

Abstract
Mitochondrial oxidative phosphorylation (OXPHOS) sustains organelle function and plays a central role in cellular energy metabolism. The OXPHOS system consists of 5 multisubunit complexes (CI–CV) that are built up of 92 different structural proteins encoded by the nuclear (nDNA) and mitochondrial DNA (mtDNA). Biogenesis of a functional OXPHOS system further requires the assistance of nDNA-encoded OXPHOS assembly factors, of which 35 are currently identified. In humans, mutations in both structural and assembly genes and in genes involved in mtDNA maintenance, replication, transcription, and translation induce ‘primary’ OXPHOS disorders that are associated with neurodegenerative diseases including Leigh syndrome (LS), which is probably the most classical OXPHOS disease during early childhood. Here, we present the current insights regarding function, biogenesis, regulation, and supramolecular architecture of the OXPHOS system, as well as its genetic origin. Next, we provide an inventory of OXPHOS structural and assembly genes which, when mutated, induce human neurodegenerative disorders. Finally, we discuss the consequences of mutations in OXPHOS structural and assembly genes at the single cell level and how this information has advanced our understanding of the role of OXPHOS dysfunction in neurodegeneration.
Keywords: mitochondria, neurodegeneration, OXPHOS
Introduction
Nearly every activity of the cell is powered by the hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP). In order to maintain ATP homeostasis and, therefore, cell integrity and function, ATP must be continuously replenished. The energy required for this process comes from the stepwise oxidation of fuel molecules originating from three different carbon sources, i.e., monosaccharides, mainly glucose (GLC) but also fructose (FRC) and galactose (GAL), fatty acids (FAs) and amino acids. Following food uptake, these fuel molecules enter the body from the intestine, where they are produced upon the enzymatic breakdown of carbohydrates, triacylglycerols (TAGs) and proteins. Their distribution throughout the body occurs via the circulatory system and cells take up the required nutrients for energy production, biosynthesis and replenishment of intracellular glycogen stores (liver cells and skeletal muscle cells) and TAGs (fat cells). Liver cells convert excess GLC to TAGs, which they package in very low density lipoprotein (VLDL) particles for transport to the fat cells. In between feeding, the blood GLC level is maintained by the liver mobilizing its glycogen stores and producing GLC from lactate (LAC), glycerol and glucogenic amino acids. At the same time, fat cells mobilize their TAG stores to release FAs and glycerol. GLC is the only fuel molecule for red blood cells and, normally, brain cells and to limit its consumption, liver cells and skeletal muscle cells at rest primarily use FAs for the production of ATP. Mature red blood cells and skeletal muscle cells at work convert GLC to LAC, which they release in the circulation. This LAC is taken up mainly by the liver, which uses ATP derived from FAs to reconvert it to GLC.
For GLC and glycerol, the stepwise oxidation process starts in the cytosol, where a series of enzymes catalyse their partial oxidation to pyruvate (PYR; Figure 1). During this process, the major part of the chemical bond energy of the fuel molecule is transferred in the form of electrons to the electron carrier nicotinamide adenine dinucleotide (NAD+) thus reducing to reduced nicotinamide adenine dinucleotide (NADH), whereas a smaller part is transferred in the form of a phosphoryl group to ADP. The latter process, referred to as substrate-level phosphorylation, uses a phosphorylated reactive intermediate as a donor. In the case of GLC, cytosolic oxidation yields two molecules each of PYR, ATP and NADH. Other contributions to the cytosolic PYR pool come from LAC and certain amino acids.
Figure 1.
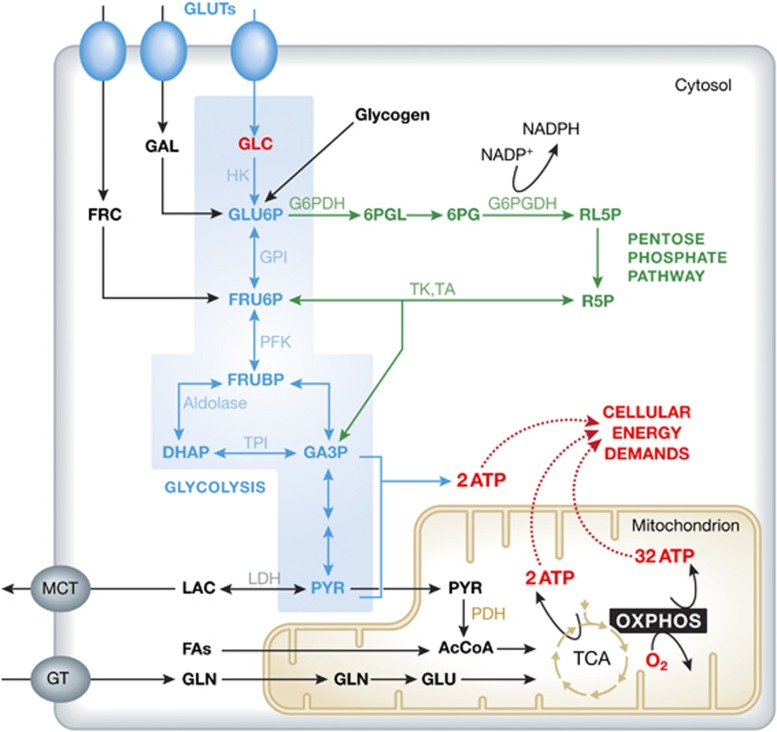
Energy metabolism in a typical mammalian cell. To meet cellular energy demands, ATP is generated by the glycolysis pathway (blue), the tricarboxylic acid (TCA) cycle and the oxidative phosphorylation (OXPHOS) system. The main energy substrate glucose (GLC) enters the cell via GLC transporters (GLUTs) and is converted into pyruvate (PYR). Alternatively, surplus GLC can be stored as glycogen for later use or enter the pentose phosphate pathway (green). PYR can have two different fates: either it is converted into lactate (LAC) that leaves the cell, or it enters the mitochondrion (yellow) to form Acetyl coenzyme A (AcCoA). The latter is processed by the TCA cycle to yield NADH and FADH2, which are substrates of the OXPHOS system. In addition to GLC also fructose (FRC), galactose (GAL), fatty acids (FAs) and glutamine (GLN) can enter the ATP producing system (see text for details). 6PG, 6-phosphogluconate; 6PGL, 6-phosphogluconolactone; DHAP, dihydroxyacetone phosphate; FRU6P, fructose 6-phosphate; FRUBP, fructose 1,6-bisphosphate; GA3P, glyceraldehyde 3-phosphate; GLU, glutamate; G6PDH, glucose 6-phosphate dehydrogenase; G6PGDH, 6-phosphogluconate dehydrogenase; GT, glutamine transporter; GPI, phosphoglycose isomerase; HK, hexokinase; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; PDH, pyruvate dehydrogenase; PFK, phospofructokinase; RL5P, ribulose 5-phosphate; R5P, ribose 5-phosphate; TA, transaldolase; TK, transketolase; TPI, triosephosphate isomerase.
PYR oxidation involves the combined action of a series of enzymes located within the mitochondrial matrix. First, PYR is oxidatively decarboxylated by pyruvate dehydrogenase (PDH), yielding one molecule each of CO2, NADH and Acetyl coenzyme A (AcCoA). Next, AcCoA is oxidized by the enzymes of the tricarboxylic acid (TCA), producing two molecules of CO2, three molecules of NADH, one molecule of the reduced form of the electron carrier flavin adenine dinucleotide (FADH2) and one molecule of GTP, by substrate-level phosphorylation.
The oxidation of FAs takes place entirely in the mitochondrial matrix by a process referred to as β oxidation. Also, this process occurs in a stepwise manner yielding one molecule each of AcCoA, NADH and FADH2 per step. The end product is either AcCoA (even-numbered FAs) or propionyl-CoA (odd-numbered FAs). The latter molecule can be converted into succinyl-CoA, which is an intermediate of the TCA cycle. Also, the oxidation of amino acids occurs entirely in the mitochondrial matrix. To this end, amino acids are first deaminated and then, depending on the type of amino acid, processed to PYR, AcCoA or an intermediate of the TCA cycle (α-ketoglutarate, succinyl-CoA, fumarate or oxaloacetate) (Lunt and Vander Heiden, 2011).
Intermediates can be withdrawn from the above oxidation processes, e.g., for the synthesis of neurotransmitters and amino acids (Dienel, 2012). Furthermore, GLC can be metabolized through the pentose phosphate pathway (PPP; Figure 1), yielding reduced nicotinamide adenine dinucleotide phosphate (NADPH) for anabolic reactions and pentoses for the synthesis of nucleotides and aromatic amino acids.
For the oxidation processes to continue, reoxidation of the reduced electron carriers (NADH and FADH2) is a prerequisite. This reoxidation can take place in the cytosol by the enzyme lactate dehydrogenase (LDH) and in the mitochondrial matrix by the combined action of the enzymes and electron carriers of the electron transport chain (ETC; Smeitink et al, 2001). During the LDH reaction, the NADH electrons are transferred to PYR, yielding LAC, whereas during the ETC reaction, the NADH and FADH2 electrons are transferred to molecular oxygen (O2), yielding H2O. The mitochondrial inner membrane (MIM) is impermeable to NADH, and under normal conditions of oxygen supply the electrons of cytosolic NADH are transferred across this membrane by shuttle systems such as the malate-aspartate shuttle and the glycerol-phosphate shuttle, yielding cytosolic NAD+ for continuation of glycolysis and mitochondrial NADH (malate-aspartate shuttle) or FADH2 (glycerol-phosphate shuttle) for reoxidation by the ETC.
Together, the enzymes of the ETC convert the oxidation energy temporarily stored in NADH and FADH2 into an electrochemical proton gradient across the MIM that is used by a proton-transporting enzyme (F1Fo-ATP synthase) to produce ATP. This process is referred to as oxidative phosphorylation (OXPHOS). Here, it is important to realize that many other MIM transporters are driven by the electrochemical proton gradient and it is for that reason that a proper electrochemical proton gradient is essential for the maintenance of mitochondrial integrity and many other aspects of mitochondrial function (apoptosis, innate immunity, redox control, calcium homeostasis and several biosynthetic processes) (Kwong et al, 2007; Wang and Youle, 2009; Koopman et al, 2010, 2012; Arnoult et al, 2011; Mammucari et al, 2011). In addition, some energy of the electrochemical proton gradient is used for thermogenesis.
The balance between cytosolic and mitochondrial ATP production depends on the type of cell and its physiological demands and environmental conditions (supply of fuel molecules and O2). Some cells depend completely on cytosolic ATP production and produce LAC to reoxidize NADH (mature red blood cells), others depend largely on the complete oxidation of GLU (brain cells) or FAs (liver cells) and use O2 as the final electron acceptor, again others oxidize mainly FAs at rest and GLU at a sudden burst of activity (skeletal muscle cells). In the latter case, LAC is produced because of a hampered supply of O2. In terms of ATP production, the maximum yield per molecule of GLU is 2 ATP in the case of oxidation to LAC and ∼30 ATP in the case of full oxidation to CO2 and H2O (Rich, 2003). Under pathological conditions, the mechanism of ATP production can change dramatically. For instance, most cancer cells oxidize GLU to LAC to produce ATP, even in the presence of O2 (Warburg effect) (Cairns et al, 2011). Other pathological conditions are caused by inborn errors of enzymes that convert energy from fuel molecules to NADH, FADH2 and ATP by substrate phosphorylation or from NADH and FADH2 to ATP by OXPHOS. Moreover, such errors can develop in time, e.g., as a consequence of insufficient control of reactive oxygen species (ROS) levels.
Neurons are high consumers of ATP and because they have no glycogen stores they depend entirely on the uninterrupted supply of GLU through the extracellular fluid. For the same reasons, neurons preferentially oxidize GLC to CO2 and H2O providing the highest yield of ATP per GLU. Therefore, maintenance of mitochondrial integrity and function is of highest priority to these cells. Mitochondria are motile organelles that exhibit fusion and fission and display a dynamic internal structure (Benard and Rossignol, 2008). The balance between these processes determines net mitochondrial (ultra)structure and distribution, which is linked to mitochondrial (dys)function and metabolism during healthy and pathological conditions including neurodegeneration (Knott et al, 2008; Lizana et al, 2008; Willems et al, 2009; Dieteren et al, 2011; Campbell et al, 2012; Court and Coleman, 2012; Kageyama et al, 2012). In humans, a (progressive) decrease in mitochondrial function in general, and of the OXPHOS system in particular, has been linked to neurodegeneration during normal ageing and many other conditions including inborn errors of energy metabolism, amyotrophic lateral sclerosis (ALS), Parkinson disease (PD), Alzheimer disease (AD), Huntington disease (HD), certain forms of (brain) cancer, diabetes, epilepsy, obesity, cognitive impairment, psychosis and anxiety (Chandra and Singh, 2011; Martin, 2011; Anglin et al, 2012; Costa and Scorrano, 2012; Finsterer and Mahjoub, 2012; Nunnari and Suomalainen, 2012; Schapira, 2012).
OXPHOS inhibition is also evoked by off-target (drug) effects, likely differentially affecting healthy individuals and patients with mitochondrial dysfunction (Wallace, 2008; Dimauro and Rustin, 2009; Cohen, 2010; Finsterer and Segall, 2010; Morán et al, 2012). For example, mice with fatal encephalomyopathy due to mitochondrial dysfunction were 2.5- to 3-fold more sensitive to the volatile anaesthetics isoflurane and halothane than wild-type (wt) mice (Quintana et al, 2012). Moreover, environmental toxins including rotenone and persistent organic pollutants (POPs) like the insecticide dichlorodiphenyltrichloroethane (DDT), the herbicide and industrial waste product 2,3,7,8-tetrachlorodibenzodioxin (TCCD) and the phenolic flame retardant tetrabromobisphenol A (TBBPA) directly or indirectly inhibit OXPHOS function (Lee et al, 2010; Schapira, 2010). During recent years, substantial progress has been made in understanding the role of mitochondrial dysfunction in neurodegeneration. We recently argued that understanding the cellular (patho)physiology of monogenic mitochondrial disorders, particularly those associated with (relatively rare) OXPHOS mutations, will not only enhance our understanding of mitochondrial (dys)function but is also therapeutically relevant for the many diseases in which OXPHOS function is disturbed (Koopman et al, 2012). Below we first provide a theoretical background regarding the OXPHOS system. This is followed by an inventory of OXPHOS genes that are, when mutated, associated with neurodegeneration in humans. Finally, we present the insights obtained from studying the consequences of mutations in OXPHOS structural and assembly genes in living cells.
The mitochondrial OXPHOS system
The OXPHOS system (Figure 2) consists of five MIM-embedded multisubunit complexes: complex I (CI or NADH:ubiquinone oxidoreductase; EC 1.6.5.3), complex II (CII or succinate:ubiquinone oxidoreductase; EC 1.3.5.1), complex III (CIII or ubiquinol:cytochrome c oxidoreductase; EC 1.10.2.2), complex IV (CIV or cytochrome-c oxidase; EC 1.9.3.1) and complex V (CV or FoF1-ATP-synthase; EC 3.6.1.34). These complexes are divided into two functional parts: (i) the four complexes (CI–CIV) of the ETC and (ii) CV that generates ATP (Distelmaier et al, 2009; Smeitink et al, 2001; Koopman et al, 2012). Genetically, 92 different genes encoding structural OXPHOS subunits have been identified (Figure 2). CII is exclusively derived from the nuclear DNA (nDNA), whereas the other OXPHOS complexes contain subunits that are encoded by nDNA and the mitochondrial DNA (mtDNA). In addition to the structural OXPHOS subunit genes, the mtDNA also contains genetic information for the 2 mitochondrial ribosomal RNAs (mt-rRNAs) and the 22 mitochondrial transfer RNAs (mt-tRNAs). All proteins involved in mtDNA repair, replication, transcription, translation and maintenance of the mitochondrial deoxynucleoside triphosphate (dNTP) pool, as well as mt-tRNA synthetases and mitochondrial ribosomal proteins, are nDNA encoded (Peralta et al, 2012). Biogenesis of a functional OXPHOS system further requires a large set (>75) of nDNA-encoded proteins (Supplementary Table 1).
Figure 2.
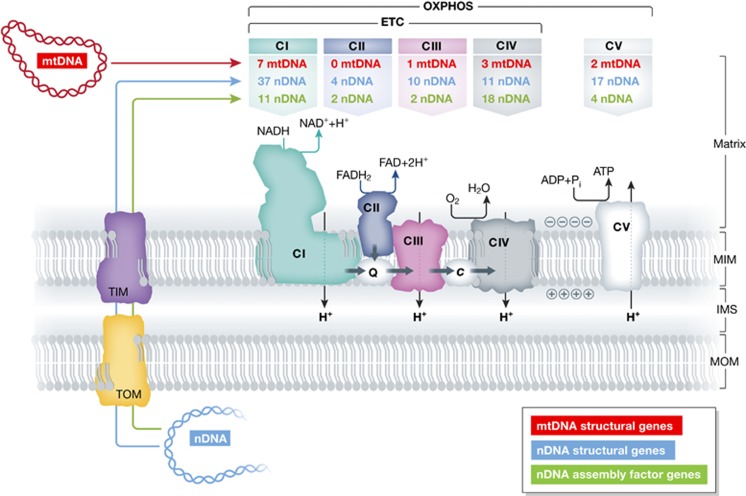
Genetic origin and functional interaction of the mitochondrial oxidative phosphorylation (OXPHOS) complexes. The mitochondrial OXPHOS system consists of five multisubunit complexes (CI–CV) that reside in the mitochondrial inner membrane (MIM). The MIM encloses the mitochondrial matrix and is surrounded by the mitochondrial outer membrane (MOM). An inter-membrane space (IMS) is located between the MIM and MOM. The subunits of CI, CIII, CIV and CV are encoded by the mitochondrial (mtDNA; red) and nuclear DNA (nDNA; blue), whereas CII exclusively consists of nDNA-encoded subunits (table at the top). OXPHOS biogenesis is mediated by nDNA-encoded assembly factors (green). The nDNA-encoded proteins are imported into the mitochondrial matrix via the TOM (translocator of the inner membrane) and TIM (translocator of the inner membrane) systems. At CI and CII, NADH and FADH2 are oxidized, respectively, and the released electrons are transported to CIII via Coenzyme Q10 (CoQ10; ‘Q’). From thereon, electrons are transported to CIV via cytochrome-c (cyt-c; ‘c’) and donated to oxygen (O2). Together, CI–CIV constitute the electron transport chain (ETC). The energy derived from the electron transport is used to expel protons (H+) from the mitochondrial matrix across the MIM. This establishes an electrochemical proton-motive force, associated with an inside-negative mitochondrial membrane potential (Δψ) and increased matrix pH. The controlled backflow of H+ is used by CV to drive the production of ATP (see text for details).
CI is the largest OXPHOS enzyme proposed to consist of 45 different subunits. Recent evidence suggests a number of 44 subunits since the NDUFA4 protein hitherto classified as a CI constituent appears to be a component of CIV (Balsa et al, 2012). Seven CI subunits (ND1, ND2, ND3, ND4, ND4L, ND5 and ND6) are encoded by the mtDNA and the remainder by the nDNA (Figure 2; Supplementary Table 1). CI oxidizes NADH to NAD+ and donates the released electrons to the electron carrier coenzyme Q10 (CoQ10, a.k.a. ubiquinone). To perform its enzymatic reactions, CI only requires a set of 14 evolutionary conserved ‘core subunits’, consisting of the 7 mtDNA-encoded ND subunits and 7 nDNA-encoded subunits (NDUFV1, NDUFV2, NDUFS1, NDUFS3, NDUFS7, NDUFS8; Koopman et al, 2010; Hirst, 2011). The remaining subunits are denoted as ‘accessory’ or ‘supernumerary’. Although the role of accessory subunits in CI biogenesis, stability and function still is incompletely understood, recent evidence in the aerobic yeast Yarrowia lipolytica suggests that they are important for CI stability (Angerer et al, 2011). Biogenesis of holo-CI is assisted by at least 11 assembly factors (NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, C8orf38, C20orf7, ACAD9, FOXRED1, ECSIT, NUBPL and OXA1L). Details about the CI assembly mechanism are provided elsewhere (e.g., Vogel et al, 2007; Dieteren et al, 2008, 2011; Koopman et al, 2010; Mckenzie and Ryan, 2010; Perales-Clemente et al, 2010; Moreno-Lastres et al, 2012; Nouws et al, 2012). In mammals, fungi and bacteria CI displays an L-shaped form consisting of a hydrophilic (matrix-protruding) and a lipophilic (MIM-embedded) arm (Clason et al, 2010). During recent years, significant progress has been made in understanding the link between electron and H+ transport in CI (Sazanov and Hinchliffe, 2006; Efremov et al, 2010; Hunte et al, 2010; Efremov and Sazanov, 2011a, 2011b). In the proposed coupling mechanism, electrons extracted from NADH are transported by a chain of iron-sulphur (Fe-S) clusters (Xu and Møller, 2011) to CoQ10 (Hinchliffe and Sazanov, 2005; Hayashi and Stuchebrukhov, 2010). This transport is linked to H+ translocation due to long-range conformational changes within the complex (Onishi, 2010; Efremov and Sazanov, 2011a, 2011b).
CII constitutes part of both the OXPHOS system and TCA cycle, oxidizes FADH2 to flavin adenine dinucleotide (FAD) and also transfers the released electrons to CoQ10 (Figure 2). CII is a heterotetrameric complex consisting of four nDNA-encoded subunits (SDHA, SDHB, SDHC and SDHD) and its assembly is assisted by two assembly factors (SDHAF1 and SDHAF2; Supplementary Table 1). Details about CII biogenesis are provided elsewhere (Rutter et al, 2010). Structurally, the SDHC and SDHC subunits are embedded in the MIM, whereas SDHA and SDHB protrude in the mitochondrial matrix (Brière et al, 2005). SDH-encoding genes are tumour suppressors, and their mutation predisposes carriers to carotid body paragangliomas and adrenal gland pheochromocytomas (Raimundo et al, 2011). In addition to CI and CII, also other enzymes can potentially donate electrons to CoQ10. These include: (i) the MIM-associated electron-transferring flavoprotein (ETF)-ubiquinone oxidoreductase, which transfers electrons generated during the flavin-linked oxidation step in the catabolism of FAs, (ii) s,n-glycerophosphate dehydrogenase and (iii) dihydroorotate dehydrogenase, present only in certain types of mitochondria (see Koopman et al, 2010 and the references therein). Electrons from CoQ10 are received by CIII and transported further to CIV by the electron carrier cytochrome-c (cyt-c). Similarly to CoQ10, cyt-c can receive electrons from an alternative source (especially in the liver) during oxidation of sulphur-containing amino acids by sulphite oxidase. However, this reaction usually occurs at a very low rate relative to other ETC inputs (see Koopman et al, 2010 and the references therein).
CIII contains 11 subunits, one of which is encoded by the mtDNA (CYB). Its assembly is described elsewhere (Smith et al, 2012) and requires the action of two identified assembly factors (BCS1L and UQCC; Supplementary Table 1). At CIV, electrons are donated to molecular oxygen (O2) to form water. About 95% of the O2 we breathe is consumed by CIV (Ferguson-Miller et al, 2012). CIV consists of 14 subunits, 3 of which are mtDNA-encoded (CO1, CO2, and CO3), and its biogenesis is assisted by at least 18 assembly factors (Supplementary Table 1), as discussed in detail elsewhere (Mick et al, 2011). At three sites in the ETC (CI, CIII and CIV), the energy released by the electron transport is used to drive the trans-MIM efflux of protons (H+) from the mitochondrial matrix. As a consequence, a trans-MIM proton motive force (PMF or Δpm) is established, which consists of an (inside negative) electric charge (Δψ) and (inside more alkaline) pH (ΔpH) difference across the MIM (Mailloux and Harper, 2012; Figure 2).
At CV, the energy released by the controlled backflow of H+ is coupled to the formation of ATP from ADP and inorganic phosphate (Pi). Experimental evidence in eukaryotes revealed that each ATP produced requires the CV-mediated backflow of 2.7 protons (Watt et al, 2010). CV is built up of 19 subunits, 2 of which are encoded by the mtDNA (ATP6 and ATP8), and its assembly requires 4 nDNA-encoded proteins (Supplementary Table 1). CV is a molecular machine composed of two mechanical rotary motors (Fo and F1), which interconvert the chemical energy of ATP hydrolysis and H+ electrochemical potential via a mechanical rotational mechanism (e.g., Okuno et al, 2011; Watanabe et al, 2011; Jonckheere et al, 2012a). This means that CV can either dissipate Δpm to generate ATP, or use ATP to fuel the trans-MIM efflux of H+. The latter condition sustains Δpm and is known as the ‘reverse-mode’ of CV (Chinopoulos and Adam-Vizi, 2010). In addition to ATP generation, the Δψ and/or ΔpH gradient is also required for mitochondrial fusion, the import of mitochondrial preproteins and the exchange of metabolite and ions with the cytosol (Figure 3), as reviewed previously (Garlid and Paucek, 2003; Kaasik et al, 2007; O’Rourke, 2007; Klingenberg, 2008; Palmieri, 2008; Koopman et al, 2010; Becker et al, 2012).
Figure 3.
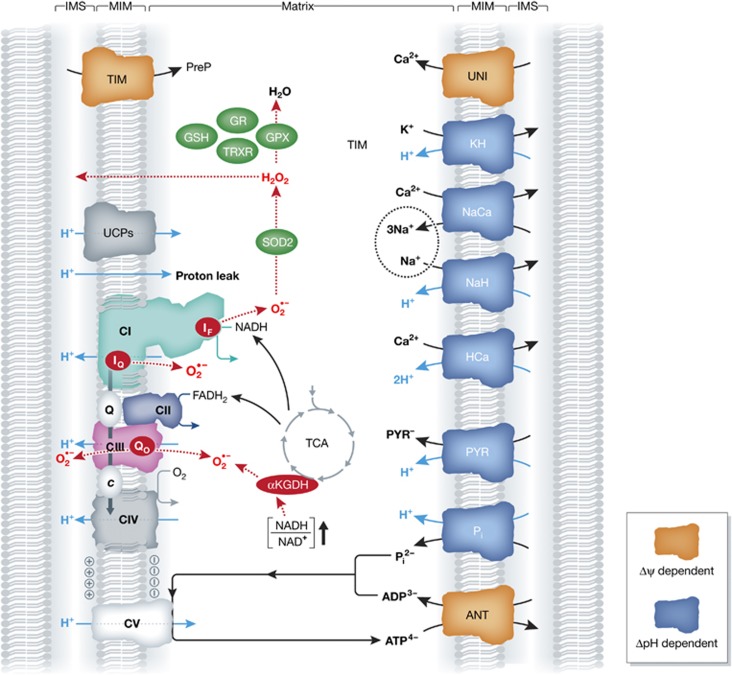
Integration of the OXPHOS system and mitochondrial metabolism. The five OXPHOS complexes, depicted on the lower left of the figure (see also Figure 2), maintain the inside-negative mitochondrial membrane potential (Δψ) and generate reactive oxygen species (ROS; red) in the form of superoxide (O2·−) and hydrogen peroxide (H2O2). ROS can also be generated by the TCA cycle enzyme α-ketoglutarate dehydrogenase (αKGDH), under conditions of elevated NADH/NAD+ ratio. ROS are removed by several antioxidant systems (green). In addition to fuelling ATP generation by CV, a sufficiently negative Δψ is also crucial for import of nDNA-encoded mitochondrial preproteins (PreP) via the TIM system. Moreover, metabolite and ion exchange across the mitochondrial inner membrane (MIM; right part of the figure) is driven by Δψ (orange) or its associated pH gradient (ΔpH; blue) (see text for details). ANT, adenine nucleotide translocase; GR, glutathione reductase; GPX, glutathione peroxidase; GSH, glutathione; HCa, proton/calcium transporter; IF, flavin site in CI; IQ, CoQ10-binding site in CI; KH, potassium/proton transporter; NaCa, sodium/calcium transporter; NaH, sodium/proton transporter; Pi, inorganic phosphate/proton transporter; PYR, pyruvate/proton transporter; Qo, CoQ10-binding site in CIII; SOD2, superoxide dismutase 2; TRXR, thioredoxin reductase; TIM, translocator of the inner membrane; UCP, uncoupling protein; UNI, mitochondrial calcium uniporter.
Supramolecular architecture of the OXPHOS system
In bovine heart mitochondria, the unit stoichiometry of the OXPHOS system equalled 1/1.3/3/6.7/0.5 for CI/CII/CIII/CIV/CV and 2–5 units of the adenine nucleotide translocase (ANT; Lenaz and Genova, 2007), which mediates the trans-MIM exchange of ADP and ATP (Figure 3). Analysis of various rat tissues (Benard et al, 2006) revealed different molecular CII/CoQ10/CIII/cyt-c/CIV ratios in heart (1:24:3:12:8), kidney (1:73:3:18:7), muscle (1:58:3:11:7), brain (1:58:3:35:8) and liver (1:135:3:9:7). This suggests that the amount of CoQ10 and cyt-c display tissue-dependent differences, whereas CII, CIII and CIV do not. Statistical analysis predicted that different tissues display different sensitivities to a pathological OXPHOS defect, with brain being more sensitive than liver and kidney tissue but less sensitive than skeletal muscle and heart tissue (Benard et al, 2006). Experimental evidence suggests that CI assembly/stability depends on its interaction with other OXPHOS complexes (Schägger et al, 2004). In addition, CIII is required to maintain CI (Acín-Pérez et al, 2004) and deficiency of CIV reduces CI function (Suthammarak et al, 2009). Moreover, in human patient cells the presence of a truncated CIV subunit destabilized not only CIV but also other ETC complexes, leading to their rapid clearance by mitochondrial quality control systems (Hornig-Do et al, 2012). These observations, supported by other experimental evidence (reviewed in Boekema and Braun, 2007; Wittig and Schägger, 2009; Dudkina et al, 2010; and Winge, 2012), are compatible with a model in which individual OXPHOS complexes are not randomly distributed but organized in supercomplexes (or ‘respirasomes’). The finding that CIII and CIV are not essential for the assembly/stability of CI in fungi (Maas et al, 2009) suggests that respirasome formation and/or stability might be species and/or tissue dependent. Although it was previously suggested that CIII interacts with CII (Chen et al, 2008), the current view is that respirasomes consist of CI, CIII and CIV (Boekema and Braun, 2007; Wittig and Schägger, 2009; Dudkina et al, 2010; Althoff et al, 2011; Winge, 2012). In order of decreasing abundance, respirasome composition in bovine heart is predicted to be I-III2-IV1, I-III2, I-III2-IV2 and I-III2-IV3–4 (Schägger and Pfeiffer, 2001; Winge, 2012). In silico evidence highlighted the involvement of lipids in the gluing together of the OXPHOS complexes at the interfaces (Dudkina et al, 2011). Based on biochemical evidence, respiratory strings of CI, CIII and CIV have been proposed meaning that respirasomes might not be the highest level of organization of the OXPHOS system (Wittig and Schägger, 2009; Dudkina et al, 2010). Also CV forms higher oligomeric structures from dimeric building blocks, thought to be involved in maintaining cristae structure (Wittig and Schägger, 2009; Dudkina et al, 2010; Davies et al, 2011). Although no live-cell data are available yet, evidence demonstrating that the activity of supercomplexes as true respirasomes has been presented (Acín-Pérez et al, 2008). This study revealed that: (i) respirasome formation requires the presence of all of its constituting complexes, (ii) there is a time-lag between assembly of the individual OXPHOS complexes and respirasome formation. These findings support a model in which individual holoenzymes are first preassembled and subsequently combined into respirasomes. This view was recently challenged by a study demonstrating that respirasome biogenesis is mediated by a CI assembly intermediate, which acts as a scaffold for the combined incorporation of CIII and CIV subunits into the respirasome (Moreno-Lastres et al, 2012). The study of the Enriquez laboratory (Acín-Pérez et al, 2008) further revealed that: (iii) isolated respirasomes mediate electron transfer from NADH to O2 and (iv) respirasomes can contain CoQ10 and cyt-c. The latter suggests that also cyt-c is associated with respiratory supercomplexes, compatible with the observation that fibroblasts from cyt-c knockout mice lacked fully assembled CI and CIV and displayed lower levels of CIII (Vempati et al, 2009), and evidence that cyt-c and CoQ10 are functionally compartmentalized (Benard et al, 2008). The first protein factor (HIGD2A) required for respirasome assembly and stability in mammals was recently identified (Chen et al, 2012; Strogolova et al, 2012; Vukotic et al, 2012). HIGD2A is a homologue of Rcf1 (respiratory supercomplex factor 1), which mediates CIII/CIV supercomplex formation in yeast (Shoubridge, 2012). Respirasomes display a reduced stability in Barth syndrome patients, which carry a mutation in the tafazzin (TAZ) gene encoding a putative phospholipid acyltransferase involved in cardiolipin (CL) remodelling (McKenzie et al, 2006). This suggests that respirasomes are stabilized by CL (Wittig and Schägger, 2009), which is a mitochondria-specific lipid dimer consisting of two phosphatidyl groups bridged by a glycerol. CL is highly unsaturated and therefore has a high susceptibility to peroxidative attack. CL is tightly bound to CI and its oxidation reduces CI activity (Paradies et al, 2002). Furthermore, CL interacts with a variety of mitochondrial proteins including the other OXPHOS complexes, cyt-c and the ANT (see Koopman et al, 2010 and the references therein). CL improves OXPHOS efficiency, stimulates mitochondrial filamentation, affects cristae morphology and provides a mitochondria-specific activation platform for caspase-8 downstream of apoptotic Fas signalling (Gonzalvez et al, 2008; Claypool and Koehler, 2012). The presence of CL is also critical for the degree of oligomerization in CV assemblies by promoting the ribbon like assembly of CV dimers and thereby the lateral organization and morphology of the cristae membrane (Acehan et al, 2011). In addition to respirasomes, structural evidence suggests the existence of ‘ATPasomes’ in the MIM (Bénit et al, 2010; Saks et al, 2010). These ATPasomes contain CV, the inorganic phosphate/proton transporter, and the ANT in a stoichiometry of 1:1:1. Their existence supports a mechanism through which mitochondrial ADP and Pi import, ATP synthesis and ATP export (Figure 3) occur in a highly localized manner. It remains to be established whether respirasomes can structurally combine with ATPasomes in vivo and if they are fixed structures or can be dynamically formed and disassembled on demand (Wittig and Schägger, 2009).
Regulation of OXPHOS function
At the cellular level, OXPHOS function can be controlled in many ways including expression regulation, post-translational modifications, metabolite-binding, second messenger systems, substrate availability and by uncoupling the ETC from CV. It is to be expected that these control mechanisms are disturbed and/or mediate adaptive responses during neurodegeneration. OXPHOS expression is controlled by the action of transcriptional activators (e.g., NRF-1 (nuclear respiratory factor 1), NRF-2, ERRa, CREB and YY1) and members of the peroxisome proliferator-activated receptor (PPAR) gamma coactivator (PGC-1) family (i.e., PGC1-α, PGC1-β and PGC-1-related coactivator; Scarpulla, 2012). Expression of these transcriptional (co)activators is regulated by other cues like temperature, nutrient availability and metabolic status (Handschin and Spiegelman, 2006; Scarpulla, 2008). With respect to OXPHOS structural proteins, several (regulatory) modifications have been described including: (i) phosphorylation (CI, CII, CIII, CIV and CV; Pagliarini and Dixon, 2006; Carlucci et al, 2008; Yadava et al, 2008; Kane and Van Eyk, 2009; Koopman et al, 2010; Hebert-Chatelain et al, 2012; Helling et al, 2012; Papa et al, 2012), (ii) acetylation (CI, CII and CV; Guan and Xiong, 2011), (iii) glycosylation (CI, CII and CV; Burnham-Marusich and Berninsone, 2012), (iv) cleavage by caspases (CI, CII and CV; Ricci et al, 2004; Martinvalet et al, 2008; Zhu et al, 2012) and (v) modification by ROS and/or reactive nitrogen species (RNS) mediated mechanisms (CI, CII, CIII, CIV and CV; Murray et al, 2003; Choksi et al, 2004; Galkin and Moncada, 2007; Chen et al, 2008; Hurd et al, 2008; Taylor and Moncada, 2009; Chinta and Andersen, 2011; Danielson et al, 2011; Wang et al, 2011). Recent evidence suggests that phosphorylation and acetylation of mitochondrial ribosomal proteins and translation factors allow for regulation of mitochondrial protein synthesis (Koc and Koc, 2012). In addition to phosphorylation, it was argued that CIV activity is (co)regulated by various biomolecules (e.g., ATP/ADP ratio, FAs and CL), as well as several of its nDNA-encoded subunits (e.g., subunit 5A and subunit 6A isoforms; Arnold, 2012). OXPHOS activity is also controlled by diverse second messenger systems (for detailed information, see Boneh, 2006 and Pagliarini and Dixon, 2006) including cyclic AMP (cAMP), Ca2+, ceramide and ROS (also see below). In adipose tissue, the coupling between electron transport (ETC action) and ATP production (CV action) is reduced (‘uncoupled’) by uncoupling protein 1 (UCP1). UCPs mediate the trans-MIM backflow of H+ and thereby bypass CV, resulting in heat generation (Divakaruni and Brand, 2011). Interestingly, analysis of live cells with a fluorescent molecular thermometer sensor revealed that the local temperature near mitochondria is higher than the temperature of the rest of the space in the cytosol (Okabe et al, 2012). Moreover, this temperature increased when cells were treated with the chemical uncoupling molecule 4-(trifluoromethoxy)phenylhydrazone (FCCP). It appears that members of the UCP family (UCP2, UCP4 and UCP5; Figure 3) are also expressed within the central nervous system (CNS; Andrews et al, 2005). Interestingly, these UCPs do not act as constitutive uncouplers, but are activated by ROS and free FAs. In this sense, neuronal UCPs can regulate mitochondrial biogenesis, Ca2+ flux, ROS production and local temperature, thereby directly affecting neurotransmission, synaptic plasticity and neurodegenerative processes (Andrews et al, 2005). ROS-induced UCP2 activation has also been implied in minimizing ROS emission from the ETC, thus providing a negative feedback loop for mitochondrial ROS production (Mailloux and Harper, 2012). Metabolic control analysis (MCA) of several tissues including brain suggests that the control over the flux through the OXPHOS system is shared by essentially all components of this system (Pathak and Davey, 2008; Korzeniewski, 2011). This means that in order to significantly increase this flux (for instance during cell activation), and at the same time maintain relatively constant concentrations of intermediate metabolites (e.g., ADP, ATP, Pi and NADH), direct activation of multiple components of the OXPHOS system is required (the ‘multistep parallel activation’ mechanism). Similarly, theoretical analysis of mathematical models of OXPHOS in heart, skeletal muscle and liver suggests that mitochondrial Ca2+ uptake during cell activation stimulates OXPHOS function at several sites (Korzeniewski, 2011). Although analysis of cardiac submitochondrial particles revealed that Ca2+ inhibits CI and thereby reduces electron transport activity (Matsuzaki and Szweda, 2007), the current consensus is that mitochondrial Ca2+ uptake from the cytosol directly or indirectly modulates the activity of mitochondrial transporters and enzymes including the glutamate/aspartate exchanger, PDH, the TCA cycle enzymes isocytrate dehydrogenase (ICD) and α-ketoglutarate dehydrogenase (αKGDH), CIII and CV (Willems et al, 2008; Gellerich et al, 2010; Glancy and Balaban, 2012). This suggests that Ca2+ might be a mediator of multistep parallel activation of the OXPHOS system during cell activation. Interestingly, experimental results in plants suggest that the activity of individual complexes within supercomplexes can be regulated (Ramírez-Aguilar et al, 2011). This process involved (reversible) dissociation of these complexes from the supercomplex depending on the O2 tension and the pH of the mitochondrial matrix.
Role of the OXPHOS system in CNS energy metabolism
The central nervous system (CNS) consists of two types of cells: neurons and glial cells (Kandel et al, 1995). Neurons can be classified based upon the number of processes that originate from the cell body as unipolar (e.g., an invertebrate neuron), bipolar (e.g., a bipolar retinal cell), pseudo-unipolar (e.g., a dorsal root ganglion) or multipolar (e.g., a spinal cord motor neuron, a hippocampal pyramidal cell or a cerebellar Purkinje cell). The principal types of glial cells in the nervous system are oligodendrocytes and astrocytes (CNS) and Schwann cells (peripheral nervous system; Kandel et al, 1995). Brain functioning requires a large amount of energy, which is highly dependent on the external supply of energy substrates delivered by the circulation and OXPHOS action, as reflected by the tight coupling between GLC and O2 delivery from the vasculature (Bélanger et al, 2011). Most evidence supports GLC as the major fuel for normal, metabolically active, brain (Dienel, 2012). In the latter condition, the majority of brain energy is consumed by synaptic transmission (Harris et al, 2012). GLC enters the brain from the circulation mediated by GLUT1 in the microvascular endothelial cells of the blood–brain barrier (BBB) and glial cells and GLUT3 in neurons (Simpson et al, 2007). Astrocytes represent the most abundant cell type in the brain and are also present in the spinal cord. Glial cells play an important role in many cellular processes including glutamate, ion and water homeostasis and ROS detoxification (Volterra and Meldolesi, 2005; Bélanger and Magistretti, 2009; Bélanger et al, 2011). Metabolically, evidence was provided that both neurons and astrocytes rely on OXPHOS for ATP generation whereas astrocytes also possess energy stores in the form of glycogen (Hertz et al, 2007; Bélanger et al, 2011). It appears that astrocytes and neurons are metabolically linked by LAC shuttling (for a critical review, see Dienel, 2012). Recent evidence (Choi et al, 2012) suggests that soluble adenylyl cyclase (sAC) in astrocytes becomes activated in response to bicarbonate (HCO3−), which enters via the electrogenic NaHCO3 cotransporter (NBC). As a consequence, astrocyte cAMP levels increase, leading to the breakdown of glycogen, stimulation of glycolysis, and release of LAC. The latter is subsequently taken up by neurons for use as an energy substrate (Choi et al, 2012).
With respect to human ageing, magnetic resonance imaging (MRI) and positron emission tomography (PET) examinations revealed that, during normal ageing, cerebral blood flow (CBF) and to a lesser extent the cerebral rate of O2 consumption (CMRO2) decreased with age in extended regions of the brain, with sparing of primary sensory-motor neurons and occipital cortices (Aanerud et al, 2012). This study further revealed significant increases of O2 extraction fraction (OEF) in frontal and parietal cortices, excluding primary motor and somatosensory regions, and in the temporal cortex. It was concluded that the increased OEF, which can compromise O2 delivery to neurons, possibly perturbs energy turnover. This suggests a possible mechanism of progression from healthy to unhealthy brain ageing, as the regions most affected by age were the areas most vulnerable to neurodegeneration. Analysis of mouse and rat brain slices suggests that gamma oscillations (i.e., neuronal network oscillations in the 30–100 Hz range that occur in the electroencephalogram) in the cerebral cortex are associated with high energy demand (Kann, 2011). The latter might explain why higher cognitive functions including sensual perception and working memory become disturbed during neurodegeneration (Kann, 2011).
ROS generation by the OXPHOS system
OXPHOS action is inherently coupled to the production of ROS. Under normal conditions, mitochondrial and cytosolic ROS levels are controlled by mitochondrial and cytosolic antioxidant systems and exert a signalling function. However, in case the antioxidant systems fail to keep these ROS levels within safe limits, lipids, proteins and DNA molecules are at risk of being damaged. The latter process may occur over the years leading to a gradual decline in mitochondrial and cellular integrity and function. ROS are chemical entities that are formed upon incomplete reduction of O2 (Forkink et al, 2010), and their generation by the mitochondrial ETC has been proposed to play a role in neurodegeneration (e.g., Abramov et al, 2010; Chinta and Andersen, 2011; Correia et al, 2012; Court and Coleman, 2012; Hedskog et al, 2012; Leuner et al, 2012). RNS-like nitric oxide (NO) can interact with ROS and also have been implicated in neurodegeneration, but this is discussed elsewhere (Calabrese et al, 2009; Brown, 2010; Nakamura et al, 2010; Doherty, 2011; Cambron et al, 2012; Ramalingam and Kim, 2012). In case of ROS, redox dysregulation and/or ROS-induced stress has been linked to various neurological presentations including AD (Hedskog et al, 2012; Leuner et al, 2012; Von Bernhardi and Eugenín, 2012), PD (Chinta and Andersen, 2008; Fato et al, 2008; Del Hoyo et al, 2010), Friedreich’s ataxia (FRX; Calabrese et al, 2005), Down syndrome (Pagano and Castello, 2012), ALS (Martin, 2011) and psychiatric conditions like schizophrenia and bipolar disorder (Clay et al, 2011; Manji et al, 2012). The association between ageing in the CNS, OXPHOS malfunction, elevated mtDNA mutation load and increased ROS-induced damage has led to the ‘vicious cycle’ theory (e.g., Bandy and Davison, 1990 and Balaban et al, 2005). The latter states that there is a feedback mechanism connecting these events in ageing and age-associated neurodegeneration. However, it is not always trivial to unequivocally determine whether ROS molecules play a damaging and/or signalling role (see below) during neurodegeneration. In this sense, it needs to be kept in mind that most evidence supporting the vicious cycle model is obtained using pharmacological inhibition of mitochondrial enzymes, which not necessary reflects the physiological situation. Moreover, recent in vivo evidence also contradicts the vicious cycle model (e.g., Fukui and Moraes, 2008 and Frenzel et al, 2010). Within mammalian cells, ROS can originate from many sources (Brown and Borutaite, 2012) including: (i) mitochondria (CI, CII, CIII, glycerol 3-phosphate dehydrogenase, the ETF:Q oxidoreductase of FA β-oxidation, αKGDH, PYR and 2-oxoglutarate dehydrogenase, p66shc), (ii) the endoplasmic reticulum (ER) (cytochrome P-450 and b5, diamine oxidase, Ero1), (iii) peroxisomes (FA oxidation, D-amino-acid oxidase, L-2-hydroxy acid oxidase and urate oxidase), (iv) the cytosol (NO synthases, lipoxygenases and PGH synthase), (v) the plasmamembrane (NADHP oxidases, lipoxygenase) and, (vi) the extracellular space (xanthine oxidase) (Boveris et al, 1972; Kukreja et al, 1986; Roy et al, 1994; O’Donnel and Azzi, 1996; McNally et al, 2003; Giorgio et al, 2005; Adam-Vizi and Chinopoulos, 2006; Gross et al, 2006; Starkov, 2008; Brand, 2010; Touyz et al, 2011; Quinlan et al, 2012). Although the relevance of each ROS source is cell-type dependent, the above suggests that mitochondria are not necessarily the main source of ROS in mammalian cells (Brown and Borutaite, 2012). However ROS generation by CI and CIII is considered relevant (Figure 3), both under physiological and pathological conditions (Adam-Vizi and Chinopoulos, 2006; Koopman et al, 2010; Pryde and Hirst, 2011; Treberg et al, 2011). In addition, recent evidence in rat skeletal muscle mitochondria suggests that CII can generate ROS at high rates under conditions that CI and CIII are inhibited and succinate concentration is low (Quinlan et al, 2012). Also αKGDH (Figure 3) is able to generate ROS in the mitochondrion when the NADH/NAD+ ratio is increased (Tretter and Adam-Vizi, 2005). The ROS family consists of a large collection of molecules, but biologically most of them are derived from superoxide (O2·−) and/or hydrogen peroxide (H2O2). The amount of cellular ROS generated, as well as its primary source, varies with the type of ROS, the type of cell, the organism from which the cells were derived, metabolic state, the (patho)physiological condition and the presence of ROS-detoxifying (or consuming) systems (Adam-Vizi and Chinopoulos, 2006; Brown and Borutaite, 2012). In case of the OXPHOS system, O2·− appears to be generated (in descending order of maximal capacity) by the CoQ10-binding sites in CI (site IQ) and CIII (site Qo) and the flavin in CI (site IF; Muller et al, 2004; Dröse and Brandt, 2008; Brand, 2010; Quinlan et al, 2011). These three sites all release O2·− into the mitochondrial matrix (Figure 3), whereas site Qo also produces O2− into the space between the MIM and MOM, the mitochondrial intermembrane space (IMS). How these individual sites contribute to ROS generation in the absence of ETC inhibitors is still unclear, but it is expected that this considerably varies with cell/tissue type, available substrates, energy demand and O2 tension (Brand, 2010).
Classically, ROS are considered as damaging entities because they can react with and thereby damage or modify many biomolecules including proteins, lipids and (mt)DNA (e.g., Ahmad et al, 2005; Dröge and Schipper, 2007; Murphy, 2009; Kourtis and Tavernarakis, 2011). In this context, cells have available an elaborate ROS detoxifying apparatus (Figure 3) consisting of enzymatic (e.g., MnSOD (manganese superoxide dismutase)/SOD2, GPX, GR, TRXR (thioredoxin reductase)) and non-enzymatic (e.g., GSH, vitamin C, vitamin E, carotenoids, and flavonoids) systems (e.g., Koopman et al, 2010; Aon et al, 2012; and Miriyala et al, 2012). Importantly, ROS are not only damaging to biomolecules but also act as (redox) signalling entities, possibly specifically affecting mitochondrial function (e.g., Thannickal and Fanburg, 2000; Dröge and Schipper, 2007; Koopman et al, 2010; Lukosz et al, 2010; Murphy et al, 2011; Distelmaier et al, 2012; Handy and Loscalzo, 2012; Murphy, 2012; and Perjés et al, 2012). In this context, evidence was provided that CIV biogenesis involves several ROS and/or redox-regulated steps (Bourens et al, 2012) and ROS signalling pathways are implicated in cell proliferation, survival, differentiation and metabolism (mediated by ASK1 (apoptosis signal-regulated kinase 1), PI3K (phosphoinositide-3-kinase), PTP (protein tyrosine phosphatase) and Shc (Src homology 2 domain-containing)), antioxidant and anti-inflammatory responses (TRX (thioredoxin), Ref1 (redox-factor 1) and Nrf2 (NF-E2-related factor 2)), iron homeostasis (IRP) and DNA-damage responses (ATM (ataxia-telangiectasia mutated); Ray et al, 2012). Moreover, an increase in ROS levels, the spatiotemporal magnitude of which by itself depends on the balance between ROS production and detoxification, often serves to activate adaptive programs that counterbalance ROS stress (Collins et al, 2012). ROS further (co)control the removal of dysfunctional mitochondria by mitophagy (Gomes and Scorrano, 2012; Lee et al, 2012; Novak, 2012; Rugarli and Langer, 2012), thereby limiting the detrimental cellular consequences of mitochondrial dysfunction and increased ROS production.
Both in healthy cells and cells from patients with an OXPHOS disorder, changes in cellular ROS levels have also been linked to mitochondrial metabolic state and net morphology (Koopman et al, 2007; Benard and Rossignol, 2008; Distelmaier et al, 2012). Mitochondrial shape is governed by the balance between mitochondrial fusion, fission and motility. These dynamics are mediated by dedicated mitochondrial fusion (e.g., mitofusins or Mfns), fission (e.g., dynamin-related protein 1 or Drp1) and motor proteins (e.g., Milton), which are controlled by cell signalling mechanisms (Lovas and Wang, 2012; Wilson et al, 2012). According to a recent conceptual model presented by Westermann (2012), mitochondrial shape and OXPHOS activity are closely linked. This model states that mitochondria exist in three states in which their net morphology appears: (i) ‘fragmented’ and OXPHOS activity is low, (ii) ‘normal’ and OXPHOS activity is normal and (iii) ‘hyperfused’ and OXPHOS activity is high. We recently provided evidence (using primary human skin fibroblasts, Chinese hamster ovary cells and immortalized mouse embryonic fibroblasts (MEFs)) suggesting that the transition between these morphological states is controlled by Mfns in an ROS-dependent manner (Distelmaier et al, 2012). This implies that cell-governed changes in ROS level (for instance by altering the balance between their production and detoxification) may allow regulation of mitochondrial morphology and function. Moreover, it was observed that inhibition of GSH synthesis by L-buthionine-(S,R)-sulphoximine (BSO; 12.5 μM, 72 h) shifts the cytosolic and mitochondrial thiol redox environment towards a fully oxidized state in human skin fibroblasts (Verkaart et al, 2007b) and that this shift is paralleled by mitochondrial shortening (Distelmaier et al, 2012). In contrast, another study reported that BSO treatment induces mitochondrial hyperfusion (Shutt et al, 2012). However in the latter experiments a different cell type (HeLa) and BSO treatment regime (100 μM, 24 h) were used, suggesting that changes in thiol redox state affect mitochondrial morphology in a time-, concentration- and cell type-dependent manner. The effect of BSO-induced GSH depletion on the mitochondrial thiol redox environment and mitochondrial shortening was counterbalanced by overexpression of BOLA1, a glutaredoxin 5 (GLRX5)-interacting protein (Willems et al, 2012). Another member of the BOLA family (BOLA3) was suggested to be involved in Fe-S cluster assembly and also bind GLRX5 (Cameron et al, 2011b).
OXPHOS mutations and neurodegeneration
Analysis of the mitochondrial proteome in 19 different mouse tissues revealed that mitochondrial functioning requires between 1100 and 1400 genes and mitochondria from different organs share ∼75% of their proteins (Mootha et al, 2003; Calvo and Mootha, 2010). In humans, ∼1000 genes have currently been identified (Human MitoCarta; www.broadinstitute.org). In principle, a mutation in any of these genes can lead to mitochondrial dysfunction and induce a ‘primary mitochondrial disorder’. When mitochondrial dysfunction occurs for another reason, this gives rise to a ‘secondary mitochondrial disorder’ (Koopman et al, 2012).
Importantly, human nDNA-encoded mutations are generally inherited in an autosomal recessive manner (Smeitink et al, 2001). From this perspective, cells from patients with nDNA-encoded mutations are well suited for microscopy imaging analysis since they all contain the genetic defect. In case of mtDNA mutations, the situation is much more complex since each cell contains many mitochondria and each mitochondrion contains many mtDNA molecules. MtDNA is generally inherited exclusively from the mother (maternal inheritance) and mtDNA mutations display ‘heteroplasmy’ (Davis and Sue, 2011; Schapira, 2012; Schiff et al, 2012). The latter means that normal and mutated mitochondrial genomes coexist in the same cell. The percentage of mutated versus normal mtDNAs needs to exceed a certain threshold to induce pathology (the ‘threshold effect’). This threshold is tissue specific leading to them being differentially sensitive to OXPHOS dysfunction (e.g., Rossignol et al, 1999). The latter might be explained by the fact that OXPHOS expression greatly differs between tissues. For instance, mitochondrial (protein) abundance was highest in mouse heart tissue and equalled 40–50% of this value in kidney, brain stem, spinal cord and skeletal muscle (Pagliarini et al, 2008). Even lower values were observed for large intestine, cerebellum, cerebrum, small intestine, stomach and liver (25–40%), and testis, adipose, thymus, placenta, fetal tissue, lung, spleen and eye (<25%). To complicate matters even further, by examining mouse chimeras with a mixture of normal and ETC-deficient neurons (‘mosaic ETC deficiency’) in cerebral cortex (Dufour et al, 2008), it was found that the presence of a low proportion of ETC-deficient neurons sufficed to induce symptoms whereas premature death occurred only at higher proportions. Interestingly, neurons with normal ETC function ameliorated disease progression and ETC-deficient neurons adversely affected normal adjacent neurons leading to trans-neuronal degeneration (Dufour et al, 2008).
In the strict sense of the word ‘neurodegeneration’ is defined as ‘any pathological condition primarily affecting neurons’ or ‘a disease process in which neurons are selectively and gradually destroyed, leading to a progressive loss of nervous system structure and function’ (Przedborski et al, 2003; Deuschl and Elble, 2009). This implies that neoplasm, oedema, haemorrhage and trauma of the nervous system are not considered to be neurodegenerative disorders. Diseases of the nervous system that implicate not neurons per se but rather their attributes, such as the myelin sheath as seen in multiple sclerosis, are not neurodegenerative disorders either, nor are pathologies in which neurons die as the result of a known cause such as hypoxia, poison or infections’ (Przedborski et al, 2003). Neurodegenerative disorders manifest with a heterogeneous clinical and pathological picture, affecting specific regions of the nervous system. They may present acutely and rapidly progressive or symptoms may be subtle and slowly progressive. The clinical course is generally unfavourable and therapeutic options are mostly not available. During recent years, several studies focused on the role of mitochondrial dysfunction in neurodegenerative disorders (e.g., Finsterer, 2006; DiMauro and Schon, 2008; McFarland et al, 2010; Schon and Przedborski, 2011). These highlighted a plethora of clinical symptoms and phenotypes. Several of them could be defined as distinct syndromes: AD, ALS, FRX, HD, cardioencephalomyopathy, Charcot-Marie Tooth disease (CMT), familial bilateral striatal necrosis (FBSN), growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis and early death (GRACILE), hereditary spastic paraparesis (HSP), Kearns-Sayre syndrome (KSS), Leber hereditary optic neuropathy (LHON), Leigh syndrome (LS), mtDNA depletion syndrome (MDS), mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy associated with ragged-red fibres (MERRF), maternally inherited diabetes and deafness (MIDD), mitochondrial neurogastrointestinal encephalopathy (MNGIE), multiple systemic lipomatosis (MSL), neuropathy, ataxia and retinitis pigmentosa (NARP), optic atrophy (OA), PD, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) and spinocerebellar ataxias (SCAs).
Mutations affecting OXPHOS function and biogenesis in human neurodegeneration
By combining information from the literature and various databases, we here present a list of nDNA- and mtDNA-encoded OXPHOS structural/biogenesis genes which, when mutated, are associated with neurodegeneration in humans (Supplementary Table 1; Figure 4). This analysis highlighted genes encoding: (i) structural OXPHOS subunits, (ii) OXPHOS assembly factors, (iv) Fe-S biogenesis enzymes, (v) enzymes involved in the synthesis of CoQ10 and cyt-c, (vi) mt-rRNAs, (vii) mt-tRNAs, (vii) mtDNA repair enzymes, (viii) mtDNA replication, transcription and translation factors, (ix) enzymes involved in the maintenance of the mitochondrial dNTP pool, (x) mitochondrial ribosomal proteins, (xi) mt-tRNA synthetases and (xii) nucleoid-associated proteins. The latter constitute mitochondrial nucleoprotein complexes consisting of mtDNA and its associated proteins involved in mtDNA organization and protection (e.g., Spelbrink, 2010; Brown et al, 2011; Cameron et al, 2011b; and He et al, 2012).
Figure 4.
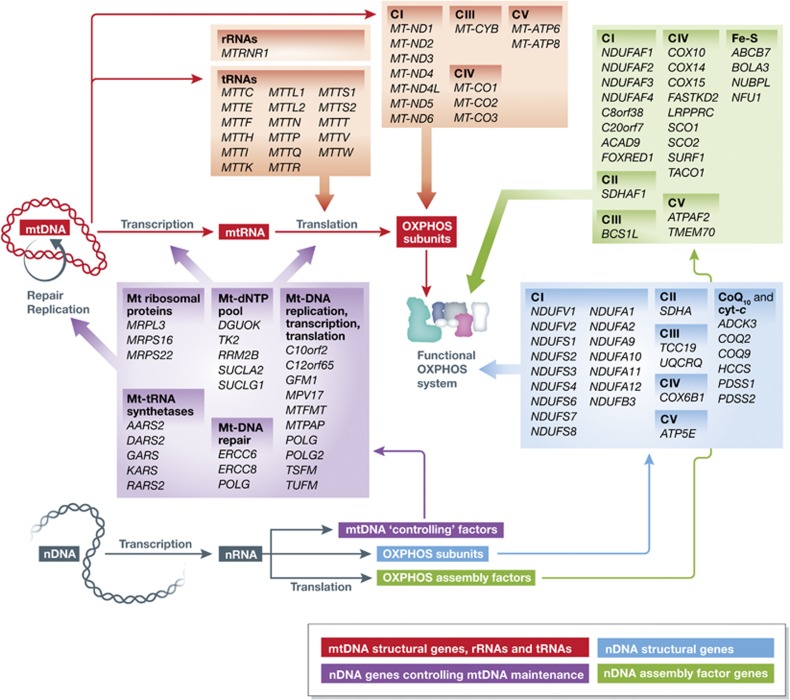
Biogenesis and neurodegeneration-associated mutations of the OXPHOS system. The mitochondrial DNA (mtDNA; red) encodes ribosomal RNAs (rRNAs), transfer RNAs (tRNAs) and OXPHOS subunits. Mitochondrial ribosomal proteins, tRNA synthetases, mtDNA repair proteins, dNTP (deoxynucleoside triphosphate) pool-maintaining proteins and proteins mediating mtDNA replication, transcription and translation are all encoded by the nuclear DNA (nDNA; purple). Also, OXPHOS assembly factors (green) and the remainder of the OXPHOS subunits (blue) are nDNA encoded. Mutated genes associated with neurodegeneration are indicated (italic). Gene names are given according to the HGNC (HUGO Gene Nomenclature Committee) standard (see text for details). cyt-c, cytochrome-c biogenesis; Fe-S, iron-sulphur cluster biogenesis; CoQ10, CoQ10 biogenesis.
Clinical aspects
From a clinical perspective, OXPHOS dysfunction presents with a wide range of neurological symptoms, including developmental regression, failure to thrive, seizures, spasticity, dystonia (movement disorder with abnormal tonicity of muscle, characterized by prolonged, repetitive involuntary muscle contractions), ataxia (loss of coordination and balance with instability of gait) and nystagmus (abnormal/oscillating eye movement). Many of these symptoms cannot be categorized into defined clinical syndromes. Especially in severe OXPHOS deficiencies, disease onset may be already neonatal, presenting with severe encephalopathy (global brain dysfunction). Apart from these ‘non-syndromic’ entities, there are several classical OXPHOS diseases, which are associated with neurodegeneration. We used the data in Supplementary Table 1 to compile a summary of neurological disorders associated with mutations in structural OXPHOS subunit and assembly factor genes (Table I). At first sight, mutations in nDNA-encoded genes are associated with different disorders than mutations in mtDNA-encoded genes. However, a certain disorder can be caused by mutations in different OXPHOS structural or assembly factor genes. Among the listed diseases, Leigh Syndrome (LS) is probably the most typical OXPHOS disease during early childhood. The main cause of LS is an isolated CI deficiency, caused by defects in its structural subunits (either mtDNA or nDNA encoded) or assembly factors. However, also mutations in other OXPHOS complexes (or in the respective assembly factors), as well as disturbances in CoQ10 metabolism or dysregulation in mitochondrial RNA/DNA maintenance may cause LS. This makes this syndrome one of the most frequent clinical entities. LS was first described by the British neuropathologist Denis Archibald Leigh (1916–1998) and is characterized by symmetrical necrotic lesions in the basal ganglia, especially in the putamen, or in variable areas within the brain stem (Leigh, 1951). However, lesions can also appear within other CNS regions such as the cerebellum, thalamus and even the spinal cord (e.g., Rossi et al, 2003; Friedman et al, 2010; Lebre et al, 2011). So far, the exact mechanism of neurodegeneration in LS is still unclear. There are no conclusive research studies, which could explain the exquisite vulnerability of circumscribed brain regions in LS patients (Wirtz and Schuelke, 2011). Classically, children with LS have a normal prenatal development and normal birth parameters. However, for respiratory chain defects in general, intrauterine growth retardation, leading to a low birth weight was reported (Yanicostas et al, 2011). In LS patients, the illness often takes a severe course within the first months of life, leading to developmental regression and failure to thrive (Distelmaier et al, 2009). As a consequence, most children die within the first years of life. The disease progression may suddenly accelerate, especially under the influence of intercurrent deleterious factors such as infection. Therapeutic options are generally unsatisfactory and palliative care is still a mainstay in the treatment of affected children.
Table 1. Mutated structural OXPHOS subunit and assembly factor genes associated with neurodegeneration in humans.

AD, Alzheimer disease; B, both nDNA and mtDNA encoded; CI, complex I; CII, complex II; CIII, complex III, CIV, complex IV; CV, complex V; LHON, Leber hereditary optic neuropathy; M, mtDNA encoded; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes; N, nDNA encoded; NARP, neuropathy, ataxia and retinitis pigmentosa; PD, Parkinson disease.
In addition to LS, LHON represents an important OXPHOS-associated neurodegenerative disease. Although LHON is already known for about 150 years, it was first linked to mutations in mtDNA genes in 1988 (Wallace et al, 1988; Newman, 2005). As summarized in Table I, LHON may be caused by defects in CI, CIII, CIV or CV. The disease typically presents with painless visual loss. Funduscopic abnormalities include hyperaemia (increased blood flow) of the optic nerve head with obscuration of the disk margins, dilation and tortuosity of posterior pole vasculature (Newman, 2005). The pathogenesis of LHON is still not fully resolved. It was suggested that the unmyelinated, prelaminar portion of the optic nerve requires a high degree of ETC activity, which might explain the specific vulnerability of this tissue (Qi et al, 2003).
Another relevant clinical phenotype (Table I) is MELAS. In addition to mutations in mtDNA-encoded CI genes, this syndrome is mostly caused by a 3243A-G mutation in the MTTL1 gene, leading to disturbed mitochondrial transcription. This frequently causes a biochemical CI and CIV deficiency (Koga et al, 2010). As a clinical syndrome, MELAS was first defined in the 1980s and is characterized by a combination of key symptoms, which are highlighted by the acronym for the disorder (Pavlakis et al, 1984). Apart from these symptoms, MELAS represents a true multisystemic disorder, potentially involving every organ, including gastrointestinal tract, heart, lungs, kidneys and skin (Sproule and Kaufmann, 2008). Almost 70% of patients present with initial symptoms between 2 and 20 years (Pavlakis et al, 1984). The pathogenesis of MELAS includes degenerative changes in small arteries and arterioles in the brain, accompanied by accumulation of mitochondria in vascular endothelial cells and smooth muscle cells. These abnormalities are thought to be involved in the genesis of non-ischaemic strokes in these patients. However, it was also suggested that the stroke-like episodes may reflect neuronal hyperexcitability with increases energy demand, causing an imbalance between energy requirements and inadequate ATP supply (Iizuka and Sakai, 2010; Koga et al, 2010).
Taken together, mutations in structural OXPHOS subunit and assembly factor genes are often associated with rare early-onset diseases displaying a devastating clinical course and some of them may present as defined clinical syndromes. Although, research is expanding our knowledge about molecular genetics and biochemistry of these diseases, numerous questions remain unsolved. Especially, the heterogeneous presentation of OXPHOS defects and the specific affection of certain tissues (e.g., the optic nerve in LHON patients, basal ganglia lesions in LS patients) in a subset of patients remain enigmatic.
Unfortunately, cell and organ material from patients is (extremely) scarce and information on brain mitochondrial function heavily relies on MRI/PET imaging and analysis of post-mortem samples. Although certainly valuable, these techniques do not provide insight into the pathophysiology of OXPHOS gene mutations at the level of single living cells. Such information is important since mitochondrial and cellular functioning are intimately linked and generally associated with submaximal metabolic rates. Moreover, the cytosolic environment allows mitochondria to communicate with the rest of the cell and other organelles (Koopman et al, 2012). In the next sections, we discuss how quantitative fluorescence microscopy techniques are applied to study the pathophysiology of OXPHOS mutations at the level of single living cells.
Quantitative live-cell microscopy
Ideally, relevant (i.e., neuronal) patient-derived cell lines should be used to study the cellular (patho)physiology of mtDNA and nDNA-encoded OXPHOS mutations during neurodegeneration. Unfortunately, these cell lines are generally unavailable and often patient-derived primary skin fibroblasts are used for genetic, diagnostic and live-cell analysis of OXPHOS disorders. Given their flat morphology, (patient) fibroblasts are ideally suited for microscopy analysis. Alternatively, primary cells and/or immortalized cell lines can be derived from appropriate mouse models or healthy cells can be treated with OXPHOS inhibitors to induce mitochondrial dysfunction. After selecting an appropriate cell model and culturing conditions, protein-based and/or chemical fluorescent reporter molecules can be introduced into the cell using transfection techniques or dedicated incubation protocols (Figure 5A). Moreover, cellular/mitochondrial autofluorescence can also be measured to monitor mitochondrial function (e.g., NAD(P)H; Verkaart et al, 2007b; Rodrigues et al, 2011). When accompanied by the proper control experiments, fluorescence microscopy/spectroscopy allows a relatively non-invasive quantification of various physiological readouts at the (sub)cellular level including ROS levels, Ca2+ dynamics, Δψ, NADH levels, thiol redox status, ATP levels, GLC levels, pH and mitochondrial dynamics and protein localization, mobility and concentration (e.g., Verkaart et al, 2007a; Benard et al, 2008; Dieteren et al, 2008, 2011; Koopman et al, 2008, 2012; Abramov et al, 2010; Dickinson et al, 2010; Digman and Gratton, 2011; Liemburg-Apers et al, 2011; Palmer et al, 2011; Distelmaier et al, 2012). Electron microscopy of fixed cells has been widely used to analyse the internal structure of the mitochondrion with the required high spatial resolution. Recently, a three-dimensional (3D) super-resolution microscopy technique (structured-illumination microscopy or SIM) was applied to visualize the temporal 3D structure of the mitochondrial matrix in living HeLa cells (Shao et al, 2011). This approach is important since it allows analysis of matrix volume, structure and topology that all appear to be linked to mitochondrial metabolic (dys)function (Hackenbrock et al, 1971; Rossignol et al, 2004; Mannella, 2008; Lizana et al, 2008; Dieteren et al, 2011). For multiparameter (‘high-content’) microscopy analysis of live cells, different reporter molecules that are spectrally compatible can be simultaneously introduced into the same cell. In case of overlapping emission spectra using multicoloured cells, spectral imaging during acquisition combined with linear unmixing of the image data can be applied (Zimmermann, 2005). The fluorescence signal(s) of the reporter molecule(s) can be quantified in space and time using live-cell fluorescence microscopy/spectroscopy and (quantitative) image analysis (Figure 5B–D; Koopman et al, 2008). Computer-controlled automated microscopy can be used to image cells cultured on multiwell plates, allowing investigation of multiple conditions in a large number of cells during a relatively short time period (‘high-throughput’; Conrad and Gerlich, 2010). The latter strategy requires extensively validated protocols for cell staining, image acquisition/processing/quantification and classification (e.g., Ljosa and Carpenter, 2009; Jain et al, 2010; Horvath et al, 2011; Shariff et al, 2011).
Figure 5.
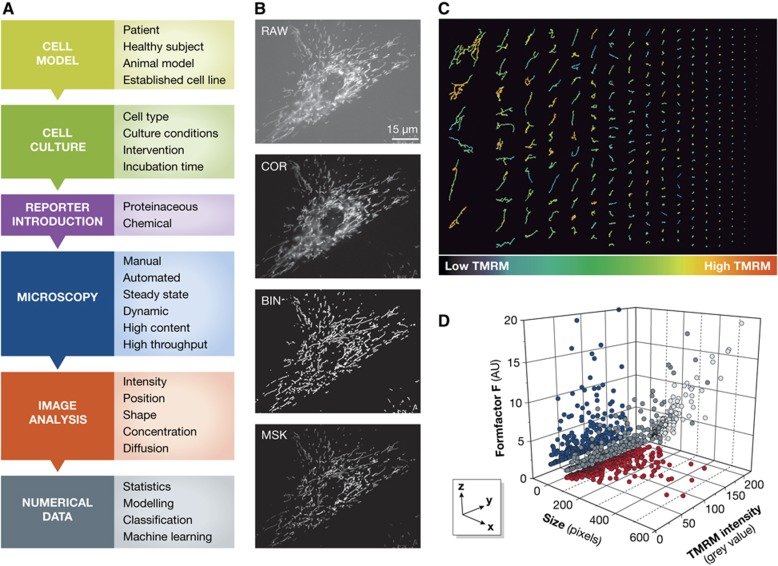
Quantitative analysis of mitochondrial (dys)function at the live-cell level. (A) Flow scheme illustrating how live-cell microscopy techniques can be applied to study OXPHOS dysfunction. Topics/decisions associated with the corresponding box are indicated at the right. (B) Image processing strategy allowing quantification of mitochondrial structure and function in a primary human skin fibroblast (#5120) from a healthy individual. Living cells were stained with the Δψ-sensitive fluorescent cation tetramethylrhodamine (TMRM) and visualized using epifluorescence microscopy. The obtained image (RAW) was corrected for background fluorescence (COR) and binarized to highlight mitochondrial structures (BIN; white objects). By masking the COR image with the BIN image information about mitochondrial structure, number and position (BIN image) were combined with TMRM intensity information from the COR image. This allows simultaneous quantification of these parameters from the MSK image. In this example, the number of mitochondrial objects equals 341, the average size of a mitochondrion equals 69±7 (s.e.m.) pixels, the average formfactor F (a combined measure of mitochondrial length and degree of filamentation) equals 2.7±0.2 (s.e.m.) arbitrary units, and the average mitochondrial TMRM fluorescence intensity equals 100±0.2 (s.e.m.) grey values. (C) Mitochondrial objects sorted (column-wise from top to bottom and from top left to lower right) based upon their size. The colour coding indicates the TMRM intensity, suggesting that Δψ is heterogeneous between individual mitochondrial objects. (D) Relationship between mitochondrial size (x axis), TMRM intensity (y axis) and formfactor (z axis) allowing multivariate analysis and multiparameter classification. Dark-grey spheres represent the original data points (each representing a mitochondrial object in the MSK figure), blue dots represent a projection of the data on the yz plane, red dots represent an projection of the data on the xy plane and light-grey dots represent a projection of the data on the xz plane). The latter reveals a linear correlation (R=0.97, P<0.001) between mitochondrial size and mitochondrial form factor F.
The live-cell consequences of mutations in OXPHOS structural and assembly genes
Live-cell fluorescence microscopy analysis has been applied to study the consequences of both mtDNA- and nDNA-encoded mutations in OXPHOS structural and assembly genes. Below we provide some typical examples that illustrate this strategy and present a framework summarizing the cellular data. In general, the effect of mtDNA mutations is analysed using ‘cytoplasmic hybrids’ (cybrids). These cells are generated by fusing non-nucleated (patient-derived) cells (cytoplasts) with mtDNA-depleted cells (ρ0 cells). The resulting cybrid cell line receives the mtDNA from the (patient) cytoplast and the nDNA from the ρ0 cell. Because the cybrid cells are derived from a common ρ0 nuclear background they have equivalent nuclear genes and biochemical and/or molecular differences are expected to reflect differences between their mtDNA content (King and Attardi, 1988, 1989; Swerdlow, 2012). In case of CI deficiency during LHON, mtDNA mutations (G3460A, G11778A and T14484C) were associated with reduced mitochondrial O2 consumption, Δψ depolarization, increased mitochondrial ROS production and reduced mitochondrial ATP production (see Pellegrini et al, 2012 and the references therein). Recently, an alternative strategy was presented to study the live-cell effects of mtDNA-encoded OXPHOS mutations. In this approach, neurons are used that are differentiated from mouse embryonic stem-cell cybrids containing mtDNA polymorphic variants or mutations (Kirby et al, 2009; Abramov et al, 2010; Trevelyan et al, 2010). These studies revealed that in neurons displaying a low residual CI activity (<10%), Δψ was hyperpolarized (i.e., more negative and likely maintained by CV reverse-mode action), ROS levels were increased and GSH was depleted. In case of CIV deficiency, a 40% residual activity was associated with a normal Δψ, increased ROS levels and normal GSH levels. This suggests that CI-deficient neurons display oxidative stress, whereas CIV-deficient neurons do not. The latter was supported by the observation that CI deficiency, but not CIV deficiency, increased neuronal death that was attenuated by ROS scavengers (Abramov et al, 2010). Analysis of Ca2+ signals in the same cell lines revealed that pathogenic mtDNA mutations did not affect the Ca2+ transient in response to single glutamatergic stimuli. However, in response to repeated stimuli, Ca2+ transients decayed more slowly in the mtDNA mutant cell lines (Trevelyan et al, 2010), suggesting insufficient fuelling of Ca2+ pumps on the ER with mitochondrial ATP (Willems et al, 2008). Although neuronal differentiation was observed, this parameter was impaired in cybrids displaying a large biochemical deficiency. Synaptic activity was detected in neurons with non-pathogenic mtDNA mutations or neurons with a mild defect of respiratory activity. However, mtDNA mutations that resulted in severe biochemical deficiency induced a marked reduction in post-synaptic events (Kirby et al, 2009). Taken together, these results suggest that neurons with a severe CI deficiency display oxidative stress, increased cell death, aberrant cytosolic Ca2+ handling due to limited mitochondrial ATP supply, impaired differentiation and a reduction in post-synaptic events. In contrast, other experimental evidence suggests that cytosolic Ca2+ clearance in cultured cerebellar granule cells during treatment with high K+ artificial cerebrospinal fluid (ACSF), is largely fuelled by glycolytic ATP and mediated by the plasma membrane Ca2+-ATPase (PMCA; Ivannikov et al, 2010). The latter study reported similar results for Purkinje cells in acutely prepared slices during electrical stimulation and further revealed that ER Ca2+ pumps are fuelled by both glycolytic and mitochondrial ATP. Mutations in the nDNA-encoded CIV assembly factor SURF1 are associated with LS in humans (Table I). Interestingly, analysis of a recombinant mouse model lacking this assembly factor (SURF1−/− mice), revealed that spontaneous neurodegeneration was absent, lifespan was markedly prolonged and animals were fully protected from kainic acid-induced Ca2+-dependent neurotoxicity (Dell’agnello et al, 2007). These results might be due to the fact that although CIV biochemical and assembly defects were present in SURF1−/− mice, they were milder than in humans. Analysis of primary neuronal cultures from SURF1−/− mice revealed that glutamate-induced cytosolic Ca2+ signals were of lower amplitude than in neurons from SURF1+/+ mice. The fact that Δψ was similar between SURF1−/− and SURF1+/+ neurons suggests that the above effects are independent of mitochondrial bioenergetics (Dell’agnello et al, 2007).
Integrating our own experimental results (largely obtained with primary skin fibroblasts from LS patients with isolated CI deficiency) with those in the literature revealed that primary monogenic mitochondrial disorders (i.e., those caused by a mutation in one of the nDNA-encoded proteins that make up the mitochondrial proteome) have only a limited number of (identified) consequences at the cellular level (see Koopman et al, 2012 and the references therein). Likely, this also holds true for defects in mtDNA/nDNA-encoded OXPHOS subunits and nDNA-encoded OXPHOS assembly factors since they represent a subset of the mitochondrial proteome. Due to the mutation, a mitochondrial protein defect is induced that is associated with its altered expression and/or activity. Subsequently, the protein defect will trigger (a combination of) ‘primary cellular consequences’ including: Δψ aberrations, altered mitochondrial shape/movement/positioning, increased ROS levels, and/or substrate accumulation. The magnitude of these changes, as well as the triggering of ‘secondary cellular consequences’ (e.g., altered ATP production, glycolysis upregulation, changes in redox state, mitophagy, ionic imbalance and mitochondrial biogenesis), depends on the nature of the mutation, the cell type, culture conditions and metabolic state (see Koopman et al, 2012 and the references therein). Importantly, both primary and secondary consequences might constitute part of an adaptive (signalling) mechanism attempting to counterbalance the consequences of the mutation. For example, the loss of a CIV assembly factor (SURF1) in fibroblasts from LS patients was associated with upregulation of CI, CIII and CV due to a post-transcriptional compensatory mechanism (Kovářová et al, 2012). Similarly, fibroblasts from patients with a mutated CV assembly factor (TMEM70), associated with reduced CV protein levels and ATP production, displayed Δψ hyperpolarization, increased ROS levels and compensatory upregulation of CIII and CIV (Havlíčková Karbanová et al, 2012). A parallel study with fibroblasts from a patient with a novel TMEM70 gene deletion revealed that reduced CI and CV activity was paralleled by mitochondrial fragmentation and aberrations in cristae structure (Jonckheere et al, 2012b). Analysis of primary fibroblasts from LS patients with isolated CI deficiency revealed increased ROS levels (Koopman et al, 2007; Verkaart et al, 2007a) but no detectable downstream effects on lipid peroxidation or thiol redox status (Verkaart et al, 2007b). Further experiments demonstrated that greatly reduced CI activity was associated with greatly increased ROS levels and mitochondrial fragmentation, whereas moderately reduced CI activity was paralleled by a minor increase in ROS levels and no effect on mitochondrial morphology (Koopman et al, 2005, 2007). The consequences of OXPHOS dysfunction with respect to ROS generation are also cell-type dependent. This is illustrated by our recent analysis of different fibroblast types derived from the NDUFS4−/− KO mouse (Kruse et al, 2008), which is the first animal model of isolated CI deficiency and LS (Roestenberg et al, 2012). In agreement with our primary patient fibroblast data, it was observed that primary mouse muscle and skin fibroblasts displayed increased ROS levels (WJHK, unpublished observation). In contrast, immortalized MEFs did not display this increase (Valsecchi et al, 2012). When the latter cells were placed in a medium containing GAL instead of GLU (to stimulate OXPHOS-mediated ATP generation; Rossignol et al, 2004), ROS levels were increased in NDUFS4−/− MEFs relative to MEFs from a wt animal (Valsecchi et al, unpublished observation) This suggests that immortalized cells and/or (high) GLU culture conditions (Marroquin et al, 2007) might not be ideal to study the (patho)physiology of OXPHOS (dys)function. Also cell differentiation can affect cellular bioenergetics and responses to oxidative stress (e.g., Schneider et al, 2011). The latter study revealed that differentiation of SH-SY5Y neuroblastoma cells to a neuronal phenotype induced Δψ hyperpolarization, increased stimulation of mitochondrial respiration by uncoupling (linked to increased CIV expression), and higher resistance to exogenous ROS application (linked to increased MnSOD expression). These results agree with the observation that real neurons rely on OXPHOS for ATP generation (Bélanger et al, 2011) and suggests that substantial changes in mitochondrial metabolism and antioxidant defences occur upon differentiation of neuroblastoma cells to a neuron-like phenotype (Schneider et al, 2011). Taken together, these results demonstrate that aberrations at the cellular level induced by OXPHOS dysfunction likely constitute a (cell-type and culture-condition dependent) convolution of primary and secondary (adaptive) effects, which requires careful interpretation. On the other hand, experimental analysis of the adaptive program will deliver valuable information about its molecular mechanism (e.g., Benard et al, 2012). In this sense, exogenous stimulation of this adaptive program might constitute a potential intervention strategy (e.g., Stranahan and Mattson, 2012).
Supplementary Material
Acknowledgments
We apologize to those authors whose articles we were unable to cite because of space limitations. This research was supported by the CSBR (Centres for Systems Biology Research) initiative from NWO (No: CSBR09/013V) and the Energy4All foundation.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aanerud J, Borghammer P, Chakravarty MM, Vang K, Rodell AB, Jónsdottir KY, Møller A, Ashkanian M, Vafaee MS, Iversen P, Johannsen P, Gjedde A (2012) Brain energy metabolism and blood flow differences in healthy aging. J Cereb Blood Flow Metab 32: 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Smulders-Srinivasan TK, Kirby DM, Acin-Perez R, Enriquez JA, Lightowlers R, Duchen MR, Turnbull DM (2010) Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain 133: 797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL, Schlame M (2011) Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J 100: 2184–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acín-Pérez R, Bayona-Bafaluy MP, Fernández-Silva P, Moreno-Loshuertos R, Pérez-Martos A, Bruno C, Moraes CT, Enríquez JA (2004) Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13: 805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA (2008) Respiratory active mitochondrial supercomplexes. Mol Cell 32: 529–539 [DOI] [PubMed] [Google Scholar]
- Adam-Vizi V, Chinopoulos C (2006) Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 27: 639–645 [DOI] [PubMed] [Google Scholar]
- Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashibuko R, Buettner G, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW, Spitz DR (2005) Mitochondrial O2·− and H2O2 mediate glucose deprivation-induced cytotoxicity and oxidative stress in human cancer cells. J Biol Chem 280: 4254–4263 [DOI] [PubMed] [Google Scholar]
- Althoff T, Mills DJ, Popot JL, Kühlbrandt W (2011) Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J 30: 4652–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Diano S, Horvath TL (2005) Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci 6: 829–840 [DOI] [PubMed] [Google Scholar]
- Angerer H, Zwicker K, Wumaier Z, Sokolova L, Heide H, Steger M, Kaiser S, Nübel E, Brutschy B, Radermacher M, Brandt U, Zickermann V (2011) A scaffold of accessory subunits links the peripheral arm and the distal proton-pumping module of mitochondrial complex I. Biochem J 437: 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin RE, Garside SL, Tarnopolsky MA, Mazurek MF, Rosebush PI (2012) The psychiatric manifestations of mitochondrial disorders: a case and review of the literature. J Clin Psychiatry 73: 506–512 [DOI] [PubMed] [Google Scholar]
- Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O’Rourke B, Paolocci N, Cortassa S (2012) Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol 139: 479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S (2012) The power of life—cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 12: 46–56 [DOI] [PubMed] [Google Scholar]
- Arnoult D, Soares F, Tattoli I, Girardin SE (2011) Mitochondria in innate immunity. EMBO Rep 12: 901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120: 483–495 [DOI] [PubMed] [Google Scholar]
- Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landázuri MO, Enríquez JA (2012) NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab 16: 378–386 [DOI] [PubMed] [Google Scholar]
- Bandy B, Davison AJ (1990) Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med 8: 523–539 [DOI] [PubMed] [Google Scholar]
- Becker T, Böttinger L, Pfanner N (2012) Mitochondrial protein import: from transport pathways to an integrated network. Trends Biochem Sci 37: 85–91 [DOI] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14: 724–738 [DOI] [PubMed] [Google Scholar]
- Bélanger M, Magistretti PJ (2009) The role of astroglia in neuroprotection. Dialogues Clin Neurosci 11: 281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Faustin B, Galinier A, Rocher C, Bellance N, Smolkova K, Casteilla L, Rossignol R, Letellier T (2008) Functional dynamic compartmentalization of respiratory chain intermediate substrates: implications for the control of energy production and mitochondrial diseases. Int J Biochem Cell Biol 40: 1543–1554 [DOI] [PubMed] [Google Scholar]
- Benard G, Faustin B, Passerieux E, Galinier A, Rocher C, Bellance N, Delage JP, Casteilla L, Letellier T, Rossignol R (2006) Physiological diversity of mitochondrial oxidative phosphorylation. Am J Physiol Cell Physiol 291: C1172–C1182 [DOI] [PubMed] [Google Scholar]
- Benard G, Rossignol R (2008) Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 10: 1313–1342 [DOI] [PubMed] [Google Scholar]
- Benard G, Trian T, Bellance N, Berger P, Lavie J, Espil-Taris C, Rocher C, Eimer-Bouillot S, Goizet C, Nouette-Gaulain K, Letellier T, Lacombe D, Rossignol R (2012) Adaptative capacity of mitochondrial biogenesis and of mitochondrial dynamics in response to pathogenic respiratory chain dysfunction. Antioxid Redox Signal (advance online publication, 19 April 2012; doi:; DOI: 10.1089/ars.2011.4244) [DOI] [PubMed] [Google Scholar]
- Bénit P, El-Khoury R, Schiff M, Sainsard-Chanet A, Rustin P (2010) Genetic background influences mitochondrial function: modeling mitochondrial disease for therapeutic development. Trends Mol Med 16: 210–217 [DOI] [PubMed] [Google Scholar]
- Boekema EJ, Braun HP (2007) Supramolecular structure of the mitochondrial oxidative phosphorylation system. J Biol Chem 282: 1–4 [DOI] [PubMed] [Google Scholar]
- Boneh A (2006) Regulation of mitochondrial oxidative phosphorylation by second messenger-mediated signal transduction mechanisms. Cell Mol Life Sci 63: 1236–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourens M, Fontanesi F, Soto IC, Liu J, Barrientos A (2012) Redox and reactive oxygen species regulation of mitochondrial cytochrome c oxidase biogenesis. Antioxid Redox Signal (advance online publication, 15 October 2012; doi:; DOI: 10.1089/ars.2012.4847) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B (1972) The cellular production of hydrogen peroxide. Biochem J 128: 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brière JJ, Favier J, El Ghouzzi V, Djouadi F, Bénit P, Gimenez AP, Rustin P (2005) Succinate dehydrogenase deficiency in human. Cell Mol Life Sci 62: 2317–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC (2010) Nitric oxide and neuronal death. Nitric oxide 23: 153–165 [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V (2012) There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 12: 1–4 [DOI] [PubMed] [Google Scholar]
- Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA (2011) Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol 31: 4994–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham-Marusich AR, Berninsone PM (2012) Multiple proteins with essential mitochondrial functions have glycosylated isoforms. Mitochondrion 12: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11: 85–95 [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA (2009) Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal 11: 2717–2739 [DOI] [PubMed] [Google Scholar]
- Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA (2005) Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J Neurol Sci 233: 145–162 [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK (2010) The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11: 25–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambron M, D’Haeseleer M, Laureys G, Clinckers R, Debruyne J, De Keyser J (2012) White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J Cereb Blood Flow Metab 32: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, Ogilvie I, Shoubridge EA, Robinson BH (2011a) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet 89: 486–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron JM, Levandovskiy V, Mackay N, Ackerley C, Chitayat D, Raiman J, Halliday WH, Schulze A, Robinson BH (2011b) Complex V TMEM70 deficiency results in mitochondrial nucleoid disorganization. Mitochondrion 11: 191–199 [DOI] [PubMed] [Google Scholar]
- Campbell GR, Ohno N, Turnbull DM, Mahad DJ (2012) Mitochondrial changes within axons in multiple sclerosis: an update. Curr Opin Neurol 25: 221–230 [DOI] [PubMed] [Google Scholar]
- Carlucci A, Lignitto L, Feliciello A (2008) Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol 18: 604–613 [DOI] [PubMed] [Google Scholar]
- Chandra D, Singh KV (2011) Genetic insights into OXPHOS defect and its role in cancer. Biochim Biophys Acta 1807: 620–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PP, Zweier JL, Chen YR (2008) Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J Biol Chem 283: 27991–28003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Taylor EB, Dephoure N, Heo JM, Tonhato A, Papandreou I, Nath N, Denko NC, Gygi SP, Rutter J (2012) Identification of a protein mediating respiratory supercomplex stability. Cell Metab 15: 348–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V (2010) Mitochondria as ATP consumers in cellular pathology (2010). Biochim Biophys Acta 1802: 221–227 [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Andersen JK (2008) Redox imbalance in Parkinson’s disease. Biochim Biophys Acta 1780: 1362–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Andersen JK (2011) Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic Res 45: 53–58 [DOI] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, Macvicar BA (2012) Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenelyl cyclase. Neuron 75: 1094–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi KB, Boylston WH, Rabek JP, Widger WR, Papaconstantinou J (2004) Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim Biophys Acta 1688: 95–101 [DOI] [PubMed] [Google Scholar]
- Clason T, Ruiz T, Schägger H, Peng G, Zickermann V, Brandt U, Michel H, Radermacher M (2010) The structure of eukaryotic and prokaryotic complex I. J Struct Biol 169: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay HB, Sillivan S, Konradi C (2011) Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci 29: 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool SM, Koehler CM (2012) The complexity of cardiolipin in health and disease. Trends Biochem Sci 37: 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BH (2010) Pharmacologic effects on mitochondrial function. Dev Disabil Res Rev 16: 189–199 [DOI] [PubMed] [Google Scholar]
- Collins Y, Chouchani ET, James AM, Menger KE, Cochemé HM, Murphy MP (2012) Mitochondrial redox signalling at a glance. J Cell Sci 125: 801–806 [DOI] [PubMed] [Google Scholar]
- Conrad C, Gerlich DW (2010) Automated microscopy for high-content RNAi screening. J Cell Biol 188: 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA (2012) Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s diseases. Adv Exp Med Biol 724: 205–221 [DOI] [PubMed] [Google Scholar]
- Costa V, Scorrano L (2012) Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J 31: 1853–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Coleman MP (2012) Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci 35: 364–372 [DOI] [PubMed] [Google Scholar]
- Danielson SR, Held JM, Oo M, Riley R, Gibson BW, Andersen JK (2011) Quantitative mapping of reversible mitochondrial Complex I cysteine oxidation in a Parkinson disease mouse model. J Biol Chem 286: 7601–7608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA 108: 14121–14126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Sue CM (2011) The genetics of mitochondrial disease. Semin Neurol 31: 519–530 [DOI] [PubMed] [Google Scholar]
- Del Hoyo P, García-Redondo A, de Bustos F, Molina JA, Sayed Y, Alonso-Navarro H, Caballero L, Arenas J, Agúndez JA, Jiménez-Jiménez FJ (2010) Oxidative stress in skin fibroblasts cultures from patients with Parkinson’s disease. BMC Neurol 10: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M (2007) Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet 16: 431–444 [DOI] [PubMed] [Google Scholar]
- Deuschl G, Elble R (2009) Essential tremor—neurodegenerative or nondegenerative disease towards a working definition of ET. Mov Disord 24: 2033–2041 [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Srikun D, Chang CJ (2010) Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr Opin Chem Biol 14: 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA (2012) Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 32: 1107–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren CE, Willems PH, Swarts HG, Fransen J, Smeitink JA, Koopman WJ, Nijtmans LG (2011) Defective mitochondrial translation differently affects the live cell dynamics of complex I subunits. Biochim Biophys Acta 1807: 1624–1633 [DOI] [PubMed] [Google Scholar]
- Dieteren CE, Willems PH, Vogel RO, Swarts HG, Fransen J, Roepman R, Crienen G, Smeitink JA, Nijtmans LG, Koopman WJ (2008) Subunits of mitochondrial complex I exist as part of matrix- and membrane-associated subcomplexes in living cells. J Biol Chem 283: 34753–34761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren CEJ, Gielen SCAM, Nijtmans LGJ, Smeitink JAM, Swarts HG, Brock R, Willems PHGM, Koopman WJH (2011) Solute diffusion is hindered in the mitochondrial matrix. Proc Natl Acad Sci USA 108: 8657–8662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Gratton E (2011) Lessons in fluctuation correlation spectroscopy. Annu Rev Phys Chem 62: 645–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimauro S, Rustin P (2009) A critical approach to the therapy of mitochondrial respiratory chain and oxidative phosphorylation diseases. Biochim Biophys Acta 1792: 1159–1167 [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA (2008) Mitochondrial disorders in the nervous system. Annu Rev Neurosci 31: 91–123 [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Koopman WJH, van den Heuvel LW, Rodenburg RJ, Mayatepek E, Willems PHGM, Smeitink JAM (2009) Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain 132: 833–842 [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Valsecchi F, Forkink M, van Emst-de Vries S, Swarts H, Rodenburg R, Verwiel E, Smeitink J, Willems P, Koopman WJ (2012) Trolox-sensitive ROS regulate mitochondrial morphology, oxidative phosphorylation and cytosolic calcium handling in healthy cells. Antioxid Redox Signal 17: 1657–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Brand MD (2011) The regulation and physiology of mitochondrial proton leak. Physiology 26: 192–205 [DOI] [PubMed] [Google Scholar]
- Doherty GH (2011) Nitric oxide in neurodegeneration: potential benefits of non-steroidal anti-inflammatories. Neurosci Bull 27: 366–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W, Schipper HM (2007) Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 6: 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröse S, Brandt U (2008) The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem 283: 21649–21654 [DOI] [PubMed] [Google Scholar]
- Dudkina NV, Kouril R, Peters K, Braun HP, Boekema EJ (2010) Structure and function of mitochondrial supercomplexes. Biochim Biophys Acta 1797: 664–670 [DOI] [PubMed] [Google Scholar]
- Dudkina NV, Kudryashev M, Stahlberg H, Boekema EJ (2011) Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc Natl Acad Sci USA 108: 15196–15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour E, Terzioglu M, Sterky FH, Sörensen L, Galter D, Olson L, Wilbertz J, Larsson NG (2008) Age-associated mosaic respiratory chain deficiency causes trans-neuronal degeneration. Hum Mol Genet 17: 1418–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA (2010) The architecture of respiratory complex I. Nature 465: 441–445 [DOI] [PubMed] [Google Scholar]
- Efremov RG, Sazanov LA (2011a) Structure of the membrane domain of respiratory complex I. Nature 476: 414–420 [DOI] [PubMed] [Google Scholar]
- Efremov RG, Sazanov LA (2011b) Respiratory complex I: ‘steam engine’ of the cell? Curr Opin Struct Biol 21: 532–540 [DOI] [PubMed] [Google Scholar]
- Fato R, Bergamini C, Leoni S, Strocchi P, Lenaz G (2008) Generation of reactive oxygen species by mitochondrial complex I: implications in neurodegeneration. Neurochem Res 33: 2487–2501 [DOI] [PubMed] [Google Scholar]
- Ferguson-Miller S, Hiser C, Liu J (2012) Gating and regulation of the cytochrome c oxidase proton pump. Biochim Biophys Acta 1817: 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J (2006) Central nervous system manifestations of mitochondrial disorders. Acta Neurol Scand 114: 217–238 [DOI] [PubMed] [Google Scholar]
- Finsterer J, Mahjoub SZ (2012) Epilepsy in mitochondrial disorders. Seizure 21: 316–321 [DOI] [PubMed] [Google Scholar]
- Finsterer J, Segall L (2010) Drugs interfering with mitochondrial disorders. Drug Chem Toxicol 33: 138–151 [DOI] [PubMed] [Google Scholar]
- Forkink M, Smeitink JA, Brock R, Willems PH, Koopman WJ (2010) Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Biochim Biophys Acta 1797: 1034–1044 [DOI] [PubMed] [Google Scholar]
- Frenzel M, Rommelspacher H, Sugawa MD, Dencher NA (2010) Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp Gerontol 45: 563–572 [DOI] [PubMed] [Google Scholar]
- Friedman SD, Shaw DWW, Ishak G, Gropman AL, Saneto RP (2010) The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev 16: 129–135 [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT (2008) The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci 31: 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin A, Moncada S (2007) S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem 282: 37448–37453 [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P (2003) Mitochondrial potassium transport: the K(+) cycle. Biochim Biophys Acta 1606: 23–41 [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, Vielhaber S, Seppet E, Striggow F (2010) The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta 1797: 1018–1027 [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233 [DOI] [PubMed] [Google Scholar]
- Glancy B, Balaban RS (2012) Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51: 2959–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Scorrano L (2012) Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta (advance online publication, 1 March 2012; doi:; DOI: 10.1016/j.bbamcr.2012.02.012) [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 183: 681–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, Fass D (2006) Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA 103: 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Xiong Y (2011) Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci 36: 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR, Rehn TG, Weinbach EC, Lemasters JJ (1971) Oxidative phosphorylation and ultrastructural transformation in mitochondria in the intact ascites tumor cell. J Cell Biol 51: 123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM (2006) Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: 728–735 [DOI] [PubMed] [Google Scholar]
- Handy DE, Loscalzo J (2012) Redox regulation of mitochondrial function. Antioxid Redox Signal 16: 1323–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75: 762–777 [DOI] [PubMed] [Google Scholar]
- Havlíčková Karbanová V, Cížková Vrbacká A, Hejzlarová K, Nůsková H, Stránecký V, Potocká A, Kmoch S, Houštěk J (2012) Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim Biophys Acta 1817: 1037–1043 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Stuchebrukhov AA (2010) Electron tunneling in respiratory complex I. Proc Natl Acad Sci USA 107: 19157–19162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Cooper HM, Reyes A, Di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A, Walker JE, Holt IJ (2012) Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res 40: 6109–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Jose C, Gutierrez Cortes N, Dupuy JW, Rocher C, Dachary-Prigent J, Letellier T (2012) Preservation of NADH ubiquinone-oxidoreductase activity by Src kinase-mediated phosphorylation of NDUFB10. Biochim Biophys Acta 1817: 718–725 [DOI] [PubMed] [Google Scholar]
- Hedskog L, Zhang S, Ankarcrona M (2012) Strategic role for mitochondria in Alzheimer’s disease and cancer. Antioxid Redox Signal 16: 1476–1491 [DOI] [PubMed] [Google Scholar]
- Helling S, Hüttemann M, Ramzan R, Kim SH, Lee I, Müller T, Langenfeld E, Meyer HE, Kadenbach B, Vogt S, Marcus K (2012) Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics 12: 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27: 219–249 [DOI] [PubMed] [Google Scholar]
- Hinchliffe P, Sazanov LA (2005) Organization of iron-sulfur clusters in respiratory complex I. Science 309: 771–774 [DOI] [PubMed] [Google Scholar]
- Hirst J (2011) Why does mitochondrial complex I have so many subunits? Biochem J 437: e1–e3 [DOI] [PubMed] [Google Scholar]
- Hornig-Do HT, Tatsuta T, Buckermann A, Bust M, Kollberg G, Rötig A, Hellmich M, Nijtmans L, Wiesner RJ (2012) Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J 31: 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Wild T, Kutay U, Csucs G (2011) Machine learning improves the precision and robustness of high-content screens: using nonlinear multiparametric methods to analyze screening results. J Biomol Screen 16: 1059–1067 [DOI] [PubMed] [Google Scholar]
- Hunte C, Zickermann V, Brandt U (2010) Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 329: 448–451 [DOI] [PubMed] [Google Scholar]
- Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP (2008) Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem 283: 24801–24815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka T, Sakai F (2010) Pathophysiology of stroke-like episodes in MELAS: neuron-astrocyte uncoupling in neuronal hyperexcitability. Future Neurol 5: 61–83 [Google Scholar]
- Ivannikov MV, Sugimori M, Llinás RR (2010) Calcium clearance and its energy requirements in cerebellar neurons. Cell Calcium 47: 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V, Seung HS, Turaga SC (2010) Machines that learn to segment images: a crucial technology for connectomics. Curr Opin Neurobiol 20: 653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Huigsloot M, Lammens M, Jansen J, van den Heuvel LP, Spiekerkoetter U, von Kleist-Retzow JC, Forkink M, Koopman WJ, Szklarczyk R, Huynen MA, Fransen JA, Smeitink JA, Rodenburg RJ (2012b) Restoration of complex V deficiency caused by a novel deletion in the human TMEM70 gene normalizes mitochondrial morphology. Mitochondrion 11: 954–963 [DOI] [PubMed] [Google Scholar]
- Jonckheere AI, Smeitink JA, Rodenburg RJ (2012a) Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis 35: 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Safiulina D, Zharkovsky A, Veksler V (2007) Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 292: C157–C163 [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H (2012) Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197: 535–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM (1995) Essentials of Neural Science and Behavior East Norwalk, CT, USA: Appleton & Lange, [Google Scholar]
- Kane LA, Van Eyk JE (2009) Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr 41: 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O (2011) The energy demand of fast neuronal network oscillations: insights from brain slice preparations. Front Pharmacol 2: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G (1988) Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell 52: 811–819 [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez JA, Trevelyan AJ, Turnbull DM, Lightowlers RN (2009) Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell Prolif 42: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M (2008) The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 1778: 1978–2021 [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc EC, Koc H (2012) Regulation of mitochondrial translation by post-translational modifications. Biochim Biophys Acta 1819: 1055–1066 [DOI] [PubMed] [Google Scholar]
- Koga Y, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T (2010) MELAS and L-arginine therapy: pathophysiology of stroke-like episodes. Ann NY Acad Sci 1201: 104–110 [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Distelmaier F, Esseling JJ, Smeitink JA, Willems PH (2008) Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods 46: 304–311 [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Verkaart S, Visch HJ, van Emst-de Vries S, Nijtmans LG, Smeitink JA, Willems PH (2007) Human NADH:ubiquinone oxidoreductase deficiency: radical changes in mitochondrial morphology? Am J Physiol Cell Physiol 293: C22–C29 [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289: C881–C890 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Nijtmans LG, Dieteren CEJ, Roestenberg P, Valsecchi F, Smeitink JAM, Willems PHGM (2010) Mammalian mitochondrial complex I: biogenesis, regulation and reactive oxygen species generation. Antioxid Redox Signal 12: 1431–1470 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Willems PHGM, Smeitink JAM (2012) Monogenic mitochondrial disorders. N Eng J Med 366: 1132–1141 [DOI] [PubMed] [Google Scholar]
- Korzeniewski B (2011) Computer-aided studies on the regulation of oxidative phosphorylation during work transitions. Prog Biophys Mol Biol 107: 274–285 [DOI] [PubMed] [Google Scholar]
- Kourtis N, Tavernarakis N (2011) Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J 30: 2520–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovářová N, Cížková Vrbacká A, Pecina P, Stránecký V, Pronicka E, Kmoch S, Houštěk J (2012) Adaptation of respiratory chain biogenesis to cytochrome c oxidase deficiency caused by SURF1 gene mutations. Biochim Biophys Acta 1822: 1114–1124 [DOI] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD (2008) Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7: 312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukreja RC, Kontos HA, Hess ML, Ellis EF (1986) PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res 59: 612–619 [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Henning MS, Starkov AA, Manfredi G (2007) The mitochondrial respiratory chain is a modulator of apoptosis. J Cell Biol 179: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebre AS, Rio M, Faivre d’Arcier L, Vernerey D, Landrieu P, Slama A, Jardel C, Laforêt P, Rodriguez D, Dorison N, Galanaud D, Chabrol B, Paquis-Flucklinger V, Grévent D, Edvardson S, Steffann J, Funalot B, Villeneuve N, Valayannopoulos V, de Lonlay P et al. (2011) A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J Med Genet 48: 16–23 [DOI] [PubMed] [Google Scholar]
- Lee HK, Cho YM, Kwak SH, Lim S, Park KS, Shim EB (2010) Mitochondrial dysfunction and metabolic syndrome—looking for environmental factors. Biochim Biophys Acta 1800: 282–289 [DOI] [PubMed] [Google Scholar]
- Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441: 523–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh D (1951) Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 14: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G, Genova ML (2007) Kinetics of integrated electron transfer in the mitochondrial respiratory chain: random collisions vs. solid state electron channeling. Am J Physiol Cell Physiol 292: C1221–C1239 [DOI] [PubMed] [Google Scholar]
- Leuner K, Schütt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, Mai S, Jendrach M, Eckert GP, Kruse SE, Palmiter RD, Brandt U, Dröse S, Wittig I, Willem M, Haass C, Reichert AS, Müller WE (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16: 1421–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liemburg-Apers DC, Imamura H, Forkink M, Nooteboom M, Swarts HG, Brock R, Smeitink JA, Willems PH, Koopman WJ (2011) Quantitative glucose and ATP sensing in mammalian cells. Pharm Res 28: 2745–2757 [DOI] [PubMed] [Google Scholar]
- Lizana L, Bauer B, Orwar O (2008) Controlling the rates of biochemical reactions and signaling networks by shape and volume changes. Proc Natl Acad Sci USA 105: 4099–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljosa V, Carpenter AE (2009) Introduction to the quantitative analysis of two-dimensional fluorescence microscopy images for cell-based screening. PLoS Comput Biol 5: e1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovas JR, Wang X (2012) The meaning of mitochondrial movement to a neuron’s life. Biochim Biophys Acta (advance online publication, 21 April 2012; doi:; DOI: 10.1016/j.bbamcr.2012.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukosz M, Jakob S, Büchner N, Zschauer TC, Altschmied J, Haendeler J (2010) Nuclear redox signaling. Antioxid Redox Signal 12: 713–742 [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464 [DOI] [PubMed] [Google Scholar]
- Maas MF, Krause F, Dencher NA, Sainsard-Chanet A (2009) Respiratory complexes III and IV are not essential for the assembly/stability of complex I in fungi. J Mol Biol 387: 259–269 [DOI] [PubMed] [Google Scholar]
- Mailloux RJ, Harper ME (2012) Mitochondrial proticity and ROS signaling: lessons from the uncoupling proteins. Trends Endocrinol Metab 23: 451–458 [DOI] [PubMed] [Google Scholar]
- Mammucari C, Patron M, Granatiero V, Rizzuto R (2011) Molecules and roles of mitochondrial calcium signaling. Biofactors 37: 219–227 [DOI] [PubMed] [Google Scholar]
- Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G (2012) Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13: 293–307 [DOI] [PubMed] [Google Scholar]
- Mannella CA (2008) Structural diversity of mitochondria. Ann NY Acad Sci 1147: 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y (2007) Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97: 539–547 [DOI] [PubMed] [Google Scholar]
- Martin LJ (2011) Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinvalet D, Dykxhoorn DM, Ferrini R, Lieberman J (2008) Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133: 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki S, Szweda LI (2007) Inhibition of complex I by Ca2+ reduces electron transport activity and the rate of superoxide anion production in cardiac submitochondrial particles. Biochemistry 46: 1350–1357 [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM (2010) A neurological perspective on mitochondrial disease. Lancet Neurol 9: 829–840 [DOI] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol 361: 462–469 [DOI] [PubMed] [Google Scholar]
- Mckenzie M, Ryan MT (2010) Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life 62: 497–502 [DOI] [PubMed] [Google Scholar]
- McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG (2003) Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol 285: H2290–H2297 [DOI] [PubMed] [Google Scholar]
- Mick DU, Fox TD, Rehling P (2011) Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat Rev Mol Cell Biol 12: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miriyala S, Spasojevic I, Tovmasyan A, Salvemini D, Vujaskovic Z, St Clair D, Batinic-Haberle I (2012) Manganese superoxide dismutase, MnSOD and its mimics. Biochim Biophys Acta 1822: 794–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M (2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115: 629–640 [DOI] [PubMed] [Google Scholar]
- Morán M, Moreno-Lastres D, Marín-Buera L, Arenas J, Martín MA, Ugalde C (2012) Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic Biol Med 53: 595–609 [DOI] [PubMed] [Google Scholar]
- Moreno-Lastres D, Fontanesi F, García-Consuegra I, Martín MA, Arenas J, Barrientos A, Ugalde C (2012) Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab 15: 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Liu Y, Van Remmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064–49073 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal 16: 476–495 [DOI] [PubMed] [Google Scholar]
- Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, Nyström T, Belousov V, Schumacker PT, Winterbourn CC (2011) Unraveling the biological roles of reactive oxygen species. Cell Metab 13: 361–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA (2003) Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem 278: 37223–37230 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Cieplak P, Cho DH, Godzik A, Lipton SA (2010) S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10: 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman NJ (2005) Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol 140: 517–523 [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO (2012) Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain 135: 12–22 [DOI] [PubMed] [Google Scholar]
- Novak I (2012) Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal 17: 794–802 [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnel VB, Azzi A (1996) High rates of extracellular superoxide generation by cultured human fibroblasts: involvement of a lipid-metabolizing enzyme. Biochem J 318: 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B (2007) Mitochondrial ion channels. Annu Rev Physiol 69: 19–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe K, Inada N, Gota C, Harada Y, Funatsu T, Uchiyama S (2012) Intracellular temperature mapping with a fluorescent polymeric thermometer and fluorescence lifetime imaging microscopy. Nat Commun 3: 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno D, Iino R, Noji H (2011) Rotation and structure of FoF1-ATP synthase. J Biochem 149: 655–664 [DOI] [PubMed] [Google Scholar]
- Onishi T (2010) Piston drives a proton pump. Nature 465: 428–429 [DOI] [PubMed] [Google Scholar]
- Pagano G, Castello G (2012) Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol 724: 291–299 [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE (2006) Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci 31: 26–34 [DOI] [PubMed] [Google Scholar]
- Palmer AE, Qin Y, Park JG, McCombs JE (2011) Design and application of genetically encoded biosensors. Trends Biotechnol 29: 144–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F (2008) Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta 1777: 564–578 [DOI] [PubMed] [Google Scholar]
- Papa S, Rasmo DD, Technikova-Dobrova Z, Panelli D, Signorile A, Scacco S, Petruzzella V, Papa F, Palmisano G, Gnoni A, Micelli L, Sardanelli AM (2012) Respiratory chain complex I, a main regulatory target of the cAMP/PKA pathway is defective in different human diseases. FEBS Lett 586: 568–577 [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Ruggiero FM (2002) Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 286: 135–141 [DOI] [PubMed] [Google Scholar]
- Pathak RU, Davey GP (2008) Complex I and energy thresholds in the brain. Biochim Biophys Acta 1777: 777–782 [DOI] [PubMed] [Google Scholar]
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 16: 481–488 [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Smeitink JAM, Willems PHGM, Koopman WJH (2012) Cellular consequences of mtDNA-encoded mutations in NADH:Ubiquinone oxidoreductase. In ‘A structural Perspective on Respiratory Complex I’ Sazanov LA (ed.) Chapter 9. pp 171–192Dordrecht: Springer, [Google Scholar]
- Perales-Clemente E, Fernández-Vizarra E, Acín-Pérez R, Movilla N, Bayona-Bafaluy MP, Moreno-Loshuertos R, Pérez-Martos A, Fernández-Silva P, Enríquez JA (2010) Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol Cell Biol 30: 3038–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta S, Wang X, Moraes CT (2012) Mitochondrial transcription: lessons from mouse models. Biochim Biophys Acta 1819: 961–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perjés A, Kubin AM, Kónyi A, Szabados S, Cziráki A, Skoumal R, Ruskoaho H, Szokodi I (2012) Physiological regulation of cardiac contractility by endogenous reactive oxygen species. Acta Physiol (Oxf) 205: 26–40 [DOI] [PubMed] [Google Scholar]
- Pryde KR, Hirst J (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem 286: 18056–18065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Vila M, Jackson-Lewis V (2003) Neurodegeneration: what is it and where are we? J Clin Invest 111: 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Hauswirth WW, Guy J (2003) Suppression of complex I gene expression induces optic neuropathy. Ann Neurol 53: 198–205 [DOI] [PubMed] [Google Scholar]
- Quinlan CL, Gerencser AA, Treberg JR, Brand MD (2011) The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem 286: 31361–31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287: 27255–27264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Morgan PG, Kruse SE, Palmiter RD, Sedensky MM (2012) Altered anesthetic sensitivity of mice lacking ndufs4, a subunit of mitochondrial complex I. PLoS ONE 7: e42904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N, Baysal BE, Shadel GS (2011) Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med 17: 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam M, Kim SJ (2012) Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J Neural Transm 119: 891–910 [DOI] [PubMed] [Google Scholar]
- Ramírez-Aguilar SJ, Keuthe M, Rocha M, Fedyaev VV, Kramp K, Gupta KJ, Rasmusson AG, Schulze WX, van Dongen JT (2011) The composition of plant mitochondrial supercomplexes changes with oxygen availability. J Biol Chem 286: 43045–43053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24: 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786 [DOI] [PubMed] [Google Scholar]
- Rich PR (2003) The molecular machinery of Keilin’s respiratory chain. Biochem Soc Trans 31: 1095–1105 [DOI] [PubMed] [Google Scholar]
- Rodrigues RM, Macko P, Palosaari T, Whelan MP (2011) Autofluorescence microscopy: a non-destructive tool to monitor mitochondrial toxicity. Toxicol Lett 206: 281–288 [DOI] [PubMed] [Google Scholar]
- Roestenberg P, Manjeri GR, Valsecchi F, Smeitink JA, Willems PH, Koopman WJ (2012) Pharmacological targeting of mitochondrial complex I deficiency: the cellular level and beyond. Mitochondrion 12: 57–65 [DOI] [PubMed] [Google Scholar]
- Rossi A, Biancheri R, Bruno C, Di Rocco M, Calvi A, Pessagno A, Tortori-Donati P (2003) Leigh syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR AM J Neuroradiol 24: 1188–1191 [PMC free article] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64: 985–993 [DOI] [PubMed] [Google Scholar]
- Rossignol R, Malgat M, Mazat JP, Letellier T (1999) Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J Biol Chem 274: 33426–33432 [DOI] [PubMed] [Google Scholar]
- Roy P, Roy SK, Mitra A, Kulkarni AP (1994) Superoxide generation by lipoxygenase in the presence of NADH and NADPH. Biochim Biophys Acta 1214: 171–179 [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31: 1336–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J, Winge DR, Schiffman JD (2010) Succinate dehydrogenase—assembly, regulation and role in human disease. Mitochondrion 10: 393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks V, Guzun R, Timohhina N, Tepp K, Varikmaa M, Monge C, Beraud N, Kaambre T, Kuznetsov A, Kadaja L, Eimre M, Seppet E (2010) Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: mitochondrial interactosome. Biochim Biophys Acta 1797: 678–697 [DOI] [PubMed] [Google Scholar]
- Sazanov LA, Hinchliffe P (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311: 1430–1436 [DOI] [PubMed] [Google Scholar]
- Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638 [DOI] [PubMed] [Google Scholar]
- Scarpulla RC (2012) Nucleus-encoded regulators of mitochondrial function: Integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta 1819: 1088–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H, de Coo R, Bauer MF, Hofmann S, Godinot C, Brandt U (2004) Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J Biol Chem 279: 36349–36353 [DOI] [PubMed] [Google Scholar]
- Schägger H, Pfeiffer K (2001) The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem 276: 37861–37867 [DOI] [PubMed] [Google Scholar]
- Schapira AH (2010) Complex I: inhibitors, inhibition and neurodegeneration. Exp Neurol 224: 331–335 [DOI] [PubMed] [Google Scholar]
- Schapira AH (2012) Mitochondrial diseases. Lancet 379: 1825–1834 [DOI] [PubMed] [Google Scholar]
- Schiff M, Bénit P, Jacobs HT, Vockley J, Rustin P (2012) Therapies in inborn errors of oxidative metabolism. Trends Endocrinol Metab 23: 488–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Giordano S, Zelickson BR, S Johnson M, A Benavides G, Ouyang X, Fineberg N, Darley-Usmar VM, Zhang J (2011) Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic Biol Med 51: 2007–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S (2011) Mitochondria: the next (neurode)generation. Neuron 70: 1033–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Kner P, Rego EH, Gustafsson MG (2011) Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods 8: 1044–1046 [DOI] [PubMed] [Google Scholar]
- Shariff A, Kangas J, Coelho LP, Quinn S, Murphy RF (2011) Automated image analysis for high-content screening and analysis. J Biomol Screen 15: 726–734 [DOI] [PubMed] [Google Scholar]
- Shoubridge EA (2012) Supersizing the mitochondrial respiratory chain. Cell Metab 15: 271–272 [DOI] [PubMed] [Google Scholar]
- Shutt T, Geoffrion M, Milne R, McBride HM (2012) The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13: 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27: 1766–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeitink J, van den Heuvel L, DiMauro S (2001) The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2: 342–352 [DOI] [PubMed] [Google Scholar]
- Smith PM, Fox JL, Winge DR (2012) Reprint of: Biogenesis of the cytochrome bc(1) complex and role of assembly factors. Biochim Biophys Acta 1817: 872–882 [DOI] [PubMed] [Google Scholar]
- Spelbrink JN (2010) Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life 62: 19–32 [DOI] [PubMed] [Google Scholar]
- Sproule DM, Kaufmann P (2008) Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann NY Acad Sci 1142: 133–158 [DOI] [PubMed] [Google Scholar]
- Starkov AA (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann NY Acad Sci 1147: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP (2012) Recruiting adaptive cellular stress responses for successful brain ageing. Nat Rev Neurosci 13: 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogolova V, Furness A, Robb-McGrath M, Garlich J, Stuart RA (2012) Rcf1 and Rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol Cell Biol 32: 1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthammarak W, Yang YY, Morgan PG, Sedensky MM (2009) Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J Biol Chem 284: 6425–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH (2012) Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal 16: 1434–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CT, Moncada S (2009) Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol 30: 643–647 [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL (2000) Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279: L1005–L1028 [DOI] [PubMed] [Google Scholar]
- Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC (2011) NOX isoforms and reactive oxygen species in vascular health. Mol Interv 11: 27–35 [DOI] [PubMed] [Google Scholar]
- Treberg JR, Quinlan CL, Brand MD (2011) Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 286: 27103–27010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V (2005) Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci 360: 2335–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Kirby DM, Smulders-Srinivasan TK, Nooteboom M, Acin-Periz R, Enriquez JA, Whittington MA, Lightowlers RN, Turnbull DM (2010) Mitochondrial DNA mutations affect calcium handling in differentiated neurons. Brain 133: 787–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsecchi F, Monge C, Forkink M, de Groof AJC, Benard G, Rossignol R, Swarts HG, van Emst-de Vries SE, Rodenburg RJ, Calvaruso MA, Nijtmans LGJ, Heeman B, Roestenberg P, Wieringa B, Smeitink JAM, Koopman WJH, Willems PHGM (2012) Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim Biophys Acta 1817: 1925–1936 [DOI] [PubMed] [Google Scholar]
- Vempati UD, Han X, Moraes CT (2009) Lack of cytochrome c in mouse fibroblasts disrupts assembly/stability of respiratory complexes I and IV. J Biol Chem 284: 4383–4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkaart S, Koopman WJ, Cheek J, van Emst-de Vries SE, van den Heuvel LW, Smeitink JA, Willems PH (2007b) Mitochondrial and cytosolic thiol redox state are not detectably altered in isolated human NADH:ubiquinone oxidoreductase deficiency. Biochim Biophys Acta 1772: 1041–1051 [DOI] [PubMed] [Google Scholar]
- Verkaart S, Koopman WJ, van Emst-de Vries SE, Nijtmans LG, van den Heuvel LW, Smeitink JA, Willems PH (2007a) Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim Biophys Acta 1772: 373–381 [DOI] [PubMed] [Google Scholar]
- Vogel RO, Dieteren CE, van den Heuvel LP, Willems PH, Smeitink JA, Koopman WJ, Nijtmans LG (2007) Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J Biol Chem 282: 7582–7590 [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6: 626–640 [DOI] [PubMed] [Google Scholar]
- Von Bernhardi R, Eugenín J (2012) Alzheimer’s disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid Redox Signal 16: 974–1031 [DOI] [PubMed] [Google Scholar]
- Vukotic M, Oeljeklaus S, Wiese S, Vögtle FN, Meisinger C, Meyer HE, Zieseniss A, Katschinski DM, Jans DC, Jakobs S, Warscheid B, Rehling P, Deckers M (2012) Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab 15: 336–347 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ II, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Wallace KB (2008) Mitochondrial off targets of drug therapy. Trends Pharmacol Sci 29: 361–366 [DOI] [PubMed] [Google Scholar]
- Wang C, Youle RJ (2009) The role of mitochondria in apoptosis. Annu Rev Genet 43: 95–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SB, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE (2011) Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res 109: 750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Okuno D, Sakakihara S, Shimabukuro K, Iino R, Yoshida M, Noji H (2011) Mechanical modulation of catalytic power on F1-ATPase. Nat Chem Biol 8: 86–92 [DOI] [PubMed] [Google Scholar]
- Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA 107: 16823–168217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B (2012) Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta 1817: 1833–1838 [DOI] [PubMed] [Google Scholar]
- Willems P, Wanschers BF, Esseling J, Szklarczyk R, Kudla U, Duarte I, Forkink M, Nooteboom M, Swarts H, Gloerich J, Nijtmans L, Koopman W, Huynen MA (2012) BOLA1 is an aerobic protein that prevents mitochondrial morphology changes induced by glutathione depletion. Antioxid Redox Signal (advance online publication, 11 September 2012; doi:; DOI: 10.1089/ars.2011.4253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems PH, Smeitink JA, Koopman WJ (2009) Mitochondrial dynamics in human NADH:ubiquinone oxidoreductase deficiency. Int J Biochem Cell Biol 41: 1773–1782 [DOI] [PubMed] [Google Scholar]
- Willems PH, Valsecchi F, Distelmaier F, Verkaart S, Visch HJ, Smeitink JA, Koopman WJ (2008) Mitochondrial Ca2+ homeostasis in human NADH:ubiquinone oxidoreductase deficiency. Cell Calcium 44: 123–133 [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Slupe AM, Strack S (2012) Cell signaling and mitochondrial dynamics: implications for neuronal function and neurodegenerative disease. Neurobiol Dis (advance online publication, 24 January 2012; doi:; DOI: 10.1016/j.nbd.2012.01.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winge DR (2012) Sealing the mitochondrial respirasome. Mol Cell Biol 32: 2647–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz S, Schuelke M (2011) Region-specific expression of mitochondrial complex I genes during murine brain development. PLoS ONE 6: e18897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Schägger H (2009) Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta 1787: 672–680 [DOI] [PubMed] [Google Scholar]
- Xu XM, Møller SG (2011) Iron-sulfur clusters: biogenesis, molecular mechanisms, and their functional significance. Antioxid Redox Signal 15: 271–307 [DOI] [PubMed] [Google Scholar]
- Yadava N, Potluri P, Scheffler IE (2008) Investigations of the potential effects of phosphorylation of the MWFE and ESSS subunits on complex I activity and assembly. Int J Biochem Cell Biol 40: 447–460 [DOI] [PubMed] [Google Scholar]
- Yanicostas C, Soussi-Yanicostas N, El-Khoury R, Bénit P, Rustin P (2011) Developmental aspects of respiratory chain from fetus to infancy. Semin Fetal Neonatal Med 16: 175–180 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Li M, Wang X, Jin H, Liu S, Xu J, Chen Q (2012) Caspase cleavage of cytochrome c1 disrupts mitochondrial function and enhances cytochrome c release. Cell Res 22: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T (2005) Spectral imaging and linear unmixing in light microscopy. Adv Biochem Eng Biotechnol 95: 245–265 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.