, Levitsky DA, Allison DB, Bray MS, de Castro JM, Clegg DJ, Clapham JC, Dulloo AG, Gruer L, Haw S, et al. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech. 2011;4:733–745. doi: 10.1242/dmm.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Magi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256:51–54. [PubMed] [Google Scholar]
- Taicher GZ, Tinsley FC, Reiderman A, Heiman ML. Quantitative magnetic resonance (QMR) method for bone and whole-body-composition analysis. Anal Bioanal Chem. 2003;377:990–1002. doi: 10.1007/s00216-003-2224-3. [DOI] [PubMed] [Google Scholar]
- Ternes P, Franke S, Zahringer U, Sperling P, Heinz E. Identification and characterization of a sphingolipid delta 4-desaturase family. J Biol Chem. 2002;277:25512–25518. doi: 10.1074/jbc.M202947200. [DOI] [PubMed] [Google Scholar]
- Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, Styrkarsdottir U, Gretarsdottir S, Thorlacius S, Jonsdottir I, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nature Genetics. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009a;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009b;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox R. Introduction to Robust Estimation and Hypothesis Testing. Elsevier/Academic Press; Amsterdam, The Netherlands: 2005. [Google Scholar]
- Xu XM, Carlson BA, Irons R, Mix H, Zhong N, Gladyshev VN, Hatfield DL. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem J. 2007;404:115–120. doi: 10.1042/BJ20070165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York B, Truett AA, Monteiro MP, Barry SJ, Warden CH, Naggert JK, Maddatu TP, West DB. Gene-environment interaction: a significant diet-dependent obesity locus demonstrated in a congenic segment on mouse chromosome 7. Mamm Genome. 1999;10:457–462. doi: 10.1007/s003359901023. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.