

Abstract
microRNA abundance has been shown to depend on the amount of the microprocessor components or, in some cases, on specific auxiliary co-factors. In this paper, we show that the FUS/TLS (fused in sarcoma/translocated in liposarcoma) protein, associated with familial forms of Amyotrophic Lateral Sclerosis (ALS), contributes to the biogenesis of a specific subset of microRNAs. Among them, species with roles in neuronal function, differentiation and synaptogenesis were identified. We also show that FUS/TLS is recruited to chromatin at sites of their transcription and binds the corresponding pri-microRNAs. Moreover, FUS/TLS depletion leads to decreased Drosha level at the same chromatin loci. Limited FUS/TLS depletion leads to a reduced microRNA biogenesis and we suggest a possible link between FUS mutations affecting nuclear/cytoplasmic partitioning of the protein and altered neuronal microRNA biogenesis in ALS pathogenesis.
Keywords: ALS, chromatin immunoprecipitation, Drosha, FUS/TLS, microRNA
Introduction
FUS/TLS (fused in sarcoma/translocated in liposarcoma, hereafter termed as FUS), belonging to the FET family of ubiquitously expressed RNA-binding proteins, is implicated in a wide range of cellular processes, including transcription and mRNA processing (Aman et al, 1996; Tan and Manley, 2009). In many human malignancies, FET genes are fused to various transcription factor genes and were suggested to be the driving forces for cancer development (Law et al, 2006; Riggi et al, 2007). More recently, mutations in the FUS gene were reported to be associated with familial forms of Amyotrophic Lateral Sclerosis (ALS) (Kwiatkowski et al, 2009; Lagier-Tourenne and Cleveland, 2009; Vance et al, 2009), thus further increasing the interest in this protein and suggesting a crucial function in neural cells.
The FUS protein contains several functionally characterized domains: an N-terminal domain (enriched in glutamine, glycine, serine and tyrosine residues) that has been recently shown to be able to form hydrogels composed of uniformly polymerized amyloid-like fibres (Han et al, 2012), a glycine-rich region, an RNA binding domain and a highly conserved C-terminus encoding for a non-classic nuclear localization signal (Iko et al, 2004). Most of the mutations found in ALS patients are clustered in the glycine-rich region and in the extreme C-terminal part of the protein (Lagier-Tourenne et al, 2010).
Recently, FUS was also shown to bind a non-coding RNA and to undergo RNA-mediated allosteric modulation, producing alternative protein interactions and transcriptional effects (Wang et al, 2008).
FUS shows predominant nuclear localization even though it is known to shuttle between the nucleus and the cytoplasm (Zinszner et al, 1997); however, ALS-linked mutations in FUS lead to a predominance of cytoplasmic versus nuclear localization. This is a particularly evident phenotype in neuronal cells (Kwiatkowski et al, 2009; Vance et al, 2009; Ito et al, 2011). Even though the exact mechanism by which this protein becomes pathogenic in ALS remains uncertain, many evidences infer that toxicity of FUS mutants is somehow related to this nucleus/cytoplasmic imbalance. Since one of the major features of the FUS protein is to bind RNA and function in several steps of gene expression, including transcription regulation and RNA maturation (Zinszner et al, 1997; Lagier-Tourenne et al, 2010; Tan et al, 2012), the altered nucleus/cytoplasmic partitioning has been proposed as a key event in ALS pathogenesis (Lagier-Tourenne et al, 2010; Yang et al, 2010). However, so far the activity of FUS in neuronal cells is still poorly defined.
One interesting observation regarding FUS function was derived from data indicating the Drosha protein as a putative FUS interactor (Gregory et al, 2004). Since Drosha is an essential component of the microprocessor complex, required for microRNA (miRNA) biogenesis, and its activity may be modulated by regulatory proteins, it has been suggested that FUS may regulate miRNA expression by modulating the activity of this processing enzyme; however, so far no data have demonstrated such a role.
In this work, we have analysed the FUS mode of action in the control of miRNA biogenesis in neuronal cells. We found that its downregulation affects the biogenesis of a large class of miRNAs. Among them, specific neuronal miRNAs known to play a crucial role in neuronal function, activity and differentiation were found. We also show that through its ability to bind pri-miRNAs, FUS is recruited to the chromatin where it facilitates Drosha loading. Moreover, we show that half the levels of FUS lead to reduced miRNA biogenesis. Altogether, these data suggest that ALS-associated mutations producing decreased nuclear levels of the protein could result in altered miRNA production, providing a possible link with the ALS pathogenesis.
Results
The expression of a subset of microRNAs is altered upon FUS knockdown
The neuroblastoma cell line SK-N-BE was utilized to test the effect of FUS downregulation on miRNA expression. Cells treated with scrambled (siScr) and anti-FUS siRNAs (siFUS) were analysed at 6 days after all-trans retinoic acid (RA)-induced differentiation. At this time point, most of the miRNAs playing a crucial role in neuronal differentiation reach the strongest upregulation while the N-MYC protein, present only in proliferating cells, is downregulated (Laneve et al, 2007; Supplementary Figure S1). Different levels of FUS downregulation were obtained in different experiments ranging from 55 to 80% (Figure 1A; Supplementary Figure S2A). FUS depletion obtained with a different siRNA, against the 3′UTR, produced the same extent of miRNA downregulation (siFUS-3′, Supplementary Figure S2B). miRNA expression profiling was carried out by high-throughput quantitative real-time PCR: out of 377 miRNAs, 166 were deregulated >15%, with the majority (90%) being downregulated (Supplementary Table I). Among these, several miRNAs known to have a crucial role in neuronal function, differentiation and synaptogenesis (miR-9, miR-125b and miR-132; Laneve et al, 2007, 2010; Packer et al, 2008; Edbauer et al, 2010; Pathania et al, 2012) were found. Notably, the protein levels of the microprocessor major components, Drosha and DGCR8, were unaffected upon FUS downregulation (Figure 1A). Figure 1B shows RT–PCR analysis on a selection of miRNAs derived from six independent experiments with similar FUS depletion (70–80%): even if the effect on accumulation was in some case small (18% for miR-9, 20% for miR-125b and 25% for miR-132), the values were very reproducible in the different experiments. Other species, not restricted to neuronal cells, were more affected, such as miR-192, miR-199a and miR-628-5p that decreased to ∼50% of control value. In contrast, miR-15a and miR-432 levels were unaffected and they have been utilized as controls in the following experiments. Notably, several of the downregulated miRNAs (such as the neuronal miR-9, miR-125b and miR-132) displayed altered expression even when FUS levels were decreased to only 45% (Supplementary Figure S2), indicating that even half the levels of FUS are sufficient to affect the accumulation of specific miRNAs.
Figure 1.
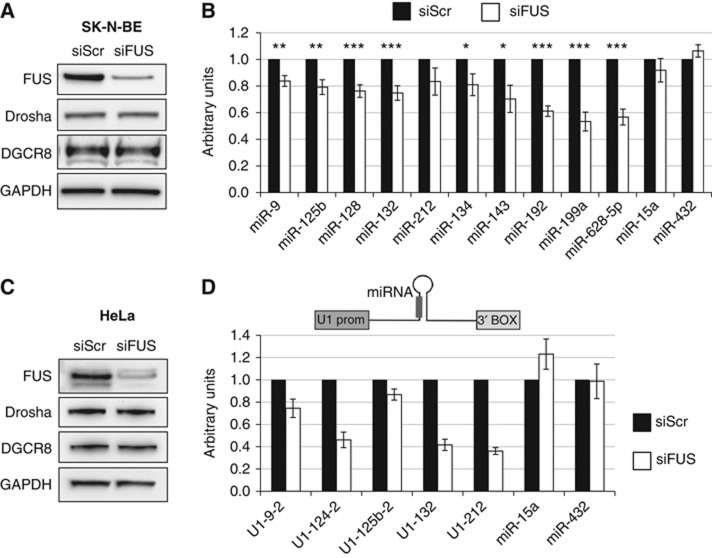
FUS affects the biogenesis of specific microRNAs. (A) SK-N-BE cells were treated with anti-FUS siRNA (siFUS) or with AllStars Negative Control siRNA (siScr) and maintained in retinoic acid (RA) for 6 days. Levels of FUS, Drosha and DGCR8 were analysed by western blot. GAPDH was used as a loading control. (B) miRNA levels from the same cells were analysed by qPCR. The histogram represents the average of six independent experiments providing an average of 75% FUS depletion (black bars—control siRNA; white bars—anti-FUS siRNA). miRNA levels were normalized for the snoRNA-U25 internal control. Significance was assessed by unpaired Student’s t-test (*P<0.05, **P<0.01, ***P<0.001). (C) Knockdown of FUS in HeLa cells. Samples were treated with siRNA as in (A) and the indicated proteins analysed by western blot. (D) Plasmid constructs carrying different pri-miRNA sequences under the control regions of the U1snRNA gene (upper panel) were transfected in HeLa cells treated with either control scrambled siRNA (black bars) or anti-FUS siRNA (white bars). Expression levels of mature microRNAs were analysed by northern blot (miR-9-2, miR-124 and miR-125b-2) or by qPCR (miR-132, miR-212, miR-15a and miR-432). For miR-15a and miR-432, the endogenous levels were measured. The values are referred to control ones set to 1. Error bars represent s.e.m. from three independent experiments. Source data for this figure is available on the online supplementary information page.
The effects of FUS downregulation were also tested in HeLa cells, where RNAi provided 85% reduction (Figure 1C). In order to test the accumulation of neuronal-specific miRNAs, expression cassettes under the control of the ubiquitous U1 snRNA promoter were produced and individually transfected. Figure 1D indicates that the accumulation of the neuronal-specific miRNAs is affected similarly to neuronal cells and in some cases at a higher degree (miR-212 and miR-132). Similarly to SK-N-BE cells, the miR-15a and miR-432 endogenous controls were unaffected. These results indicate that FUS regulates specific miRNA levels independently from their promoters, acting at some post-transcriptional stage in miRNA biogenesis.
Notably, high-throughput analysis using a Taqman array real-time PCR revealed that in HeLa cells a lower proportion of miRNA species were negatively affected with respect to neuronal cells (Supplementary Figure S1B).
FUS binds specific pri-miRNA transcripts
Binding of a recombinant GST-FUS protein to different labelled pri-miRNAs was tested in vitro by band shift analysis. Figure 2A shows that those miRNAs affected by FUS depletion are able to interact with it, maintaining a considerable amount of binding even in the presence of 250-fold excess of cold tRNA competitor. Notably, the control miR-15a, unaffected by FUS depletion, does not show any specific interaction. The only exception, among the tested miRNAs, was pri-miR-628 that, even if affected by FUS depletion, did not show, in our experimental conditions, any specific binding. Moreover, titration of FUS protein in an in vitro binding assay revealed that pri-miR-9-2/FUS interaction is concentration dependent (Figure 2B).
Figure 2.
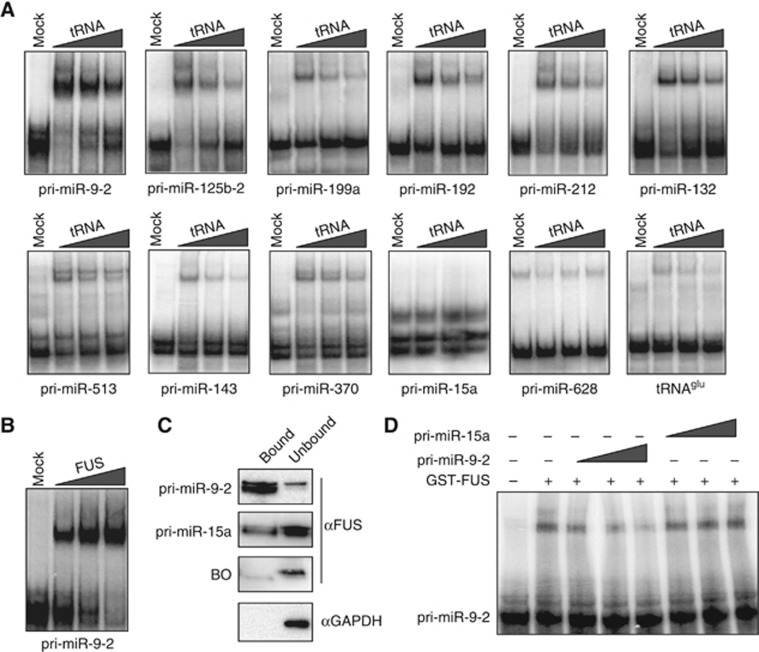
FUS binds in vitro specific pri-miRNA transcripts. (A) Band shift assays with recombinant GST-FUS using in vitro32P-labelled pri-miRNAs in the presence of increasing amounts of cold tRNA competitor (50-, 100- and 250-fold molar excess). Mock samples with the GST peptide were used as control (lanes Mock). (B) Band shift analysis with increasing amounts of GST-FUS (75, 150 and 300 ng) using pri-miR-9-2. (C) Streptavidin-conjugated magnetic beads bound to biotinylated pri-miRNA transcripts were loaded with nuclear extract from SK-N-BE cells. The bound and unbound fractions were tested for FUS binding by western analysis. GAPDH detection and beads-only (BO) samples were used as negative controls. (D) Band shift assay with recombinant GST-FUS using in vitro32 P-labelled pri-miR-9-2 in the presence of increasing amounts (10, 100 and 500-fold molar excess) of cold pri-miR-9-2 or pri-miR-15a. Source data for this figure is available on the online supplementary information page.
Specificity of binding was also analysed in extracts of SK-N-BE cells loaded on streptavidin columns pre-bound with in vitro transcribed biotinylated pri-miR-9-2 or pri-miR-15a. Figure 2C shows that FUS is strongly enriched in the bound fraction of pri-miR-9-2 at difference with pri-miR-15a. Further confirmation of binding specificity is shown in Figure 2D, where pri-miR-9-2, and not pri-miR-15a, competes for FUS binding on its own primary transcript.
Previous analyses on several pri-miRNA binding proteins indicated that the highly conserved terminal loops can act as platforms for trans-acting factors (Michlewski et al, 2008, 2010). In this regard, sequence comparison of the loops of the affected miRNAs did not show any obvious consensus (Dini Modigliani, personal communication). However, since the miR-9-2 loop contains a GU-rich sequence that was suggested to represent an FUS recognition element (Iko et al, 2004), we tested the effect of its mutation on FUS binding. The three G residues of the loop were substituted by C nucleotides and the resulting construct (miR-9-2 mut) was tested for in vitro binding (Figure 3A). Such mutation produced a decrease of 50% in FUS interaction, indicating a consistent contribution of the terminal loop in binding specificity. However, it is possible that the stem provides the remaining binding specificity, as shown by FUS global RNA targets analysis (Hoell et al, 2011). A similar feature was also demonstrated for HnRNP A1 where two binding regions were found: a primary one corresponding to the terminal loop of pri-miR-18a and a secondary site at the bottom of the stem (Michlewski et al, 2008). Moreover, the existence of different apparently disparate binding motifs of FUS has been already observed and suggested to be due to multiple distinct nucleic acid-binding domains, which may function independently or in combination (Tan et al, 2012).
Figure 3.
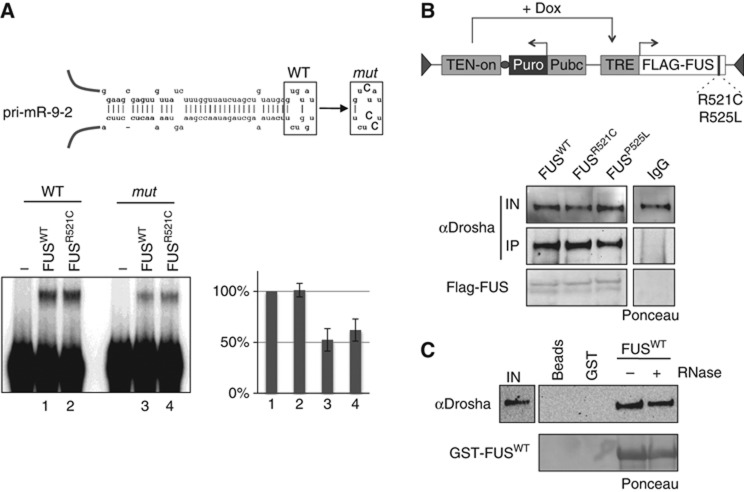
Mutations in the C-terminal domain of FUS do not alter pri-miRNA and Drosha binding. (A) Upper panel: schematic representation of pri-miR-9-2. The wild-type sequence (WT) is shown together with the base substitutions made in the loop of the mutant derivative (mut). Lower panel: band shift assay with GST peptide alone (lanes -) or with 300 ng of wild-type (FUSWT) and mutant (FUSR521C) GST fusions using in vitro32P-labelled pri-miR-9-2 in presence of 250-fold excess of cold tRNA competitor. A densitometric analysis of the shifted bands is also shown (numbers refer to the band shift samples). Error bars represent s.e.m. from three independent experiments. Quantification of the GST-fusion proteins is shown in Supplementary Figure S3. (B) Upper panel: schematic representation of the constructs expressing flagged version of FUS wild type and of its mutant derivatives (R521C and P525L). Lower panel: nuclear extracts from stable SK-N-BE cell lines expressing either flagged FUSWT or the mutant derivatives (FUSR521C and FUSP525L) were immunoprecipitated with anti-FLAG antibodies and analysed by western blot for Drosha interaction. Input samples (IN) are 10% of total nuclear extract. The relative Ponceau and IgG control samples are also shown. (C) Pull down of GST-FUS incubated with SK-N-BE nuclear extracts treated (+) or not (−) with RNase A. Input samples (IN) are 10% of total nuclear extract. The relative Ponceau and beads control samples are also shown. Source data for this figure is available on the online supplementary information page.
We next tested the RNA binding ability of the FUSR521C mutant derivative, one of the most common mutation linked to the ALS pathology shown to provide a severe phenotype (Belzil et al, 2009; Kwiatkowski et al, 2009). Recombinant GST-FUSR521C was tested for pri-miR-9-2 binding (Figure 3A). Interestingly, this derivative provided the same binding activity of the wild-type protein both on pri-miR-9-2 (WT) and on its mutant derivative (mut), indicating that the C-terminal region is not involved in miRNA recognition. Coomassie staining of the purified GST-FUS fusions is shown in Supplementary Figure S3.
Mutations in the C-terminal region have been described to produce cytoplasmic delocalization of the protein (Chiò et al, 2009; Kwiatkowski et al, 2009; Vance et al, 2009; Dormann et al, 2010). In fact, FUSR521C as well as FUSP525L, another common ALS-associated mutation, was shown to delocalize in the cytoplasm in HeLa-transfected cells (Dormann et al, 2010) as well as in post-mortem motor neurons, where they form aggregates (Kwiatkowski et al, 2009; Vance et al, 2009).
In order to test FUS localization in our cellular system, stable clones of SK-N-BE cells, expressing recombinant FUSR521C and FUSP525L fused to the Green Fluorescent Protein under a Doxycycline (Dox) inducible promoter, were generated. Each cell line contained also wild-type FUS fused to the Red Fluorescent Protein (see schematic representation in Supplementary Figure S4A). Supplementary Figure S4B shows that both mutant proteins display altered cellular localization with respect to the WT form: EGFP-FUSP525L, which corresponds to a very severe and juvenile form of ALS, is highly delocalized in the cytoplasm 3 days after Dox induction and produces a large number of aggregates. On the contrary, the cytoplasmic delocalization of EGFP-FUSR521C, which is a more common mutation and correlates with an adult form of ALS, is less pronounced than EGFP-FUSP525L. Notably, neither EGFP-FUSR521C nor EGFP-FUSP525L affected the cellular localization of the co-expressed RFP-FUSWT which remained confined to the nucleus.
Since FUS was described as a Drosha interactor, we next tested the ability of the two FUS mutants (FUSR521C and FUSP525L), to form complexes with Drosha in SK-N-BE cells expressing FLAG-tagged FUS constructs (see schematic representation in Figure 3B). Co-IP experiments indicated that both Flag-FUSR521C and Flag-FUSP525L are complexed with Drosha similarly to the wild type (Figure 3B) while a GST-pull down assay demonstrated that FUS–Drosha interaction is resistant to RNase treatment (Figure 3C).
These data indicate that the C-terminal mutations of FUS do not affect either miRNA or Drosha binding. This, together with the finding that even 50% depletions of FUS (Supplementary Figure S2A) alter miRNA biogenesis, suggests that the cytoplasmic delocalization observed with the FUSR521C and FUSP525L mutants could affect the cellular repertoire of miRNAs by decreasing the levels of the protein available in the nucleus.
Exogenous FUS rescues miRNA accumulation in RNAi-FUS-treated cells
We next checked to what extent wild-type and mutant FUS proteins were able to rescue miRNA biogenesis in RNAi-treated cells. SK-N-BE cell lines, carrying integrated copies of wild-type or mutant Flag-FUS cDNAs with an unrelated 3′UTR and under the control of Dox, were utilized (see Figure 3B). Upon treatment with siRNAs specific for the FUS 3′UTR, efficient depletion of the endogenous FUS protein was obtained (Figure 4A) and, upon Dox induction, exogenous Flag-FUS expression was induced (Figure 4B).
Figure 4.
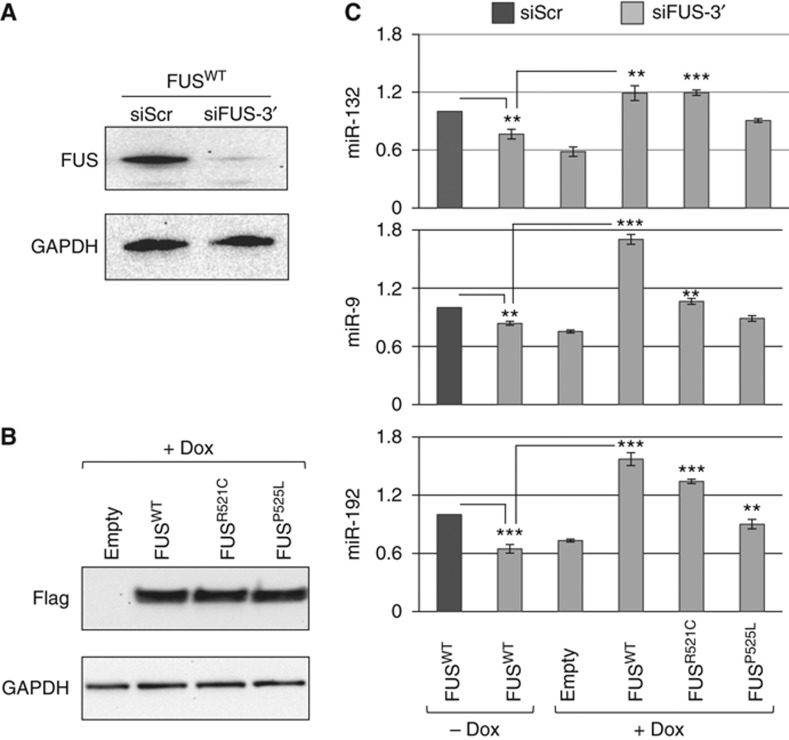
Exogenous FUS can rescue the effects of endogenous FUS depletion. (A) Western blot with FUS and GAPDH antibodies of protein samples from SK-N-BE cell lines carrying the construct FLAG-FUSwt indicated in Figure 3B. These cells were treated with siRNA against the 3′UTR of FUS (siFUS-3′) for 6 days in RA and in the absence of Doxycycline. Scrambled siRNAs (siScr) were used as control. (B) Western blot of protein samples from SK-N-BE cell lines carrying the constructs indicated in Figure3B (FLAG-FUSwt, FLAG-FUSR521C and FLAG-FUSP525L), together with a construct with no cDNA insertion (empty) treated as in (A) in the presence of Doxycycline. Exogenous FLAG-FUS expression was tested using Flag antibodies while GAPDH antibodies were utilized as control. (C) The histogram show the miRNA levels from cells treated as described in (A) and (B), analysed by RT–qPCR. Error bars represent s.e.m. from three independent measurements and the significance was assessed by Unpaired Student’s t-test (*P<0.05, **P<0.01, ***P<0.001). Source data for this figure is available on the online supplementary information page.
The experiments of Figure 4C indicate that miR-132, miR-9 and miR-192 levels are decreased in cells treated with siRNA against FUS in the absence of Dox and are rescued upon activation of the exogenous wild-type FUS. The results with the two FUS mutants are consistent with their delocalization phenotype: FUSR521C, which displays only a slight cytoplasmic delocalization, is able to rescue miRNAs at levels similar to control, while FUSP525L, which has a stronger delocalization phenotype, has a lower rescue activity. It is important to note that also FUSP525L provides sufficient rescue activity since, due to the overexpression conditions utilized, considerable amount of protein is still present in the nucleus (see Supplementary Figure S4B).
In conclusion, these experiments demonstrate a direct involvement of FUS on miRNA biogenesis and again indicate a direct correlation with the amount of FUS localized in the nucleus.
FUS cooperates with co-transcriptional Drosha recruitment
Since it has been shown that the microprocessor complex acts co-transcriptionally (Ballarino et al, 2009), we examined whether FUS is associated with the chromatin and whether it participates in Drosha recruitment. Chromatin immunoprecipitation (ChIP) assays were performed on chromatin from SK-N-BE cells treated with RA for 6 days.
Figure 5 shows that FUS is bound to the chromatin of miR-9-2 and miR-125b-2 coding loci, and that this association is lost after RNase treatment. Upon RNAi-mediated downregulation (Figure 6A), FUS association to the chromatin was consistently reduced (Figure 6B, panels FUS). Moreover, specific association was found on those pri-miRNA loci for which specific FUS binding was identified, whereas very low levels were detected on the pri-miR-15a locus. These findings suggest that chromatin recruitment of FUS at specific miRNA loci occurs during transcription and that it requires binding to nascent pri-miRNAs.
Figure 5.
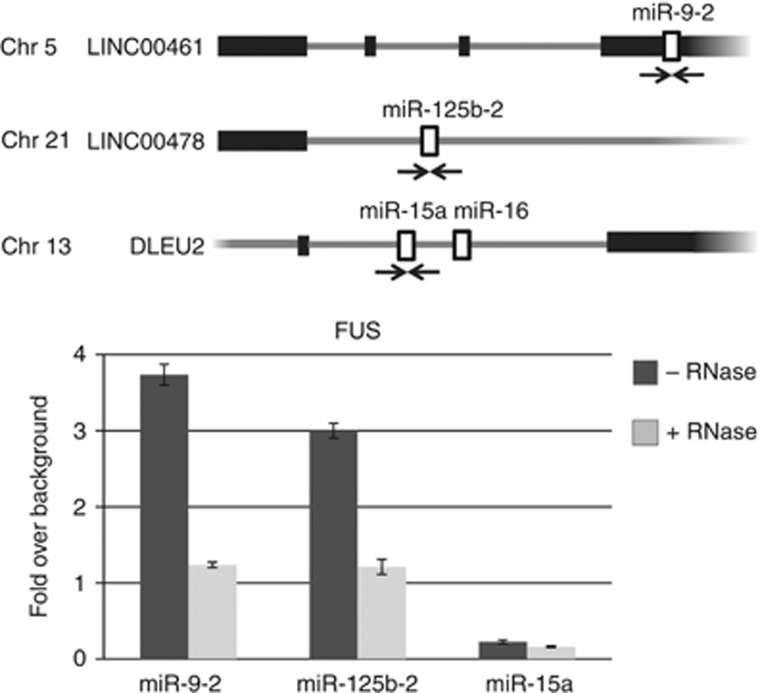
FUS is associated to the chromatin. Upper panel: schematic representation of miR-9-2, miR-125b-2 and miR-15a gene organization. Arrows indicate the positions of the PCR primers used. Lower panel: ChIP analysis with anti-FUS antibodies using chromatin of SK-N-BE cells treated with retinoic acid (RA) for 6 days (black bars). Before immunoprecipitation, half of the sample was treated with RNase (grey bars). Co-amplifications were carried out with miRNA- and tRNA-specific primers. The histograms show the values of FUS immunoprecipitation on miRNA loci normalized for the tRNA signal and expressed as enrichment over background (IgG). Error bars represent s.e.m. from three independent experiments.
Figure 6.
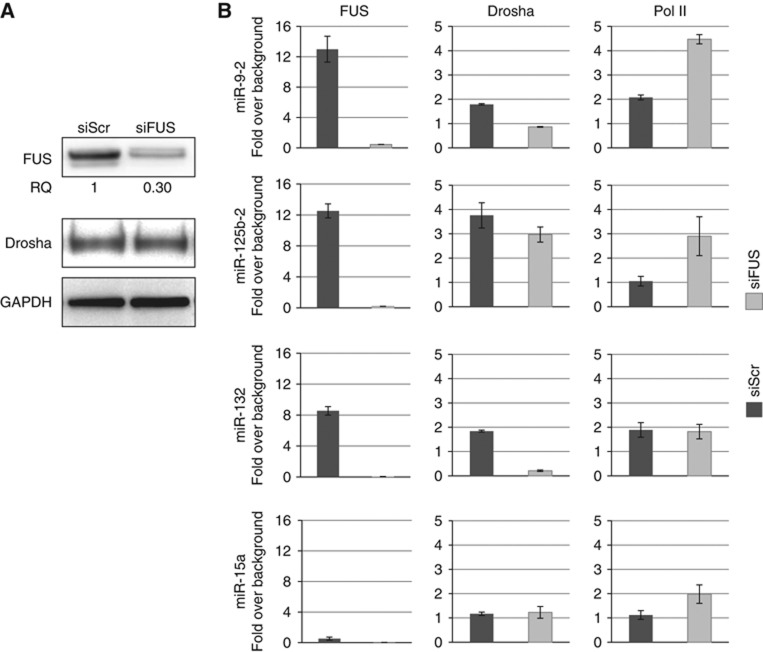
FUS affects co-transcriptional Drosha recruitment. SK-N-BE cells were treated with anti-FUS siRNA (siFUS) or with control siRNA (siScr) and maintained in retinoic acid (RA) for 6 days. (A) Western blot analysis of FUS, Drosha and GAPDH. (B) Histograms showing the results of ChIP analyses with antibodies against FUS, Drosha and Pol II. Chromosomal regions coding for miR-9-2, miR-125b-2, miR-132 and miR-15a were analysed. Co-amplifications were carried out with miRNA- and chromosome IV intergenic region-specific primers. The histograms show the IP values on miRNA loci normalized for the intergenic region and expressed as enrichment over background signals (IgG). Error bars represent s.e.m. from three independent experiments. Source data for this figure is available on the online supplementary information page.
ChIP with Drosha antibodies indicated that this protein was present on all miRNA loci. Upon FUS depletion, even though Drosha cellular levels were unaffected (Figure 6A), its association was reduced on those miRNA loci where FUS–pri-miRNA interaction was found (Figure 6B, panels Drosha). In fact, Drosha recruitment was not affected in the case of miR-15a that neither binds FUS nor is affected by its depletion. The decrease in Drosha recruitment on FUS-dependent miRNA loci was not due to defects in transcription since no decrease in RNA polymerase II loading was detected (Figure 6B, panels Pol II). Instead, a slight increase in PolII recruitment was observed upon FUS depletion for both miR-9-2 and miR125b-2. In consideration of previous data on FUS affecting transcription, with both positive and negative effects (Wang et al, 2008; Tan et al, 2012), it cannot be excluded that the alterations of PolII loading upon FUS depletion on miRNA loci could be due to a secondary effect of FUS on transcription elongation or polymerase release and recycling.
These data allowed us to conclude that FUS interaction is required for efficient recruitment of Drosha at specific pri-miRNA sites at early stages of transcription. These data, together with the observation that the FUS–Drosha interaction does not require RNA, allow us to suggest that the binding of FUS to nascent pri-miRNA molecules cooperates with efficient subsequent Drosha recruitment at the same sites.
In vitro processing extracts were produced from SK-N-BE cells treated with either scrambled or anti-FUS siRNAs. Supplementary Figure S5 shows that reduced FUS levels do not affect in vitro processing of several neuronal pri-miRNAs. Due to the low efficiency of these processing extracts and to the small modulation produced by FUS on miRNA biogenesis, it is possible that the in vitro conditions are not appropriate to reproduce the stoichiometry and architecture of the events occurring on the nascent transcripts on the chromatin. It cannot be excluded also that the pri-miRNA portions utilized have a context different from the primary transcript.
Discussion
The FUS protein has been recently linked to familial forms of ALS, a severe age-dependent disorder causing degeneration of motor neurons in the brain and spinal cord. Since mutations compatible with life seem to mainly affect the nucleus/cytoplasmic distribution of the protein, it has been suggested that these mutations may have a dual effect: (i) loss of function in the nucleus and (ii) toxic gain of function in the cytoplasm. Therefore, dosage alteration of the protein in the two compartments can provide a hint for understanding such neuronal-restricted pathology. FUS has been attributed a large number of functions in the nucleus mainly related to transcription and RNA processing, whereas cytoplasmic aggregated forms have been suggested to cause alteration in neuronal plasticity, or in nuclear RNA maturation and transport (Belly et al, 2005; Polymenidou et al, 2012).
Among the large repertoire of nuclear functions, we focused on the observation that FUS was described as a Drosha interactor. Here, we demonstrate that the FUS protein has a dual function of interacting with specific pri-miRNA sequences and with Drosha. Moreover, we show that FUS binds to nascent pri-miRNA molecules and helps Drosha recruitment on the chromatin allowing efficient miRNA processing.
We also show that, among the others, FUS affects the biogenesis of miRNAs with a relevant role in neuronal function, differentiation and synaptogenesis such as miR-9, miR-125b and miR-132 (Laneve et al, 2007, 2010; Packer et al, 2008; Edbauer et al, 2010; Pathania et al, 2012).
Notably, we observed that the accumulation levels of these miRNAs were lowered even when the residual amount of FUS was half with respect to control. These data could explain why mutations affecting FUS nuclear dosage could have a remarkable negative effect on miRNA homeostasis, thus providing a possible correlation with the ALS pathogenesis. Due to the fact that ubiquitous miRNAs are affected by FUS downregulation, one should envisage a more general toxic effect not restricted to the nervous system. However, several considerations could explain a higher susceptibility of neuronal cells: (i) the miRNA downregulation is limited and only neuronal cells could be affected by such tiny changes; (ii) the neuronal miRNA species identified play non-redundant essential functions; (iii) protein delocalization and aggregate formation could be partially compensated in proliferating cells, while in post-mitotic neuronal cells these processes would have additive effects. The progressive accumulation and aggregation is indeed a phenomenon common to other neuro-degenerative diseases due to proteins having the ability of forming amyloid-like fibres (Yamamoto and Simonsen, 2011; Han et al, 2012).
It is important to underline that FUS plays multiple roles in the nucleus and in particular during transcription. ChIP and promoter microarrays have identified a large number of target genes regulated by this factor (Tan et al, 2012), thus indicating that miRNA biogenesis may represent only part of FUS activity. Due to the fact that the pathological effects of FUS mutations are mainly restricted to neuronal cells, it is possible that FUS threshold becomes critical only in these cells, and that miRNA biogenesis is part of the molecular mechanisms whose deregulation may have a relevant role in ALS pathogenesis.
Materials and methods
Oligonucleotides used in this study
Oligonucleotide sequences are listed in Supplementary Table II.
Cell cultures and treatments
SK-N-BE cells were cultured in RPMI medium 1640 (Gibco), supplemented with 10% fetal bovine serum (FBS), 1-L-glutamine, and penicillin/streptomycin, and induced to differentiate by 10 μM all-trans-Retinoic acid (RA, Sigma). SK-N-BE plasmid transfection was carried out as previously described (Laneve et al, 2007) while siRNAs targeting FUS coding region (Hs_FUS_4 FlexiTube siRNA, SI00070518, Qiagen) or 3′UTR (see Supplementary Table I) were transfected using HiPerfect Transfection Reagent (Qiagen) according to manufacturer’s instructions.
For the generation of stable SK-N-BE cells expressing FUS protein, upon plasmid transfection (epB-Puro-TT derived plasmids and epiggyBac transposase vector), the cells were selected by Puromycin (1 μg/ml) treatment and the expression of the different forms of FUS protein was induced by adding Dox (0.2 μg/ml) to the culture medium.
For the rescue experiments, stable SK-N-BE cells expressing FLAG-FUSwt, FLAG-FUSR521C and FLAG-FUSP525L were treated with siRNA against the 3′UTR of FUS (siFUS-3′; see Supplementary Table II) for 6 days in RA. The last 2 days the cells were treated or not with Dox (0.02 μg/ml final concentration).
HeLa cells were cultured and transfected as previously described (Morlando et al, 2008).
Plasmid construction
To generate the constructs overexpressing miRNAs, the genomic fragments containing pri-miR-9-2, pri-miR-124-2, pri-miR-212 e pri-miR-132 were PCR amplified (oligonucleotides are listed in Supplementary Table II) and cloned using BglII and XhoI restriction sites of U1snRNA expression cassette (Denti et al, 2004). Plasmid overexpressing pri-mir-125b-2 is described in Laneve et al (2007). The vectors were transfected in combination with a plasmid carrying a modified snRNA U1 gene (U1#23; Denti et al, 2006) to measure the efficiency of transfection.
For generating GST fused FUS protein, FUS cDNA was PCR amplified from vector pCMV6-AC (SC320263, OriGene Technologies) with the oligolucleotides FUS FW and FUS REV and inserted in BamHI and XhoI restriction sites of pGEX-4T-1 (Amersham Biosciences) raising FUSWT vectors. The mutant form FUSR521C was obtained by inverse PCR amplification on FUSWT vectors using the oligonucleotides FUS R521C fw and FUS R521C rev.
For the generation of the transposable element vectors for inducible expression of FUS, cDNA from vector pCMV6-AC was amplified using the Flag-FUS FW, FUS WT REV, FUS R521C REV and FUS P525L REV and inserted into the epB-Puro-TT vector (see Supplementary Methods) generating the Flag-FUSWT, Flag-FUSR521C and Flag-FUSP525L plasmids.
Protein extraction and western blot
SK-N-BE and HeLa protein extracts and western blot analysis were performed as previously described (Laneve et al, 2010). The immunoblots were incubated with the following antibodies: anti-FUS/TLS (sc-47711, Santa Cruz), anti-DGCR8 (ab90579, Abcam), anti-Drosha (ab12286, Abcam), anti-N-Myc (sc-56729, Santa Cruz), anti-FlagM2 (Sigma) and anti-GAPDH (sc-32233, Santa Cruz) as a loading control. The densitometric analysis was performed using Image Lab software (Bio-Rad).
RNA preparation and analysis
Total RNA was isolated using miRNeasy Mini Kit according to manufacturer’s instructions (Qiagen).
For the Northern blot assay, 5 μg of total RNA was analysed on 10% polyacrylamide denaturing gel as described in Laneve et al (2010). DNA oligonucleotides complementary to the sequence of mature miR-9, miR-124, miR-125b, miR-132, U1#23 and to 5.8S-rRNA were 32P-labelled and used as probes. Densitometric analysis was performed using Typhoon Imager (GE Healthcare) and ImageQuant software (Molecular Dynamics).
Quantitative real-time PCR analysis
cDNA generation was carried out using the miScript Reverse Transcription Kit (Qiagen). The real-time PCR detection of miRNAs was performed using miScript SYBR-Green PCR Kit and DNA oligonucleotides by Qiagen, on a 7500 Fast Real-Time PCR (Applied Biosystems). The values obtained were normalized for snoRNA-U25 and were analysed by the unpaired Student’s t-test. P-values were calculated for samples from three independent experiments unless otherwise indicated.
miRNAs high-throughput analysis
In all, 700 ng of total RNA extracted from SK-N-BE cells was retrotranscribed using the TaqMan MicroRNA RT Kit (Applied Biosystems). The real-time detection of the miRNA levels was performed using the TaqMan® Human MicroRNA Array A (Applied Biosystems) according to manufacturer’s instructions. The values obtained were normalized for snoRNA-U44.
ChIP assay
ChIP analyses were performed on chromatin extracts from SK-N-BE cells according to manufacturer’s specifications of MAGnify Chromatin Immunoprecipitation System kit (Invitrogen). Sheared chromatin was immunoprecipitated with the following antibodies: anti-FUS/TLS (sc-47711, Santa Cruz), anti-Drosha (ab12286, Abcam) and anti-Pol II (sc-889, Santa Cruz). The occupancy of the immunoprecipitated factor on miRNA loci was estimated by normalizing for the occupancy on tRNA coding region or chromosome IV intergenic region and expressed as enrichment over background (IgG) (Chakrabarti et al, 2002). Densitometric analysis was performed using Typhoon Imager (GE Healthcare) and ImageQuant software (Molecular Dynamics). RNase treatment of the chromatin and the occupancy of the immunoprecipitated factor on miRNA loci were carried out as described in Morlando et al (2008). Oligonucleotide used for PCR amplifications is listed in Supplementary Table II.
Band shift
Band shift assays were carried out as previously described (Song et al, 2012) with minor modifications. Purified in vitro labelled transcripts were incubated with 300 ng of recombinant wild-type and mutant GST-FUS in the presence of increasing amount of cold tRNA competitor, from 50 to 250 molar excess. The complexes were separated by a 4% acrylamide non-denaturing gel. Densitometric analysis was performed using Typhoon Imager (GE Healthcare) and Optiquant software.
Biotin pull-down
Binding of biotinylated transcripts to paramagnetic streptavidin Dynabeads (Dynal) and incubation with nuclear lysate were carried out as described in Figueroa et al (2003). Biotinylated transcripts were obtained from PCR-generated templates (oligonucleotides are listed in Supplementary Table II) using 0.35 mM Biotin-16-UTP (Roche) as described previously (Dye and Proudfoot, 1999).
GST-FUS purification
FUSWT and FUSR521C were transfected in BL21 cells and induced with 0.5 mM IPTG for 4 h at 28°C. Cell pellets were resuspended in 5 ml of NET-N buffer (Tris–HCl pH 8 20 mM, NaCl 100 mM, NP-40 0.5%, EDTA 0.5 mM) supplemented with a cocktail of protease inhibitor (Roche). After sonication, the supernatant fractions were loaded onto Glutathione-Agarose resin (G4510, Sigma) and incubated for 1 h at 4°C and then washed once with NET-N buffer and twice with NET (Tris–HCl pH 8 20 mM, NaCl 100 mM, EDTA 0.5 mM). The recombinant GST proteins were eluted with the elution buffer containing 20 mM L-Glutathione reduced and 100 mM Tris–HCl pH 8.
Co-immunoprecpitation and GST-pull down
Co-immunoprecipitation was performed using Immunoprecipitation kit—Dynabeads Protein G (Invitrogen) according to manufacturer’s instructions. To obtain the nuclear extracts, the cell pellets were resuspended with Buffer A (Tris–HCl pH 8 20 mM, NaCl 10 mM, MgCl2 3 mM, Igepal 0.1%, glycerol 10%, EDTA 0.2 mM) supplemented with protease inhibitor (Roche) and after centrifugation the nuclei were resuspended in Buffer C (Tris–HCl pH 8 20 mM, NaCl 400 mM, glycerol 20%, DTT 1 mM) supplemented with protease inhibitor (Roche). After three cycles of incubation in liquid nitrogen followed by incubation at 37°C the nuclear extract was recovered by centrifugation.
The GST-pull down experiments were carried out as described in Morlando et al (2004) with minor modification. In all, 50 μg of SK-N-BE nuclear extract was used instead of in vitro translated Drosha protein and the RNase treatment was carried out with RNase A (Sigma) at 20 mg/ml final concentration.
Supplementary Material
Acknowledgments
We thank A Fatica and M Ballarino for helpful discussion, James Hughes for reading the manuscript and M Marchioni for technical support. AR was recipient of an HFSP fellowship. This work was partially supported by grants from: Telethon (GGP07049), Parent Project Italia, AIRC, IIT ‘SEED’, FIRB, PRIN and BEMM.
Author contributions: MM did ChIP experiments, band shift and in vitro processing analysis; SDM took care of RNAi experiments, miRNA profiles and RT–PCR analysis; GT prepared recombinant proteins and performed Co-IP; AR made the fusion constructs and tested their expression; VDC handled neuronal cell differentiation; EC did mutant analysis; IB coordinated the research and wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aman P, Panagopoulos I, Lassen C, Fioretos T, Mencinger M, Toresson H, Höglund M, Forster A, Rabbits TH, Ron D, Mandahl N, Mitelman F (1996) Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37: 1–8 [DOI] [PubMed] [Google Scholar]
- Ballarino M, Pagano F, Girardi E, Morlando M, Cacchiarelli D, Marchioni M, Proudfoot NJ, Bozzoni I (2009) Coupled RNA processing and transcription of intergenic primary microRNAs. Mol Cell Biol 29: 5632–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y (2005) Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett 379: 152–157 [DOI] [PubMed] [Google Scholar]
- Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, Gauthier J, Hince P, Desjarlais A, Bouchard JP, Lacomblez L, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2009) Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 73: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti SK, James JC, Mirmira RG (2002) Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem 277: 13286–13293 [DOI] [PubMed] [Google Scholar]
- Chiò A, Restagno G, Brunetti M, Ossola I, Calvo A, Mora G, Sabatelli M, Monsurro MR, Battistini S, Mandrioli J, Salvi F, Spataro R, Schymick J, Traynor BJ, La Bella V (2009) Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging 30: 1272–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denti MA, Rosa A, Sthandier O, De Angelis FG, Bozzoni I (2004) A new vector, based on the PolIII promoter of siRNAs in mammalian cells. Mol Ther 10: 191–199 [DOI] [PubMed] [Google Scholar]
- Denti MA, Rosa A, D’Antona G, Sthandier O, De Angelis FG, Nicoletti C, Allocca M, Pansarasa O, Parente V, Musarò A, Auricchi A, Bottinelli R, Bozzoni I (2006) Chimeric adeno-associated virus/antisense U1 small nuclear RNA effectively rescues dystrophin synthesis and muscle function by local treatment of mdx mice. Hum Gene Ther 17: 565–574 [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than M, Mackenzie I, Capell A, Schmid B, Newmann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transortin-mediated nuclear import. EMBO J 29: 2841–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye MJ, Proudfoot NJ (1999) Terminal exon definition occurs cotranscriptionally and promotes termination of RNA polymerase II. Mol Cell 3: 371–378 [DOI] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, Tada T, Dolan BM, Sharp PA, Sheng M (2010) Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65: 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat EG, Muñoz-Canoves P, Gorospe M, Muñoz A (2003) Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol 23: 4991–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan K, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shlekhattar R (2004) The microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240 [DOI] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149: 768–779 [DOI] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T (2011) RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18: 1428–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iko Y, Kodama TS, Kasai N, Oyama T, Morita EH, Muto T, Okumura M, Fujii R, Takumi T, Tate S, Morikawa K (2004) Domain architectures and characterization of an RNA-binding protein, TLS. J Biol Chem 279: 44834–44840 [DOI] [PubMed] [Google Scholar]
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuzi N (2011) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69: 152–162 [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136: 1001–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19: R46–R64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneve P, Di Marcotullio L, Gioia U, Fiori ME, Ferretti E, Gulino A, Bozzoni I, Caffarelli E (2007) The interplay between microRNAs and the neurotrophin receptor tropomyosin-related kinase C controls proliferation of human neuroblastoma cells. Proc Natl Acad Sci USA 104: 7957–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneve P, Gioia U, Andriotto A, Moretti F, Bozzoni I, Caffarelli E (2010) A minicircuitry involving REST and CREB controls miR-9-2 expression during human neuronal differentiation. Nucleic Acids Res 38: 6895–6905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law WJ, Cann KL, Hicks GG (2006) TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic 5: 8–14 [DOI] [PubMed] [Google Scholar]
- Michlewski G, Guil S, Semple CA, Cáceres JF (2008) Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell 32: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlewski G, Guil S, Cáceres JF (2010) Stimulation of pri-miR-18a processing by hnRNP A1. Adv Exp Med Biol 700: 28–35 [DOI] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Greco P, Caffarelli E, Dichtl B, Bozzoni I (2004) Coupling between snoRNP assembly and 3' processing controls box C/D snoRNA biosynthesis in yeast. EMBO J 23: 2392–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ (2008) Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15: 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL (2008) The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J Neurosci 28: 14341–14346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, Torres-Reveron J, Yan L, Kimura T, Lin TV, Gordon V, Teng ZQ, Zhao X, Fulga TA, Van Vactor D, Bordey A (2012) miR-132 enhances dendritic morphogenesis, spine density, synaptic integration, and survival of newborn olfactory bulb neurons. PLoS ONE 7: e38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Bennett CF, Cleveland DW, Yeo GW (2012) Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res 1462: 3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi N, Cironi L, Suvà ML, Stamenkovic I (2007) Sarcomas: genetics, signalling, and cellular origins. J Pathol 213: 4–20 [DOI] [PubMed] [Google Scholar]
- Song X, Wang X, Arai S, Kurokawa R (2012) Promoter-associated noncoding RNA from the CCND1 promoter. Methods Mol Biol 809: 609–622 [DOI] [PubMed] [Google Scholar]
- Tan AY, Manley JL (2009) The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1: 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL (2012) TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci USA 109: 6030–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323: 1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454: 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Warraich ST, Nicholson GA, Blair IP (2010) Fused in sarcoma/translocated in liposarcoma: a multifunctional DNA/RNA binding protein. Int J Biochem Cell Biol 42: 1408–1411 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Simonsen A (2011) The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 43: 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110: 1741–1750 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.