

Abstract
Cancer stem cells have been described in various cancers including squamous tumours of the skin by their ability to reform secondary tumours upon transplantation into immunodeficient mice. Here, we used transplantation of limiting dilution of different populations of FACS-isolated tumour cells from four distinct mouse models of squamous skin tumours to investigate the frequency of tumour propagating cells (TPCs) at different stages of tumour progression. We found that benign papillomas, despite growing rapidly in vivo and being clonogenic in vitro, reformed secondary tumours upon transplantation at very low frequency and only when tumour cells were co-transplanted together with tumour-associated fibroblasts or endothelial cells. In two models of skin squamous cell carcinoma (SCC), TPCs increased with tumour invasiveness. Interestingly, the frequency of TPCs increased in CD34HI but not in CD34LO SCC cells with serial transplantations, while the two populations initially gave rise to secondary tumours with the same frequency. Our results illustrate the progressive increase of squamous skin TPCs with tumour progression and invasiveness and reveal that serial transplantation may be required to define the long-term renewal potential of TPCs.
Keywords: cancer stem cells, carcinoma, papilloma, skin cancers
Introduction
Different models have been proposed to explain tumour growth and heterogeneity. In the stochastic model of tumour growth, all cancer cells have the same intrinsic properties to contribute to tumours growth and choose between self-renewal and differentiation in a stochastic manner (Shackleton et al, 2009; Nguyen et al, 2012). In contrast, in the cancer stem cell (CSC) model, tumours are hierarchally organized, with only some tumour cells, called CSCs, presenting greater renewing potential that sustain long-term tumour growth (Reya et al, 2001; Shackleton et al, 2009; Nguyen et al, 2012). Many recent studies using prospective isolation of a fraction of tumour cells followed by their transplantation into immunodeficient mice have demonstrated that certain population of tumour cells contains cells with higher probability to reform secondary tumour upon transplantation, supporting the existence of CSC (Reya et al, 2001; Pardal et al, 2003; Lobo et al, 2007; Shackleton et al, 2009; Nguyen et al, 2012). Furthermore, the existence of CSCs in different types of primary solid tumours, including skin squamous tumours, has recently been confirmed by lineage-tracing experiments (Driessens et al, 2012; Schepers et al, 2012).
Skin squamous cell carcinoma (SCC) is the second most frequent skin cancer in human and affects about 500 000 new patients per year worldwide (Alam and Ratner, 2001). Mouse models for skin squamous tumours resemble human skin cancers and offer an ideal model to study cancer initiation and growth (Owens and Watt, 2003; Perez-Losada and Balmain, 2003). The most extensively used mouse cancer model is the multistage chemically induced skin tumours (Kemp, 2005; Abel et al, 2009). In the first step (called ‘initiation’), mice are treated with a low dose of the mutagen 9,10-dimethyl-1,2-benzanthracene (DMBA). In the second step (called ‘promotion’), mice are treated with 12-O-tetradecanoyl phorbol-13-acetate (TPA), a drug that stimulates epidermal proliferation. During promotion, benign tumours (papilloma) arise, some of which progress to invasive SCC. Genetic mouse models of skin squamous tumours have been developed by combining conditional expression KRasG12D mutant and p53 deletion in different population of epidermal cells (Lapouge et al, 2011; White et al, 2011). These models allow the initiation of either benign tumours (expression of KRasG12D alone) or aggressive SCC (combining oncogenic KRasG12D expression and p53 deletion) and genetically control their p53 status, which plays a key role in skin tumour progression (Kemp et al, 1993). Skin SCCs arising in these genetic mouse models present features of very invasive tumours including spindle shape and expression of EMT markers (Lapouge et al, 2011; White et al, 2011).
Recently, CD34-expressing tumour propagating cells (TPCs) with increased clonogenic potential and the ability to form secondary tumours upon transplantation into immunodeficient mice have been isolated from DMBA/TPA-induced skin SCC (Malanchi et al, 2008). However, it was later shown that both CD34HI and CD34LO tumour epithelial cell (TECs) populations from DMBA/TPA-induced skin SCC cultured in vitro 4 days before their transplantation present similar ability to reform secondary tumour upon transplantation into immunodeficient mice (Schober and Fuchs, 2011). These two conflicting studies raise questions to whether CD34 expression can be used as marker to enrich for TPCs in skin SCCs. Moreover, it is not clear from these studies what is the exact frequency of TPCs in primary murine SCC and whether this frequency changes with tumour progression.
In this study, we investigate the frequency of TPCs along tumour progression in different models of squamous skin tumours. We found that papilloma, although proliferating intensively in primary tumours in vivo and during in vitro culture, present a very low tumour propagating potential in this assay, and only when co-transplanted together with tumour endothelial cells or tumour-associated fibroblasts. In contrast, we found that the frequency of TPCs massively increased with tumour progression, invasiveness and serial transplantation. These data demonstrate the importance of the tumour microenvironment and other intrinsic tumour features such as the loss of p53 in dictating the ability of TPCs to reform secondary tumour upon transplantation into immunodeficient mice, and which may not necessarily reflect the actual growing rate of the primary tumours or their CSC content.
Results
CD34 is expressed in papilloma and carcinoma from different mouse models of skin tumours
We first examined the expression of CD34, a previously reported CSC marker of human SCC and mouse DMBA/TPA-induced squamous skin tumours (Malanchi et al, 2008), in different mouse models of skin squamous tumours that differed by their stage of tumour progression and invasiveness (Figure 1A). Immunofluorescence analysis revealed that Lin−/α6+/Epcam+/CD34+ TECs were located mostly basally (K5 positive), close to the endothelial cells (endoglin positive cells) in both KRasG12D and DMBA/TPA-induced skin papillomas (Figure 1B) (Beck et al, 2011). CD34 is more widely expressed in tumour cells of invasive SCCs irrespective of the mouse model used to induce cancer formation (Figure 1B). We used FACS analysis to quantify the proportion of CD34+ in Lin−/α6+/Epcam+ TECs in benign papilloma and invasive SCC induced by DMBA/TPA-induced carcinogenesis and in Lin−/α6+/YFP+ TECs genetic mouse model involving the combination of oncogenic KRasG12D expression and p53 deletion. Irrespective of whether papilloma are induced by DMBA/TPA carcinogenesis or by oncogenic KRasG12D expression, about 20% of Lin−/α6+/Epcam+ basal TECs expressed CD34 (Figure 1C and D). As tumours progress, the proportion of CD34+ TECs significantly increased, reaching at least 60–70% of basal Lin−/α6+/Epcam+ or YFP+ TECs in invasive SCC from both DMBA/TPA and genetically induced SCC (Figure 1C and D). Altogether, these data show that CD34 is expressed by a fraction of squamous skin tumours including benign papilloma and malignant SCC and the proportion of Lin−/α6+/Epcam+ or YFP+/CD34+ TECs increased during tumour progression.
Figure 1.
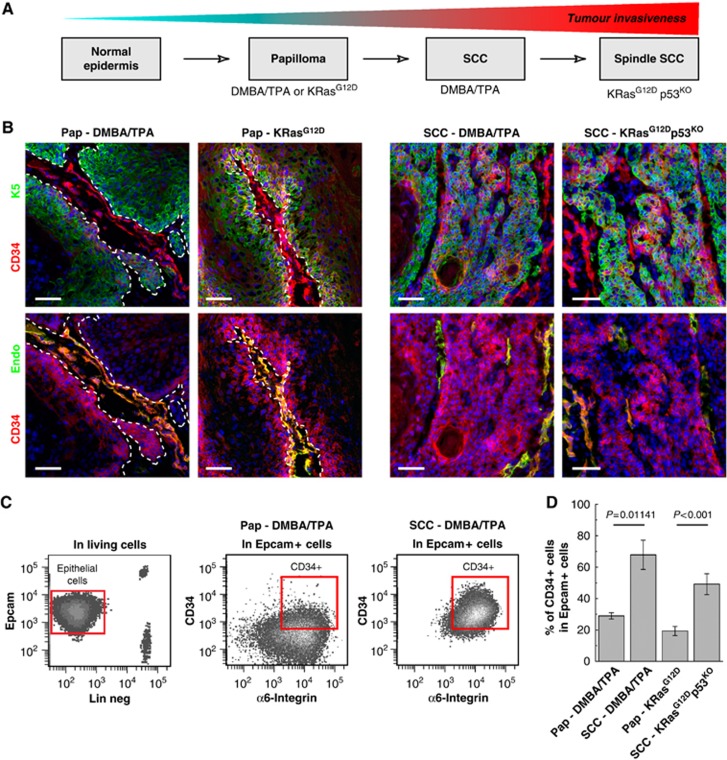
CD34 is expressed in papilloma and carcinoma from different mouse models of skin tumours. (A) Scheme representing the progression of mouse skin tumours and the model used to study them. Adapted from Frame et al (1998). (B) Immunostaining for CD34 and K5 (upper panel) or CD34 and endothelial cell marker endoglin (endo—lower panel) showing the presence of CD34-expressing cells in all types of squamous tumours. (C) FACS analysis of the proportion of Lin−/α6+/Epcam+/CD34+ TEC population in benign papilloma and malignant SCC. (D) Histogram showing the percentage of Lin−/α6+/Epcam+/CD34+ cells in benign tumours (papilloma) arising from DMBA/TPA mice and in K19CREER::KRasG12D mice and in malignant carcinoma (SCC) arising from DMBA/TPA-treated mice and following TAM administration in K14CREER::KRasG12D::p53fl/fl mice (n=6 mice/condition). Pap, Papilloma. Scale bars represents 50 μm.
TECs from benign papilloma cannot be propagated into immunodeficient mice without their tumour stroma
During DMBA/TPA-induced carcinogenesis, papillomas arise around 10 weeks after the first administration of DMBA/TPA and grow steady thereafter upon TPA treatment (Figure 2A). Similarly, papilloma arising from oncogenic KRasG12D expression in the basal epidermis (K14CREER/KRasG12D) or in the bulge SCs (K19CREER/KRasG12D and Lgr5CREER/KRasG12D) developed 2−4 months after TAM administration and grew at a slightly higher rate than the DMBA/TPA-induced papillomas (Figure 2A). Analysis of cell proliferation in DMBA/TPA and KRasG12D induced papillomas demonstrated the basal cells were highly proliferative and about 30% of basal TECs incorporate ethynyl deoxyuridine (EdU) after a 4-h pulse (Figure 2B and C).
Figure 2.
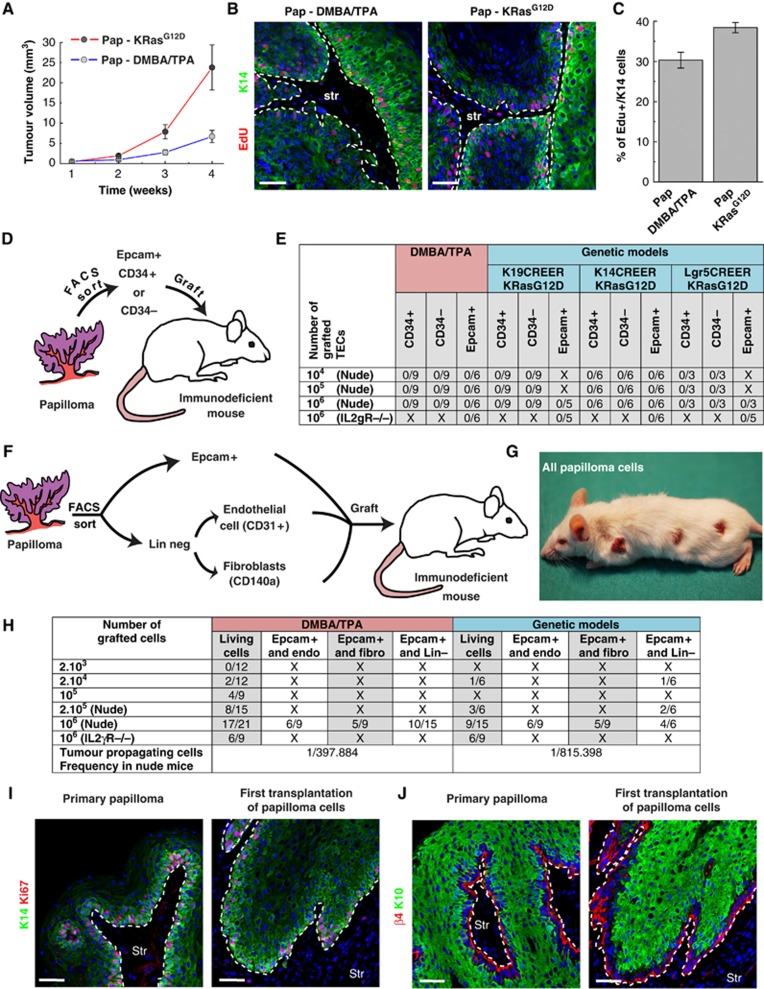
TECs from benign papilloma cannot be propagated into immunodeficient mice without tumour stromal cells. (A) Tumour volume of chemically (DMBA/TPA, blue) and genetically (KRasG12D, red) induced papilloma over time (n=32 tumours from three different mice per condition). Values are presented as mean values±s.e.m. (B) Immunostaining of EdU and K14 in DMBA/TPA and genetically induced papilloma, 4 h after an EdU pulse. (C) Proliferation of papilloma TECs. Quantification of percentage of EdU+/K5+ cells in papilloma arising from DMBA/TPA-treated and K19CREER::KRasG12D mice (D) Scheme representing the strategy used to isolate and grafted TECs from DMBA/TPA and KRasG12D papillomas. (E) Table summarizing the frequency of secondary tumour formation after subcutaneous injection of Lin−/α6+/Epcam+/CD34+, Lin−/α6+/Epcam+/CD34− or Lin−/α6+/Epcam+ epithelial cells from DMBA/TPA and genetically induced papilloma, into immunodeficient Swiss Nude and NOD/SCID/IL2Rγ null mice. (F) Scheme representing the strategy used to isolate and co-graft Lin−/α6+/Epcam+ cells with tumour endothelial cells (CD31+), tumour fibroblasts (CD140a+) or all tumour stromal cells (Lin neg). (G) Picture of a mouse grafted with 106 total living cells from DMBA/TPA-induced papilloma 6 weeks after grafting. (H) Table summarizing the frequency of secondary tumour formation after subcutaneous injection of Lin−/α6+/Epcam+ epithelial cells co-grafted with tumour endothelial cells, tumour fibroblasts or tumour stromal cells (Lin−) from DMBA/TPA and genetically induced papilloma, into immunodeficient Swiss Nude and NOD/SCID/IL2Rγ null mice. (I, J) Immunostaining of K14 and Ki67 (I) as well as β4 integrin and K10 (J) in primary papilloma and secondary (first transplantation) tumours arising from the transplantation of total tumour cells into immunodeficient mice. Scale bars represents 50 μm, str means stroma, Endo means Endothelial cells, fibro means fibroblast, Lin− means Lineage negative. Hoechst is represented in blue.
We first investigated the renewal potential of TECs from papillomas by testing their ability to form colonies in vitro. FACS-isolated Lin−/α6+/Epcam+/CD34+ and Lin−/α6+/Epcam+/CD34− TECs from DMBA/TPA and KRasG12D induced papillomas were cultured on feeder layers and their ability to form proliferative colonies was assessed during the first primary culture, to avoid the confounding factors of chromosomal abnormalities that can arise after multiple passages of murine keratinocytes. The colony forming efficiency as well as the total cell output during the 2 weeks of primary culture were comparable between Lin−/α6+/Epcam+/CD34+ and Lin−/α6+/Epcam+/CD34− TECs isolated from DMBA/TPA and genetically induced tumours (Supplementary Figure S1A and B). The TECs from papillomas proliferated very rapidly in vitro, as demonstrated by the high percentage of EdU-positive cells after a short pulse and were able to undergo terminal differentiation as shown by the presence of few K10-positive cells in the centre of the colonies (Supplementary Figure S1C and D).
We next investigated the ability of Lin−/α6+/Epcam+/CD34+ and Lin−/α6+/Epcam+/CD34− TECs from papillomas to reform the parental tumour upon transplantation into immunodeficient mice (Figure 2D). Very surprisingly, one million of FACS purified Lin−/α6+/Epcam+/CD34+ and Lin−/α6+/Epcam+/CD34− TECs which correspond to the total number of Lin−/α6+/Epcam+/CD34+ cells found in multiple papillomas induced by DMBA/TPA treatment or by oncogenic KRasG12D expression in different cell lineages of the skin epidermis did not induce secondary tumour formation upon transplantation into immunodeficient mice (Figure 2E). To investigate whether the degree of immunosupression affect the formation of secondary tumour following transplantation of Lin−/α6+/Epcam+ TECs from skin papilloma, as it has been proposed for human melanoma (Quintana et al, 2008), we transplanted one million of total Lin−/α6+/Epcam+ TECs into the most severe immunodeficient mice (NOD/SCID/Il2Rγ null mice). Despite the complete absence of T, B and NK cells in these mice (Shultz et al, 2005), no secondary tumour could be obtained following the transplantation of papilloma cells (Figure 2E).
To determine whether the absence of secondary tumour formation upon the transplantation of FACS purified Lin−/α6+/Epcam+ TECs was due to the absence of tumour stromal cells essential for tumour initiation, we transplanted one million of unpurified cells from different papillomas containing immune cells, blood vessel and fibroblasts presented in the primary tumours into NOD/SCID/Il2Rγ null mice (Figure 2F–H). In contrast to the transplantation of purified Epcam+ populations, the transplantation of one million of unsorted tumour cells containing TECs, as well as immune, endothelial and fibroblast tumour-associated cells from DMBA/TPA and genetically induced papillomas gave rise to secondary tumours upon transplantation into Swiss Nude and NOD/SCID/IL2RγKO immunodeficient mice (Figure 2G and H). These secondary tumours presented a similar histology, proliferation (Figure 2I) and differentiation (Figure 2J) as compared with the primary tumours.
To identify which cells within the tumour stroma is necessary for the propagation of Lin−/α6+/Epcam+ TECS from skin papilloma into immunodeficient mice, we co-transplanted FACS-isolated Lin−/α6+/Epcam+ TECs with either tumour-associated fibroblasts (CD140a+ cells) or tumour endothelial cells (CD31+ cells). Interestingly, both tumour-associated fibroblasts and endothelial cells were able to promote secondary tumour formation following the transplantation of papilloma Lin−/α6+/Epcam+ TECs (Figure 2H). Taken together, these results show that despite the high proliferation of papilloma Lin−/α6+/Epcam+ TECs in vivo and their ability to form rapidly growing colonies in vitro, these cells are not clonogenic on their own, and rely on the presence of stromal cells such as endothelial cells and tumour-associated fibroblasts to initiate secondary tumour formation upon transplantation in immunodeficient mice.
Low frequency of TPCs within CD34HI and CD34LO populations in DMBA/TPA-induced SCC
Different studies have already demonstrated that Lin−/α6+/Epcam+/CD34+ TECs from mouse DMBA/TPA-induced SCC can reform secondary tumour upon transplantation into immunodeficient mice (Malanchi et al, 2008; Beck et al, 2011; Schober and Fuchs, 2011). However, it is not clear from these studies what is the frequency of TPCs within mouse primary SCC and whether CD34LO cells can directly, without prior in vitro culture, reform the parental tumour upon transplantation into immunodeficient mice.
To define precisely the frequency of TPCs in mouse primary SCC, we isolated Lin−/Epcam+/α6+/CD34HI and Lin−/Epcam+/α6+/CD34LO cells from SCC induced by DMBA/TPA treatment (Figure 3A–C), and assess the frequency of tumour formation after the transplantation of decreasing amount of FACS purified cells (Figure 3D). Using limiting dilution analysis (O'Brien et al, 2007; Quintana et al, 2008; Hu and Smyth, 2009), we found that around 1/6000 of primary Lin−/Epcam+/α6+/CD34HI directly transplanted after FACS isolation is able to initiate tumour formation upon transplantation into immunodeficient mice. (Figure 3D; Table I). The overall histology of these secondary tumours was similar to the primary SCC as shown by the expression of basal epithelial markers (e.g., K14) and the almost absence of cells expressing K8 (Figure 3E), a marker of more invasive and undifferentiated skin tumours (Caulin et al, 1993). However, the secondary tumours contain more differentiated cells expressing K10 (Figure 3E). Interestingly, Lin−/Epcam+ and Lin−/Epcam+/α6+/CD34LO cells were also able to reform secondary tumour with similar efficiency (1/5000) and histology as Lin−/Epcam+/α6+/CD34HI cells (Figure 3D and E; Table I). However, while these three different SCC TECs populations gave rise to formation of secondary tumour with similar frequency, Lin−/Epcam+/α6+/CD34HI cells grew faster than Lin−/Epcam+ and Lin−/Epcam+/α6+/CD34LO cells as determined the tumour size 8 weeks after transplantation (Figure 3F). Interestingly, tumour arising from the transplantation of Lin−/Epcam+/α6+/CD34LO gave rise to secondary tumour expressing CD34 with the same frequency as the primary tumours or tumours arising from the transplantation of Lin−/Epcam+/α6+/CD34HI cells (Figure 3G), demonstrating the plasticity of CD34 expression in TECs. qRT–PCR analysis revelled the preferential expression of EMT regulators and a decrease in E-cadherin expression in Lin−/α6+/Epcam+/CD34HI compared with Lin−/α6+/Epcam+/CD34LO TECs (Figure 3H).
Figure 3.
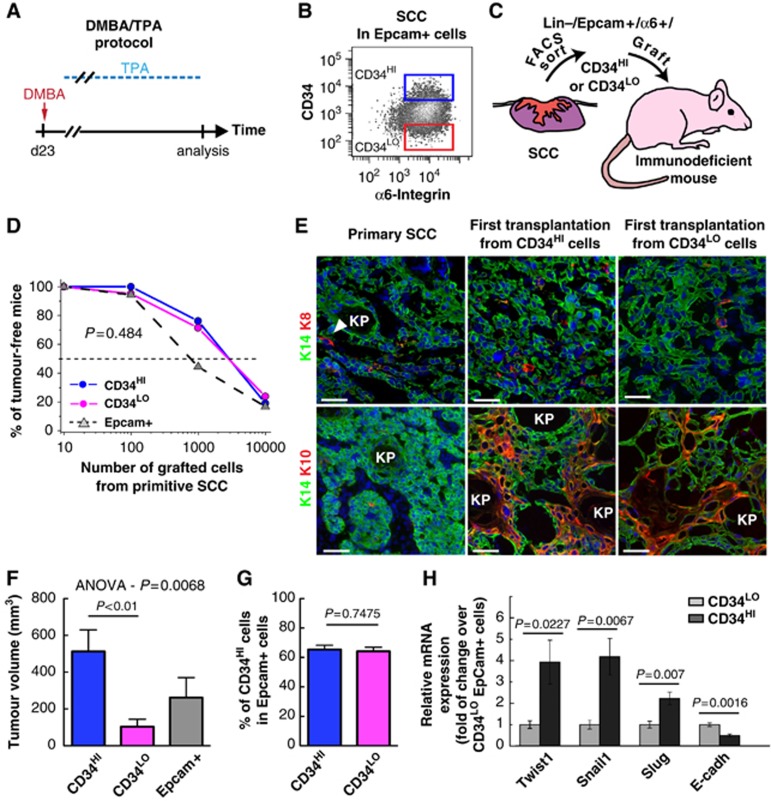
Low frequency of TPCs in primary CD34HI and CD34LO cell populations from DMBA/TPA-induced SCC. (A) Scheme representing the DMBA/TPA protocol to induce SCC formation. (B) FACS plot illustrating the gating of the Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO cells in DMBA/TPA-induced SCC. (C) Scheme representing the strategy used to isolate and graft Lin−/α6+/Epcam+/CD34HI, Lin−/α6+/Epcam+/CD34LO and Lin−/α6+/Epcam+ cells into immunodeficient mice. (D) Graph representing the percentage of tumour-free mice after subcutaneous injection of different concentrations of Lin−/α6+/Epcam+ cells into immunodeficient mice 10 weeks after transplantation (for details see Table I). Values are calculated from the sum of all biological replicates. (E) Immunostaining of K14 and K8 (upper panel) or K14 and K10 (lower panel) in the primary SCC and in the secondary (first transplantation) tumours originating from Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO cells. (F) Average volume of secondary tumours derived from Lin−/α6+/Epcam+/CD34HI (n=12), Lin−/α6+/Epcam+/CD34LO (n=11) or Lin−/α6+/Epcam+ cells (n=5). (G) FACS quantification of the proportion of Lin−/α6+/Epcam+/CD34HI amongst Lin−/α6+/Epcam+ cells in secondary tumours originating from Lin−/α6+/Epcam+/CD34HI (n=19) and Lin−/α6+/Epcam+/CD34LO (n=16) cells. (H) Expression of EMT-related genes by qRT–PCR in Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO TECs from DMBA/TPA-induced SCC (n=5). Data were normalized by gene expression in Lin−/α6+/Epcam+/CD34LO cells. Scale bars represent 50 μm.
Table 1. Summary of the tumour propagating frequency estimated by the transplantation of limiting dilution of CD34HI and CD34LO populations isolated from DMBA/TPA and KRasG12D p53KO-induced SCC.
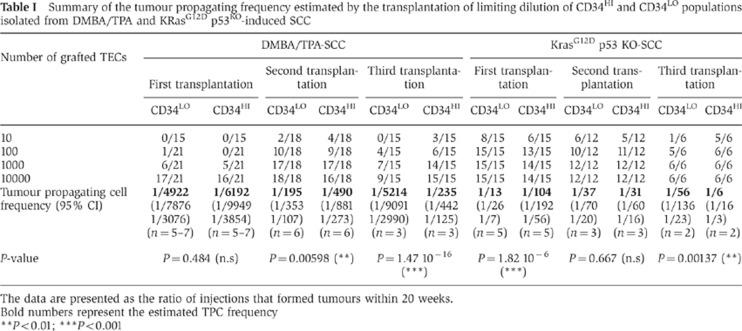
The data are presented as the ratio of injections that formed tumours within 20 weeks.
Bold numbers represent the estimated TPC frequency
**P<0.01; ***P<0.001
Increased frequency of TPCs in more aggressive genetically induced SCC
To assess whether the invasiveness of SCCs found in genetic mouse model of sporadic skin SCC mediated by KRasG12D expression and p53 deletion is associated with increased frequency of tumour propagation, we assessed the frequency of TPCs in Lin−/YFP+ TECs from invasive SCC from KRasG12Dp53KOYFP+ induced mice (Figure 4A–C), which present features of aggressive SCC with spindle shape cells expressing K8 and EMT markers (Lapouge et al, 2011; White et al, 2011a), upon transplantation into immunodeficient mice. Interestingly, transplantation of FACS purified Lin−/YFP+/CD34HI and Lin−/YFP+/CD34LO TECs gave rise to the formation of secondary tumours with a much greater efficiency compared with the same population of cells in the DMBA/TPA-induced SCC. Limiting dilution experiments indicated that around 1 cell among 100 Lin−/YFP+/CD34HI is able to initiate secondary SCCs, which is one order of magnitude higher that in the DMBA/TPA-induced SCC (Figure 4C; Table I). Lin−/YFP+/CD34LO TECs gave rise to secondary SCCs with even a higher frequency than Lin−/YFP+/CD34HI TECs (Figure 4C; Table I) that grew at similar rate (Figure 4D), suggesting that CD34 expression does not enrich for TPCs in KRasG12Dp53KO SCC in primary transplantation assay. Moreover, both primary and secondary tumours shared features of aggressive SCC characterized by heterogeneous expression K8 and K14 expression (Figure 4E and F). Both Lin−/YFP+/CD34HI and Lin−/YFP+/CD34LO populations gave rise to CD34-expressing cells, which represent the majority of primary and secondary tumour TECs as shown by FACS analysis and co-staining of CD34 with YFP (Figure 4G and I). qRT–PCR analysis revealed the increased expression of EMT regulators and signs of invasiveness in Lin−/YFP+/CD34HI cells from genetically induced SCC compared with DMBA/TPA-induced SCC (Figure 4J).
Figure 4.
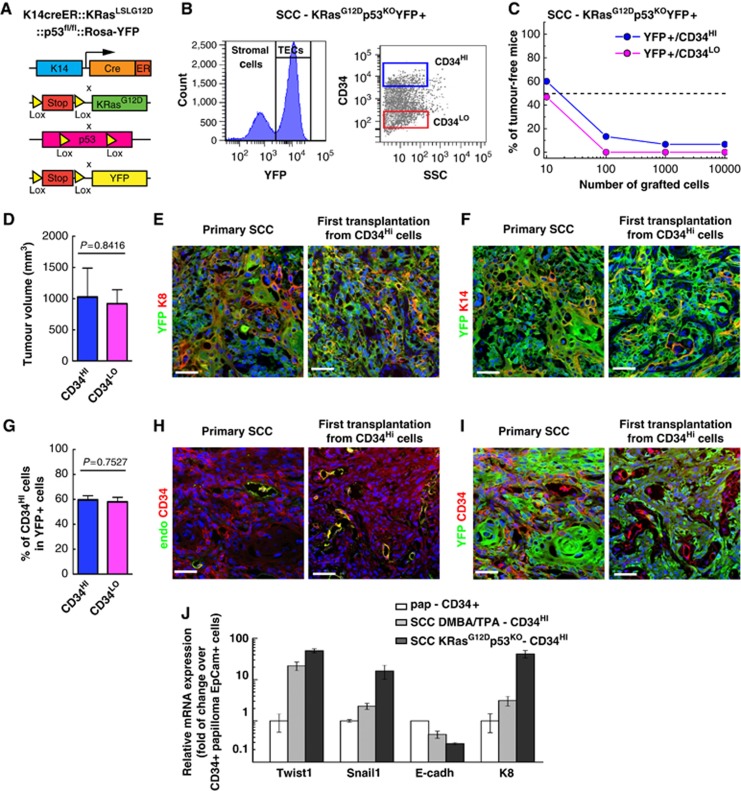
Increased TPCs in more invasive genetically induced SCCs. (A) Scheme summarizing the genetic model used to induce SCC and study the frequency of TPCs in more highly invasive SCC. (B) FACS plot representing the strategy used to isolate Lin−/YFP+/CD34HI and Lin−/YFP+/CD34LO TECs based on YFP expression from K14CREER::KRasG12D::p53fl/fl::RosaYFP Tamoxifen-treated mice. (C) Graph representing the percentage of tumour-free mice 10 weeks after subcutaneous injection of different concentrations of Lin−/YFP+/CD34HI and Lin−/YFP+/CD34LO cells into immunodeficient mice (for details see Table I). (D) FACS quantification of the proportion of CD34 in Lin−/YFP+/CD34HI cells in secondary tumours originating from Lin−/YFP+/CD34HI (n=7) and from Lin−/YFP+/CD34LO cells (n=7). (E, F) Immunostaining of YFP and K8 (E) or YFP and K14 (F) in the primary KRasG12Dp53KOYFP+ SCC and the first transplantation from Lin−/YFP+/CD34HI cells. (G) Average volume of secondary tumours derived from Lin−/YFP+/CD34HI (n=8) or Lin−/YFP+/CD34LO cells (n=8). (H, I) Immunostaining of CD34 and endoglin (H) or YFP and CD34 (I) in the primary KRasG12Dp53KOYFP+ SCC and the secondary (first transplantation) carcinoma from Lin−/YFP+/CD34HI cells. (J) Expression of EMT-related genes by qRT–PCR in Lin−/YFP+/CD34HI epithelial cells from benign papilloma, DMBA/TPA (SCC DMBA) and genetically (KRasG12Dp53KO- SCC) induced SCC (n=5). Data were normalized to gene expression in Lin−/α6+/Epcam+/CD34+ papilloma cells. Scale bars represent 50 μm.
Increased frequency of CD34HI but not CD34LO TPCs upon serial transplantation
To further characterize the long-term renewal potential of TPCs from skin SCC, we assessed the frequency of TPCs following serial transplantations into immunodeficient mice.
Interestingly, the frequency of tumour initiating cells TPCs in Lin−/Epcam+/α6+/CD34HI population from DMBA/TPA-induced SCC considerably increased during serial transplantation rising from 1/6000 cells after the first transplantation to 1/200 cells after the third transplantation (Figure 5A; Table I). Strikingly, while the initial frequency of TPCs was similar between Lin−/Epcam+/α6+/CD34HI and Lin−/Epcam+/α6+/CD34LO TECs after the second round of serial transplantation, the frequency of TPCs continued to increase in Lin−/Epcam+/α6+/CD34HI population, while tumour propagating TPC frequency dramatically decreased in Lin−/Epcam+/α6+/CD34LO population after the third round of serial transplantation (Figure 5B; Table I). Importantly, in addition to the increased frequency of TPCs, the latency before tumour appearance decreased with serial transplantation of Lin−/Epcam+/α6+/CD34HI TECs (Figure 5C). Moreover, tumour arising from Lin−/Epcam+/α6+/CD34HI TECs grew more rapidly than those derived from Lin−/Epcam+/α6+/CD34LO TECs after the third transplantation (Figure 5D–F). Nevertheless, the overall histology of tumours did not change with serial transplantation (Figure 5G). Like primary tumours, tumours from the third transplantation of DMBA/TPA-induced SCC present virtually no cells expressing K8 (Figure 5G) and like the tumour arising from the first transplantation, tumours from the third transplantation contained more cells expressing the differentiation marker K10 compared with the primary tumours (Figure 5G). Tumour propagating TPC frequency during serial transplantation of genetically induced KRasG12Dp53KO SCC increased in Lin−/YFP+/CD34HI TECs population and decreased Lin−/YFP+/CD34LO TECs population and as few as 1/6 Lin−/YFP+/CD34HI TECs were able to initiate tumour formation after the third transplantation (Figure 6A–C; Table I). Tumours arising from the third transplantation presented the same histology and differentiation as the primary tumours, characterized by a loss of K14 expression in some cells and the expression of K8 (Figure 6D). Tumour latency also decreased between the first and second transplantation of Lin−/YFP+/CD34HI TECs (Figure 6C).
Figure 5.
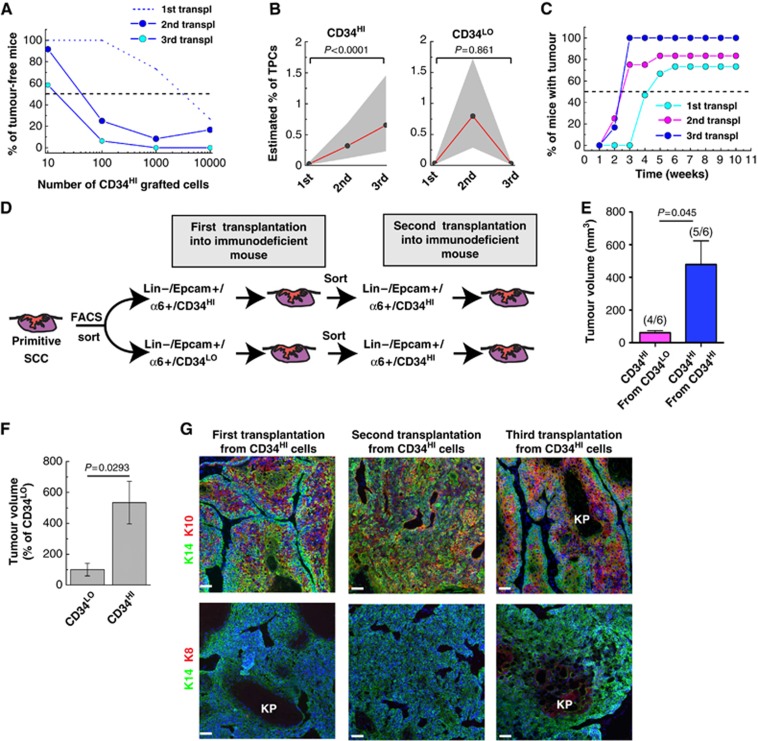
Increased frequency of TPCs upon serial transplantation in DMBA/TPA SCC. (A) Graph representing the percentage of tumour-free mice 10 weeks after subcutaneous injection of different concentrations of Lin−/α6+/Epcam+/CD34HI SCC cells from DMBA/TPA-treated SCC into immunodeficient mice after the first (dash line, data are presented in Figure 3D) (n=15 replicates), second (blue) (n=12 replicates) and the third (light blue) (n=6 replicates) transplantations. (B) Graph representing the estimated percentage of TPCs in Lin−/α6+/Epcam+/CD34HI (left) and Lin−/α6+/Epcam+/CD34LO (right) from DMBA/TPA-induced SCC during serial transplantations. Dots represent the estimated percentage of TPCs at each transplantation (detailed in Table I). Grey area represents the 95% confidence interval of the estimation. (C) Graph representing the percentage of mice with tumour at different time following subcutaneous injection of 104 Lin−/α6+/Epcam+/CD34HI cells from DMBA/TPA-induced SCC after the first, second and third serial transplantation of Lin−/α6+/Epcam+/CD34HI cells (see Table I for different transplanted of replicates). (D) Scheme summarizing the transplanted strategy to assess the conversion of Lin−/α6+/Epcam+/CD34LO into Lin−/α6+/Epcam+/CD34HI cells during serial transplantations of DMBA/TPA induced SCC. (E) Average volume of tertiary tumours (second transplantation) after the transplantation of 103 Lin−/α6+/Epcam+/CD34HI cells isolated from secondary DMBA/TPA induced SCC (first transplantation) tumours derived from Lin−/α6+/Epcam+/CD34HI grafted cells (n=5) or Lin−/α6+/Epcam+/CD34LO (n=4) cells. The values between brackets show the efficiency of tumour propagation for each population. (F) Graph representing the average size of quaternary tumours after the third round of serial transplantation of Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO cells from DMBA/TPA-induced SCC (n=6). (G) Immunostaining of K14 and K10 (upper panel) or K14 and K8 (lower panel) after the first, the second and the third transplantation of DMBA/TPA tumour cells. Scale bars represent 50 μm. KP, Keratin pearl.
Figure 6.
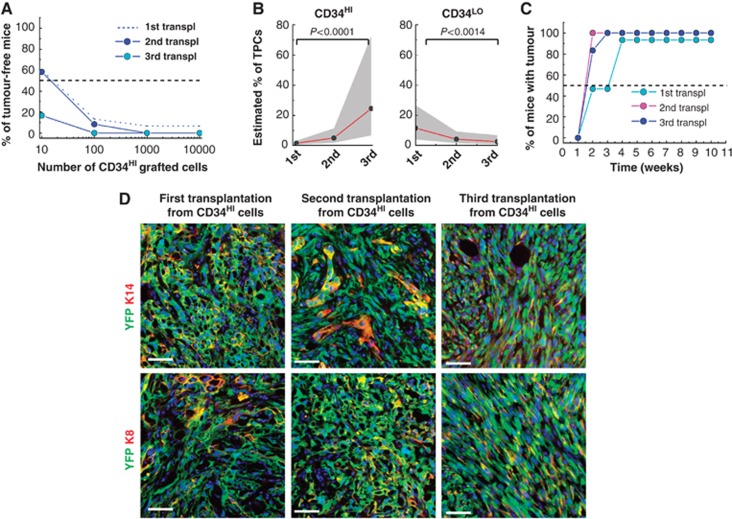
Increased frequency of TPCs upon serial transplantation in CD34HI but not inCD34LO genetically induced SCC. (A) Graph representing the percentage of tumour-free mice 10 weeks after subcutaneous injection of different concentrations of Lin−/YFP+/CD34HI SCC cells into immunodeficient mice cells after the first (dash line, data are presented in Figure 4C) (n=15 replicates), second (blue) (n=12 replicates) and the third (light blue) (n=6 replicates) transplantations from KRasG12Dp53KOYFP+ SCCs. (B) Graph representing the estimated percentage of TPCs in Lin−/YFP+/CD34HI (left) and Lin−/YFP+/CD34LO (right) from KRasG12Dp53KOYFP+ induced SCC during serial transplantation. Dots represent the estimated frequency of TPCs (detailed in Table I). Grey area represents the 95% confidence interval of the estimation. (C) Graph representing the percentage of mice with tumour at different time following subcutaneous injection of 104 Lin−/YFP+/CD34HI cells from KRasG12Dp53KOYFP+ induced SCC after the first, the second and the third serial transplantation of Lin−/YFP+/CD34HI cells. (Number of replicates detailed in Table I). (D) Immunostaining of K14 and YFP (upper panel) or K8 and YFP (lower panel) in the first, the second and the third transplantation of genetically induced SCC. Scale bars represent 50 μm.
These data indicate that Lin−/YFP+/CD34HI TEC population presents the ability to massively self-renew during serial transplantation and the frequency of TPCs becomes very frequent after the third round of serial transplantation. In contrast, the frequency of tumour propagating declines in Lin−/YFP+/CD34LO cells, suggesting that Lin−/YFP+/CD34LO TECs present a reduced long-term self-renewal capacity compared with Lin−/YFP+/CD34HI TPCs.
Tumour propagating frequency of mouse SCC does not increase in immunodeficient mice
Several immunodeficient mice models are routinely used for tumour transplantation assays (Quintana et al, 2008). The Swiss Nude mice are one of the most commonly used immunodeficient mice and lack functional thymus leading essentially to T (and B) cell deficiency (Gershwin et al, 1975). NOD/SCID/Il2Rγ null mice in contrast lack NK cells in addition to B and T cells (Shultz et al, 2005). It has been previously reported that primary human melanoma grew much more efficiently in the most immunodeficient NOD/SCID/Il2Rγ null mice (Quintana et al, 2008). To determine whether the degree of immunodeficiency underestimated the frequency of tumour initiating cells in mouse SCC, we assessed the frequency of tumour formation in syngenic (FVB/N) and NOD/SCID/Il2Rγ null mice following the transplantation of limiting dilution of Lin−/α6+/Epcam+ cells from DMBA/TPA-induced SCC. No significant difference in the tumour propagating frequency of Lin−/α6+/Epcam+ population was observed when SCC cells were transplanted into mice presenting different degree of immunosupression (Figure 7A). Similarly, transplantation of Lin−/YFP+ TECs population into Swiss Nude or NOD/SCID/Il2Rγ null mice gave rise to the same frequency of TPCs (Figure 7B). These data demonstrate that the degree of immunodeficiency of recipient mice is not a limiting factor for the propagation of mouse invasive SCCs.
Figure 7.
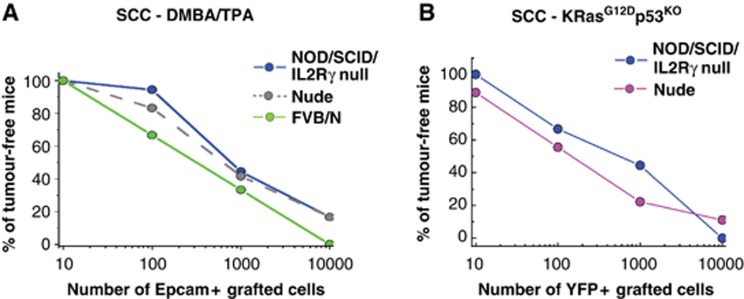
Transplantation into more severely immunodeficient mice does not increase the frequency of mouse SCC propagating cells. (A) Graph representing the percentage of tumour-free mice 10 weeks after subcutaneous injection of different concentrations of Lin−/α6/Epcam+ SCC cells from DMBA/TPA SCC into Swiss Nude mice (grey dash line, data are presented in Figure 3D), NOD/SCID/Il2Rγ null mice (blue line, n=9 replicates for each dilution) and FVB/N mice (green line, n=6 replicates for each dilution) 10 weeks after transplantation. (B) Graph representing the percentage of tumour-free mice 10 weeks after subcutaneous injection of different concentrations of Lin−/YFP+ SCC cells from KRasG12Dp53KOYFP+ SCC into Swiss Nude mice (pink line, n=9 replicates for each dilution), NOD/SCID/Il2Rγ null mice (blue line, n=9 replicates for each dilution) 10 weeks after transplantation.
Discussion
The relative frequency of TPCs can vary from being extremely frequent (1/4 cells of late stage or metastatic melanoma can be propagated into NOD/SCID/Il2Rγ null mice) (Quintana et al, 2008) to very rare in other type of solid tumours such as colon cancer (<1/1000) (O’Brien et al, 2007). Recent studies have demonstrated the existence of two populations of TPCs in DMBA/TPA-induced SCC (Malanchi et al, 2008; Schober and Fuchs, 2011). However, the precise frequency of TPCs in primary murine SCC remains unclear. More importantly, conflicting results have been obtained to which extend CD34+ cells are encriched for TPCs in primary SCCs (Malanchi et al, 2008; Schober and Fuchs, 2011). Some technical differences between these two studies including the culture of cells before transplantation (Schober and Fuchs, 2011) or the transplantation of crude population of CD34+ cells isolated by magnetic beads separation, which included endothelial cells as well as TECs (Malanchi et al, 2008) render difficult the comparison between these two studies.
Here, we used different models of skin tumours to assess the relative frequency of TPCs during skin tumour progression by directly transplanting freshly isolated tumour cells with high purity. Both Lin−/α6+/Epcam+/CD34+ and Lin−/α6+/Epcam+/CD34− TECs from benign tumours proliferated rapidly in vivo leading to tumour growth and were able to form large and rapidly proliferative colonies in vitro. Surprisingly, neither the Lin−/α6+/Epcam+/CD34+, nor the Lin−/α6+/Epcam+/CD34− TECs were able to give rise to secondary tumours when transplanted into immunodeficient mice. This could at least be partially explained by the interruption of TPA treatment after transplantation in DMBA/TPA-induced papilloma. However, genetically induced papillomas grow quickly in vivo, do not require exogenous drugs to sustain tumour promotion, and fail to form secondary tumours. Interestingly, the co-grafting of tumour stromal cells, such as tumour-associated fibroblasts or tumour endothelial cells together with papilloma Lin−/α6+/Epcam+ TECs confer the ability of Lin−/α6+/Epcam+ TECs to form secondary tumour formation upon transplantation, although at very low frequency, indicating the apparent inability of papilloma Lin−/α6+/Epcam+ TECs to propagate tumours in transplantation assays is the direct consequence of their strong dependence to their tumour microenvironment, which is particularly important in skin papilloma, as recently shown by the critical role of the perivascular niche in regulating stemness of skin papilloma (Beck et al, 2011). The very low frequency of TPC in skin papilloma demonstrates the limit of the transplantation assay to assess the renewal potential of certain types of tumours as well as their absolute dependence of tumour stroma and greatly underestimates their stem cell content. Indeed, using lineage-tracing and clonal analysis, we have recently demonstrated that papilloma contain a high frequency of tumour stem cells (around 20%) during unperturbed tumour growth (Driessens et al, 2012), suggesting that novel approach such as lineage tracing should be undertaken to decipher the proliferation hierarchy in tumours that are highly dependent on their microenvironment.
Our limiting dilution transplantation experiments directly after FACS isolation reveal that only one out of several thousands of DMBA/TPA-induced primary SCC cells are capable of tumour propagation. Moreover, the frequency of TPCs in more invasive SCC, where oncogenic KRasG12D is expressed and p53 genetically deleted, is much higher with one cell out of 100 is capable of forming secondary tumours upon transplantation, clearly revealing a strong correlation between the frequency of tumour initiating cells and the invasiveness of SCC, as TECs proliferation and in vivo growth of the DMBA/TPA and genetic SCCs are not so different to explain the major difference in the frequency of TPCs. In addition, serial transplantations into immunodeficient mice massively increase the frequency of TPCs as well as accelerate the appearance of tumour after transplantation, with as few as 1 cell out of 6 are TPCs after the third serial transplantation of genetically induced SCC, a number similar to that was recently reported for human melanoma transplanted into NOD/SCID/Il2Rγ null mice (Quintana et al, 2008), suggesting that transplantation may select for tumour cells particularly adapted to growth into immunodeficient mice and/or the self-renewal of TPCs increased with serial transplantation, leading to a progressive enrichment of TPCs overtime. In DMBA/TPA and genetically induced skin tumours, there is a good correlation between the frequency of primary TPCs and their stromal dependence with their p53 status, as p53 is well known to be lost during the progression of papilloma to invasive SCC (Kemp et al, 1993) and p53 is deleted in the primary genetically induced SCC (Lapouge et al, 2011; White et al, 2011), suggesting that p53 indeed may play a critical role in the ability of SCCs to propagate in the absence of stromal cells upon transplantation. However, the increased frequency of TPCs during serial transplantation is unlikely to be the consequence of the p53 status, since p53 is already deleted in primary SCCs. The increase in the frequency of TPCs during transplantation into immunodeficient mice have been previously reported for human primary melanoma that have been first grown into immunodeficient mice before being transplanted at limiting dilution (Boiko et al, 2010), suggesting that the increase of TPCs with serial transplantations could be an hallmark of tumour propagation assay.
While it was initially reported that CD34HI murine SCC presented a thousand-fold increase in the frequency of TPCs compared with CD34LO TECs (Malanchi et al, 2008), it was latter suggested that both CD34HI and CD34LO TECs populations contain similar frequency of TPCs (Schober and Fuchs, 2011), although these experiments have been performed by culturing primary SCC cells for a short period of time before their transplantation. Our data clearly demonstrate that indeed both Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO and both Lin−/YFP+/CD34LO and Lin−/YFP+/CD34HI− TECs contain similar frequency of TPCs during the first two round of serial transplantations, and suggest that the culture condition prior transplantation did not modify the intrinsic ability of these two populations of tumour cells to reform tumour upon transplantation (Schober et al, 2011).
However, while CD34HI and CD34LO give rise to tumour containing the same proportion of CD34HI and CD34LO cells, clearly demonstrating that there is an interconversion between CD34HI and CD34LO during transplantation of SCC cells as previously described (Schober and Fuchs, 2011), the long-term renewal potential of these two populations is different. Indeed, the frequency of TPCs of Lin−/α6+/Epcam+/CD34HI and Lin−/YFP+/CD34HI SCCs continued to increase after the third serial transplantation, while there is a progressive decrease in the frequency of TPCs after the third round of transplantation in Lin−/α6+/Epcam+/CD34LO and Lin−/YFP+/CD34LO SCCs in DMBA/TPA-induced SCC and in genetically induced SCC, as well as the rate of tumour growth, suggesting that Lin−/α6+/Epcam+/CD34LO and Lin−/YFP+/CD34LO SCC cells present reduced long-term self-renewal capacities compared with Lin−/α6+/Epcam+/CD34HI and Lin−/YFP+/CD34HI SCC cells. Future studies will be needed to determine whether other markers beside CD34 can be used to isolate a population of tumour cells with higher tumour propagating capacity, already detectable after the first transplantation. The discrepancy between the long-term renewal capacity and the robust tumour contribution of the Lin−/α6+/Epcam+/CD34HI and CD34LO SCC cells in the secondary transplant is reminiscent of the situation recently described in Id1 low and Id1 high in glioblastoma (Barrett et al, 2012).
The precise molecular determinants responsible for the progressive increase of TPCs during cancer progression and serial transplantation remain to be characterized. However, among other factors EMT regulators appear as likely candidates to contribute to this characteristic in skin SCC. Indeed, Twist1 and EMT that have been previously associated by tumour stemness (Mani et al, 2008) and long-term renewal of tumour cells upon oncogenic KRasG12D expression (Ansieau et al, 2008), and many very well-known EMT regulators including Twist1, Snail1 are upregulated between Lin−/α6+/Epcam+/CD34HI and Lin−/α6+/Epcam+/CD34LO as well as between DMBA/TPA and KRasG12D/p53 null SCC. Further studies would be required to elucidate the functional role of these factors during tumour initiation and progression.
Materials and methods
Mice
FVB/N, Swiss Nude and NOD/SCID/Il2Rγ null mice were obtained from Charles River. K14CREER (Vasioukhin et al, 1999), K19CREER (Means et al, 2008), Lgr5CREER (Barker et al, 2007), KRasLSL-G12D (Tuveson et al, 2004), p53fl/fl mice (Jonkers et al, 2001) have been previously described. Mouse colonies were maintained in a certified animal facility in accordance with the European guidelines.
DMBA/TPA carcinogenesis
Mice were treated with DMBA and TPA as previously described (Beck et al, 2011).
Tamoxifen administration for oncogenic KRas model
K19CREER::KRasLSL-G12D, Lgr5CREER::KRasLSL-G12D were treated with 10 mg of tamoxifen from 23 to 25 days after birth. K14CREER::KRasLSL-G12D and K14CREER::KRasLSL-G12D::p53fl/fl::Rosa-YFP were, respectively, treated with 1 and 5 mg of tamoxifen at 28 and 29 days after birth. Papillomas and carcinomas were harvested from 1 month to 4 months after tamoxifen administration.
Measurement of papilloma growth
Skin tumours were measured using a precision calliper allowing to discriminate size modifications >0.1 mm. Tumour volumes were measured the first day of treatment and every week until the end of the experiments with the formula V=π × [d2 × D]/6, where d is the minor tumour axis and D is the major tumour axis.
Histology, immunostaining and imaging
Tumours mice were either embedded in OCT and sections were fixed in 4% PAF for 10 min at room temperature, or tumours pre-fixed for 2 h in 4% PAF and embedded in OCT. Samples were sectioned at 4–6 μm sections using CM3050S Leica cryostat (Leica Microsystems GmbH).
The following primary antibodies were used: anti-CD34 (rat, clone RAM34, 1:100, BD), anti-K5 (polyclonal rabbit, 1:2000, Covance), anti-β4 (rat, clone 346-11A, 1:200, BD), anti-KI67 (polyclonal rabbit, 1:200, Abcam), anti-K10 (polyclonal rabbit, 1:2000, Covance), anti-endoglin (polyclonal goat, 1:500, R&D), anti-K14 (polyclonal chicken, 1:1000, covance) anti-K8 (polyclonal rat, 1:1000, hybridoma bank), anti-E-Cadherin (rat, clone ECCD-2, 1:1000, invitrogen), anti-YFP (polyclonal rabbit, 1:1.000, invitrogen). Sections were incubated in blocking buffer (PBS/NDS 5%, BSA 1%, Triton 0.2%) for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C. Sections were rinsed three times in PBS and incubated with secondary antibodies diluted at 1:400 for 1 h at room temperature. The following secondary antibodies were used: anti-rabbit, anti-rat, anti-goat, anti-chicken conjugated to AlexaFluor488 (Molecular Probes), to rhodamine Red-X (JacksonImmunoResearch) or to Cy5 (Jackson ImmunoResearch). Nuclei were stained in Hoechst solution (4 μM) and slides were mounted in DAKO mounting medium supplemented with 2.5% Dabco (Sigma).
Pictures were acquired using Axio Imager M1 Microscope, AxioCamMR3 camera and using Axiovision software (Carl Zeiss Inc.).
EdU detection
EdU staining in culture cells
Cells from colony papilloma were incubated 2 h with the EdU (Invitrogen) and are fixed and permeabilized with 4% PAF (pH 7.4) for 10 min. After three washes in PBS, the cells were incubated 1 h with PBS/BSA1%/HS5%/Triton 0.2%. Then the detection of EdU-positive cells was performed following the protocol provided by the manufacturer for Click-IT™ EdU Imaging Kits (Invitrogen, Molecular Probes).
EdU staining in papilloma
Mice were treated with 100 μl EdU solution (5 mg/kg). After 4 h of EdU incubation, papilloma were harvested, embedded in OCT and cut in slides of 6 μm. The slides were fixed in 4% PAF and stained with K14 antibody and anti-rabbit conjugated with Alexa488 as described above. Just after the secondary antibody and the three PBS washes, the detection of EdU was performed as described by the manufacturer (Invitrogen, Molecular Probes).
Nuclei were stained in Hoechst solution (4 μM) and the slides were mounted in DAKO mounting medium supplemented with 2.5% Dabco (Sigma). Pictures were acquired using Axio Imager M1 Microscope, AxioCamMR3 camera and using Axiovision software (Carl Zeiss Inc.).
Isolation of TECs
Tumours from FVB/N mice and mice from each oncogenic KRas line were digested in collagenase I (Sigma) for 2 h at 37°C on a rocking plate. Collagenase I activity was blocked by addition of EDTA (5 mM) and then rinsed in PBS supplemented with 2% FCS. After tumour digestion, cells were blocked for 15 min at room temperature. Immunostaining was performed using biotin-conjugated anti-CD34 (clone RAM34; BD Pharmingen), FITC-conjugated anti-α6-integrin (clone GoH3; BD Pharmingen), PE-conjugated anti-CD45 (clone 30F11, eBiosciences), PE-conjugated anti CD31 (clone MEC13.3; BD Pharmingen), PE-conjugated anti-CD140a (clone APA5; eBiosciences), APC-Cy7-conjugated anti-Epcam (clone G8.8; Biolegend) by incubation for 30 min on ice. Cells were washed and stained using APC-conjugated Streptavidin (BD Pharmingen) for 20 min on ice. Living tumour cells were selected by forward scatter, side scatter and doublet discrimination by Hoechst dye exclusion. Fluorescence-activated cell sorting analysis was performed using FACSAria and FACSDiva software (BD Biosciences). Sorted cells were collected either in culture medium for in vitro culture or for in vivo transplantation experiments and into lysis buffer for RNA extraction.
Culture of TECs
The culture of TECs were performed as described previously(Blanpain et al, 2004). In brief, viability of FACS-isolated adult keratinocytes was assessed by trypan blue (Sigma) staining, and the cell numbers were determined by the hemocytometer. Equal numbers of live cells were plated onto mitomycin-treated 3T3 fibroblasts in E-media (Rheinwald and Green, 1977) supplemented with 15% serum and 0.3 mM calcium. After 14 days in vitro, cells were trypsinized and counted (Coulter counter; Beckman).
Transplantation assays in immunodeficient mice
Lin−/α6/Epcam+/CD34HI, Lin−/α6/Epcam+/CD34LO and Lin−/α6/Epcam+ tumour cells were isolated by FACS from papillomas and carcinoma arising from DMBA-TPA mice, K14CreER::KRasLSL-G12D::p53fl/fl::RosaYFP, K14CreER::KRasLSL-G12D, K19CreER::KRasLSL-G12D and Lgr5CreER::KRasLSL-G12D. Cells were harvested in 4°C medium supplemented with 30% serum. Cells were then washed in PBS complemented with 2% FCS and resuspended in matrigel (50 μl, E1270, 970 mg/ml; Sigma). Different dilution of Lin−/α6/Epcam+/CD34HI, Lin−/α6/Epcam+/CD34LO and Lin−/α6/Epcam+ tumour cells resuspended in 50 μl of Matrigel were injected subcutaneously to NOD/SCID/Il2Rγ null mice, Swiss Nude mice or FVB/N mice (Charles River, France). Technical triplicates injections per mouse were performed. The Lin−/α6/Epcam+/CD34HI cells were grafted in the same mouse than Lin−/α6/Epcam+/CD34LO cells, as the Lin−/YFP+/ CD34HI and Lin−/YFP+/CD34LO cells. For co-grafting, fibroblasts (CD140+) or endothelial cells (CD31+) were co-transplanted with TECs (Epcam+) at a ratio of 1/5. Secondary tumours were detected by palpation every week and their size monitored until tumour reached an ethical size (1 cm3) or mice present sign of distress and the experiments were terminated. The tumours volume was calculated with the formula V=π × [d2 × D]/6, where d is the minor tumour axis and D is the major tumour axis.
Estimation of the relative frequency of tumour propagating cells
Estimation of the relative frequency of cancer propagating cells was performed using the extreme limiting dilution analysis (ELDA) as described by Hu and Smyth (2009) and calculated online using the ELDA software online ( http://bioinf.wehi.edu.au/software/elda/).
Statistics
Statistical and graphical data analyses were performed using Origin 7 (OriginLab) and Prism 5 (Graphpad) software.
RNA extraction, quantitative real-time PCR
The protocol used was previously described (Beck et al, 2011). Briefly, total RNA extraction and DNase treatment were performed using the RNeasy micro kit (Qiagen) according to the manufacturer’s recommendations. After nanodrop RNA quantification, purified RNA was used to synthesize the first-strand cDNA in a 50-μl final volume, using Superscript II (Invitrogen) and random hexamers (Roche). Control of genomic contaminations was measured for each sample by performing the same procedure with or without reverse transcriptase. Quantitative PCR analyses were performed with 2 ng of cDNA reaction as template, using a SYBRGreen mix (Applied Bioscience) and an Agilent Technologies Stratagene Mx3500P real-time PCR system. Relative quantitative RNA was normalized using the housekeeping genes β-actin and Hprt. Primers were designed using Lasergene 7.2 software (DNAStar) and are presented below. Analysis of the results was performed using Mxpro software (Stratagene) and relative quantification was performed using the DDCt method using β-actin as a reference. The entire procedure was repeated in three biologically independent samples. Error bars represent standard error of the mean (s.e.m.). Results were presented as the ratio of Lin−/α6+/Epcam+/CD34LO in DMBA/TPA SCC or as the ratio of Lin−/α6+/Epcam+/CD34HI in papillomas.
List of primer used in qRT–PCR
Twist1 mRNA forward 5′-agctacgccttctccgtct-3′, reverse 5′-tccttctctggaaacaatgaca-3′, Snail1 mRNA forward 5′-cttgtgtctgcacgacctgt-3′, reverse 5′-caggagaatggcttctcacc-3′, Slug mRNA forward 5′-cattgccttgtgtctgcaag-3′, reverse 5′-agaaaggcttttccccagtg-3′, E-Cadh mRNA forward 5′-atcctcgccctgctgatt-3′, reverse 5′-accaccgttctcctccgta-3′, K8 mRNA forward 5′-agttcgcctccttcattgac-3′, reverse 5′-gctgcaacaggctccact-3′.
Supplementary Material
Acknowledgments
CB is investigator of WELBIO. BB is a chargé de recherche of the FRS/FNRS. This work was supported by the FNRS, the program d’excellence CIBLES of the Wallonia Region, a research grant from the Fondation Contre le Cancer, the ULB foundation and the fond Gaston Ithier, a starting grant of the European Research Council (ERC) and the EMBO Young Investigator Program.
Author contributions: CB, BB and GL designed the experiments and performed data analysis. BB, GL and DN performed all the experiments. SD and CD performed FACS sorting experiments. CB, GL and BB wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J (2009) Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 4: 1350–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M, Ratner D (2001) Cutaneous squamous-cell carcinoma. N Engl J Med 344: 975–983 [DOI] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A (2008) Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14: 79–89 [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007 [DOI] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, Nam HS, Benezra R (2012) Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell 21: 11–24 [DOI] [PubMed] [Google Scholar]
- Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, Mascre G, Drogat B, Dekoninck S, Haigh JJ, Carmeliet P, Blanpain C (2011) A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478: 399–403 [DOI] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E (2004) Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118: 635–648 [DOI] [PubMed] [Google Scholar]
- Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, Longaker MT, Weissman IL (2010) Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466: 133–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin C, Bauluz C, Gandarillas A, Cano A, Quintanilla M (1993) Changes in keratin expression during malignant progression of transformed mouse epidermal keratinocytes. Exp Cell Res 204: 11–21 [DOI] [PubMed] [Google Scholar]
- Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C (2012) Defining the mode of tumour growth by clonal analysis. Nature 488: 527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Crombie R, Liddell J, Stuart D, Linardopoulos S, Nagase H, Portella G, Brown K, Street A, Akhurst R, Balmain A (1998) Epithelial carcinogenesis in the mouse: correlating the genetics and the biology. Philos Trans R Soc Lond B Biol Sci 353: 839–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershwin ME, Merchant B, Gelfand MC, Vickers J, Steinberg AD, Hansen CT (1975) The natural history and immunopathology of outbred athymic (nude) mice. Clin Immunol Immunopathol 4: 324–340 [DOI] [PubMed] [Google Scholar]
- Hu Y, Smyth GK (2009) ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347: 70–78 [DOI] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A (2001) Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29: 418–425 [DOI] [PubMed] [Google Scholar]
- Kemp CJ (2005) Multistep skin cancer in mice as a model to study the evolution of cancer cells. Semin Cancer Biol 15: 460–473 [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, Balmain A (1993) Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74: 813–822 [DOI] [PubMed] [Google Scholar]
- Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, Blanpain C (2011) Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci USA 108: 7431–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo NA, Shimono Y, Qian D, Clarke MF (2007) The biology of cancer stem cells. Annu Rev Cell Dev Biol 23: 675–699 [DOI] [PubMed] [Google Scholar]
- Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, Huelsken J (2008) Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 452: 650–653 [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means AL, Xu Y, Zhao A, Ray KC, Gu G (2008) A CK19(CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis 46: 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LV, Vanner R, Dirks P, Eaves CJ (2012) Cancer stem cells: an evolving concept. Nat Rev Cancer 12: 133–143 [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106–110 [DOI] [PubMed] [Google Scholar]
- Owens DM, Watt FM (2003) Contribution of stem cells and differentiated cells to epidermal tumours. Nat Rev Cancer 3: 444–451 [DOI] [PubMed] [Google Scholar]
- Pardal R, Clarke MF, Morrison SJ (2003) Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 3: 895–902 [DOI] [PubMed] [Google Scholar]
- Perez-Losada J, Balmain A (2003) Stem-cell hierarchy in skin cancer. Nat Rev Cancer 3: 434–443 [DOI] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ (2008) Efficient tumour formation by single human melanoma cells. Nature 456: 593–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105–111 [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Green H (1977) Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265: 421–424 [DOI] [PubMed] [Google Scholar]
- Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H (2012) Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337: 730–735 [DOI] [PubMed] [Google Scholar]
- Schober M, Fuchs E (2011) Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci USA 108: 10544–10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, Morrison SJ (2009) Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138: 822–829 [DOI] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174: 6477–6489 [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387 [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E (1999) The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA 96: 8551–8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, Lowry WE (2011) Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci USA 108: 7425–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.